 Tamara Salloum
Tamara Salloum Sima Tokajian
Sima Tokajian Robert P. Hirt
Robert P. Hirt- 1Department of Natural Sciences, School of Arts and Sciences, Lebanese American University, Byblos, Lebanon
- 2Faculty of Medical Sciences, Biosciences Institute, Newcastle University, Newcastle upon Tyne, United Kingdom
Leishmaniasis is a vector-borne disease caused by a protozoa parasite from over 20 Leishmania species. The clinical manifestations and the outcome of the disease vary greatly. Global RNA sequencing (RNA-Seq) analyses emerged as a powerful technique to profile the changes in the transcriptome that occur in the Leishmania parasites and their infected host cells as the parasites progresses through their life cycle. Following the bite of a sandfly vector, Leishmania are transmitted to a mammalian host where neutrophils and macrophages are key cells mediating the interactions with the parasites and result in either the elimination the infection or contributing to its proliferation. This review focuses on RNA-Seq based transcriptomics analyses and summarizes the main findings derived from this technology. In doing so, we will highlight caveats in our understanding of the parasite’s pathobiology and suggest novel directions for research, including integrating more recent data highlighting the role of the bacterial members of the sandfly gut microbiota and the mammalian host skin microbiota in their potential role in influencing the quantitative and qualitative aspects of leishmaniasis pathology.
Introduction
Leishmaniasis is caused by a parasitic protozoan carried by over 90 sandfly species which are known to transmit more than 20 species of Leishmania parasites to humans, through either zoonotic or anthroponotic infection cycles (Ready, 2013; Rostamian and Niknam, 2019; World Health Organization [WHO], 2019). Three main forms of the disease exist and range in severity from mutilating cutaneous leishmaniases (CL) causing skin lesions and ulcers, mucocutaneous leishmaniasis (MCL) leading to a partial or total destruction of mucous membranes of the nose, mouth and throat and visceral leishmaniasis (VL), also known as kala-azar, affecting the spleen and liver and causing mortality in over 95% of untreated cases (World Health Organization [WHO], 2019).
Leishmania parasites, when alternating between the sandfly vector and the mammalian host, have two major corresponding life stages: promastigotes living in the sandfly’s gut and amastigotes residing inside mammalian macrophages (Figure 1). Following a blood meal, the sandfly ingests a macrophage containing Leishmania amastigotes. Once liberated from the macrophage inside the sandfly midgut, amastigotes differentiate into procyclic promastigotes. Then, the procyclic promastigotes become nectomonad promastigotes, which are able to cross the protective peritrophic matrix of the sandfly gut and attach to the microvilli of the epithelial cells of the midgut (Bates, 1994; Gossage et al., 2003; Bates, 2007; Sunter and Gull, 2017). From there, they can migrate to the thoracic midgut and stomodeal valve where they differentiate into leptomonad promastigotes. The leptomonad promastigotes can eventually differentiate into either haptomonad promastigotes, which attach to the stomodeal valve, or metacyclic promastigotes, which are the mammalian infective form. Metacyclic promastigotes are transmitted to the host by the sandfly during the next blood meal (Bates, 1994; Sacks et al., 2000; Rogers et al., 2002; Bates, 2007; Dillon et al., 2015a).
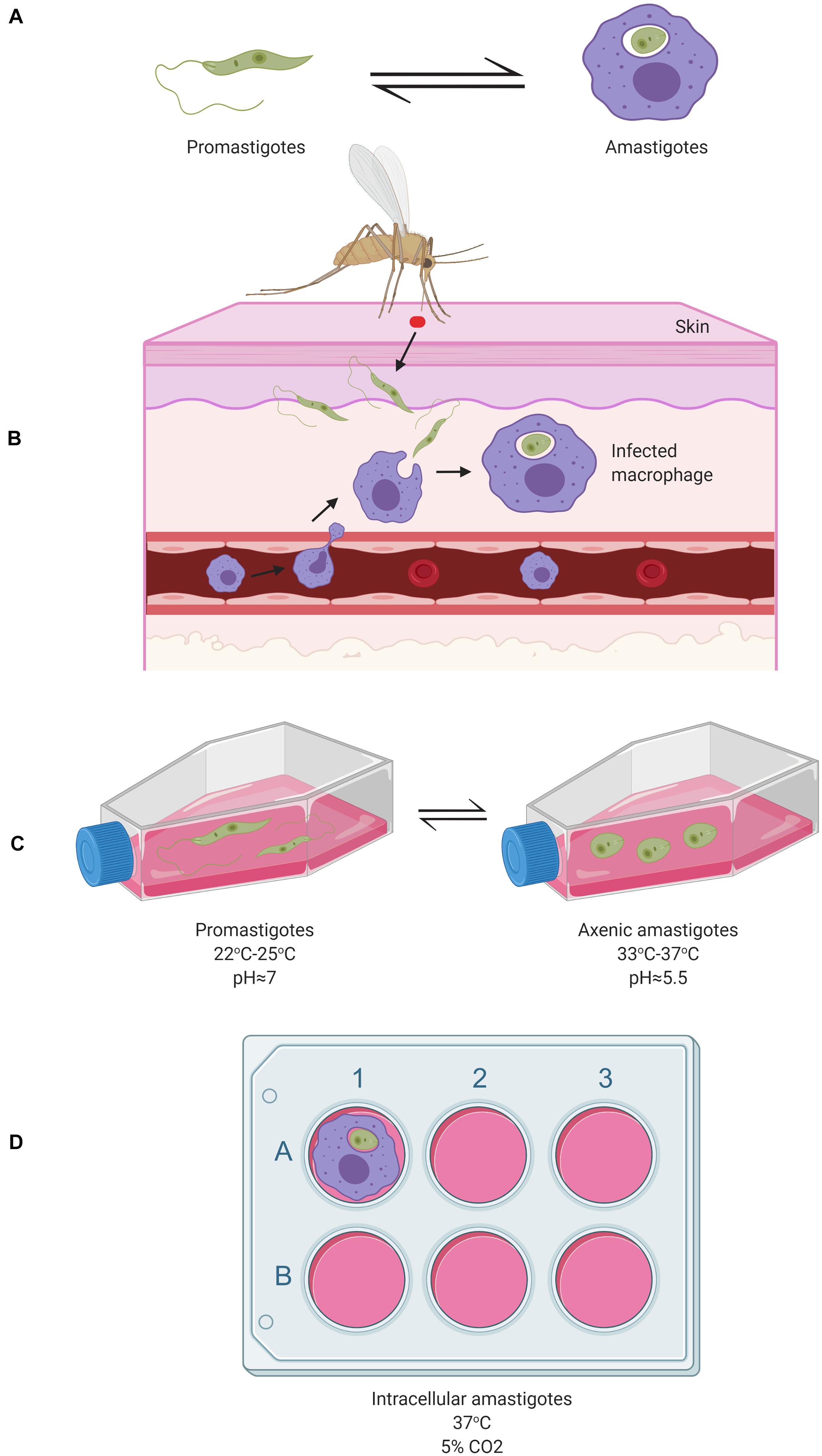
Figure 1. Schematic representation of the two major Leishmania cellular forms during their digenic life cycle in vivo and their study in corresponding in vitro model systems. (A) The flagellated motile promastigotes form (left) in the gut of a sandfly and the round intracellular amastigotes form residing inside infected mammalian macrophages (right), represent the two major cellular forms of Leishmania species. (B) Under in vivo conditions, sandfly vectors inject Leishmania promastigotes into the host’s skin following a blood meal which are then taken up by macrophages where they differentiate into amastigotes; (C) Under in vitro conditions, axenic promastigotes can be maintained in cell culture flasks in a rich, 10%- Fetal Bovine Serum (FBS)-supplemented medium (e.g., RPMI 1640, Schneider, Grace, or M199) at 22–25°C (left). These can be differentiated in vitro into axenic amastigotes in a medium at an acidic pH∼5.5 (Achieved through the addition of HCl) at 33–37°C (right) (Teixeira et al., 2002); (D) Macrophages (e.g., from human or mouse), can be seeded in 6-well cell plates in RPMI media supplemented with 10% FBS at 37°C to which the parasites are added resulting in macrophages infection within 4 to 24 h. More detailed aspects of the Leishmania life cycle are reviewed by Sunter and Gull (2017). Image created with BioRender.com.
The diverse pathologies associated with Leishmania infections develop following excessive inflammatory responses by the infected tissues due to dis-regulated immune responses, which are increasingly appreciated to be triggered by complex network of interactions between the parasites, host immunocytes, the bacteria from the human skin microbiota and sandfly vector gut microbiota in addition to Leishmania RNA viruses (LRV) known to infect some Leishmania species (Ives et al., 2011; Brodskyn and de Oliveira, 2012; Gimblet et al., 2017; Cruz and Freitas-Castro, 2019) (Figure 2). Understanding the complex interplay between these factors involved in the host-parasite-microbiota interactions during Leishmania infection is crucial to refine the design of in vivo and in vitro Leishmania infection models and assays to study the biology of these parasites in more details and to eventually develop more effective prophylactic and therapeutic strategies with minimal or no side effects (Figure 2).
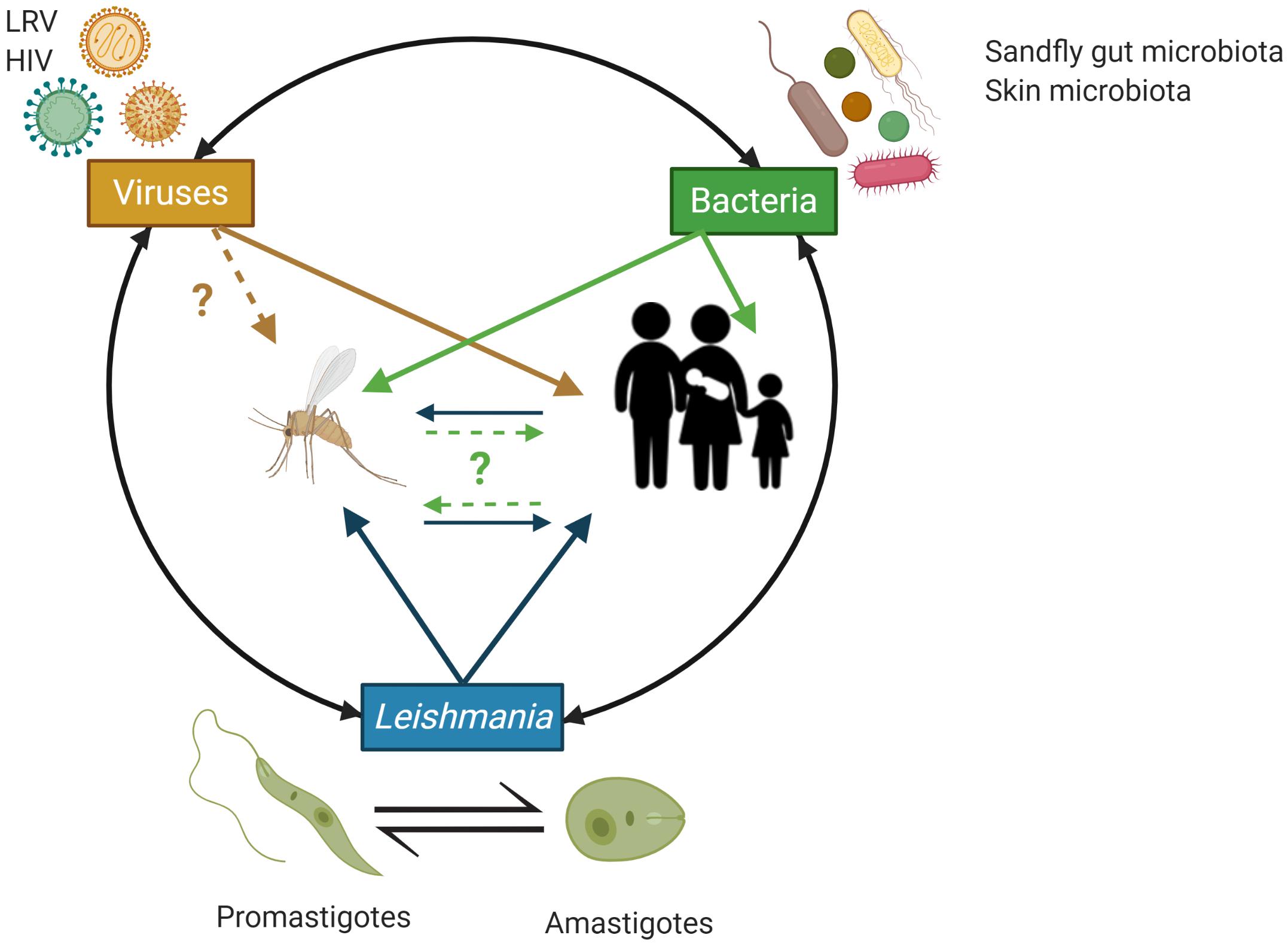
Figure 2. Schematic overview of the potential trans-kingdom cross talk between Leishmania, viruses (including Leishmania RNA virus – LRV) and bacteria from the implicated animal hosts microbiota and their impact on the human host. Animal hosts (here, sand flies and humans) are increasingly recognized to be supra-organisms made of animal cells/tissues/organs and their associated microbiota (e.g., associated with the human skin and mucosal surfaces) that through complex, continuous and highly dynamic interactions contribute to the host phenotype in both health and disease. In the context of infections by Leishmania, initiated by the bite of a sand fly, bacteria from the insect gut microbiota, the human skin microbiota and Leishmania RNA virus (LRV), which can infect Leishmania, have all be implicated in regulating the inflammatory tone associated with the infection by Leishmania and by doing so to the various pathobiologies associated with Leishmania infections. HIV is also indicated as AIDS can facilitate/worsen the pathobiology or Leishmania infections through weakening the host immune response and represent an important case of co-infections with negative impact on the human host. Image created with BioRender.com.
