
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 13 October 2021
Sec. Stem Cell Research
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.709923
This article is part of the Research Topic Hematopoiesis: Learning from in vitro and in vivo Models View all 10 articles
 Shuang Wu1†
Shuang Wu1† Kai Chen1†
Kai Chen1† Tao Xu1,2†
Tao Xu1,2† Ke Ma1
Ke Ma1 Lei Gao1Cong Fu1Wenjuan Zhang1
Lei Gao1Cong Fu1Wenjuan Zhang1 Changbin Jing1
Changbin Jing1 Chunguang Ren1Min Deng1Yi Chen3Yi Zhou4
Chunguang Ren1Min Deng1Yi Chen3Yi Zhou4 Weijun Pan1*
Weijun Pan1* Xiaoe Jia1,5*
Xiaoe Jia1,5*Vertebrate erythropoiesis involves nuclear and chromatin condensation at the early stages of terminal differentiation, which is a unique process to distinguish mature erythrocytes from erythroblasts. However, the underlying mechanisms of chromatin condensation during erythrocyte maturation remain elusive. Here, we reported a novel zebrafish mutantcas7 with erythroid maturation deficiency. Positional cloning showed that a single base mutation in tprb gene, which encodes nucleoporin translocated promoter region (Tpr), is responsible for the disrupted erythroid maturation and upregulation of erythroid genes, including ae1-globin and be1-globin. Further investigation revealed that deficient erythropoiesis in tprbcas7 mutant was independent on HIF signaling pathway. The proportion of euchromatin was significantly increased, whereas the percentage of heterochromatin was markedly decreased in tprbcas7 mutant. In addition, TPR knockdown in human K562 cells also disrupted erythroid differentiation and dramatically elevated the expression of globin genes, which suggests that the functions of TPR in erythropoiesis are highly conserved in vertebrates. Taken together, this study revealed that Tpr played vital roles in chromatin condensation and gene regulation during erythroid maturation in vertebrates.
The zebrafish (Danio rerio) is a well-established model organism to study hematopoietic development due to its unique advantages (Jong and Zon, 2005; Paik and Zon, 2010). Similar to mammalian hematopoiesis, zebrafish hematopoiesis occurs in two distinct waves. The first primitive wave of hematopoiesis occurs in two different regions: the posterior lateral mesoderm that forms the intermediate cell mass later and the anterior lateral mesoderm, which generate primitive erythroid cells and myeloid cells, respectively (Chen and Zon, 2009; Paik and Zon, 2010). With the start of the heartbeat at 24 h post-fertilization (hpf), these primitive blood cells enter circulation throughout the embryo. The second definitive wave of hematopoiesis is characterized by the formation of self-renewing hematopoietic stem cells (HSCs) in the aorta–gonad–mesonephros region (Bertrand et al., 2010; Kissa and Herbomel, 2010). Then the nascent HSCs migrate to the caudal hematopoietic tissue and colonize in kidney marrow finally (Murayama et al., 2006; Li et al., 2018).
Almost in all vertebrates, erythropoiesis is a stepwise process that involves differentiation of HSCs to committed burst forming unit-erythroid (BFU-E) followed by colony forming unit-erythroid (CFU-E) (Dzierzak and Philipsen, 2013; Palis, 2014). In terminal erythropoiesis, CFU-Es differentiate to mature erythrocytes and can be divided into four morphologically distinguishable cell types: proerythroblasts, basophilic erythroblasts, polychromatophilic erythroblasts, and orthochromatophilic erythroblasts (Schwartz, 2016). Also, many progressive characteristics are involved in terminal erythropoiesis including several cell divisions, decrease in cell size, nuclear and chromatin condensation, and extrusion of mitochondria and even the nucleus (Palis, 2014).
Although nuclear chromatin condensation is a common feature to define morphologically distinctive erythroblasts at different developmental stages, enucleation is unique to mammals, and nuclei are still retained in mature circulating erythrocytes in fish and avian (Ji et al., 2011). Chromatin condensation is also required for terminal erythropoiesis, as defects in this process are associated with anemia and myelodysplastic syndrome (Wickramasinghe and Wood, 2005; Menon and Ghaffari, 2021). Previous studies have identified several architectural factors including linker histone H5 and nuclear serpin MENT that promote avian erythroid chromatin condensation (Verreault and Thomas, 1993; Istomina et al., 2003). At the same time, histone deacetylation and caspase-3 are necessary for mammalian chromatin condensation (Zermati et al., 2001; Popova et al., 2009; Zhao et al., 2016). However, the detailed mechanisms of chromatin condensation are unclear.
Nucleoporin Tpr (translocated promoter region) is an architectural component of nuclear pore complex (NPC), which locates on the nucleoplasmic side of the pore to form a nuclear basket structure (Cordes et al., 1997; Krull et al., 2004). It exists as a homodimer via the N-terminal coiled-coil domain to be one of the eight basket filaments, whereas the C-terminal domain appears unfolded and flexible into the nucleus (Hase et al., 2001). Tpr has been implicated in a variety of nuclear functions, including chromatin organization and transcriptional regulation, nuclear transport, and mitosis. Megator (Drosophila Tpr) and its binding partner Nup153 have been shown to bind to 25% of the genome in continuous domains that are transcriptionally active (Vaquerizas et al., 2010). Also, Tpr was shown to be required for heterochromatin exclusion zones (HEZs) at nuclear pores after poliovirus infection (Krull et al., 2010) and maintain an open chromatin state favorable for HIV replication (Lelek et al., 2015). Recently, Tpr has been reported to be necessary for formation and maintenance of senescence-associated heterochromatin foci (SAHFs) during oncogenic Ras-induced senescence (Boumendil et al., 2019). However, Tpr-mediated chromatin organization during developmental processes in vivo remains uncertain.
In this study, we reported an essential role of Tpr in terminal erythropoiesis by characterizing a zebrafish mutantcas7 with a missense mutation in tprb gene. The homozygous tprb mutant manifested erythroid cell maturation blockage and abnormal erythroid gene expression. Tpr deficiency did not induce the activation of HIF signaling pathway, and hif-2a knockdown could not rescue abnormal erythroid phenotypes in tprbcas7 mutant. The elevated erythroid gene expression including ae1-globin and be1-globin in tprbcas7 mutants was due to their enhanced transcriptional activity, rather than the increased number of erythrocytes. Further investigation revealed that nuclear chromatin condensation and organization were strikingly disrupted specifically in erythrocytes in tprbcas7 mutant, but not in other tissue cells. In addition, in human K562 cells, TPR knockdown also caused blocking of erythroid differentiation and a dramatic increase in α globin and β globin gene expression, indicating that the function of TPR in erythropoiesis is highly conserved in mammalian. Our findings not only revealed the critical role of Tpr in chromatin condensation and gene regulation but also highlighted the importance of nuclear pore proteins in erythroid maturation.
In a large-scale ENU mutagenesis screen for erythroid development mutations, we obtained cas7, a novel mutant with erythrocyte development defects and recessive lethality. The mutantcas7 embryos were morphologically indistinguishable from wild-type siblings before 3 dpf, with normal blood flow and heart beats, and survived to about 6 dpf (Figures 1A,B). Whole-mount in situ hybridization (WISH) revealed that the expressions of erythroid markers (ae1-globin, alas2, and band3) were all increased abnormally (Figures 1C–H). Consistent with WISH analysis, real-time qPCR also showed the same result (Figure 1L). During the time course of αe1-globin WISH analysis, we did not detect any difference between sibling and mutantcas7 before 3 dpf (Supplementary Figures 1A–D). The marginal decrease of αe1-globin expression was detectable until 3 dpf (Supplementary Figures 1E,F), while the mutantcas7 embryos were morphologically distinguishable from wild-type siblings after 4 dpf.
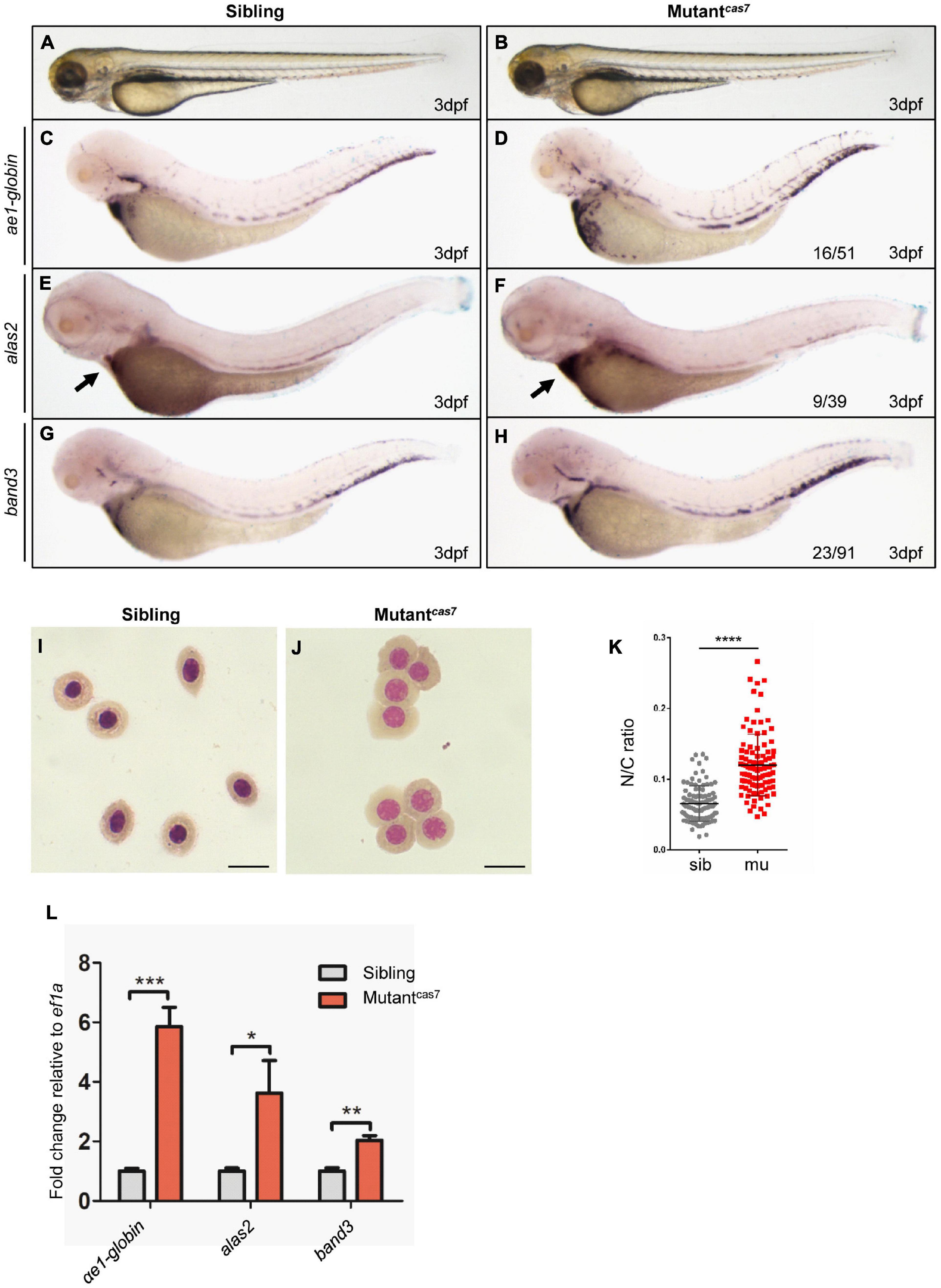
Figure 1. Erythroid maturation is disrupted in zebrafish mutantcas7. (A,B) Light microscope images of zebrafish wild-type (WT) sibling and mutantcas7 embryos at 3 dpf. (C–H) WISH analysis of ae1-globin (C,D), alas2 (E,F), and band3 (G,H) expression in sibling and mutantcas7 embryos at 3 dpf. After WISH and photographing, all embryos were extracted for genomic DNA and genotyped by sequencing, then the mutant percentage was evaluated. The number of embryos in a het-het incross clutch with the expression pattern is shown in the mutantcas7 column and corresponding percentages are listed inside each panel. Arrow in panel (E,F) indicates the alas2 staining in heart region. (I,J) Giemsa staining for erythrocytes at 4 dpf in sibling and mutantcas7. Relative to the number of orthochromatophilic erythroblasts, mutantcas7 displayed a marked increase in circulating polychromatophilic erythroblasts compared with sibling. Scale bars represent 10 μm. (K) Statistical analysis of the nucleus-to-cytoplasm (N/C) ratio in sibling and mutantcas7. N = 100, error bars represent SEM. ****p ≤ 0.0001. (L) The relative expression of αe1-globin, alas2, band3 in sibling and mutantcas7 embryos at 3 dpf. Error bars represent SEM. *p ≤ 0.05; **p ≤ 0.01; *** p ≤ 0.001.
