 Sumaiah S. Al-Asmari1,2
Sumaiah S. Al-Asmari1,2 Aleksandra Rajapakse3
Aleksandra Rajapakse3 Tomalika R. Ullah1,2
Tomalika R. Ullah1,2 Geneviève Pépin1,2
Geneviève Pépin1,2 Laura V. Croft3*†
Laura V. Croft3*† Michael P. Gantier1,2*†
Michael P. Gantier1,2*†- 1Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, VIC, Australia
- 2Department of Molecular and Translational Science, Monash University, Clayton, VIC, Australia
- 3School of Biomedical Sciences, Centre for Genomics and Personalised Health, Cancer and Ageing Research Program at the Translational Research Institute, Queensland University of Technology (QUT), Brisbane, QLD, Australia
Activation of the STING pathway upon genotoxic treatment of cancer cells has been shown to lead to anti-tumoral effects, mediated through the acute production of interferon (IFN)-β. Conversely, the pathway also correlates with the expression of NF-κB-driven pro-tumorigenic genes, but these associations are only poorly defined in the context of genotoxic treatment, and are thought to correlate with a chronic engagement of the pathway. We demonstrate here that half of the STING-expressing cancer cells from the NCI60 panel rapidly increased expression of pro-tumorigenic IL-6 upon genotoxic DNA damage, often independent of type-I IFN responses. While preferentially dependent on canonical STING, we demonstrate that genotoxic DNA damage induced by camptothecin (CPT) also drove IL-6 production through non-canonical STING signaling in selected cancer cells. Consequently, pharmacological inhibition of canonical STING failed to broadly inhibit IL-6 production induced by CPT, although this could be achieved through downstream ERK1/2 inhibition. Finally, prolonged inhibition of canonical STING signaling was associated with increased colony formation of MG-63 cells, highlighting the duality of STING signaling in also restraining the growth of selected cancer cells. Collectively, our findings demonstrate that genotoxic-induced DNA damage frequently leads to the rapid production of pro-tumorigenic IL-6 in cancer cells, independent of an IFN signature, through canonical and non-canonical STING activation; this underlines the complexity of STING engagement in human cancer cells, with frequent acute pro-tumorigenic activities induced by DNA damage. We propose that inhibition of ERK1/2 may help curb such pro-tumorigenic responses to DNA-damage, while preserving the anti-proliferative effects of the STING-interferon axis.
Introduction
Upon activation by cytoplasmic DNA, cyclic guanosine monophosphate–adenosine monophosphate (cGAMP) synthase (cGAS) synthesizes cGAMP, which binds to the adaptor protein STING (stimulator of interferon [IFN] genes) (Zhang et al., 2013). This results in STING translocation from the ER to the Golgi, where it is palmitoylated to recruit TANK-binding kinase 1 (TBK1) and the inhibitor of nuclear factor kappa-B kinase subunit epsilon (IKKε) (Mukai et al., 2016; Balka et al., 2020). This in turn activates IRF3 and NF-κB transcriptional programs, culminating in the production of IFN-β and pro-inflammatory cytokines such as IL-6 and TNFα, respectively.
In addition to its immune function in the sensing of cytosolic pathogenic DNA, cGAS can initiate immune responses to endogenous nuclear and mitochondrial DNA (Dou et al., 2017; Wu et al., 2019). Such cGAS sensing of cytosolic DNA arising from genome instability promotes senescence and replicative crisis, aimed at eliminating pre-cancerous cells (Dou et al., 2017; Glück et al., 2017; Nassour et al., 2019). Accordingly, since cancer cells have deregulated cell cycle checkpoints they frequently harbor cytoplasmic DNA, which is increased further upon genotoxic damage and radiotherapy exposure, and can lead to cGAS-STING activation (Chen et al., 2017; Dou et al., 2017; Harding et al., 2017; Mackenzie et al., 2017; Bakhoum et al., 2018; Nassour et al., 2019; Carozza et al., 2020); (Marcus et al., 2018; Schadt et al., 2019; Carozza et al., 2020).
While DNA damage-driven GAS-STING cell-intrinsic engagement in cancer cells has been shown to be involved in the recruitment of immune cells to promote anti-cancer activities, through the engagement of the IRF3/IFN-β arm (Ho et al., 2016; Takashima et al., 2016; Harding et al., 2017; Vanpouille-Box et al., 2017; Yamazaki et al., 2020; Suter et al., 2021; Tian et al., 2021), there is also evidence that chronic activation of the pathway can drive tumorigenesis and metastasis (Ahn et al., 2014; Lemos et al., 2016; Bakhoum et al., 2018). The latter is aligned with a correlation between cGAS-STING expression in human cancers and pro-inflammatory NF-κB signatures, including the expression of IL-6 (Dou et al., 2017; Bakhoum et al., 2018). Such NF-κB signals can fuel the resistance to the DNA damage (Didonato et al., 2012), and directly contribute to the growth of cancer cells (Chen et al., 2016; Bakhoum et al., 2018). As such, IL-6 production results in autocrine and paracrine activation of STAT3 signaling that promotes survival of cancer cells in response to DNA damage and pro-apoptotic mediators such as TNFα (Li et al., 2012; Yun et al., 2012). Further, IL-6 directly inhibits the IRF3/IFN-β arm of STING signaling in selected cancer cells, alleviating the tumor suppressive effects of the pathway in vivo (Wu et al., 2017; Suter et al., 2021).
Albeit currently proposed to be associated with chronic STING activation (Decout et al., 2021), little is known of the mechanisms regulating the engagement of STING-dependent pro-inflammatory NF-κB factors in the context of acute genotoxic treatment of cancer cells. A recent study reported the existence of a non-canonical STING pathway, rapidly driving IL-6 production with minimal IFN-β production upon DNA damage resulting from topoisomerase-2 inhibition in HaCaT keratinocytes (Dunphy et al., 2018). This non-canonical STING pathway was independent of cGAS/cGAMP/TBK1 and did not require translocation from the ER to the Golgi (Dunphy et al., 2018). However, whether this non-canonical STING pathway is involved in the response to acute genotoxic treatment of cancer cells is currently unknown.
Following on the observation that pharmacological inhibition of STING reduced IL-6 production upon topoisomerase 1 inhibition in mouse TC-1 cancer cells, we decided to broadly interrogate the role of STING signaling in the IL-6 response to acute DNA damage in human cancer cells. Our results collectively support a direct role for STING signaling in the frequent IL-6 production in response to genotoxic treatment of cancer cells, most often independent of a marked IRF3 signature. As such, we demonstrate that both canonical and non-canonical STING signaling can participate in the rapid IL-6 production seen upon DNA damage in different cancer cells, indicating that the pro-tumorigenic activities of the pathway are not limited to its chronic engagement. We also provide evidence that ERK1/2 pharmacological inhibition may provide therapeutic opportunities to limit production of IL-6 upon genotoxic treatment, while preserving the anti-proliferative effects of the STING-interferon axis.
Materials and Methods
Cell Culture and Treatments
Human osteosarcoma MG-63 and HOS cells were purchased from ATCC (#CRL-1427 and #CRL-1543, respectively) and grown in ATCC-formulated Eagle’s Minimum Essential Medium, supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific) and 1 × antibiotic/antimycotic (Thermo Fisher Scientific). PC-3 cells purchased from ATCC (#CRL-1435) and BT-549 breast ductal carcinoma cells (a kind gift from Prof S. Lakhani) were grown in Roswell Park Memorial Institute (RPMI) 1,640 plus L-glutamine medium (Life Technologies) complemented with 1x antibiotic/antimycotic and 10% heat inactivated fetal bovine serum (referred to as complete RPMI). TC-1 cells (kind gift from Prof. N. McMillan) and HaCaT cells (wild type–kind gift from Prof. S.M. Jane) were cultured in Dulbecco’s modified Eagle’s medium plus L-glutamine supplemented with 1 × antibiotic/antimycotic (Thermo Fisher Scientific) and 10% heat-inactivated fetal bovine serum (referred to as complete DMEM). SK-OV-3 ovarian carcinoma cells (a kind gift from Prof J. Hooper) were cultured in McCoy’s medium (Thermo Fisher Scientific) plus L-glutamine and 10% heat inactivated fetal bovine serum. MDA-MD-231 and HS-578T breast carcinoma cells (a kind gift from Prof S. Lakhani) were cultured in complete DMEM. HaCaT, MDA-MD-231, SK-OV-3 and BT-549 were authenticated using the GenePrint® 10 System kit from Promega. All the cells were cultured at 37°C with 5% CO2. Cell lines were passaged 2–3 times a week and tested for mycoplasma contamination on a routine basis by PCR. For clonogenic assays, ∼1,500 cells were added per well of a 6-well plate, and the drugs/medium changed every 2–3 days. After the indicated times, cells were fixed with 10% formalin and stained with 0.1% crystal violet (w/v) in 20% ethanol, before several thorough H2O washes.
