 Dan Yi
Dan Yi Huan Yu1,2
Huan Yu1,2 Ke Lu
Ke Lu Changhai Ding
Changhai Ding Di Chen
Di Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 22 June 2021
Sec. Stem Cell Research
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.696602
This article is part of the Research Topic Cartilage: from Developmental to Translational Biology View all 18 articles
The adenosine monophosphate (AMP)–activated protein kinase (AMPK) was initially identified as an enzyme acting as an “energy sensor” in maintaining energy homeostasis via serine/threonine phosphorylation when low cellular adenosine triphosphate (ATP) level was sensed. AMPK participates in catabolic and anabolic processes at the molecular and cellular levels and is involved in appetite-regulating circuit in the hypothalamus. AMPK signaling also modulates energy metabolism in organs such as adipose tissue, brain, muscle, and heart, which are highly dependent on energy consumption via adjusting the AMP/ADP:ATP ratio. In clinics, biguanides and thiazolidinediones are prescribed to patients with metabolic disorders through activating AMPK signaling and inhibiting complex I in the mitochondria, leading to a reduction in mitochondrial respiration and elevated ATP production. The role of AMPK in mediating skeletal development and related diseases remains obscure. In this review, in addition to discuss the emerging advances of AMPK studies in energy control, we will also illustrate current discoveries of AMPK in chondrocyte homeostasis, osteoarthritis (OA) development, and the signaling interaction of AMPK with other pathways, such as mTOR (mechanistic target of rapamycin), Wnt, and NF-κB (nuclear factor κB) under OA condition.
Advanced technology and medical therapy in health care are highly demanded in the era after the Millennium. World Health Organization introduced the concept of “healthy aging” based on the statistics demonstrating that (1) the faster pace of the growing of aging population in the past 30 years; (2) by 2050, the proportion of the world population older than 60 years will reach nearly 22%; (3) the occurrence of chronic and metabolic diseases, such as cardiovascular disease, Alzheimer disease, diabetes mellitus, and cancer, has been dramatically increased in the upper- to middle-income and high-income countries between 2000 and 20191. So, the improvements in medical care are key mission required in the future to reduce social–economic burden. Regarding to this situation, chronic metabolic diseases have been gained significant attention in the research field where scientists are aiming to increase life quality and extend life expectancy via controlling the initiation and/or the progress of chronic metabolic diseases (Aburto et al., 2020). Previous studies have indicated that there is a negative correlation between metabolic diseases and energy expenditure. It illustrated that low energy expenditure is associated with high risk of obesity (Zurlo et al., 1990; Lam and Ravussin, 2016). High energy expenditure levels and low body weight may protect people from potentially atherogenic diet (Mbalilaki et al., 2010; Ross et al., 2020).
Brown et al. (1975) found a cytosolic factor could inactivate 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGR) in the presence of adenosine triphosphate (ATP) and ADP, as shown in Figure 1A (Steinberg and Carling, 2019). Thereafter, several studies have been focused on the origin and function of this factor. They found that a self-regulated phosphorylation of HMGR kinase could form a protein kinase cascade, alone with adenosine monophosphate (AMP) activating a protein kinase but inactivating acetyl-CoA carboxylase (ACC). It has been demonstrated that ACC and HMGR could be purified separately, and the ACC and HMGR are regulated by AMP (Figure 1B). When the scientists discovered and named molecule AMPK in 1990s, Crute et al., and Bateman described the structure of AMP-activated protein kinase (AMPK) comprised an α1 catalytic subunit with a functional domain and a β1 subunit. Most importantly, it has been reported that AMPK is allosterically activated by AMP to phosphorylate and inactivate ACC at Ser79, implying the function of AMPK in an allosteric and covalent activation manner (Figure 1C). This laid the foundation for all the future studies on exploring the functional activation of AMPK. Meanwhile, a few attempts were conducted and tried to study the functions of AMPK in tissue distribution associated with metabolic physiological conditions: glycogen synthase in skeletal muscle related