Erythroid maturation is a stepwise process typically including dramatically decrease in cell size, and the N/C (nucleus-to-cytoplasm) ratio is a robust parameter for terminal differentiation, which had been reported previously (Fraser et al., 2007). Therefore, we can use the N/C ratio as a quantitative marker for erythroid maturation. Comparison of circulating erythrocytes from sibling and mutantcas7 embryos showed impaired maturation in the latter, for the latter contains more polychromatophilic erythroblasts while the former was abundant in orthochromatophilic erythroblasts at 4 dpf (Figures 1I,J; Qian et al., 2007). Also, the N/C ratio was higher in mutantcas7, confirming the impaired erythrocyte maturation (Figure 1K).
To examine other hematopoietic lineages, we firstly performed WISH analysis of definitive hematopoietic cells markers including cmyb (hematopoietic stem and progenitor cell marker), mpx (granulocyte marker), lyz (macrophage marker), and rag-1 (lymphocyte marker). The expression of these markers was identical between sibling and mutantcas7 at 3 or 4 dpf, which indicated that the definitive hematopoiesis was normal (Supplementary Figures 2A–H). Consistent with WISH results, the real-time qPCR suggested that the expressions of these definitive hematopoietic cell markers (cmyb, mpo, lyz, and rag-1) were unchanged (Supplementary Figure 2I). However, the expression levels of erythroid genes were not all increased, such as sdhb, fth1a (iron-related protein gene), and hebp2 (heme binding protein gene) (Supplementary Figure 2I). Then, we investigated the primitive hematopoiesis and vascular morphogenesis in mutantcas7. WISH analysis showed that the expression of scl (hematopoietic progenitor marker), gata1 (erythrocyte progenitor marker), pu.1 (myeloid progenitor marker), mpx, and kdrl (endothelial cell marker) were also normal in mutantcas7 at 22 or 26 hpf (Supplementary Figure 3). Taken together, these results suggested that erythroid maturation was specifically disrupted in zebrafish mutantcas7.
To elucidate the mechanism of erythroid maturation failure in mutantcas7, we carried out positional cloning. The mutantcas7 was first mapped on chromosome 20 by bulk segregation analysis (BSA). A high-resolution sequence length polymorphism (SSLP)–based mapping approach established the mutation within a 225-kb region between two markers 294-04 and 297-01 (Figure 2A). After sequencing all three candidate genes in this region, we found a C to G missense mutation in tprb gene in mutantcas7 (Figure 2B). Also, genomic sequencing of tprb gene also confirmed this result. This mutation yielded the leucine to valine at ninth amino acid (L9V) of Tpr protein and the leucine was highly conserved from human to fruit fly (Figure 2C). Among five commonly used laboratory zebrafish strains (Tu, WIK, AB, Shanghai, and Longfin), this point mutation was not found (Supplementary Figure 4), excluding the possibility of single-nucleotide polymorphism (SNP). The tprb locus on zebrafish chromosome 20 is syntenic to a region of human chromosome 1 that contains the TPR gene, based on the conserved location of neighboring orthologous gene pairs (PRG4, PDC, PTGS2, PLA2G4A, and HMCN1), suggesting that zebrafish tprb gene is an ortholog of human TPR (Figure 2D).
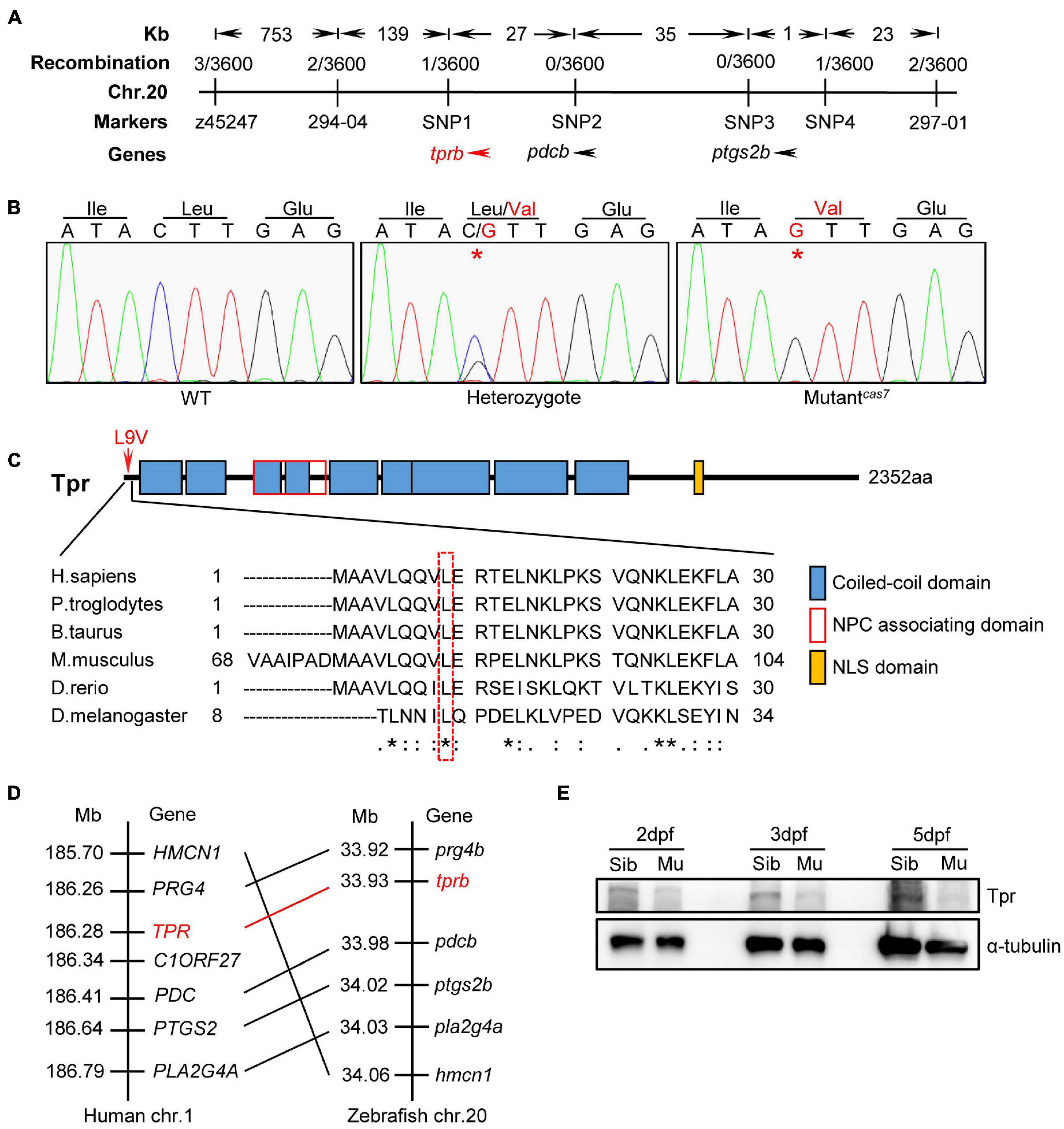
Figure 2. Positional cloning of mutantcas7. (A) Genetic mapping of the cas7 region on chromosome 20. Bulk segregation analysis positioned cas7 mutation to Chr. 20. Fine mapping using SSLPs narrowed down the region between markers SNP1 and SNP4, including tprb and two other genes as indicated. (B) There is a C-to-G missense mutation in tprb gene in mutantcas7, which leads to leucine to valine (L9V) at ninth amino acid in Tpr protein. (C) The ninth amino acid leucine of Tpr is highly conserved from human to fruit fly (* marked). (D) Comparison of the gene syntenic relationship between zebrafish tprb and human TPR loci. Seven genes, including TPR, are located within a genomic region on human chromosome 1. (Right) Seven zebrafish homologs of these genes are listed according to their map positions on chromosome 20 (Ensembl website). Mb, mega base. (E) Western blotting images of Tpr protein in whole embryo lysates of sibling and mutantcas7 embryos at the indicated time. All embryos were extracted for genomic DNA and genotyped by sequencing.
To explore the consequence of the point mutation to tprb gene, we customized an anti-Tpr antibody to examine the endogenous Tpr protein level. Western blot results showed that the expression of Tpr protein was markedly decreased over time in mutantcas7, and became undetectable at 5 dpf, suggesting that this point mutation may disrupt the function of tprb gene (Figure 2E).
To confirm the point mutation in tprb gene is responsible for mutantcas7 phenotypes, we injected a validated tprb ATG morpholino (MO) into one-cell-stage wild-type embryos to block the translation of endogenous tprb mRNA. Also, immunoblotting analysis confirmed the Tpr protein was poorly detectable in tprb morphants (Supplementary Figure 5A). WISH results showed that erythroid markers including ae1-globin, alas2, and band3 were all upregulated (Figures 3A–F), while the other hematopoietic lineages were barely unchanged in tprb morphants (Supplementary Figures 5B–I). Consistent with WISH results, the real-time qPCR suggested the expression levels of ae1-globin, alas2, and band3 were all enhanced, while other definitive hematopoietic cell markers (cmyb, mpo, lyz, rag, and kdrl) were unchanged (Supplementary Figure 5J).
