 Anna Chanou
Anna Chanou Stephan Hamperl
Stephan Hamperl- Chromosome Dynamics and Genome Stability, Institute of Epigenetics and Stem Cells, Helmholtz Zentrum München, Munich, Germany
Besides the basic organization in nucleosome core particles (NCPs), eukaryotic chromatin is further packed through interactions with numerous protein complexes including transcription factors, chromatin remodeling and modifying enzymes. This nucleoprotein complex provides the template for many important biological processes, such as DNA replication, transcription, and DNA repair. Thus, to understand the molecular basis of these DNA transactions, it is critical to define individual changes of the chromatin structure at precise genomic regions where these machineries assemble and drive biological reactions. Single-molecule approaches provide the only possible solution to overcome the heterogenous nature of chromatin and monitor the behavior of individual chromatin transactions in real-time. In this review, we will give an overview of currently available single-molecule methods to obtain mechanistic insights into nucleosome positioning, histone modifications and DNA replication and transcription analysis—previously unattainable with population-based assays.
Introduction
The remarkable length and complexity of eukaryotic genomes poses several challenges to the cell. The limited 3D space in the nucleus requires a high degree of DNA compaction, while maintaining sufficient accessibility for gene expression. In order to overcome this challenge, cells package the genome into a nucleoprotein complex termed chromatin (Igo-Kemenes et al., 1982). Chromatin plays a central role for all DNA-dependent transactions, such as replication, transcription, recombination, and DNA repair, thus requiring tight regulation (Kornberg and Lorch, 2020).
The nucleosome is the basic unit of chromatin, each occupying 147 bp of DNA wrapped around histone octamers. Each octamer consists of two histone H3-H4 dimers that complexes with two histone H2A-H2B dimers (McGhee and Felsenfeld, 1980; Luger et al., 1997). Individual nucleosome core particles (NCPs) are separated by short 15–50 bp of linker DNA, giving chromatin the typical appearance of “beads on a string,” as shown by classical electron microscopy studies (Olins and Olins, 1978). In general, the tight interaction of DNA and octamers is inhibitory to most nuclear processes and thus, nucleosomes must be dynamically repositioned to allow binding or repel effector proteins from specific regulatory DNA regions. Apart from intrinsically favorable or unfavorable DNA sequences for nucleosome formation (Travers et al., 2010), other cellular components, such as transcription factors and ATP-dependent chromatin remodeling machines contribute to the chromatin landscape in vivo (Dou and Gorovsky, 2000; Jenuwein, 2001; Lusser and Kadonaga, 2003; Heintzman et al., 2009; Bell et al., 2011). In addition, the unstructured N-terminal tails of histones protrude from the nucleosomal core, presenting platforms of discrete epitopes that can be targeted for addition or removal of distinct post-translational modifications (PTMs). More than a hundred histone modifications are described including the acetylation of lysines, the methylation of arginines and lysines, and the phosphorylation of serines and threonines (Tyler et al., 1999; Strahl and Allis, 2000; Jenuwein, 2001; Lusser and Kadonaga, 2003; Gibney and Nolan, 2010). Combining these diverse PTMs in a combinatorial way greatly increases the heterogeneity of distinct chromatin states that can co-exist in a given genome. Broadly, two major categories of chromatin can be distinguished: highly condensed heterochromatin that is transcriptionally inert and more open, transcriptionally active euchromatin.
It is clear that the dynamics, structure and composition of chromatin have a major influence on the transcriptional and replicative output of a genome. To understand these complex processes in more mechanistic details, it is critical to obtain a comprehensive overview of the composition and histone PTM patterns in correlation with the functional state of a locus of interest. Various population-based techniques have been developed to investigate the architecture of chromatin. The genome can be surveyed for exposed regions accessible for nuclease (DNAse-seq, MNase-seq, FAIRE-seq) (Schones et al., 2008; Bianco et al., 2015; Ishii et al., 2015) or transposase attack (ATAC-seq) to distinguish between open regulatory regions of chromatin vs. those protected by nucleosomes (Buenrostro et al., 2015).
To probe for more specific protein-DNA interactions, chromatin immunoprecipitation (ChIP) has been a powerful tool to determine genome-wide binding profiles of transcription factors, histone PTMs or variants and other chromatin-bound factors on the one-dimensional genetic sequence (Schmid et al., 2004; Wal and Pugh, 2012). In recent years, the Cleavage Under Targets & Release Using Nuclease (CUT&RUN) method has emerged as a powerful alternative to ChIP, as the antibody-mediated tethering of MNase releases directly DNA-protein complexes into the supernatant in situ without the need for fragmentation and solubilization of total chromatin, which represents a major drawback of ChIP (Meers et al., 2019). However, the binding profiles generated by both methods oversimplify the activities of regulatory elements like enhancers, silencers, insulators, and boundaries, that can affect their corresponding target genes over megabase distances by 2D and 3D chromatin looping (Bonev and Cavalli, 2016). Thus, to characterize the global contacts in 3D physical space, several Hi-C chromosome conformation capture protocols have been developed providing detailed interaction maps up to kilobase pair resolution (Rao et al., 2014). More recently, Micro-C, where MNase is used for fragmentation prior to re-ligation, was shown to provide interaction maps at nucleosome resolution in yeast and several human and murine cell lines (Hsieh et al., 2016).
Despite their usefulness and great insights into the chromatin structure and spatial organization of chromosomes, it is important to realize that all of these genomic methods describe the properties of enormous numbers of molecules, averaging the measured parameters over a large population of molecules and cells. Thus, the behavior of individual molecules with distinct conformations and properties cannot be observed over time. In addition, bulk methods fail to detect rare events that occur only in a small subpopulation of molecules. Therefore, single-molecule methods provide the only possible solution to detect the functional differences and uncover intermolecular heterogeneities, particularly important in the context of chromatin. In this review, we will give an overview of currently available single-molecule methods that provide mechanistic insight into chromatin structure and processes, such as chromatin accessibility, histone modifications, replication and transcription (Supplementary Table 1). For each technique, we start with a brief description of the general principles and provide selected examples how these methods have contributed to our understanding of chromatin which would not have been possible by using bulk and ensemble studies.
Single-Molecule Approaches to Detect Nucleosome Occupancy and Positioning
Electron Microscopy After Psoralen Crosslinking
Nucleosomal arrays were first detected by electron microscopy (EM) in 1974 (Olins and Olins, 1974) revealing a “beads on a string” structure (Kornberg, 1974). In 1976, photochemical crosslinking of DNA with derivates of psoralen was used for nucleosome visualization under EM (Hanson et al., 1976; Sogo et al., 1984). Psoralen is a furanocoumarin compound with the ability to intercalate into double-stranded DNA. After irradiation with ultraviolet (UV)-light at 366 nm, psoralen creates covalent crosslinks between pyrimidines of opposite DNA stands (Cimino et al., 1985). The crosslinking occurs in linker DNA, whereas the nucleosomal DNA is protected, which allows to distinguish whether a DNA region had been occupied by a nucleosome or not (Figure 1). After deproteinization, the crosslinked DNA shows a characteristic shift in native agarose gel electrophoresis and can be analyzed by Southern blot analysis (Cimino et al., 1985; Wellinger and Sogo, 1998; Lucchini, 2001). Such experiments could for example demonstrate the co-existence of two major populations of ribosomal DNA (rDNA) genes, a nucleosome-free open state and a nucleosomal closed state (Wittner et al., 2011). However, this bulk analysis could only determine qualitative differences and not inform on the individual nucleosome configurations and heterogeneity among individual molecules.
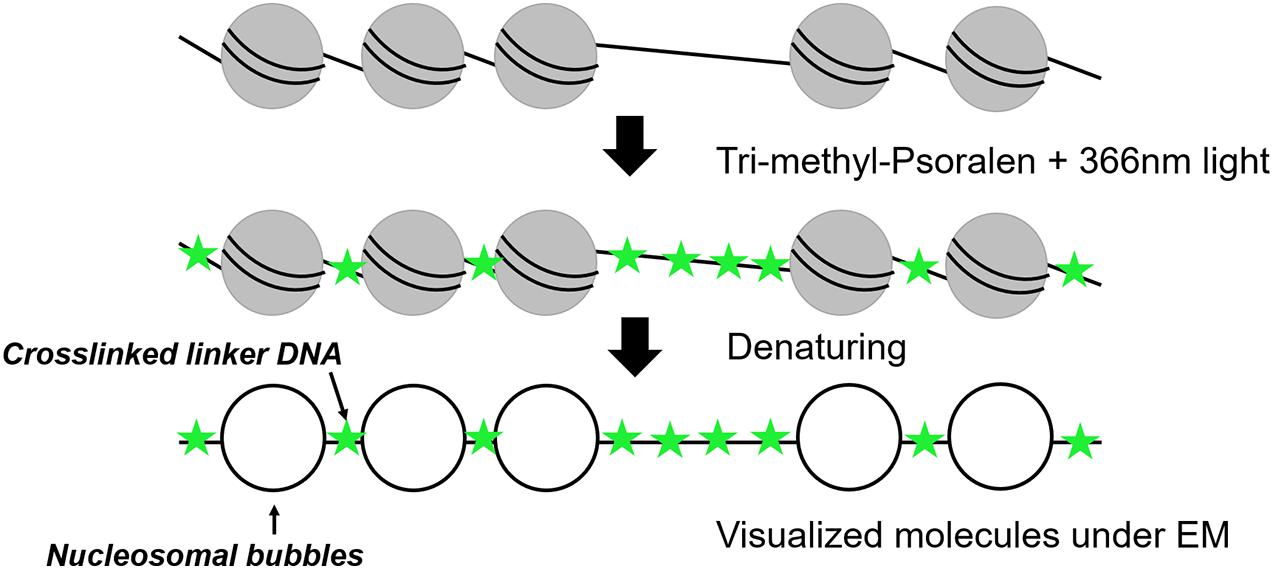
Figure 1. Schematic representation of psoralen crosslinking on a nucleosomal template.
Thus, the combination of psoralen-crosslinking and visualization by EM can be considered as one of the pioneering tools to determine nucleosome positioning at the single-molecule level. After denaturation of the DNA strands, linker DNA is observed double-stranded, whereas nucleosomal DNA appears as single-stranded DNA bubbles (Brown and Boeger, 2014; Figure 1). Inspection of yeast rDNA chromatin by this technique clearly demonstrated the co-existence of the two classes of rRNA genes at the single-molecule level (Dammann et al., 1993). More recently, this technique was applied to single-gene molecules encompassing the PHO5 promoter in yeast under inducing and non-inducing conditions. Interestingly, both the induced as well as non-induced PHO5 promoter displayed with different frequencies every single combinatorial possibility to occupy the three PHO5 promoter nucleosome positions (23 = 8). This finding suggests that individual PHO5 promoters pass, in a certain sequence, through alternative nucleosome configurations (Brown et al., 2013). Such a large heterogeneity in promoter nucleosome configuration—uncovered by a single-molecule technique—can thus explain gene expression fluctuations and suggest a structural basis for transcriptional bursting. Recently, integration of these single-molecule with other existing data on chromatin accessibility and histone turnover revealed that only few models that include directional sliding and a regulated assembly instead of disassembly process was compatible with the experimental data and suggests PHO5 promoter chromatin opening by binding competition (Wolff et al., 2021). Psoralen crosslinking has also been used to determine the nucleosome positioning at 5S rDNA molecules. The molecules were clustered into 12 different groups exhibiting high level of heterogeneity among the cell population, but the number of molecules with nucleosome-free 5S sequences could recapitulate the expected accessibility and transcriptional activity of 5S rRNA genes (Hamperl et al., 2014). One drawback of this method is the need for special equipment and the costly and time-consuming experimental setup limiting this application to more specialized labs. However, EM remains the only available tool offering sufficient resolution to directly visualize individual nucleosomal footprints at the single-molecule level.
Methylation Footprinting
Similar to psoralen only intercalating into accessible, nucleosome-free regions of chromatin, bulk chromatin can be treated with specific DNA methyltransferases that target unprotected cytosines of CpG dinucleotides within free DNA but not the DNA in complex with a nucleosome or a transcription factor (Kladde and Simpson, 1994; Miranda et al., 2010). Upon bisulfite conversion, unmethylated cytosine residues are deaminated to uracil but methylated cytosine (5-mC) residues remain inert to this reaction (Figures 2A,B). The methylated DNA fragment is amplified by PCR and cloned into a vector for amplification in E. coli. By comparison to a reference or untreated DNA sequence, methylation events can therefore be detected directly by traditional Sanger or Illumina sequencing, providing a single-molecule readout with base-pair resolution that is highly quantitative (Miranda et al., 2010; Li and Tollefsbol, 2011).