Although numerous studies have employed microarrays and RT-PCR to profile gene expression changes in Leishmania and host macrophages following infection (McNicoll et al., 2006; Srividya et al., 2007; Guerfali et al., 2008; Lin et al., 2008; Kumari et al., 2008; y Fortéa et al., 2009; Alcolea et al., 2010, 2011; Adaui et al., 2011; Probst et al., 2012; Rabhi et al., 2012, 2013; Beattie et al., 2013), global RNA sequencing (RNA-Seq) analyses emerged as a powerful methodology to generate a more global and systematic analyses of the transcriptome of the parasite and relevant cells from the concomitant insect vector and mammalian host. This review focuses on RNA-Seq based analyses of the transcriptome of Leishmania-hosts interactions and summarizes the main findings extracted using this technology. This includes the more detailed understanding of the interactions between human macrophages and Leishmania parasites and the differentiation of the parasite along its various cellular forms characteristic of its complex life cycle. By doing so we will highlight current caveats in our understanding of the parasite’s pathobiology and speculate on how RNA-Seq investigations will be able to further contribute at reducing our knowledge gap on the molecular and cellular basis of host-parasite interactions among these fascinating and complex parasites along with their microbial endosymbionts (e.g., LRV) and microbial neighbors (insect gut and mammalian skin microbiotas). This will integrate the more recent insights into the potential role played by other microbes associated with Leishmania environment, including bacteria members of the insect gut microbiota and the mammalian host skin microbiota.
Control of Gene Expression in Leishmania
Leishmania and related trypanosomatids have unusual mechanisms to control gene expression (Clayton, 2016). Little transcriptional regulation appears to exist as non-functionally related genes arranged in large clusters are constantly transcribed into long polycistronic precursor RNAs (Martıìnez-Calvillo et al., 2003). Large polycistronic transcription units (PTUs) may be comprised of more than hundred genes with no obvious/apparent functional relationship (Beverley, 2003; Ivens et al., 2005). Kinetoplastid parasites rely almost exclusively on post-transcriptional gene regulation because of their constitutive transcription of RNA polymerase II (pol II)-driven polycistronic gene arrays (de Pablos et al., 2016). Accordingly, RNA binding proteins (RBPs) act as primary gene regulators and are overrepresented in the proteome (de Pablos et al., 2019). RNA binding proteins dynamically bind to mRNA forming ribonucleoprotein complexes (mRNPs) and regulate the trafficking and processing of mRNA molecules from synthesis to decay (Gehring et al., 2017). Moreover, it has been shown that in Leishmania mexicana parasites, mRNA levels are not a strong predictor of whole cell RNA expression or RNA binding potential of encoded proteins (de Pablos et al., 2019). Thus, the mechanisms mRNA stability, decay or translation are controlled in near absence of transcriptional regulation are still not fully elucidated (de Pablos et al., 2016).
Following transcription, precursor transcripts are processed into mature mRNAs coding for individual proteins through an unusual process known as trans-splicing (Matthews et al., 1994; Liang et al., 2003; Kramer and Carrington, 2011). During trans-splicing at specific positions, a polycistronic precursor is removed and a 39 bp long mini-exon is added to the 5’end of all mRNAs. The added mini-exon is also known as the spliced leader (SL) (LeBowitz et al., 1993). Then, a poly-A tail is added to the 3′-end of the mRNA. As such, the polyadenylation of the upstream gene is directed by and coupled to the trans-splicing of the downstream gene (LeBowitz et al., 1993). The mRNA is then exported to the cytoplasm where it can be recognized by the translation machinery by a highly modified 5′-cap structure on the mini-exon part (Freire et al., 2017). Notably, despite of the polycistronic transcription approach being used, adjacent genes often do not have the same levels of gene expression (Rastrojo et al., 2013). Indeed, some collinear mRNAs can have highly distinct steady-state expression levels (Rastrojo et al., 2013).
Despite these challenging and interesting/unusual features of the Leishmania transcriptome, variation in mRNA abundance has been observed between different life stages of the digenic infection cycles where mRNA levels were not considered as a strong predictor of the whole cell expression and protein levels (Lahav et al., 2011; de Pablos et al., 2016, 2019).
Advantages and Limitations of RNA-Seq Technology
Global RNA sequencing and transcriptomics analysis illustrates significant variations in gene expression occurring during host infection with Leishmania, its survival inside the sandfly vector gut and during its differentiation between its two life forms as amastigotes and promastigotes. The implementation of RNA-Seq for studying gene expression in Leishmania culminated in a rapid expansion of the available knowledge with regard to the host parasite interactions, the existing variations, the relationship between the parasite and its vector and the differentiation of the parasite (Shadab et al., 2019). Haydock et al. (2015) developed the RNA-Seq-based protocol to exploit the 39 bp SL found at the 5′-end of all Leishmania mRNA transcripts (Haydock et al., 2015).
Numerous studies on gene expression profiling were performed in Leishmania under various conditions and on its different life stages (McNicoll et al., 2006; Srividya et al., 2007; Guerfali et al., 2008; Lin et al., 2008; Kumari et al., 2008; y Fortéa et al., 2009; Alcolea et al., 2010, 2011; Adaui et al., 2011; Probst et al., 2012; Rabhi et al., 2012, 2013; Beattie et al., 2013). RNA-Seq provided, however, several advantages over other less advanced gene expression profiling approaches such as RT-PCR and microarrays (Nowrousian, 2010). As opposed to hybridization-based methodologies, RNA-Seq can be used without the availability of previous genomic data, which is particularly useful in studying non-model organisms (Vera et al., 2008). It is also less dependent on complete gene annotations as it can identify new genes missed in initial annotations of the genomes of interest (Dillon et al., 2015a). Additionally, RNA-Seq has very low background signal when compared to DNA microarrays having a large dynamic range of expression levels without an upper limit for quantification (Wang et al., 2009). It also provides more accurate quantification of expression levels which could be confirmed through qPCR (Nagalakshmi et al., 2008). Finally, less starting material, total RNA, is required and the results obtained can be highly reproducible for technical and biological replicates (Nagalakshmi et al., 2008; Wang et al., 2009). The most frequently used parameter to measure mRNA abundance based on RNA-Seq data is fragments–or reads-per kilobase of transcript per million mapped reads (FPKM). FPMK represents transcript abundance by considering the RNA length and the total read number in the measurement (Mortazavi et al., 2008). However, other units have been shown to be more effective at contrasting mRNA abundance between samples, including transcripts per millions reads (TPM) (Wagner et al., 2012)
With the vast amount of data generated, there are many opportunities to “translate” this data into forms that can be exploited for drug and vaccine development. The comparison of results between various RNA-seq studies has been hindered by differences in the parasite host, developmental stages of the parasite and experiment lab conditions (for instance, hours of infection) (Dillon et al., 2015a) as such various variables must be addressed and standardized during experimental design to allow for accurate comparative studies of differential gene expression.
Notably, RNA-Seq experiments can also be used to perform metatranscriptomics analyses, reviewed below, at the infection sites and covering the variations in host skin microbiota and insect gut microbiota can provide additional insights into the complex relationships between the bacterial members of the sandfly gut microbiota and the mammalian host skin microbiota in shaping the disease outcome as well as the potential role of LRV present in at least some species of Leishmania.
Sandfly Vector Transcriptomics
Isolating promastigotes from the natural microenvironment (i.e., the vector host) is one potentially useful approach but technically challenging (Alcolea et al., 2016). Previously, in vitro infectivity and differential gene expression have been studied in Leishmania infantum promastigotes isolated from the stomodeal valve of the sandfly Phlebotomus perniciosus (Alcolea et al., 2016). Differential gene expression was determined by RNA shotgun genome microarray hybridization analysis and showed that most differentially expressed genes were involved in regulation of gene expression, intracellular signaling, amino acid metabolism and biosynthesis of surface molecules (Alcolea et al., 2016). Microarray hybridization analysis, however, cannot account for the presence of bacterial or viral genes (Schulze and Downward, 2001). Thus, metatranscriptomics analysis using RNA-Seq on Leishmania promastigotes isolated from the inside of the stomodeal valve of the sandfly of the vector could potentially provide more information on the complex relationship existing between parasites, viruses and bacteria co-existing in the vector.
The diversity of the natural gut microbiota of sandflies is acquired from several sources, including feeding on their respective animal and plant sources of blood and sugar, or re-colonization of the gut by the microbes ingested by the terrestrial dwelling larval stages (Dillon et al., 1996; Hillesland et al., 2008; Mukhopadhyay et al., 2012; Peterkova-Koci et al., 2012; Sant’Anna et al., 2012).
A recent 16S rRNA sequence profiling of the gut microbiota from Lutzomyia longipalpis, the primary vector of VL in Brazil, revealed 13 distinct bacterial genera (Bacillus, Enterococcus, Erwinia, Enterobacter, Escherichia, Klebsiella, Lysinibacillus, Pseudocitrobacter, Providencia, Pseudomonas, Serratia, Staphylococcus, and Solibacillus) (Campolina et al., 2020). Following a co-cultivation of the identified bacteria with various Leishmania species in in vitro conditions, a growth reduction in all tested parasites was observed suggesting a potential role of the gut microbiota in hindering parasite transmission by the sandfly vector (Campolina et al., 2020). In contrast, the bacterial communities naturally present in the Phlebotomus duboscqi sandfly midgut were shown to be essential for the colonization of the midgut by infective stage, metacyclic Leishmania major promastigotes (Louradour et al., 2017).
Although insects are hosts to a vast variety of viruses (Telleria et al., 2018), relatively little is known about viruses infecting sandflies (Depaquit et al., 2010). These include phleboviruses such as Toscana viruses (TOSV) (Depaquit et al., 2010). As such, a recent metatranscriptomics analysis of individual mosquitoes unveiled their blood meal sources and uncovered a rich microbial cargo consisting of eukaryotes, prokaryotes, and a high frequency of viral co-infection with 70 known and novel viral species (Batson et al., 2020).
The salivary transcriptome of the Nyssomyia neivai sandfly, one of the main vectors of tegumentary leishmaniasis in Brazil, has also been recently performed and highlighted the abundances of several N. neivai salivary proteins which can be used as biomarkers of N. neivai (Vernal et al., 2020). Thus, looking into the microbial diversity of sandfly vectors gut is of particular interest. Such experiments could also compare and contrast various species of sandfly vectors.
Findings in Leishmania Global Transcriptomics
The first RNA-Seq global gene expression profiling was performed on L. major in 2013 (Rastrojo et al., 2013). A time-based evolution of RNA-Seq experiments conducted on Leishmania parasites is presented in Figure 3. Rastrojo et al. (2013) investigated the transcriptome of L. major axenic promastigotes and identified a total of 10,285 transcripts including 1,884 novel transcripts that did not match genes previously annotated. They observed and reported the presence of extensive heterogeneity in the SL and polyadenylation addition sites. A sequence comparison of ribosomal protein L23 showed that both genes have identical coding regions, but marked differences both in length and sequence in the 3′-UTRs., which could be linked to the efficiency of the mRNA translation (Rastrojo et al., 2013). Most abundant transcripts included cytosolic heat shock protein 70 (HSP70), various ribosomal proteins, nucleoside transporters, histone H4, peptidases, cyclophilin, Leishmania-activated C-kinase antigen (LACK), amastin-like surface protein and alpha tubulin (Rastrojo et al., 2013).
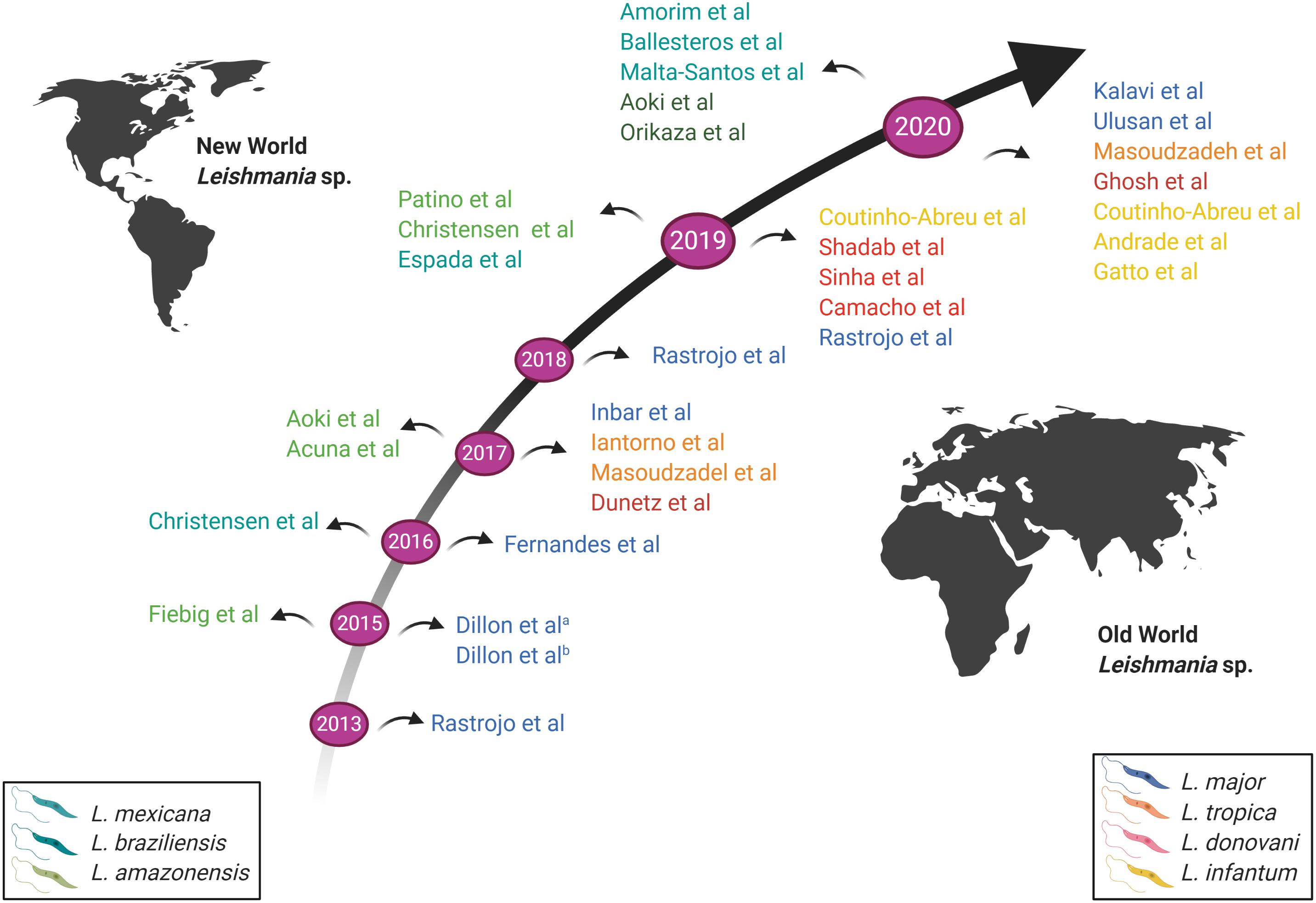
Figure 3. Schematic representation of the time-based evolution of RNA-seq conducted on Leishmania parasites. Studies above the arrow represent RNA-Seq conducted on New World Leishmania species; studies below the arrow represent RNA-Seq conducted on Old World Leishmania species. The various Leishmania species studied are indicated by different colors, as shown in the legend. New World Leishmania species are shown on the left; Old World Leishmania species are shown on the right. Image created with BioRender.com.