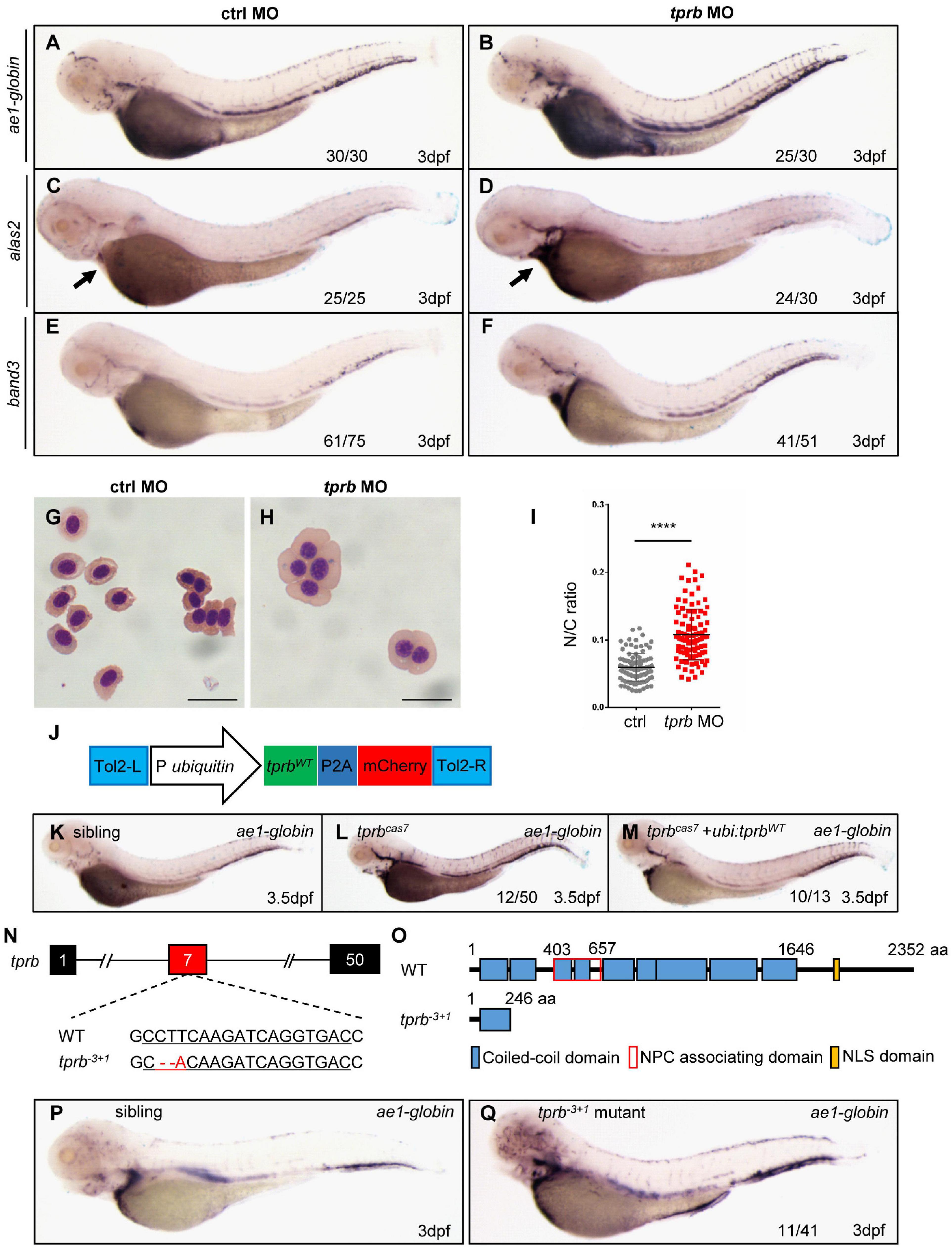
Figure 3. Mutation in tprb gene is responsible for mutantcas7 phenotypes. (A–I) Morpholino knocking down of tprb mutantcas7 phenocopies. WISH results of ae1-globin (A,B), alas2 (C,D), and band3 (E,F) expression in control and tprb morphants at 3 dpf. The percentages of indicated phenotype are listed at the bottom of each panel. Arrow in panel (E,F) indicates the alas2 staining in heart region. (G,H) Giemsa staining for erythrocytes at 4 dpf in control and tprb morphants. More immature erythrocytes in tprb morphants are shown. Scale bars represent 10 μm. (I) Statistical analysis of the nucleus-to-cytoplasm (N/C) ratio in control and tprb morphants. N = 100, error bars represent SEM. ****p < 0.0001. (J) Construction of the plasmid used in Tol2-transposease-mediated rescue assay. (K–M) WISH analysis of ae1-globin expression in sibling, tprbcas7 mutants, and rescued embryos at 3.5 dpf. After WISH and photographing, all embryos were extracted for genomic DNA and genotyped by sequencing, then the rescue percentage was evaluated. The percentage of fully rescued mutant embryos is about 77% (10 out of 13 mutant embryos), while the rest are rescued partially. (N,O) Generation of tprb– 3+1 mutant via CRISPR-Cas9 technique. The alignment of WT and mutated sequences is listed. The underlined sequence is gRNA target site. The sequencing result of tprb genomic DNA shows a 3-bp deletion and 1-bp addition at exon 7 (N), which caused a premature stop codon leading to the production of a truncated 246-amino-acid Tpr protein (O). (P,Q) WISH results of ae1-globin expression in sibling and tprb– 3+1 mutant at 3 dpf. After WISH and photographing, all embryos were extracted for genomic DNA and genotyped by sequencing, then the mutant percentage was evaluated. The number of embryos in tprb– 3+1 mutant incross clutch with the expression pattern as shown in the tprb– 3+1 column and corresponding percentages are listed inside each panel.
To check whether and when the erythroid maturation was impaired, we have performed a different time course of Giemsa staining in circulating erythroid cells in control and tprb morphants. The result showed that before 2 dpf, the N/C ratio was not significantly changed between control and tprb morphants. Beginning at 3 dpf, the N/C ratio of tprb morphants was obviously increased, indicating that erythroid cell differentiation was inhibited from 3 dpf (Supplementary Figure 6B). The bigger nuclear area in erythroid cells and immature erythrocytes in tprb morphants could be observed at 3 and 4 dpf (Supplementary Figures 6A,C–J). Together with these data, Giemsa staining showed more immature erythrocytes in tprb morphants by morphological assessment and nucleo-cytoplasmic ratio computation (Figures 3G–I), indicating impaired erythroblast maturation in tprb morphants.
To further demonstrate our findings, rescue assay was performed by ectopic expression of wild-type tprb under the ubiquitin promoter in mutantcas7 (Figure 3J). The result showed that when Tpr protein was rescued in tprb mutants, the upregulated erythroid markers could be also largely rescued by wild-type tprb overexpression (Figures 3K–M and Supplementary Figure 7).
Moreover, we generated a second zebrafish tprb–3+1 mutant by CRISPR-Cas9, which carried a frameshift mutation and premature stop codon in exon 7 and led to a truncated Tpr protein (Figures 3N,O). Consistent with phenotype of mutantcas7, the embryonic development of tprb–3+1 mutant was normal before 3 dpf but died at 5–6 dpf, and WISH analysis of ae1-globin level was also significantly increased (Figures 3P,Q). Thus, results from MO phenocopy assay, wild-type tprb rescue assay, and same phenotype of tprb–3+1 mutant strongly suggested that the C-to-G mutation led to tprb gene loss of function, which was causative for phenotype of mutantcas7. Hence, we rename the mutant as tprbcas7.
Given that Tpr is a structural protein that exists in every cell theoretically, we examined the temporal and spatial expression of tprb by WISH analysis. The result showed that tprb transcript was expressed as maternal mRNA and ubiquitously expressed during zebrafish embryogenesis (Supplementary Figure 8). To answer whether the localization of Tpr is in erythroid cells, we performed the Tpr immunofluorescence in tprb mutants and morphant erythroid cells at 3 dpf (Supplementary Figure 9). In erythroid cells from sibling and control morphant, Tpr protein is indeed located on the nuclear membrane (Supplementary Figures 9A,B,E,F). While in tprb mutants and morphants there was no fluorescence signal of Tpr protein in erythroid cells (Supplementary Figures 9C,D,G,H), indicating the endogenous Tpr protein was missing in tprb mutant or morphant, consisting with the WB result in Figure 2E.
It has been reported that HIF signaling pathway plays an important role in erythropoiesis, especially in erythrocytosis (Lee and Percy, 2011). Von Hippel-Lindau tumor suppressor (VHL) was the negative regulator of HIFs. The zebrafish vhl mutant displayed a hypoxia response, severe polycythemia, and immature erythropoiesis (van Rooijen et al., 2009). Also, results revealed global upregulation of both red and white hematopoietic lineages. Interestingly, we found that the phenotype of upregulation of erythroid genes and immature erythrocytes in tprbcas7 mutant was similar to reported vhl zebrafish mutant. Therefore, to reveal the molecular mechanism of erythroid maturation defect in tprbcas7 mutant, we explored whether the HIF signaling pathway was activated. First, flow cytometry analysis showed that the number of erythrocytes was not significantly changed in tprb morphants compared with control (Figure 4A and Supplementary Figure 10). Also, mRNA levels of epo, a HIF target gene involved in erythropoiesis (Bunn, 2013), and vegfaa were not elevated in tprb morphants (Figure 4B).
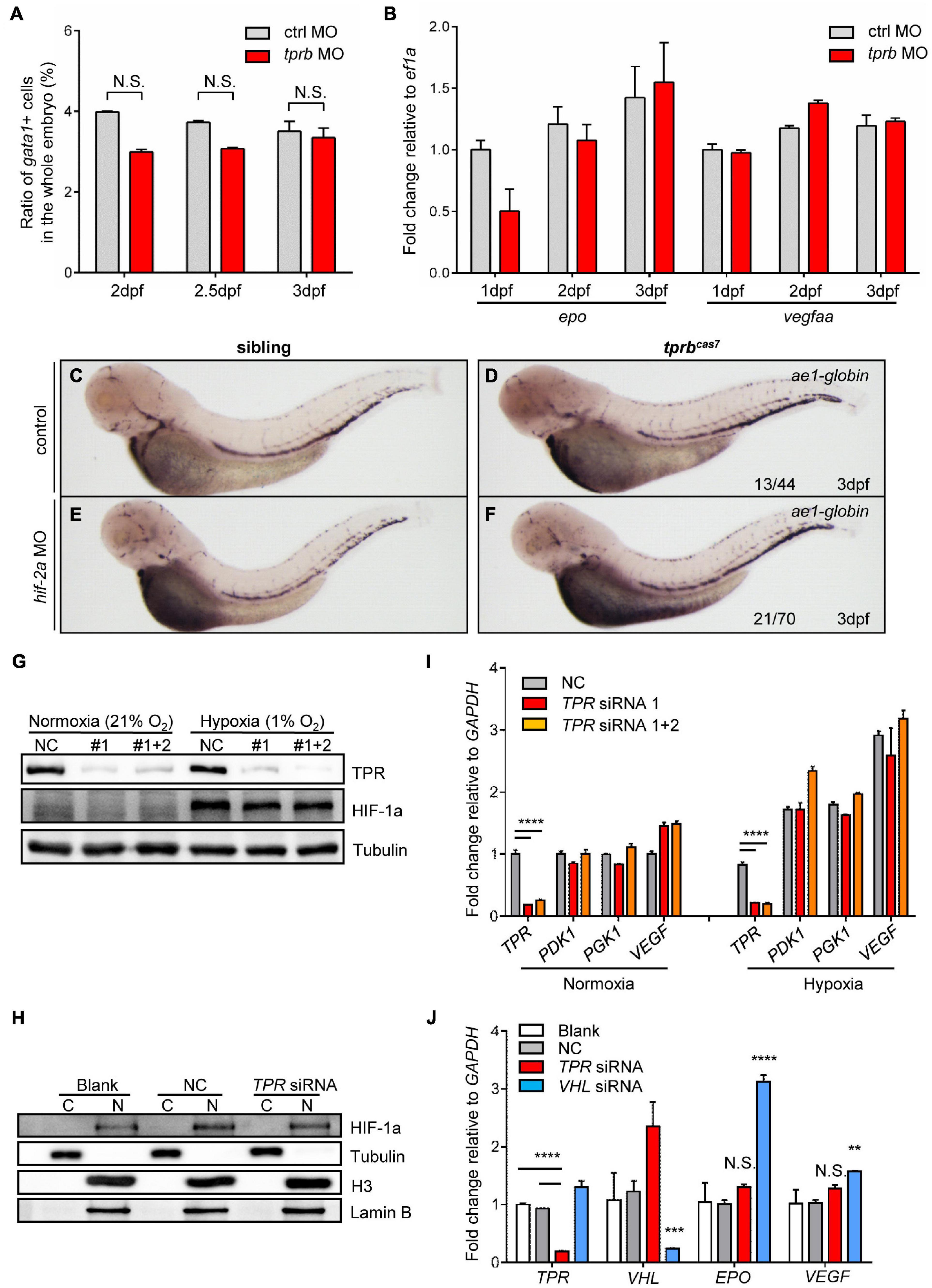
Figure 4. HIF signaling is not activated in tprbcas7 mutant. (A) The statistic results of the ratio of gata1+ cells sorted by flow cytometer in the whole embryo of control and tprb morphants by Tg(gata1: DsRed; kdrl: EGFP) transgenic line at different time points. (B) Quantitative PCR results of epo and vegfaa in control and tprb morphants at different time points. (C–F) WISH analysis of ae1-globin expression in sibling and tprbcas7 mutants at 3 dpf under the control and hif-2a morpholino (E,F) injection. (G) Immunoblotting analysis of TPR and HIF-1a protein level in whole HEK293T cells under normoxia and hypoxia conditions during TPR knockdown through siRNA. (H) Immunoblotting analysis of HIF-1a protein level in the nuclei of HEK293T cells under hypoxia condition after nuclear and cytoplasmic separation. C, cytoplasma; N, nucleus. (I) Quantitative PCR results of HIF target genes including TPR, PDK1, PGK1, and VEGF in HEK293T cells during TPR knockdown through siRNA. (J) Quantitative PCR results of HIF target genes including EPO and VEGF in Hep3B cells during TPR or VHL knockdown through siRNA under hypoxia condition. NC: negative control. #1: TPR siRNA 1; #1 + 2: TPR siRNA 1 + 2. Error bars represent SEM. N.S.: not significant; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
Furthermore, to compare the difference of phenotype in tprbcas7 mutant with embryos activated by HIF signaling, we established a vhl mutant by CRISPR-Cas9 as our positive control (Supplementary Figures 11A,B). To our surprise, WISH result of ae1-globin level was much more increased in vhl mutant than tprbcas7 mutant (Supplementary Figures 11C,D). Given that HIF-2a is mainly involved in the regulation of polycythemia (Lee and Percy, 2011; Metelo et al., 2015), we considered rescue experiment to alleviate the degree of polycythemia by using zebrafish hif-2a MO. WISH results showed that hif-2a MO could partially rescue the increased expression of ae1-globin in vhl mutant, which indicated the effectiveness of the hif-2a MO (Supplementary Figures 11E,F), but not in tprbcas7 mutant (Figures 4C–F). These results suggested that loss of tprb gene most likely did not cause the activation of HIF signal pathway.