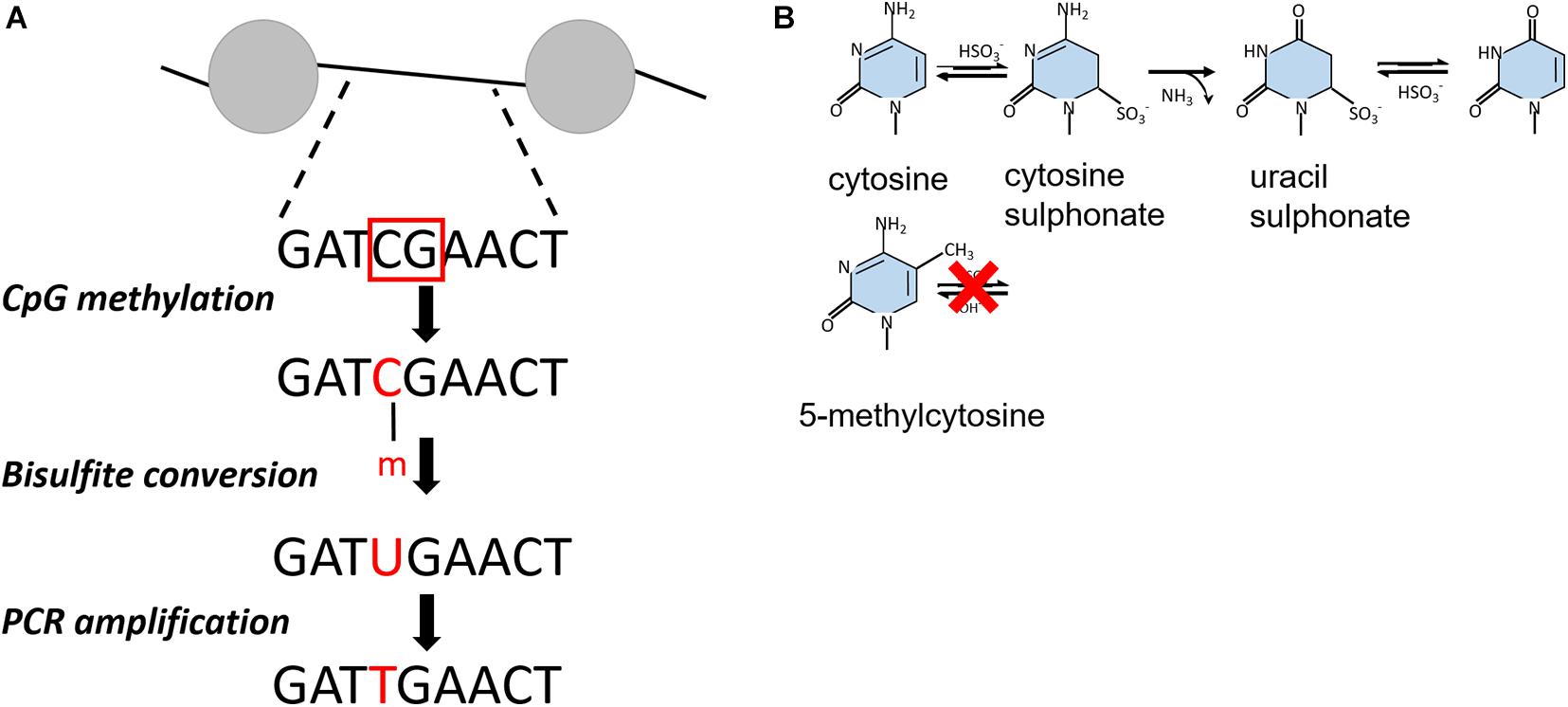
Figure 2. (A) Outline of bisulfite conversion on an example DNA fragment. The red squares indicate non-nucleosomal regions recognized by CpG methyltranferases. After bisulfite conversion, cytosines are presented as uracil, which are converted into thymidine. Nucleosomal regions are discriminated from non-nucleosomal by comparing the resulting PCR product with the initial DNA sequence. (B) Chemical reaction of bisulfite-catalyzed conversion of cytosine to uracil.
Unlike yeast and Drosophila, mammalian genomes show endogenous DNA methylation at CpG dinucleotides (Stadler et al., 2011), preventing the use of exogenous CpG methyltransferases in this sequence context. By using GCH (H = A, T, or C) cytosines to track nucleosome occupancy and HCG for endogenous DNA methylation, this technique could be adjusted to provide a simultaneous readout of nucleosome positioning and DNA methylation from the same DNA molecule across the human genome (Kelly et al., 2012). At gene promoters, nucleosome occupancy and DNA methylation patterns were consistent with transcription potential. The active promoters showed an unmethylated profile with a defined nucleosome free region. The inactive promoters were unmethylated as well but occupied by nucleosomes, whereas silent promoters were both methylated and fully nucleosomal. These findings demonstrate that only nucleosome positioning, or DNA methylation state of promoters as exclusive factors cannot predict the transcriptional state of a gene (Kelly et al., 2012).
In more recent years, third generation sequencing platforms including the Single-Molecule Real-Time sequencing (SMRT-seq) and Nanopore sequencing emerged (Clarke et al., 2009; Eid et al., 2009). For both techniques, sequencing library preparation does not include an amplification step, therefore enabling single-molecule sequencing. Importantly, these methods allow direct detection of modified bases in natural DNA samples without bisulfite conversion. In addition, single read lengths can reach the megabase range, giving the opportunity to accurately assemble and define repetitive elements and epigenetic modifications on variable chromatin stages and domains (Jain et al., 2016; Ardui et al., 2018).
The SMRT-seq approach by PacBio is based on fluorescently labeled nucleotides that are incorporated by DNA polymerase into the complementary DNA strand. The arrival times and durations of the individual fluorescent nucleotides provides information on the polymerase kinetics and the direct detection of covalently modified nucleotides including N6-methyladenine and 5-hydroxymethylcytosine (Figure 3). Each modification shows a unique kinetic signature and therefore allows to distinguish between them on the same DNA molecule (Flusberg et al., 2010). In the beginning, the accurate discrimination between a methylated and non-methylated base necessitated many controls and optimization steps. For that reason, a SMRT-BS method was reported combining bisulfite conversion with third-generation single-molecule real-time (SMRT) sequencing. The conceptual idea was to use the bisulfite conversion for accurate detection of CpG methylation on 1.5 kb regions and in parallel take advantage of SMRT sequencing with minimal clonal PCR artifacts (Yang et al., 2015).
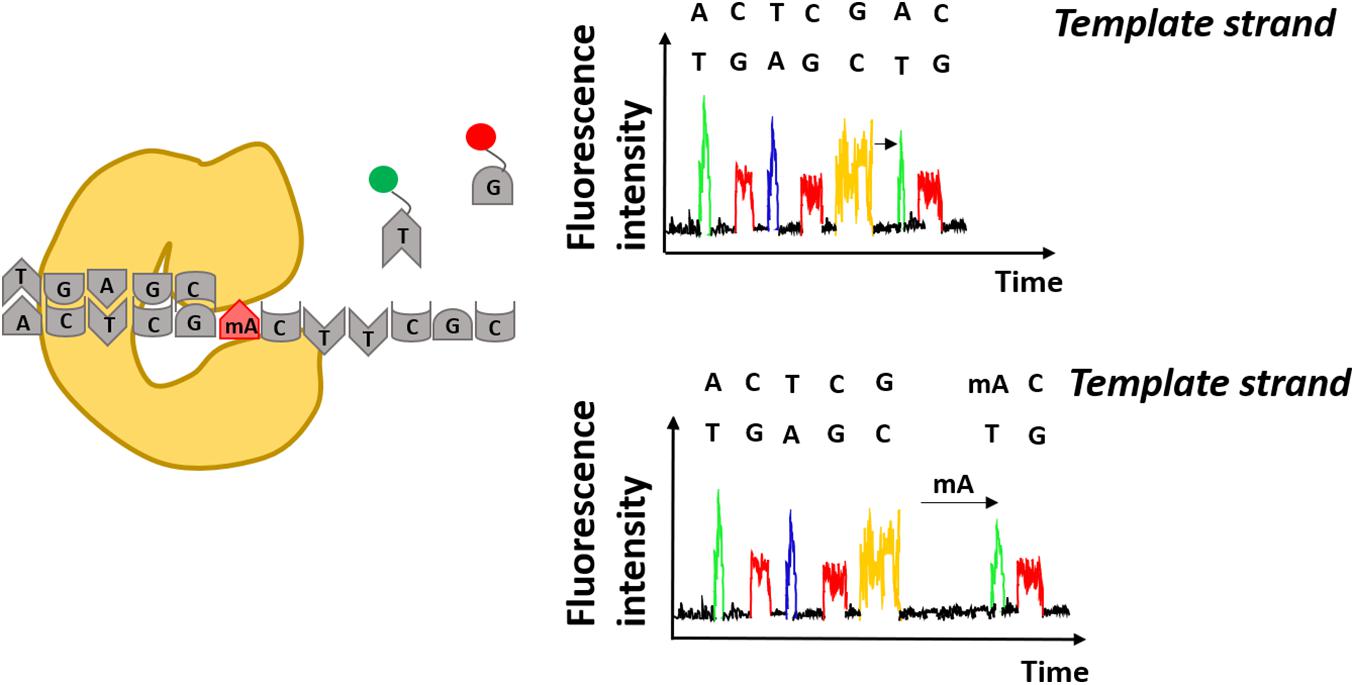
Figure 3. Detection of modified adenines using Single-Molecule Real Time sequencing. Specific fluorescence underlies incorporation of modified nucleotides by DNA polymerase. Differences in arrival times of the same nucleotides e.g., thymidine (T) indicate presence of modified nucleotides on the DNA template strand e.g., methylated adenine (mA).
Recently, using SMRT-seq only, two techniques named Fiber-seq and Single-Molecule-Adenine Methylated Oligonucleosome Sequencing Assay (SAMOSA) were developed using an unspecific N6-Methyladenine (m6A) DNA-methyltransferase for nucleosome detection in higher resolution (Abdulhay et al., 2020; Stergachis et al., 2020). Eukaryotic genomes are devoid of m6A but at the same time, adenines are present on double stranded DNA with an average rate of one every two DNA bases, providing unprecedented coverage of methylation sites and therefore higher resolution compared to CpG methylation.
Using this methodology, Fiber-seq could determine the primary architecture of single chromatin fibers at basepair resolution (Stergachis et al., 2020). Firstly, Drosophila melanogaster S2 cells were used to prove that DNA methyltranferases are unable to modify DNA wrapped in NCPs. The aim was to investigate whether the presence or absence of nucleosomes is the exclusive and most essential factor for the actuation of the regulatory elements or if the possible chromatin stages are more diverse and complex due to intermediate configurations. The results suggest that co-actuation exist on tightly clustered elements, meaning that high DNA accessibility of regulatory sequences at a given distal element can also increase accessibility of neighboring elements in a distance-dependent manner. Moreover, the obtained data demonstrate that DNA sequence alone might not define nucleosome positioning but it is mostly dependent on the actuation of regulatory DNA, since well-positioned nucleosomes were preferentially derived from fibers in which the regulatory element is in an actuated state (Stergachis et al., 2020). Similarly, SAMOSA was first applied as a proof of concept on in vitro assembled chromatin confirming its ability to track nucleosome positioning. As a next step, oligonucleosomes of K562 cells were used showing impartial nucleosome mapping on both euchromatin and heterochromatin regions (Abdulhay et al., 2020). Surprisingly, this study revealed elevated heterogeneity in both actively transcribed euchromatin and constitutive heterochromatin regions, the latter being typically viewed as a conformationally more static epigenomic domain.
Apart from SMRT-seq, nanopore sequencing provided by Oxford Nanopore Technologies (ONT) comprises another powerful tool for defining nucleosome positioning and chromatin accessibility. The idea of nanopore sequencing was developed by the observation that the different ionic current of DNA bases passing through a biological pore could allow discrimination of the different DNA bases (Kasianowicz et al., 1996). Apart from the canonical four DNA bases, different ionic current is also observed for methylated vs. non-methylated bases (Figure 4). In 2019, this property was exploited by Methyltransferase treatment at GpC sites (by M.CviPI) on haploid Saccharomyces cerevisiae followed by Single-Molecule Long-Read sequencing (MeSMLR-seq) to probe long-range chromatin accessibility and nucleosome mapping at single DNA molecules (Wang Y. et al., 2019). The authors focused their analysis on transcription start sites (TSSs) and showed that silent genes show larger heterogeneity in nucleosome positioning compared to transcriptionally active genes. Additionally, MeSMLR-seq reads fully capture the coupled differences of chromatin accessibility of two genes in close neighborhood, providing the ability to uncover the heterogeneity not only within a cell population, but also at adjacent regions within the same molecule. Although bulk nucleosome mapping methods of the CLN2 promoter showed significant accessibility, it remained unknown if the open promoter conformation is uniform in the sense that the length of the accessible region among CLN2 promoters is identical. MeSMLR-seq revealed three distinct profiles of accessibility on CLN2 promoters: closed, narrowly opened and widely opened. This precise and accurate quantitative analysis expanded our current view on the different chromatin states and their heterogeneity at promoter elements (Wang Y. et al., 2019).