Later Fiebig et al. (2015) used RNA-Seq to characterize and compare the transcriptomes of L. mexicana promastigotes, axenic and intracellular amastigotes. A comparative analysis of gene expression between promastigotes and amastigotes revealed 3,832 significantly differentially expressed among 9,169 protein-coding genes. Genes associated with the motility of the flagellum were mainly downregulated while those linked to cell surface proteins, transporters, peptidases, and a number of uncharacterized proteins were upregulated in amastigotes. Around 936 novel transcripts were identified providing the first evidence of a link between the whole chromosome duplication event and adaptation to the vertebrate host in this group. Gene ontology-term (GO-term) enrichment analyses exploit the GO system of classification to link genes based on functional characteristics (Bauer et al., 2008). This approach revealed that unfolded protein binding, protein folding, aminoacyl-tRNA ligase activity and tRNA aminoacylation (for protein translation and microtubule motor activity) were preferentially expressed in promastigotes compared to intracellular amastigotes in L. mexicana. Genes related to the cytoplasm, calmodulin binding, microtubule-based flagellum, protein folding, and aminoacyl-tRNA ligase activity were preferentially expressed in promastigotes when compared to axenic amastigotes. Chromosome 30 carried genes the expression of which was upregulated in amastigotes suggesting a link between chromosome duplication, gene expression, and parasite host adaptation. Finally, genes having a GO-term annotation related to nucleosome, nucleosome assembly, DNA binding, and DNA replication were preferentially expressed in axenic amastigotes versus intracellular amastigotes (Fiebig et al., 2015).
The differences in gene expression occurring as Leishmania acquires its infectivity in vitro were also assessed. Dillon et al. (2015a) conducted a global transcriptomic analysis on L. major as it gained its infectivity comparing non-infective procyclic promastigotes and infective metacyclic promastigotes. In doing so, they detected 3,138 differentially expressed genes that included iron/zinc transporters, histone H4, adenine amino hydrolysases and 1,044 novel open reading frames (ORFs). Using GO analysis, they revealed that ATP synthesis coupled proton transport and cytoplasm related genes were among the downregulated genes in metacyclic promastigotes. On the other hand, genes encoding for protein kinases and ATP binding were upregulated during differentiation from non-infective procyclic promastigotes and infective metacyclic promastigotes. A widespread alternative trans-splicing and polyadenylation was also detected in 8,981 genes (94.2% of a total of 9,530 genes). 5′ and 3′ UTR boundaries for a majority of genes were also identified. No association was found between stage-specific preferential trans-splicing or polyadenylation sites and differentially expressed genes. As a result, differences in gene expression levels could not be attributed to stage-regulated alternative mRNA processing (Dillon et al., 2015a).
Furthermore, Iantorno et al. (2017) combined genome-wide high-throughput DNA sequencing with RNA-seq and could identify correlation between chromosome aneuploidy (somy), CNVs, and SNPs across 14 Leishmania tropica isolates with total gene expression at the promastigotes stage grown in vitro. They found that gene dosage, at the level of individual genes or chromosomal “somy” resulted in more than 85% of total gene expression variation in genes with a two-fold or greater change in expression. Genes encoding membrane-bound transporters were among the most highly upregulated genes. Biopterin transporter BT1 and folate transporter FT1, aquaglyceroporin genes responsible for transport of trivalent antimonials into the cell and the LiMT/LiRos3 transport system, were all previously linked to drug resistance (Marquis et al., 2005; Mandal et al., 2015; Imamura et al., 2016). Notably, in L. tropica LRC-L810 recovered from a distinct sandfly vector (Phlebotomus arabicus rather than the more commonly known vector Phlebotomus sergenti) (Soares et al., 2004) showed a very distinct expression profile (Iantorno et al., 2017). The most highly upregulated gene in isolate LRC-L810 encoded for a receptor-type adenylate cyclase (Iantorno et al., 2017) that has been previously linked to differentiation in African trypanosomes (Fraidenraich et al., 1993), inhibition of the host immune response in Trypanosoma brucei (Salmon et al., 2012), and motility in the insect stages (Lopez et al., 2015). L. tropica species remains understudied using RNA-seq. As opposed to other Leishmania species such as L. major (Rastrojo et al., 2013; Dillon et al., 2015a), L. mexicana (Fiebig et al., 2015; Patino et al., 2019) and others (Figure 3), very few studies addressed differences in gene expression in L. tropica (Iantorno et al., 2017).
Dumetz et al. (2017) also correlated chromosome aneuploidy to changes in gene expression in Leishmania donovani during their adaptation to in vivo conditions while mimicking natural vertebrate and invertebrate host environments. To mimic the various life stages of the parasite, they used Syrian golden hamsters and sandflies to investigate in vivo amastigote and promastigote stages, respectively, and compared the results to in vitro-maintained parasites. Aneuploidy was highly variable and correlated with the corresponding transcript levels although aneuploidy-independent regulation of gene expression was also observed. A total of 589 genes were upregulated in promastigotes compared to amastigotes. These mainly belonged to carbon, lipid, and fatty acid metabolism, translation, protein modification, membrane transport, DNA replication and nucleosome assembly, and the function of the flagellum. On the other hand, only 261 genes encoding for amastins and amastin-like proteins, among others were upregulated in amastigotes (Dumetz et al., 2017).
Camacho et al. (2019) also generated the transcriptome of L. donovani HU3 promastigotes and identified 2,410 novel transcripts 1,513 of which were homologs to transcripts annotated in L. major (Rastrojo et al., 2013). Histone transcripts, alpha tubulin genes, ribosomal proteins, HSP70, and kinetoplastid membrane protein 11 (KMP11) were found to be among the most abundant transcripts (Camacho et al., 2019).
Inbar et al. (2017) described the transcriptome dynamics of L. major as it develops inside its natural vector P. duboscqi by examining global gene expression in procyclic (PP), nectomonad (NP), and metacyclic promastigotes (MP) compared to BALB/c mice lesion-derived amastigotes (AM) and culture-derived metacyclic promastigotes (CMP). Notably, the greatest number of differentially expressed genes was observed during early transformation from AM to PP in the blood-fed midgut where amastins were down-regulated while multiple cell surface proteins, sugar and amino acid transporters, and genes related to glucose metabolism and cell cycle progression were upregulated. The NP stage revealed changes in genes acting in cell cycle arrest and the upregulation of genes associated with starvation and stress, including autophagic pathways of protein recycling. Maturation to the infective MP showed amastigote-like profiles of surface proteins and metabolism-related genes. The results obtained from comparing gene expression profiles of sandfly derived and culture-derived MP revealed similar results except for an upregulation of transcripts associated with nutrient stress in vivo (Inbar et al., 2017).
Coutinho-Abreu et al. (2020b) performed RNA-seq on unaltered midguts of infected L. longipalpis sandflies, the vector of L. infantum. The sequences obtained from procyclic, nectomonad, leptomonad or metacyclic promastigote stages were grouped into distinct populations based on principal component analysis, with the procyclic stage being the most distinct. A total of 836 genes were differentially expressed between procyclic and nectomonad promastigotes, 113 between nectomonad and leptomonad promastigotes and 302 between leptomonad and metacyclic promastigotes. Most of the differentially expressed genes were uniquely expressed in each stage and not in other stages. Stage-specific markers included genes encoding a zinc transporter in procyclics, a beta-fructofuranidase in nectomonads, a surface antigen-like protein in leptomonads, and an amastin-like surface protein in metacyclics (Coutinho-Abreu et al., 2020b). Conversely, the presence of L. infantum in L. longipalpis sandflies produced a limited change in the sandfly’s transcript expression profile (Coutinho-Abreu et al., 2020a). As such, the parasite appeared to modulate gene expression early on in the developmental cycle (Days 1 to 4) in order to overcome the barriers imposed by the midgut of the insect, then most of the differentially expressed transcripts were up-regulated with small fold changes at later time points (Day 6 and onward) with only slight changes observed in midgut gene expression (Coutinho-Abreu et al., 2020a). The most abundant transcripts in promastigotes and amastigotes of various Leishmania species are summarized in Tables 1, 2, respectively.
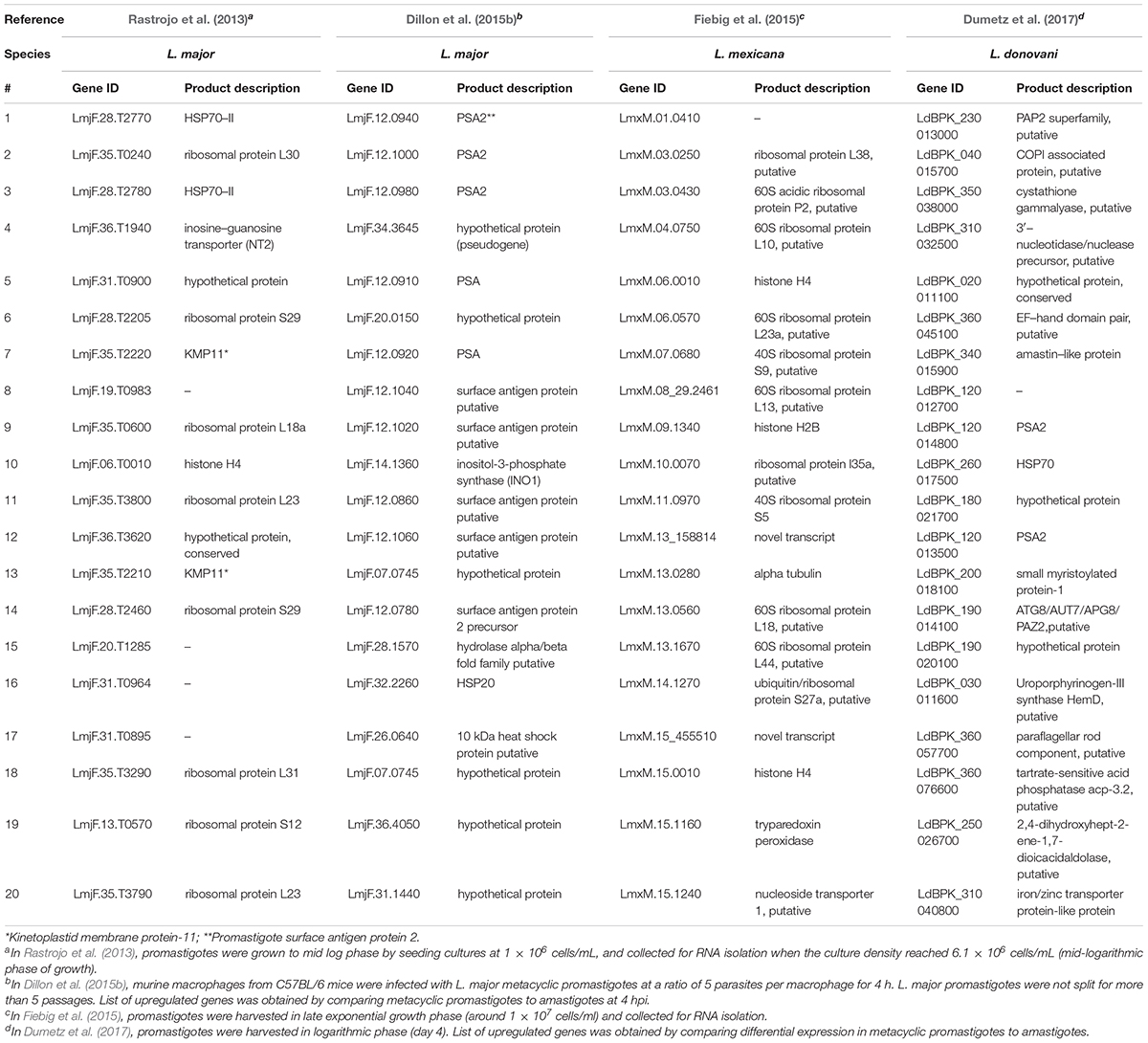
Table 1. Top twenty most abundant transcripts in promastigotes of various Leishmania species.
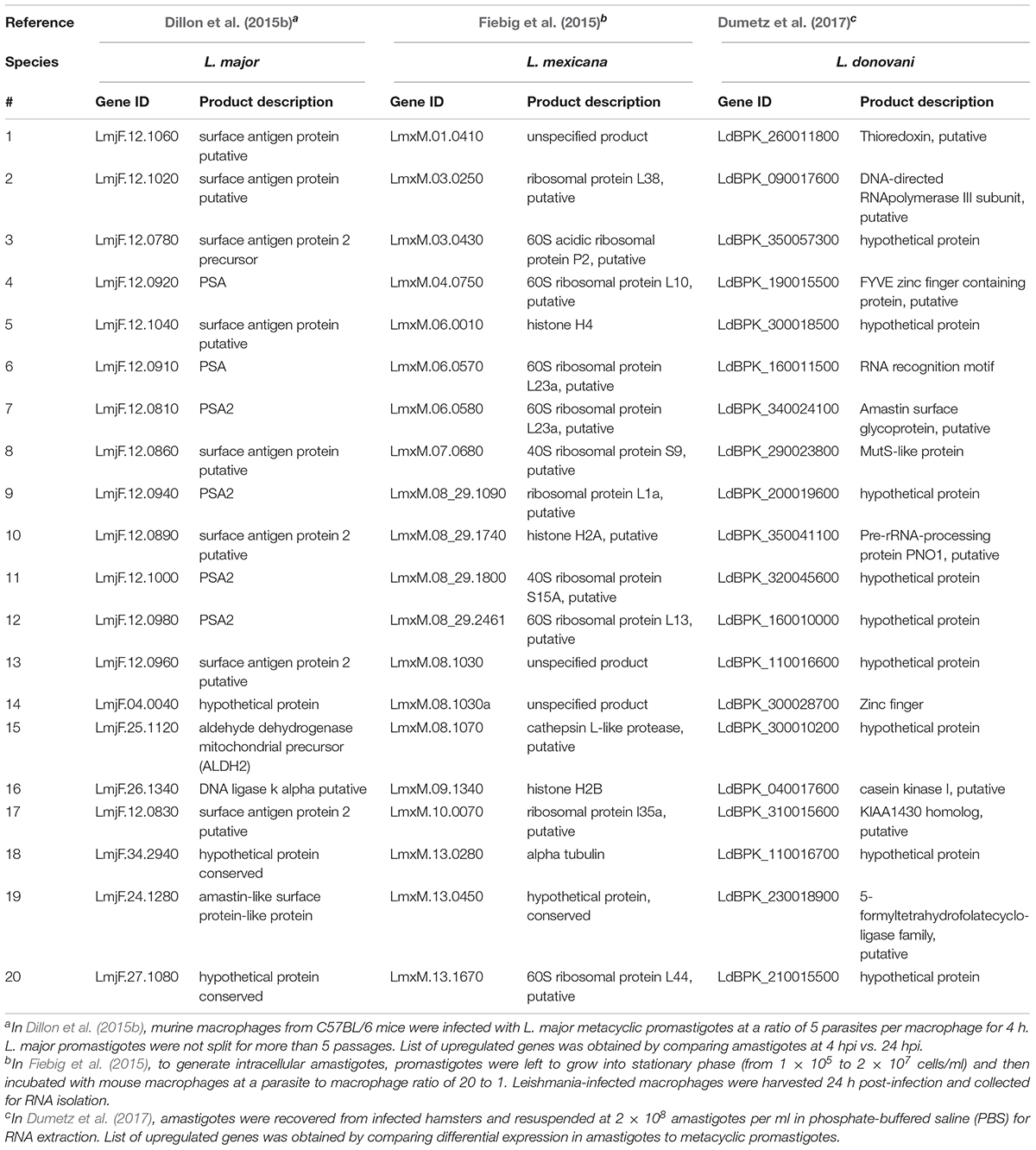
Table 2. Top twenty most abundant transcripts in amastigotes of three major Leishmania species.