Previous studies have demonstrated that HIF-1a protein is stabilized and enters the nucleus under hypoxic conditions, which is essential for hypoxic response (Koh and Powis, 2012). To further directly confirm whether loss of Tpr protein could result in the activation of HIF signaling, we analyzed the accumulation of HIF-1a protein and the expression of HIF target genes in human cultured cells under hypoxia condition. Western blot analysis in HEK293T cells showed that neither the amount of HIF-1a protein from whole cells nor nucleus was barely changed as TPR knock-down (Figures 4G,H). At the same time, the expression of several important HIF target genes including PDK1, PGK1, and VEGF also showed no significant change with or without TPR protein (Figure 4I). Similarly, we tested in human hepatocyte Hep3B cells and designed VHL siRNA as the positive control. Under hypoxic conditions, knocking down VHL could upregulate the expression of EPO and VEGF dramatically, whereas knocking down TPR had no significant change (Figure 4J). Collectively, the defective erythroid phenotype of tprbcas7 mutant was indeed independent on the activation of HIF signaling.
As described previously, the number of erythrocytes in tprbcas7 mutant did not alter, leading us to investigate whether the increased staining of erythroid markers by WISH analysis in tprbcas7 mutant was caused by the upregulation of genes in every erythrocyte. To test this hypothesis, we injected the control and tprb MO into one-cell-stage embryos of Tg(gata1: DsRed) transgenic line separately. Then we sorted the same number of erythrocytes through Tg(gata1: DsRed) transgenic line and carried out real-time qPCR to analyze the expression levels of erythroid genes. As expected, the ae1-globin and be1-globin expression levels were dramatically increased in tprb morphants, especially for ae1-globin (Figure 5A).
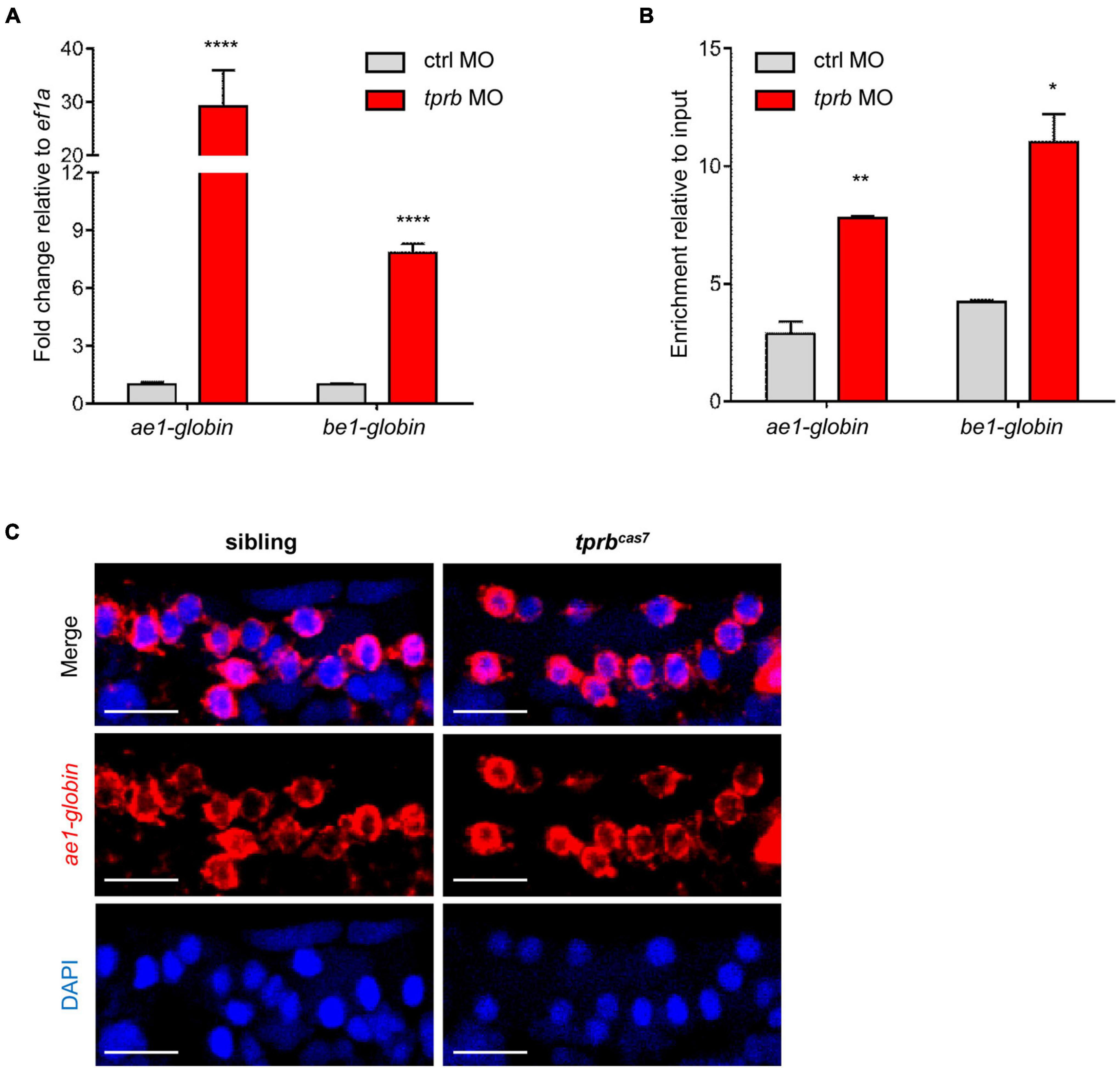
Figure 5. tprb inactivation enhances the transcriptional activity of erythroid genes. (A) Quantitative PCR results of ae1-globin, be1-globin, and alas2 in erythrocytes after sorting an equal number of gata1+ cells in control and tprb morphants at 3 dpf. (B) Quantitative qChIP results of the binding enrichment of RNA polymerase II (Pol II) in ae1-globin, be1-globin, and alas2 after sorting equal number of gata1+ cells. (C) Fluorescence in situ hybridization (FISH) results of ae1-globin mRNA localization in sibling and tprbcas7 mutants at 3 dpf. The nuclei were stained with DAPI (blue). Error bars represent SEM. N.S., not significant; *p < 0.05; **p < 0.01; ****p < 0.0001. Scale bars represent 20 μm.
To determine how Tpr deficiency triggered erythroid genes expression abnormally, we checked the transcriptional activity of these genes by quantitative chromatin immunoprecipitation (qChIP) experiment through RNA polymerase II (Pol II) antibody. ChIP results showed that the relative abundance of Pol II in the promoter region of ae1-globin and be1-globin in tprb morphants was dramatically higher than that of in control morphants (Figure 5B).
In yeast, Mlp1 and Mlp2 proteins (ortholog of mammalian Tpr protein) contribute to RNA surveillance by retention of unspliced mRNA in nucleus (Green et al., 2003; Galy et al., 2004) and downregulate gene expression in response to mRNA export defect (Vinciguerra et al., 2005). To analyze whether the mRNA export is blocked in erythrocytes in tprbcas7 mutant, we tested the cellular localization of ae1-globin mRNA. Fluorescent in situ hybridization result showed that the ae1-globin mRNA was exported out of the nucleus normally and distributed throughout the cytoplasm in both sibling and tprbcas7 mutant, indicating the mRNA export was normal (Figure 5C). These results taken together suggested that loss of Tpr can upregulate erythroid genes expression by enhancing their transcriptional activity, rather than the number of erythrocytes.
Chromatin condensation is essential for terminal erythropoiesis and erythroid maturation. To further explore how Tpr regulate the transcriptional activity of erythroid genes, we performed transmission electron microscopy to analyze the ultrastructural changes in the nucleus. It was found that at 4 dpf, there were distinct heterochromatin regions in most of the nuclei of erythrocytes in siblings, and the separation between euchromatin and heterochromatin was obvious (Figures 6A–C). Also, in tprbcas7 mutant, the boundary between them was hard to define precisely, presenting a “homogeneous” chromatin state (Figures 6D–F). Relying on the chromatin aggregation degree within the nuclei of erythrocytes, statistical analysis result showed that the proportion of euchromatin was significantly increased, whereas the heterochromatin percentage markedly decreased in tprbcas7 mutant (Figure 6G). Moreover, we also found the disruption of HEZs near NPCs in tprbcas7 mutant as previously reported (Krull et al., 2010). The aforementioned results indicated that the chromatin condensation in erythroid cells was impaired in tprbcas7 mutant.