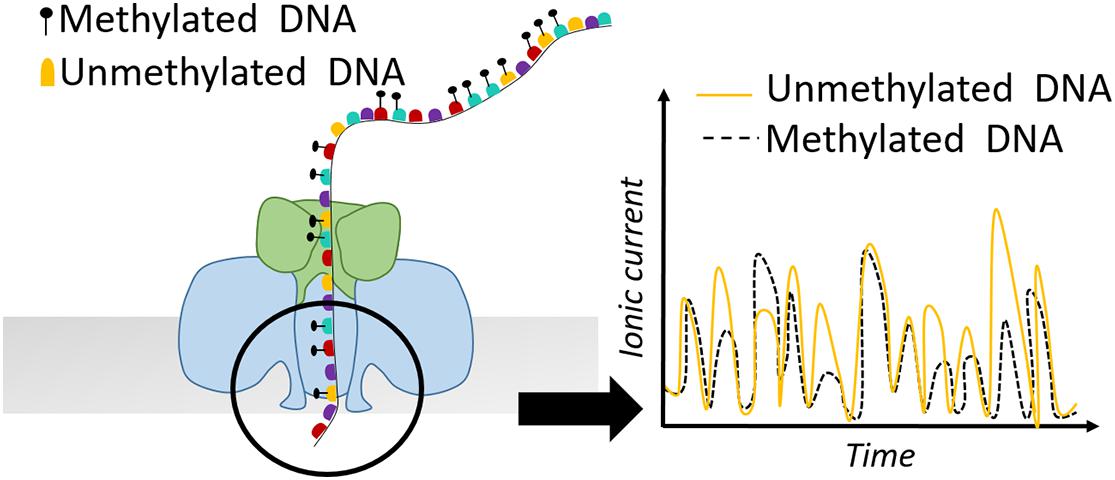
Figure 4. Schematic representation of a nanopore detecting ionic current of a DNA sequence. Methylated bases show different electric current signal compared to unmethylated residues.
Single-Molecule long-read Accessible Chromatin mapping sequencing (SMAC-seq) is another recently developed approach sharing the same concept with previous studies, but in parallel providing much higher resolution of the nucleosome landscape due to the simultaneous use of three different methyltransferases M.CviPI (GpC-5mC), M.SssI (CpG-5mC), and EcoGII (A at any sequence context-m6A) (Shipony et al., 2020). Given that this analysis has not been performed before, a combination of three different bioinformatic tools to detect methylation was necessary. Albacore was applied for the raw base calling, Tombo for m6A methylation detection, and Nanopolish for detection of CpG and GpC methylation. The findings of this study confirmed well-positioned nucleosomal patterns on yeast centromeric DNA in agreement with previous data. Moreover, it was shown that SMAC-seq is able to reveal binary chromatin states on rDNA, where approximately 25% of the individual molecules present full accessibility of the transcribed 35S region compared to the non-transcribed intergenic sequence (Shipony et al., 2020).
Taking the advantage of long read sequencing, ONT was also applied to four different human cell lines revealing the epigenetic status of single chromatin molecules based on endogenous CpG methylation and the chromatin accessibility measured by exogenous GpC methylation labeling (Lee et al., 2020). As a proof of concept, it was observed that regions closer to TSS exhibit higher CpG methylation status and lower GpC accessibility when H3K27me3 marks are present. However, after repression of H3K27me3, TSS promoter regions showed lower CpG methylation level and higher GpC accessibility. Thus, combining the two features- methylation and accessibility- and focusing on the same single reads around TSS could reveal characteristic epigenetic profiles, that are highly correlated with their corresponding expression profile (Lee et al., 2020).
Single-Molecule Approaches to Monitor Histone PTMs and Effector Proteins
Besides the positioning of nucleosomes allowing or denying access to regulatory elements in the genome, the presence or absence of specific histone PTMs, histone variants, or other chromatin factors play pivotal roles for the functional state of the associated DNA. Ideally, one would need to measure multiple histone modifications at defined genomic loci and follow their dynamics live over time in single cells. Although such combinatorial in-depth analysis of histone PTMs still needs to be established at the single cell level, recent advances in imaging and sequencing techniques have started to uncover the dynamics of distinct histone modifications in various ways at the single cell level.
Imaging Techniques
Imaging techniques emerge as powerful tools to detect histone PTMs at single cell and even single-molecule resolution. For example, High-throughput Histone Mapping (HiHiMap) allowed rapid and iterative quantification of histone PTMs as well as total histone levels in single cells throughout the cell cycle. Several histone modifications showed differences between normal and cancer cells, suggesting a potential role in oncogenic transformation (Zane et al., 2017). Another multicolor immunofluorescence technique used directly labeled modification-specific antibodies and profiled the dynamics of up to four histone modifications during the cell cycle (Hayashi-Takanaka et al., 2020). As epigenetic instability is a hallmark of cancer cells and many other human diseases, these high-throughput microscopy methods to quantify epigenetic marks also promise great potential to detect rare subpopulation of cells in patient samples and may inform on improved therapeutic strategies.
Förster resonance energy transfer (FRET) is a well-established method for studying structural and dynamic changes of nucleosomes at both the ensemble and the single-molecule level including PTMs (Simon et al., 2011) and presence of histone variants (Park et al., 2004; Hoch et al., 2007). These studies led to important insights, for example that mononucleosomes unwrap in a time window of ∼250 ms but rewrap more rapidly in 10–50 ms (Li et al., 2005). In addition, FRET based job plot experiments could reveal the stoichiometry of the trimeric CAF-1 histone chaperone leading to mechanistic insights how CAF-1 can deposit an (H3-H4)2 tetramer on substrate DNA in the first step of nucleosome assembly (Mattiroli et al., 2018; Sauer et al., 2018). More recently, single-molecule FRET (smFRET) of reconstituted, site-specifically labeled chromatin fibers revealed that NPCs undergo stacking interactions, which turnover on the micro- to millisecond timescale. Interestingly, binding of the heterochromatin protein HP1α transiently stabilizes stacked nucleosomes, resulting in a more compact albeit dynamic chromatin state (Kilic et al., 2018). In another smFRET study, the effect of histone acetylation and phosphorylation on nucleosome dynamics was analyzed (Brehove et al., 2015). Tyrosine 41 and threonine 45 phosphorylation of the H3 nucleosomal core resulted in enhanced DNA accessibility, similar to lysine 56 acetylation in the same core histone region. Remarkably, simultaneous phosphorylation and acetylation showed an additive effect and increased DNA accessibility more than 10-fold. These studies demonstrate the great potential of single-molecule fluorescence techniques to visualize nucleosome dynamics depending on histone PTMs and the binding of effector proteins. The vast amount of identified histone PTMs and other effector proteins await more single-molecule studies and promise great advance to our understanding of nucleosome structure and dynamics. However, interpretation of fluorescence microscopy data must always take into account the photophysical properties of the fluorophores regarding photostability, size, redox properties, and several other factors (Ha and Tinnefeld, 2012).
Force spectroscopy methods overcome these limitations as they detect the light scattered by micron-size particles. Thus, atomic force microscopy (AFM) has been extensively used to analyze chromatin at multiple levels, ranging from large chromatin fibers down to NCPs (Kalle and Strappe, 2012). In AFM, interactions between atoms of the tip and atoms of the sample leads to deflection that can be converted into topographic images of the sample (Zlatanova and van Holde, 2006). For example, high-speed time-lapse AFM demonstrated that nucleosomes undergo heterogenous and spontaneous disassembly and sliding events at the timescale of milliseconds, highlighting an important role of electrostatic interactions in chromatin dynamics (Miyagi et al., 2011). Optical and magnetic tweezers are two force manipulation methods that have been used to investigate nucleosome dynamics, histone DNA interactions as well as higher-order structural properties of chromatin (Killian et al., 2012). For example, magnetic tweezers experiments revealed that certain histone PTMs in vicinity to the dyad axis decrease the stability and promote eviction of nucleosomes (Simon et al., 2007). In an independent approach, single native chromatin molecules were subjected to nanochannels allowing the simultaneous high-throughput detection of fluorescent signatures of both the DNA and histone proteins within the chromatin. This single-chromatin analysis at the nanoscale (SCAN) method was used to determine the relationship of H3K9me3, H3K27me3, and CpG methylation in normal and cancer cells (Cipriany et al., 2010). Further studies employing such nano-platforms (Li et al., 2020) have sufficient throughput and high potential to provide a comprehensive picture of chromatin structure and may therefore also provide translational insights, such as cancer cell plasticity or the development of chemoresistance.
Sequencing Techniques
The development of single cell ChIP-seq (scChIP-seq) technology provides histone modification detection at the genome-wide, single-cell level (Rotem et al., 2015). A droplet microfluidics-based procedure is necessary to limit possible sonication and sequencing library preparation problems. Although the low number (less than a thousand) reads per cell is still a significant challenge hampering broader applications, the authors reported H3K4me2 and H3K4me3 patterns in several cell lines, such as mouse ES cells, embryonic fibroblasts and hematopoietic progenitors that correlated well with bulk ChIP-Seq data. In addition, this paved the way for three new immunoprecipitation-free epigenomic profiling methods relying on in situ (inside nuclei) reactions. Single cell chromatin integration labeling followed by sequencing (scChIL-seq) (Harada et al., 2019) and the single cell cleavage under targets and tagmentation (scCUT&Tag) (Kaya-Okur et al., 2019) methods use specific antibodies against histone modifications and integrate a sequencing tag into double stranded DNA via transposase. This method was recently adapted to scalable nanowell and droplet-based single-cell platforms to profile polycomb group (PcG) silenced regions marked by H3K27me3 in tissue culture as well as patient samples (Wu et al., 2021). Similarly, combinatorial barcoding and targeted chromatin release (COBATCH) uses the enrichment of a Protein A-Tn5 transposase fusion protein to genomic regions by specific antibodies and produced for H3K27 acetylation marks ∼12,000 unique non-duplicated reads per cell (Wang Q. et al., 2019). The successful implementation and further developments of the Cleavage Under Targets & Release Using Nuclease (CUT&RUN) method by the Henikoff lab have now clearly revolutionized chromatin profiling methods as the use of MNase avoids sonication, material loss and background issues and thus overcomes many of the drawbacks of conventional and widely used chromatin immunoprecipitation (ChIP) methods (Meers et al., 2019). CUT&RUN-based single-cell chromatin immunocleavage sequencing (scChIC-Seq) uses an MNase-conjugated antibody which cleaves the non-nucleosomal DNA regions (Ku et al., 2019). Together, these latest developments of CUT&RUN-based methods toward single-cell applications promise great advances in epigenome profiling and insights into the heterogeneity of epigenetic marks even at the single cell resolution.
Single-Molecule Approaches to Monitor Transcription
The different steps of the RNA Polymerase II (RNAP) transcription cycle have been studied in great detail by a combination of structural, biochemical and single-molecule approaches. Eukaryotic transcription is initiated by the binding of RNAP together with general transcription factors (GTF) onto the promoter region, the double stranded DNA is melted, and the transcription bubble is generated. When RNAP and GTF—TFIIA, TFIIB, TFIID, TFIIE, TFIIF, and TFIIH—assemble at promoters, they form a pre-initiation complex (PIC) to direct accurate transcription initiation. Although many of these key factors have been identified using bulk methods (Hofmann et al., 2004), important questions regarding the dynamic aspects or the mechanism of DNA opening and transcription start site (TSS) scanning required single-molecule approaches to provide sufficient resolution and to avoid population-averaging effects. Single-molecule approaches were also required to study transcription elongation, given that RNAP does not transcribe all DNA templates uniformly and at constant speeds (Kassavetis et al., 1978; Galburt et al., 2009). Instead, transcription elongation is interrupted by several pause and release states, such as promoter proximal escape (Muse et al., 2007), binding of transcription factors like TFIIS (Fish and Kane, 2002) nucleosomes (Kireeva et al., 2005) and association of splicing factors (Wachutka et al., 2019), which could not be revealed by population based methods.
Force Manipulation Methods (Optical and Magnetic Tweezers)
For this purpose, the development of optical tweezer technologies was instrumental towards a greater understanding of the dynamics of eukaryotic transcription on a single-molecule level. This technology was initiated in 1986 by the observation that interaction of a laser beam and small particles results in the formation of a three-dimensional trap with restoring forces on the order of piconewtons (Dziedzic et al., 1986). Regarding transcription, optical tweezers have been initially used for studies on elongation (Galburt et al., 2007, 2009; Larson et al., 2012; Schweikhard et al., 2014) and later they were also applied on transcription initiation (Fazal et al., 2015). In general, the experimental set-up is based on a “dumbbell” construction, consisting of two beads held in separate optical traps, connected by a segment of DNA. RNAP enzyme is attached to one bead and the opposite end of the DNA template is attached to another optical bead (Figure 5; Galburt et al., 2009; Fazal et al., 2015). T he force that is applied on the enzyme either helps or hampers transcription. Transcription elongation and nucleosome arrangement have been broadly investigated in the helping force mode. In this setup, one DNA molecule is labeled upstream of RNAP which results in increasing the distance between the two beads and therefore decreasing the generating force (Fitz et al., 2016). On the other hand, in the opposing mode, the tagged DNA is anchored downstream of RNAP. In this way, transcription reduces the distance between the two beads resulting in increased force that can be measured. For example, this setup was instrumental to determine the dynamics and frequency of pausing events of RNAP (Figure 5; Lisica and Grill, 2017).
