 Frederic Abou Azar
Frederic Abou Azar Gareth E. Lim
Gareth E. Lim
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 09 September 2021
Sec. Signaling
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.709823
This article is part of the Research Topic Wnt Signaling at the Plasma Membrane: Activation, Regulation and Disease Connection View all 16 articles
The canonical Wnt signaling pathway is ubiquitous throughout the body and influences a diverse array of physiological processes. Following the initial discovery of the Wnt signaling pathway during wing development in Drosophila melanogaster, it is now widely appreciated that active Wnt signaling in mammals is necessary for the development and growth of various tissues involved in whole-body metabolism, such as brain, liver, pancreas, muscle, and adipose. Moreover, elegant gain- and loss-of-function studies have dissected the tissue-specific roles of various downstream effector molecules in the regulation of energy homeostasis. This review attempts to highlight and summarize the contributions of the Wnt signaling pathway and its downstream effectors on whole-body metabolism and their influence on the development of metabolic diseases, such as diabetes and obesity. A better understanding of the Wnt signaling pathway in these tissues may aid in guiding the development of future therapeutics to treat metabolic diseases.
During embryogenesis, the Wnt signaling pathway has active and critical roles in tissue development, including, but not limited to, axon guidance, body segmentation, limb development, and stem cell differentiation (Yang, 2003; Dunty et al., 2008; Geetha-Loganathan et al., 2008; Nusse, 2008; Stanganello et al., 2019). While its importance during embryogenesis cannot be understated, the influence of active Wnt signaling on the postnatal growth of tissues and their physiological functions are now gaining wide appreciation. Some of the lesser appreciated functions of the Wnt signaling pathway are its influence on systemic, organ, and tissue metabolism and energy homeostasis.
Whole-body metabolism is regulated by various tissues, including brain, liver, pancreas, muscle and adipose, and each contributes significantly to carbohydrate and/or lipid metabolism. The brain requires a constant uptake of oxygen and glucose, where astrocytes convert glucose to lactate and neurons use glucose and lactate in an oxidative manner (Deitmer et al., 2019). The liver is a critical metabolic tissue, as it is capable of glucose and glycogen metabolism, gluconeogenesis, fatty acid metabolism, and drug metabolism (Rui, 2014). The pancreas is responsible for secreting digestive enzymes and hormones, most notably insulin and glucagon, to control glucose homeostasis (Roder et al., 2016). Skeletal muscles require glucose and fatty acids to function, and can themselves be metabolized and converted to amino acids for use by other tissues. Insulin promotes glucose uptake in muscle where it can also be stored as glycogen (McPherron et al., 2013). Adipose tissue not only functions as energy reservoirs, but also secretes key hormones and metabolites to help control systemic energy balance (Choe et al., 2016). For example, it releases leptin to promote satiety (McDuffie et al., 2004). Thus, the goal of this review is aimed at highlighting the important contributions of the Wnt signaling pathway and its effectors within these metabolic tissues.
The naming of Wnt is derived from the gene wingless (wg) in Drosophila melanogaster, where its mutation resulted in a wingless phenotype (Sharma and Chopra, 1976). Its vertebrate homolog Int1 encodes for a retrovirus docking site and was first discovered in mice (Nusse and Varmus, 1982). Wnts are thought to primarily mediate their effects via autocrine and paracrine mechanisms, as the hydrophobic palmitate group near the N-terminus serves to tether the protein to the plasma membrane of its secreting cell (Kaemmerer and Gassler, 2016). However, recent findings have detected Wnt within exosomes, which suggests that Wnts could exert effects in distal sites in the body (Gross et al., 2012; Beckett et al., 2013).
The initiation of the canonical Wnt signaling pathway, sometimes referred to as the Wnt/β-catenin pathway, occurs by the binding of Wnt proteins to complexes consisting of the cell-surface Frizzled (Fz) receptor and lipoprotein-related proteins (LRP5/6). This leads to a cascade of downstream events, that culminate in the dissociation of the β-catenin “destruction complex” comprised of Glycogen Synthase Kinase 3 (GSK-3), Axin, Casein-kinase 1 α (CK1-α), and Adenomatosis Polypsis Coli (APC) (Figure 1; Thompson and Williams, 2018). In the absence of Wnt signaling, GSK-3 binds β-catenin, the transcriptional co-activator integral to Wnt signaling, to promote its phosphorylation. The interaction of β-catenin with GSK-3 is initiated by CK1-α-mediated phosphorylation of Ser47, which allows GSK-3 to bind and subsequently phosphorylate additional serine and threonine residues (Thompson and Williams, 2018), leading to the eventual ubiquitination and degradation of β-catenin. Disheveled (DSH) is also recruited to the Fz-LRP5/6 complex and recruits the components of the destruction complex to the cell membrane (MacDonald and He, 2012). This aids in the stabilization and accumulation of β-catenin in the cytoplasm. Once β-catenin accumulates in the cytosol, it translocates to the nucleus where it binds the TCF family of transcription factors that influence downstream Wnt target genes and those that exert negative- or positive-feedback effects on the Wnt signaling pathway itself (Jho et al., 2002). The mechanism by which β-catenin enters and exits the nucleus is currently unknown, although some studies suggest the involvement of the scaffold protein 14-3-3ζ (Li et al., 2008).
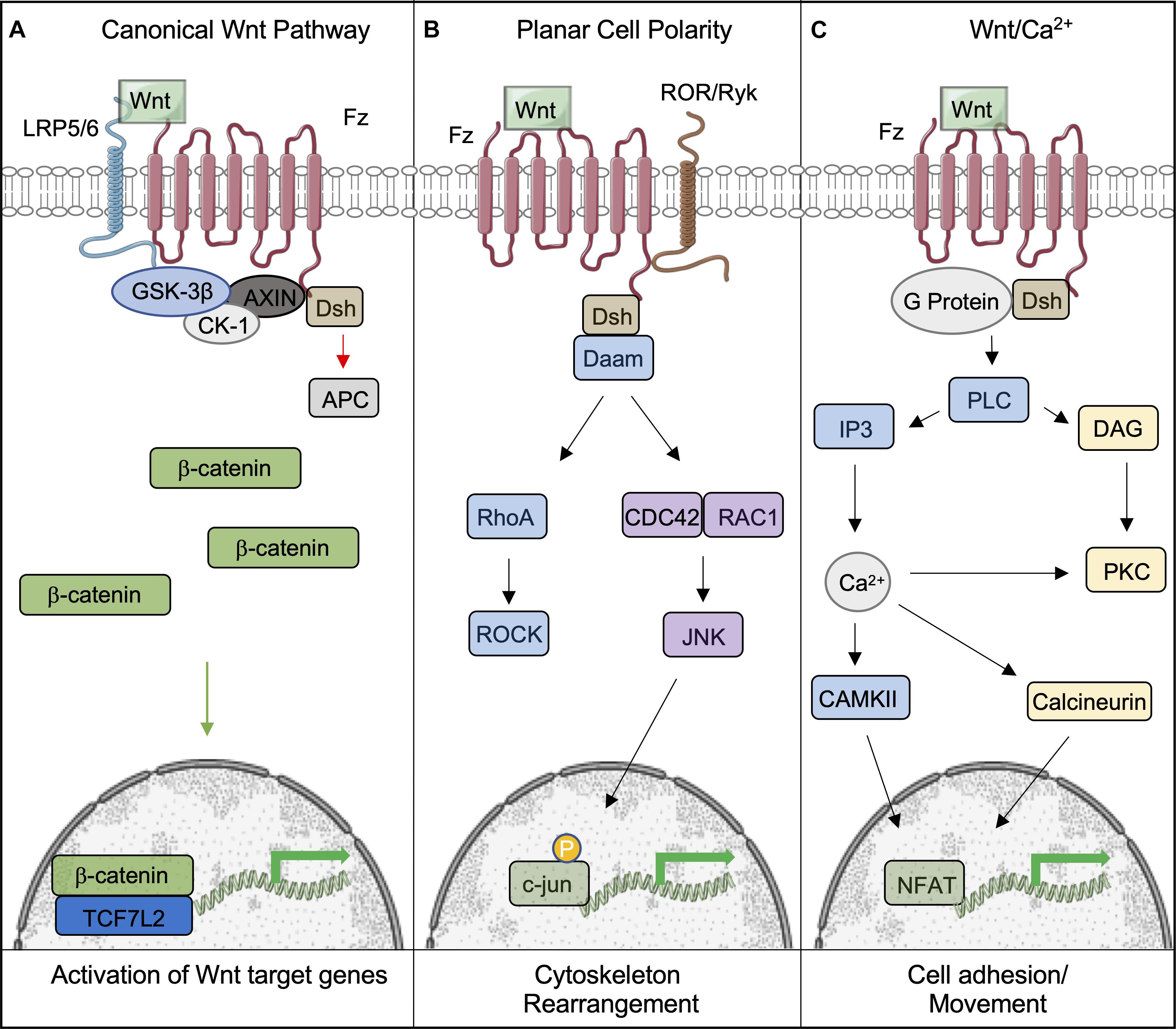
Figure 1. Overview of the canonical and non-canonical Wnt signaling pathways. (A) Following binding of Wnt to the Frizzled-LRP5/6 complex and the activation of the signaling pathway, the destruction complex (comprising of GSK-3β, CK-1, AXIN, and APC) is tethered to the plasma membrane, which permits β-catenin to accumulate and translocate to the nucleus, where it bind to TCF7L2 to activate downstream genes. (B,C) Wnts can also signal through non-canonical pathways to induce cell responses. (B) The Planar Cell Polarity pathway is activated upon Wnt binding to the Fz receptor. Dsh is subsequently activated, initiating the activites of Rho and Rac through Daam1. This leads to the eventual activation of ROCK and Jnk, which phosphorylates c-jun within the nucleus and allows it to initiate the transcription of target genes. (C) The Wnt/Ca2+ pathways is also activated by Fz; however, it is coupled to a G-protein, leading to intracellular Ca2+ release through PLC/IP3 activity. Ca2+ promotes the activity of CAMKII and Calcineurin to provoke the accumulation of the transcription factor NFAT in the nucleus. This figure was made with modified images from Servier Medical Art (Creative Commons Attribution 3.0 Unported License).
Wnts can also bind to Fz and activate the non-canonical pathway, also known as the β-catenin-independent pathway. Non-canonical Wnts include Wnt1, Wnt2, Wnt5a, Wnt5b, and Wnt11 (Nie et al., 2020). This sub-grouping includes two distinct pathways called the Planar Cell Polarity (PCP) and Wnt/Ca2+ pathways (Komiya and Habas, 2008). PCP was discovered through loss of function studies of Fz and Dvl in Drosophila. The ensuing phenotype displayed randomized orientations of epithelial structures (Seifert and Mlodzik, 2007). This pathway has also been shown to be involved in cytoskeletal reorganization and coordination of cellular polarization (Komiya and Habas, 2008). It is activated by Wnt binding Dsh and Fz, which activates Rho through Daam1, activating the Rho kinase. Daam1 also activates Rac and JNK to initiate transcription via the actions of c-jun (Le Floch et al., 2005). The Wnt/Ca2+ pathway is activated by the same cytoplasmic receptors; however, it is coupled to a G-protein, leading to intracellular Ca2+ release by the actions of PLC and IP3 (Kohn and Moon, 2005). Collectively, the actions of CAMKII and Calcineurin lead to the accumulation of NFAT in the nucleus to initiate the transcription of target genes (Saneyoshi et al., 2002). This pathway is believed to affect early gastrulation cell movements and in some cases may inhibit the canonical Wnt pathway (Gilland et al., 1999; Wallingford et al., 2001).
Additional negative and positive modulators of the canonical pathway have been identified. For example, Frizzled-related protein family (SFRP) proteins and Wnt inhibitory factors, such as Dickkopf (DKK1), bind LRP5/6 directly to inhibit its association with the Fz receptor (Niehrs, 2006). On the other hand, antagonists like Cerberus bind Wnts extracellularly, inhibiting their ability to complex with the Fz/LRP5/6 receptor complex (Piccolo et al., 1999). Corepressors like Groucho/TLE, MTGR1 or COOP exert inhibitory effects by dissociating β-Catenin from TCFs (Cavallo et al., 1998; Moore et al., 2008; Song et al., 2010; Valenta et al., 2012). Nuclear APC has also been reported to sequester β-Catenin and prevent its interactions with TCFs (Hamada and Bienz, 2004). Positive modulators like serine-threonine phosphatase 2A (PP2A) increase β-Catenin’s activity by antagonizing the effects of GSK3 through de-phosphorylation of β-Catenin (Kimelman and Xu, 2006). This allows β-Catenin to translocate into the nucleus without being degraded. PP2A is also able to dephosphorylate Axin and prevent its association with β-Catenin (Willert et al., 1999). When taken together, multiple endogenous mechanisms exist that exert inhibitory effects on Wnt-β-catenin signaling.
Through the use of genetic approaches, the contributions of different cell types and tissues, such as adipose tissue, skeletal muscle, brain, pancreas, and liver, to overall metabolic homeostasis have been identified (Kitamura et al., 2003; von Maltzahn et al., 2012; Kubota et al., 2017), and Wnt signaling, which influences spatial patterning and organogenesis, is required for the development of these metabolic tissues (Tables 1–3 and Figure 2).

Table 1. Developmental phenotype resulting from over-expression or deletion of Wnt signaling effectors in Drosophila.
Numerous mammalian Wnt proteins have been identified, with each isoform having distinct physiological functions in different tissues [for a detailed list, please refer to van Amerongen and Berns (2006)]. Deletion of one allele of a given Wnt does not necessarily lead to an overt phenotype, as demonstrated by the outcome of homo- and hetero-zygous Wnt1 deletion on brain development and viability (Thomas and Capecchi, 1990; van Amerongen and Berns, 2006). The Wnt ortholog in Drosophila, wg, has been shown to be important for brain development (Table 1; Richter et al., 1998), as deletion of wg in fly embryos resulted in partial development of the protocerebrum. Loss of function of wg also led to enhanced apoptosis in the ventral ganglia, and gain-of-function experiments using a hsp70 promoter exhibited an increase in CNS development in flies, with a two-fold increase in cerebral mass (Richter et al., 1998).