to GLUT4 translocation and lipolysis and lipogenesis in isolated rat adipocytes with AICAR (Steinberg and Carling, 2019). The limitation of these studies may be due to the shortage of scientific knowledge and appropriate measurement techniques. After the Millennium, with the great leap forward of science and technology, the scientists have ushered in a prosperous age that advanced technology applied to explore comprehensive molecular mechanisms in detail. In this transition period, gamma subunit of AMPK had been revealed and implied its function in mediating energy; thus, peroxisome proliferator-activated receptor δ (PPARδ) agonists were applied in few studies. In addition, besides the depth in understanding the activation and deactivation of AMP, depending on allosteric stimulation, autoinhibitory effects of AMPK also pointed out by Pang et al. To expand the understanding of signaling pathway of AMPK, several factors have been identified (Figure 1D). Liver kinase B1 (LKB1) and Ca2+/calmodulin-dependent protein kinase kinases (CaMKK2) are the upstream kinases phosphorylating AMPK at Thr172. Sanders et al., illustrated that A-769662 was a valuable tool for AMPK activation, and ADaM site activators activate AMPK at Ser108 at the β1 subunit. Leptin and adiponectin both stimulate fatty acid oxidation by activating AMPK. Berberine and metformin acted as antidiabetic effects via activating AMPK activities. On the basis of this, the idea of APMK-allied food intake has leaped into public attention. AMPK has been shown to be involved in glucose transport in skeletal muscle, orexigenic neuropeptide inhibition, mitochondrial biogenesis, circadian clock regulation, and promotion of autophagy. Furthermore, the idea “energy sensor” of AMPK has been pointed out as the response to the changes of intracellular AMP, ADP, and ATP. In 2010s, the structural domains of AMPK were fully characterized, including an α subunit with a catalytic domain, β-regulatory subunits, and a γ-regulatory subunit. Moreover, Xiao et al., thought the high efficiency of AMPK activator 991 and A769662 was due to their high potency in tight binding to carboxy-terminal (CBM, C-interacting helix) of the β1 subunit (Steinberg and Carling, 2019). Apart from only explaining the activity of AMPK involved in certain pathophysiological conditions, in these time periods, researchers were eager to clarify the underlying mechanism in regulation of energy homeostasis to treat metabolic diseases. Carling et al., have defined AMPK as an energy sensor through phosphorylation of both ACC and HMG-CoA reductase in response to the changes in ADP/ATP concentration. Other groups have been working on observing activities of AMPK in aforementioned conditions such as glucose uptake and lipid homeostasis and identified several novel molecules (Figure 1E). For instance, AMPK regulated glucose uptake via GLUT1, controlling lipid homeostasis via phosphorylation of ACC1 and ACC2, and downregulation of their activity in tumor cells by Akt phosphorylation. AMPK also increased autophagy and mitophagy through phosphorylation of ULK1 and BECN1 at threonine 388. Moreover, the role of AMPK in mitochondrial homeostasis was well-established as regulation of PGC-1α expression in mouse adipose tissue and skeletal muscle. Next, Price et al., found that resveratrol (an AMPK activator) functions as an antioxidant through activation of AMPK in mitochondria. In addition, salicylate, biguanides, and canagliflozin were also found as AMPK activators in controlling metabolic process including glucose transformation, fatty acid oxidation, and cancer cell proliferation. A big step at this stage was that AMPK has been shown to play a key role in interorgan interaction. Reduced AMPK-ACC and mechanistic target of rapamycin (mTOR) signaling were reported in muscle of aged male in mankind. AMPK and hypoxia-inducible factor (HIF) together regulated adipogenesis via miR-455 (Steinberg and Carling, 2019). AMPK also interacts with nuclear factor κB (NF-κB) to increase p53 activity in liver cancer cells (Steinberg and Carling, 2019). Under extensive studies of AMPK functions in the past three decades, knowledge has been gained regarding AMPK structure and functional activities and signaling pathways in certain pathological conditions; however, limited information has been gained in recent years regarding roles of cardiovascular diseases, diabetes mellitus, and cancers, etc. In addition, studies on the functional activities and metabolic balance of AMPK in other organs are also needed.