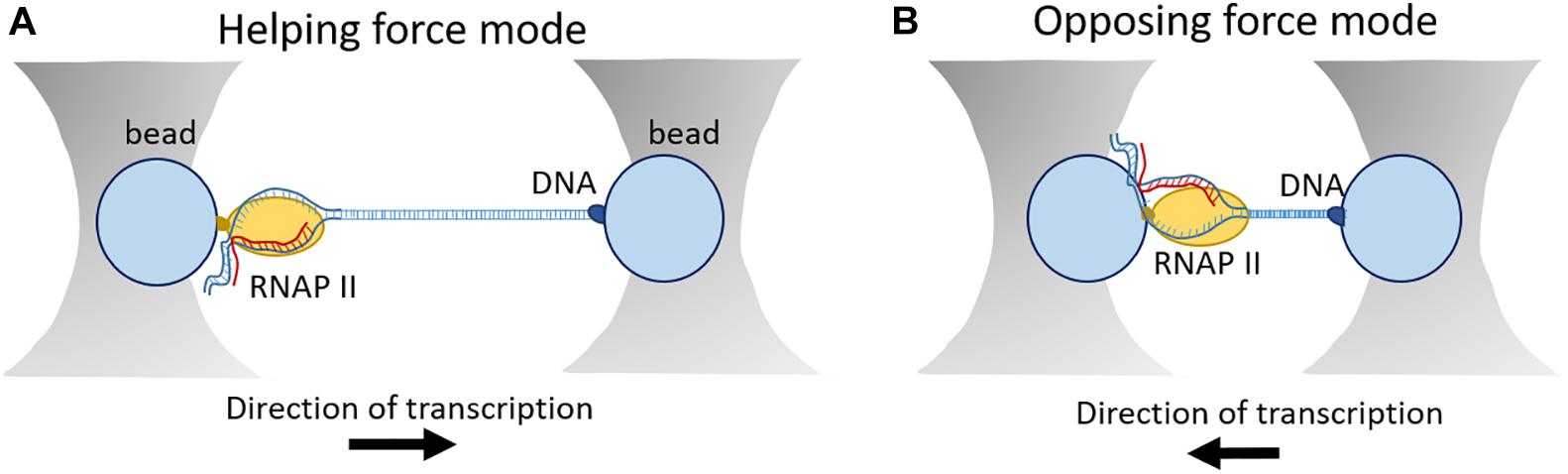
Figure 5. Outline of an optical tweezer set-up. (A) In the helping force mode, DNA is labeled upstream of RNAP leading to increased distance between the two beads and decreased force generation. (B) In the assisting force mode, DNA is labeled downstream of RNAP resulting in shortened distance between the two beads and increased force generation.
Other applications of optical tweezer assays revealed that yeast RNAP II is unable to transcribe once the force of optical beads exceeds 8 pN. However, the addition of a single transcription factor (TFIIS) allows the polymerase to continue transcription against forces up to 20 pN (Galburt et al., 2007). An interesting interpretation of these data might be that the eukaryotic polymerase has evolved alongside with mechanical gene regulatory processes that control the ability of the polymerase to perform work (Galburt et al., 2009). In a large biochemical effort, a 32-protein, 1.5-megadalton yeast PIC could be assembled in an optical tweezer setup which gave important insights into the TFIIH-dependent DNA scanning mode that results in rapid DNA unwinding and in an extended transcription bubble formation with an average size of 85 bp (Fazal et al., 2015).
Complimentary to optical tweezer technology, magnetic tweezers were invented to allow single-molecule manipulation of paramagnetic beads using a magnetic field gradient. The first application of magnetic tweezers was in 1996 (Strick et al., 1996), who investigated the elasticity of supercoiled DNA. In a typical experiment, one side of a DNA molecule is attached to a paramagnetic bead and the opposite side is immobilized via streptavidin to a glass surface. Fixed magnets produce a magnetic field which pulls the bead in the direction of the field gradient. The magnets can be moved in a way to stretch or twist the single DNA molecule and these changes are recorded in real-time (Sarkar and Rybenkov, 2016). This approach offered great insight into the kinetics of promoter unwinding and clearance during transcription initiation (Revyakin et al., 2012). Positive or negative supercoiled DNA structure is formed by clockwise or anti-clockwise twists, respectively (Figure 6). After the binding to promoter DNA, RNAP unwinds the DNA template which leads to loss of one turn in the negatively supercoiled structure and to formation of one additional turn in the positively supercoiled DNA template. Magnetic tweezers were also used to test different models for PIC-mediated DNA opening and start site selection. Measuring the DNA bubble sizes generated by yeast PIC assembly showed that ATP hydrolysis by the subunit Ssl2 of TFIIH opens an initial 6 bp bubble that is extended by RNAP transcription to a larger 13 bp bubble (Tomko et al., 2017).
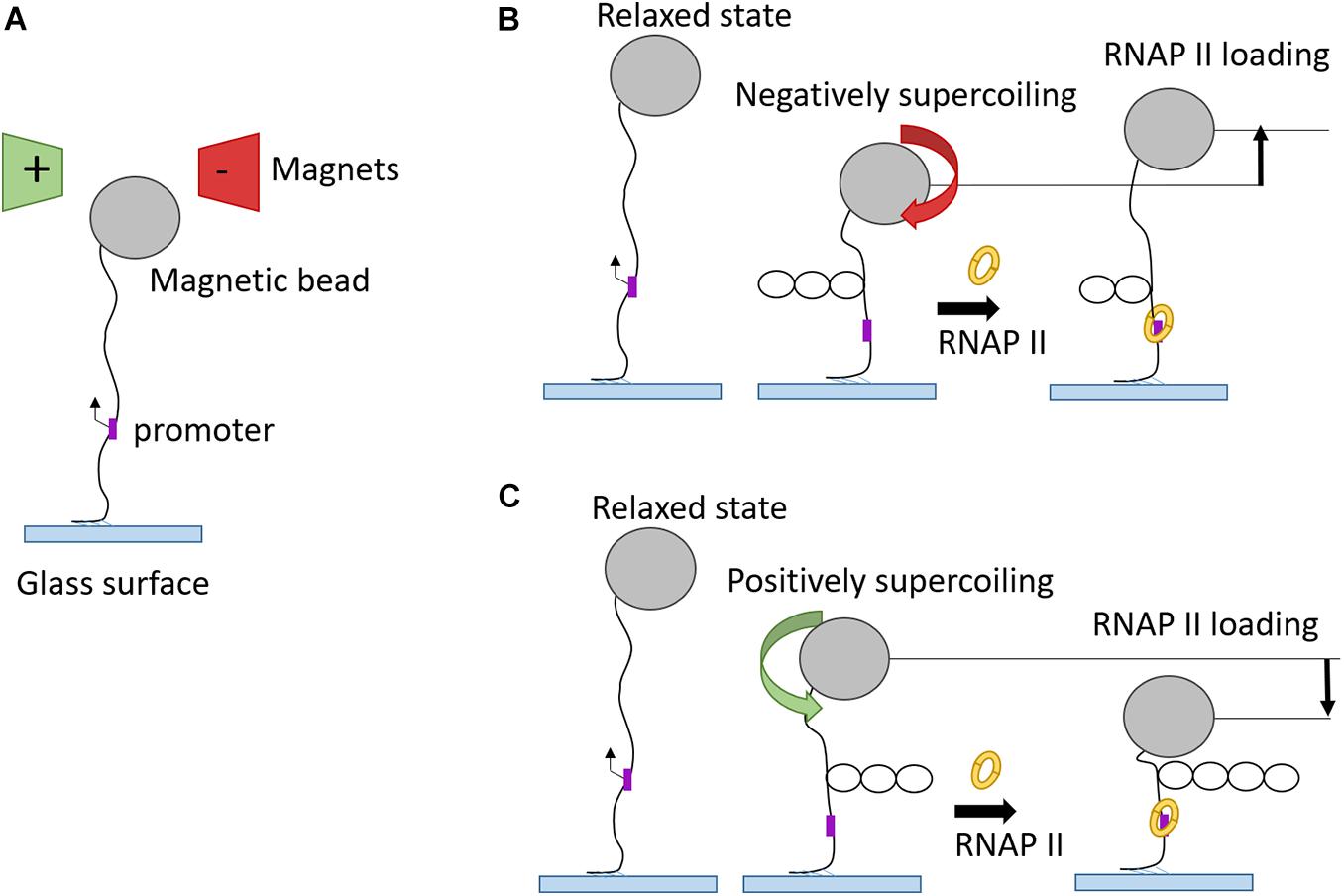
Figure 6. Magnetic tweezers used in transcription elongation studies. (A) Schematic representation of magnetic tweezers set-up in “relaxed” phase. (B) RNAP binding onto promoter of negatively supercoiled DNA substrate leads to unwinding of one turn and, therefore, to higher magnetic bead position. (C) RNAP binding onto promoter of positively supercoiled DNA substrate gives rise to one additional turn and, therefore, to a drop of magnetic bead position.
Fluorescence-Based Methods for Transcription Initiation and Elongation
Another significant barrier to RNAP progression and therefore transcription elongation are nucleosomes, making their removal ahead of the transcription complex a necessity for productive transcription (Kulaeva et al., 2010). Optical tweezers have uncovered the crucial role of transcription factors TFIIH and TFIIF in avoiding transcription elongation pauses at nucleosomal barriers (Bintu et al., 2012; Ishibashi et al., 2014). However, one disadvantage of these methods could be the applied mechanical force, since it might lead to acceleration of transcription elongation or even to the eviction of individual nucleosomal barriers. A recently developed single-molecule FRET method could overcome these possible limitations and provided more accurate transcription kinetic measurements (Lee et al., 2019). The experimental set-up consists of a reconstituted nucleosome assembled on a DNA template containing yeast RNAP II that is attached to a polyethylenglycol coated glass surface via an Rpb1 C-terminal domain (CTD) antibody and a Protein A-Streptavidin complex. A FRET pair of labeled nucleotides that are in close proximity when wrapped in a nucleosomal core particle, but not in free DNA can then be used to detect the nucleosomal dynamics during transcription elongation (Lee et al., 2019). Using this system, it was found that the transcription elongation factors Spt4/5 are essential members of the elongation complex, bridging the “jaws” of RNAPII with nucleic acids in the transcription scaffold to allow nucleosomal transcription (Crickard et al., 2017).
The analysis of promoter-specific transcription initiation in a reconstituted human RNAP system was achieved at single-molecule resolution by fluorescence video microscopy (Revyakin et al., 2012). Fluorescently labeled templates with promoter DNA were immobilized on a polysiloxane-coated glass coverslip and incubated with NTPs and purified human transcription factors. The PIC assembly was monitored through specific interactions between transcription factors and DNA templates at sub-second time resolution and based on spatial colocalization of the two fluorescence signals recorded in two fluorescence optical channels (Revyakin et al., 2012). In this way, the authors could not only directly visualize interactions between transcription factors and DNA, but also track and directly count RNA production by individual promoters in real time.
Single-Molecule Approaches to Monitor DNA Replication
The faithful transmission of epigenetic information undergoes a major challenge during DNA replication. Nucleosomes in front of the replication fork are disassembled and the corresponding parental histones with their PTMs need to be redeposited on the two daughter strands in order to maintain gene expression and cellular identities. Therefore, understanding how nascent chromatin is established, maintained or changed with the replication program is of paramount importance. Single-molecule approaches have provided valuable solutions to simultaneously observe replication fork proteins and DNA synthesis, furthering our understanding of replication fork dynamics.
DNA Fiber Stretching and Molecular Combing to Detect Replication Fork Progression
Classic methods to detect cells undergoing DNA replication is based on measuring the incorporation of thymidine analogs like BrdU (5-bromo-2′-deoxyuridine) or EdU (5-ethynyl-2′-deoxyuridine) into newly synthesized DNA. The labeled DNA can then be quantified by flow cytometry or immunofluorescence (Pozarowski and Darzynkiewicz, 2004; Harris et al., 2018). Although these methods are widely used as a tool for quantitative analysis of replication profiles in a cell population, this approach is not able to resolve the heterogeneity and the complexity of individual replication forks undergoing DNA replication.