Aside from roles in brain development, Wnt ligands also have essential roles in primitive streak formation and at various stages of embryogenesis (van Amerongen and Berns, 2006). For example, Wnt7a mediates the formation of ventral cell types during limb development in the central nervous system of mice, whereas Wnt4 is involved in the formation of the anterior-posterior axis during spinal cord development and is responsible for directing the migration of neurons and their axons (Parr et al., 1993; Hollis and Zou, 2012).
Orthologs of Wnt also exist in different species, where they exhibit evolutionarily conserved functions. For example, in C. elegans, the orthologous Wnt gene, mom-2 is responsible for maintaining polarity of the embryo during development (Bischoff and Schnabel, 2006). In mammals, several Wnt proteins have also been found to have similar roles in maintaining cell polarity and the development of various metabolic organs, as seen by deletion of Wnt3 that results in the failure of Anterior-Posterior axis formation in the developing vertebrate embryo (Liu et al., 1999). Deletion of Wnt proteins are also associated with defects in organogenesis, as demonstrated by the failure of mesenchymal stem cells to differentiate into epithelial cells in kidneys of mice deficient in Wnt4. This results in the inability to form nephrons (Stark et al., 1994).
In the early stages of Xenopus embryo development (Table 2), inhibition of the Wnt signaling pathway appears to be essential, as injection of mRNA for dkk-1, the Wnt inhibitor, into blastomeres of Xenopus embryos led to anteriorized embryos displaying large heads, enlarged cement glands, and short trunks (Glinka et al., 1998). On the other hand, embryos injected with antibodies targeting dkk-1 exhibited the opposite phenotype of microcephaly accompanied with a lack of cement glands. The embryonic axis remained unaffected, highlighting the importance of Wnt inhibition for Xenopus brain development. Toward the later stages of development, Wnt signaling is necessary for the downregulation of BMP-4 expression to induce the formation of the dorsal ectoderm (Baker et al., 1999). Antagonizing BMP-4 further allows for neural induction, a function found to be mediated by TCF7L2. For a more detailed anaylsis of the Wnt signaling pathway in the developing Xenopus brain, please see the review by Patapoutian and Reichardt (Patapoutian and Reichardt, 2000).

Table 2. Developmental phenotype resulting from over-expression or deletion of Wnt signaling effectors in Xenopus.
Wnt proteins are highly expressed in neurons in the posterior segment of the Planarian brain (Adell et al., 2009). They plays an inhibitory role on brain expansion, as evidenced by the increase in brain cells following RNAi mediated suppression of wnt11-6 (Kobayashi et al., 2007; Hill and Petersen, 2015). Effectors of the Wnt signaling pathway can also affect organogenesis independent of β-catenin, as RNAi-mediated silencing of Dvl in Planarians compromised the development of the posterior ectopic brain (Almuedo-Castillo et al., 2011). Knockdown of β-catenin led to trunk loss, lack of tail identity, and loss of gut anterior-posterior polarity. However, brain development appeared to be phenotypically normal, indicating Dvl exerts its effects through a non- canonical Wnt pathway (Gurley et al., 2008).
During embryogenesis, various Wnt genes exhibit a biphasic pattern of expression. Some family members are upregulated before embryonic day-15 (e15) in mouse embryos, followed by their rapid downregulation by e16 (Heller et al., 2002). Wnt1 over-expression under the control of the Pdx1 promoter disrupted spleen development and caused pancreas agenesis (Heller et al., 2002; Table 3). Similarly, over-expression of Wnt5a, the non-canonical Wnt, also altered pancreatic development, resulting in reduced pancreas size and the production of atypical endocrine structures (Heller et al., 2002), showing that both Wnt pathways can share common functions. Another example of the requirement of Wnt signaling in pancreatic development has been observed in mice expressing a dominant-negative Fz receptor (Pdx1-Fz8CRD) (Papadopoulou and Edlund, 2005). Transgenic mice expressing the dominant-negative FZ receptor in PDX1-positive cells displayed reduced pancreatic mass, but did not develop hyperglycemia or diabetes. β-cells of transgenic mice had a 50% increase in insulin content, suggesting a compensatory mechanism for the loss of pancreatic mass. The reduction in pancreatic mass of transgenic mice is partially attributed to decreased pancreatic cell proliferation (Papadopoulou and Edlund, 2005).

Table 3. Developmental phenotype resulting from the over-expression or deletion of Wnt signaling effectors in mice.
In the liver, Wnt ligands have been found to influence liver growth and regeneration, as well as regulating cell proliferation and apoptosis (Monga, 2011). Various Wnt ligands, are expressed in the liver during development (Lade and Monga, 2011), and while it is not known if all Wnts have roles in liver development, Wnt2b has been implicated in liver organogenesis during the embryogenesis (Ober et al., 2006). The conservation of Wnt2b in liver development has also been observed in zebrafish embryos, as morpholinos against wnt2bb led to agenesis of hepatic tissue (Ober et al., 2006). The Wnt signaling pathway is active in Xenopus blastula as well; however, its activity decreases during gastrulation, suggesting it is not essential for early stage organogenesis (Schohl and Fagotto, 2002). Development of the pancreas, liver and intestine can be impaired by injection of Wnt8 plasmids into D1 cells of late stage Xenopus embryos. Moreover, micro-injection of stabilized forms of β-catenin mRNA into blastomeres were sufficient to supress foregut markers and recapitulated the phenotypes observed under Wnt8 administration. Administering DKK1 enhanced expression of for1 and pdx1, markers of the liver and pancreas, respectively. Enlarged liver and pancreas buds were observed at stage 42 of embryogenesis. Similarly, overexpression GSK3-β enhanced expression of for1 and pdx1 (Schohl and Fagotto, 2002).
One critical function of the hepatic Wnt signaling pathway is its role in liver zonation, which is the distribution of proteins along a protocentral axis defined by periportal (PP) and perivenous (PV) hepatocytes (Sekine et al., 2006; Yang et al., 2014). PP hepatocytes are situated near the portal vein and are exposed to high concentrations of oxygen and nutrients when compared to PV hepatocytes. Thus, enzymes requiring a high demand of oxygen and nutrients, such as gluconeogenic enzymes, are predominantly expressed in in PP hepatocytes, as they are more aerobic than PV hepatocytes (Jungermann and Katz, 1989; Braeuning et al., 2006). Meanwhile, enzymes involved in glutamine synthesis are enriched in PV hepatocytes, which provide glutamine to PP hepatocytes for urea biosynthesis (Jungermann and Katz, 1989).
Claudin-2 is a tight junction protein important for the regulation of bile composition and flow (Ma, 2020), and deletion of Claudin-2 in mice results in decreased bilary flow and impaired generation of the osmotic water gradient in bile ducts (Matsumoto et al., 2014). While it is predominantly located in PV hepatocytes, its expression pattern can be altered by inhibiting Wnt exocytosis by deleting Wls, the gene encoding the Wnt receptor GPR177, in murine endothelial cells (Ma, 2020). This results in decreased Claudin-2 expression in adult PV hepatocytes (Ma, 2020). However, more recent studies suggest Wnt signaling may also play a role in liver zonation of newborn mice (Boj et al., 2012). Microarray analysis of hepatocytes isolated from whole-body TCF7L2 knockout mice revealed differential expression of liver zonation genes (Boj et al., 2012). In particular, Glu1 (Glutamine synthase) and Rnase4 (Ribonuclease 4) were found to be upregulated at the neonatal stage, with no differences during the embryonic phase (Boj et al., 2012). These studies highlight the importance of the Wnt signaling pathway in postnatal liver development and zonation, as well as demonstrating cellular crosstalk between hepatocytes and endothelial cells to regulate Wnt activity in adult hepatocytes.
Myogenesis is the development of muscle tissue, originating in precursor satellite cells expressing PAX3/7. Satellite cells undergo commitment and differentiation into myoblasts, followed by differentiation to myotubes. The fusion of myotubes leads to the formation of specific muscle fibers (Buckingham et al., 2003; Ganassi et al., 2018; Ultimo et al., 2018). Each step of myogenesis requires the input of multiple signaling pathways that control the expression of myogenic regulatory factors, such as Myogenin, MyoD and Myf5 (Zammit, 2017), and the Wnt signaling pathway influenced the expression of these myogenic factors (Ridgeway et al., 2000; Vertino et al., 2005). Whole-body deletion of Wnt10b in mice is associated with increased myogenic potential, as defined by higher levels of MyoD and Myogenin, and this is partially mediated by an upregulation of Wnt7b (Vertino et al., 2005).
Xenopus myogenesis requires enhanced Wnt signaling activity, as dominant-negative Wnt-8 supresses induction of MyoD in embryos. Gain of function experiments in the dorsal marginal zone of the gastrula showed ectopic expression of Wnt8 is sufficient to induce MyoD expression (Hoppler et al., 1996). GSK3-β mRNA injections in the lateral region of embryos diminished expression of MyoD and Myf5, independent of their expression in the dorsal mesoderm (Shi et al., 2002). These studies suggest Wnt has local, specific effects on myogenesis during embryogenesis.
Formation of neuromuscular junctions, which are responsible for the contraction of skeletal muscle, are also dependent on Wnt ligands. Acetylcholine receptors (AChRs) are highly enriched within neuromuscular junctions (Cisternas et al., 2019), and over-expression of Wnt3 in chick wing muscles increased AChR aggregates by up to 70% (Henriquez et al., 2008). In contrast, inhibition of the Wnt signaling pathway due to over-expression of Sfrp1 decreased the number of AChR aggregates and reduced the formation of neuromuscular junctions (Henriquez et al., 2008). The actions of Wnt3 were mediated by a non-canonical Wnt signaling mechanism that required the actions of the Rho GTPase, Rac (Henriquez et al., 2008). Collectively, these findings indicate Wnt ligands are required for skeletal muscle formation and function.
β-Catenin is a member of the Armadillo protein family and has been shown to have pleiotropic functions, including cell adhesion to gene transcription. One of the earliest, identified functions of β-catenin is its involvement in cell-cell adhesion via its association with E-cadherin to form adherens junctions (Valenta et al., 2012). β-catenin has also been reported to serve as a transcriptional co-activator of the Wnt signaling pathway (Sineva and Pospelov, 2014). In early embryonic development, β-catenin is involved in establishing the body axis and is critical for tissue and organ development, including but not limited to enamel, lung, kidney, and cartilage (Eberhart and Argani, 2001; Grigoryan et al., 2008; Prakash and Swaminathan, 2015). In the postnatal period, β-catenin contributes to cell renewal, as it has been shown to mediate the regeneration of hair follicles and retinal cells (Grigoryan et al., 2008; Steinhart and Angers, 2018).
β-catenin is characterized by the presence of 12 Armadillo repeats, which help to form a rigid center with flexibility at the C- and N- termini (Huber et al., 1997). Armadillo repeats generally share 30% homology, and this provides β-catenin with the ability to have a wide range of binding partners, including proteins associated with its destruction complex, inhibitors of the Wnt pathway, and transcription factors, such as BCL9, CBP, and Pontin-52 (Peifer et al., 1994; Xu and Kimelman, 2007). Depending on its role in the cell, β-Catenin can undergo conformational changes or dimerize. For instance, dimerization with α-catenin occurs in the context of cell-cell adhesion, but with gene transcription, a monomeric, backfolded conformation is required (Pokutta and Weis, 2000; Gottardi and Gumbiner, 2004). The C-terminal domain has been found to be integral for signaling activities, whereas its Helix-C motif is required for adhesion (Xing et al., 2008). In fact, most of β-catenin’s binding partners, such as 14-3-3ζ, require a Helix-C motif recognition sequence (Fang et al., 2007; Mosimann et al., 2009). Lastly, the motif recognized by GSK3-β, which marks β-catenin for degradation, is located on the N-terminal domain (Wu et al., 2003). Mutations in this region of the N-terminal domain leads to a constitutively active form β-catenin, which is found in multiple cancers (Buendia, 2002; Nejak-Bowen and Monga, 2011; Lade et al., 2012).
Proteins related to β-catenin include plakoglobin (γ-catenin), δ-catenin, and α-catenin, and like β-catenin, they associate with E-cadherin (Zhao et al., 2011). Among the related proteins, only plakoglobin exhibits a high degree of homology to β-Catenin, and it also contains 12 Armadillo repeats. Evolutionary analyses have revealed that plakoglobin is derived from a gene duplication of β-Catenin in vertebrates (Zhao et al., 2011), and despite the high degree of homology, plakoglobin, unlike β-catenin, is primarily localized to desmosomes (Lewis et al., 1997). Instead of armadillo repeats, α-catenin contains 3 vinculin domains, which has led to its classification as its own catenin subfamily (Zhao et al., 2011). Although δ-catenin has ten Armadillo repeats, it belongs to its own subfamily of catenins, named the p120 family. δ-catenin’s primary binding partner is the NF-kB transcription factor (Perez-Moreno et al., 2006).
Deletion of β-catenin in PDX1-positive cells in the pancreas revealed that β-catenin is necessary for acinar development, but not for the development of endocrine cells. Knockout mice exhibited early lethality and were smaller in size than wildtype mice, and they developed pancreatic hypoplasia (Dessimoz et al., 2005; Murtaugh et al., 2005; Wells et al., 2007). Histological analysis of pancreata from β-catenin-deficient, PDX1-positive cells resembled hepatocytes, suggesting that trans-differentiation had occurred (Wells et al., 2007). Conversely, transgenic mice over-expressing a degradation-resistant form of β-Catenin displayed a hypoplastic pancreas that lacked endocrine and exocrine tissue when compared to wild-type controls (Heiser et al., 2006). Immunohistological analysis of pancreata revealed that these mice displayed significant reductions in PDX1-positive pancreatic progenitor cells and increased expression of the hedgehog signaling pathway receptor, PTCH1, which is known to regulate postnatal pancreatic β-cell mass (Heiser et al., 2006; Nakayama et al., 2008). Thus, modulating intracellular levels of β-catenin in the pancreas during embryogenesis can having lasting effects on the formation of a functioning pancreas.