Host Cell’s Response to Infection
Another advantage of using RNA-Seq is to be able to look into gene expression changes in the host cell following parasitic infections in so called dual RNA-Seq experiments (Dillon et al., 2015b; Fernandes et al., 2016). Dillon et al. (2015b) aimed at identifying global changes in gene expression using murine macrophages from C57BL/6 mice and L. major at 4, 24, 48, and 72 h post-infection (hpi). The most significant response to infection by the macrophage was observed at 4 hpi, with 6,897 mouse genes significantly differentially expressed between uninfected and infected cells. Genes related to both pro- and anti-inflammatory immune responses and glycolysis were substantially upregulated, and genes related to lipid metabolism, biogenesis, and Fc gamma receptor-mediated phagocytosis were downregulated in the murine macrophages. Genes linked to the mitigation of oxidative stress by the host immune system were upregulated while genes related to translation, cell signaling, fatty acid biosynthesis, and flagellum structure were downregulated in L. major amastigotes. KEGG enrichment analysis revealed that the differentially expressed genes in macrophages were related to immune response (cytokine-cytokine receptor interactions and arginine and proline metabolism) and glycolysis. The immunoregulatory activity detected in macrophages infected with L. major has been previously also observed in macrophages stimulated with lipopolysaccharide (LPS) (Fleming et al., 2015). Dillon et al. (2015b) also observed a total of 2,962 genes that were differentially expressed in metacyclic promastigotes compared to amastigotes. These were mostly thought, based on their annotation, to be involved in reducing the effects of an oxidative stress response exerted on the parasite by the innate immune response taking place in the phagosome (Zhang et al., 2010). Heat shock proteins, especially HSP83, multiple tryparedoxin peroxidase family members, and multiple cyclophilins were all upregulated upon entry of metacyclic promastigotes into host cells (Dillon et al., 2015b). Human monocytes infected with L. major also revealed an upregulation of pro-inflammatory cytokine and cytokines receptors including IL1A, IL1RN, IL6, and IL6R (Kalavi et al., 2020).
It’s noteworthy that Fernandes et al. (2016) used a dual transcriptome approach to profile gene expression in human CD14+ monocytes infected by L. major and Leishmania amazonensis at different time points (4, 24, 48, and 72 hpi). A temporal expression pattern was observed in both the macrophage and the parasite as macrophage response and parasite transformation stabilized shortly (4 hpi) after entry of the Leishmania into the host. No significant difference was observed in the parasite species transcriptomes or the macrophage response between the two different Leishmania species. Following infection with L. major, 5,713 human genes were differentially expressed between uninfected and infected macrophages. Similar results were obtained with L. amazonensis, mainly detecting genes encoding for inflammatory cytokines (IL-1β, TNF, TNF superfamily members, and IL-6) and a number of immunomodulators. Immunomodulators included: prostaglandin-endoperoxide synthase 2 (PTGS2), colony-stimulating factors 1 and 2 (CSF1 and CSF2), and superoxide dismutase 2 (SOD2). SOD2 was upregulated only following infection with L. major. Of interest were the host metallothionein 1 family members with their role in the disease outcome requiring further attention. On the parasite side, genes encoding for amastin, gp63, kinesin, flagellar attachment zone protein, AAT family members, dynein and cysteine peptidase B were all upregulated post-infection in both Leishmania species (Fernandes et al., 2016).
Moreover, Shadab et al. (2019) investigated host-specific and parasite-specific factors modulating the host-parasite interaction in L. donovani AG83 strain. They infected murine peritoneal macrophages with either the virulent or a non-virulent variant of the parasite derived from the AG83 strain. Infection with virulent L. donovani strains revealed suppression of many important cellular processes, including protein synthesis. Genes encoding virulence factors and those important for parasite survival were significantly upregulated in the intracellular virulent amastigotes. In contrast, genes involved in the immune stimulations and negative regulation of the cell cycle and transcriptional regulation were also all upregulated in the non-virulent strains. The non-virulent L. donovani AG83 strain was generated by Sinha et al. (2018) through continuous in vitro passages and revealed global changes in the genome and transcriptome of the serially passaged L. donovani AG83 promastigotes. No massive transcript expression changes between the virulent and non-virulent AG83 promastigotes were observed and among the few differentially expressed genes were the one coding for acid phosphatase playing a role in virulence and endosome sorting (Katakura and Kobayashi, 1988), and cyclin-dependent kinase pho85-like protein. Homologs of the latter gene was previously implicated environmental signaling in yeast in response to stressful environmental conditions (Carroll and O’Shea, 2002).
Dual RNA sequencing of both enucleated fibroblasts (cytoplasts) and intracellular L. amazonesis has been recently performed to gain further insights in the parasite’s control over the host cell (Orikaza et al., 2020). This experimental system demonstrated that the parasite multiplication and biogenesis of large parasitophorous vacuoles were independent of the host cell nucleus (Orikaza et al., 2020). Infected cytoplast transcripts were enriched in phagolysosome-related pathway, pro-survival, and SerpinB-mediated immunomodulation compared to control non-infected cytoplasts. Notably, these results suggested that a parasite-mediated control of host cell transcripts half-life was beneficial to the parasite’s intracellular multiplication and evasion of the host immune response (Orikaza et al., 2020).
L. infantum did not activate the inflammasomes in the THP-1 human macrophage infected cells (Gatto et al., 2020). In fact, L. infantum triggered a gene expression pattern more similar to non-infected THP-1 cells yet very different from LPS-stimulated cells. Some of the most up-regulated genes in L. infantum-infected cells were related to: cell cycle (CDC20, CDKN2C, CNNL2), glycolysis/gluconeogenesis (ENO1), metabolism of carbohydrates (AGRN), signal transduction (RIT1, RPS6KA1, ILR6, SFPQ), MAPK signaling (RPS6KA1), gene expression (NOTCH2), ubiquitin mediated proteolysis (CDC20) and interleukin signaling (TNFRSF14, TNFRSF1B, ILR6, CSF1). Genes associated with the inflammasome signaling pathway were not differentially expressed and caspase-1 activation and IL-1β production were absent following infection (Gatto et al., 2020). Genes significantly upregulated or downregulated in macrophages following infection with various Leishmania species are summarized in Tables 3, 4, respectively.
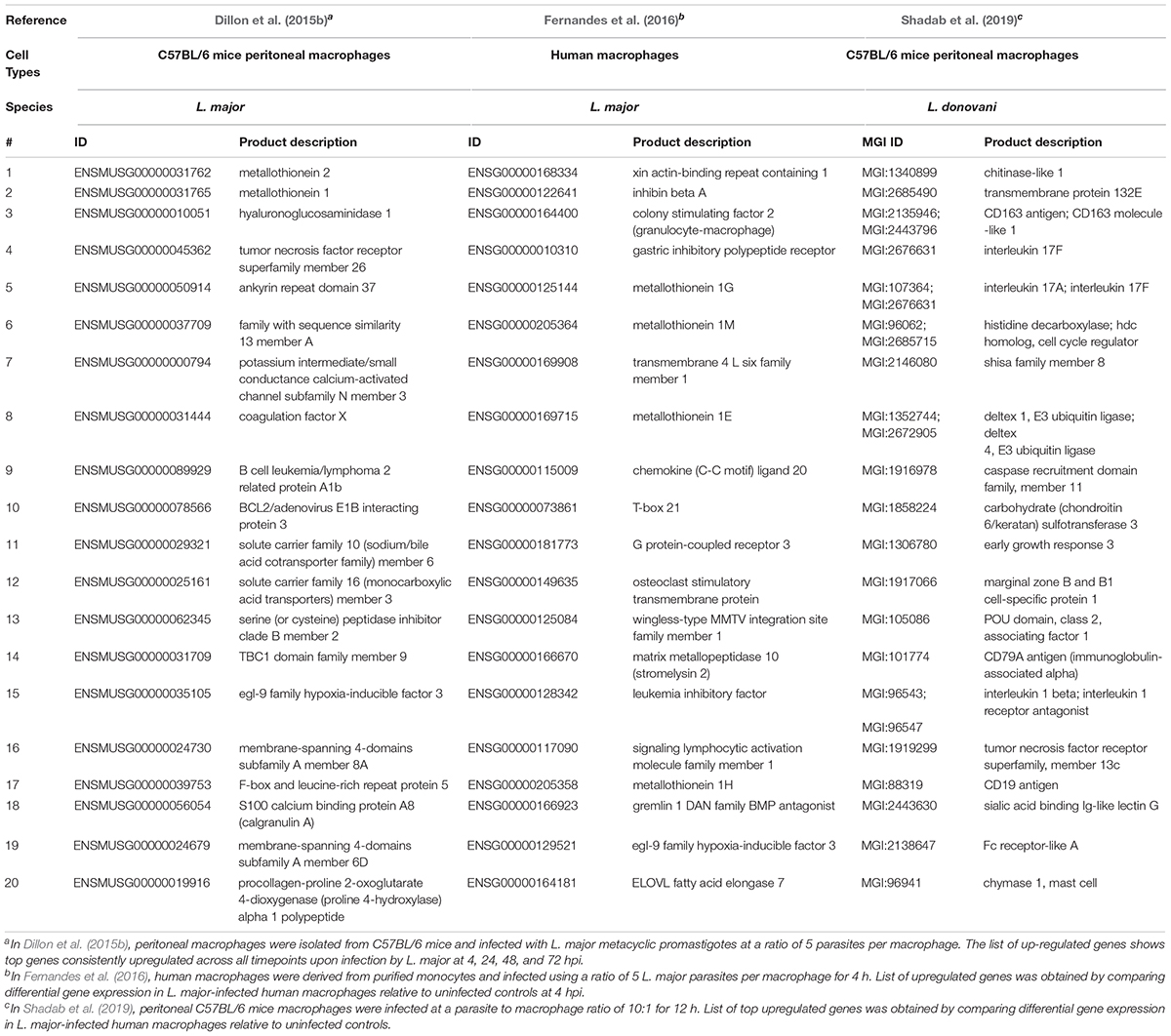
Table 3. Top twenty upregulated genes in macrophages following infection with various Leishmania species.
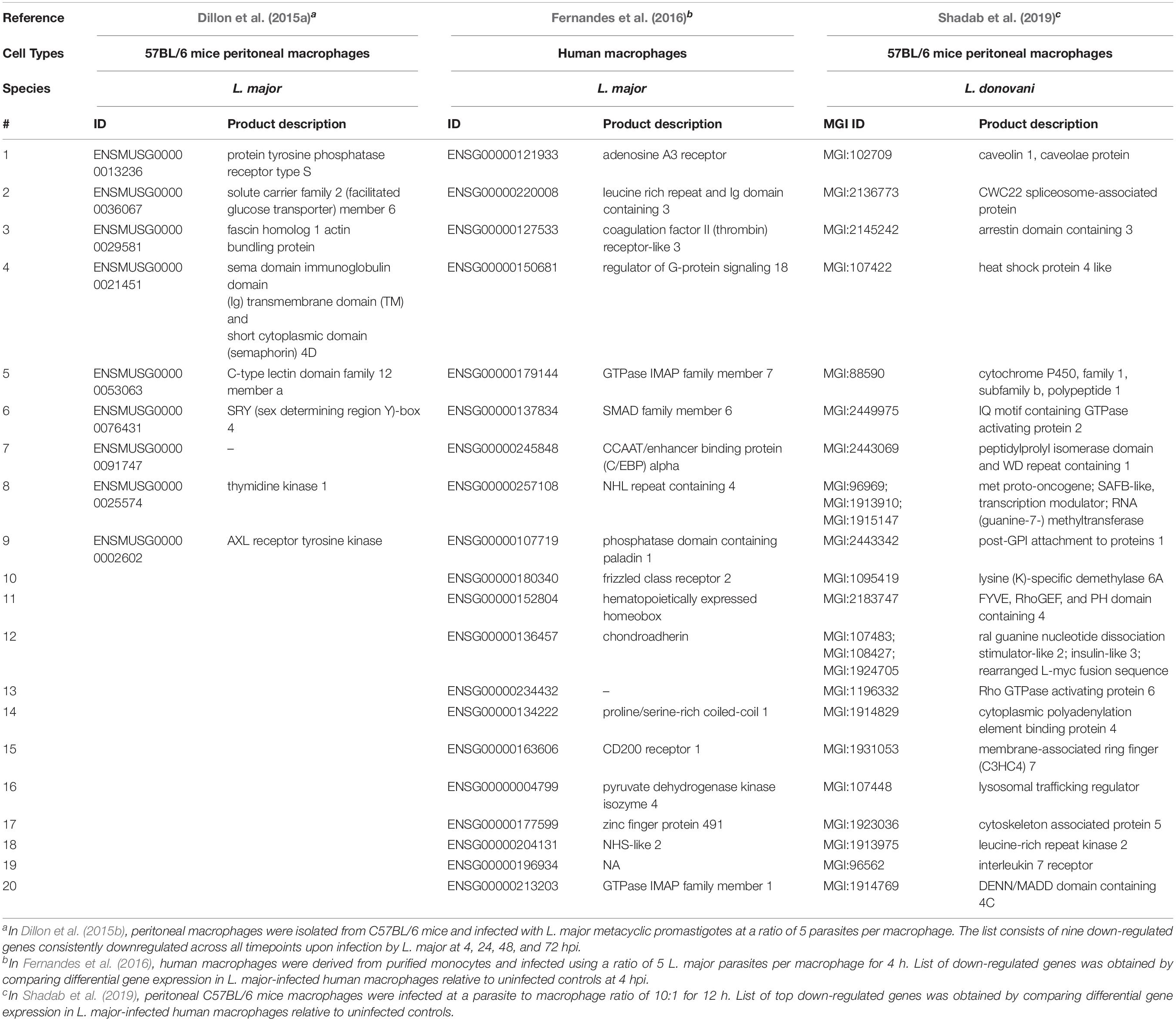
Table 4. Top twenty down-regulated genes in macrophages following infection with various Leishmania species.