Analysis of DNA fibers stretched either by DNA spreading or by molecular combing proved to be powerful tools to shed light into the dynamics of DNA and chromatin replication (Bensimon et al., 1994; Michalet, 1997; Tuduri et al., 2010). Both techniques share the same concept of two subsequent pulse labels with the different thymidine analogs iodo-deoxy-uridine IdU and chloro-deoxy-uridine CldU (Técher et al., 2013). The labeled DNA replication tracts are stained with antibodies directed against the halogenated nucleotides and the microscope slides with stretched DNA molecule are mounted for fluorescence microscopy. In contrast to DNA fiber spreading, molecular combing employs gentle lysis and slow stretching of DNA fibers on glass slides and enables the analysis of DNA replication on much larger DNA molecules up to 12 Mb in length (Kaykov et al., 2016). Each pulse (IdU/CldU) usually lasts 20–30 min and, consequently, an ongoing fork is represented as tracks of red and green labeled DNA (Nieminuszczy et al., 2016). The use of two different analogs gives the opportunity not only to detect the active replication forks in different chromosomes at a specific time-point (Czajkowsky et al., 2008), but also to extract additional parameters of DNA replication, including the replication fork speed, inter-origin distance, as well as initiation and termination events (Figure 7; Bialic et al., 2015; Nieminuszczy et al., 2016; Ray Chaudhuri et al., 2016; Vujanovic et al., 2017). The fork speed is calculated by the ratio between the fork length and the pulse time. Initiation events are represented as forks progressing symmetrically in opposite directions and termination events are merging forks.
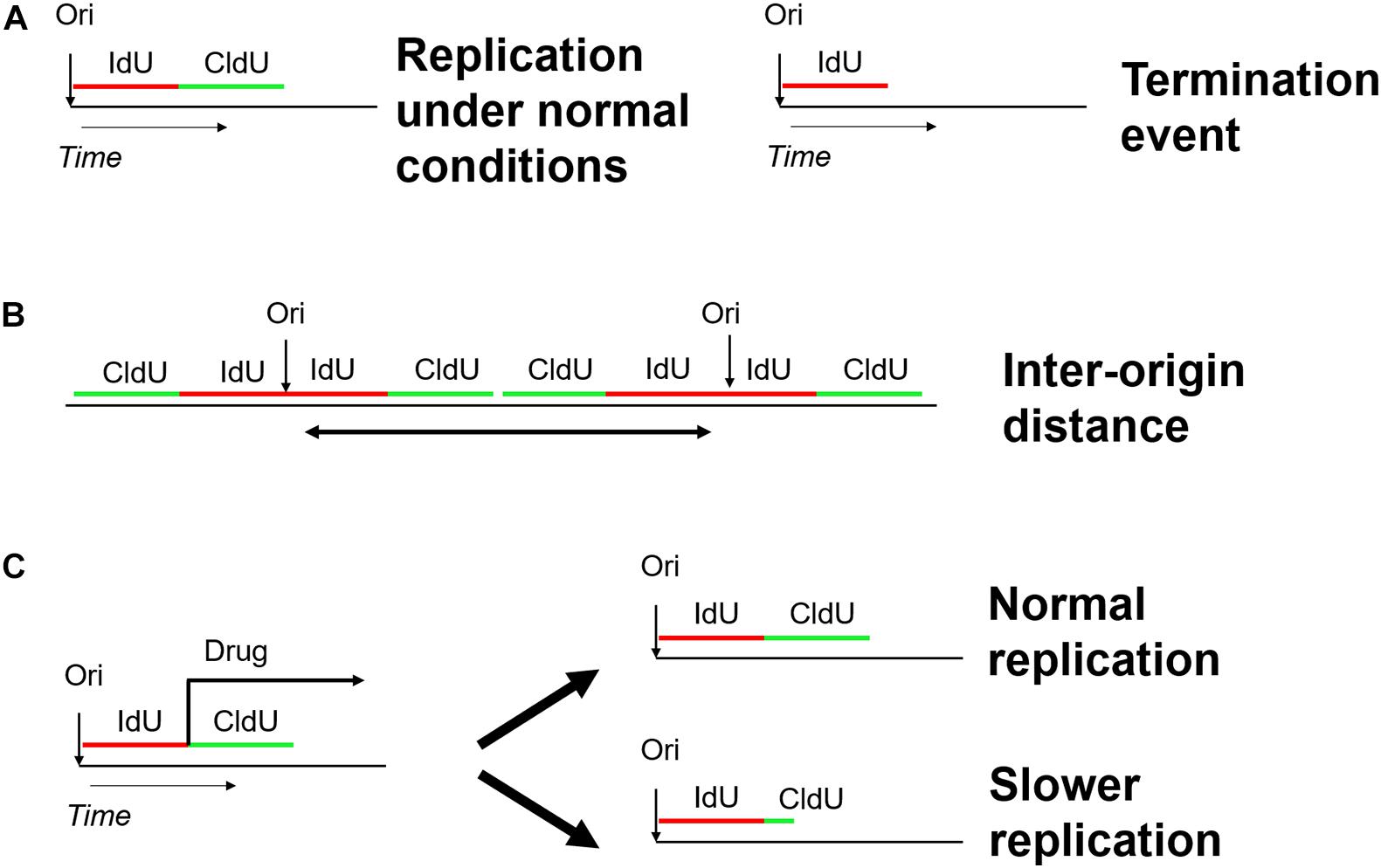
Figure 7. Schematic overview of the DNA fiber approach to analyze distinct replication events. Two subsequent pulses of the thymidine analogs IdU (5-iodo-2–deoxy-uridine) and CldU (5-chloro-2-deoxy-uridine) are used to label active DNA synthesis. (A) Study of replication initiation, elongation, and termination events. (B) Determining the distance between two neighboring replication origins. (C) Investigating the effect of genotoxic stress-inducing agents on replication progression.
Under normal conditions, this strategy has been used to quantify the progression of DNA replication forks in many organisms and cell culture systems, including chicken DT40 cells, Xenopus and mammalian cells (Blow et al., 2001; Schwab et al., 2010; Guilbaud et al., 2011). When combined with fluorescence in situ hybridization (FISH), this technique has even the potential to provide insights into the replication landscape at genomic loci of interest, such as Common Fragile Sites (Palumbo et al., 2010). DNA fiber analysis in different mutant cell lines could also reveal important interaction partners essential for replication initiation, e.g., the interaction of RECQ4 with MCM10 at replication origins (Kliszczak et al., 2015).
Under conditions of replication stress, fork speed is typically reduced as seen by DNA fiber analysis and this assay has been extensively used to assess the function of numerous proteins in replication fork progression and genome instability. A variation of the assay includes the treatment of cells with common replication inhibitors like hydroxyurea or aphidicolin, and monitor nucleotide resection following replication fork stalling by specific endonucleases. Successful implementation of this approach include the identification of the role of human RIF1 and Protein Phosphatase 1 in preventing over-degradation and accumulation of DNA breakage by replication stalling (Garzón et al., 2019; Mukherjee et al., 2019). Apart from the information regarding the overall slower rate of DNA replication or the fork degradation under specific conditions, the double labeling strategy is used to determine the frequency of fork stalling by measuring the ratio between two sister forks. In case of global slower rate of DNA polymerization, the sister forks would show the same length, as they are equally affected (ratio = 1). In case of fork stalling, one of the second tracts is typically shortened resulting in an asymmetric ratio< 1. This approach has been recently used to describe a mechanistic model of transcription-replication conflict resolution. It was shown that the restart of semi-conservative DNA replication is achieved by a fork cleavage mediated by the endonuclease MUS81/EME1 and religation cycle catalyzed by LIG4/XRCC4 (Chappidi et al., 2020).
Although DNA fiber assays are easy to implement in the laboratory and successfully used by many labs, one drawback is that the analysis of the molecules is labor-intense and low throughput. To improve this bottleneck, alternative methods use nanochannels for DNA stretching (Lacroix et al., 2016; De Carli et al., 2018). For example, an optical DNA mapping device (Bionano Genomics Irys) was repurposed to visualize DNA replication in Xenopus laevis egg extracts (De Carli et al., 2018). As a proof of principle, bacteriophage λ DNA was replicated in Xenopus egg extracts complemented with fluorescent deoxyuridine triphosphate (dUTP). Then, DNA was purified and labeled at specific restriction sites with a nicking endonuclease (NE) with another fluorescent nucleotide to allow optical alignment of the DNA molecules with the reference genome. Using this approach, the authors could confirm with high statistical significance that replication initiation in this system is not sequence-dependent (De Carli et al., 2018). Similar tools are currently developed in mammalian systems (Wang et al., 2020) and we envision that such methods may pave the way to DNA replication analysis at the genome-wide and single-molecule level.
Psoralen Crosslinking-EM to Visualize Replication Forks
Psoralen crosslinking and EM have been used as complimentary approach to DNA fiber/combing to give structural insights into DNA replication process under normal and stressed conditions (Vindigni and Lopes, 2017; Zellweger and Lopes, 2018). Many different replication intermediates were captured under EM revealing the mechanistic role of many factors controlling replication. For example, absence of RAD53 kinase could lead to extensive single stranded gaps and to accumulation of Holiday junctions through fork reversal (Sogo, 2002). Moreover, DNA2-depletion and RECQ1-depletion are followed by increased fork reversal events demonstrating the important role of both factors in fork progression (Thangavel et al., 2015). To date, EM remains the only method allowing direct visualization of fork reversal, an important four-way junction that forms frequently upon replication perturbation (Neelsen and Lopes, 2015).
Fluorescent and Force Manipulation Methods for DNA Replication Monitoring
In vitro reconstituted bacterial replisomes have been instrumental for the development of single-molecule techniques to visualize DNA replication. Using a rolling-circle amplification assay that flow-stretches the newly replicated DNA product on a functionalized glass coverslip (Tanner et al., 2008; Yao et al., 2009), important dynamics of individual components of the replication fork could be uncovered that were not possible by in vitro bulk biochemical experiments. For example, it was shown that T7 bacteriophage replisomes undergo frequent translocation in and out of replisomes in the process of DNA synthesis (Loparo et al., 2011), which could also be confirmed in vivo (Beattie et al., 2017). Another important aspect of the replisome concerns the coordination of leading and lagging-strand synthesis, as the continuous and discontinuous modes of replication at the two strands require strong coordination of the different sets of enzymatic reactions. Simultaneous Real-Time Imaging of Leading and Lagging Strand Synthesis showed that this coordination is regulated by both pausing of the leading-strand synthesis machinery and looping of the ssDNA (Duderstadt et al., 2016). Another single-molecule technique called DNA molecular curtains is based on the principle to align arrays of parallel nucleic acid strands in a flow chamber where the interaction with fluorescently labeled proteins can be observed in real time (Fazio et al., 2008). This high-throughput technique was used to study the transiently occurring intermediates of homologous recombination (HR) and provided for example crucial insights into the properties of individual Rad51 presynaptic complexes as one of the key intermediates in HR (Qi and Greene, 2016). With the successful biochemical reconstitution of eukaryotic in vitro DNA and chromatin replication in recent years (Yeeles et al., 2015; Kurat et al., 2017), we expect many more mechanistic insights also into eukaryotic replication fork and repair pathways using similar fluorescent single-molecule approaches.
Force manipulation methods are also suited for DNA replication studies. For example, using magnetic tweezers, the function of two different DNA polymerases (Sequenase and Klenow) was investigated on a single-strand and double-strand DNA (ssDNA)/(dsDNA) template. Sequenase is considered as a fast and Klenow as a slow polymerase (Maier et al., 2000). One end of the ssDNA was attached on a glass surface together with a primer allowing the binding of DNA polymerase and the other end was connected to a magnetic bead via streptavidin. The data shows that pausing of replication occurs at certain sites with both enzymes. However, there is no sequence specificity to these events, but the rate of replication was dependent on the stretching force applied to the DNA template. Low forces tend to increase the replication rate and forces stronger than 4 pN tend to decrease it. Pausing events were observed after a force greater than 20 pN is applied to the ssDNA. These events occur on multiple base pairs and follow the Arrhenius law. These results are consistent with other studies using optical tweezers set-up and demonstrate an increased probability of fork pausing events upon maximum tension of the DNA template (Wuite et al., 2000).
Third Generation Sequencing to Detect DNA Replication
Nanopore sequencing technology was used for the first time to map DNA replication at a single-molecule level monitoring the incorporation of the thymidine analog BrdU (Hennion et al., 2018). After defining the characteristic signal current of BrdU on MinION nanopore device using an in vitro system, a basecalling algorithms was developed to recognize the BrdU-specific shift in electric current as a fifth DNA base. To train the analysis pipeline, a modified yeast strain was used that fully depends on exogenous addition of thymidine or BrdU for replication. This allowed accurate mapping of early replication origins in S phase (Hennion et al., 2018). The resolution and accuracy of this approach was further improved by D-Nascent (detecting nucleotide analog signal currents on extremely long nanopore traces) (Müller et al., 2019) using a software based on Hidden Markov Model to detect BrdU incorporation. Application of this technology in yeast and mammalian cells also revealed pausing events of replication forks at single-molecule resolution (Georgieva et al., 2020; Hennion, 2020).