β-catenin’s role in myogenic differentiation is not well-defined, as different groups have reported different effects of over-expression or depletion on the initiation of myogenesis (Cossu et al., 1996; Tajbakhsh et al., 1998; Goichberg et al., 2001; Mermelstein et al., 2007; Brack et al., 2008). Retroviral over-expression of β-catenin in C2 and L8 cells were sufficient to inhibit myogenesis by reducing the expression of myogenin (Goichberg et al., 2001), and this has been proposed to be mediated by adherens junctions, as myogenin levels were rescued through the co-transfection of N-cadherin (Goichberg et al., 2001). This observation was recently supported by the finding that dimerization of β-catenin with α-catenin at adherens junctions is necessary for myogenesis (Cui et al., 2019). Moreover, RNA sequencing of control and β-catenin-null myocytes treated with Wnt3a revealed an upregulation of myogenic genes in control cells but not in knockout cells (Cui et al., 2019). It should be noted that expression of a β-catenin mutant unable to bind TCF7L2 did not impair myogenesis, suggesting that β-catenin exerts its myogenic effects via a TCF7L2-independent manner (Cui et al., 2019). In contrast, depletion of β-catenin in C2C12 cells inhibited myotube differentiation (Kim et al., 2008), and β-catenin depletion in 10T1/2 fibroblasts was also found to reduce the expression of MHC, a marker of muscle cell differentiation, indicating an impairment in myogenesis (Kim et al., 2008). Critical to β-catenin’s role in myogenesis was the finding that interactions of β-Catenin to MyoD via its C-terminal domain was necessary for the transcriptional activity of MyoD (Kim et al., 2008).
Human CD56+ muscle cell progenitors express low levels of nuclear β-catenin prior to the induction of myogenesis, followed by a marked increase post-differentiation (Agley et al., 2017). Pharmacological inhibition of GSK-3β with BIO, CHIR, or LiCl was sufficient to inhibit differentiation, demonstrating inhibitory effects of active β-catenin myogenesis (Agley et al., 2017). Over-expression of a dominant-negative TCF7L2 in CD56+ cells also reduced intracellular levels of β-catenin and impaired the onset of myogenesis, which suggests that TCF7L2 is required for β-catenin-mediated myogenesis (Agley et al., 2017).
In addition to its roles as a tumor suppressor and a component of the β-catenin destruction complex, APC is also responsible for regulating chromosome formation, DNA replication, cell cycle progression, and apoptosis (Groden et al., 1995; Morin et al., 1996; Green et al., 2005). Nonsense mutations in the APC gene, which are generally associated with the generation of truncated forms of APC, contribute to the development of colon cancer (Powell et al., 1992; Laken et al., 1999; Fearnhead et al., 2001; Hankey et al., 2018).
Liver-specific deletion of APC produces an inverted expression of β-catenin along the proto-central axis and is associated with hepatomegaly and increased mortality in mice (Benhamouche et al., 2006). Knockout mice were also found to have increased circulating ammonia and elevated glutamine in the brain, which is suggestive of hepatic encephalopathy (Benhamouche et al., 2006).
In contrast to β-catenin, which is responsible for the development of the exocrine pancreas, APC appears to be important for both endocrine and exocrine cells (Sharon et al., 2019). Deletion of APC in cells expressing NEUROG3, an endocrine marker in the pancreas, impaired pancreatic endocrine cell development, as insulin- and glucagon-containing β-cells and α-cells, respectively, failed to develop (Sharon et al., 2019). In contrast, ablation of APC in exocrine cells led to an enlargement of the pancreas, characterized by acinar cell hyperplasia, and no changes in glucose homeostasis, or insulin and glucagon content were detected (Strom et al., 2007). Overall, APC is able to regulate the development of both exocrine and endocrine cells of the pancreas.
TCF7L2 (also known as TCF4) is an important component of the Wnt signaling pathway, as it binds to β-catenin to mediate the expression of critical Wnt-target genes. Genome-wide association studies have revealed a strong association with SNPs close to the TCF7L2 locus and the risk of developing of type 2 diabetes (Grant et al., 2006; Boj et al., 2012). These SNPs have been associated with impaired insulin secretion and increased hepatic glucose production under periods of fasting (Florez et al., 2006; Saxena et al., 2006; Lyssenko et al., 2007; Boj et al., 2012).
Systemic deletion of TCF7L2 (TCF7L2KO) in mice results in early lethality, as newborns were considerably hypoglycemic 3 h postpartum (Boj et al., 2012). Notably, embryonic development of the endocrine pancreas was not affected, but glycogen storage, triglyceride synthesis, fatty acid oxidation, and ketone body synthesis were diminished in livers of TCF7L2KO mice (Boj et al., 2012). Indeed, Gys2 (Glycogen Synthase 2) mRNA levels were decreased, while mRNA levels of Pck1, G6pc, and Fbp1 were increased, which may account for changes in glycogen synthesis and gluconeogenesis at birth (Boj et al., 2012).
Postnatal deletion of TCF7L2 in β-cells was not associated with any effects on body weight or glucose homeostasis in mice fed normal chow or high-fat diets (Boj et al., 2012). Histological examination of pancreata did not reveal any differences in β-cell mass or β-cell proliferation, suggesting that TCF7L2-dependent Wnt signaling was not required for pancreatic β-cell development (Boj et al., 2012).
Deletion of TCF7L2 in hepatocytes (LTCF7L2KO) resulted in mild fasting hypoglycemia, similar to what was observed with systemic TCF7L2KO mice (Boj et al., 2012). Microarray analysis revealed diminished expression of genes involved in glycolysis, fatty acid metabolism, and in the Wnt signaling pathway in livers of LTCF7L2KO mice (Boj et al., 2012). Under high-fat diet conditions, LTCF7L2KO mice displayed a more severe hypoglycemic phenotype, as hepatocytes from LTCF7L2KO mice demonstrated impaired gluconeogenesis following glucagon or IBMX treatment and reduced mRNA levels of gluconeogenic genes G6pc and Aldh3a2 (Boj et al., 2012). Opposite to the LTCF7L2KO mice, adenoviral-mediated over-expression of TCF7L2 increased fasting glucose concentrations, in addition to enhanced hepatic glucose production following pyruvate injections (Boj et al., 2012). Genes associated with gluconeogenesis, such as Pck1, G6pc, and Fbp1, require functional TCF7L2, as hepatic over-expression of a dominant-negative TCF7L2 mutant under the control of the Alb promoter, resulted in their upregulation (Ip et al., 2015). Moreover, over-expression of the TCF7L2 mutant was associated with a progressive worsening in gluconeogenesis and glucose tolerance, despite no changes in insulin sensitivity (Ip et al., 2015).
In skeletal muscle, TCF7L2 expression is low when compred to other tissues (Osmark et al., 2009); however, TCF7L2 appears to be highly expressed in muscle connective tissue fibroblasts (Mathew et al., 2011). Fibroblast-specific deletion of TCF7L2 in mice was associated with reduced expression of Myh7, a marker of myogenic maturation, in slow-muscle fibers. However, its expression was upregulated in the skeletal muscle of adult mice, with the exception of the soleus muscle, suggesting that TCF7L2 exerts developmental effects on myogenic maturation. Deletion of TCF7L2 in fetal myogenic cells using a Pax7-Cre driver was associated with reduced Myh7 and Myh2 gene expression in hind limb muscles and the diaphragm, supporting the hypothesis that TCF7L2 is important for myogenesis (Mathew et al., 2011).
Since establishing the roles of Wnt signaling and its downstream effectors in the development of metabolic tissues and organs, the individual contributions of effectors to cellular and whole-body metabolism is now gaining wide appreciation. This includes their metabolic contributions in various metabolic organs and tissues, including liver, brain, pancreas, and adipose tissue (Tables 4, 5 and Figure 2).
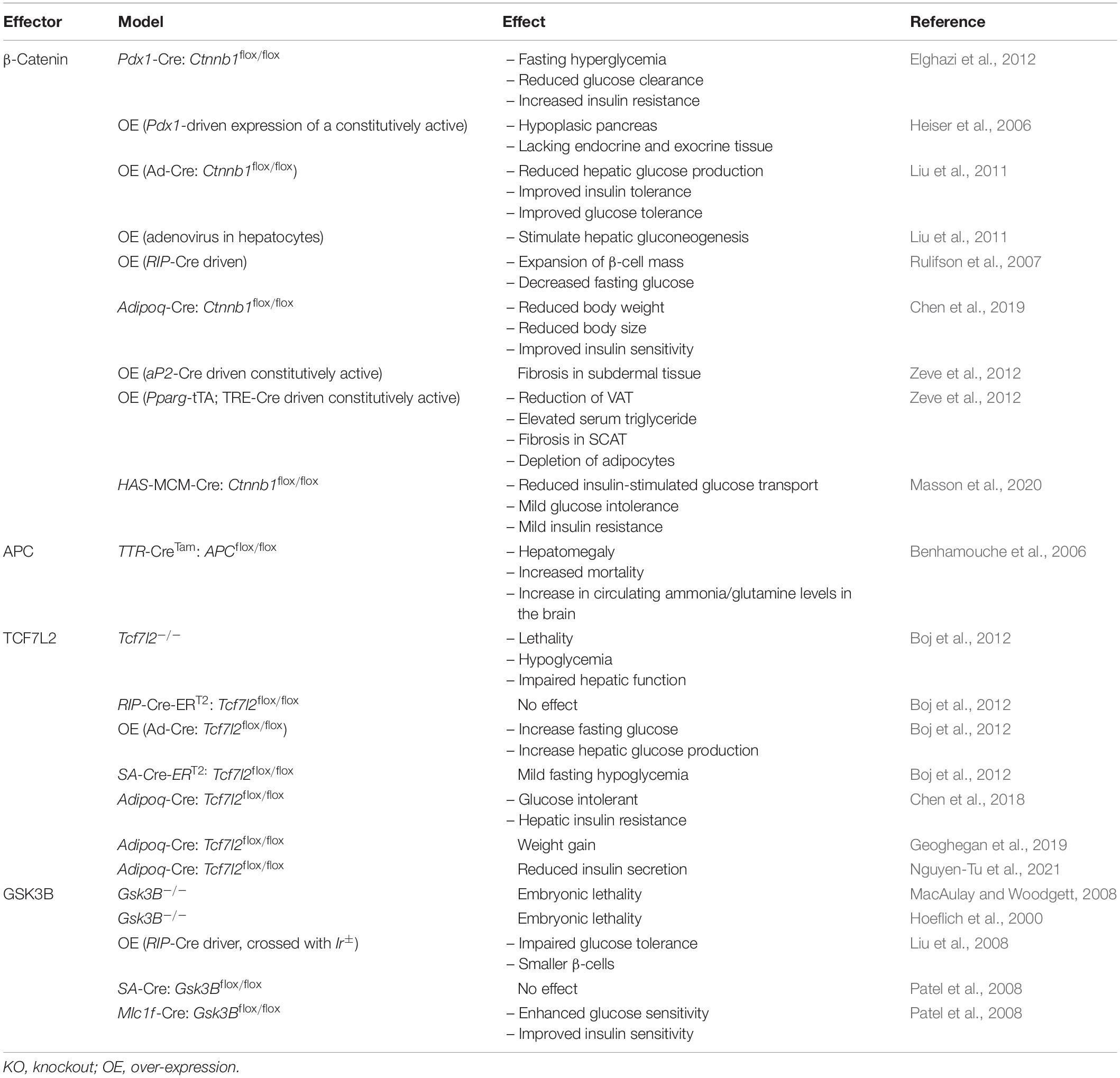
Table 4. Metabolic phenotype resulting from the over-expression or deletion of Wnt signaling effectors in mice.

Table 5. Metabolic phenotype resulting from over-expression or deletion of Wnt signaling effectors in Drosophila.
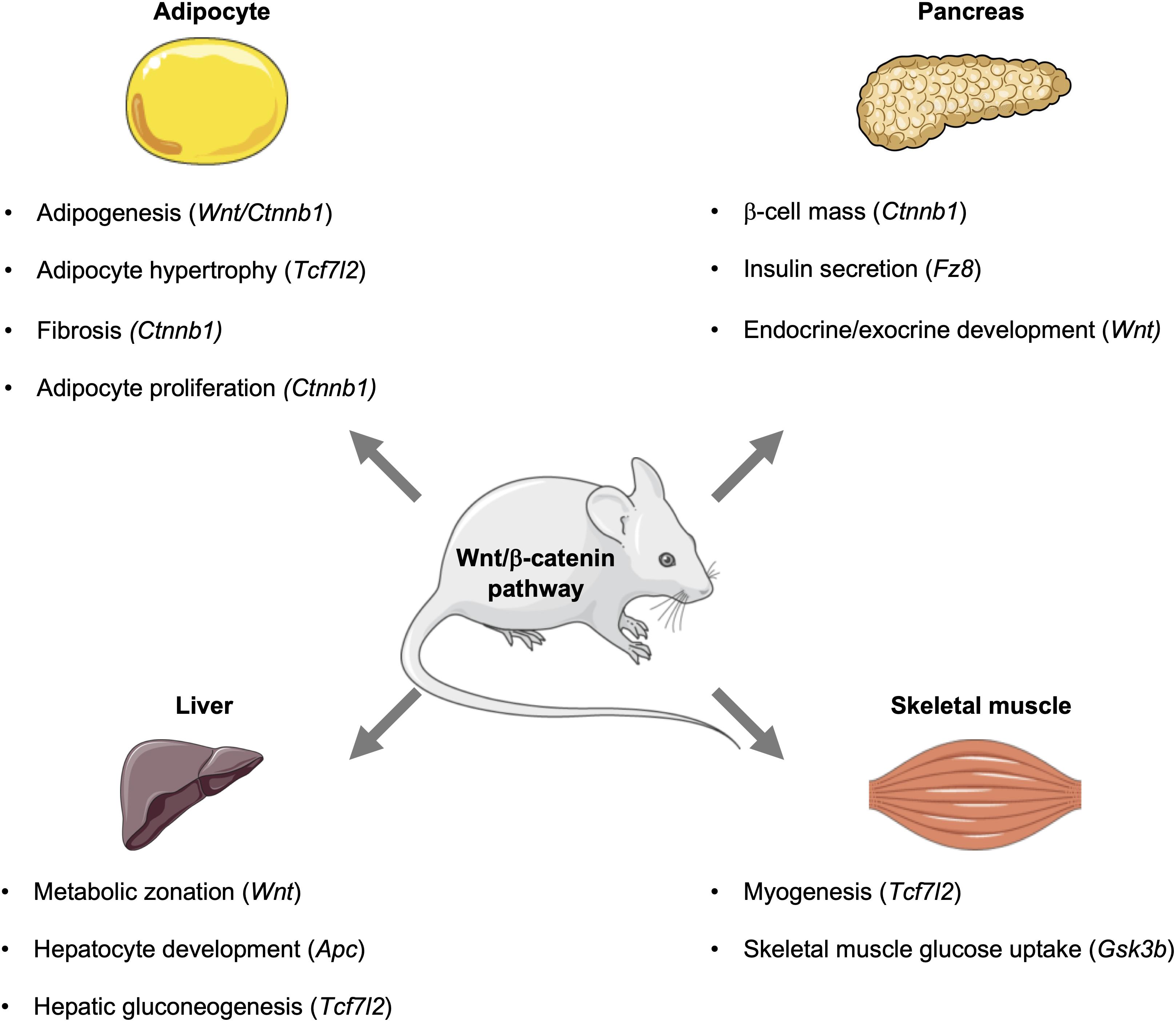
Figure 2. Influence of the Wnt signaling pathway and its downstream effectors in metabolic tissues. In mice, the Wnt signaling pathway influences metabolic events in key tissues, including the pancreas, adipose, liver, and skeletal muscle. Activation of this pathway can influence the development of tissues and organs, as well as signaling events that control glucose homeostasis and metabolism. This figure was made with modified images from Servier Medical Art (Creative Commons Attribution 3.0 Unported License).