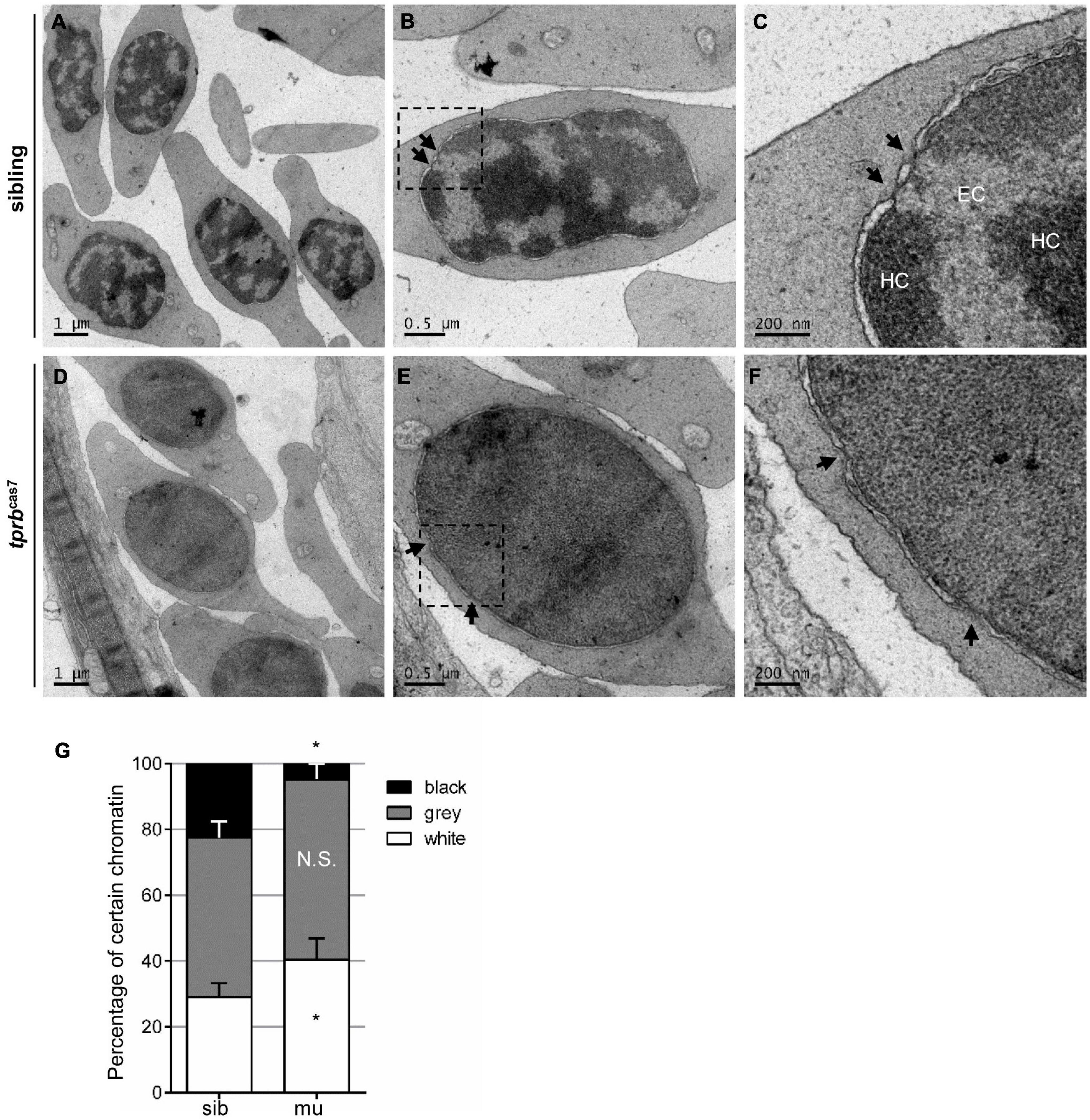
Figure 6. Chromatin condensation is defective in tprbcas7 mutant erythroid cells. (A–F) Representative transmission electron microscopy (TEM) images of erythrocytes from sibling (A–C) and tprbcas7 mutants (D–F) at 4 dpf. The images in panels (C,F) showed a higher magnification of the corresponding boxes (B,E). Arrows indicate nuclear pores. EC, euchromatin; HC, heterochromatin. Scale bars are shown in each image. (G) Quantifications of certain chromatin with classified different color in sibling and tprbcas7 mutants at 4 dpf. Error bars represent SEM. N.S., not significant; *p < 0.05.
Given Tpr is a structural protein, we analyzed chromatin structures in other tissue cells to test whether chromatin organization abnormality was a common phenomenon. The results showed that such heterochromatin blockage surprisingly only existed in erythrocytes and some hematopoietic cells, not in other tissue cells, such as neural cells, endothelial cells, and skeletal muscular cells (Supplementary Figure 12). Also, there was no significant difference in chromatin morphology in those tissue cells between sibling and tprbcas7 mutant (Supplementary Figure 12). Overall, these results suggested that Tpr deficiency might specifically disrupt the chromatin condensation and later on maturation in erythrocytes.
To test whether Tpr is also important for mammalian hematopoiesis, we knocked down TPR in human K562 cells (Supplementary Figure 13). The TPR shRNA efficiently knocked down TPR expression (Supplementary Figures 13A,B). The K562 line is composed of undifferentiated blast cells that are rich in glycophorin and may be induced to produce fetal and embryonic hemoglobin in the presence of hemin. The K562 cell membrane glycoproteins show many similarities with that of erythrocytes and, in particular, the cells synthesize glycophorin A (CD235a, also named GPA), which is found exclusively in human erythrocytes (Drexler et al., 2004). Therefore, the K562 cell appears to be an excellent tool for the study of human erythroid differentiation and globin gene expression.
We induced K562 erythroid differentiation by treated with hemin, to find TPR functions in erythroid differentiation. Expression of the glycophorin A (CD235a) on the surface of K562 cells was determined by direct immunofluorescence staining and flow cytometer. When treated with hemin for 48 h, the expression of CD235a on the surface showed a significant increase in scramble shRNA K562 cells; however, the expression of CD235a in TPR knock-down K562 cells was not obviously enhanced (Supplementary Figures 13C,D). We also checked globin expression when TPR is knocked down. The α globin and β globin upregulated in TPR knock-down cells, while γ globin and δ globin was not obviously changed (Supplementary Figure 13E). When treated with hemin, the expressions of α globin, β globin, and γ globin were all increased (Supplementary Figure 13E). These data indicated that TPR knock-down caused blocking of erythroid differentiation, although the globin expression increased largely in K562 cell line.
Here, we reported a novel zebrafish tprbcas7 mutant with defect in erythroid maturation. Genetic analysis revealed that the missense mutation in tprb gene leads to Tpr protein loss of function, which was causative for tprbcas7 mutant phenotypes. Further studies found that Tpr was essential for the organization and maintenance of chromatin condensation and erythroid gene regulation during terminal erythropoiesis, and the function of TPR in erythropoiesis is highly conserved in mammalian.
In this study, we confirmed that the point mutation in tprb gene would lead to Tpr protein loss of function by tprb knockdown, rescue, and knockout experiments. Zebrafish Tpr protein contains 2,352 amino acids. It is interesting to find how the single base mutation results in whole protein inactivation. By detecting the amount of Tpr protein at different stages, we found that the level of Tpr was gradually decreased in tprbcas7 mutant and almost disappeared from 3 dpf (Figure 2E). This result indicates that the missense mutation might affect the stability of Tpr proteins and accelerate their degradation, leading to the loss of function finally. In addition, the erythroid phenotype of tprbcas7 heterozygotes was checked, and no defect was observed. We propose that the mutation type of Tpr (L9V) has no dominant negative effect.
The increased staining of WISH generally shows the increased expression level of the genes. We found that tprb knockdown did not change the number of erythrocytes compared with control, although the expressions of erythroid markers (ae1-globin, alas2, and band3) were all increased by WISH staining. Thus, our results showed that loss of Tpr could upregulate the expression of erythroid genes by enhancing their transcriptional activity.
Also, we confirmed that the phenotype of tprbcas7 mutant was independent on HIF signaling pathway mainly through four reasons: (1) the number of erythrocytes was not increased; (2) the expression of HIF target genes was not upregulated; (3) hif-2a MO could partially rescue the WISH result of ae1-globin in vhl mutant, rather than in tprbcas7 mutant; (4) in human cells, there was no significant change in the accumulation of HIF-1a protein and expression of HIF target genes as TPR knockdown.
Although the expression of NPC as the structural protein has no tissue specificity, its tissue-specific function has been widely reported (Raices and D’Angelo, 2012). In this study, we found that the chromatin condensation and organization were specifically disrupted in erythrocytes in tprbcas7 mutant. On the one hand, this may be the process of nuclear condensation and globally chromatin aggregation specific to erythrocytes, and erythroid terminal differentiation highly depends on this. Therefore, the erythroid cells in tprbcas7 mutant are more sensitive to chromatin changes. On the other hand, studies have identified different molecules involved in chromatin reorganization during terminal differentiation in different types of cells. Such as in terminally differentiating myotubes, methyl CpG–binding protein MeCP2 and MBD2 are important for the aggregation of pericentric heterochromatin (Brero et al., 2005). Our finding that the chromatin condensation in muscular cells was not affected in tprbcas7 mutant is consistent with the previous research.
In different species, there are various molecular mechanisms involved in chromatin condensation during terminal erythropoiesis. In chicken erythrocytes, two architectural factors, linker histone H5 and nuclear serpin MENT, accumulate at repressed chromatin domains and promote chromatin condensation (Verreault and Thomas, 1993; Istomina et al., 2003). Also, in mammalian erythrocytes, histone deacetylation and caspase-3 may be the greatest players (Zermati et al., 2001; Popova et al., 2009; Ji et al., 2011; Zhao et al., 2016). In this study, we first showed that nucleoporin Tpr, which faces the nucleoplasm, was essential for chromatin condensation and erythroid maturation in zebrafish embryogenesis. Also, we found that the human TPR was important for K562 erythroid cell maturation. It is still intriguing to explore whether the histones and their modifications are involved in this process and how they can corporate with Tpr. One clue is that Tpr can specifically interact with histone H1.1 and H1.2 to regulate the stability of these replication-dependent linker histones (Zhang et al., 2016).
At present, there are some human diseases associated with TPR. In several different cancers, TPR is present through fusion genes, which is formed by coiled-coil motif in TPR with some kinase partner genes such as MET (gastric cancer) (Soman et al., 1991), NTRK1 (thyroid carcinoma) (Greco et al., 1992), FGFR1 (myeloproliferative syndromes) (Li et al., 2012), and ALK (lung adenocarcinoma) (Choi et al., 2014). In all fusion proteins, TPR N-terminal coiled-coil domain is maintained and fused to the partner kinase domain, resulting to allow dimerization that aberrantly activates kinases and drives the cancer progression. In addition, patients with Hutchinson–Gilford Progeria Syndrome (HGPS) premature aging had abnormal cytoplasm localization of TPR protein. HGPS is caused by a mutation in LMNA gene that generates a mutant lamin A protein, which is a major component of the nuclear lamina. Fibroblasts from HGPS patients have defects in the Ran GTPase system that cause a nuclear import defect in TPR and mislocalized in the cytoplasm, not the nuclear side of the nuclear pore complex (Snow et al., 2013). However, it is not clear how the abnormal localization of TPR affects the development of the HGPS.
Previous studies have reported that the chromatin structure can determine the expression of specific genes (Deng et al., 2012; Deng et al., 2014). During terminal erythropoiesis, nuclear and chromatin are gradually condensed and associated with wide downregulation of gene expression (Wong et al., 2011). In tprbcas7 mutant, chromatin condensation was destroyed, accompanied by a significant increase in euchromatin ratio. This was consistent with the increased total mRNA amount as tprb knockdown (data not shown). However, the relationship between chromatin organization and gene expression is still unclear in tprbcas7 mutant. It is necessary to investigate how the chromatin state and the transcriptome changed.