Conclusion and Perspectives
In this review, we summarized currently available approaches for chromatin analysis on a single-molecule level. The complex nature of chromatin with a myriad of dynamic interactions, large inter- and intramolecular heterogeneity and fast conformational changes among different states implies large stochasticity and variability on individual molecules that undergo replication, transcription and other nuclear processes. For this reason, single-molecule readouts are becoming increasingly important and at the same time feasible with the latest technological improvements in microscopy and third-generation sequencing.
These efforts have rewarded us with valuable insights into the dynamics of nucleosome positioning, transcription and replication dynamics that were not within our reach when only measuring bulk population of cells. Although many of these findings already proof to be consistent with bulk in vivo assays, we foresee that further development of live-cell imaging approaches will be needed to bridge the gap between the single-molecule in vitro assays and the behavior of the molecules in living cells.
Another aspect is that many of the single-molecule assays using nucleosomal templates rely on artificial, in vitro reconstituted mononucleosome or nucleosomal arrays that lack the majority of histone PTMs or in vivo positioning of nucleosomes. Purifying native chromatin from single loci would provide an interesting avenue for future single-molecule studies as such templates likely reflect better the in vivo situation (Hermans et al., 2017).
Third generation sequencing approaches might be the most promising technological development in recent years. Even though in the present review we mostly focus on applications in chromatin accessibility, their potential in other epigenetic profiling assays is enormous. Their revolutionized technology regarding the simple workflow, high throughput and long reads can also make them a useful diagnostic tool and with these new toolkits in hand, we expect many more important insights into basic and translational research in the next few years.
Author Contributions
AC searched the literature, created the figures, and wrote and edited the manuscript. SH provided the guidance and wrote and edited the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
SH was supported by the Helmholtz Association and by a Starting Grant from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program (Grant Agreement No. 852798).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all the scientists whose results we discussed in this review and apologize to all scientists whose work we could not cite due to space limitations. We thank all members of the Hamperl lab for their inspiring input, helpful discussions, and continuous support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.699771/full#supplementary-material
References
Abdulhay, N. J., McNally, C. P., Hsieh, L. J., Kasinathan, S., Keith, A., Estes, L. S., et al. (2020). Massively multiplex single-molecule oligonucleosome footprinting. eLife 9:e59404. doi: 10.7554/eLife.59404
Ardui, S., Ameur, A., Vermeesch, J. R., and Hestand, M. S. (2018). Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 46, 2159–2168. doi: 10.1093/nar/gky066
Beattie, T. R., Kapadia, N., Nicolas, E., Uphoff, S., Wollman, A. J., Leake, M. C., et al. (2017). Frequent exchange of the DNA polymerase during bacterial chromosome replication. eLife 554:e21763. doi: 10.7554/eLife.21763
Bell, O., Tiwari, V. K., Thomä, N. H., and Schübeler, D. (2011). Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 12, 554–564. doi: 10.1038/nrg3017
Bensimon, A., Simon, A., Chiffaudel, A., Croquette, V., Heslot, F., and Bensimon, D. (1994). Alignment and sensitive detection of DNA by a moving interface. Science 265, 2096–2098. doi: 10.1126/science.7522347
Bialic, M., Coulon, V., Drac, M., Gostan, T., and Schwob, E. (2015). “Analyzing the Dynamics of DNA Replication in Mammalian Cells Using DNA Combing,” in DNA Replication Methods in Molecular Biology, eds S. Vengrova and J. Dalgaard (New York, NY: Springer), 67–78. doi: 10.1007/978-1-4939-2596-4_4
Bianco, S., Rodrigue, S., Murphy, B. D., and Gévry, N. (2015). “Global Mapping of Open Chromatin Regulatory Elements by Formaldehyde-Assisted Isolation of Regulatory Elements Followed by Sequencing (FAIRE-seq),” in DNA-Protein Interactions Methods in Molecular Biology, eds B. P. Leblanc and S. Rodrigue (New York, NY: Springer), 261–272. doi: 10.1007/978-1-4939-2877-4_17
Bintu, L., Ishibashi, T., Dangkulwanich, M., Wu, Y.-Y., Lubkowska, L., Kashlev, M., et al. (2012). Nucleosomal Elements that Control the Topography of the Barrier to Transcription. Cell 151, 738–749. doi: 10.1016/j.cell.2012.10.009
Blow, J. J., Gillespie, P. J., Francis, D., and Jackson, D. A. (2001). Replication Origins in Xenopus Egg Extract Are 5–15 Kilobases Apart and Are Activated in Clusters That Fire at Different Times. J. Cell Biol. 152, 11.
Bonev, B., and Cavalli, G. (2016). Organization and function of the 3D genome. Nat. Rev. Genet. 17, 661–678. doi: 10.1038/nrg.2016.112
Brehove, M., Wang, T., North, J., Luo, Y., Dreher, S. J., Shimko, J. C., et al. (2015). Histone Core Phosphorylation Regulates DNA Accessibility. J. Biol. Chem. 290, 22612–22621. doi: 10.1074/jbc.M115.661363
Brown, C. R., and Boeger, H. (2014). Nucleosomal promoter variation generates gene expression noise. Proc. Natl. Acad. Sci. 111, 17893–17898. doi: 10.1073/pnas.1417527111
Brown, C. R., Mao, C., Falkovskaia, E., Jurica, M. S., and Boeger, H. (2013). Linking Stochastic Fluctuations in Chromatin Structure and Gene Expression. PLoS Biol. 11:e1001621. doi: 10.1371/journal.pbio.1001621
Buenrostro, J. D., Wu, B., Chang, H. Y., and Greenleaf, W. J. (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 109. doi: 10.1002/0471142727.mb2129s109
Chappidi, N., Nascakova, Z., Boleslavska, B., Zellweger, R., Isik, E., Andrs, M., et al. (2020). Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-transcriptional R-Loops. Mol. Cell 77, 528–541.e5. doi: 10.1016/j.molcel.2019.10.026
Cimino, D. G., Howard, B., Gamper, I., Stephen, T., and Hearst, E. J. (1985). Psoralens as Photoactive Probes of Nucleic Acid Structure and Function: Organic Chemistry. Photochem. Biochem. Ann. Rev. Biochem. 54, 1151–1193.
Cipriany, B. R., Zhao, R., Murphy, P. J., Levy, S. L., Tan, C. P., Craighead, H. G., et al. (2010). Single Molecule Epigenetic Analysis in a Nanofluidic Channel. Anal. Chem. 82, 2480–2487. doi: 10.1021/ac9028642
Clarke, J., Wu, H.-C., Jayasinghe, L., Patel, A., Reid, S., and Bayley, H. (2009). Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 4, 265–270. doi: 10.1038/nnano.2009.12
Crickard, J. B., Lee, J., Lee, T.-H., and Reese, J. C. (2017). The elongation factor Spt4/5 regulates RNA polymerase II transcription through the nucleosome. Nucleic Acids Res. 45, 6362–6374. doi: 10.1093/nar/gkx220
Czajkowsky, D. M., Liu, J., Hamlin, J. L., and Shao, Z. (2008). DNA Combing Reveals Intrinsic Temporal Disorder in the Replication of Yeast Chromosome VI. J. Mol. Biol. 375, 12–19. doi: 10.1016/j.jmb.2007.10.046
Dammann, R., Lucchini, R., Koller, T., and Sogo, J. M. (1993). Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res. 21, 2331–2338. doi: 10.1093/nar/21.10.2331
De Carli, F., Menezes, N., Berrabah, W., Barbe, V., Genovesio, A., and Hyrien, O. (2018). High-Throughput Optical Mapping of Replicating DNA. Small Methods 2:1800146. doi: 10.1002/smtd.201800146
Dou, Y., and Gorovsky, M. A. (2000). Phosphorylation of Linker Histone H1 Regulates Gene Expression In Vivo by Creating a Charge Patch. Mol. Cell 6, 225–231. doi: 10.1016/S1097-2765(00)00024-1
Duderstadt, K. E., Geertsema, H. J., Stratmann, S. A., Punter, C. M., Kulczyk, A. W., Richardson, C. C., et al. (2016). Simultaneous Real-Time Imaging of Leading and Lagging Strand Synthesis Reveals the Coordination Dynamics of Single Replisomes. Mol. Cell 64, 1035–1047. doi: 10.1016/j.molcel.2016.10.028
Dziedzic, J. M., Bjorkholm, J. E., and Chu, S. (1986). Observation of a Single-Beam Gradient Force Optical Trap for Dielectric Particles. Opt. Lett. 11, 288–290.
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Fazal, F. M., Meng, C. A., Murakami, K., Kornberg, R. D., and Block, S. M. (2015). Real-time observation of the initiation of RNA polymerase II transcription. Nature 525, 274–277. doi: 10.1038/nature14882
Fazio, T., Visnapuu, M.-L., Wind, S., and Greene, E. C. (2008). DNA Curtains and Nanoscale Curtain Rods: High-Throughput Tools for Single Molecule Imaging. Langmuir 24, 10524–10531. doi: 10.1021/la801762h
Fish, R. N., and Kane, C. M. (2002). Promoting elongation with transcript cleavage stimulatory factors. Biochim. Biophys. Acta 1577, 287–307. doi: 10.1016/S0167-4781(02)00459-1
Fitz, V., Shin, J., Ehrlich, C., Farnung, L., Cramer, P., Zaburdaev, V., et al. (2016). Nucleosomal arrangement affects single-molecule transcription dynamics. Proc. Natl. Acad. Sci. U. S. A. 113, 12733–12738. doi: 10.1073/pnas.1602764113
Flusberg, B. A., Webster, D. R., Lee, J. H., Travers, K. J., Olivares, E. C., Clark, T. A., et al. (2010). Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 7, 461–465. doi: 10.1038/nmeth.1459
Galburt, E. A., Grill, S. W., and Bustamante, C. (2009). Single molecule transcription elongation. Methods 48, 323–332. doi: 10.1016/j.ymeth.2009.04.021
Galburt, E. A., Grill, S. W., Wiedmann, A., Lubkowska, L., Choy, J., Nogales, E., et al. (2007). Backtracking determines the force sensitivity of RNAP II in a factor-dependent manner. Nature 446, 820–823. doi: 10.1038/nature05701
Garzón, J., Ursich, S., Lopes, M., Hiraga, S., and Donaldson, A. D. (2019). Human RIF1-Protein Phosphatase 1 Prevents Degradation and Breakage of Nascent DNA on Replication Stalling. Cell Rep. 27, 2558–2566.e. doi: 10.1016/j.celrep.2019.05.002
Georgieva, D., Liu, Q., Wang, K., and Egli, D. (2020). Detection of Base Analogs Incorporated During DNA Replication by Nanopore Sequencing. Nucleic Acids Res. 48:88. doi: 10.1093/nar/gkaa517
Gibney, E. R., and Nolan, C. M. (2010). Epigenetics and gene expression. Heredity 105, 4–13. doi: 10.1038/hdy.2010.54
Guilbaud, G., Rappailles, A., Baker, A., Chen, C.-L., Arneodo, A., Goldar, A., et al. (2011). Evidence for Sequential and Increasing Activation of Replication Origins along Replication Timing Gradients in the Human Genome. PLoS Comput. Biol. 7:e1002322. doi: 10.1371/journal.pcbi.1002322
Ha, T., and Tinnefeld, P. (2012). Photophysics of Fluorescent Probes for Single-Molecule Biophysics and Super-Resolution Imaging. Annu. Rev. Phys. Chem. 63, 595–617. doi: 10.1146/annurev-physchem-032210-103340
Hamperl, S., Brown, C. R., Garea, A. V., Perez-Fernandez, J., Bruckmann, A., Huber, K., et al. (2014). Compositional and structural analysis of selected chromosomal domains from Saccharomyces cerevisiae. Nucleic Acids Res. 42:20. doi: 10.1093/nar/gkt891
Hanson, C., Shen, C., and Hearst, J. (1976). Cross-linking of DNA in situ as a probe for chromatin structure. Science 193, 62–64. doi: 10.1126/science.935855