Further methods are available in Supplementary Materials and Methods.
Results
Pharmacological Inhibition of Canonical STING Signaling Decreases CPT-Induced IL-6 in Mouse TC-1 Cells
We have recently reported that expression of the simian virus 40 (SV40) large T antigen could lead to potentiation of cGAS-STING engagement in cells treated with low-dose topoisomerase 1 inhibition with camptothecin (CPT) treatment (Pépin et al., 2017a). To broaden our observations to other viral oncogenes, we initially investigated whether CPT could induce STING-dependent signaling in mouse epithelial TC-1 cancer cells, which were co-transformed with HPV-16 E6 and E7 and c-Ha-ras oncogenes (Lin et al., 1996). Focusing on IL-6 and IP-10 production as surrogate markers of the NF-κB and IRF3 branches of STING activation, respectively (Pépin et al., 2017b; Dunphy et al., 2018), we first showed that low-dose CPT significantly induced the production of both cytokines in TC-1 cells (Figure 1A).
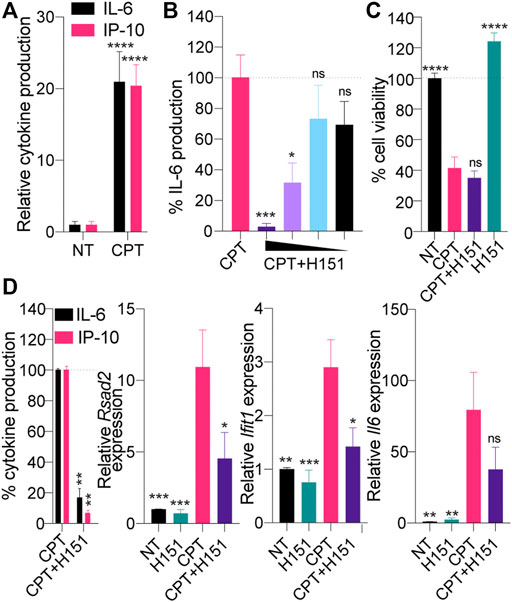
FIGURE 1. (A) TC-1 cells were treated with 0.5 μM CPT for 48 h, and IL-6 and IP-10 levels in supernatants were determined by ELISA. Cytokine levels were normalized to the non-treated (“NT”) condition after background correction with the NT condition. Data shown are averaged from three independent experiments in biological replicate (±s.e.m. and ordinary two-way ANOVA with Sidak’s multiple comparison tests). (B, C) TC-1 cells were treated with 0.5 μM CPT in the presence of decreasing amount of H151 [7.2, 3.6, 1.8 or 0.9 μM for (B), 3.6 μM for (C)] for 48 h and IL-6 levels in supernatants were determined by ELISA (B) or cell viability assessed with resazurin assay (C). B) IL-6 levels were normalized to the “CPT only” and are shown as percentages. Data shown are averaged from two independent experiments in biological triplicate (±s.e.m. and ordinary one-way ANOVA with Dunnett’s multiple comparison tests to the “CPT only” condition). (C) Data were normalized to the NT condition, after background correction with blank condition. Data shown are averaged from three independent experiments in biological replicate (±s.e.m. and ordinary one-way ANOVA with Dunnett’s multiple comparison tests to the “CPT only” condition). (D) TC-1 cells were treated with 0.5 μM CPT with or without 3.6 μM H151 for 48 h, and IL-6 and IP-10 levels in supernatants were determined by ELISA, while cell lysates were processed for RNA purification. Left: cytokine levels were normalized to the “CPT only” condition and are shown as percentages. Right: Expression of the panel of 3 mouse IFN-driven genes was analyzed by RT-qPCR. Expression of the indicated genes was reported relative to 18 s expression and divided further by the mean of the NT condition. Data shown are averaged from three independent experiments in biological replicate. Left: ± s.e.m. and Mann-Whitney U tests are shown; right: ± s.e.m. and ordinary one-way ANOVA with Dunnett’s multiple comparison tests to the “CPT only” condition. *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001 and “ns” is non-significant.
To implicate STING directly in this response to CPT, we repeated the experiments above using a recently reported pharmacological inhibitor of canonical STING, by preventing its palmitoylation, referred to as H151 (Haag et al., 2018). CPT-driven IL-6 production by TC-1 cells was significantly inhibited by H151 in a dose-dependent manner, without increasing further the cell death induced by CPT (Figures 1B,C). Accordingly, while H151 decreased production of IP-10 and IL-6 protein by ELISA, we also observed a decrease in expression of interferon-stimulated genes (ISGs) Rsad2 and Ifit1, along with Il-6 at the mRNA level by RT-qPCR (Figure 1D).
Divergent Induction of IL-6 and ISGs in Response to DNA Damage in Human Cancer Cells
This concurrent induction of Il-6, Rsad2 and Ifit1 by CPT in TC-1 cells prompted us to broadly assess whether such convergent induction of the NF-κB and IRF3 branches was a frequent response to DNA damage in cancer cells. For this purpose, we relied on a published dataset comparing the time-dependent transcriptional responses of cancer cells from the NCI60 panel, treated with several genotoxic agents (Monks et al., 2018). Forty-two cell lines in this panel significantly expressed STING based on the Cancer Cell Line Encyclopedia (Barretina et al., 2012), and were used for our in silico studies (Supplementary Table S1). Transcriptional analyses of IL-6, RSAD2, IFIT1 and IFNB1 following treatment with the CPT analogue topotecan (Top) suggested that 15 and 21 out of 42 human cancer cell lines expressing STING showed increased IL-6 expression >2 fold after 6 and 24 h Top treatment, respectively (Figure 2A and Supplementary Table S1).