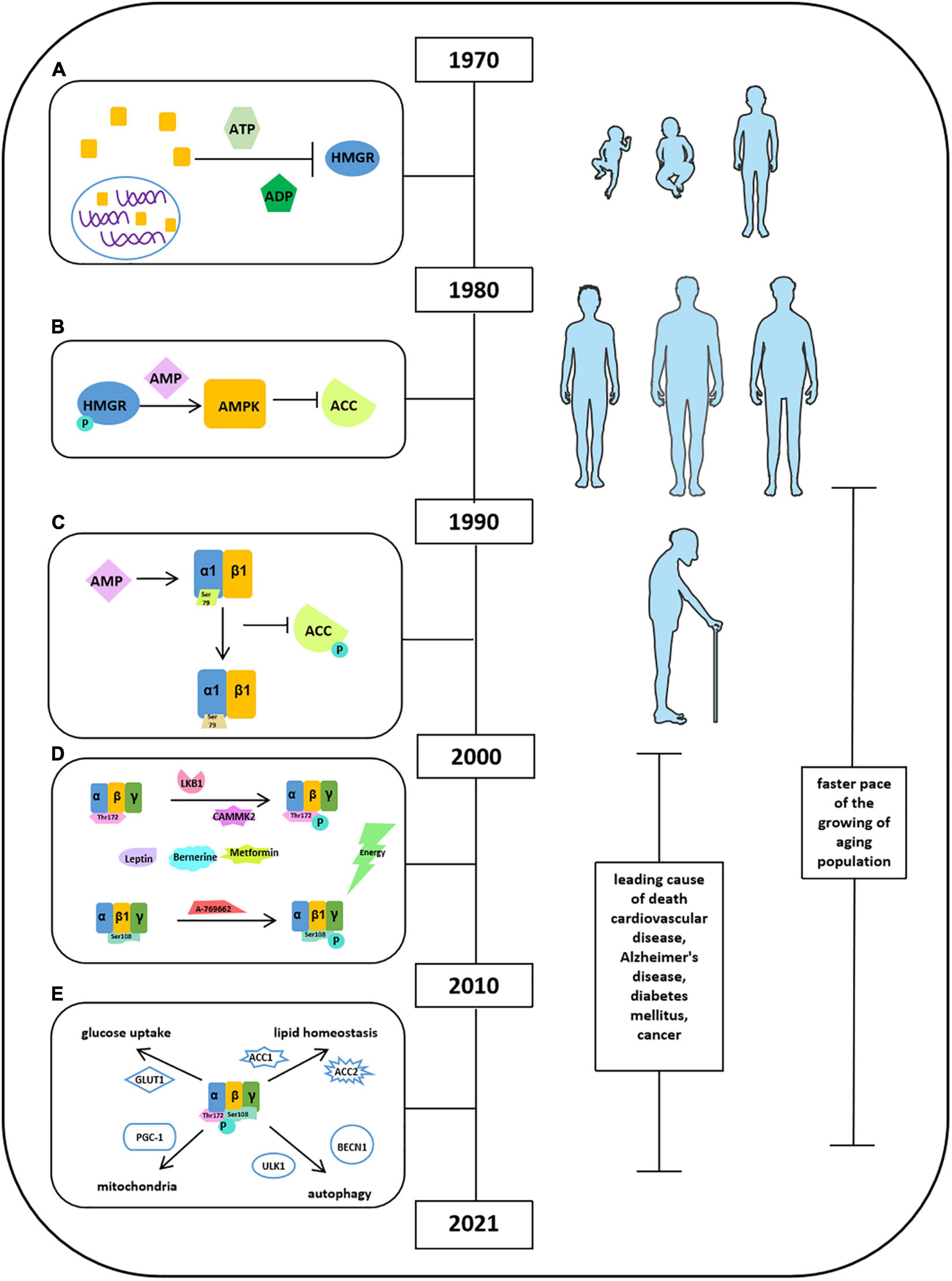
Figure 1. Milestones of discoveries of AMPK protein. (A) Cytosolic factor could inactivate 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGR) in the presence of ATP and ADP. (B) ACC and HMGR could be purified separately, and the ACC and HMGR are regulated by AMP. (C) The structure of AMPK comprised an α1 catalytic subunit with a functional domain and a β1 subunit. AMPK is allosterically activated by AMP to phosphorylate and inactivate ACC at Ser79. (D) Factors reported to stimulate AMPK activation. (E) Activities of AMPK in glucose uptake, lipid homeostasis, autophagy, and mitochondria.
Energy balance refers to the equilibrium between energy intake and energy expenditure, accompanied by energy storage, and this concept was first introduced in 1996 (Figure 2A; Hill and Commerford, 1996; Hill and Saris, 2003; Hill et al., 2013). In the following 15 years, in pursing high life quality and the encouragement of healthy diet by social media, energy balance has been linked with obesity, diet, and exercise due to a high prevalence of obesity in public epidemic worldwide (Hall et al., 2011; Hill et al., 2013; Ng et al., 2014; Manore et al., 2017). The well-balanced energy is critical in maintaining a stable body weight illustrated by several groups since 1999 (Figure 2B; van Baak, 1999; Webber, 2003; Hill et al., 2012; Hill et al., 2013; Manore et al., 2017), whereas the imbalanced energy resulted in weight loss, which could be used as the intervention of obesity (Hall et al., 2011). It is known that half of the body energy was obtained from glucose metabolism after energy intake (Figure 2C; Murphy and Bloom, 2006). When it comes to energy balance, energy intake refers to catabolic pathways (glycolysis, fatty acid oxidation, and mitochondrial biogenesis) and energy expenditure regarded as anabolic pathways (gluconeogenesis, glycogen, fatty acid, and protein synthesis) (Figure 2D; Lim et al., 2010). The quantification of energy was measured by the unit “calorie” (Figure 2E; Halliday et al., 2011). The generated energy produced by food intake could be stored as body fat, triglycerides, or glycogen in the form of ATP and dissipated as heat (Figure 2F; Hill and Saris, 2003; Halliday et al., 2011; Keith, 2013). Moyes et al. (2010) thought that there is a link between negative energy balance and impaired immune response. Gerber and Corpet (1999) proposed that prevention of visceral obesity could potentially prevent cancer via balancing caloric intake and caloric expenditure. In addition, other studies have demonstrated that energy balance is critical in neuronal activity and leptin and ghrelin control energy balance in neurons (Strassburg et al., 2008; Dietrich et al., 2010; Brown et al., 2017).