Activation of Wnt signaling has been found to increase pancreatic β-cell proliferation and the expression of cell cycle genes via the actions of the transcription factor PITX2 (Table 4; Rulifson et al., 2007). β-cell-specific over-expression of a constituitively active form of β-catenin was found to stimulate β-cell expansion. Conversely, inhibition of Wnt signaling by over-expression of the Wnt signaling inhibitor, AXIN, led to β-cell hypoplasia and dysregulated islet architecture (Rulifson et al., 2007). Increasing or decreasing Wnt signaling was associated with enhanced or defective insulin secretion, respectively, along with changes in glucose tolerance (Rulifson et al., 2007).
Adipogenesis, or adipocyte differentiation, in white adipose tissue is a two-step process by which mesenchymal stem cells commit to preadipocytes, followed by differentiation into mature adipocytes. The differentiation of preadipocytes is governed by the transcription factors PPARγ and CEBP-α, which drive committed preadipocytes toward terminal differentiation (Cristancho and Lazar, 2011). In general, activation of the Wnt-β-catenin pathway in pre-adipocytes inhibits adipogenesis with the exception of the non-canonical Wnt5b-related signaling, which potentiates adipocyte differentiation of 3T3-L1 preadipocytes (Kanazawa et al., 2005; Cristancho and Lazar, 2011). Moreover, some Wnt proteins, such as Wnt6 and Wnt10, can also inhibit the maturation of thermogenic brown adipocytes (Tseng et al., 2005). Wnt proteins have also been reported to increase leptin production, as acute administration of Wnt3a to differentiated 3T3-L1 cells is sufficient to enhance leptin mRNA levels (Chen et al., 2015).
Wingless (wg) has also been implicated in obesity in fruit flies. Abdominal fat body mass was increased following knockdown of wg using a Mef2-Gal4 driver (Table 5; Lee et al., 2014). Mhc-Gal4 and Tin-Gal4 drivers were also used to supress wg in the muscle and heart, respectively, and in both models, whole body triglyceride content was increased (Lee et al., 2014). Overexpression of wg in muscle exhibited the opposite phenotype, with a decrease in total fat accumulation, indicating Wingless can function as a repressor of adipogenesis (Riechmann et al., 1998). Indeed, deletion of wg in embryos showed a complete absence of fat in the ventral section. Conversely, the dorsolateral segment displayed high expression of adipocyte progenitors. Thus, wg appears to be essential for ventral adipogenesis and is required to supress dorsolateral mesodermal fat development. Gonadal agenesis was also reported in the mutant embryos, suggesting all dorsolateral cells were converted to adipocyte progenitors (Riechmann et al., 1998).
Non-canonical Wnt pathways have also been implicated in metabolic diseases. Wnt5A was found to be upregulated in visceral adipose tissue (VAT) of obese individuals compared to subcutaneous adipose tissue (SCAT) (Fuster et al., 2015), and Wnt5a inhibition in mice fed a high-fat diet prevented the development of metabolic dysfunction, with a reduction in insulin and glucose levels (Fuster et al., 2015). However, no changes in body weight or body fat percentage were detected, suggesting no effects on adipogenesis. Interestingly, proinflammatory cytokines such as TNF-α and CCL2/MCP-1 were downregulated, indicating a potential reduction in inflammation. sFRP5, which sequesters and antagonizes Wnt5a was also found to produce anti-inflammatory effects in mice (Ouchi et al., 2010). Deletion of sFRP5 in leptin-deficient Ob/Ob mice on a high-fat diet elevated glucose and triglyceride levels. Additionally, increases in F4/80 cells were detected in epididymal white adipose tissue, indicating increased inflammation (Ouchi et al., 2010).
LRP6 is essential for Wnt binding to Fz in the canonical pathway, but it can also inhibit the non-canonical Wnt signaling pathway (Bryja et al., 2009). LRP6 mutant mice, with nucleotide substitutions at 10443 and 10445, were found to have an enhanced levels of cytokines, MPO, IL-6, and CD68, in the liver (Wang et al., 2015). Histological analysis revealed mutant mouse livers to have a greater accumulation of lipid droplets, indicative of hepatic steatosis. Modest reductions in β-catenin were detected in the liver, with increased expression of RhoA and ROCK2 mRNA, suggesting enhanced activity of the non-canonical pathway. Fibrosis was also noted in the liver of mutant mice following high-fat diet feeding. Notably, recombinant Wnt3a administration was able to rescue the mutant phenotype and reduced inflammation (Wang et al., 2015). These findings demonstrate contrasting and opposing functions of the canonical and non-canonical Wnt pathways within the same tissue.
Gain- and loss-of-function approaches have revealed that β-catenin can influence hepatic metabolism (Liu et al., 2011). Adenovrial-mediated depletion of β-catenin in hepatocytes attenuated hepatic gluconeogenesis, resulting in hypoglycemia. Conversely, over-expression of β-catenin had opposite effects on hepatic glucose production. Levels of mRNA corresponding to the rate-limiting gluconeogenic enzymes Phosphoenolpyruvate carboxykinase (Pck) and Glucose-6-phosphatase (G6p) in vivo, as well as in Hepa1-6 hepatoma cells, were decreased or increased following depletion or over-expression of β-catenin, respectively (Liu et al., 2011). Under high-fat diet feeding conditions, depletion of β-catenin in hepatocytes was associated with improved glucose tolerance and reduced hepatic gluconeogenesis (Liu et al., 2011).
In pancreatic endocrine cells, β-catenin is essential for β-cell function and survival. Targeted deletion of β-catenin in murine β-cells during embryogenesis resulted in severe hypoglycemia and premature death. Moreover, β-cells from knockout mice displayed impaired insulin release (Dabernat et al., 2009). In various in vitro β-cell models, acute depletion of β-catenin has also been shown to impair glucose-stimulated insulin secretion (GSIS) due to negative effects on re-modeling of the actin cytoskeleton (Sorrenson et al., 2016). Deletion of β-catenin in the exocrine pancreas resulted in mice that were smaller in size and weight when compared to controls, in addition to impaired development of exocrine tissues (Elghazi et al., 2012). Deletion of β-catenin in exocrine tissue was also associated with fasting hyperglycemia, in addition to lower rates of glucose clearance and insulin sensitivity (Elghazi et al., 2012). Depletion of β-catenin also led to impaired pancreatic function and growth, as knockout mice displayed reduced β-cell proliferation and β-cell mass (Wells et al., 2007; Elghazi et al., 2012).
Recently, β-catenin levels have been found to be elevated in subcutaneous white adipose tissue from individuals with obesity (Chen et al., 2019). Similarly in mice, over-feeding was sufficient to increase β-catenin expression in murine adipose tissue, suggesting pro-adipogenic or -obesogenic roles in adipose tissues (Chen et al., 2019). Indeed deletion of β-catenin in adipocytes was found to be protective against high-fat diet-induced weight gain and improved insulin sensitivity. The attenuation of fat mass expansion was associated with significant reductions in proliferating PDGFRA-positive pre-adipocytes (Chen et al., 2019).
Over-expression of β-catenin in mature adipocytes under the control of the aP2 promoter was associated with fibrosis in subdermal tissues, with no other significant changes under normal chow diet conditions (Zeve et al., 2012). In contrast, β-catenin over-expression in adipocyte progenitor cells led to significant reductions in visceral white adipose tissue and circulating levels of leptin and adiponectin. Interestingly, over-expression of β-catenin in adipocyte progenitor cells was not associated with glucose intolerance or insulin resistance, as would be expected from a lipodystrophic phenotype (Zeve et al., 2012).
Inhibitors of the Wnt/β-catenin pathway have been shown to promote adipogenesis. For example, the β-catenin antagonist Chibby, which removes unphosphorylated β-catenin from the nucleus (Killoran et al., 2015), is necessary for adipogenesis. Depletion or ectopic over-expression of Chibby can inhibit or promote spontaneous adipogenesis, respectively (Li et al., 2007). Similarly, the Wnt antagonist, Dickkopf1 (DKK1), has similar effects in promoting adipogenesis. DKK1 inhibits the Wnt pathway by binding the receptors LRP5/6, and in vitro assays using human pre-adipocytes have shown that DKK1 expression increases during the early stages of adipogenesis (Christodoulides et al., 2006). In conditions associated with sub-optimial differentiation of 3T3-L1 adipocytes, ectopic expression of human DKK1 could restore differentiation to the same extent as complete adipogenic conditions due to inhibition of Wnt signaling (Christodoulides et al., 2006).
Skeletal muscle is responsible for 70–90% of glucose uptake from the circulation, and this process is impaired in individuals with type 2 diabetes (Evans et al., 2019). Although the Wnt signaling pathway is known to be involved in myogenesis, it has also been found to participate in skeletal muscle glucose metabolism. Deletion of β-catenin in skeletal muscle was found to diminish insulin-stimulated glucose transport in mice, and this was also associated with mild glucose intolerance and insulin resistance (Masson et al., 2020). In vitro studies with isolated muscle and myocytes demonstrate that knockdown of β-catenin can also reduce GLUT4 protein abundance upon insulin stimulation, demonstrating impaired insulin action (Masson et al., 2020).
Removal of exon 11 of Axin (Axn) in Drosophila and its replacement with a repetitive heterochromatin sequence resulted in a hyperactive Wnt signaling response, as Armadillo (B -catenin’s Drosophila ortholog) expression increased (Zhang et al., 2017). Homozygous Axn mutants displayed decreased accumulation of abdominal adipocytes at mid to late larval stages, along with lower levels of diacylglycerol, free fatty acid, and triglycerides. Furthermore, RNAi-mediated knock down of Armadillo successfully rescued the phenotype (Zhang et al., 2017).
The axin ortholog in C.elegans, PRY-1, was recently shown to be involved in lipid metabolism (Ranawade et al., 2018). Worms with a nonsense pry-1 mutation had enhanced Wnt activity, demonstrating that β-catenin activity on lipid metabolism is conserved across species. Mutants exhibited a reduction in lipid content, egg laying and survival following starvation. Genetic studies revealed a downregulation in vits and fats, the genes responsible for yolk lipoproteins and fatty acid desaturases (Ranawade et al., 2018).
Ck1-α emerged as a candidate gene with high association to hyperglycemia in Drosophila (Ugrankar et al., 2015). RNAi-mediated knockdowns in adipose tissue or muscle of third instar larvae led to an increase in haemolymph glucose levels. Most notably, CSNK1a1 knockouts in murine pre-adipocytes recapitulated the hyperglycemic phenotype, indicating a species-conserved function of CK1-α (Ugrankar et al., 2015).
GSK-3 is an important regulator of glycogen synthesis, as it is responsible for inhibiting glycogen synthase to attenuate glycogenolysis and glucose production (MacAulay and Woodgett, 2008). GSK-3 has two paralogs, GSK-3α and GSK-3β, and while systemic knockouts of GSK-3β are embryonically lethal due to enhanced TNF-α-associated hepatocyte apoptosis, systemic GSK-3α knockouts were found to be viable (Hoeflich et al., 2000; MacAulay and Woodgett, 2008). In addition, improved glucose tolerance, as well as higher insulin sensitivity, were observed, which may be attributed to increased mRNA levels of Glycogen Synthase and Insulin Receptor Substrate-1 (MacAulay and Woodgett, 2008).
Deletion of the insulin receptor (IR) in mice is known to cause perinatal lethality due to diabetic ketoacidosis, and mice heterozygous for Ir display mild insulin resistance and compensatory increases in β-cell mass. Partial deletion of the insulin receptor is also associated with significant hyperglycemia and hyperinsulinemia, despite exhibiting no changes in insulin signaling (Bruning et al., 1997). GSK3β is involved in the insulin signaling pathway, as it inactivates glycogen synthase by phosphorylating serine residues on its C-terminal domain. GSK3β can also inhibit the insulin pathway by phosphorylating the insulin receptor substrate-1 (IRS-1) directly (Liberman and Eldar-Finkelman, 2005). Mice heterozygous for Gsk3b (Gsk-3β±) exhibited decreased fed and fasting insulin levels, and when bred with mice heterozygous for Ir, haploinsufficiency of GSK3-β was able to inprove the metabolic phenotype of heterozygous Ir mice (Tanabe et al., 2008). Previous studies have shown that whole body-IRS2 knockout mice resemble insulin receptor knockout mice, as they displayed insulin resistance, hyperglycemia, reduced β-cell mass, and decreased body weight (Uchida et al., 2005). When compared to systemic IRS2 knockout mice, GSK-3β haploinsufficient mice on an Irs2-null background did not exhibit loss of β-cell mass due to increased β-cell proliferation and reduced rates of β-cell apoptosis (Uchida et al., 2005). Expansion of β-cell mass was regulated by reductions in the Cyclin-dependent kinase inhibitor, p27Kip1, a rate limiting factor in cell proliferation (Uchida et al., 2005; Rachdi et al., 2006).