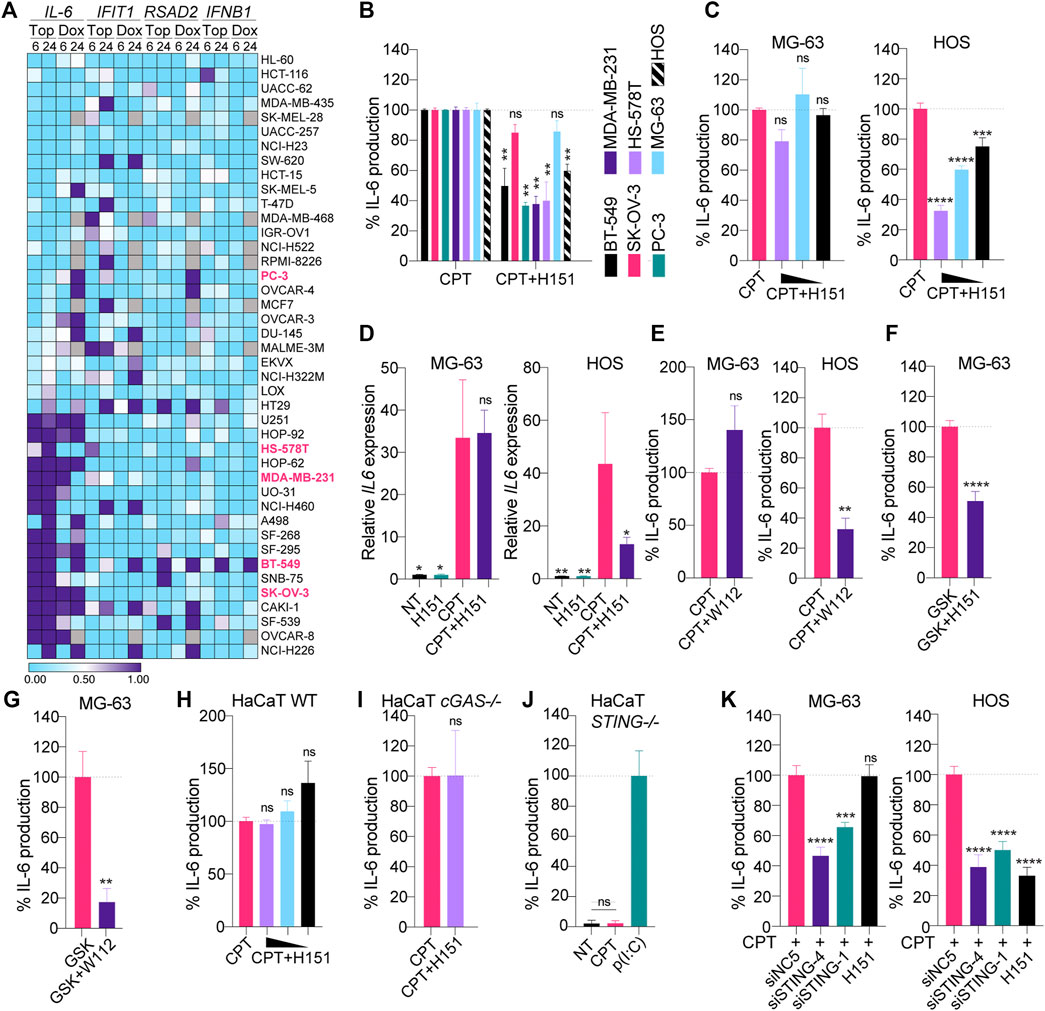
FIGURE 2. (A) Selected NCI-60 cell lines expressing STING (see Supplementary Table S1) were treated for 6 or 24 h with 1 μM topotecan (Top) or doxorubicin (Dox) and analyzed by microarray as reported in the NCI Transcriptional Pharmacodynamics Workbench (Monks et al., 2018). The heatmap shows the log2 fold change to NT condition (the values below 0.5 are blue and the values above are purple). Missing values are shown in grey. (B) Indicated cell lines were treated with CPT (see Materials and Methods for dosage used) with or without 3.6 μM H151 for 24 (BT-549, HS-578T, MDA-MB-231, PC-3 and SK-OV-3 cells) or 48 h (MG-63 and HOS cells), and IL-6 levels in supernatants were determined by ELISA. IL-6 levels were normalized to the “CPT only” condition and are shown as percentages. Data shown are averaged from three independent experiments in biological replicate (±s.e.m. and Mann-Whitney U tests are shown). (C) MG-63 and HOS were treated with CPT with or without decreasing concentrations of H151 (3.6, 1.8 and 0.9 μM) for 48 h, and IL-6 levels in supernatants were determined by ELISA. IL-6 levels were normalized to the “CPT only” condition and are shown as percentages. Data shown are averaged from three independent experiments in biological replicate (±s.e.m. and ordinary one-way ANOVA with Dunnett’s multiple comparison tests to the “CPT only” condition). (D) MG-63 and HOS were treated with CPT with or without 3.6 μM H151 for 48 h, and cell lysates were processed for RNA purification and RT-qPCR analyses. IL-6 levels were reported relative to 18S expression and divided further by the mean of the NT condition. Data shown are averaged from three independent experiments (±s.e.m. and ordinary one-way ANOVA with Dunnett’s multiple comparison tests to the “CPT only” condition). (E) MG-63 and HOS were treated with CPT with or without 200 nM WEHI-122 for 48 h and IL-6 levels in supernatants were determined by ELISA. IL-6 levels were normalized to the “CPT only” condition and are shown as percentages. Data shown are averaged from three (MG-63) or two (HOS) independent experiments in biological replicate (±s.e.m. and Mann-Whitney U tests are shown). (F, G) MG-63 were treated overnight with 100 nM GSK#3, with or without 3.6 μM H151 (F) or 200 nM WEHI-112 (G), and IL-6 levels in supernatants were determined by ELISA. IL-6 levels were normalized to the “GSK only” condition and are shown as percentages. Data shown are averaged from three (F) or two (G) independent experiments in biological replicate (±s.e.m. and Mann-Whitney U tests are shown). (H, I, J) Wild-type (WT) (H), cGAS-deficient (I) and STING-deficient (J) HaCaT cells were treated with 0.2 μM CPT in the presence of decreasing amounts of H151 (3.6, 1.8 or 0.9 μM) (H) or 3.6 μM (I) for 24 h, and IL-6 levels in supernatants were determined by ELISA. (J) Cells were treated with poly(I:C) [p(I:C)] at 1 μg/ ml, where indicated. IL-6 levels were normalized to the “CPT only” (H, I) or “p(I:C)” (J) condition and are shown as percentages. Data shown are averaged from two (I) or three (H, J) independent experiments in biological replicate [±s.e.m. and ordinary one-way ANOVA with Dunnett’s multiple comparison tests to the “CPT only” condition (H), and Mann-Whitney U tests are shown (I, J)]. (K) MG-63 and HOS were transfected with 10 nM of the indicated siRNAs for 24 or 48 h, respectively, prior to CPT treatment for 48 or 24 h, respectively, and IL-6 levels in supernatants were determined by ELISA. IL-6 levels were normalized to the “CPT + siNC5” condition and are shown as percentages (±s.e.m. and ordinary one-way ANOVA with Dunnett’s multiple comparison tests to the “CPT + siNC5” condition). *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001 and “ns” is non-significant.
Critically, the induction of IFIT1/RSAD2 and IFNB1 was mostly divergent from that of IL-6, while being more restricted. As an example, 14/42 cell lines showed >2-fold increase in IFIT1 expression at 24 h, but only five also displayed increased IL-6 levels (Figure 2A). A similar observation was made with Doxorubicin (Dox)-driven topoisomerase 2 inhibition; albeit some of the cells that induced IL-6 > 2 fold differed from those treated with Topotecan. Nonetheless, Dox treatment induced IL-6 in 23/35 cells lines at 24 h with a 2-fold threshold, versus 16/35 for IFIT1—with only five cell lines showing increases in both genes (Figure 2A). Collectively, these analyses revealed that while IL-6 was rapidly induced in 50% of cancer cells by DNA damage, this induction was often independent of that of ISGs.
Inhibition of STING Palmitoylation Does Not Reduce IL-6 in MG-63 and SK-OV-3 Cells
To confirm the potential involvement of STING signaling in this rapid IL-6 production upon DNA damage, we selected a subset of five cancer cell lines from this panel to which we had access (BT-549, HS-578T, MDA-MB-231, PC-3 and SK-OV-3 cells), that exhibited various profiles of IL-6/ISGs responses. For example, SK-OV-3 cells induced high amounts of IL-6 but not ISG, while BT-549 cells robustly induced both IL-6/ISG responses (Figure 2A). MDA-MB-231 and HS-578T cells had more variable responses to Dox and Top but did induce IL-6 and IFIT1 >2 fold with Top, while PC3 displayed a stronger IFIT1 induction than IL-6 with Top. We also tested two STING-expressing osteosarcoma lines we had previously found to produce IL-6 upon CPT treatment (HOS and MG-63 cells).