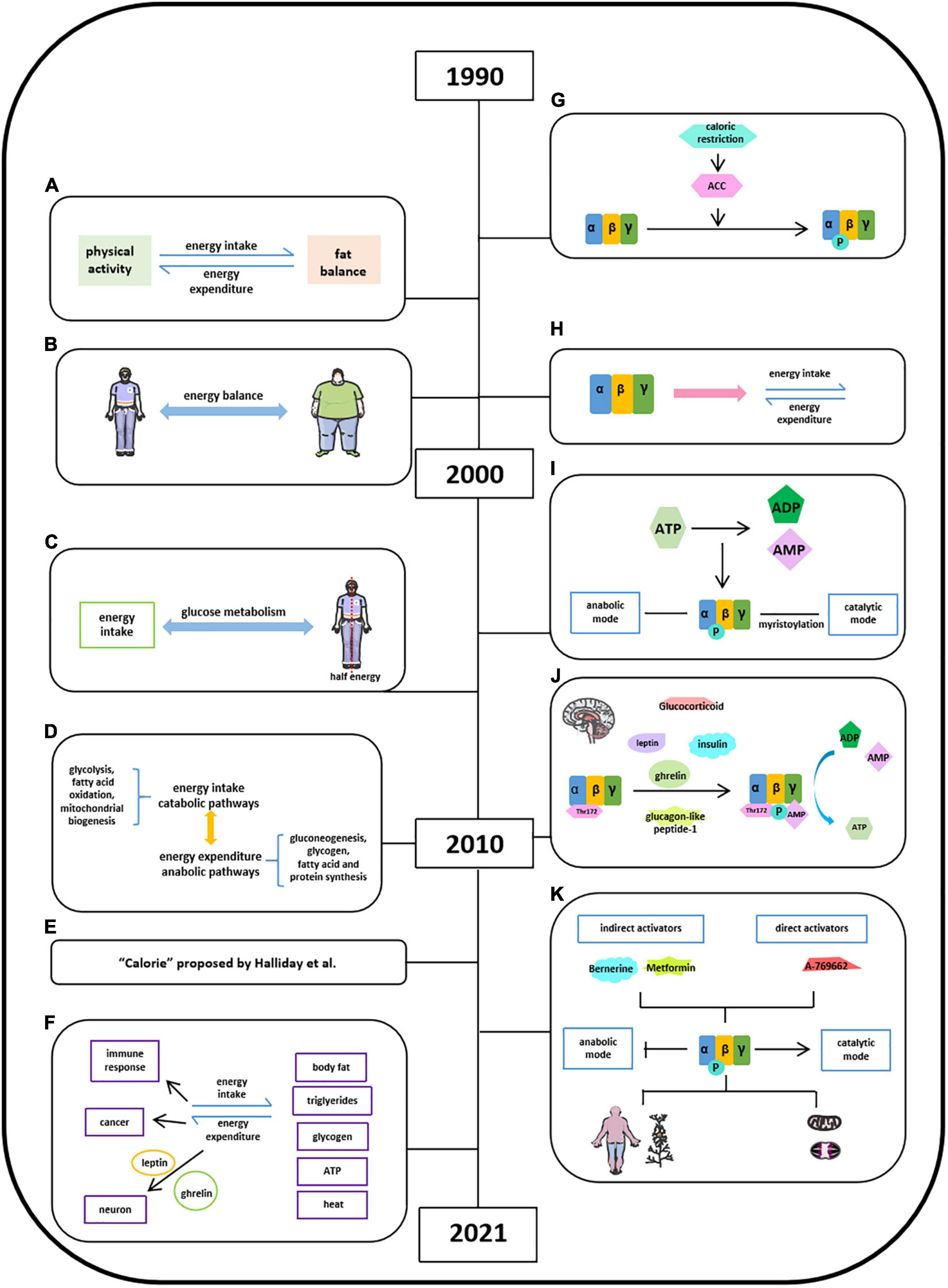
Figure 2. Milestones of discoveries of energy balance and the role of AMPK in energy balance. (A) Concept of energy balance. (B) Well-balanced energy is critical in maintaining a stable body weight. (C) Half of the body energy was obtained from glucose metabolism after energy intake. (D) Concept of energy intake (catabolic pathways) and energy expenditure (anabolic pathways). (E) The quantification of energy was measured by the unit “calorie” proposed by Halliday et al. (2011). (F) Format of energy store and energy balance involved activities. (G) AMPK is activated via inducing ACC by calorie restriction. (H) AMPK protein structure has been studied in energy balance. (I), Energy imbalance between anabolic and catabolic process in some metabolic syndromes is due to ATP consumption. (J) Factors reported to stimulate AMPK activation in hypothalamus. (K) AMPK may act as the key regulator in energy balance in a specific organ or tissue.