Assessment of Metabolic Pathways
RNA-Seq was also used to assess the importance of L-arginine pathway in L. amazonensis promastigotes and axenic amastigotes (Aoki et al., 2017). The L-arginine pathway in Leishmania is important during the parasite life cycle and infection of the mammalian macrophages (Cunningham, 2002). Nitric oxide (NO) production is a defense mechanism used by macrophages being produced by nitric oxide synthase 2 (NOS2) in the presence of the amino acid L-arginine as substrate (Cunningham, 2002). Arginase on the other hand, reduces NO production by limiting the availability of L-arginine and favoring the survival of Leishmania in the macrophage (Camargo et al., 1978; Aoki et al., 2020). Aoki et al. (2017) studied gene expression differences between L. amazonensis wild-type (La-WT) and L. amazonensis arginase knockout (La-arg–) promastigotes and axenic amastigotes. In doing so, they identified a total of 8,253 transcripts in both strains of which 60% encoded hypothetical proteins, 443 were novel transcripts that did not match any previously annotated genes, and 85% were constitutively expressed. WT amastigotes had lower levels of arginase and higher levels of glutamate-5-kinase compared to WT promastigotes. La-arg– promastigotes had increased levels of pyrroline 5-carboxylate reductase, but decreased levels of arginosuccinate synthase, pyrroline 5-carboxylate dehydrogenase, acetylornithine deacetylase and spermidine synthase compared to the WT. Thus, arginase activity is important in Leishmania gene expression modulation during its differentiation and adaptation to environmental changes. Acuña et al. (2017) also described an arginase-dependent NOS-like activity in L. amazonensis which could be important to trigger parasite differentiation during infection of macrophages. They suggested that NO production could be arginase-dependent and higher levels of NO were produced in axenic amastigotes compared to promastigotes. Aoki et al. (2019) further expanded the study of the role of arginase in the modulation of virulence factors involved in parasite recognition, growth and differentiation in L. amazonensis. Arginase was found to upregulate membrane markers that affect parasite recognition, autophagy-related genes implicated in parasite differentiation and amastins that impact amastigote replication and survival, while modulating oxidative-stress related genes (Aoki et al., 2019). The parasite’s arginase activity also appeared to modulate gene expression in the host macrophage including in the immune response and amino acid transport and metabolism (Aoki et al., 2020).
Recently, Malta-Santos et al. (2020) investigated the importance of the polyamine biosynthetic pathway in CL lesions. Diffuse CL was associated with higher concentrations of amino acids, polyamines and its substrate transporters compared to MCL or localized CL. The most upregulated transcripts in diffuse CL lesions were CAT2A (isoform encoded by SLC7A2), ARG1 and SMS compared to MCL lesions acting in the regulation of arginine availability. A differential gene expression of polyamine metabolism-related genes of patients’ lesions was associated with parasite loads and the leishmaniasis disease outcome (Malta-Santos et al., 2020).
Direct Application to Skin Biopsies
RNA-Seq can also be directly applied on human tissues such as skin biopsies. Christensen et al. (2016, 2019) studied gene expression changes in the host and the parasite using skin biopsies from Leishmania braziliensis-infected patients with, respectively, localized cutaneous leishmaniasis (LCL) and diffuse cutaneous leishmaniasis (DCL). In Christensen et al. (2016), detectable parasite transcripts in only one group of patients were found in which an increase in B lymphocyte-specific and immunoglobulin transcripts in the lesions, and an upregulation of immune inhibitory molecules was observed. Patients negative for parasite transcripts had a decrease in B cell activation, but increased expression of antimicrobial genes and genes encoding skin barrier functions, and which were different from transcripts expressed in human macrophages in vitro. Therefore, the differences in gene expression observed in vitro in macrophages does not accurately reflect the one observed from skin lesions, as expected as in the skin biopsies one collects multiple cells. Hence it is not possible/very difficult to compare the transcriptome of macrophages in cell culture versus mix of cells, including macrophages, from a biopsy. Highly expressed genes in the parasite included those encoding for cysteine peptidase and synthase, glycerophosphoryl phosphodiesterase family protein and transport protein SEC13 among others. Upregulated genes in lesions with detectable parasite transcripts encoded immunoglobulin fragments, CXCL8 (encoding chemokine IL-8, granulocyte and neutrophil chemotaxis), IL-21 (B cell proliferation), and granulysin (cellular cytotoxicity) encoding genes (Christensen et al., 2016).
Skin lesions from BALB/c mice infected with L. major showed an upregulation of FCGR4, CCL4, CXCL9, Arg1, and IL-1β and an enrichment of the Triggering Receptor Expressed on Myeloid Cells 1 (TREM1) signaling pathway (Ulusan et al., 2020).
To date, very few metatranscriptomics studies have been performed on L. tropica. These included RNA-Seq performed on skin biopsies in L. tropica infected patients (Masoudzadeh et al., 2017). Unfortunately, the usefulness and application of this study for downstream comparative analysis remains limited due to the unavailability of reads from L. tropica which were not analyzed nor deposited in a public database. Notably, transcription profiling of L. tropica-infected skin identified over 5,000 differentially regulated human genes compared to uninfected controls (Masoudzadeh et al., 2017). Gene ontology enrichment analysis indicated that upregulated genes were mostly involved in immune response activation, extracellular matrix degradation, inflammatory cell recruitment, cytotoxicity and antimicrobial peptides (Masoudzadeh et al., 2017). Similarly to the previous report by Christensen et al. (2016), they observed an increase in B lymphocyte-specific (immunoglobulin lambda-like polypeptide 5, IGLL5) and immunoglobulin receptor (Fc Fragment of IgG Receptor Ia, FCGR1A) transcripts. These findings highlight the role of B cells at the infection site. Downregulated genes on the other hand, encoded mainly for structural proteins associated with muscle contraction (e.g., myosin 2, alpha 1 actin, myosin 1, and troponin C type 2) (Masoudzadeh et al., 2017). Furthermore, shared and unique functional transcriptional pathways in L. tropica infected skin lesions of ulcerative CL (UCL) and non-ulcerative CL (NUCL) patients were investigated using RNA-Seq (Masoudzadeh et al., 2020a). Inflammatory cytokines and chemokines were differentially expressed in the UCL and NUCL lesions. Transcriptional pathways for Fcγ receptor dependent phagocytosis were enriched in both conditions (Masoudzadeh et al., 2020a).
Notably, Christensen et al. (2019) using meta-transcriptomic analysis of biopsies from patients with DCL, demonstrated an infiltration of atypical B cells producing a dominance of the IgG4 isotype in these L. amazonensis infected patients. They additionally, revealed the absence of cytotoxic and TH2 cell responses. A regulatory phenotype was observed in macrophages with some genes being actively expressed such as an ATP-binding cassette, subfamily B, member 5 (ABCB5), dendritic cell specific transmembrane protein (DC-STAMP), and a secreted phosphoprotein 1 (SPP1) among others. Gene expression in DCL lesions resembled patterns obtained from in vitro parasite growth in resting macrophages. Furthermore, 336 genes in L. amazonensis were upregulated in LCL compared to DCL. Among these upregulated genes, seven potential virulence factors and four stress response genes were detected, including GP63, proteins containing leucine-rich repeats, a cyclophilin protein, a protein containing a RmlC-like jelly roll fold domain, and a Betv1-like superfamily protein.
The blood of L. braziliensis infected patients was also recently analyzed using RNA-seq to determine systemic responses that might be influencing the disease (Amorim et al., 2020). A strong interferon stimulated gene (ISG) signature has been identified which was correlated with an increase in circulating monocytes and macrophages (Amorim et al., 2020). A cytotoxicity signature was correlated with an increase in cytolytic cells (Amorim et al., 2020).
Similarly, the blood of VL-HIV patients was studied through transcriptional profiling (Adriaensen et al., 2020). A downregulation of genes associated with host cellular activity and immunity, and upregulation of antimicrobial peptide activity in phagolysosomes was observed in patients that were successfully treated with AmBisome or AmBisome/miltefosine, in contrast, no pathway enrichment among differentially expressed genes was observed in treatment failure patients (Adriaensen et al., 2020).
Assessing Drug Resistance
The appearance of drug resistance phenotypes is a major concern hindering available anti-leishmanial treatment (Sundar and Singh, 2016). Rastrojo et al. (2018) identified genomic and transcriptomic alterations associated with experimental resistance in L. donovani, L. infantum and L. major promastigotes and amastigotes in response to common drugs used against VL including: trivalent antimony (S line), amphotericin B (A line), miltefosine (M line), and paromomycin (P line). In total, 1,006 differentially expressed transcripts were identified in the S line, 379 in the A line, 146 in the M line, and 129 in the P line. Changes in chromosomal aneuploidy and amplification/deletion of particular regions were correlated with resistance. A series of genes were identified as possible drivers of the resistance phenotype. The S line included peptidyl dipeptidase, amastin, and amastin-like surface proteins among others. The M line included protein kinase, carboxypeptidase and amino acid transporter AAT1.4 among others. The P line included flavoprotein subunit-like protein, amastin-like protein, protein associated with differentiation and iron/zinc transporter protein-like protein (LIT1) among others. Finally, the A line included a sterol 24-c- methyltransferase and phosphatidylinositol 4- kinase alpha among others.
RNA-Seq experiments became more frequently used in 2019 where shifts in transcriptional responses were measured in various Leishmania species and under more variable conditions. Patino et al. (2019) used RNA-Seq to analyze transcriptome profiles between antimony-resistant and antimony-sensitive L. amazonensis promastigotes. They identified a total of 723 differentially expressed genes between lines that were resistant or sensitive to trivalent sodium stibogluconate (SbIII). These genes encoded proteins associated with various biological processes, like adhesion, metabolism, cell cycle, autophagy, structural organization and stress response. Some mRNA encoding amastin proteins were overexpressed in resistant lines, whereas the downregulated protein-encoding genes encoded putative superoxide dismutase, other subset of the amastin family and transporter proteins, which included a folate/biopterin transporter, pteridine transporter and an ABC transporter (Patino et al., 2019).
Differential gene expression analysis between wild-type and SbIII-resistant L. infantum lines revealed 933 differentially expressed transcript including 837 upregulated and 96 downregulated transcripts (Andrade et al., 2020). Upregulated transcripts in the SbIII-resistant line were associated with protein phosphorylation, microtubule-based movement, ubiquitination, host–parasite interaction, cellular process, and other categories while downregulated transcripts were associated with ribonucleoprotein complex, ribosome biogenesis, rRNA processing, nucleosome assembly and translation (Andrade et al., 2020).
Global transcriptomics studies were similarly performed in L. braziliensis promastigotes and intracellular amastigotes. Espada et al. (2019) investigated the pathways related to intrinsic miltefosine tolerance in L. braziliensis clinical isolates and found upregulated Ros3 mRNA levels in sensitive strains compared to resistant ones and suggested that miltefosine transporter (MT)-Ros3 was responsible for the observed drug sensitivity in L. braziliensis. Drug efflux and compartmentalization were similar in resistant and sensitive strains and drug susceptibility did not correlate with SNPs in the MT-Ros3 (Espada et al., 2019).
Mechanisms underlying artemisinin resistance in L. donovani have been recently investigated using comparative genomic and transcriptomic analyses (Ghosh et al., 2020). A dependency on lipid and amino acid metabolism, a reduced DNA and protein synthesis leading to parasites in the quiescence state, and an active drug efflux have been observed in resistant isolates. Upregulated genes included those encoding cathepsin-L like protease, amastin-like surface protein, and amino acid transporter, while downregulated genes include ABCG2, pteridine receptor, adenylatecyclase-type receptor, phosphoaceylglucosamine mutase, and a collection of hypothetical proteins (Ghosh et al., 2020).
Assessing the Effects of Environmental Variations
Leishmania parasites alternate between poikilothermic and homoeothermic hosts. Sudden temperature variations are natural events in their life cycles as the transmission from insect vector to the mammalian host includes a drastic increase over ambient temperature by more than 10 °C, which is in addition to the variation in temperature the insects experience in their habitats (Requena et al., 2012). The transcriptome of L. major promastigotes following a moderate heat shock from 26°C to 37°C was determined (Rastrojo et al., 2019). Following a moderate heat shock, Rastrojo et al. (2019) observed that the upregulated transcripts were heat shock proteins, amastigote-specific proteins and several hypothetical proteins. Downregulated transcripts, however, were associated with transporters, proteins involved in RNA metabolism or translational factors. Putative long non-coding RNAs were also identified among the differentially expressed transcripts and temperature-dependent changes in the selection of the SL addition sites were observed. Accordingly, alternative trans-splicing was suggested as an additional mechanism altering gene expression in Leishmania (Rastrojo et al., 2019).
Moreover, minor temperature shifts from 26°C to 24°C, 28°C, and 30°C) did not affect chromosomal ploidy but caused transcriptomic changes in L. braziliensis promastigotes grown in vitro (Ballesteros et al., 2020). Amastin surface-like proteins were downregulated under the three temperatures compared with the control (Ballesteros et al., 2020).
Future Directions
Global RNA-Seq and transcriptome analysis has gained significant attention as a tool to unveil Leishmania species molecular biology and their pathogenicity, differentiation into various life stages and interplay with the host immune response (Dillon et al., 2015b; Shadab et al., 2019). The simultaneous analysis of the gene expression profiles of parasites and hosts is a critical step in our understanding of the disease (Dillon et al., 2015b; Masoudzadeh et al., 2020b). However, with the colossal amount of data generated, there is a need to “translate” these complex datasets into forms that can be exploited for drug and vaccine development.