Low-dose CPT increased IL-6 production that was significantly inhibited by H151 in five cell lines (BT-549, HS-578T, MDA-MB-231, PC-3 and HOS cells), independent of increased cell death, supporting a direct contribution of canonical STING signaling in the pro-inflammatory response to CPT in these cells (Figure 2B; Supplementary Figures S1A,B). This aligned with the detection of IFIT1/ISG induction upon genotoxic treatment in our transcriptional analyses for BT-549, HS-578T, MDA-MB-231 and PC-3 cells (Figure 2A). Conversely, H151 failed to significantly reduce IL-6 production in MG-63 and SK-OV-3 cells (Figure 2B). Consistently with this, pharmacological inhibition of canonical STING or TBK1 failed to reduce the CPT-driven IL-6 induction at the mRNA and protein levels in MG-63, while it did in HOS cells (Figures 2C–E). MG-63, however, did produce IL-6 in response to a human synthetic STING agonist (referred to as GSK#3 herein - (Ramanjulu et al., 2018)), and this was significantly reduced by H151 or TBK1 inhibition (Figures 2F,G), confirming the capacity of MG-63 cells to also produce IL-6 through canonical STING signaling.
Pharmacological Inhibition of STING Palmitoylation Does Not Impact Non-Canonical STING Signaling
Non-canonical STING signaling does not require translocation from the ER to the Golgi and as such is not impacted by TBK1 inhibition (Dunphy et al., 2018). Since STING palmitoylation occurs at the Golgi, we speculated that the lack of inhibitory activity of H151 in MG-63 cells could relate to non-canonical STING signaling being at play in these cells upon CPT treatment. We first confirmed that H151 could not inhibit CPT-driven IL-6 production stemming from non-canonical STING signaling in wild-type and cGAS-deficient HaCaT cells (Figures 2H,I, Material and Methods, and Supplementary Figures S1A,S2). Importantly, STING deficiency entirely abolished CPT-driven IL-6 production in HaCaT cells, confirming the reliance on STING for this non-canonical response (Figure 2J) (Dunphy et al., 2018). In agreement with this, RNA interference mediated down-regulation of STING significantly decreased CPT-driven IL-6 production in both MG-63 and HOS cells, demonstrating the dependence on STING in both cell lines (Figure 2K; Supplementary Figures S1C,D). Collectively, these results demonstrated that the inhibitory activity of H151 was limited to canonical STING signaling and supported the engagement of non-canonical STING signaling upon genotoxic DNA damage in select cancer cell lines.
Inhibition of Downstream MAP Kinases Broadly Suppresses CPT-Driven IL-6
The lack of activity of H151 on non-canonical STING signaling led us to investigate whether targeting of downstream mediators of NF-κB signaling could help broadly dampen CPT-driven IL-6 production, independent of the type of STING signaling engaged. Non-canonical STING has been shown to rely on TRAF6 activity (Dunphy et al., 2018). While the signaling components operating downstream of TRAF6 to control STING-driven IL-6 have not been characterized to date, we posited a role for mitogen-activated protein kinases (p38 and ERK1/2) based on their known involvement in DNA-damage responses and control of IL-6 expression (Craig et al., 2000; Phong et al., 2010; Wei et al., 2011; Dainichi et al., 2019). Inhibition of ERK1/2 with SCH772984 (Morris et al., 2013) and p38 with SB202190 were initially assessed with dose responses on canonical STING signaling induced with the GSK#3 STING agonist in MG-63 cells (Figures 3A,B). p38 and ERK1/2 inhibition both significantly reduced STING-driven IL-6 production in these cells (Figures 3A,B), although the effect was more potent with ERK1/2 inhibition. In agreement with a selective effect on NF-κB signaling downstream of canonical STING signaling, ERK1/2 inhibition did not reduce but rather increased IP-10 production upon GSK#3 stimulation - consistent with the prior findings that ERK1/2 inhibit type-I IFN production (Figure 3B) (Janovec et al., 2018).
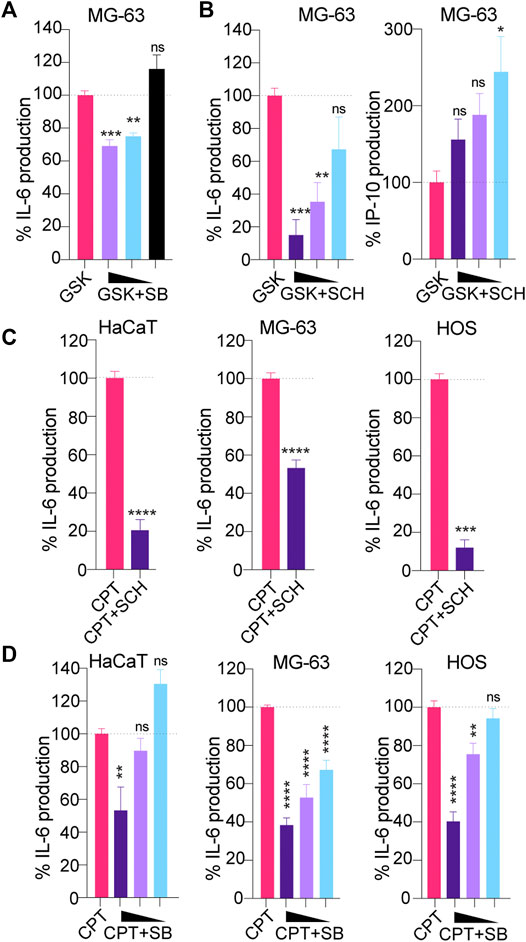
FIGURE 3. (A, B) MG-63 were treated overnight with 100 nM of the STING agonist GSK#3 with or without decreasing concentrations of the p38 inhibitor SB202190 [SB] (at 3, 1.5 and 0.75 μM) (A) or decreasing concentrations of the ERK1/2 inhibitor SCH772984 [SCH] (at 1.25, 0.63 and 0.313 μM) (B), and IL-6/IP-10 levels in supernatants were determined by ELISA. IL-6 and IP-10 levels were normalized to the “GSK only” condition and are shown as percentages. (A, B) Data shown are averaged from two independent experiments in biological replicate (±s.e.m. and ordinary one-way ANOVA with Dunnett’s multiple comparison tests to the “GSK only” condition). (C) HaCaT WT, MG-63 and HOS were treated with CPT for 24 (HaCaT) or 48 h (MG-63 and HOS), with or without 1 μM SCH, and IL-6 levels in supernatants were determined by ELISA. IL-6 levels were normalized to the “CPT only” condition and are shown as percentages. Data shown are averaged from a minimum of three independent experiments in biological replicate (±s.e.m. and Mann-Whitney U tests are shown). (D) HaCaT WT, MG-63 and HOS were treated with CPT for 24 (HaCaT) or 48 h (MG-63 and HOS), with or without decreasing concentrations of SB (at 3, 1.5 and 0.75 μM) and IL-6 levels in supernatants were determined by ELISA. IL-6 levels were normalized to the “CPT only” condition and are shown as percentages. Data shown are averaged from a minimum of three independent experiments in biological replicate (±s.e.m. and ordinary one-way ANOVA with Dunnett’s multiple comparison tests to the “CPT only” condition). *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001 and “ns” is non-significant.
We next studied the effect of p38 and ERK1/2 inhibition in MG-63/HaCaT cells (non-canonical STING) and HOS cells (canonical STING) treated with low-dose CPT. Both inhibitors lead to a significant reduction of CPT-driven IL-6 in the three cell models, without impacting further cell viability (Figures 3C,D, Supplementary Figure S1A), suggesting that they may be suitable to control the production of pro-tumorigenic factors upon DNA damage.