Witters et al. (1994) found AMPK is activated via inducing ACC by calorie restriction in varying nutritional states (Figure 2G; Steinberg and Carling, 2019). Hardie et al. (1999) found that AMPK monitored cellular energy changes through regulation of nucleotide concentration. Based on scattered analyses, in 2000s, with the information of AMPK protein structure, significant progress has been made in the understanding of the role of AMPK in energy balance (Figure 2H). Hardie (2004) concluded that the energy imbalance between anabolic and catabolic process in some metabolic syndromes is due to ATP consumption disorder, and several AMPK activators have promising therapeutic effects via activating AMPK phosphorylation. The AMPK isoforms α (α1, α2), β (β1, β2), and γ (γ1, γ2, and γ3) were identified in humans and rodents and were encoded by distinct genes (Figure 2I). And a year after, Steinberg and Kemp summarized a comprehensive story about the structure and regulation of AMPK in metabolism. The ratio of [ATP]/[ADP]:[AMP] was regarded to metabolic coupling of anabolic and catabolic pathways of AMPK activities known as “adenylate charge hypothesis.” Steinberg and Kemp also suggest that the metabolic stress could sense the change of AMPK levels in yeast. Moreover, the functional roles of AMPK in carbohydrate metabolism (glucose), lipid metabolism (fatty acid, mitochondrial, and cholesterol), protein synthesis, cell growth, cell apoptosis, cell polarity, and ion flux were well-established (Steinberg and Kemp, 2009). Lim et al. (2010) further empathized that the γ subunit of AMPK could tightly bind AMP in low energy states to activate AMPK with persistent phosphorylation of Thr172 residue in maintaining energy balance (Figure 2J). Leptin stimulates hypothalamosympathetic axis in an AMPK-independent manner. Adiponectin could activate and stimulate AMPK activity both in vivo and in vitro, thus leading to excitation of glucose uptake, fatty acid oxidation, and PEPCK annexation in liver and muscle. Resistin decreases fatty acid uptake and oxidation in skeletal muscle via inhibiting AMPK activity (He et al., 2018). Ghrelin and cannabinoids stimulate AMPK activity in hypothalamus and heart, while inhibiting AMPK activity in adipose tissue and liver. Insulin, as an anorectic hormone, stimulates glucose uptake and inhibits hypothalamic AMPK activity. Glucagon-like peptide-1 inhibited fasting-induced increase of hypothalamic AMPK activity as an anorectic effect. And glucocorticoids activate AMPK activity in hypothalamus via stimulating endocannabinoid synthesis (Lim et al., 2010). By 2021, accumulated evidence suggest that AMPK may act as the key regulator in energy balance in a specific organ or tissue to a certain extent (Figure 2K; Hardie, 2014; Lopez et al., 2016; Garcia and Shaw, 2017; Herzig and Shaw, 2018; Steinberg and Carling, 2019; Wang et al., 2020b).
Hardie provided the insights into stimulatory effects of AMPK on catabolic pathway and inhibitory effects of AMPK on anabolic pathways with direct targets and also listed a number of natural products, which have been reported to activate AMPK pathway and underlying mechanisms. Lopez’s group suggests that hypothalamic AMPK signaling pathway regulates energy homeostasis by integrating peripheral signals, including hormones and metabolites, with neuronal networks. Garcia and Shaw showed that AMPK, as a cellular energy sensor, restores metabolic balance, together with upstream and downstream factors. Herzig and Shaw were focusing on serine/threonine kinase AMPK complex in guarding mitochondrial homeostasis, including mitophagy and autophagy. Steinberg and Carling summarized pharmacological agents of AMPK activators, including berberine, metformin, and A769662, in the treatment of metabolic syndrome based on the information of AMPK structure and regulation. Generally speaking, the core of AMPK controls energy balance and acts as a critical bioenergy sensor, regulating anabolic and catabolic pathways under different physiological conditions, which are highly sensitive to the changes of AMP and ATP levels.