Harada, A., Maehara, K., Handa, T., Arimura, Y., Nogami, J., Hayashi-Takanaka, Y., et al. (2019). A chromatin integration labelling method enables epigenomic profiling with lower input. Nat. Cell Biol. 21, 287–296. doi: 10.1038/s41556-018-0248-3
Harris, L., Zalucki, O., and Piper, M. (2018). BrdU/EdU dual labeling to determine the cell-cycle dynamics of defined cellular subpopulations. J. Mol. Histol. 49, 229–234. doi: 10.1007/s10735-018-9761-8
Hayashi-Takanaka, Y., Kina, Y., Nakamura, F., Becking, L. E., Nakao, Y., Nagase, T., et al. (2020). Histone modification dynamics as revealed by multicolor immunofluorescence-based single-cell analysis. J. Cell Sci. 133:jcs243444. doi: 10.1242/jcs.243444
Heintzman, N. D., Hon, G. C., Hawkins, R. D., Kheradpour, P., Stark, A., Harp, L. F., et al. (2009). Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112. doi: 10.1038/nature07829
Hennion, M. (2020). FORK-seq: replication landscape of the Saccharomyces cerevisiae genome by nanopore sequencing. Genome Biol. 21:125. doi: 10.1186/s13059-020-02013-3
Hennion, M., Arbona, J.-M., Cruaud, C., Proux, F., Tallec, B. L., Novikova, E., et al. (2018). Mapping DNA replication with nanopore sequencing. Genomics 42:858. doi: 10.1101/426858
Hermans, N., Huisman, J. J., Brouwer, T. B., Schächner, C., van Heusden, G. P. H., Griesenbeck, J., et al. (2017). Toehold-enhanced LNA probes for selective pull down and single-molecule analysis of native chromatin. Sci. Rep. 7:16721. doi: 10.1038/s41598-017-16864-7
Hoch, D. A., Stratton, J. J., and Gloss, L. M. (2007). Protein–Protein Förster Resonance Energy Transfer Analysis of Nucleosome Core Particles Containing H2A and H2A.Z. J. Mol. Biol. 371, 971–988. doi: 10.1016/j.jmb.2007.05.075
Hofmann, W. A., Stojiljkovic, L., Fuchsova, B., Vargas, G. M., Mavrommatis, E., Philimonenko, V., et al. (2004). Actin is part of pre-initiation complexes and is necessary for transcription by RNA polymerase II. Nat. Cell Biol. 6, 1094–1101. doi: 10.1038/ncb1182
Hsieh, T.-H. S., Fudenberg, G., Goloborodko, A., and Rando, O. J. (2016). Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat. Methods 13, 1009–1011. doi: 10.1038/nmeth.4025
Ishibashi, T., Dangkulwanich, M., Coello, Y., Lionberger, T. A., Lubkowska, L., Ponticelli, A. S., et al. (2014). Transcription factors IIS and IIF enhance transcription efficiency by differentially modifying RNA polymerase pausing dynamics. Proc. Natl. Acad. Sci. U. S. A 111, 3419–3424. doi: 10.1073/pnas.1401611111
Ishii, H., Kadonaga, J. T., and Ren, B. (2015). MPE-seq, a new method for the genome-wide analysis of chromatin structure. Proc. Natl. Acad. Sci. U. S. A 112, E3457–E3465. doi: 10.1073/pnas.1424804112
Jain, M., Olsen, H. E., Paten, B., and Akeson, M. (2016). The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 17:239. doi: 10.1186/s13059-016-1103-0
Jenuwein, T. (2001). Translating the Histone Code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Kalle, W., and Strappe, P. (2012). Atomic force microscopy on chromosomes, chromatin and DNA: A review. Micron 43, 1224–1231. doi: 10.1016/j.micron.2012.04.004
Kasianowicz, J. J., Brandin, E., Branton, D., and Deamer, D. W. (1996). Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. U. S. A 93, 13770–13773. doi: 10.1073/pnas.93.24.13770
Kassavetis, G. A., Kaya, K. M., and Chamberlin, M. J. (1978). Escherichia coli RNA polymerase-rifampicin complexes bound at promoter sites block RNA chain elongation by Escherichia coli/RNA polymerase and T7-specific RNA polymerase. Biochemistry 17, 5798–5804. doi: 10.1021/bi00619a029
Kaya-Okur, H. S., Wu, S. J., Codomo, C. A., Pledger, E. S., Bryson, T. D., Henikoff, J. G., et al. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10:1930. doi: 10.1038/s41467-019-09982-5
Kaykov, A., Taillefumier, T., Bensimon, A., and Nurse, P. (2016). Molecular Combing of Single DNA Molecules on the 10 Megabase Scale. Sci. Rep. 6:19636. doi: 10.1038/srep19636
Kelly, T. K., Liu, Y., Lay, F. D., Liang, G., Berman, B. P., and Jones, P. A. (2012). Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 22, 2497–2506. doi: 10.1101/gr.143008.112
Kilic, S., Felekyan, S., Doroshenko, O., Boichenko, I., Dimura, M., Vardanyan, H., et al. (2018). Single-molecule FRET reveals multiscale chromatin dynamics modulated by HP1α. Nat. Commun. 9:235. doi: 10.1038/s41467-017-02619-5
Killian, J. L., Li, M., Sheinin, M. Y., and Wang, M. D. (2012). Recent advances in single molecule studies of nucleosomes. Curr. Opin. Struct. Biol. 22, 80–87. doi: 10.1016/j.sbi.2011.11.003
Kireeva, M. L., Hancock, B., Cremona, G. H., Walter, W., Studitsky, V. M., and Kashlev, M. (2005). Nature of the Nucleosomal Barrier to RNA Polymerase II. Mol. Cell 18, 97–108. doi: 10.1016/j.molcel.2005.02.027
Kladde, M. P., and Simpson, R. T. (1994). Positioned nucleosomes inhibit Dam methylation in vivo. Proc. Natl. Acad. Sci. U. S. A. 91, 1361–1365. doi: 10.1073/pnas.91.4.1361
Kliszczak, M., Sedlackova, H., Pitchai, G. P., Streicher, W. W., Krejci, L., and Hickson, I. D. (2015). Interaction of RECQ4 and MCM10 is important for efficient DNA replication origin firing in human cells. Oncotarget 6, 40464–40479. doi: 10.18632/oncotarget.6342
Kornberg, R. D. (1974). Chromatin Structure: A Repeating Unit of Histones and DNA. Science 184, 868–871. doi: 10.1126/science.184.4139.868
Kornberg, R. D., and Lorch, Y. (2020). Primary Role of the Nucleosome. Mol. Cell 79, 371–375. doi: 10.1016/j.molcel.2020.07.020
Ku, W. L., Nakamura, K., Gao, W., Cui, K., Hu, G., Tang, Q., et al. (2019). Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat. Methods 16, 323–325. doi: 10.1038/s41592-019-0361-7
Kulaeva, O. I., Hsieh, F.-K., and Studitsky, V. M. (2010). RNA polymerase complexes cooperate to relieve the nucleosomal barrier and evict histones. Proc. Natl. Acad. Sci. U. S. A. 107, 11325–11330. doi: 10.1073/pnas.1001148107
Kurat, C. F., Yeeles, J. T. P., Patel, H., Early, A., and Diffley, J. F. X. (2017). Chromatin Controls DNA Replication Origin Selection, Lagging-Strand Synthesis, and Replication Fork Rates. Mol. Cell 65, 117–130. doi: 10.1016/j.molcel.2016.11.016
Lacroix, J., Pélofy, S., Blatché, C., Pillaire, M.-J., Huet, S., Chapuis, C., et al. (2016). Analysis of DNA Replication by Optical Mapping in Nanochannels. Small 12, 5963–5970. doi: 10.1002/smll.201503795
Larson, M. H., Zhou, J., Kaplan, C. D., Palangat, M., Kornberg, R. D., Landick, R., et al. (2012). Trigger loop dynamics mediate the balance between the transcriptional fidelity and speed of RNA polymerase II. Proc. Natl. Acad. Sci. U. S. A. 109, 6555–6560. doi: 10.1073/pnas.1200939109
Lee, I., Razaghi, R., Gilpatrick, T., Molnar, M., Gershman, A., Sadowski, N., et al. (2020). Simultaneous profiling of chromatin accessibility and methylation on human cell lines with nanopore sequencing. Nat. Methods 17, 1191–1199. doi: 10.1038/s41592-020-01000-7
Lee, J., Crickard, J. B., Reese, J. C., and Lee, T.-H. (2019). Single-molecule FRET method to investigate the dynamics of transcription elongation through the nucleosome by RNA polymerase II. Methods 159–160, 51–58. doi: 10.1016/j.ymeth.2019.01.009
Li, G., Levitus, M., Bustamante, C., and Widom, J. (2005). Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 12, 46–53. doi: 10.1038/nsmb869
Li, Y., Eshein, A., Virk, R., Eid, A., VanDerway, D., Frederick, J., et al. (2020). Nanoscale Chromatin Imaging and Analysis (nano-ChIA) Platform Bridges 4-D Chromatin Organization with Molecular Function. Microsc. Microanal. 26, 1046–1050. doi: 10.1017/S1431927620016785
Li, Y., and Tollefsbol, T. O. (2011). ““DNA Methylation Detection: Bisulfite Genomic Sequencing Analysis,”,” in Epigenetics Protocols Methods in Molecular Biology, ed. T. O. Tollefsbol (Totowa, NJ: Humana Press), 11–21. doi: 10.1007/978-1-61779-316-5_2
Lisica, A., and Grill, S. W. (2017). Optical tweezers studies of transcription by eukaryotic RNA polymerases. Biomol. Concepts 8, 1–11. doi: 10.1515/bmc-2016-0028
Loparo, J. J., Kulczyk, A. W., Richardson, C. C., and van Oijen, A. M. (2011). Simultaneous single-molecule measurements of phage T7 replisome composition and function reveal the mechanism of polymerase exchange. Proc. Natl. Acad. Sci. U. S. A. 108, 3584–3589. doi: 10.1073/pnas.1018824108
Lucchini, R. (2001). Nucleosome positioning at the replication fork. EMBO J. 20, 7294–7302. doi: 10.1093/emboj/20.24.7294
Luger, K., Mäder, W. A., Richmond, K. R., Sargent, F. D., and Richmod, J. T. (1997). Crystal Structure of the Nucleosome Core Particle at 2.8 Å Resolution. Nature 389, 251–260. doi: 10.1038/38444
Lusser, A., and Kadonaga, J. T. (2003). Chromatin remodeling by ATP-dependent molecular machines. BioEssays 25, 1192–1200. doi: 10.1002/bies.10359
Maier, B., Bensimon, D., and Croquette, V. (2000). Replication by a single DNA polymerase of a stretched single-stranded DNA. Proc. Natl. Acad. Sci. U. S. A. 97, 12002–12007. doi: 10.1073/pnas.97.22.12002
Mattiroli, F., Gu, Y., and Luger, K. (2018). FRET-based Stoichiometry Measurements of Protein Complexes in vitro. BIO Protoc. 8:713. doi: 10.21769/BioProtoc.2713
Meers, M. P., Bryson, T. D., Henikoff, J. G., and Henikoff, S. (2019). Improved CUT&RUN chromatin profiling tools. eLife 8:e46314. doi: 10.7554/eLife.46314
Michalet, X. (1997). Dynamic Molecular Combing: Stretching the Whole Human Genome for High-Resolution Studies. Science 277, 1518–1523. doi: 10.1126/science.277.5331.1518
Miranda, T. B., Kelly, T. K., Bouazoune, K., and Jones, P. A. (2010). Methylation-Sensitive Single-Molecule Analysis of Chromatin Structure. Curr. Protoc. Mol. Biol. 89, 21.17.1–21.17.16. doi: 10.1002/0471142727.mb2117s89
Miyagi, A., Ando, T., and Lyubchenko, Y. L. (2011). Dynamics of Nucleosomes Assessed with Time-Lapse High-Speed Atomic Force Microscopy. Biochemistry 50, 7901–7908. doi: 10.1021/bi200946z
Mukherjee, C., Tripathi, V., Manolika, E. M., Heijink, A. M., Ricci, G., Merzouk, S., et al. (2019). RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 10:3287. doi: 10.1038/s41467-019-11246-1
Müller, C. A., Boemo, M. A., Spingardi, P., Kessler, B. M., Kriaucionis, S., Simpson, J. T., et al. (2019). Capturing the dynamics of genome replication on individual ultra-long nanopore sequence reads. Nat. Methods 16, 429–436. doi: 10.1038/s41592-019-0394-y
Muse, G. W., Gilchrist, D. A., Nechaev, S., Shah, R., Parker, J. S., Grissom, S. F., et al. (2007). RNA polymerase is poised for activation across the genome. Nat. Genet. 39, 1507–1511. doi: 10.1038/ng.2007.21
Neelsen, K. J., and Lopes, M. (2015). Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 16, 207–220. doi: 10.1038/nrm3935
Nieminuszczy, J., Schwab, R. A., and Niedzwiedz, W. (2016). The DNA fibre technique – tracking helicases at work. Methods 108, 92–98. doi: 10.1016/j.ymeth.2016.04.019
Olins, A. L., and Olins, D. E. (1974). Spheroid Chromatin Units (ngr Bodies). Science 183, 330–332. doi: 10.1126/science.183.4122.330
Olins, D. E., and Olins, A. L. (1978). Nucleosomes: The Structural Quantum in Chromosomes: Virtually all the DNA of eukaryotic cells is organized into a repeating array of nucleohistone particles called nucleosomes. These chromatin subunits are close-packed into higher-order fibers and are modified during chromosome expression. Am. Sci. 66, 704–711.