NPCs have emerged as a crucial regulator of chromatin organization and gene expression (Ibarra and Hetzer, 2015). As an architectural component of NPCs, although Tpr can directly bind to 25% of the genome, most functions of Tpr depend on its interaction with Nup153 to locate at the NPC (Vaquerizas et al., 2010). It is believed that the nuclear envelope is associated with heterochromatin, whereas NPCs are surrounded by euchromatin. The maintenance of these heterochromatin exclusion zones was shown to involve Tpr (Krull et al., 2010). And recently, Tpr is reported to be necessary for SAHF formation, which also relies on its association with the NPC (Boumendil et al., 2019). Nup62 is an element of central channel of NPC (Grossman et al., 2012). We found that the WISH result of ae1-globin was increased in zebrafish nup62l mutant (Supplementary Figure 14), which was identical to tprbcas7 mutant. It was suggested that the function of Tpr in regulation of erythroid gene expression might be associated with NPC. It will be interesting to clarify the precise mechanism of Tpr in maintenance of chromatin condensation and gene expression. Also, our study would not only uncover the important role of Tpr in chromatin condensation but also facilitate ex vivo production of erythrocytes and regulation of chromatin status to change the cell fate.
The zebrafish facility and study were approved by the Animal Research Advisory Committee of Institute of Nutrition and Health, SIBS, CAS, and zebrafish were maintained according to the guidelines of the Institutional Animal Care and Use Committee. Wild-type (WT) zebrafish strains Tubingen (Vinciguerra et al., 2005) and WIK, the transgenic zebrafish line Tg(gata1: DsRed) (Traver et al., 2003), and Tg(kdrl: EGFP) (Cross et al., 2003) were described previously. To prevent melanin pigment formation, embryos were incubated in egg water containing 0.045% 1-phenyl-2-thiourea (PTU; Sigma) after 1 dpf and the egg water was changed every day. The embryos were collected at the desired stages (Kimmel et al., 1995).
ENU mutagenesis and positional cloning were performed as previously described (Bahary et al., 2004). The cas7 (Tu background) allele was mapped by out-crossing Tu background heterozygous fish into polymorphic WIK background wild-type strain. The mutantcas7 was first mapped on chromosome 20 by linked SSLP markers by bulk segregation analysis (BSA) (Knapik et al., 1998; Shimoda et al., 1999). Simple sequence-length polymorphism (SSLP) markers used for BSA were selected from the Massachusetts General Hospital Zebrafish Server website (MGH)1. Fine mapping using mainly SSLP markers was carried out to narrow down the genetic interval within a 225-kb region between two markers 294-04 and 297-01. The cDNAs of three candidate genes in the interval were cloned and sequenced from mutants. The sequence of tprb gene in mutantscas7 had a C to G missense mutation, while other genes were normal. Also, the C to G missense mutation was confirmed by sequencing genomic DNA. All primers used for this study are provided in Supplementary Table.
Morpholino oligonucleotides (MOs) used for microinjection were ordered from Gene Tools. The MOs were microinjected into one-cell-stage embryos as previously described (Nasevicius and Ekker, 2000). Transient tprb transgene construct within Tol2 vectors (40 pg) was microinjected into one-cell-stage embryos with Tol2 transposase mRNA (50 pg). CRISPR-Cas9–mediated generation of zebrafish mutants (tprb–3+1 and vhl) were performed as previously described (Xiao et al., 2013). The gRNAs (50 pg) and Cas9 protein (500 pg) were co-microinjected into one-cell-stage embryos.
The zebrafish cDNA of tprb gene was amplified from reverse transcription products and cloned into pCS2+ vector with BamHI and EcoRI restriction sites. To construct Tol2 transgenesis vectors, the ubiquitin promoter followed by P2A and in-frame mCherry was cloned into modified Tol2 backbone. Also, the zebrafish tprbWT was amplified from tprb-pCS2+ plasmid and inserted between the ubiquitin promoter and P2A with BamHI restriction site. We changed several bases of the first 30 bases in the tprbWT gene, to mismatch the tprb ATG MO sequence. Changes in these bases would not affect the tprb amino acid sequence, for these bases are all changes in synonymous amino acids.
The antisense RNA probes were transcribed in vitro by T3 or T7 polymerase (Ambion) with Digoxigenin RNA Labelling Mix (Roche). Conventional and fluorescence whole-mount in situ hybridization (WISH and FISH) was described previously (Jowett and Lettice, 1994). In WISH, NBT/BCIP (Sigma) was used for staining and images were captured by Olympus SZX16 microscope. In FISH, embryos were stained with Cy5 (TSA system; Perkin Elmer) and DAPI (1:500; Beyotime) and imaged by Olympus FV1000 confocal microscope.
Zebrafish embryos at indicated time points were anesthetized in calcium-free and magnesium-free PBS (pH 7.4) containing 0.02% tricaine (Sigma) and 1% BSA. Circulating blood cells were collected by cutting tails and cytospined onto slides by centrifugation at 1,000 rpm for 3 min. Then the cells were air-dried and stained by Giemsa solutions (Nanjing Jiancheng) according to the manufacturer’s protocol. Images were finally captured by Zeiss Axio Imager A2 microscope. Nuclear area, and nuclear and cytoplasmic diameters of at least 100 randomly selected erythrocytes were measured by using ImageJ software, and nucleo-cytoplasmic ratio was calculated through spherical volume formula.
The analysis and sorting of gata1+ erythrocytes from Tg(gata1: DsRed; kdrl: EGFP) embryos were carried out with flow cytometer (Beckman) as previously described (Traver et al., 2003). For total RNA extraction, 10–20 zebrafish embryos or the sorted erythrocytes or cultured human cells were dissolved by TRIzol (Invitrogen), and then transcribed into cDNA by PrimerScript RT Master Mix (TaKaRa). The SYBR Green Real-time PCR Master Mix (TOYOBO) was used to perform quantitative PCR system with ABI Q7 real-time PCR instrument. The primers are listed in Supplementary Table.
HEK293T and Hep3B cells were cultured in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. O2 (1%) was generated by flushing a 94% N2/5% CO2 mixture into the incubator and sustained for 48 h until cell confluence. For siRNA transfection, Lipofectamine RNAiMAX Reagent (Invitrogen) was used following the manufacturer’s protocol.
To extract the protein from zebrafish embryos, the embryos were de-membraned and de-yolked first, then homogenized in lysis buffer [20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 10% glycerol, 0.1% Triton X-100, and protease inhibitor cocktail] and boiled with 2 × SDS buffer for 10 min. To obtain the protein from cultured cells, the cells were homogenized directly with 2 × SDS buffer and boiled for 10 min. Nuclear and cytoplasmic extracts were prepared from cultured cells with Nuclear and Cytoplasmic Extraction Kit (CWBIO) according to the manufacturer’s instruction. The immunoblotting was carried out as previously described (Gao et al., 2015), with rabbit anti-Tpr antibody (immunized by 461-710 amino acids of zebrafish Tpr protein; Abclonal), mouse anti-a-tubulin (Sigma), mouse anti-HIF-1a (NOVUS), rabbit anti-TPR (human) (Bethyl Laboratory), goat anti-Lamin B (Santa Cruz), mouse anti-H3 (CST), and goat-anti-rabbit/mouse and donkey-anti-goat secondary antibodies (LIANKE).
Zebrafish embryos at indicated time points were anesthetized in calcium-free and magnesium-free PBS (pH 7.4) containing 0.02% tricaine (Sigma) and 1% BSA. Circulating blood cells were collected by cutting tails and cytospined onto slides by centrifugation at 1,000 rpm for 3 min. Immunofluorescence was carried out as previously described (Gao et al., 2015). The cells were stained with rabbit anti-Tpr antibody, fluorescence was detected with goat-anti-rabbit Alexa Fluor 488 secondary antibody (Invitrogen) and DAPI (Beyotime) was used for nucleus staining.
Quantitative ChIP was performed as previously described (Wardle et al., 2006; Wong et al., 2011) with modifications. Briefly, about 300,000 gata1+ erythrocytes from Tg(gata1: DsRed) embryos at 3 dpf were sorted and cross-linked with 1% formaldehyde for 10 min at room temperature, followed by glycine (0.125 M) treatment for 5 min. Then the cells were homogenized in lysis buffer [50 mM Tris–HCl (pH8.0), 10 mM EDTA (pH8.0), 0.2% SDS, and Cocktail proteinase inhibitor] and sonicated by Bioruptor to yield fragments. Each sample was incubated with RNA Pol II antibody (clone CTD4H8, Millipore) overnight at 4°C, followed by washing and elution with magnetic beads, and reverse cross-linked for 6 h at 65°C. DNA was purified using MinElute PCR Purification Kit (QIAGEN). The primers used in qChIP are shown in Supplementary Table.
For sample preparation, the tails of zebrafish embryos at 4 dpf were fixed with PBS solution containing 2% paraformaldehyde and 2.5% glutaraldehyde overnight at 4°C, followed by 2% osmium tetroxide. The tails were dehydrated in ethanol and infiltrated in acetone, then embedded in epon 812, and sections were analyzed using a FEI Tecnai G2 Spirit electron microscope. Quantification of certain chromatin was performed as previously described (Baarlink et al., 2017) with modifications. Briefly, each nucleus of erythrocyte was manually segmented and generated a binary mask by ImageJ. Then, the certain chromatin state was classified by the different gray threshold values. Finally, the proportion of certain chromatin was subsequently analyzed by measuring the chromatin area and nucleus area.
The shRNA sequences for human TPR gene knockdown and the scrambled control shRNA sequence are supplied in Supplementary Table. The shRNA sequences were cloned into pLSLG lentiviral vector, which contained the EGFP for detecting transfection efficiency. The respective lentiviral vectors and helper vectors were transfected to 293T cells for viral packaging. Then 60 h after transfection, virus was collected to infect K562 cells in the presence of 10 mg/ml polybrene (Sigma-Aldrich). After 4 days, the cells were used in the experiments as described.
The K562 cells were treated with hemin (Sigma) at 0.04 mM for 48 h to induce erythroid differentiation. For immune-fluorescence staining, 1 × 106 cells suspended into PBS were simultaneously incubated for 30 min of anti-APC-conjugated CD235a at 4°C avoiding light. The cells were then washed in PBS to remove unbound antibody for immediate flow cytometry analysis.
Statistical analysis was performed by GraphPad Prism 6 software using two-tailed Student’s t-test and p value of less than 0.05 was considered significant. Error values were calculated by SEM.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by the Animal Research Advisory Committee of Institute of Nutrition and Health, SIBS, CAS.
SW, KC, WP, and XJ designed the research, analyzed the data, and wrote the manuscript. SW, KC, and TX performed the experiments. KM, LG, CF, WZ, CJ, CR, MD, and YC assisted with the experiments. CJ, XJ, LG, KM, YC, and MD contributed to the forward genetics screen. WP and YZ gave suggestions for experiment design. All authors contributed to the article and approved the submitted version.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key R&D Program of China (2018YFA0800203 to WP), National Natural Science Foundation of China (81901918, 81660204 to XJ, 31925014, 31571505 to WP) and Chinese Academy of Sciences (CAS) Research Program of Frontier Sciences (ZDBS-LY-SM010) to WP; and Inner Mongolia Science Foundation (2019MS08060); and Innovative and Entrepreneurial Talents in the “Prairie Talents” Project of Inner Mongolia (Q2017047), local science and technology projects guided by the central government (2020ZY0040), and CAS “Light of West China” Program to XJ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Jun Zhu for hif-2a morpholino; Ying Cao for zebrafish nup62l mutant; and Xiaolan Peng, Zhiwei Dong, Tingxi Liu, Jiang Zhu, Jun Zhu, and Lan Wang for technical support and suggestions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.709923/full#supplementary-material
Supplementary Figure 1 | Developmental serial analysis of αe1-globin expression in mutantcas7. Embryos were fixed at time points as indicated, followed by WISH analysis of αe1-globin expression. Expression of αe1-globin is shown in siblings and mutants at 36 hpf (A,B), 2 dpf (C,D), 3 dpf (E,F), and 4 dpf (G,H). The number and percentage of het-het incross embryos with the in situ pattern are listed at the bottom of the mutantcas7 panels.