Chondrocytes are derived from mesenchymal stem cells and form “hyaline cartilage” (Lee et al., 2013; Rim et al., 2020). It is well-established that chondrocytes contribute to endochondral ossification in the embryonic cartilage, thus helping bone elongation in skeletal development via self-proliferation and self-hypertrophy (Rim et al., 2020). The initial studies of “chondrocyte” could retrospect to the mid-20th century when Eichelberger et al. (1951) proposed that chondrocytes are the primary cells forming cartilage (Figure 3A). Okada (1960) found that chondrocytes had ability in generating cartilaginous matrix, and Godman and Porter applied electron microscope to reveal the evolution of chondrogenesis from chondroblasts to chondrocytes (Figure 3B; Okada, 1959; Godman and Porter, 1960). After experiencing a decade of research blank, by 1980, studies of chondrocytes back to the researchers’ scope and chondrogenesis were emerging as well. Searls’ group first showed that cartilage-forming region could be detected by light microscope (Searls et al., 1972). Vasan and Lash (1975) and Lewis et al. (1978) found that vitamin A could inhibit chondrogenesis by restraining cell proliferation, limiting extracellular space with thick and banded collagen fibrils but no proteoglycan granules (Figure 3C). Green found that chondrocytes isolated and cultured in vitro could repair a large articular defect when allografting took place in rabbit knee (Green, 1977). And later, in 1980s, Bruckner’s group suggests that the transition from resting chondrocytes to proliferative and hypertrophic chondrocytes depends on cell-seeded densities and fetal bovine serum (FBS) concentrations (Figure 3D; Bruckner et al., 1989). Moreover, Radomska et al. (1989) implied that chondrocytes could be used as the target in labeling cell-mediated cytotoxicity with immune organs. In vitro chondrocyte cultures were highly advocated in this period because of its obvious repairing capacity (Grande et al., 1989; Wakitani et al., 1989; Robinson et al., 1990). Kawamura et al. (1998) invented that the gel–chondrocyte composite could be used to treat cartilage defects better than rabbit articular cartilage. Aulthouse’s group successfully cultured human chondrocytes with ultrastructural features presented in vitro (Aulthouse et al., 1989). Tumor growth factor β (TGF-β) potentially modulates chondrocyte proliferation and matrix synthesis in endochondral calcification in an autocrine manner (Rosier et al., 1989; Li et al., 2010). The role of mechanical load in the development of osteoarthritis (OA) and the tissue engineering in cartilage repair are the focus of research in the last decade before the 21st century (Figure 3E; Mauck et al., 2000; Wilkins et al., 2000). Besides TGF-β, Studer et al. (2000) also indicated that chondrocytes are insensitive to anabolic actions in the presence of nitric oxide (NO), which is partially due to the inhibition of insulin-like growth factor I (IGF-I) receptor self-autophosphorylation (Figure 3E). Other studies also suggested some upstream mediators, such as interleukin 1β (IL-1β), electric fields, and thyroid hormones in the regulation of chondrocyte activities (Chao et al., 2000; Nerucci et al., 2000; Robson et al., 2000). After millennium, studies about chondrocytes were still focused on its role in bone homeostasis. Ichinose et al. (2010) well-depicted the differences among chondrocytes, bone marrow–mesenchymal stem cells (MSCs), and synovium–MSCs in cellular morphology, aggregation, and differentiation during in vitro chondrogenesis. Articular cartilage repair and OA remain to be a hot topic when exploring the function of chondrocytes. In addition, Dreier suggested that OA is initiated from vascularization and focal calcification of joint cartilage (Dreier, 2010). Kishimoto suggested oxidized low-density lipoprotein leads hypertrophic chondrocyte-like phenotype in OA through oxidative stress induction (Figure 3F; Kishimoto et al., 2010). Parathyroid hormone–related protein (PTHrP) and fibroblast growth factor (FGF) were found to be involved in chondrocyte proliferation and differentiation (Figure 3F; MacLean et al., 2004; Yamaoka et al., 2010). Bohensky et al. (2010) indicated that HIF-1 regulates chondrocyte autophagy via AMPK and mTOR signaling pathway (Figure 3F). By now, studies of chondrocytes were moving forward from macroregulation of cartilage integrity to the systemic mechanisms. Sharifi and Gharravi (2019) summarized a general properties of articular chondrocytes, such as matrix synthesis and its response to shear stimuli. Xing et al. (2019) indicated that osterix could mediate early postnatal growth during the formation of secondary ossification center. Bougault et al. (2014) indicated frizzled-related protein B could modulate matrix metalloproteinase induction in mouse chondrocytes via regulation of Wnt/β-catenin pathway (Figure 3G). Li and Dong (2016) conclude that bone morphogenetic protein (BMP), PTHrP, Ihh, FGFR3, Sox9, β-catenin, O2 tension, and reactive oxygen species (ROS) signaling pathways mediate chondrocyte formation, differentiation, maturation, and hypertrophic differentiation (Figure 3G). Other studies also mentioned the utilization of modern technique to explore the function of chondrocytes. For instance, Kim et al. (2020) found that equine bone marrow–derived cells with nanoparticles could promote the growth of chondrocytes and reduce cytokine-induced apoptosis on chondrocytes. Several other groups also illustrated that chondrocyte apoptosis was induced by NO from endoplasmic reticulum (ER) stress (Takada et al., 2011, 2013; Yamabe et al., 2013).