Deletion of GSK-3β in β-cells is sufficient to partially rescue the diabetic phenotype of IRS2 knockout mice, as mice were normoglycemic despite being hyperinsulinemic (Tanabe et al., 2008). This was attributed to the deletion of GSK-3β resulting in reducing the incidence of apoptosis and promoting β-cell proliferation (Tanabe et al., 2008). Over-expression of a constitutively active GSK3-β (RIP-GSK3βCA) mutant in β-cells led to impaired glucose tolerance, which was only observed in male mice. Histological analysis of pancreata revealed that transgenic mice had decreased β-cell area, as well as impaired β-cell proliferation. Moreover, over-expression of a constituitively active GSK3-β mutant led to defects in insulin secretion and resulted in impaired glucose tolerance (Liu et al., 2008). In conjunction, these studies demonstrate the importance of GSK-3β in regulating β-cell mass and proliferation.
In liver-specific GSK-3β knockout mice, no significant metabolic changes were observed, thereby suggesting that GSK-3β in the liver is dispensable for whole-body metabolism (Patel et al., 2008). Skeletal muscle-specific knockout of GSK-3β mediated by Mlc1f-promoter driven Cre-mediated recombination results in mice with enhanced glucose tolerance. Morover, these mice had increased glycogen synthase, resulting in improved insulin signaling and actions, and this highlights GSK-3β’s importance in regulating insulin action in a tissue specific manner (Patel et al., 2008).
In the context of obesity, GSK-3β inhibition has been found to have beneficial metabolic effects. Administration of the GSK3 inhibitors SB216763 or CHIR99021 to high-fat diet-induced obese mice improved insulin sensitivity without any changes in fat mass or body weight (Wang et al., 2018). Furthermore, GSK-3 inhibition was found to influence macrophage polarization such that increased numbers of anti-inflammatory M2 macrophages could be detected (Wang et al., 2018). Additionally, in vitro studies performed on human adipose- derived stem cells showed that GSK3 inhibitors, such as LiCl and BIO, have the ability to inhibit cell proliferation, and BIO-treated human adipose-derived stem cells were unable to differentiate to adipocytes in the presence of adipogenic stimuli (Zaragosi et al., 2008).
Glucagon-like peptide-1 (GLP-1), which is derived from intestinal endocrine L cells, potentiates glucose-stimulated insulin secretion through actions on the GLP-1R on the surface of β-cells (Lim and Brubaker, 2006). In L cells, TCF7L2 participates in GLP-1 synthesis by regulating the transcription of Gcg, thereby representing an alternative mechanism whereby the Wnt signaling pathway can influence insulin secretion from β-cells (Shao et al., 2013). Indeed, targeted transgenic expression of a dominant-negative TCF7L2 mutant in neurons and enteroendocrine cells led to significant reductions in Gcg-positive neurons and GLP-1-positive cells, respectively (Shao et al., 2013, 2015). Although these mutant mice were found to be glucose intolerant and insulin resistant, compensatory increases in β-cell mass were detected (Shao et al., 2013). In adult mice, GLP-1 was found to induce the phosphorylation of β-catenin by cAMP/PKA or insulin/PAK-1 signaling pathways to facilitate β-cell proliferation (Xiong et al., 2012; Shao et al., 2013).
To examine TCF7L2’s effects on type 2 diabetes in zebrafish, heterozygous knockouts were generated by introducing a mutation in intron 1, which produced a truncated protein (Facchinello et al., 2017). Adult fish displayed postprandial hyperglycemia. Reduction of the overall size of the fish was observed, as well as reductions in the sizes of the endocrine and exocrine pancreas. Interestingly, the mutant exocrine pancreas was also found to have an accumulation of adipose tissue, along with a decrease in number of β-cells, suggesting a mechanism by which deficiency in TCF7L2 may lead to a diabetic phenotype protein (Facchinello et al., 2017).
TCF7L2 has been demonstrated to have important roles in adipogenesis, but it is not without controversy (Figure 3). One study suggests its expression increases during adipogenesis, which may seem contradictory as activated Wnt signaling is known to inhibit adipocyte differentiation (Cristancho and Lazar, 2011; Chen et al., 2018). Indeed, knockdown of TCF7L2 via short-hairpin RNA in 3T3-L1 cells inhibited adipogenesis and downregulated Slc2a4 (GLUT4) mRNA. Adipocyte-specific TCF7L2 knockout mice were glucose intolerant and showed signs of hepatic insulin resistance that was associated with increased gluconeogenesis. Under high-fat diet conditions, knockout mice gained more weight and showed increased hypertrophy of inguinal white adipose tissue, and gene expression analysis of inguinal white adipose tissue displayed an increase in the Wnt target gene, Axin2, suggesting that the reduction of TCF7L2 activated the Wnt signaling pathway to initiate adipocyte hypertrophy.
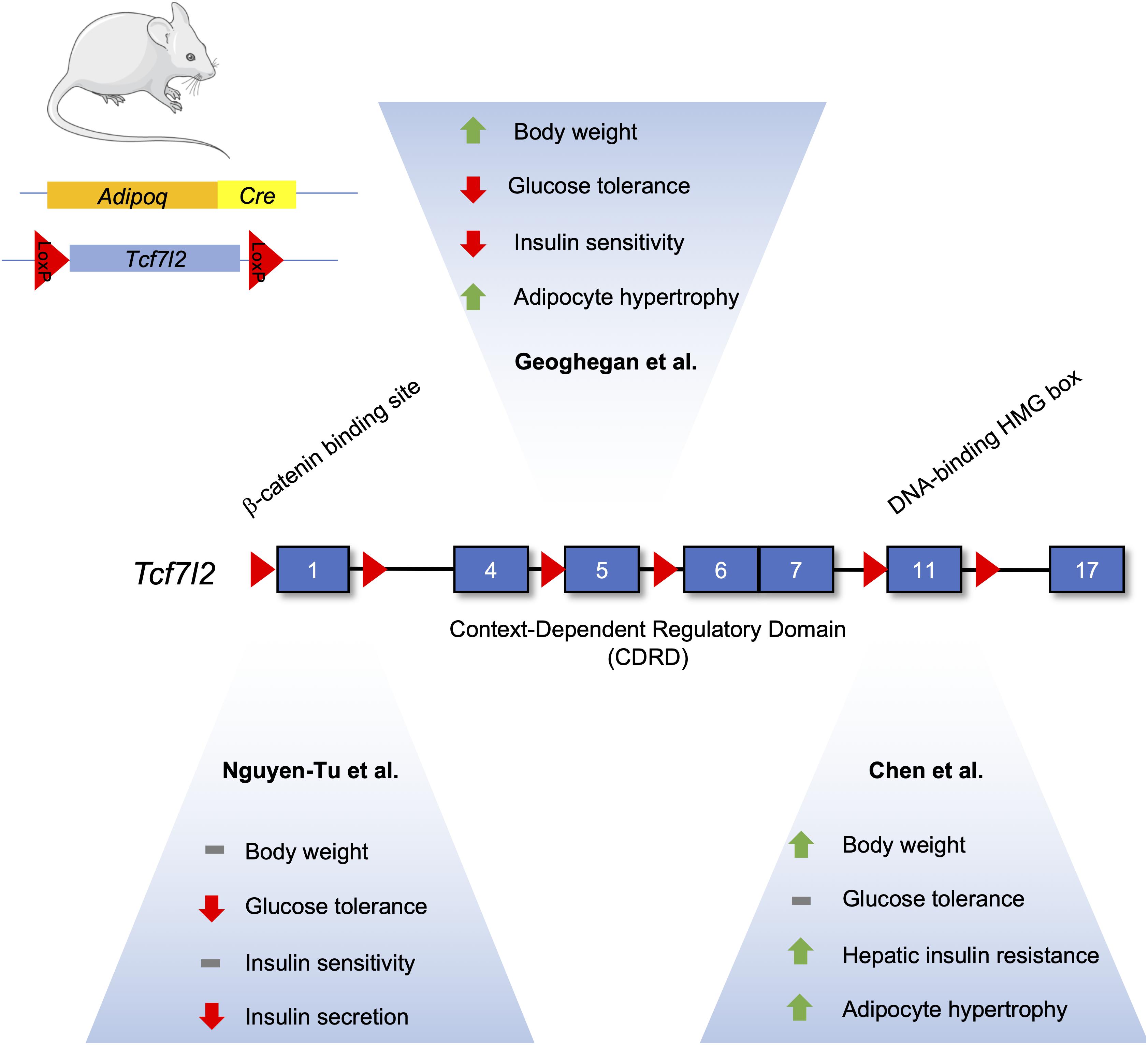
Figure 3. Differences in the metabolic phenotype of adipocyte-specific TCF7L2 knockout mouse models. The Tcf7l2 gene contains 17 exons, as denoted by numerated blue rectangles. Recombination of floxed alleles for Exon 1 (Nguyen-Tu et al., 2021), Exon 5 (Geoghegan et al., 2019), and Exon 11 (Chen et al) by Cre recombinase under the control of the Adipoq promoter results in different metabolic phenotypes. This figure was made with modified images from Servier Medical Art (Creative Commons Attribution 3.0 Unported License).
In contrast, Geoghegan et al. (2019) have reported that TCF7L2 expression is reduced during adipogenesis in both in vitro and in vivo models. Mice fed a high fat diet exhibited an increased abundance of TCF7L2 and adiponectin expression in epididymal white adipose tissue, and this increase in TCF7L2 abundance could be recapitulated in adipose tissue from ob/ob and db/db mice (Geoghegan et al., 2019). Adipocyte-specific deletion of TCF7L2 led to weight gain when compared to control mice fed a normal chow diet. On a high-fat diet, knockout mice displayed significantly higher weight gain, in addition to impaired glucose tolerance and insulin resistance. Histological analysis revealed hypertrophy in inguinal WAT depots and lipolytic genes, such as Tgh and Tgh2, were downregulated. Adipocyte-specific knockout of TCF7L2 was associated with increased lipid storage, due in part to changes in Tgh1 (triacylglycerol hydrolase) and Tgh2 gene expression, and adipocyte hypertrophy (Geoghegan et al., 2019).
Further studies done by Nguyen-Tu et al. (2021) on TCF7L2 knockout mice using an Adipoq-Cre driver recapitulated the hyperglycemic phenotype seen by Chen et al. (2018). However, this phenotype was only seen in 16-week old males, and they were not insulin resistant. Insulin secretion was abrogated in isolated islet ex vivo, possibly due to the decreased islet expression of Glut2 mRNA. No differences in insulin secretion were seen in vivo under normal chow-fed conditions, possibly due to compensatory increases in circulating levels of NEFAs that can potentiate glucose-stimulated insulin secretion. Furthermore, under high-fat diet, no weight changes were seen but TCFL2 knockout diminished insulin secretion (Nguyen-Tu et al., 2021).
All three studies examining TCF7L2’s effects on adipocyte function and adipogenesis were performed on male mice using different Adipocyte-specific Cre driver mice. However, the exons targeted for recombination were also different for all three studies. Chen et al., floxed exon 11, known to encode the DNA binding HMG box (Hansson et al., 2010). On the other hand, Geoghegan et al. (2019) used mice with lox-p sites flanking exon 5, while Nguyen-Tu et al. (2021) used exon 1 floxed mice. Both found TCF7L2 to have an inhibitory role on adipogenesis. Taken together, the common and differing phenotypes associated with TCF7L2 deletion demonstrate the complexity of the biological actions of the Wnt signalng pathway in adipocytes.
Although the Wnt signaling pathway is conserved across species and is necessary for embryogenesis and development, it is now clear that it also has essential metabolic roles in mammals. Moreover, aberrant expression or activity of various downstream effectors in the Wnt signaling pathway is associated with the development of chronic diseases, including diabetes and obesity. Given the pathway’s wide ranging effects and multiple downstream targets, more research is required to further our understanding of its involvement across different metabolic diseases.
FAA wrote the manuscript. GEL edited the manuscript and the guarantor of this work. Both authors contributed to the article and approved the submitted version.
This work was supported by CIHR Project (PJT-153144) and a Discovery Award from the Banting Research Foundation. GEL holds the Canada Research Chair in Adipocyte Development.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to apologize the authors whose work was not cited in this review due to space limitations. The authors would also like to thank Les Laboratoires Servier for the helpful images from the Servier Medical Art collection (Creative Commons Attribution 3.0 Unported License).
APC, adenomatous polyposis coli; GSK, glycogen synthase kinase; TCF/LEF, transcription like factor lymphocyte enhancer factor; LRP, lipoprotein receptor-related protein; CK, casein kinase; Mtgr1, myeloid translocation gene related-1; PP, periportal hepatocytes; PV, perivenous hepatocytes; MyoD, myoblast determination protein; Myf, myogenic factor; NMJ, neuromuscular junction; AChR, acetylcholine receptor; BCL9, B-cell CLL/lymphoma protein 9; CBP, CREB-binding protein; NF-kB, nuclear factor kB; PTCH1, protein patched homolog 1; MHC, myosin heavy chain; LTCF7L2KO, liver specific TCF7L2 knockout; IBMX, isobutylmethylxanthine; PITX2, paired like homeodomain 2; MIN6, mouse INsulinoma 6 cells; CDK4, cyclin dependent kinase 4; PPAR, peroxisome proliferater activated receptor; CEBP, CCAT enhancer binding proteins; Brdu, bromodeoxyuridine; PDGFRa, platelet derived growth factor; DDR2, discoidin domain receptor; FSP-1, fibroblast specific protein 1; DKK1, Dickopf; TNF, tumor necrosis factor; IRS, Insulin receptor substrate; LiCl, lithium chloride; BIO, 6-bromoindirubin; GLP, glucagon like peptide; PKA, protein kinase A; cAMP, cyclic adenosine monophosphate; WAT, white adipose tissue; NEFA, non-esterified fatty acid; FABP, Fatty acid binding protein; HMG, high mobility group; DIO, diet induced obesity.