Various species such as L. tropica remain understudied using RNA-seq, which mostly include data derived from in vitro cultures. Also, in order to perform an accurate comparative analysis using global RNA-Seq data, experimental design should be reproducible and cover various Leishmania species grown in vitro under similar well-defined conditions and infected in human macrophages similarly maintained under constant growth conditions and followed up in time course experiments to gain insights into the dynamic process of the infection. The outcome of such analysis should be further enriched and validated in vivo with transcriptomics data derived from promastigotes isolated directly from the sandfly gut and from amastigotes collected from induced lesions in mice models.
Reports suggesting only a moderate correlation between transcript abundance and cellular protein levels in Leishmania species requires further attention (de Pablos et al., 2019). Proteomics allow the analysis of expression levels, post-translational modifications, interactions, structure and subcellular distribution of proteins (Cox and Mann, 2007). Proteomics have been widely used to generate a proteome map providing an overall picture of gene expression at a given point in time (Sundar and Singh, 2016). In Leishmania, the comparison of proteomes has been employed to provide a better understanding of the parasite’s life cycle, host–pathogen interactions, protein–protein interactions and drug resistance mechanisms for different Leishmania species (Capelli-Peixoto et al., 2019). Several key candidates for vaccine development have also been identified through proteomics investigations (Sundar and Singh, 2018; Sundar et al., 2019).
Despite the low observed correlation between RNA and protein abundance in Leishmania (Leifso et al., 2007; de Pablos et al., 2019), proteomic technologies could complement genomics and transcriptome profiling studies to characterize specific gene products (Sundar and Singh, 2016). Furthermore, complementing genome sequence with transcriptomics and proteomics data enable more accurate assembly and annotation of newly sequenced genomes (Cox and Mann, 2007; Prasad et al., 2017).
Moreover, the control of mRNA stability, decay and translation in the near absence of transcriptional regulation is not well understood (de Pablos et al., 2016). Also, the dynamics and regulation of messenger ribonucleoprotein complexes (mRNPs) in Leishmania species or differences in translational regulation from ribosomal profiling of the mRNAs from each life cycle stage are not yet elucidated (de Pablos et al., 2016).
Potentially sinefungin could be used to examine gene transcription. Sinefungin is a naturally occurring nucleoside isolated from Streptomyces griseolus and S. incarnates bacteria with structural similarities with S-adenosyl-1-methionine (Bachrach et al., 1980; Dube et al., 1983; McNally and Agabian, 1992). Sinefungin has antitrypanosomal activity and inhibits SL trans-splicing in these organisms (Bachrach et al., 1980; Dube et al., 1983; McNally and Agabian, 1992; Pandarakalam et al., 2019).
The role of adenylate cyclase in Leishmania parasites residing inside insect hosts requires further investigations (Iantorno et al., 2017). Also, more studies should focus on the role of amastins in infection, survival and host-parasite interactions.
Another interesting target is to investigate the roles of non-coding RNAs (ncRNAs) in regulating Leishmania mRNA transcription (Castro et al., 2017; Ruy et al., 2019). ncRNAs emerge as key players in a variety of regulatory processes including mRNA processing, mRNA stability in addition to DNA replication, chromosome maintenance and transcriptional regulation in various organisms (Mattick, 2005; Morris and Mattick, 2014). In L. major and L. donovani, 26 and 30 putative ncRNAs were identified, respectively, with the majority arising from UTRs (Castro et al., 2017). The biological function of these ncRNA and their regulatory roles in transcription deserve further investigation.
Moreover, a detailed metatranscriptomics analysis in situ in the skin and in the insect vector covering the insect gut microbiota and the human skin microbiota, respectively, could aid in enriching our understanding of the factors that govern the complex host–parasite-microbiota interplay and their potential impact and relationship with the different forms of leishmaniasis, CL, VL, and MCL. Skin microbiota dysbiosis associated with CL requires further attention modulating the pathobiology of Leishmania infections and appears, importantly, as a potential novel therapeutic target (Gimblet et al., 2017). Notably, the mammalian skin and insect gut microbiota are also potentially relevant to trigger and modulate the shift from CL to VL (Gimblet et al., 2017). Moreover, the impact of LRV and other viruses, such as the Lymphocytic choriomeningitis virus (LCMV), on the pathobiology of various Leishmania species would also be of great interest to investigate with more integrative approaches and model systems (Ives et al., 2011; Crosby et al., 2015). Lymphocytic choriomeningitis virus, for instance, is a rodent-borne, Old World arenavirus that causes asymptomatic or mild, self-limited illness in otherwise healthy humans (Zhou et al., 2012). It is a known cause of aseptic meningitis and rarely fatal (Fischer et al., 2006). It has been reported that an increased recruitment of neutrophils and more severe lesions were observed in mice co-infected with L. major and LCMV (Crosby et al., 2015). More recently, a ssRNA Leishmania-infecting leishbunyavirus (LmarLBV1) has been isolated from Leishmania martiniquensis (Grybchuk et al., 2020). Thus, the diversity and implications of viruses on leishmaniasis outcome and disease progression are other lines of research of great interest to investigate.
Gut microbes from the sandfly are egested into the host skin alongside Leishmania parasites at the bite sites (Dey et al., 2018). These have been shown to augment the severity of VL caused by L. donovani via inflammasome-derived IL-1β production (Dey et al., 2018).
Although much remains to be elucidated on the effects of skin microbiota and sandfly gut microbes on the progression of leishmaniasis, understanding the complex interactions between Leishmania, skin microbiota and the sandfly gut microbes in disease outcome appears as a potential therapeutic target and an important parameter to untangle the complex immunological cross-talks occurring between the parasite and the host skin and/or sandfly gut microbiota.
Author Contributions
TS: conceptualization, investigation, and writing—original draft and review and editing. TS, ST, and RH: data curation, investigation, and writing—original draft and review and editing. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Acuña, S. M., Aoki, J. I., Laranjeira-Silva, M. F., Zampieri, R. A., Fernandes, J. C., Muxel, S. M., et al. (2017). Arginase expression modulates nitric oxide production in Leishmania (Leishmania) amazonensis. PLoS One 12:e0187186. doi: 10.1371/journal.pone.0187186
Adaui, V., Maes, I., Huyse, T., Van den Broeck, F., Talledo, M., Kuhls, K., et al. (2011). Multilocus genotyping reveals a polyphyletic pattern among naturally antimony-resistant Leishmania braziliensis isolates from Peru. Infect. Genet. Evol. 11, 1873–1880. doi: 10.1016/j.meegid.2011.08.008
Adriaensen, W., Cuypers, B., Cordero, C. F., Mengasha, B., Blesson, S., Cnops, L., et al. (2020). Host transcriptomic signature as alternative test-of-cure in visceral leishmaniasis patients co-infected with HIV. EBioMedicine 55:102748. doi: 10.1016/j.ebiom.2020.102748
Alcolea, P. J., Alonso, A. N., and Larraga, V. (2011). Proteome profiling of Leishmania infantum promastigotes. J. Eukaryot. Microbiol. 58, 352–358. doi: 10.1111/j.1550-7408.2011.00549.x
Alcolea, P. J., Alonso, A., and Larraga, V. (2016). Rationale for selection of developmentally regulated genes as vaccine candidates against Leishmania infantum infection. Vaccine 34:5474. doi: 10.1016/j.vaccine.2016.08.081
Alcolea, P. J., Alonso, A., Gómez, M. J., Moreno, I., Domínguez, M., Parro, V., et al. (2010). Transcriptomics throughout the life cycle of Leishmania infantum: high down-regulation rate in the amastigote stage. Int. J. Parasitol. 40, 1497–1516. doi: 10.1016/j.ijpara.2010.05.013
Amorim, C. F., Novais, F. O., Nguyen, B. T., Nascimento, M. T., Lago, J., Lago, A. S., et al. (2020). Localized skin inflammation during cutaneous leishmaniasis drives a chronic, systemic IFN-γ signature. medRxiv [Preprint]. doi: 10.1101/2020.12.03.20240978
Andrade, J. M., Gonçalves, L. O., Liarte, D. B., Lima, D. A., Guimarães, F. G., de Melo Resende, D., et al. (2020). Comparative transcriptomic analysis of antimony resistant and susceptible Leishmania infantum lines. Parasit. Vectors 13:600. doi: 10.1186/s13071-020-04486-4
Aoki, J. I., Muxel, S. M., Laranjeira-Silva, M. F., Zampieri, R. A., Müller, K. E., Nerland, A. H., et al. (2020). Dual transcriptome analysis reveals differential gene expression modulation influenced by Leishmania arginase and host genetic background. Microb. Genom. 6:mgen000427. doi: 10.1099/mgen.0.000427
Aoki, J. I., Muxel, S. M., Zampieri, R. A., Laranjeira-Silva, M. F., Müller, K. E., Nerland, A. H., et al. (2017). RNA-seq transcriptional profiling of Leishmania amazonensis reveals an arginase-dependent gene expression regulation. PLoS Negl. Trop. Dis. 11:e0006026. doi: 10.1371/journal.pntd.0006026
Aoki, J. I., Muxel, S. M., Zampieri, R. A., Müller, K. E., Nerland, A. H., and Floeter-Winter, L. M. (2019). Differential immune response modulation in early Leishmania amazonensis infection of BALB/c and C57BL/6 macrophages based on transcriptome profiles. Sci. Rep. 9, 1–4. doi: 10.1038/s41598-019-56305-1
Bachrach, U., Schnur, L. F., El-On, J., Greenblatt, C., Pearlman, E., Robert-Gero, M., et al. (1980). Inhibitory activity of Sinefungin and SIB A on the growth of promastigotes and amastigotes of different species of Leishmania. FEBS Lett. 121, 287–291. doi: 10.1016/0014-5793(80)80364-4
Ballesteros, N., Vásquez, N. M., Patiño, L. H., Cruz-Saavedra, L., and Ramírez, J. D. (2020). Minor temperature shifts do not affect chromosomal ploidy but cause transcriptomic changes in Leishmania braziliensis promastigotes in vitro. Mem. I. Oswaldo. Cruz 115:e190413. doi: 10.1590/0074-02760190413
Bates, P. A. (1994). Complete developmental cycle of Leishmania mexicana in axenic culture. Parasitology 108, 1–9. doi: 10.1017/s0031182000078458
Bates, P. A. (2007). Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int. J. Parasitol. 37, 1097–1106. doi: 10.1016/j.ijpara.2007.04.003
Batson, J., Kistler, A., Dudas, G., Li, L., Ratnasari, K., Haas-Stapleton, E., et al. (2020). Single mosquito metatranscriptomics recovers mosquito species, blood meal sources, and microbial cargo, including viral dark matter. BioRxiv [Preprint]. doi: 10.1101/2020.02.10.942854
Bauer, S., Grossmann, S., Vingron, M., and Robinson, P. N. (2008). Ontologizer 2.0—a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics 24, 1650–1651. doi: 10.1093/bioinformatics/btn250
Beattie, L., Hermida, M. D., Moore, J. W., Maroof, A., Brown, N., Lagos, D., et al. (2013). A transcriptomic network identified in uninfected macrophages responding to inflammation controls intracellular pathogen survival. Cell Host Microbe 14, 357–368. doi: 10.1016/j.chom.2013.08.004
Beverley, S. M. (2003). Protozomics: trypanosomatid parasite genetics comes of age. Nat. Rev. Gen. 4:11. doi: 10.1038/nrg980
Brodskyn, C. I., and de Oliveira, C. I. (2012). The immunobiology of Leishmania braziliensis infection. Front. Immunol. 3:145. doi: 10.3389/fimmu.2012.00145
Camacho, E., González-de la Fuente, S., Rastrojo, A., Peiró-Pastor, R., Solana, J. C., Tabera, L., et al. (2019). Complete assembly of the Leishmania donovani (HU3 strain) genome and transcriptome annotation. Sci. Rep. 9:6127. doi: 10.1038/s41598-019-42511-4
Camargo, E. P., Coelho, J. A., Moraes, G., and Figueiredo, E. N. (1978). Trypanosoma spp., Leishmania spp. and Leptomonas spp.: enzymes of ornithine-arginine metabolism. Exp. Parasitol. 46, 141–144. doi: 10.1016/0014-4894(78)90125-x
Campolina, T. B., Villegas, L. E. M., Monteiro, C. C., Pimenta, P. F. P., and Secundino, N. F. C. (2020). Tripartite interactions: Leishmania, microbiota and Lutzomyia longipalpis. PLoS Neglect. Trop. Dis. 14:e0008666. doi: 10.1371/journal.pntd.0008666
Capelli-Peixoto, J., Mule, S. N., Tano, F. T., Palmisano, G., and Stolf, B. S. (2019). Proteomics and leishmaniasis: potential clinical applications. Proteom. Clin. Appl. 13:1800136. doi: 10.1002/prca.201800136
Carroll, A. S., and O’Shea, E. K. (2002). Pho85 and signaling environmental conditions. Trends Biochem. Sci. 27, 87–93. doi: 10.1016/j.bbalip.2019.158557