Pharmacological Inhibition of STING Palmitoylation can Lead to Increased Cancer Cell Growth
Having demonstrated the capacity of H151 to block CPT-induced canonical STING signaling in selected cell lines, we next assessed its impact on cancer cell proliferation, independent of DNA damage, compared to p38 and ERK1/2 inhibition. MG-63, HOS, and TC-1 cells were grown in the continuous presence of H151, SB202190 or SCH772984 for 7–12 days in clonogenic assays. Surprisingly, H151 and SB202190 robustly increased clone formation in MG-63 cells (Figures 4A,B). This positive effect of H151 on clone formation was limited to MG-63 cells and reflected by increased growth curves (Figure 4C). However, SB202190 also potentiated the growth of HOS cells and, to a lesser extent, TC-1 cells (Figures 4A,B). Conversely, ERK1/2 inhibition with SCH772984 strongly limited the expansion of MG-63 and TC-1 cells, and modestly impacted that of HOS cells (Figures 4A,B). Having previously shown that MG-63 had a functional cGAS-STING response (Valentin et al., 2021), we reasoned that H151 may block canonical STING signaling basally engaged in these cells, normally restraining their growth. Accordingly, the basal expression of several ISGs (RSAD2, IFIT1, IFIT2, IFIT3) was significantly decreased by H151 treatment in MG-63 cells (Figure 4D). In addition, treatment of MG-63 cells with increasing amounts of type-I IFN significantly decreased the growth of the cells (Figure 4E), supporting the concept that H151 increased cell proliferation through the inhibition of constitutive anti-proliferative interferon signaling.
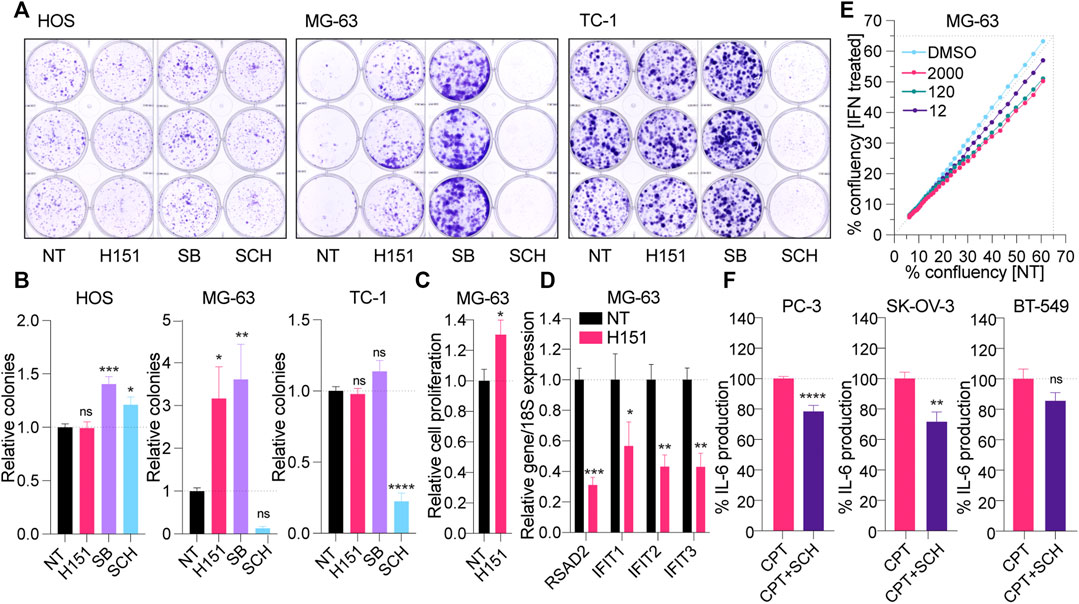
FIGURE 4. (A, B) HOS, MG-63 and TC-1 cells were plated at low density and treated with 3.6 μM H151, 3 μM of the p38 inhibitor SB202190 [SB] or 1 μM of the ERK1/2 inhibitor SCH772984 [SCH] for 10, 12, and 7 days, respectively (see Materials and Methods). The clones formed were stained with crystal violet after fixing (A) and counted manually (B). (B) The number of colonies were reported to the NT condition. The data shown are representative (A) or averaged (B) from three independent experiments (in biological triplicate) (±s.e.m. and ordinary one-way ANOVA with Dunnett’s multiple comparison tests to the “NT” condition). (C) Proliferation of MG-63 cells treated with 3.6 μM H151 was measured by xCELLigence real-time monitoring over 48 h of treatment. Cell proliferation slopes were calculated and normalized to the NT condition. Data shown are averaged from three independent experiments in biological triplicate (±s.e.m. and Mann-Whitney U tests are shown). (D) MG-63 cells were treated or not with 3.6 μM H151 for 48 h, and cell lysates were processed for RNA purification and RT-qPCR analyses. Expression of the indicated genes was reported relative to 18S expression. Data shown are averaged from three independent experiments in biological duplicate and normalized to the NT condition (±s.e.m. and ordinary two-way ANOVA with Sidak’s multiple comparison tests). (E) Relative confluency of MG-63 cells with or without (NT) increasing concentrations of recombinant IFN (12, 120, 2000 IU/ ml) was assessed over 48 h with Incucyte. Data shown are averaged from 6 wells per condition, and trends are representative of three independent experiments. (F) PC-3, SK-OV-3 and BT-549 cells were treated with CPT for 24 h with or without 1 μM SCH, and IL-6 levels in supernatants were determined by ELISA. IL-6 levels were normalized to the “CPT only” condition and are shown as percentages. Data shown are averaged from a minimum of three independent experiments in biological replicate (±s.e.m. and Mann-Whitney U tests are shown). *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001 and “ns” is non-significant.
Finally, since it appeared to limit CPT-driven inflammation from both canonical and non-canonical STING signaling, without promoting cancer cell proliferation, we also tested the effect of ERK1/2 inhibition with SCH772984 on CPT-treated PC-3, SK-OV-3 and BT-549 cells (Figure 4F). Although less potent than in the other cells, SCH772984 significantly reduced CPT-driven IL-6 in PC-3 and SK-OV-3 cells, supporting its broad anti-inflammatory effect independent of how STING is activated (noting that there was no significant effect of SCH772984 on cell viability–Supplementary Figure S1E). Nonetheless, SCH772984 did not significantly reduce IL-6 production in BT-549.
Discussion
Well before its description as a selective agonist of murine Sting (Gao et al., 2013), the small molecule 5,6-dimethylxanthenone-4-acetic acid (DMXAA, or vadimezan) had been characterized as a strong anti-cancer drug that potentiated anti-cancer activities promoted by radio and chemotherapies in syngeneic murine cancer models (recently reviewed in (Le Naour et al., 2020)). Accordingly, several human STING agonists have been developed in recent years by academic and pharmaceutical industry laboratories (Ramanjulu et al., 2018; Chin et al., 2020; Pan et al., 2020), and clinical trials are underway to assess their efficacy against cancers in combination with immune checkpoint inhibitors (Le Naour et al., 2020). While it is clear that STING activation of the immune-cell compartment of the tumor microenvironment can have strong anti-tumoral activities, owing to production of anti-proliferative IFN-β (Parker et al., 2016) and the ensuing recruitment of CD8+T cells (Diamond et al., 2011), the cell-intrinsic role of STING signaling on the growth of cancer cells remains poorly defined.
In the current work, we investigated the cell-intrinsic effects of genotoxic DNA damage on STING signaling in human cancer cells. Our analyses of a dataset of 42 STING-expressing cancer cell lines demonstrated the frequent induction of IL-6 upon topoisomerase 1 and 2 inhibition in
Critically, we showed that IL-6 induced by DNA damage was partially dependent on STING signaling in all the cell lines we tested–as revealed by a significant decrease in IL-6 production with pharmacological inhibition or down-regulation of STING expression. Given that up to 85% (819/934) of the cancer cell lines in the Cancer Cell Line Encyclopedia expressed STING, we speculate that STING-dependent IL-6 induction in response to DNA damage is very frequent in cancer cells. This aligns with the literature supporting that IL-6 and its activation of STAT3 counteracts the effects of radio- and chemotherapy in many cancers (Yang et al., 2020). It remains possible, however, that CPT-driven IL-6 production in select cancer cells is independent of STING, and reliant on alternative pathways involving other innate immune sensors detecting DNA damage from the nucleus or the mitochondria (Burleigh et al., 2020; Tigano et al., 2021). While warranting further studies in larger datasets of cancer cells, this constitutes, to our knowledge, the first direct evidence that cell-intrinsic canonical STING signaling frequently contributes to the production of pro-tumorigenic IL-6 upon DNA damage in cancer cells.