Adell, T., Salo, E., Boutros, M., and Bartscherer, K. (2009). Smed-Evi/Wntless is required for beta-catenin-dependent and -independent processes during planarian regeneration. Development 136, 905–910. doi: 10.1242/dev.033761
Agley, C. C., Lewis, F. C., Jaka, O., Lazarus, N. R., Velloso, C., Francis-West, P., et al. (2017). Active GSK3beta and an intact beta-catenin TCF complex are essential for the differentiation of human myogenic progenitor cells. Sci. Rep. 7:13189. doi: 10.1038/s41598-017-10731-1
Almuedo-Castillo, M., Salo, E., and Adell, T. (2011). Dishevelled is essential for neural connectivity and planar cell polarity in planarians. Proc. Natl. Acad. Sci. U.S.A. 108, 2813–2818. doi: 10.1073/pnas.1012090108
Baker, J. C., Beddington, R. S., and Harland, R. M. (1999). Wnt signaling in Xenopus embryos inhibits bmp4 expression and activates neural development. Genes Dev. 13, 3149–3159. doi: 10.1101/gad.13.23.3149
Beckett, K., Monier, S., Palmer, L., Alexandre, C., Green, H., Bonneil, E., et al. (2013). Drosophila S2 cells secrete wingless on exosome-like vesicles but the wingless gradient forms independently of exosomes. Traffic 14, 82–96. doi: 10.1111/tra.12016
Benhamouche, S., Decaens, T., Godard, C., Chambrey, R., Rickman, D. S., Moinard, C., et al. (2006). Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev. Cell 10, 759–770. doi: 10.1016/j.devcel.2006.03.015
Bischoff, M., and Schnabel, R. (2006). A posterior centre establishes and maintains polarity of the Caenorhabditis elegans embryo by a Wnt-dependent relay mechanism. PLoS Biol. 4:e396. doi: 10.1371/journal.pbio.0040396
Boj, S. F., van Es, J. H., Huch, M., Li, V. S., Jose, A., Hatzis, P., et al. (2012). Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 151, 1595–1607. doi: 10.1016/j.cell.2012.10.053
Brack, A. S., Conboy, I. M., Conboy, M. J., Shen, J., and Rando, T. A. (2008). A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell 2, 50–59. doi: 10.1016/j.stem.2007.10.006
Braeuning, A., Ittrich, C., Kohle, C., Hailfinger, S., Bonin, M., Buchmann, A., et al. (2006). Differential gene expression in periportal and perivenous mouse hepatocytes. FEBS J. 273, 5051–5061. doi: 10.1111/j.1742-4658.2006.05503.x
Bruning, J. C., Winnay, J., Bonner-Weir, S., Taylor, S. I., Accili, D., and Kahn, C. R. (1997). Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell 88, 561–572. doi: 10.1016/s0092-8674(00)81896-6
Bryja, V., Andersson, E. R., Schambony, A., Esner, M., Bryjova, L., Biris, K. K., et al. (2009). The extracellular domain of Lrp5/6 inhibits noncanonical Wnt signaling in vivo. Mol. Biol. Cell 20, 924–936. doi: 10.1091/mbc.E08-07-0711
Buckingham, M., Bajard, L., Chang, T., Daubas, P., Hadchouel, J., Meilhac, S., et al. (2003). The formation of skeletal muscle: from somite to limb. J. Anat. 202, 59–68. doi: 10.1046/j.1469-7580.2003.00139.x
Buendia, M. A. (2002). Genetic alterations in hepatoblastoma and hepatocellular carcinoma: common and distinctive aspects. Med. Pediatr. Oncol. 39, 530–535. doi: 10.1002/mpo.10180
Cavallo, R. A., Cox, R. T., Moline, M. M., Roose, J., Polevoy, G. A., Clevers, H., et al. (1998). Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature 395, 604–608. doi: 10.1038/26982
Chen, M., Lu, P., Qinyun, M., and Yanan, C. (2019). CTNNB1/β-catenin dysfunction contributes to adiposity by regulating the cross-talk of mature adipocytes and preadipocytes. Sci. Adv. 6:eaax9605.
Chen, X., Ayala, I., Shannon, C., Fourcaudot, M., Acharya, N. K., Jenkinson, C. P., et al. (2018). The diabetes gene and Wnt pathway effector TCF7L2 regulates adipocyte development and function. Diabetes 67, 554–568. doi: 10.2337/db17-0318
Chen, Z. L., Shao, W. J., Xu, F., Liu, L., Lin, B. S., Wei, X. H., et al. (2015). Acute Wnt pathway activation positively regulates leptin gene expression in mature adipocytes. Cell. Signal. 27, 587–597. doi: 10.1016/j.cellsig.2014.12.012
Choe, S. S., Huh, J. Y., Hwang, I. J., Kim, J. I., and Kim, J. B. (2016). Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front. Endocrinol. (Lausanne) 7:30. doi: 10.3389/fendo.2016.00030
Christodoulides, C., Laudes, M., Cawthorn, W. P., Schinner, S., Soos, M., O’Rahilly, S., et al. (2006). The Wnt antagonist Dickkopf-1 and its receptors are coordinately regulated during early human adipogenesis. J. Cell Sci. 119(Pt 12), 2613–2620. doi: 10.1242/jcs.02975
Cisternas, P., Zolezzi, J. M., Martinez, M., Torres, V. I., Wong, G. W., and Inestrosa, N. C. (2019). Wnt-induced activation of glucose metabolism mediates the in vivo neuroprotective roles of Wnt signaling in Alzheimer disease. J. Neurochem. 149, 54–72. doi: 10.1111/jnc.14608
Cossu, G., Kelly, R., Tajbakhsh, S., Di Donna, S., Vivarelli, E., and Buckingham, M. (1996). Activation of different myogenic pathways: myf-5 is induced by the neural tube and MyoD by the dorsal ectoderm in mouse paraxial mesoderm. Development 122, 429–437.
Cristancho, A. G., and Lazar, M. A. (2011). Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 12, 722–734. doi: 10.1038/nrm3198
Cui, S., Li, L., Yu, R. T., Downes, M., Evans, R. M., Hulin, J. A., et al. (2019). β-Catenin is essential for differentiation of primary myoblasts via cooperation with MyoD and alpha-catenin. Development 146:dev167080. doi: 10.1242/dev.167080
Dabernat, S., Secrest, P., Peuchant, E., Moreau-Gaudry, F., Dubus, P., and Sarvetnick, N. (2009). Lack of beta-catenin in early life induces abnormal glucose homeostasis in mice. Diabetologia 52, 1608–1617. doi: 10.1007/s00125-009-1411-y
Deitmer, J. W., Theparambil, S. M., Ruminot, I., Noor, S. I., and Becker, H. M. (2019). Energy dynamics in the brain: contributions of astrocytes to metabolism and pH homeostasis. Front. Neurosci. 13:1301. doi: 10.3389/fnins.2019.01301
Dessimoz, J., Bonnard, C., Huelsken, J., and Grapin-Botton, A. (2005). Pancreas-specific deletion of beta-catenin reveals Wnt-dependent and Wnt-independent functions during development. Curr. Biol. 15, 1677–1683. doi: 10.1016/j.cub.2005.08.037
Dunty, W. C. Jr., Biris, K. K., Chalamalasetty, R. B., Taketo, M. M., Lewandoski, M., and Yamaguchi, T. P. (2008). Wnt3a/beta-catenin signaling controls posterior body development by coordinating mesoderm formation and segmentation. Development 135, 85–94. doi: 10.1242/dev.009266
Eberhart, C. G., and Argani, P. (2001). Wnt signaling in human development: beta-catenin nuclear translocation in fetal lung, kidney, placenta, capillaries, adrenal, and cartilage. Pediatr. Dev. Pathol. 4, 351–357. doi: 10.1007/s10024001-0037-y
Elghazi, L., Gould, A. P., Weiss, A. J., Barker, D. J., Callaghan, J., Opland, D., et al. (2012). Importance of beta-Catenin in glucose and energy homeostasis. Sci. Rep. 2:693. doi: 10.1038/srep00693
Evans, P. L., McMillin, S. L., Weyrauch, L. A., and Witczak, C. A. (2019). Regulation of skeletal muscle glucose transport and glucose metabolism by exercise training. Nutrients 11:2432. doi: 10.3390/nu11102432
Facchinello, N., Tarifeno-Saldivia, E., Grisan, E., Schiavone, M., Peron, M., Mongera, A., et al. (2017). Tcf7l2 plays pleiotropic roles in the control of glucose homeostasis, pancreas morphology, vascularization and regeneration. Sci. Rep. 7:9605. doi: 10.1038/s41598-017-09867-x
Fang, D., Hawke, D., Zheng, Y., Xia, Y., Meisenhelder, J., Nika, H., et al. (2007). Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229. doi: 10.1074/jbc.M611871200
Fearnhead, N. S., Britton, M. P., and Bodmer, W. F. (2001). The ABC of APC. Hum. Mol. Genet. 10, 721–733. doi: 10.1093/hmg/10.7.721
Florez, J. C., Jablonski, K. A., Bayley, N., Pollin, T. I., de Bakker, P. I., Shuldiner, A. R., et al. (2006). TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N. Engl. J. Med. 355, 241–250. doi: 10.1056/NEJMoa062418
Fuster, J. J., Zuriaga, M. A., Ngo, D. T., Farb, M. G., Aprahamian, T., Yamaguchi, T. P., et al. (2015). Noncanonical Wnt signaling promotes obesity-induced adipose tissue inflammation and metabolic dysfunction independent of adipose tissue expansion. Diabetes 64, 1235–1248. doi: 10.2337/db14-1164
Ganassi, M., Badodi, S., Ortuste Quiroga, H. P., Zammit, P. S., Hinits, Y., and Hughes, S. M. (2018). Myogenin promotes myocyte fusion to balance fibre number and size. Nat. Commun. 9:4232. doi: 10.1038/s41467-018-06583-6
Geetha-Loganathan, P., Nimmagadda, S., and Scaal, M. (2008). Wnt signaling in limb organogenesis. Organogenesis 4, 109–115. doi: 10.4161/org.4.2.5857
Geoghegan, G., Simcox, J., Seldin, M. M., Parnell, T. J., Stubben, C., Just, S., et al. (2019). Targeted deletion of Tcf7l2 in adipocytes promotes adipocyte hypertrophy and impaired glucose metabolism. Mol. Metab. 24, 44–63. doi: 10.1016/j.molmet.2019.03.003
Gilland, E., Miller, A. L., Karplus, E., Baker, R., and Webb, S. E. (1999). Imaging of multicellular large-scale rhythmic calcium waves during zebrafish gastrulation. Proc. Natl. Acad. Sci. U.S.A. 96, 157–161. doi: 10.1073/pnas.96.1.157
Glinka, A., Wu, W., Delius, H., Monaghan, A. P., Blumenstock, C., and Niehrs, C. (1998). Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 391, 357–362. doi: 10.1038/34848
Goichberg, P., Shtutman, M., Ben-Ze’ev, A., and Geiger, B. (2001). Recruitment of beta-catenin to cadherin-mediated intercellular adhesions is involved in myogenic induction. J. Cell Sci. 114(Pt 7), 1309–1319.
Gottardi, C. J., and Gumbiner, B. M. (2004). Distinct molecular forms of beta-catenin are targeted to adhesive or transcriptional complexes. J. Cell Biol. 167, 339–349. doi: 10.1083/jcb.200402153
Grant, S. F., Thorleifsson, G., Reynisdottir, I., Benediktsson, R., Manolescu, A., Sainz, J., et al. (2006). Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 38, 320–323. doi: 10.1038/ng1732
Green, R. A., Wollman, R., and Kaplan, K. B. (2005). APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol. Biol. Cell 16, 4609–4622. doi: 10.1091/mbc.e05-03-0259
Grigoryan, T., Wend, P., Klaus, A., and Birchmeier, W. (2008). Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 22, 2308–2341. doi: 10.1101/gad.1686208
Groden, J., Joslyn, G., Samowitz, W., Jones, D., Bhattacharyya, N., Spirio, L., et al. (1995). Response of colon cancer cell lines to the introduction of APC, a colon-specific tumor suppressor gene. Cancer Res. 55, 1531–1539.