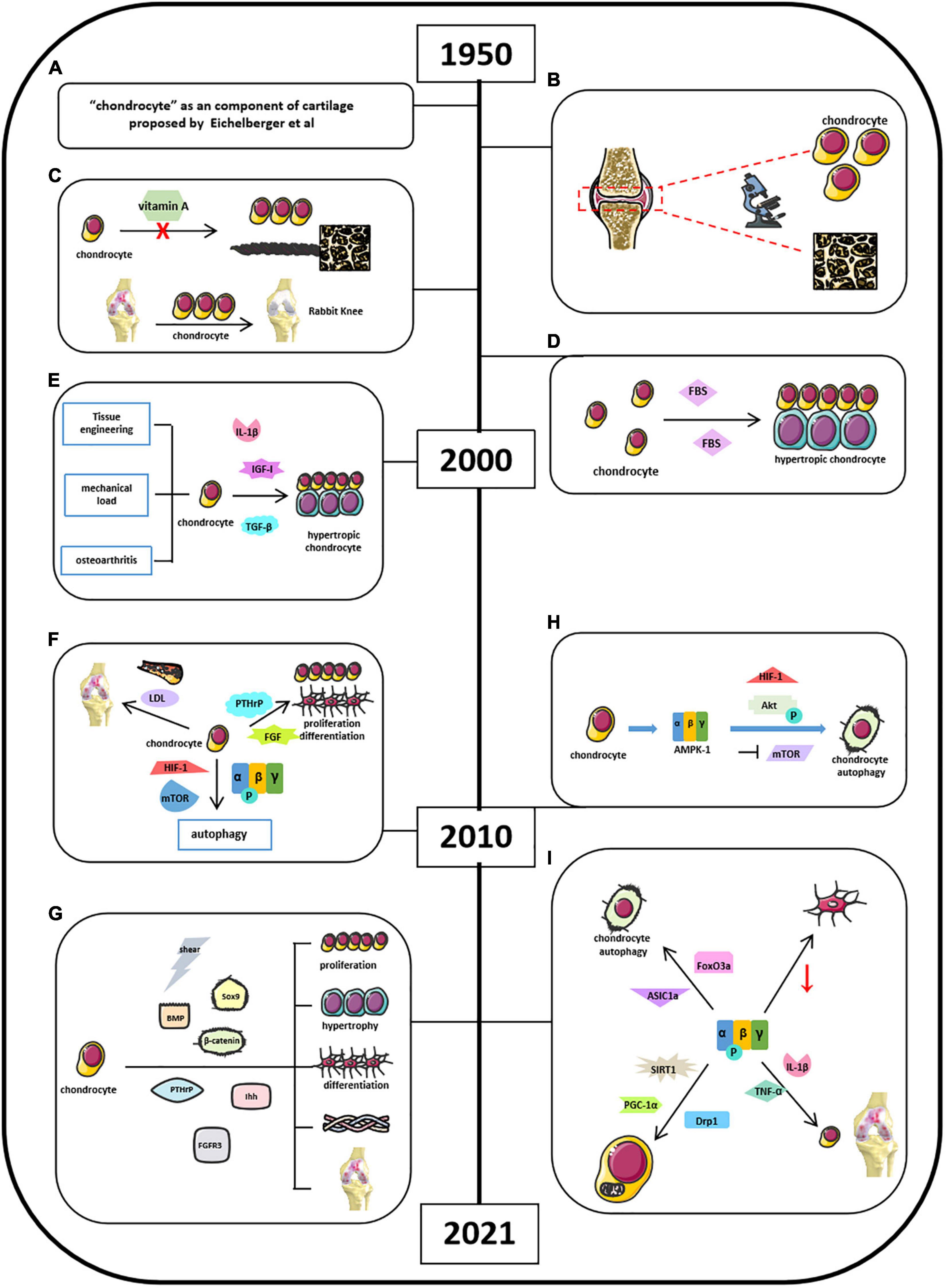
Figure 3. Milestones of discoveries of chondrocytes and the role of AMPK in cartilage chondrocytes. (A) Eichelberger et al. (1951) proposed “chondrocyte” as a component of cartilage. (B) Godman and Porter applied electron microscope to reveal the evolution of chondrogenesis. (C) Vitamin A could inhibit chondrogenesis and limiting extracellular space. (D) Cell density and FBS concentrations determine the transitional fate of resting chondrocytes to proliferative and hypertrophic chondrocyte. (E) Understanding of chondrocytes associated with osteoarthritis and upstream mediators in regulating chondrocyte activities. (F) Factors involved in cartilage chondrocytes’ homeostasis by 2010. (G) Current signaling pathways mediate chondrocyte activities. (H) Mouse chondrocytes expressed energy sensor AMPK-1, which promotes chondrocyte autophagy associated with Akt activation, mTOR suppression, and HIF-1 expression. (I) Current AMPK-related signaling pathways in mediating chondrocyte homeostasis.