Supplementary Figure 2 | WISH analysis of hematopoietic lineages at 3/4 dpf. WISH analysis of cmyb (A,B), mpx (C,D), lyz (E,F), and rag-1 (G,H′) expression shows no significant difference between sibling and mutantcas7 at 3/4 dpf. After WISH and photographing, all embryos were extracted for genomic DNA and genotyped by sequencing, then the mutant percentage was evaluated. The percentages of embryos in a het-het incross clutch with the expression pattern as shown in the mutantcas7 column is listed at the bottom of each panel. (I) The relative expression of αe3-globin, βe1-globin, βe2-globin, hebp2, sdhb, fth1a, cmyb, mpo, lyz, kdrl, and rag1 in sibling and mutantcas7 embryos at 3 dpf. Error bars represent SEM. ns, not significant; *p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001.
Supplementary Figure 3 | Normal primitive hematopoiesis and vascular morphogenesis in mutantcas7. Sibling and mutantcas7 embryos were fixed at 22 or 26 hpf, followed by WISH analysis of scl, gata1, pu.1, mpx, and kdrl expression. Expressions of scl (A,B), gata1 (C,D), pu.1 (E,F), mpx (G,H), and kdrl (I,J) show no significant difference between wild-type siblings and mutants. Insets in panels (E,F) show dorsal views of the head staining region. After WISH and photographing, all embryos were extracted for genomic DNA and genotyped by sequencing, then the mutant percentage was evaluated. The number and percentage of het-het incross embryos with the in situ pattern are listed at the bottom of the mutantcas7 panels.
Supplementary Figure 4 | The point mutation (C-G) in mutantcas7 was not found in all five wild-type strains, indicating that C-G transition at the site is not a SNP. Sequencing electropherogram of the mutation site of tprb genomic DNA from five wild-type strains. Genomic DNA were extracted each from 20 to 30 embryos of TU, AB, WIK, Longfin, and Shanghai strains, and then sequenced for tprb gene. This point mutation (C-G) in mutantcas7 was not found in all five wild-type strains.
Supplementary Figure 5 | WISH analysis of hematopoietic lineages in tprb morphants at 3/4 dpf. (A) Representative immunoblotting images of Tpr in control and tprb morphants. WISH analysis of cmyb (B,C), mpx (D,E), lyz (F,G), and rag-1 (H,I′) expression show no significant difference between control and tprb morphants at 3/4 dpf. After WISH and photographing, all embryos were extracted for genomic DNA and genotyped by sequencing, then the mutant percentage was evaluated. The percentage of indicated phenotype is listed at the bottom of each panel. (J) The relative expression of αe1-globin, alas2, band3, αe3-globin, βe1-globin, βe2-globin, gata1, hebp2, sdhb, fth1a, cmyb, mpo, lyz, kdrl, and rag1 at 3 dpf in tprb morphants. Error bars represent SEM. ns, not significant; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001.
Supplementary Figure 6 | Giemsa staining in circulating erythroid cells in control and tprb morphants at different time stages. (A) Quantitative analysis of each erythroid cell nuclear area. (B) Quantitative analysis of nucleus-to-cytoplasm (N/C) ratio. Error bars represent SEM. ns, not significant; ****p ≤ 0.0001; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. (C–G) Representative images of Giemsa staining in circulating erythroid cells at different stages: 30 hpf (C,D), 2 dpf (E,F), 3 dpf (G,H), and 4 dpf (I,J). Scale bars represent 20μm.
Supplementary Figure 7 | The result of tprbcas7 rescue experiment at 4 dpf. Transient expression of wild-type tprb under the ubiquitin promoter in mutantcas7 could rescue the phenotypes. Mutantcas7 embryos at one-cell stage were injected the tol2 plasmid with tprbWT expression driven by ubiquitin promoter. Then embryos with mCherry fluorescence would be selected for downstream analysis. At 4 dpf, every embryo was cut off in head for genotyping, then the trunk and tail of the embryo was taken for RNA or protein extraction. After genotyping the mutant embryos, we pooled three to five of the same phenotype embryo RNA extraction and took the qPCR experiments. (A) The relative expression of αe1-globin, alas2, band3, αe3-globin, βe1-globin, and βe2-globin at 4 dpf. Error bars represent SEM. ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. (B) Representative immunoblotting images of Tpr in tprb mutants rescued by tol2 plasmid with tprbWT expression.
Supplementary Figure 8 | Expression pattern of tprb mRNA during zebrafish embryogenesis. (A–J) WT (TU strain) embryos were fixed at the time points as indicated in each panel, followed by WISH analysis of tprb mRNA expression.
Supplementary Figure 9 | The endogenous Tpr protein is located on the nuclear membrane and Tpr protein is missing in tprb mutant or morphant. Representative images of Tpr immunofluorescence in sibling (A,B) and mutants (C,D) at 3 dpf. All embryos were extracted for genomic DNA and genotyped by sequencing. Representative images of Tpr immunofluorescence in control morphants (E,F) and tprb morphants (G,H) at 3 dpf. Scale bars represent 20 μm.
Supplementary Figure 10 | Flow cytometry analysis of erythrocytes from control and tprb morphants. Flow cytometry analysis of gata1+ cells from Tg(gata1: DsRed; kdrl: EGFP) transgenic line in control and tprb morphants at different time points. The ratio of gata1+ erythrocytes between control and tprb morphants was similar.
Supplementary Figure 11 | Generation and phenotype characterization of zebrafish vhl mutant. (A,B) Generation of vhl mutant via CRISPR-Cas9 technique. The alignment of WT and mutated sequences is listed. The underlined sequence is vhl gRNA target site. The sequencing result of vhl genomic DNA showed changed bases (shown in red) at exon 1 (A), which caused a premature stop codon leading to the production of a truncated 31-amino-acid Vhl protein (B). (C–F) WISH analysis of ae1-globin expression in sibling and vhl mutants at 3 dpf under the control and hif-2a morpholino (E,F) injection, indicating that the hif-2a MO can partially rescue the increased expression of ae1-globin in vhl mutant.
Supplementary Figure 12 | The other tissue cells in tprbcas7 mutant have normal chromatin organization. (A–F) Representative TEM images of chromatin organization within the tissue cells show no obvious difference between sibling and tprbcas7 mutants at 4 dpf, including neural cells (A,B), endothelial cells (C,D), and muscular cells (E,F).
Supplementary Figure 13 | TPR is also important for erythroid differentiation in mammalian. (A) Representative immunoblotting images of TPR shRNA and scramble shRNA in K562 cell line. Tubulin serves as the loading control. (B) The relative expression of TPR in different groups. (C) Representative FACS images of the ratio of CD235a-positive cells with or without hemin treatment. (D) Quantitative analysis of panel (C). (E) The relative expression of α/β/γ/δ-globins with or without hemin treatment. Error bars represent SEM. ns, not significant; *p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001.
Supplementary Figure 14 | Zebrafish nup62l mutants represent similar phenotype with tprbcas7 mutants. (A,B) WISH analysis of ae1-globin show the increased expression in nup62l mutants compared with sibling at 3.5 dpf. The number and percentage of het-het incross embryos with the in situ pattern in a clutch are listed at the bottom of the mutant panels.
Baarlink, C., Plessner, M., Sherrard, A., Morita, K., Misu, S., Virant, D., et al. (2017). A transient pool of nuclear F-actin at mitotic exit controls chromatin organization. Nat. Cell Biol. 19, 1389–1399. doi: 10.1038/ncb3641
Bahary, N., Davidson, A., Ransom, D., Shepard, J., Stern, H., Trede, N., et al. (2004). The zon laboratory guide to positional cloning in zebrafish. Methods Cell Biol. 77, 305–329. doi: 10.1016/s0091-679x(04)77017-x
Bertrand, J. Y., Chi, N. C., Santoso, B., Teng, S., Stainier, D. Y., and Traver, D. (2010). Haematopoietic stem cells derive directly from aortic endothelium during development. Nature 464, 108–111. doi: 10.1038/nature08738
Boumendil, C., Hari, P., Olsen, K. C. F., Acosta, J. C., and Bickmore, W. A. (2019). Nuclear pore density controls heterochromatin reorganization during senescence. Genes Dev. 33, 144–149. doi: 10.1101/gad.321117.118
Brero, A., Easwaran, H. P., Nowak, D., Grunewald, I., Cremer, T., Leonhardt, H., et al. (2005). Methyl CpG–binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 169, 733–743. doi: 10.1083/jcb.200502062
Bunn, H. F. (2013). Erythropoietin. Cold Spring Harb. Perspect. Med. 3:a011619. doi: 10.1101/cshperspect.a011619
Chen, A. T., and Zon, L. I. (2009). Zebrafish blood stem cells. J. Cell Biochem. 108, 35–42. doi: 10.1002/jcb.22251
Choi, Y.-L., Lira, M. E., Hong, M., Kim, R. N., Choi, S.-J., Song, J.-Y., et al. (2014). A novel fusion of TPR and ALK in lung adenocarcinoma. J. Thorac. Oncol. 9, 563–566. doi: 10.1097/jto.0000000000000093
Cordes, V. C., Reidenbach, S., Rackwitz, H. R., and Franke, W. W. (1997). Identification of protein p270/Tpr as a constitutive component of the nuclear pore complex-attached intranuclear filaments. J. Cell Biol. 136, 515–529. doi: 10.1083/jcb.136.3.515
Cross, L. M., Cook, M. A., Lin, S., Chen, J.-N., and Rubinstein, A. L. (2003). Rapid analysis of angiogenesis drugs in a live fluorescent zebrafish assay. Arterioscler. Thromb. Vasc. Biol. 23, 911–912. doi: 10.1161/01.ATV.0000068685.72914.7E
Deng, W., Lee, J., Wang, H., Miller, J., Reik, A., Gregory, P. D., et al. (2012). Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 149, 1233–1244. doi: 10.1016/j.cell.2012.03.051
Deng, W., Rupon, J. W., Krivega, I., Breda, L., Motta, I., Jahn, K. S., et al. (2014). Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell 158, 849–860. doi: 10.1016/j.cell.2014.05.050
Drexler, H. G., Matsuo, Y., and MacLeod, R. A. F. (2004). Malignant hematopoietic cell lines: in vitro models for the study of erythroleukemia. Leuk. Res. 28, 1243–1251. doi: 10.1016/j.leukres.2004.03.022
Dzierzak, E., and Philipsen, S. (2013). Erythropoiesis: development and differentiation. Cold Spring Harb. Perspect. Med. 3:a011601. doi: 10.1101/cshperspect.a011601
Fraser, S. T., Isern, J., and Baron, M. H. (2007). Maturation and enucleation of primitive erythroblasts during mouse embryogenesis is accompanied by changes in cell-surface antigen expression. Blood 109, 343–352. doi: 10.1182/blood-2006-03-006569
Galy, V., Gadal, O., Fromont-Racine, M., Romano, A., Jacquier, A., and Nehrbass, U. (2004). Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell 116, 63–73. doi: 10.1016/s0092-8674(03)01026-2
Gao, L., Li, D., Ma, K., Zhang, W., Xu, T., Fu, C., et al. (2015). TopBP1 governs hematopoietic stem/progenitor cells survival in zebrafish definitive hematopoiesis. PLoS Genet. 11:e1005346. doi: 10.1371/journal.pgen.1005346
Greco, A., Pierotti, M. A., Bongarzone, I., Pagliardini, S., Lanzi, C., and Della Porta, G. (1992). TRK-T1 is a novel oncogene formed by the fusion of TPR and TRK genes in human papillary thyroid carcinomas. Oncogene 7, 237–242.