Gross, J. C., Chaudhary, V., Bartscherer, K., and Boutros, M. (2012). Active Wnt proteins are secreted on exosomes. Nat. Cell Biol. 14, 1036–1045. doi: 10.1038/ncb2574
Gurley, K. A., Rink, J. C., and Sanchez Alvarado, A. (2008). Beta-catenin defines head versus tail identity during planarian regeneration and homeostasis. Science 319, 323–327. doi: 10.1126/science.1150029
Hamada, F., and Bienz, M. (2004). The APC tumor suppressor binds to C-terminal binding protein to divert nuclear beta-catenin from TCF. Dev. Cell 7, 677–685. doi: 10.1016/j.devcel.2004.08.022
Hankey, W., Frankel, W. L., and Groden, J. (2018). Functions of the APC tumor suppressor protein dependent and independent of canonical WNT signaling: implications for therapeutic targeting. Cancer Metastasis Rev. 37, 159–172. doi: 10.1007/s10555-017-9725-6
Hansson, O., Zhou, Y., Renstrom, E., and Osmark, P. (2010). Molecular function of TCF7L2: consequences of TCF7L2 splicing for molecular function and risk for type 2 diabetes. Curr. Diab. Rep. 10, 444–451. doi: 10.1007/s11892-010-0149-8
Heiser, P. W., Lau, J., Taketo, M. M., Herrera, P. L., and Hebrok, M. (2006). Stabilization of beta-catenin impacts pancreas growth. Development 133, 2023–2032. doi: 10.1242/dev.02366
Heller, R. S., Dichmann, D. S., Jensen, J., Miller, C., Wong, G., Madsen, O. D., et al. (2002). Expression patterns of Wnts, Frizzleds, sFRPs, and misexpression in transgenic mice suggesting a role for Wnts in pancreas and foregut pattern formation. Dev. Dyn. 225, 260–270. doi: 10.1002/dvdy.10157
Henriquez, J. P., Webb, A., Bence, M., Bildsoe, H., Sahores, M., Hughes, S. M., et al. (2008). Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. Proc. Natl. Acad. Sci. U.S.A. 105, 18812–18817. doi: 10.1073/pnas.0806300105
Hill, E. M., and Petersen, C. P. (2015). Wnt/Notum spatial feedback inhibition controls neoblast differentiation to regulate reversible growth of the planarian brain. Development 142, 4217–4229. doi: 10.1242/dev.123612
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90. doi: 10.1038/35017574
Hollis, E. R. II, and Zou, Y. (2012). Expression of the Wnt signaling system in central nervous system axon guidance and regeneration. Front. Mol. Neurosci. 5:5. doi: 10.3389/fnmol.2012.00005
Hoppler, S., Brown, J. D., and Moon, R. T. (1996). Expression of a dominant-negative Wnt blocks induction of MyoD in Xenopus embryos. Genes Dev. 10, 2805–2817. doi: 10.1101/gad.10.21.2805
Huber, A. H., Nelson, W. J., and Weis, W. I. (1997). Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell 90, 871–882. doi: 10.1016/s0092-8674(00)80352-9
Ip, W., Shao, W., Song, Z., Chen, Z., Wheeler, M. B., and Jin, T. (2015). Liver-specific expression of dominant-negative transcription factor 7-like 2 causes progressive impairment in glucose homeostasis. Diabetes 64, 1923–1932. doi: 10.2337/db14-1329
Jho, E. H., Zhang, T., Domon, C., Joo, C. K., Freund, J. N., and Costantini, F. (2002). Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 22, 1172–1183. doi: 10.1128/mcb.22.4.1172-1183.2002
Jungermann, K., and Katz, N. (1989). Functional specialization of different hepatocyte populations. Physiol. Rev. 69, 708–764. doi: 10.1152/physrev.1989.69.3.708
Kaemmerer, E., and Gassler, N. (2016). Wnt lipidation and modifiers in intestinal carcinogenesis and cancer. Cancers (Basel) 8:69. doi: 10.3390/cancers8070069
Kanazawa, A., Tsukada, S., Kamiyama, M., Yanagimoto, T., Nakajima, M., and Maeda, S. (2005). Wnt5b partially inhibits canonical Wnt/beta-catenin signaling pathway and promotes adipogenesis in 3T3-L1 preadipocytes. Biochem. Biophys. Res. Commun. 330, 505–510. doi: 10.1016/j.bbrc.2005.03.007
Killoran, R. C., Fan, J., Yang, D., Shilton, B. H., and Choy, W. Y. (2015). Structural analysis of the 14-3-3zeta/Chibby interaction involved in Wnt/beta-Catenin signaling. PLoS One 10:e0123934. doi: 10.1371/journal.pone.0123934
Kim, C. H., Neiswender, H., Baik, E. J., Xiong, W. C., and Mei, L. (2008). Beta-catenin interacts with MyoD and regulates its transcription activity. Mol. Cell. Biol. 28, 2941–2951. doi: 10.1128/MCB.01682-07
Kimelman, D., and Xu, W. (2006). beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene 25, 7482–7491. doi: 10.1038/sj.onc.1210055
Kitamura, T., Kahn, C. R., and Accili, D. (2003). Insulin receptor knockout mice. Annu. Rev. Physiol. 65, 313–332. doi: 10.1146/annurev.physiol.65.092101.142540
Kobayashi, C., Saito, Y., Ogawa, K., and Agata, K. (2007). Wnt signaling is required for antero-posterior patterning of the planarian brain. Dev. Biol. 306, 714–724. doi: 10.1016/j.ydbio.2007.04.010
Kohn, A. D., and Moon, R. T. (2005). Wnt and calcium signaling: beta-catenin-independent pathways. Cell Calcium 38, 439–446. doi: 10.1016/j.ceca.2005.06.022
Komiya, Y., and Habas, R. (2008). Wnt signal transduction pathways. Organogenesis 4, 68–75. doi: 10.4161/org.4.2.5851
Kubota, T., Kubota, N., and Kadowaki, T. (2017). Imbalanced insulin actions in obesity and Type 2 diabetes: key mouse models of insulin signaling pathway. Cell Metab. 25, 797–810. doi: 10.1016/j.cmet.2017.03.004
Lade, A. G., and Monga, S. P. (2011). Beta-catenin signaling in hepatic development and progenitors: which way does the WNT blow? Dev. Dyn. 240, 486–500. doi: 10.1002/dvdy.22522
Lade, A., Ranganathan, S., Luo, J., and Monga, S. P. (2012). Calpain induces N-terminal truncation of beta-catenin in normal murine liver development: diagnostic implications in hepatoblastomas. J. Biol. Chem. 287, 22789–22798. doi: 10.1074/jbc.M112.378224
Laken, S. J., Papadopoulos, N., Petersen, G. M., Gruber, S. B., Hamilton, S. R., Giardiello, F. M., et al. (1999). Analysis of masked mutations in familial adenomatous polyposis. Proc. Natl. Acad. Sci. U.S.A. 96, 2322–2326. doi: 10.1073/pnas.96.5.2322
Le Floch, N., Rivat, C., De Wever, O., Bruyneel, E., Mareel, M., Dale, T., et al. (2005). The proinvasive activity of Wnt-2 is mediated through a noncanonical Wnt pathway coupled to GSK-3beta and c-Jun/AP-1 signaling. FASEB J. 19, 144–146. doi: 10.1096/fj.04-2373fje
Lee, J. H., Bassel-Duby, R., and Olson, E. N. (2014). Heart- and muscle-derived signaling system dependent on MED13 and Wingless controls obesity in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 111, 9491–9496. doi: 10.1073/pnas.1409427111
Lewis, J. E., Wahl, J. K. III, Sass, K. M., Jensen, P. J., Johnson, K. R., and Wheelock, M. J. (1997). Cross-talk between adherens junctions and desmosomes depends on plakoglobin. J. Cell Biol. 136, 919–934. doi: 10.1083/jcb.136.4.919
Li, F. Q., Mofunanya, A., Harris, K., and Takemaru, K. (2008). Chibby cooperates with 14-3-3 to regulate beta-catenin subcellular distribution and signaling activity. J. Cell Biol. 181, 1141–1154. doi: 10.1083/jcb.200709091
Li, F. Q., Singh, A. M., Mofunanya, A., Love, D., Terada, N., Moon, R. T., et al. (2007). Chibby promotes adipocyte differentiation through inhibition of beta-catenin signaling. Mol. Cell. Biol. 27, 4347–4354. doi: 10.1128/MCB.01640-06
Liberman, Z., and Eldar-Finkelman, H. (2005). Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J. Biol. Chem. 280, 4422–4428. doi: 10.1074/jbc.M410610200
Lim, G. E., and Brubaker, P. (2006). Glucagon-Like peptide 1 secretion by the L-cell. Am. Diabetes Assoc. 55, S70–S77.
Liu, H., Fergusson, M. M., Wu, J. J., Rovira, I. I., Liu, J., Gavrilova, O., et al. (2011). Wnt signaling regulates hepatic metabolism. Sci. Signal. 4:ra6. doi: 10.1126/scisignal.2001249
Liu, P., Wakamiya, M., Shea, M. J., Albrecht, U., Behringer, R. R., and Bradley, A. (1999). Requirement for Wnt3 in vertebrate axis formation. Nat. Genet. 22, 361–365. doi: 10.1038/11932
Liu, Z., Tanabe, K., Bernal-Mizrachi, E., and Permutt, M. A. (2008). Mice with beta cell overexpression of glycogen synthase kinase-3beta have reduced beta cell mass and proliferation. Diabetologia 51, 623–631. doi: 10.1007/s00125-007-0914-7
Lyssenko, V., Lupi, R., Marchetti, P., Del Guerra, S., Orho-Melander, M., Almgren, P., et al. (2007). Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Invest. 117, 2155–2163. doi: 10.1172/JCI30706
Ma, R. E. A. (2020). Metabolic and non-metabolic liver zonation is established non-synchronously and requires sinusoidal Wnts. ELife 9:e46206. doi: 10.7554/eLife.46206
MacAulay, K., and Woodgett, J. R. (2008). Targeting glycogen synthase kinase-3 (GSK-3) in the treatment of Type 2 diabetes. Expert Opin. Ther. Targets 12, 1265–1274. doi: 10.1517/14728222.12.10.1265
MacDonald, B. T., and He, X. (2012). Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb. Perspect. Biol. 4:a007880. doi: 10.1101/cshperspect.a007880
Masson, S. W. C., Sorrenson, B., Shepherd, P. R., and Merry, T. L. (2020). beta-catenin regulates muscle glucose transport via actin remodelling and M-cadherin binding. Mol. Metab. 42:101091. doi: 10.1016/j.molmet.2020.101091
Mathew, S. J., Hansen, J. M., Merrell, A. J., Murphy, M. M., Lawson, J. A., Hutcheson, D. A., et al. (2011). Connective tissue fibroblasts and Tcf4 regulate myogenesis. Development 138, 371–384. doi: 10.1242/dev.057463
Matsumoto, K., Imasato, M., Yamazaki, Y., Tanaka, H., Watanabe, M., Eguchi, H., et al. (2014). Claudin 2 deficiency reduces bile flow and increases susceptibility to cholesterol gallstone disease in mice. Gastroenterology 147, 1134–1145.e1110. doi: 10.1053/j.gastro.2014.07.033
McDuffie, J. R., Riggs, P. A., Calis, K. A., Freedman, R. J., Oral, E. A., DePaoli, A. M., et al. (2004). Effects of exogenous leptin on satiety and satiation in patients with lipodystrophy and leptin insufficiency. J. Clin. Endocrinol. Metab. 89, 4258–4263. doi: 10.1210/jc.2003-031868
McPherron, A. C., Guo, T., Bond, N. D., and Gavrilova, O. (2013). Increasing muscle mass to improve metabolism. Adipocyte 2, 92–98. doi: 10.4161/adip.22500
Mermelstein, C. S., Portilho, D. M., Mendes, F. A., Costa, M. L., and Abreu, J. G. (2007). Wnt/beta-catenin pathway activation and myogenic differentiation are induced by cholesterol depletion. Differentiation 75, 184–192. doi: 10.1111/j.1432-0436.2006.00129.x
Monga, S. P. (2011). Role of Wnt/beta-catenin signaling in liver metabolism and cancer. Int. J. Biochem. Cell Biol. 43, 1021–1029. doi: 10.1016/j.biocel.2009.09.001
Moore, A. C., Amann, J. M., Williams, C. S., Tahinci, E., Farmer, T. E., Martinez, J. A., et al. (2008). Myeloid translocation gene family members associate with T-cell factors (TCFs) and influence TCF-dependent transcription. Mol. Cell. Biol. 28, 977–987. doi: 10.1128/MCB.01242-07
Morin, P. J., Vogelstein, B., and Kinzler, K. W. (1996). Apoptosis and APC in colorectal tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 93, 7950–7954. doi: 10.1073/pnas.93.15.7950
Mosimann, C., Hausmann, G., and Basler, K. (2009). Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 10, 276–286. doi: 10.1038/nrm2654
Murtaugh, L. C., Law, A. C., Dor, Y., and Melton, D. A. (2005). Beta-catenin is essential for pancreatic acinar but not islet development. Development 132, 4663–4674. doi: 10.1242/dev.02063
Nakayama, S., Arakawa, M., Uchida, T., Ogihara, T., Kanno, R., Ikeda, F., et al. (2008). Dose-dependent requirement of patched homologue 1 in mouse pancreatic beta cell mass. Diabetologia 51, 1883–1892. doi: 10.1007/s00125-008-1080-2
Nejak-Bowen, K. N., and Monga, S. P. (2011). Beta-catenin signaling, liver regeneration and hepatocellular cancer: sorting the good from the bad. Semin. Cancer Biol. 21, 44–58. doi: 10.1016/j.semcancer.2010.12.010
Nguyen-Tu, M. S., Martinez-Sanchez, A., Leclerc, I., Rutter, G. A., and da Silva Xavier, G. (2021). Adipocyte-specific deletion of Tcf7l2 induces dysregulated lipid metabolism and impairs glucose tolerance in mice. Diabetologia 64, 129–141. doi: 10.1007/s00125-020-05292-4
Nie, X., Liu, H., Liu, L., Wang, Y. D., and Chen, W. D. (2020). Emerging roles of Wnt ligands in human colorectal cancer. Front. Oncol. 10:1341. doi: 10.3389/fonc.2020.01341
Niehrs, C. (2006). Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene 25, 7469–7481. doi: 10.1038/sj.onc.1210054
Nusse, R. (2008). Wnt signaling and stem cell control. Cell Res. 18, 523–527. doi: 10.1038/cr.2008.47
Nusse, R., and Varmus, H. E. (1982). Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 31, 99–109. doi: 10.1016/0092-8674(82)90409-3
Ober, E. A., Verkade, H., Field, H. A., and Stainier, D. Y. (2006). Mesodermal Wnt2b signalling positively regulates liver specification. Nature 442, 688–691. doi: 10.1038/nature04888
Osmark, P., Hansson, O., Jonsson, A., Ronn, T., Groop, L., and Renstrom, E. (2009). Unique splicing pattern of the TCF7L2 gene in human pancreatic islets. Diabetologia 52, 850–854. doi: 10.1007/s00125-009-1293-z
Ouchi, N., Higuchi, A., Ohashi, K., Oshima, Y., Gokce, N., Shibata, R., et al. (2010). Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 329, 454–457. doi: 10.1126/science.1188280
Papadopoulou, S., and Edlund, H. (2005). Attenuated Wnt signaling perturbs pancreatic growth but not pancreatic function. Diabetes 54, 2844–2851. doi: 10.2337/diabetes.54.10.2844
Parr, B. A., Shea, M. J., Vassileva, G., and McMahon, A. P. (1993). Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development 119, 247–261.