The recent study by Dunphy et al. suggested the existence of a cGAS-independent, non-canonical STING signaling, activated upon DNA damage with the topoisomerase 2 inhibitor Dox in human immortalized and primary keratinocytes (Dunphy et al., 2018). Although the study did not define whether this pathway was frequently invoked upon DNA damage in cancer cells (beyond the case of PMA-differentiated THP-1 cells), it is noteworthy that this alternative STING pathway favored the activation of NF-κB driven pro-inflammatory factors such IL-6, with limited IRF3 signaling (Dunphy et al., 2018).
Here we confirmed the observation from Dunphy et al. that HaCaT cells lacking cGAS can produce IL-6 upon DNA damage, in a STING-dependent manner (Dunphy et al., 2018). In support of the concept of a non-canonical STING signaling pathway, we demonstrated that pharmacological inhibition of STING palmitoylation did not impact CPT-driven IL-6 in these cells. Critically, we provide evidence that the occurrence of non-canonical STING signaling is not limited to keratinocytes, and that it can also be activated by DNA damage in cancer cells such as MG-63 cells. While IL-6 production was dependent on STING expression in MG-63 cells, pharmacological inhibition of canonical STING/TBK1 signaling did not reduce CPT-driven IL-6 in these cells. Although additional experiments would be required to confirm that the STING-dependent responses to DNA damage seen in HaCaT operate the same way in MG-63 cells, the hallmarks of the responses in both cell lines support the concept that they share key similarities. How frequently this non-canonical STING signaling is engaged in human cancers remains to be determined, but the fact that it can be engaged independently of cGAS suggests that it could be relatively common. For example, analyses of TCGA datasets indicate that >30% of high expressing STING lung adenocarcinoma or testicular cancer tumors have low cGAS expression (Supplementary Table S2).
Importantly, albeit failing to respond to H151 inhibition, MG-63 and HaCaT cells both responded to transfected DNA through canonical cGAS-STING signaling (Supplementary Figure S2) (Valentin et al., 2021). Perhaps most surprisingly, we demonstrated that prolonged exposure to H151 increased the growth of MG-63 cells, concurrently with a significant decrease of a basal ISG signature (noting that MG-63 cells are known to produce high levels of type-I IFN) (Billiau et al., 1977). These observations support a basal anti-proliferative activity of STING in MG-63 cells, through the IRF3/IFN arm of the pathway, supported by the reduced growth of the cells cultured in the presence of type-I IFN. This points to the capacity of MG-63 cells to rapidly switch between steady-state canonical STING signaling, most likely resulting from low levels of cytoplasmic DNA, to non-canonical STING signaling activated by acute DNA damage.
The results collectively obtained in MG-63 cells crystalize the duality of the pathway in cancer cells, which can rapidly shift from anti-proliferative to pro-tumorigenic in the context of DNA damage. Given how frequently rapid induction of IL-6 was observed in cancer cells, the current concept that pro-tumorigenic activities of the pathway would be limited to its chronic engagement clearly needs revision (Decout et al., 2021). These findings are also important to our understanding of how to best apply STING agonists in cancer immunotherapy involving DNA damage, since IL-6 was found to inhibit the anti-tumoral effects of STING activation in vivo (Suter et al., 2021).
With the aim of inhibiting the pro-tumorigenic NF-κB branch of STING signaling, but retaining that of IRF3/IFN-β, we discovered that inhibition of ERK1/2 was able to reduce IL-6 production upon canonical and non-canonical STING activation. Critically, ERK1/2 inhibition did not compromise the IRF3 branch of STING signaling, as seen with preserved IP-10 levels in MG-63 cells treated with a human STING agonist. Accordingly, in addition to its own anti-cancer activities (Kidger et al., 2018), pharmacological ERK1/2 inhibition may be a viable strategy to broadly decrease IL-6 production upon DNA damage, while retaining the anti-proliferative effects of the pathway, seen in MG-63 cells. Although further studies are warranted, this is the first description, to our knowledge, that ERK1/2 participate in the production of pro-inflammatory factors downstream of STING. Note that ERK1/2 phosphorylation has been reported in mouse embryonic fibroblasts stimulated with DMXAA (Abe and Barber, 2014). Nevertheless, ERK1/2 inhibition may not universally limit IL-6 production driven by DNA damage in cells where the IRF3 branch of STING signaling dominates the response to DNA damage, as suggested by our results in BT-549 cells.
In conclusion, we demonstrate here that STING is an important contributor to the rapid IL-6 production frequently seen upon DNA damage in cancer cells. Our results collectively indicate that targeting of signaling components operating downstream of STING to modulate NF-κB activity may be more useful than direct STING inhibitors to help prevent production of pro-tumorigenic factors such as IL-6. We propose that pharmacological targeting of ERK1/2, which is already investigated in cancer patients with oncogenic RAS-dependent tumors (Lu et al., 2020), may also help attenuate the resistance to radio- and chemotherapy treatments mediated in part by STING-dependent pro-inflammatory factors, while retaining the anti-tumor activity of the IRF3/IFN-β branch of the pathway.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
SA-A designed, performed and analyzed experiments. AR and TU performed and analyzed experiments. GP performed CRISPR/Cas9 experiments to generate HaCaT mutant cell lines. MG conceived the study and drafted the manuscript. LC and MG coordinated the study, and assisted in design and analysis of experiments. All authors reviewed the results and approved the final version of the manuscript.
Funding
This work was supported by the funding from the Australian National Health and Medical Research Council (1081167 and 1124485 to MG); the Australian Research Council (140100594 Future Fellowship to MG); the Quebec Fonds de Recherche du Québec (FRSQ)—Santé (35071 to GP); Noxopharm Ltd and the Victorian Government’s Operational Infrastructure Support Program. Queensland Government (Advance Queensland Industry Research Fellowship to LVC).