Green, D. M., Johnson, C. P., Hagan, H., and Corbett, A. H. (2003). The C-terminal domain of myosin-like protein 1 (Mlp1p) is a docking site for heterogeneous nuclear ribonucleoproteins that are required for mRNA export. Proc. Natl. Acad. Sci. U.S.A. 100, 1010–1015. doi: 10.1073/pnas.0336594100
Grossman, E., Medalia, O., and Zwerger, M. (2012). Functional architecture of the nuclear pore complex. Annu. Rev. Biophys. 41, 557–584. doi: 10.1146/annurev-biophys-050511-102328
Hase, M. E., Kuznetsov, N. V., and Cordes, V. C. (2001). Amino acid substitutions of coiled-coil protein Tpr abrogate anchorage to the nuclear pore complex but not parallel, in-register homodimerization. Mol. Biol. Cell 12, 2433–2452. doi: 10.1091/mbc.12.8.2433
Ibarra, A., and Hetzer, M. W. (2015). Nuclear pore proteins and the control of genome functions. Genes Dev. 29, 337–349. doi: 10.1101/gad.256495.114
Istomina, N. E., Shushanov, S. S., Springhetti, E. M., Karpov, V. L., Krasheninnikov, I. A., Stevens, K., et al. (2003). Insulation of the chicken β-globin chromosomal domain from a chromatin-condensing protein, MENT. Mol. Cell. Biol. 23, 6455–6468. doi: 10.1128/mcb.23.18.6455-6468.2003
Ji, P., Murata-Hori, M., and Lodish, H. F. (2011). Formation of mammalian erythrocytes: chromatin condensation and enucleation. Trends Cell Biol. 21, 409–415. doi: 10.1016/j.tcb.2011.04.003
Jong, J. L. O. D., and Zon, L. I (2005). Use of the zebrafish system to study primitive and definitive hematopoiesis. Annu. Rev. Genet. 39, 481–501. doi: 10.1146/annurev.genet.39.073003.095931
Jowett, T., and Lettice, L. (1994). Whole-mount in situ hybridization on zebrafish embryos using a mixture of digoxigenin- and fluorescein-labelled probes. Trends Genet. 10, 73–74. doi: 10.1016/0168-9525(94)90220-8
Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., and Schilling, T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310. doi: 10.1002/aja.1002030302
Kissa, K., and Herbomel, P. (2010). Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature 464, 112–115. doi: 10.1038/nature08761
Knapik, E. W., Goodman, A., Ekker, M., Chevrette, M., Delgado, J., Neuhauss, S., et al. (1998). A microsatellite genetic linkage map for zebrafish (Danio rerio). Nat. Genet. 18, 338–343. doi: 10.1038/ng0498-338
Koh, M. Y., and Powis, G. (2012). Passing the baton: the HIF switch. Trends Biochem. Sci. 37, 364–372. doi: 10.1016/j.tibs.2012.06.004
Krull, S., Dorries, J., Boysen, B., Reidenbach, S., Magnius, L., Norder, H., et al. (2010). Protein Tpr is required for establishing nuclear pore-associated zones of heterochromatin exclusion. EMBO J. 29, 1659–1673. doi: 10.1038/emboj.2010.54
Krull, S., Thyberg, J., Bjorkroth, B., Rackwitz, H. R., and Cordes, V. C. (2004). Nucleoporins as components of the nuclear pore complex core structure and Tpr as the architectural element of the nuclear basket. Mol. Biol. Cell 15, 4261–4277. doi: 10.1091/mbc.E04-03-0165
Lee, F. S., and Percy, M. J. (2011). The HIF pathway and erythrocytosis. Annu. Rev. Pathol. 6, 165–192. doi: 10.1146/annurev-pathol-011110-130321
Lelek, M., Casartelli, N., Pellin, D., Rizzi, E., Souque, P., Severgnini, M., et al. (2015). Chromatin organization at the nuclear pore favours HIV replication. Nat. Commun. 6:6483. doi: 10.1038/ncomms7483
Li, D., Xue, W., Li, M., Dong, M., Wang, J., Wang, X., et al. (2018). VCAM-1(+) macrophages guide the homing of HSPCs to a vascular niche. Nature 564, 119–124. doi: 10.1038/s41586-018-0709-7
Li, F., Zhai, Y.-P., Tang, Y.-M., Wang, L.-P., and Wan, P.-J. (2012). Identification of a novel partner gene, TPR, fused to FGFR1 in 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer 51, 890–897. doi: 10.1002/gcc.21973
Menon, V., and Ghaffari, S. (2021). Erythroid enucleation: a gateway into a “bloody” world. Exp. Hematol. 95, 13–22. doi: 10.1016/j.exphem.2021.01.001
Metelo, A. M., Noonan, H. R., Li, X., Jin, Y., Baker, R., Kamentsky, L., et al. (2015). Pharmacological HIF2α inhibition improves VHL disease–associated phenotypes in zebrafish model. J. Clin. Invest. 125, 1987–1997. doi: 10.1172/JCI73665
Murayama, E., Kissa, K., Zapata, A., Mordelet, E., Briolat, V., Lin, H. F., et al. (2006). Tracing hematopoietic precursor migration to successive hematopoietic organs during zebrafish development. Immunity 25, 963–975. doi: 10.1016/j.immuni.2006.10.015
Nasevicius, A., and Ekker, S. C. (2000). Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 26, 216–220. doi: 10.1038/79951
Paik, E. J., and Zon, L. I. (2010). Hematopoietic development in the zebrafish. Int. J. Dev. Biol. 54, 1127–1137. doi: 10.1387/ijdb.093042ep
Palis, J. (2014). Primitive and definitive erythropoiesis in mammals. Front. Physiol. 5:3. doi: 10.3389/fphys.2014.00003
Popova, E. Y., Krauss, S. W., Short, S. A., Lee, G., Villalobos, J., Etzell, J., et al. (2009). Chromatin condensation in terminally differentiating mouse erythroblasts does not involve special architectural proteins but depends on histone deacetylation. Chromosome Res. 17, 47–64. doi: 10.1007/s10577-008-9005-y
Qian, F., Zhen, F., Xu, J., Huang, M., Li, W., and Wen, Z. (2007). Distinct functions for different scl isoforms in zebrafish primitive and definitive hematopoiesis. PLoS Biol. 5:e132. doi: 10.1371/journal.pbio.0050132
Raices, M., and D’Angelo, M. A. (2012). Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nat. Rev. Mol. Cell Biol. 13, 687–699. doi: 10.1038/nrm3461
Schwartz, T. U. (2016). The structure inventory of the nuclear pore complex. J. Mol. Biol. 428, 1986–2000. doi: 10.1016/j.jmb.2016.03.015
Shimoda, N., Knapik, E. W., Ziniti, J., Sim, C., Yamada, E., Kaplan, S., et al. (1999). Zebrafish genetic map with 2000 microsatellite markers. Genomics 58, 219–232. doi: 10.1006/geno.1999.5824
Snow, C. J., Dar, A., Dutta, A., Kehlenbach, R. H., and Paschal, B. M. (2013). Defective nuclear import of Tpr in Progeria reflects the Ran sensitivity of large cargo transport. J. Cell Biol. 201, 541–557. doi: 10.1083/jcb.201212117
Soman, N. R., Correa, P., Ruiz, B. A., and Wogan, G. N. (1991). The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc. Natl. Acad. Sci. U.S.A. 88, 4892–4896. doi: 10.1073/pnas.88.11.4892
Traver, D., Paw, B. H., Poss, K. D., Penberthy, W. T., Lin, S., and Zon, L. I. (2003). Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat. Immunol. 4, 1238–1246. doi: 10.1038/ni1007
van Rooijen, E., Voest, E. E., Logister, I., Korving, J., Schwerte, T., Schulte-Merker, S., et al. (2009). Zebrafish mutants in the von Hippel-Lindau tumor suppressor display a hypoxic response and recapitulate key aspects of Chuvash polycythemia. Blood 113, 6449–6460. doi: 10.1182/blood-2008-07-167890
Vaquerizas, J. M., Suyama, R., Kind, J., Miura, K., Luscombe, N. M., and Akhtar, A. (2010). Nuclear pore proteins nup153 and megator define transcriptionally active regions in the Drosophila genome. PLoS Genet. 6:e1000846. doi: 10.1371/journal.pgen.1000846
Verreault, A., and Thomas, J. O. (1993). Chromatin structure of the beta-globin chromosomal domain in adult chicken erythrocytes. Cold Spring Harb. Symp. Quant. Biol. 58, 15–24. doi: 10.1101/sqb.1993.058.01.005
Vinciguerra, P., Iglesias, N., Camblong, J., Zenklusen, D., and Stutz, F. (2005). Perinuclear Mlp proteins downregulate gene expression in response to a defect in mRNA export. EMBO J. 24, 813–823. doi: 10.1038/sj.emboj.7600527
Wardle, F. C., Odom, D. T., Bell, G. W., Yuan, B., Danford, T. W., Wiellette, E. L., et al. (2006). Zebrafish promoter microarrays identify actively transcribed embryonic genes. Genome Biol. 7:R71. doi: 10.1186/gb-2006-7-8-r71
Wickramasinghe, S. N., and Wood, W. G. (2005). Advances in the understanding of the congenital dyserythropoietic anaemias. Br. J. Haematol. 131, 431–446. doi: 10.1111/j.1365-2141.2005.05757.x
Wong, P., Hattangadi, S. M., Cheng, A. W., Frampton, G. M., Young, R. A., and Lodish, H. F. (2011). Gene induction and repression during terminal erythropoiesis are mediated by distinct epigenetic changes. Blood 118, e128–e138. doi: 10.1182/blood-2011-03-341404
Xiao, A., Wang, Z., Hu, Y., Wu, Y., Luo, Z., Yang, Z., et al. (2013). Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 41:e141. doi: 10.1093/nar/gkt464
Zermati, Y., Garrido, C., Amsellem, S., Fishelson, S., Bouscary, D., Valensi, F., et al. (2001). Caspase activation is required for terminal erythroid differentiation. J. Exp. Med. 193, 247–254. doi: 10.1084/jem.193.2.247
Zhang, P., Branson, O. E., Freitas, M. A., and Parthun, M. R. (2016). Identification of replication-dependent and replication-independent linker histone complexes: Tpr specifically promotes replication-dependent linker histone stability. BMC Biochem. 17:18. doi: 10.1186/s12858-016-0074-9
Keywords: erythrocytes, erythroid maturation, chromatin condensation, zebrafish, Tpr
Citation: Wu S, Chen K, Xu T, Ma K, Gao L, Fu C, Zhang W, Jing C, Ren C, Deng M, Chen Y, Zhou Y, Pan W and Jia X (2021) Tpr Deficiency Disrupts Erythroid Maturation With Impaired Chromatin Condensation in Zebrafish Embryogenesis. Front. Cell Dev. Biol. 9:709923. doi: 10.3389/fcell.2021.709923
Received: 14 May 2021; Accepted: 08 September 2021;
Published: 13 October 2021.
Edited by:
Yiyue Zhang, South China University of Technology, ChinaReviewed by:
Rui Monteiro, University of Birmingham, United KingdomCopyright © 2021 Wu, Chen, Xu, Ma, Gao, Fu, Zhang, Jing, Ren, Deng, Chen, Zhou, Pan and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weijun Pan, d2VpanVucGFuQHNpYnMuYWMuY24=; Xiaoe Jia, ZXZhbmdlbGluZTIwMDRAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.