Patapoutian, A., and Reichardt, L. F. (2000). Roles of Wnt proteins in neural development and maintenance. Curr. Opin. Neurobiol. 10, 392–399. doi: 10.1016/s0959-4388(00)00100-8
Patel, S., Doble, B. W., MacAulay, K., Sinclair, E. M., Drucker, D. J., and Woodgett, J. R. (2008). Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol. Cell. Biol. 28, 6314–6328. doi: 10.1128/MCB.00763-08
Peifer, M., Berg, S., and Reynolds, A. B. (1994). A repeating amino acid motif shared by proteins with diverse cellular roles. Cell 76, 789–791. doi: 10.1016/0092-8674(94)90353-0
Perez-Moreno, M., Davis, M. A., Wong, E., Pasolli, H. A., Reynolds, A. B., and Fuchs, E. (2006). p120-catenin mediates inflammatory responses in the skin. Cell 124, 631–644. doi: 10.1016/j.cell.2005.11.043
Piccolo, S., Agius, E., Leyns, L., Bhattacharyya, S., Grunz, H., Bouwmeester, T., et al. (1999). The head inducer Cerberus is a multifunctional antagonist of Nodal, BMP and Wnt signals. Nature 397, 707–710. doi: 10.1038/17820
Pokutta, S., and Weis, W. I. (2000). Structure of the dimerization and beta-catenin-binding region of alpha-catenin. Mol. Cell 5, 533–543. doi: 10.1016/s1097-2765(00)80447-5
Powell, S. M., Zilz, N., Beazer-Barclay, Y., Bryan, T. M., Hamilton, S. R., Thibodeau, S. N., et al. (1992). APC mutations occur early during colorectal tumorigenesis. Nature 359, 235–237. doi: 10.1038/359235a0
Prakash, S., and Swaminathan, U. (2015). beta catenin in health: a review. J. Oral Maxillofac. Pathol. 19, 230–238. doi: 10.4103/0973-029X.164537
Rachdi, L., Balcazar, N., Elghazi, L., Barker, D. J., Krits, I., Kiyokawa, H., et al. (2006). Differential effects of p27 in regulation of beta-cell mass during development, neonatal period, and adult life. Diabetes 55, 3520–3528. doi: 10.2337/db06-0861
Ranawade, A., Mallick, A., and Gupta, B. P. (2018). PRY-1/Axin signaling regulates lipid metabolism in Caenorhabditis elegans. PLoS One 13:e0206540. doi: 10.1371/journal.pone.0206540
Richter, S., Hartmann, B., and Reichert, H. (1998). The wingless gene is required for embryonic brain development in Drosophila. Dev. Genes Evol. 208, 37–45. doi: 10.1007/s004270050151
Ridgeway, A. G., Petropoulos, H., Wilton, S., and Skerjanc, I. S. (2000). Wnt signaling regulates the function of MyoD and myogenin. J. Biol. Chem. 275, 32398–32405. doi: 10.1074/jbc.M004349200
Riechmann, V., Rehorn, K. P., Reuter, R., and Leptin, M. (1998). The genetic control of the distinction between fat body and gonadal mesoderm in Drosophila. Development 125, 713–723.
Roder, P. V., Wu, B., Liu, Y., and Han, W. (2016). Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 48:e219. doi: 10.1038/emm.2016.6
Rui, L. (2014). Energy metabolism in the liver. Compr. Physiol. 4, 177–197. doi: 10.1002/cphy.c130024
Rulifson, I. C., Karnik, S. K., Heiser, P. W., ten Berge, D., Chen, H., Gu, X., et al. (2007). Wnt signaling regulates pancreatic beta cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 104, 6247–6252. doi: 10.1073/pnas.0701509104
Saneyoshi, T., Kume, S., Amasaki, Y., and Mikoshiba, K. (2002). The Wnt/calcium pathway activates NF-AT and promotes ventral cell fate in Xenopus embryos. Nature 417, 295–299. doi: 10.1038/417295a
Saxena, R., Gianniny, L., Burtt, N. P., Lyssenko, V., Giuducci, C., Sjogren, M., et al. (2006). Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes 55, 2890–2895. doi: 10.2337/db06-0381
Schohl, A., and Fagotto, F. (2002). Beta-catenin, MAPK and Smad signaling during early Xenopus development. Development 129, 37–52.
Seifert, J. R., and Mlodzik, M. (2007). Frizzled/PCP signalling: a conserved mechanism regulating cell polarity and directed motility. Nat. Rev. Genet. 8, 126–138. doi: 10.1038/nrg2042
Sekine, S., Lan, B. Y., Bedolli, M., Feng, S., and Hebrok, M. (2006). Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 43, 817–825. doi: 10.1002/hep.21131
Shao, W., Wang, D., Chiang, Y. T., Ip, W., Zhu, L., Xu, F., et al. (2013). The Wnt signaling pathway effector TCF7L2 controls gut and brain proglucagon gene expression and glucose homeostasis. Diabetes 62, 789–800. doi: 10.2337/db12-0365
Shao, W., Xiong, X., Ip, W., Xu, F., Song, Z., Zeng, K., et al. (2015). The expression of dominant negative TCF7L2 in pancreatic beta cells during the embryonic stage causes impaired glucose homeostasis. Mol. Metab. 4, 344–352. doi: 10.1016/j.molmet.2015.01.008
Sharma, R. P., and Chopra, V. L. (1976). Effect of the Wingless (wg1) mutation on wing and haltere development in Drosophila melanogaster. Dev. Biol. 48, 461–465. doi: 10.1016/0012-1606(76)90108-1
Sharon, N., Vanderhooft, J., Straubhaar, J., Mueller, J., Chawla, R., Zhou, Q., et al. (2019). Wnt signaling separates the progenitor and endocrine compartments during pancreas development. Cell Rep. 27, 2281–2291.e2285. doi: 10.1016/j.celrep.2019.04.083
Shi, D. L., Bourdelas, A., Umbhauer, M., and Boucaut, J. C. (2002). Zygotic Wnt/beta-catenin signaling preferentially regulates the expression of Myf5 gene in the mesoderm of Xenopus. Dev. Biol. 245, 124–135. doi: 10.1006/dbio.2002.0633
Sineva, G. S., and Pospelov, V. A. (2014). beta-Catenin in pluripotency: adhering to self-renewal or Wnting to differentiate? Int. Rev. Cell Mol. Biol. 312, 53–78. doi: 10.1016/B978-0-12-800178-3.00002-6
Song, H., Goetze, S., Bischof, J., Spichiger-Haeusermann, C., Kuster, M., Brunner, E., et al. (2010). Coop functions as a corepressor of Pangolin and antagonizes Wingless signaling. Genes Dev. 24, 881–886. doi: 10.1101/gad.561310
Sorrenson, B., Cognard, E., Lee, K. L., Dissanayake, W. C., Fu, Y., Han, W., et al. (2016). A critical role for beta-catenin in modulating levels of insulin secretion from beta-cells by regulating actin cytoskeleton and insulin vesicle localization. J. Biol. Chem. 291, 25888–25900. doi: 10.1074/jbc.M116.758516
Stanganello, E., Zahavi, E. E., Burute, M., Smits, J., Jordens, I., Maurice, M. M., et al. (2019). Wnt signaling directs neuronal polarity and axonal growth. iScience 13, 318–327. doi: 10.1016/j.isci.2019.02.029
Stark, K., Vainio, S., Vassileva, G., and McMahon, A. P. (1994). Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372, 679–683. doi: 10.1038/372679a0
Steinhart, Z., and Angers, S. (2018). Wnt signaling in development and tissue homeostasis. Development 145:dev146589. doi: 10.1242/dev.146589
Strom, A., Bonal, C., Ashery-Padan, R., Hashimoto, N., Campos, M. L., Trumpp, A., et al. (2007). Unique mechanisms of growth regulation and tumor suppression upon Apc inactivation in the pancreas. Development 134, 2719–2725. doi: 10.1242/dev.02875
Tajbakhsh, S., Borello, U., Vivarelli, E., Kelly, R., Papkoff, J., Duprez, D., et al. (1998). Differential activation of Myf5 and MyoD by different Wnts in explants of mouse paraxial mesoderm and the later activation of myogenesis in the absence of Myf5. Development 125, 4155–4162.
Tanabe, K., Liu, Z., Patel, S., Doble, B. W., Li, L., Cras-Meneur, C., et al. (2008). Genetic deficiency of glycogen synthase kinase-3beta corrects diabetes in mouse models of insulin resistance. PLoS Biol. 6:e37. doi: 10.1371/journal.pbio.0060037
Thomas, K. R., and Capecchi, M. R. (1990). Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature 346, 847–850. doi: 10.1038/346847a0
Thompson, J. J., and Williams, C. S. (2018). Protein phosphatase 2A in the regulation of Wnt signaling, stem cells, and cancer. Genes (Basel) 9:121. doi: 10.3390/genes9030121
Tseng, Y. H., Butte, A. J., Kokkotou, E., Yechoor, V. K., Taniguchi, C. M., Kriauciunas, K. M., et al. (2005). Prediction of preadipocyte differentiation by gene expression reveals role of insulin receptor substrates and necdin. Nat. Cell Biol. 7, 601–611. doi: 10.1038/ncb1259
Uchida, T., Nakamura, T., Hashimoto, N., Matsuda, T., Kotani, K., Sakaue, H., et al. (2005). Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat. Med. 11, 175–182. doi: 10.1038/nm1187
Ugrankar, R., Berglund, E., Akdemir, F., Tran, C., Kim, M. S., Noh, J., et al. (2015). Drosophila glucome screening identifies Ck1alpha as a regulator of mammalian glucose metabolism. Nat. Commun. 6:7102. doi: 10.1038/ncomms8102
Ultimo, S., Zauli, G., Martelli, A. M., Vitale, M., McCubrey, J. A., Capitani, S., et al. (2018). Influence of physical exercise on microRNAs in skeletal muscle regeneration, aging and diseases. Oncotarget 9, 17220–17237. doi: 10.18632/oncotarget.24991
Valenta, T., Hausmann, G., and Basler, K. (2012). The many faces and functions of beta-catenin. EMBO J. 31, 2714–2736. doi: 10.1038/emboj.2012.150
van Amerongen, R., and Berns, A. (2006). Knockout mouse models to study Wnt signal transduction. Trends Genet. 22, 678–689. doi: 10.1016/j.tig.2006.10.001
Vertino, A. M., Taylor-Jones, J. M., Longo, K. A., Bearden, E. D., Lane, T. F., McGehee, R. E. Jr., et al. (2005). Wnt10b deficiency promotes coexpression of myogenic and adipogenic programs in myoblasts. Mol. Biol. Cell 16, 2039–2048. doi: 10.1091/mbc.e04-08-0720
von Maltzahn, J., Chang, N. C., Bentzinger, C. F., and Rudnicki, M. A. (2012). Wnt signaling in myogenesis. Trends Cell Biol. 22, 602–609. doi: 10.1016/j.tcb.2012.07.008
Wallingford, J. B., Ewald, A. J., Harland, R. M., and Fraser, S. E. (2001). Calcium signaling during convergent extension in Xenopus. Curr. Biol. 11, 652–661. doi: 10.1016/s0960-9822(01)00201-9
Wang, L., Wang, Y., Zhang, C., Li, J., Meng, Y., Dou, M., et al. (2018). Inhibiting glycogen synthase kinase 3 reverses obesity-induced white adipose tissue inflammation by regulating apoptosis inhibitor of macrophage/CD5L-mediated macrophage migration. Arterioscler. Thromb. Vasc. Biol. 38, 2103–2116. doi: 10.1161/ATVBAHA.118.311363
Wang, S., Song, K., Srivastava, R., Dong, C., Go, G. W., Li, N., et al. (2015). Nonalcoholic fatty liver disease induced by noncanonical Wnt and its rescue by Wnt3a. FASEB J. 29, 3436–3445. doi: 10.1096/fj.15-271171
Wells, J. M., Esni, F., Boivin, G. P., Aronow, B. J., Stuart, W., Combs, C., et al. (2007). Wnt/beta-catenin signaling is required for development of the exocrine pancreas. BMC Dev. Biol. 7:4. doi: 10.1186/1471-213X-7-4
Willert, K., Shibamoto, S., and Nusse, R. (1999). Wnt-induced dephosphorylation of axin releases beta-catenin from the axin complex. Genes Dev. 13, 1768–1773. doi: 10.1101/gad.13.14.1768
Wu, G., Xu, G., Schulman, B. A., Jeffrey, P. D., Harper, J. W., and Pavletich, N. P. (2003). Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol. Cell 11, 1445–1456. doi: 10.1016/s1097-2765(03)00234-x
Xing, Y., Takemaru, K., Liu, J., Berndt, J. D., Zheng, J. J., Moon, R. T., et al. (2008). Crystal structure of a full-length beta-catenin. Structure 16, 478–487. doi: 10.1016/j.str.2007.12.021
Xiong, X., Shao, W., and Jin, T. (2012). New insight into the mechanisms underlying the function of the incretin hormone glucagon-like peptide-1 in pancreatic beta-cells: the involvement of the Wnt signaling pathway effector beta-catenin. Islets 4, 359–365. doi: 10.4161/isl.23345
Xu, W., and Kimelman, D. (2007). Mechanistic insights from structural studies of beta-catenin and its binding partners. J. Cell Sci. 120(Pt 19), 3337–3344. doi: 10.1242/jcs.013771
Yang, J., Mowry, L. E., Nejak-Bowen, K. N., Okabe, H., Diegel, C. R., Lang, R. A., et al. (2014). beta-catenin signaling in murine liver zonation and regeneration: a Wnt-Wnt situation! Hepatology 60, 964–976. doi: 10.1002/hep.27082
Yang, Y. (2003). Wnts and wing: Wnt signaling in vertebrate limb development and musculoskeletal morphogenesis. Birth Defects Res. C Embryo Today 69, 305–317. doi: 10.1002/bdrc.10026
Zammit, P. S. (2017). Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 72, 19–32. doi: 10.1016/j.semcdb.2017.11.011
Zaragosi, L. E., Wdziekonski, B., Fontaine, C., Villageois, P., Peraldi, P., and Dani, C. (2008). Effects of GSK3 inhibitors on in vitro expansion and differentiation of human adipose-derived stem cells into adipocytes. BMC Cell Biol. 9:11. doi: 10.1186/1471-2121-9-11
Zeve, D., Seo, J., Suh, J. M., Stenesen, D., Tang, W., Berglund, E. D., et al. (2012). Wnt signaling activation in adipose progenitors promotes insulin-independent muscle glucose uptake. Cell Metab. 15, 492–504. doi: 10.1016/j.cmet.2012.03.010
Zhang, T., Hsu, F. N., Xie, X. J., Li, X., Liu, M., Gao, X., et al. (2017). Reversal of hyperactive Wnt signaling-dependent adipocyte defects by peptide boronic acids. Proc. Natl. Acad. Sci. U.S.A. 114, E7469–E7478. doi: 10.1073/pnas.1621048114
Keywords: Wnt signaling, β-catenin, TCF7L2, GSK-3β, metabolism, embryogenesis, organogenesis
Citation: Abou Azar F and Lim GE (2021) Metabolic Contributions of Wnt Signaling: More Than Controlling Flight. Front. Cell Dev. Biol. 9:709823. doi: 10.3389/fcell.2021.709823
Received: 14 May 2021; Accepted: 16 August 2021;
Published: 09 September 2021.
Edited by:
Gunes Ozhan, Dokuz Eylül University, TurkeyReviewed by:
Evin Iscan, Dokuz Eylül University, TurkeyCopyright © 2021 Abou Azar and Lim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gareth E. Lim, Z2FyZXRoLmxpbUB1bW9udHJlYWwuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.