Conflict of Interest
MG receives funding from Noxopharm Ltd. to study the activity of STING inhibitors in cancer. MG does not personally own any shares/equity in Noxopharm Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank N. McMillan for TC-1 cells, S.M. Jane for HaCaT cells, S. Lakhani for BT-549, MDA-MD-231 and HS-578T cells, J. Hooper for SK-OV-3 cells; S. Chu and F. Sharafath for help with the xCELLigence experiments; The Cancer Therapeutics CRC for the GSK human STING agonist; D. De Nardo for the WEHI-112 TBK1 inhibitor; and Frances Cribbin for editorial assistance; we also acknowledge the Monash Health Translational Precinct Research Platforms for access to the RT-qPCR instruments and cell line sequencing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.709618/full#supplementary-material
References
Abe, T., and Barber, G. N. (2014). Cytosolic-DNA-Mediated, STING-dependent Proinflammatory Gene Induction Necessitates Canonical NF- B Activation through TBK1. J. Virol. 88, 5328–5341. doi:10.1128/jvi.00037-14
Ahn, J., Xia, T., Konno, H., Konno, K., Ruiz, P., and Barber, G. N. (2014). Inflammation-driven Carcinogenesis Is Mediated through STING. Nat. Commun. 5, 5166. doi:10.1038/ncomms6166
Bakhoum, S. F., Ngo, B., Laughney, A. M., Cavallo, J.-A., Murphy, C. J., Ly, P., et al. (2018). Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 553, 467–472. doi:10.1038/nature25432
Balka, K. R., Louis, C., Saunders, T. L., Smith, A. M., Calleja, D. J., D’Silva, D. B., et al. (2020). TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-Κb Responses in Myeloid Cells. Cel Rep. 31, 107492. doi:10.1016/j.celrep.2020.03.056
Barretina, J., Caponigro, G., Stransky, N., Venkatesan, K., Margolin, A. A., Kim, S., et al. (2012). The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 483, 603–607. doi:10.1038/nature11003
Billiau, A., Edy, V. G., Heremans, H., Van Damme, J., Desmyter, J., Georgiades, J. A., et al. (1977). Human Interferon: Mass Production in a Newly Established Cell Line, MG-63. Antimicrob. Agents Chemother. 12, 11–15. doi:10.1128/aac.12.1.11
Burleigh, K., Maltbaek, J. H., Cambier, S., Green, R., Gale, M., James, R. C., et al. (2020). Human DNA-PK Activates a STING-independent DNA Sensing Pathway. Sci. Immunol. 5, eaba4219. doi:10.1126/sciimmunol.aba4219
Carozza, J. A., Böhnert, V., Nguyen, K. C., Skariah, G., Shaw, K. E., Brown, J. A., et al. (2020). Extracellular cGAMP Is a Cancer-Cell-Produced Immunotransmitter Involved in Radiation-Induced Anticancer Immunity. Nat. Cancer 1, 184–196. doi:10.1038/s43018-020-0028-4
Chen, Q., Boire, A., Jin, X., Valiente, M., Er, E. E., Lopez-Soto, A., et al. (2016). Carcinoma-astrocyte gap Junctions Promote Brain Metastasis by cGAMP Transfer. Nature 533, 493–498. doi:10.1038/nature18268
Chen, Y.-A., Shen, Y.-L., Hsia, H.-Y., Tiang, Y.-P., Sung, T.-L., and Chen, L.-Y. (2017). Extrachromosomal Telomere Repeat DNA Is Linked to ALT Development via cGAS-STING DNA Sensing Pathway. Nat. Struct. Mol. Biol. 24, 1124–1131. doi:10.1038/nsmb.3498
Chin, E. N., Yu, C., Vartabedian, V. F., Jia, Y., Kumar, M., Gamo, A. M., et al. (2020). Antitumor Activity of a Systemic STING-Activating Non-nucleotide cGAMP Mimetic. Science 369, 993–999. doi:10.1126/science.abb4255
Craig, R., Larkin, A., Mingo, A. M., Thuerauf, D. J., Andrews, C., Mcdonough, P. M., et al. (2000). p38 MAPK and NF-Κb Collaborate to Induce Interleukin-6 Gene Expression and Release. J. Biol. Chem. 275, 23814–23824. doi:10.1074/jbc.m909695199
Dainichi, T., Matsumoto, R., Mostafa, A., and Kabashima, K. (2019). Immune Control by TRAF6-Mediated Pathways of Epithelial Cells in the EIME (Epithelial Immune Microenvironment). Front. Immunol. 10, 1107. doi:10.3389/fimmu.2019.01107
Decout, A., Katz, J. D., Venkatraman, S., and Ablasser, A. (2021). The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat. Rev. Immunol. 21, 548–569. doi:10.1038/s41577-021-00524-z
Diamond, M. S., Kinder, M., Matsushita, H., Mashayekhi, M., Dunn, G. P., Archambault, J. M., et al. (2011). Type I Interferon Is Selectively Required by Dendritic Cells for Immune Rejection of Tumors. J. Exp. Med. 208, 1989–2003. doi:10.1084/jem.20101158
Didonato, J. A., Mercurio, F., and Karin, M. (2012). NF-κB and the Link between Inflammation and Cancer. Immunological Rev. 246, 379–400. doi:10.1111/j.1600-065x.2012.01099.x
Dou, Z., Ghosh, K., Vizioli, M. G., Zhu, J., Sen, P., Wangensteen, K. J., et al. (2017). Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 550, 402–406. doi:10.1038/nature24050
Dunphy, G., Flannery, S. M., Almine, J. F., Connolly, D. J., Paulus, C., Jønsson, K. L., et al. (2018). Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-Κb Signaling after Nuclear DNA Damage. Mol. Cel 71, 745–760. e745. doi:10.1016/j.molcel.2018.07.034
Gao, P., Ascano, M., Zillinger, T., Wang, W., Dai, P., Serganov, A. A., et al. (2013). Structure-Function Analysis of STING Activation by c[G(2′,5′)pA(3′,5′)p] and Targeting by Antiviral DMXAA. Cell 154, 748–762. doi:10.1016/j.cell.2013.07.023
Glück, S., Guey, B., Gulen, M. F., Wolter, K., Kang, T.-W., Schmacke, N. A., et al. (2017). Innate Immune Sensing of Cytosolic Chromatin Fragments through cGAS Promotes Senescence. Nat. Cel Biol 19, 1061–1070. doi:10.1038/ncb3586
Haag, S. M., Gulen, M. F., Reymond, L., Gibelin, A., Abrami, L., Decout, A., et al. (2018). Targeting STING with Covalent Small-Molecule Inhibitors. Nature 559, 269–273. doi:10.1038/s41586-018-0287-8
Harding, S. M., Benci, J. L., Irianto, J., Discher, D. E., Minn, A. J., and Greenberg, R. A. (2017). Mitotic Progression Following DNA Damage Enables Pattern Recognition within Micronuclei. Nature 548, 466–470. doi:10.1038/nature23470
Ho, S. S. W., Zhang, W. Y. L., Tan, N. Y. J., Khatoo, M., Suter, M. A., Tripathi, S., et al. (2016). The DNA Structure-specific Endonuclease MUS81 Mediates DNA Sensor STING-dependent Host Rejection of Prostate Cancer Cells. Immunity 44, 1177–1189. doi:10.1016/j.immuni.2016.04.010
Janovec, V., Aouar, B., Font-Haro, A., Hofman, T., Trejbalova, K., Weber, J., et al. (2018). The MEK1/2-ERK Pathway Inhibits Type I IFN Production in Plasmacytoid Dendritic Cells. Front. Immunol. 9, 364. doi:10.3389/fimmu.2018.00364
Kidger, A. M., Sipthorp, J., and Cook, S. J. (2018). ERK1/2 Inhibitors: New Weapons to Inhibit the RAS-Regulated RAF-Mek1/2-Erk1/2 Pathway. Pharmacol. Ther. 187, 45–60. doi:10.1016/j.pharmthera.2018.02.007
Le Naour, J., Zitvogel, L., Galluzzi, L., Vacchelli, E., and Kroemer, G. (2020). Trial Watch: STING Agonists in Cancer Therapy. OncoImmunology 9, 1777624. doi:10.1080/2162402x.2020.1777624
Lemos, H., Mohamed, E., Huang, L., Ou, R., Pacholczyk, G., Arbab, A. S., et al. (2016). STING Promotes the Growth of Tumors Characterized by Low Antigenicity via Ido Activation. Cancer Res. 76, 2076–2081. doi:10.1158/0008-5472.can-15-1456
Li, S., Wang, N., and Brodt, P. (2012). Metastatic Cells Can Escape the Proapoptotic Effects of TNF-α through Increased Autocrine IL-6/STAT3 Signaling. Cancer Res. 72, 865–875. doi:10.1158/0008-5472.can-11-1357
Lin, K. Y., Guarnieri, F. G., Staveley-O'carroll, K. F., Levitsky, H. I., August, J. T., Pardoll, D. M., et al. (1996). Treatment of Established Tumors with a Novel Vaccine that Enhances Major Histocompatibility Class II Presentation of Tumor Antigen. Cancer Res. 56, 21–26.