Castro, F. F., Ruy, P. C., Zeviani, K. N., Santos, R. F., Toledo, J. S., and Cruz, A. K. (2017). Evidence of putative non-coding RNAs from Leishmania untranslated regions. Mol. Biochem. Parasit. 214, 69–74. doi: 10.1016/j.molbiopara.2017.04.002
Christensen, S. M., Belew, A. T., El-Sayed, N. M., Tafuri, W. L., Silveira, F. T., and Mosser, D. M. (2019). Host and parasite responses in human diffuse cutaneous leishmaniasis caused by L. amazonensis. PLoS Negl. Trop. Dis. 13:e0007152. doi: 10.1371/journal.pntd.0007152
Christensen, S. M., Dillon, L. A., Carvalho, L. P., Passos, S., Novais, F. O., Hughitt, V. K., et al. (2016). Meta-transcriptome profiling of the human-Leishmania braziliensis cutaneous lesion. PLoS Negl. Trop. Dis. 10:e0004992. doi: 10.1371/journal.pntd.0005588
Clayton, C. E. (2016). Gene expression in kinetoplastids. Curr. Opin. Microbiol. 32, 46–51. doi: 10.1016/j.mib.2016.04.018
Coutinho-Abreu, I. V., Serafim, T. D., Meneses, C., Kamhawi, S., Oliveira, F., and Valenzuela, J. G. (2020a). Distinct gene expression patterns in vector-residing Leishmania infantum identify parasite stage-enriched markers. PLoS Neglect. Trop. Dis. 14:e0008014. doi: 10.1371/journal.pntd.0008014
Coutinho-Abreu, I. V., Serafim, T. D., Meneses, C., Kamhawi, S., Oliveira, F., and Valenzuela, J. G. (2020b). Leishmania infection induces a limited differential gene expression in the sand fly midgut. BMC Genomics 21:608. doi: 10.1186/s12864-020-07025-8
Cox, J., and Mann, M. (2007). Is proteomics the new genomics? Cell 130, 395–398. doi: 10.1016/j.cell.2007.07.032
Crosby, E. J., Clark, M., Novais, F. O., Wherry, E. J., and Scott, P. (2015). Lymphocytic choriomeningitis virus expands a population of NKG2D+ CD8+ T cells that exacerbates disease in mice coinfected with Leishmania major. J. Immunol. 195, 3301–3310. doi: 10.4049/jimmunol.1500855
Cruz, A. K., and Freitas-Castro, F. (2019). Genome and transcriptome analyses of Leishmania spp.: opening Pandora’s box. Curr. Opin. Microbiol. 52, 64–69. doi: 10.1016/j.mib.2019.05.004
Cunningham, A. C. (2002). Parasitic adaptive mechanisms in infection by Leishmania. Exp. Mol. Pathol. 72, 132–141. doi: 10.1006/exmp.2002.2418
de Pablos, L. M., Ferreira, T. R., and Walrad, P. B. (2016). Developmental differentiation in Leishmania lifecycle progression: post-transcriptional control conducts the orchestra. Curr. Opin. Microbiol. 34, 82–89. doi: 10.1016/j.mib.2016.08.004
de Pablos, L. M., Ferreira, T. R., Dowle, A. A., Forrester, S., Parry, E., Newling, K., et al. (2019). The mRNA-bound proteome of Leishmania mexicana: novel genetic insight into an ancient parasite. Mol. Cell. Proteomics 18, 1271–1284. doi: 10.1074/mcp.RA118.001307
Depaquit, J., Grandadam, M., Fouque, F., Andry, P. E., and Peyrefitte, C. (2010). Arthropod-borne viruses transmitted by Phlebotomine sandflies in Europe: a review. Eurosurveillance 15:19507. doi: 10.2807/ese.15.10.19507-en
Dey, R., Joshi, A. B., Oliveira, F., Pereira, L., Guimarães-Costa, A. B., Serafim, T. D., et al. (2018). Gut microbes egested during bites of infected sand flies augment severity of leishmaniasis via inflammasome-derived IL-1β. Cell Host Microbe 23, 134–143.e6. doi: 10.1016/j.chom.2017.12.002
Dillon, L. A., Okrah, K., Hughitt, V. K., Suresh, R., Li, Y., Fernandes, M. C., et al. (2015a). Transcriptomic profiling of gene expression and RNA processing during Leishmania major differentiation. Nucleic Acids Res. 43, 6799–6813. doi: 10.1093/nar/gkv656
Dillon, L. A., Suresh, R., Okrah, K., Bravo, H. C., Mosser, D. M., and El-Sayed, N. M. (2015b). Simultaneous transcriptional profiling of Leishmania major and its murine macrophage host cell reveals insights into host-pathogen interactions. BMC Genomics 16:1108. doi: 10.1186/s12864-015-2237-2
Dillon, R. J., el Kordy, E., Shehata, M., and Lane, R. P. (1996). The prevalence of a microbiota in the digestive tract of Phlebotomus papatasi. Ann. Trop. Med. Parasitol. 90, 669–673. doi: 10.1080/00034983.1996.11813102
Dube, D. K., Mpimbaza, G., Allison, A. C., Lederer, E., and Rovis, L. (1983). Antitrypanosomal activity of sinefungin. Am. J. Trop. Med. Hyg. 32, 31–33. doi: 10.4269/ajtmh.1983.32.31
Dumetz, F., Imamura, H., Sanders, M., Seblova, V., Myskova, J., Pescher, P., et al. (2017). Modulation of aneuploidy in Leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. MBio 8, e00599–17. doi: 10.1128/mBio.00599-17
Espada, C. R., Magalhães, R. M., Cruz, M. C., Machado, P. R., Schriefer, A., Carvalho, E. M., et al. (2019). Investigation of the pathways related to intrinsic miltefosine tolerance in Leishmania (Viannia) braziliensis clinical isolates reveals differences in drug uptake. Int. J. Parasitol. 11, 139–147. doi: 10.1016/j.ijpddr.2019.02.005
Fernandes, M. C., Dillon, L. A., Belew, A. T., Bravo, H. C., Mosser, D. M., and El-Sayed, N. M. (2016). Dual transcriptome profiling of Leishmania-infected human macrophages reveals distinct reprogramming signatures. MBio 7, e00027–16. doi: 10.1128/mBio.00027-16
Fiebig, M., Kelly, S., and Gluenz, E. (2015). Comparative life cycle transcriptomics revises Leishmania mexicana genome annotation and links a chromosome duplication with parasitism of vertebrates. PLoS Pathog. 11:e1005186. doi: 10.1371/journal.ppat.1005186
Fischer, S. A., Graham, M. B., Kuehnert, M. J., Kotton, C. N., Srinivasan, A., Marty, F. M., et al. (2006). Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 354, 2235–2249. doi: 10.1056/NEJMoa053240
Fleming, B. D., Chandrasekaran, P., Dillon, L. A., Dalby, E., Suresh, R., Sarkar, A., et al. (2015). The generation of macrophages with anti−inflammatory activity in the absence of STAT6 signaling. J. Leukoc. Biol. 3, 395–407. doi: 10.1189/jlb.2A1114-560R
Fraidenraich, D., Pena, C., Isola, E. L., Lammel, E. M., Coso, O., Añel, A. D., et al. (1993). Stimulation of Trypanosoma cruzi adenylyl cyclase by an alpha D-globin fragment from Triatoma hindgut: effect on differentiation of epimastigote to trypomastigote forms. Proc. Natl. Acad. Sci. U.S.A. 21, 10140–10144. doi: 10.1073/pnas.90.21.10140
Freire, E. R., Sturm, N. R., Campbell, D. A., and de Melo Neto, O. P. (2017). The role of cytoplasmic mRNA cap-binding protein complexes in Trypanosoma brucei and other trypanosomatids. Pathogens 6:55. doi: 10.3390/pathogens6040055
Gatto, M., Borim, P. A., Wolf, I. R., Fukuta, da Cruz, T., Ferreira Mota, G. A., et al. (2020). Transcriptional analysis of THP-1 cells infected with Leishmania infantum indicates no activation of the inflammasome platform. PLoS Neglect. Trop. Dis. 14:e0007949. doi: 10.1371/journal.pntd.0007949
Gehring, N. H., Wahle, E., and Fischer, U. (2017). Deciphering the mRNP code: RNA-bound determinants of post-transcriptional gene regulation. Trends Biochem. Sci. 42, 369–382. doi: 10.1016/j.tibs.2017.02.004
Ghosh, S., Verma, A., Kumar, V., Pradhan, D., Selvapandiyan, A., Salotra, P., et al. (2020). Genomic and transcriptomic analysis for identification of genes and interlinked pathways mediating artemisinin resistance in Leishmania donovani. Genes 11:1362. doi: 10.3390/genes11111362
Gimblet, C., Meisel, J. S., Loesche, M. A., Cole, S. D., Horwinski, J., Novais, F. O., et al. (2017). Cutaneous leishmaniasis induces a transmissible dysbiotic skin microbiota that promotes skin inflammation. Cell Host Microbe 22, 13–24. doi: 10.1016/j.chom.2017.06.006
Gossage, S. M., Rogers, M. E., and Bates, P. A. (2003). Two separate growth phases during the development of Leishmania in sand flies: implications for understanding the life cycle. Int. J. Parasitol. 33, 1027–1034. doi: 10.1016/S0020-7519(03)00142-5
Grybchuk, D., Macedo, D. H., Kleschenko, Y., Kraeva, N., Lukashev, A. N., Bates, P. A., et al. (2020). The first non-LRV RNA virus in Leishmania. Viruses 12:168. doi: 10.3390/v12020168
Guerfali, F. Z., Laouini, D., Guizani-Tabbane, L., Ottones, F., Ben-Aissa, K., Benkahla, A., et al. (2008). Simultaneous gene expression profiling in human macrophages infected with Leishmania major parasites using SAGE. BMC Genomics 9:238. doi: 10.1186/1471-2164-9-238
Haydock, A., Terrao, M., Sekar, A., Ramasamy, G., Baugh, L., and Myler, P. J. (2015). “RNA-seq approaches for determining mRNA abundance in Leishmania,” in Parasite Genomics Protocols, ed. C. Peacock (New York, NY: Human Press), 207–219. doi: 10.1007/978-1-4939-1438-8_12
Hillesland, H., Read, A., Subhadra, B., Hurwitz, I., McKelvey, R., Ghosh, K., et al. (2008). Identification of aerobic gut bacteria from the kala azar vector, Phlebotomus argentipes: a platform for potential paratransgenic manipulation of sand flies. Am. J. Trop. Med. Hyg. 79, 881–886. doi: 10.4269/ajtmh.2008.79.881
Iantorno, S. A., Durrant, C., Khan, A., Sanders, M. J., Beverley, S. M., Warren, W. C., et al. (2017). Gene expression in Leishmania is regulated predominantly by gene dosage. MBio 8, e01393–17. doi: 10.1128/mBio.01393-17
Imamura, H., Downing, T., Van den Broeck, F., Sanders, M. J., Rijal, S., Sundar, S., et al. (2016). Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. Elife 5:e12613. doi: 10.7554/eLife.12613
Inbar, E., Hughitt, V. K., Dillon, L. A., Ghosh, K., El-Sayed, N. M., and Sacks, D. L. (2017). The transcriptome of Leishmania major developmental stages in their natural sand fly vector. MBio 8, e00029–e17.
Ivens, A. C., Peacock, C. S., Worthey, E. A., Murphy, L., Aggarwal, G., Berriman, M., et al. (2005). The genome of the kinetoplastid parasite, Leishmania major. Science 309, 436–442. doi: 10.1126/science.1112680
Ives, A., Ronet, C., Prevel, F., Ruzzante, G., Fuertes-Marraco, S., Schutz, F., et al. (2011). Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 331, 775–778. doi: 10.1126/science.1199326
Kalavi, K., Jorjani, O., Faghihi, M. A., and Mowla, S. J. (2020). Cytokine gene expression alterations in human macrophages infected by Leishmania major. Cell J. 22, 476–481. doi: 10.22074/cellj.2021.6524
Katakura, K., and Kobayashi, A. K. (1988). Acid phosphatase activity of virulent and avirulent clones of Leishmania donovani promastigotes. Infect. Imm. 56, 2856–2860. doi: 10.1128/iai.56.11.2856-2860.1988
Kramer, S., and Carrington, M. (2011). Trans-acting proteins regulating mRNA maturation, stability and translation in trypanosomatids. Trends Parasitol. 27, 23–30. doi: 10.1016/j.pt.2010.06.011
Kumari, S., Kumar, A., Samant, M., Singh, N., and Dube, A. (2008). Discovery of novel vaccine candidates and drug targets against visceral leishmaniasis using proteomics and transcriptomics. Curr. Drug Targets 9, 938–947. doi: 10.2174/138945008786786091
Lahav, T., Sivam, D., Volpin, H., Ronen, M., Tsigankov, P., Green, A., et al. (2011). Multiple levels of gene regulation mediate differentiation of the intracellular pathogen Leishmania. FASEB J. 25, 515–525. doi: 10.1096/fj.10-157529
LeBowitz, J. H., Smith, H. Q., Rusche, L., and Beverley, S. M. (1993). Coupling of poly (A) site selection and trans-splicing in Leishmania. Gene Dev. 7, 996–1007. doi: 10.1101/gad.7.6.996
Leifso, K., Cohen-Freue, G., Dogra, N., Murray, A., and McMaster, W. R. (2007). Genomic and proteomic expression analysis of Leishmania promastigote and amastigote life stages: the Leishmania genome is constitutively expressed. Mol. Biochem. Parasitol. 152, 35–46. doi: 10.1016/j.molbiopara.2006.11.009
Liang, X. H., Haritan, A., Uliel, S., and Michaeli, S. (2003). Trans and cis splicing in trypanosomatids: mechanism, factors, and regulation. Euk. Cell 2, 830–840. doi: 10.1128/ec.2.5.830-840.2003
Lin, C. Y., Chin, C. H., Wu, H. H., Chen, S. H., Ho, C. W., and Ko, M. T. (2008). Hubba: hub objects analyzer—a framework of interactome hubs identification for network biology. Nucleic Acids Res. 36, W438–W443. doi: 10.1093/nar/gkn257
Lopez, M. A., Saada, E. A., and Hill, K. L. (2015). Insect stage-specific adenylate cyclases regulate social motility in African trypanosomes. Euk. Cell 14, 104–112. doi: 10.1128/EC.00217-14
Louradour, I., Monteiro, C. C., Inbar, E., Ghosh, K., Merkhofer, R., Lawyer, P., et al. (2017). The midgut microbiota plays an essential role in sand fly vector competence for Leishmania major. Cell Microbiol. 19:e12755. doi: 10.1111/cmi.12755
Malta-Santos, H., França-Costa, J., Macedo, A., Queiroz, A. T., Fukutani, K. F., Muxel, S. M., et al. (2020). Differential expression of polyamine biosynthetic pathways in skin lesions and in plasma reveals distinct profiles in diffuse cutaneous leishmaniasis. Sci. Rep. 10:10543. doi: 10.1038/s41598-020-67432-5
Mandal, G., Mandal, S., Sharma, M., Charret, K. S., Papadopoulou, B., Bhattacharjee, H., et al. (2015). Species-specific antimonial sensitivity in Leishmania is driven by post-transcriptional regulation of AQP1. PLoS Negl. Trop. Dis. 9:e0003500. doi: 10.1371/journal.pntd.0003500
Marquis, N., Gourbal, B., Rosen, B. P., Mukhopadhyay, R., and Ouellette, M. (2005). Modulation in aquaglyceroporin AQP1 gene transcript levels in drug−resistant Leishmania. Mol. Microbiol. 57, 1690–1699. doi: 10.1111/j.1365-2958.2005.04782.x