Palumbo, E., Matricardi, L., Tosoni, E., Bensimon, A., and Russo, A. (2010). Replication dynamics at common fragile site FRA6E. Chromosoma 119, 575–587. doi: 10.1007/s00412-010-0279-4
Park, Y.-J., Dyer, P. N., Tremethick, D. J., and Luger, K. (2004). A New Fluorescence Resonance Energy Transfer Approach Demonstrates That the Histone Variant H2AZ Stabilizes the Histone Octamer within the Nucleosome. J. Biol. Chem. 279, 24274–24282. doi: 10.1074/jbc.M313152200
Pozarowski, P., and Darzynkiewicz, Z. (2004). “Analysis of Cell Cycle by Flow Cytometry,” in Checkpoint Controls and Cancer, ed. A. H. Schönthal (New Jersey: Humana Press), 301–312. doi: 10.1385/1-59259-811-0:301
Qi, Z., and Greene, E. C. (2016). Visualizing recombination intermediates with single-stranded DNA curtains. Methods 105, 62–74. doi: 10.1016/j.ymeth.2016.03.027
Rao, S. S. P., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Ray Chaudhuri, A., Callen, E., Ding, X., Gogola, E., Duarte, A. A., Lee, J.-E., et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. doi: 10.1038/nature18325
Revyakin, A., Zhang, Z., Coleman, R. A., Li, Y., Inouye, C., Lucas, J. K., et al. (2012). Transcription initiation by human RNA polymerase II visualized at single-molecule resolution. Genes Dev. 26, 1691–1702. doi: 10.1101/gad.194936.112
Rotem, A., Ram, O., Shoresh, N., Sperling, R. A., Goren, A., Weitz, D. A., et al. (2015). Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 33, 1165–1172. doi: 10.1038/nbt.3383
Sarkar, R., and Rybenkov, V. V. (2016). A Guide to Magnetic Tweezers and Their Applications. Front. Phys. 4:48. doi: 10.3389/fphy.2016.00048
Sauer, P. V., Gu, Y., Liu, W. H., Mattiroli, F., Panne, D., Luger, K., et al. (2018). Mechanistic insights into histone deposition and nucleosome assembly by the chromatin assembly factor-1. Nucleic Acids Res. 46, 9907–9917. doi: 10.1093/nar/gky823
Schmid, M., Durussel, T., and Laemmli, U. K. (2004). ChIC and ChEC: Genomic Mapping of Chromatin Proteins. Mol. Cell 16, 147–157. doi: 10.1016/j.molcel.2004.09.007
Schones, D. E., Cui, K., Cuddapah, S., Roh, T.-Y., Barski, A., Wang, Z., et al. (2008). Dynamic Regulation of Nucleosome Positioning in the Human Genome. Cell 132, 887–898. doi: 10.1016/j.cell.2008.02.022
Schwab, R. A., Blackford, A. N., and Niedzwiedz, W. (2010). ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 29, 806–818. doi: 10.1038/emboj.2009.385
Schweikhard, V., Meng, C., Murakami, K., Kaplan, C. D., Kornberg, R. D., and Block, S. M. (2014). Transcription factors TFIIF and TFIIS promote transcript elongation by RNA polymerase II by synergistic and independent mechanisms. Proc. Natl. Acad. Sci. U. S. A. 111, 6642–6647. doi: 10.1073/pnas.1405181111
Shipony, Z., Marinov, G. K., Swaffer, M. P., Sinnott-Armstrong, N. A., Skotheim, J. M., Kundaje, A., et al. (2020). Long-range single-molecule mapping of chromatin accessibility in eukaryotes. Nat. Methods 17, 319–327. doi: 10.1038/s41592-019-0730-2
Simon, M., North, J. A., Shimko, J. C., Forties, R. A., Ferdinand, M. B., Manohar, M., et al. (2011). Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl. Acad. Sci. U. S. A. 108, 12711–12716. doi: 10.1073/pnas.1106264108
Simon, M. D., Chu, F., Racki, L. R., de la Cruz, C. C., Burlingame, A. L., Panning, B., et al. (2007). The Site-Specific Installation of Methyl-Lysine Analogs into Recombinant Histones. Cell 128, 1003–1012. doi: 10.1016/j.cell.2006.12.041
Sogo, J. M. (2002). Fork Reversal and ssDNA Accumulation at Stalled Replication Forks Owing to Checkpoint Defects. Science 297, 599–602. doi: 10.1126/science.1074023
Sogo, J. M., Ness, P. J., Widmer, R. M., Parish, R. W., and Koller, and Th. (1984). Psoralen-crosslinking of DNA as a probe for the structure of active nucleolar chromatin. J. Mol. Biol. 178, 897–919. doi: 10.1016/0022-2836(84)90318-8
Stadler, M. B., Murr, R., Burger, L., Ivanel, R., Lienert, F., Schöler, A., et al. (2011). DNA-Binding Factors Shape the Mouse Methylome at Distal Regulatory Regions. Nature 480, 490–495. doi: 10.1038/nature10716
Stergachis, A. B., Debo, B. M., Haugen, E., Churchman, L. S., and Stamatoyannopoulos, J. A. (2020). Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science 368, 1449–1454. doi: 10.1126/science.aaz1646
Strahl, B. D., and Allis, C. D. (2000). The Language of Covalent Histone Modifications. Nature 403, 41–45. doi: 10.1038/474125
Strick, T. R., Allemand, J.-F., Bensimon, D., Bensimon, A., and Croquette, V. (1996). The Elasticity of a Single Supercoiled DNA Molecule. Science 271, 1835–1837. doi: 10.1126/science.271.5257.1835
Tanner, N. A., Hamdan, S. M., Jergic, S., Loscha, K. V., Schaeffer, P. M., Dixon, N. E., et al. (2008). Single-molecule studies of fork dynamics in Escherichia coli DNA replication. Nat. Struct. Mol. Biol. 15, 170–176. doi: 10.1038/nsmb.1381
Técher, H., Koundrioukoff, S., Azar, D., Wilhelm, T., Carignon, S., Brison, O., et al. (2013). Replication Dynamics: Biases and Robustness of DNA Fiber Analysis. J. Mol. Biol. 425, 4845–4855. doi: 10.1016/j.jmb.2013.03.040
Thangavel, S., Berti, M., Levikova, M., Pinto, C., Gomathinayagam, S., Vujanovic, M., et al. (2015). DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 208, 545–562. doi: 10.1083/jcb.201406100
Tomko, E. J., Fishburn, J., Hahn, S., and Galburt, E. A. (2017). TFIIH generates a six-base-pair open complex during RNAP II transcription initiation and start-site scanning. Nat. Struct. Mol. Biol. 24, 1139–1145. doi: 10.1038/nsmb.3500
Travers, A., Hiriart, E., Churcher, M., Caserta, M., and Di Mauro, E. (2010). The DNA Sequence-dependence of Nucleosome Positioning in vivo and in vitro. J. Biomol. Struct. Dyn. 27, 713–724. doi: 10.1080/073911010010524942
Tuduri, S., Tourrière, H., and Pasero, P. (2010). Defining replication origin efficiency using DNA fiber assays. Chromosome Res. 18, 91–102. doi: 10.1007/s10577-009-9098-y
Tyler, J. K., Adams, C. R., Chen, S.-R., Kobayashi, R., Kamakaka, R. T., and Kadonaga, J. T. (1999). The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 402, 555–560. doi: 10.1038/990147
Vindigni, A., and Lopes, M. (2017). Combining electron microscopy with single molecule DNA fiber approaches to study DNA replication dynamics. Biophys. Chem. 225, 3–9. doi: 10.1016/j.bpc.2016.11.014
Vujanovic, M., Krietsch, J., Raso, M. C., Terraneo, N., Zellweger, R., Schmid, J. A., et al. (2017). Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 67, 882–890.e. doi: 10.1016/j.molcel.2017.08.010
Wachutka, L., Caizzi, L., Gagneur, J., and Cramer, P. (2019). Global donor and acceptor splicing site kinetics in human cells. eLife 8:e45056. doi: 10.7554/eLife.45056
Wal, M., and Pugh, B. F. (2012). Genome-Wide Mapping of Nucleosome Positions in Yeast Using High-Resolution MNase ChIP-Seq. Methods Enzymol. 513, 233–250. doi: 10.1016/B978-0-12-391938-0.00010-0
Wang, Q., Xiong, H., Ai, S., Yu, X., Liu, Y., Zhang, J., et al. (2019). CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol. Cell 76, 206–216.e. doi: 10.1016/j.molcel.2019.07.015
Wang, Y., Wang, A., Liu, Z., Thurman, A. L., Powers, L. S., Zou, M., et al. (2019). Single-molecule long-read sequencing reveals the chromatin basis of gene expression. Genome Res. 29, 1329–1342. doi: 10.1101/gr.251116.119
Wang, W., Klein, K., Proesmans, K., Yang, H., Marchal, C., Zhu, X., et al. (2020). Genome-Wide Mapping of Human DNA Replication by Optical Replication Mapping Supports a Stochastic Model of Eukaryotic Replication. bioRxiv [preprint]. doi: 10.1101/2020.08.24.263459
Wellinger, R. E., and Sogo, J. M. (1998). In Vivo Mapping of Nucleosomes Using Psoralen–DNA Crosslinking and Primer Extension. Nucleic Acids Res. 26, 1544–1545. doi: 10.1093/nar/26.6.1544
Wittner, M., Hamperl, S., Stöckl, U., Seufert, W., Tschochner, H., Milkereit, P., et al. (2011). Establishment and Maintenance of Alternative Chromatin States at a Multicopy Gene Locus. Cell 145, 543–554. doi: 10.1016/j.cell.2011.03.051
Wolff, M. R., Schmid, A., Korber, P., and Gerland, U. (2021). Effective dynamics of nucleosome configurations at the yeast PHO5 promoter. eLife 10:e58394. doi: 10.7554/eLife.58394
Wu, S. J., Furlan, S. N., Mihalas, A. B., Kaya-Okur, H. S., Feroze, A. H., Emerson, S. N., et al. (2021). Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat. Biotechnol. 39. doi: 10.1038/s41587-021-00865-z [Epub ahead of print].
Wuite, G. J. L., Smith, S. B., Young, M., Keller, D., and Bustamante, C. (2000). Single-molecule studies of the effect of template tension on T7 DNA polymerase activity. Nature 404, 103–106. doi: 10.1038/35003614
Yang, Y., Sebra, R., Pullman, B. S., Qiao, W., Peter, I., Desnick, R. J., et al. (2015). Quantitative and multiplexed DNA methylation analysis using long-read single-molecule real-time bisulfite sequencing (SMRT-BS). BMC Genomics 16:350. doi: 10.1186/s12864-015-1572-7
Yao, N. Y., Georgescu, R. E., Finkelstein, J., and O’Donnell, M. E. (2009). Single-molecule analysis reveals that the lagging strand increases replisome processivity but slows replication fork progression. Proc. Natl. Acad. Sci. U. S. A. 106, 13236–13241. doi: 10.1073/pnas.0906157106
Yeeles, J. T. P., Deegan, T. D., Janska, A., Early, A., and Diffley, J. F. X. (2015). Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 519, 431–435. doi: 10.1038/nature14285
Zane, L., Chapus, F., Pegoraro, G., and Misteli, T. (2017). HiHiMap: single-cell quantitation of histones and histone posttranslational modifications across the cell cycle by high-throughput imaging. Mol. Biol. Cell 28, 2290–2302. doi: 10.1091/mbc.e16-12-0870
Zellweger, R., and Lopes, M. (2018). “Dynamic Architecture of Eukaryotic DNA Replication Forks In Vivo, Visualized by Electron Microscopy,” in Genome Instability Methods in Molecular Biology, eds M. Muzi-Falconi and G. W. Brown (New York, NY: Springer), 261–294. doi: 10.1007/978-1-4939-7306-4_19
Keywords: single-molecule techniques, chromatin accessibility, electron microscopy, third-generation sequencing, histone modifications, magnetic/optical tweezers, chromatin replication/transcription, DNA fiber analysis
Citation: Chanou A and Hamperl S (2021) Single-Molecule Techniques to Study Chromatin. Front. Cell Dev. Biol. 9:699771. doi: 10.3389/fcell.2021.699771
Received: 24 April 2021; Accepted: 26 May 2021;
Published: 05 July 2021.
Edited by:
Anastassiia Vertii, University of Massachusetts Medical School, United StatesReviewed by:
Laura Manelyte, University of Regensburg, GermanyJessica Tyler, Cornell University, United States
Copyright © 2021 Chanou and Hamperl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephan Hamperl, c3RlcGhhbi5oYW1wZXJsQGhlbG1ob2x0ei1tdWVuY2hlbi5kZQ==