Lu, Y., Liu, B., Liu, Y., Yu, X., and Cheng, G. (2020). Dual Effects of Active ERK in Cancer: A Potential Target for Enhancing Radiosensitivity (Review). Oncol. Lett. 20, 993–1000. doi:10.3892/ol.2020.11684
Mackenzie, K. J., Carroll, P., Martin, C.-A., Murina, O., Fluteau, A., Simpson, D. J., et al. (2017). cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature 548, 461–465. doi:10.1038/nature23449
Marcus, A., Mao, A. J., Lensink-Vasan, M., Wang, L., Vance, R. E., and Raulet, D. H. (2018). Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763. e754. doi:10.1016/j.immuni.2018.09.016
Monks, A., Zhao, Y., Hose, C., Hamed, H., Krushkal, J., Fang, J., et al. (2018). The NCI Transcriptional Pharmacodynamics Workbench: A Tool to Examine Dynamic Expression Profiling of Therapeutic Response in the NCI-60 Cell Line Panel. Cancer Res. 78, 6807–6817. doi:10.1158/0008-5472.can-18-0989
Morris, E. J., Jha, S., Restaino, C. R., Dayananth, P., Zhu, H., Cooper, A., et al. (2013). Discovery of a Novel ERK Inhibitor with Activity in Models of Acquired Resistance to BRAF and MEK Inhibitors. Cancer Discov. 3, 742–750. doi:10.1158/2159-8290.cd-13-0070
Mukai, K., Konno, H., Akiba, T., Uemura, T., Waguri, S., Kobayashi, T., et al. (2016). Activation of STING Requires Palmitoylation at the Golgi. Nat. Commun. 7, 11932. doi:10.1038/ncomms11932
Nassour, J., Radford, R., Correia, A., Fusté, J. M., Schoell, B., Jauch, A., et al. (2019). Autophagic Cell Death Restricts Chromosomal Instability during Replicative Crisis. Nature 565, 659–663. doi:10.1038/s41586-019-0885-0
Pan, B. S., Perera, S. A., Piesvaux, J. A., Presland, J. P., Schroeder, G. K., Cumming, J. N., et al. (2020). An Orally Available Non-nucleotide STING Agonist with Antitumor Activity. Science 369, eaba6098. doi:10.1126/science.aba6098
Parker, B. S., Rautela, J., and Hertzog, P. J. (2016). Antitumour Actions of Interferons: Implications for Cancer Therapy. Nat. Rev. Cancer 16, 131–144. doi:10.1038/nrc.2016.14
Pépin, G., Nejad, C., Ferrand, J., Thomas, B. J., Stunden, H. J., Sanij, E., et al. (2017a). Topoisomerase 1 Inhibition Promotes Cyclic GMP-AMP Synthase-dependent Antiviral Responses. mBio 8, e01611–01617. doi:10.1128/mBio.01611-17
Pépin, G., Nejad, C., Thomas, B. J., Ferrand, J., Mcarthur, K., Bardin, P. G., et al. (2017b). Activation of cGAS-dependent Antiviral Responses by DNA Intercalating Agents. Nucleic Acids Res. 45, 198–205. doi:10.1093/nar/gkw878
Phong, M. S., Van Horn, R. D., Li, S., Tucker-Kellogg, G., Surana, U., and Ye, X. S. (2010). p38 Mitogen-Activated Protein Kinase Promotes Cell Survival in Response to DNA Damage but Is Not Required for the G 2 DNA Damage Checkpoint in Human Cancer Cells. Mol. Cel Biol 30, 3816–3826. doi:10.1128/mcb.00949-09
Ramanjulu, J. M., Pesiridis, G. S., Yang, J., Concha, N., Singhaus, R., Zhang, S.-Y., et al. (2018). Design of Amidobenzimidazole STING Receptor Agonists with Systemic Activity. Nature 564, 439–443. doi:10.1038/s41586-018-0705-y
Schadt, L., Sparano, C., Schweiger, N. A., Silina, K., Cecconi, V., Lucchiari, G., et al. (2019). Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cel Rep. 29, 1236–1248. e1237. doi:10.1016/j.celrep.2019.09.065
Suter, M. A., Tan, N. Y., Thiam, C. H., Khatoo, M., Macary, P. A., Angeli, V., et al. (2021). cGAS-STING Cytosolic DNA Sensing Pathway Is Suppressed by JAK2-STAT3 in Tumor Cells. Sci. Rep. 11, 7243. doi:10.1038/s41598-021-86644-x
Takashima, K., Takeda, Y., Oshiumi, H., Shime, H., Okabe, M., Ikawa, M., et al. (2016). STING in Tumor and Host Cells Cooperatively Work for NK Cell-Mediated Tumor Growth Retardation. Biochem. Biophysical Res. Commun. 478, 1764–1771. doi:10.1016/j.bbrc.2016.09.021
Tian, J., Zhang, D., Kurbatov, V., Wang, Q., Wang, Y., Fang, D., et al. (2021). 5‐Fluorouracil Efficacy Requires Anti‐tumor Immunity Triggered by Cancer‐cell‐intrinsic STING. EMBO J. 40, e106065. doi:10.15252/embj.2020106065
Tigano, M., Vargas, D. C., Tremblay-Belzile, S., Fu, Y., and Sfeir, A. (2021). Nuclear Sensing of Breaks in Mitochondrial DNA Enhances Immune Surveillance. Nature 591, 477–481. doi:10.1038/s41586-021-03269-w
Valentin, R., Wong, C., Alharbi, A. S., Pradeloux, S., Morros, M. P., Lennox, K. A., et al. (2021). Sequence-dependent Inhibition of cGAS and TLR9 DNA Sensing by 2′-O-Methyl Gapmer Oligonucleotides. Nucleic Acids Res. 49, 6082–6099. doi:10.1093/nar/gkab451
Vanpouille-Box, C., Alard, A., Aryankalayil, M. J., Sarfraz, Y., Diamond, J. M., Schneider, R. J., et al. (2017). DNA Exonuclease Trex1 Regulates Radiotherapy-Induced Tumour Immunogenicity. Nat. Commun. 8, 15618. doi:10.1038/ncomms15618
Wei, F., Yan, J., and Tang, D. (2011). Extracellular Signal-Regulated Kinases Modulate DNA Damage Response - A Contributing Factor to Using MEK Inhibitors in Cancer Therapy. Cmc 18, 5476–5482. doi:10.2174/092986711798194388
Wu, X., Yang, J., Na, T., Zhang, K., Davidoff, A. M., Yuan, B.-Z., et al. (2017). RIG-I and IL-6 Are Negative-Feedback Regulators of STING Induced by Double-Stranded DNA. PLoS One 12, e0182961. doi:10.1371/journal.pone.0182961
Wu, Z., Oeck, S., West, A. P., Mangalhara, K. C., Sainz, A. G., Newman, L. E., et al. (2019). Mitochondrial DNA Stress Signalling Protects the Nuclear Genome. Nat. Metab. 1, 1209–1218. doi:10.1038/s42255-019-0150-8
Yamazaki, T., Kirchmair, A., Sato, A., Buqué, A., Rybstein, M., Petroni, G., et al. (2020). Mitochondrial DNA Drives Abscopal Responses to Radiation that Are Inhibited by Autophagy. Nat. Immunol. 21, 1160–1171. doi:10.1038/s41590-020-0751-0
Yang, P.-L., Liu, L.-X., Li, E.-M., and Xu, L.-Y. (2020). STAT3, the Challenge for Chemotherapeutic and Radiotherapeutic Efficacy. Cancers 12, 2459. doi:10.3390/cancers12092459
Yun, U. J., Park, S. E., Jo, Y. S., Kim, J., and Shin, D. Y. (2012). DNA Damage Induces the IL-6/STAT3 Signaling Pathway, Which Has Anti-senescence and Growth-Promoting Functions in Human Tumors. Cancer Lett. 323, 155–160. doi:10.1016/j.canlet.2012.04.003
Keywords: STING, IL-6, cancer, DNA damage, STING inhibitor, ERK1/2, Non-canonical STING
Citation: Al-Asmari SS, Rajapakse A, Ullah TR, Pépin G, Croft LV and Gantier MP (2022) Pharmacological Targeting of STING-Dependent IL-6 Production in Cancer Cells. Front. Cell Dev. Biol. 9:709618. doi: 10.3389/fcell.2021.709618
Received: 14 May 2021; Accepted: 20 December 2021;
Published: 11 January 2022.
Edited by:
James A. L. Brown, University of Limerick, IrelandReviewed by:
Niamh Buckley, Queen’s University Belfast, United KingdomLei Huang, Newcastle University, United Kingdom
Copyright © 2022 Al-Asmari, Rajapakse, Ullah, Pépin, Croft and Gantier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura V. Croft, bGF1cmEuY3JvZnRAcXV0LmVkdS5hdQ==; Michael P. Gantier, bWljaGFlbC5nYW50aWVyQGh1ZHNvbi5vcmcuYXU=
†These authors share senior authorship