Martıìnez-Calvillo, S., Yan, S., Nguyen, D., Fox, M., Stuart, K., and Myler, P. J. (2003). Transcription of Leishmania major Friedlin chromosome 1 initiates in both directions within a single region. Mol. Cell 11, 1291–1299. doi: 10.1016/s1097-2765(03)00143-6
Masoudzadeh, N., Mizbani, A., and Rafati, S. (2020a). Transcriptomic profiling in cutaneous Leishmaniasis patients. Expert Rev. Proteomic 17, 533–541. doi: 10.1080/14789450.2020.1812390
Masoudzadeh, N., Mizbani, A., Taslimi, Y., Mashayekhi, V., Mortazavi, H., Sadeghipour, P., et al. (2017). Leishmania tropica infected human lesions: whole genome transcription profiling. Acta tropica 176, 236–241. doi: 10.1016/j.actatropica.2017.08.016
Masoudzadeh, N., Östensson, M., Persson, J., Goyonlo, V. M., Agbajogu, C., Taslimi, Y., et al. (2020b). Molecular signatures of anthroponotic cutaneous leishmaniasis in the lesions of patients infected with Leishmania tropica. Sci. Rep. 10:16198. doi: 10.1038/s41598-020-72671-7
Matthews, K. R., Tschudi, C., and Ullu, E. (1994). A common pyrimidine-rich motif governs trans-splicing and polyadenylation of tubulin polycistronic pre-mRNA in trypanosomes. Genes Dev. 8, 491–501. doi: 10.1101/gad.8.4.491
Mattick, J. S. (2005). The functional genomics of noncoding RNA. Science 309, 1527–1528. doi: 10.1126/science.1117806
McNally, K. P., and Agabian, N. I. (1992). Trypanosoma brucei spliced-leader RNA methylations are required for trans splicing in vivo. Mol. Cell. Biol. 12, 4844–4851. doi: 10.1128/mcb.12.11.4844
McNicoll, F., Drummelsmith, J., Müller, M., Madore, É, Boilard, N., Ouellette, M., et al. (2006). A combined proteomic and transcriptomic approach to the study of stage differentiation in Leishmania infantum. Proteomics 6, 3567–3581. doi: 10.1002/pmic.200500853
Morris, K. V., and Mattick, J. S. (2014). The rise of regulatory RNA. Nat. Rev. Genet. 15, 423–437. doi: 10.1038/nrg3722
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621. doi: 10.1038/nmeth.1226
Mukhopadhyay, J., Braig, H. R., Rowton, E. D., and Ghosh, K. (2012). Naturally occurring culturable aerobic gut flora of adult Phlebotomus papatasi, vector of Leishmania major in the Old World. PLoS One 7:e35748. doi: 10.1371/journal.pone.0035748
Nagalakshmi, U., Wang, Z., Waern, K., Shou, C., Raha, D., Gerstein, M., et al. (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349. doi: 10.1126/science.1158441
Nowrousian, M. (2010). Next-generation sequencing techniques for eukaryotic microorganisms: sequencing-based solutions to biological problems. Euk. Cell 9, 1300–1310. doi: 10.1128/EC.00123-10
Orikaza, C. M., Pessoa, C. C., Paladino, F. V., Florentino, P. T., Barbiéri, C. L., Goto, H., et al. (2020). Dual host-intracellular parasite transcriptome of enucleated cells hosting Leishmania amazonensis: control of half-life of host cell transcripts by the parasite. Infect. Immun. 88, e00261–20. doi: 10.1128/IAI.00261-20
Pandarakalam, G. C., Speake, M., McElroy, S., Alturkistani, A., Philippe, L., Pettitt, J., et al. (2019). A high-throughput screen for the identification of compounds that inhibit nematode gene expression by targeting spliced leader trans-splicing. Int. J. Parasitol. Drugs Drug Res. 10, 28–37. doi: 10.1016/j.ijpddr.2019.04.001
Patino, L. H., Imamura, H., Cruz-Saavedra, L., Pavia, P., Muskus, C., Méndez, C., et al. (2019). Major changes in chromosomal somy, gene expression and gene dosage driven by Sb III in Leishmania braziliensis and Leishmania panamensis. Sci. Rep. 9:9485. doi: 10.1038/s41598-019-45538-9
Peterkova-Koci, K., Robles-Murguia, M., Ramalho-Ortigao, M., and Zurek, L. (2012). Significance of bacteria in oviposition and larval development of the sand fly Lutzomyia longipalpis. Parasit. Vectors 5:145. doi: 10.1186/1756-3305-5-145
Prasad, T. K., Mohanty, A. K., Kumar, M., Sreenivasamurthy, S. K., Dey, G., Nirujogi, R. S., et al. (2017). Integrating transcriptomic and proteomic data for accurate assembly and annotation of genomes. Genome Res. 27, 133–144. doi: 10.1101/gr.201368.115
Probst, C. M., Silva, R. A., Menezes, J. P., Almeida, T. F., Gomes, I. N., Dallabona, A. C., et al. (2012). A comparison of two distinct murine macrophage gene expression profiles in response to Leishmania amazonensis infection. BMC Microbiol. 12:22. doi: 10.1186/1471-2180-12-22
Rabhi, I., Rabhi, S., Ben-Othman, R., Aniba, M. R., Trentin, B., Piquemal, D., et al. (2013). Comparative analysis of resistant and susceptible macrophage gene expression response to Leishmania major parasite. BMC Genomics 14:723. doi: 10.1186/1471-2164-14-723
Rabhi, I., Rabhi, S., Ben-Othman, R., Rasche, A., Daskalaki, A., Trentin, B., et al. (2012). Transcriptomic signature of Leishmania infected mice macrophages: a metabolic point of view. PLoS Negl. Trop. Dis. 6:e0001763. doi: 10.1371/journal.pntd.0001763
Rastrojo, A., Carrasco-Ramiro, F., Martín, D., Crespillo, A., Reguera, R. M., Aguado, B., et al. (2013). The transcriptome of Leishmania major in the axenic promastigote stage: transcript annotation and relative expression levels by RNA-seq. BMC Genomics 14:223. doi: 10.1186/1471-2164-14-223
Rastrojo, A., Corvo, L., Lombraña, R., Solana, J. C., Aguado, B., and Requena, J. M. (2019). Analysis by RNA-seq of transcriptomic changes elicited by heat shock in Leishmania major. Sci. Rep. 9:6919. doi: 10.1038/s41598-019-43354-9
Rastrojo, A., García-Hernández, R., Vargas, P., Camacho, E., Corvo, L., Imamura, H., et al. (2018). Genomic and transcriptomic alterations in Leishmania donovani lines experimentally resistant to antileishmanial drugs. Int. J. Parasitol. 8, 246–264. doi: 10.1016/j.ijpddr.2018.04.002
Ready, P. D. (2013). Biology of phlebotomine sandflies as vectors of disease agents. Ann. Rev. Entomol. 58, 227–250. doi: 10.1146/annurev-ento-120811-153557
Requena, J. M., Chicharro, C., García, L., Parrado, R., Puerta, C. J., and Cañavate, C. (2012). Sequence analysis of the 3’-untranslated region of HSP70 (type I) genes in the genus Leishmania: its usefulness as a molecular marker for species identification. Parasit. Vector 5:87. doi: 10.1186/1756-3305-5-87
Rogers, M. E., Chance, M., and Bates, P. (2002). The role of promastigote secretory gel in the origin and transmission of the infective stage of Leishmania mexicana by the sandfly Lutzomyia longipalpis. Parasitology 124, 495–507. doi: 10.1017/s0031182002001439
Rostamian, M., and Niknam, H. M. (2019). Leishmania tropica: what we know from its experimental models. Adv. Parasit. 104, 1–38. doi: 10.1016/bs.apar.2018.11.001
Ruy, P. D., Monteiro-Teles, N. M., Miserani Magalhaes, R. D., Freitas-Castro, F., Dias, L., Aquino Defina, T. P., et al. (2019). Comparative transcriptomics in Leishmania braziliensis: disclosing differential gene expression of coding and putative noncoding RNAs across developmental stages. RNA Biol. 16, 639–660. doi: 10.1080/15476286.2019.1574161
Sacks, D. L., Modi, G., Rowton, E., Späth, G., Epstein, L., Turco, S. J., et al. (2000). The role of phosphoglycans in Leishmania–sand fly interactions. Proc. Natl. Acad. Sci. U.S.A. 97, 406–411. doi: 10.1073/pnas.97.1.406
Salmon, D., Vanwalleghem, G., Morias, Y., Denoeud, J., Krumbholz, C., Lhommé, F., et al. (2012). Adenylate cyclases of Trypanosoma brucei inhibit the innate immune response of the host. Science 337, 463–466. doi: 10.1126/science.1222753
Sant’Anna, M. R., Darby, A. C., Brazil, R. P., Montoya-Lerma, J., Dillon, V. M., Bates, P. A., et al. (2012). Investigation of the bacterial communities associated with females of Lutzomyia sand fly species from South America. PLoS One 7:e42531. doi: 10.1371/journal.pone.0042531
Schulze, A., and Downward, J. (2001). Navigating gene expression using microarrays — a technology review. Nat. Cell Biol. 3, E190–E195. doi: 10.1038/35087138
Shadab, M., Banerjee, A., Sinha, R., Das, S., Asad, M., Maji, M., et al. (2019). RNA-seq revealed expression of many novel genes associated with Leishmania donovani persistence and clearance in the host macrophage. Front. Cell Infect. Microbiol. 9:17. doi: 10.3389/fcimb.2019.00017
Sinha, R., Malar, C., Kumar, R., Das, S., Das, S., Shadab, M., et al. (2018). Genome plasticity in cultured Leishmania donovani: comparison of early and late passages. Front. Microbiol. 9:1279. doi: 10.3389/fmicb.2018.01279
Soares, R. P., Barron, T., McCoy-Simandle, K., Svobodova, M., Warburg, A., and Turco, S. J. (2004). Leishmania tropica: intraspecific polymorphisms in lipophosphoglycan correlate with transmission by different Phlebotomus species. Exp. Parasitol. 107, 105–114. doi: 10.1016/j.exppara.2004.05.001
Srividya, G., Duncan, R., Sharma, P., Raju, B. V., Nakhasi, H. L., and Salotra, P. (2007). Transcriptome analysis during the process of in vitro differentiation of Leishmania donovani using genomic microarrays. Parasitology 134, 1527–1539. doi: 10.1017/S003118200700296X
Sundar, S., and Singh, A. (2016). Recent developments and future prospects in the treatment of visceral leishmaniasis. Ther. Adv. Inf. Dis. 3, 98–109. doi: 10.1177/2049936116646063
Sundar, S., and Singh, B. (2018). Understanding Leishmania parasites through proteomics and implications for the clinic. Expert Rev. Proteom. 15, 371–390. doi: 10.1080/14789450.2018.1468754
Sundar, S., Agrawal, N., and Singh, B. (2019). Exploiting knowledge on pharmacodynamics-pharmacokinetics for accelerated anti-leishmanial drug discovery/development. Expert Opin. Drug Metab. Toxicol. 15, 595–612. doi: 10.1080/17425255.2019.1629417
Sunter, J., and Gull, K. (2017). Shape, form, function and Leishmania pathogenicity: from textbook descriptions to biological understanding. Open Biol. 7:170165. doi: 10.1098/rsob.170165
Teixeira, M., de Jesus Santos, R., Sampaio, R., Pontes-de-Carvalho, L., and dos-Santos, W. L. (2002). A simple and reproducible method to obtain large numbers of axenic amastigotes of different Leishmania species. Parasitol. Res. 88, 963–968. doi: 10.1007/s00436-002-0695-3
Telleria, E. L., Martins-da-Silva, A., Tempone, A. J., and Traub-Csekö, Y. M. (2018). Leishmania, microbiota and sand fly immunity. Parasitology 145, 1336–1353. doi: 10.1017/S0031182018001014
Ulusan, Ö, Mert, U., Sadıqova, A., Öztürk, S., and Caner, A. (2020). Identification of gene expression profiles in Leishmania major infection by integrated bioinformatics analyses. Acta Tropica 208:105517. doi: 10.1016/j.actatropica.2020.105517
Vera, J. C., Wheat, C. W., Fescemyer, H. W., Frilander, M. J., Crawford, D. L., Hanski, I., et al. (2008). Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol. Ecol. 17, 1636–1647. doi: 10.1111/j.1365-294X.2008.03666.x
Vernal, S., Oliveira, F., Oliveira, W. H., Goulart, T. M., Oristian, J., Calvo, E., et al. (2020). RNA-sequencing of the Nyssomyia neivai sialome: a sand fly-vector from a Brazilian endemic area for tegumentary leishmaniasis and pemphigus foliaceus. Sci. Rep. 10:17664. doi: 10.1038/s41598-020-74343-y
Wagner, G. P., Kin, K., and Lynch, V. J. (2012). Measurement of mRNA abundance using RNA-seq data: rpkm measure is inconsistent among samples. Theor. Biosci. 131, 281–285. doi: 10.1007/s12064-012-0162-3
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Gen. 10:57. doi: 10.1038/nrg2484
World Health Organization [WHO] (2019). WHO Fact sheets, Leishmaniasis, 14 March 2019. Available online at: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (Accessed November 11, 2019)
y Fortéa, J. O., de La Llave, E., Regnault, B., Coppée, J. Y., Milon, G., Lang, T., et al. (2009). Transcriptional signatures of BALB/c mouse macrophages housing multiplying Leishmania amazonensis amastigotes. BMC Genomics 10:119. doi: 10.1186/1471-2164-10-119
Zhang, S., Kim, C. C., Batra, S., and McKerrow, J. H. (2010). Delineation of diverse macrophage activation programs in response to intracellular parasites and cytokines. PLoS Negl. Trop. Dis. 4:e648. doi: 10.1371/journal.pntd.0000648
Keywords: Leishmania, RNA-Seq, gene expression, promastigotes, amastigotes, macrophages, skin lesions, metatranscriptomics
Citation: Salloum T, Tokajian S and Hirt RP (2021) Advances in Understanding Leishmania Pathobiology: What Does RNA-Seq Tell Us? Front. Cell Dev. Biol. 9:702240. doi: 10.3389/fcell.2021.702240
Received: 29 April 2021; Accepted: 30 July 2021;
Published: 01 September 2021.
Edited by:
Adrian Hehl, University of Zurich, SwitzerlandReviewed by:
Markus Engstler, Julius Maximilian University of Würzburg, GermanyDerek Prosser, Virginia Commonwealth University, United States
Copyright © 2021 Salloum, Tokajian and Hirt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tamara Salloum, dHNhbGxvdW1AYndoLmhhcnZhcmQuZWR1
†Present address: Tamara Salloum, Jeff and Penny Vinik Center for Allergic Disease Research, Division of Allergy and Clinical Immunology, Brigham and Women’s Hospital, Boston, MA, United States; Department of Medicine, Harvard Medical School, Boston, MA, United States