Chondrocyte is the only cell type in articular cartilage. AMPK signaling mainly affects chondrocyte functions during OA development, but not other cells. The roles of AMPK in chondrocyte function have been reported since 2009. It started from understanding the autophagy in chondrocytes. This might be related to the previous findings that AMPK activation was induced by ER stress leading to chondrocyte apoptosis. Srinivas et al. (2009) and Bohensky et al. (2010) found that mouse chondrocytes expressed energy sensor AMPK-1, which promotes chondrocyte autophagy associated with Akt activation, mTOR suppression, and HIF-1 expression (Figure 3H). AMPK/FoxO3a pathway was found to be involved in ASIC1α-mediated articular chondrocyte autophagy in rats (Figure 3I; Dai et al., 2017; Zhao et al., 2018; Ge et al., 2019). Bandow’s group indicated that significant decrease in phosphorylation of catalytic AMPK α subunit was found during chondrogenic differentiation of primary chondrocyte precursors (Bandow et al., 2015). Terkeltaub et al. (2011) suggest that AMPK restrained matrix degradation in chondrocytes in the presence of inflammatory cytokines IL-1β and tumor necrosis factor α (TNF-α) in both human and mouse (Figure 3I). Zheng et al. (2020) showed that activation of AMPK/Drp1/mitochondrial fission pathway mediates chondrocyte death and migration injury (Figure 3I). Moreover, AMPK participated in chondrocyte dysfunction, hypertrophy, and fibrotic differentiation (Liu N. et al., 2020; Liu Z. et al., 2020). Petursson et al. (2013) and Zhou et al. (2017) showed that downregulation of AMPK signaling resulted in inhibition of matrix catabolic responses in articular chondrocytes during OA development. Ma et al. (2018) stated that SIRT1/AMPK/PGC-1α signaling leads to mitochondrial dysfunction in chondrocytes with increased oxidative stress and apoptosis, which might be the etiology of OA (Figure 3I). Although these studies suggest that AMPK participates in the regulation of chondrocyte activity, more in-depth studies are required to further clarify the molecular mechanisms of AMPK in the regulation of chondrocyte homeostasis.
Osteoarthritis is characterized as a degenerative joint disease and influenced 303 million people globally, as reported in 2017 (Chen et al., 2017; Kloppenburg and Berenbaum, 2020). OA has gained great attention due to its high prevalence worldwide. Studies about OA could retrospect back to the 18th century, and physicians did not recognize the inflammatory process of OA until the 18th century (Figure 4A; Suri et al., 2012; Hardcastle et al., 2015; Peter et al., 2015; Wallace et al., 2017). Bollet suggests that the pathogenesis of OA was related to cartilage degeneration via activation of proteolytic enzyme, decrease in matrix components, and increase in penetration of synovial fluid. Our recent studies demonstrated that AMPK expression was significantly reduced in articular cartilage tissues in OA mouse model. Inflammatory cytokines were found in synovial fluid and serum in patients with RA and OA since 1970 (Figures 4B,C; Gervis, 1949; Suri et al., 2012; Hardcastle et al., 2015; Peter et al., 2015). Physicians and researchers were still trying to exploring a better way to treat OA; thus, non-steroidal anti-inflammatory drugs (NSAIDs), and some herbal medicines were used to treat OA or relieve OA-related pain, which was the major complain in clinical OA (Figures 4D,E; Long et al., 2001; Seed et al., 2009; Hu et al., 2020; Yu et al., 2021). Meanwhile, other researchers were aiming to demonstrate the potential pathobiology of OA. Besides the well-known risk factors such as aging and obesity, Abramson indicated cartilage chondrocytes as mechanosensors and osmosensors to negatively charge cartilaginous extracellular matrix in response to mechanical and osmotic stresses. And degeneration of articular cartilage leads to the activation of cartilage anabolic factors, such as BMP, IGF-I, TGF-β, and FGFs, which were associated with the degradation of both proteoglycans and collagen type II and cleaving matrix metalloproteinase (Figure 4F; Abramson and Attur, 2009). By 2021, accumulated knowledge of OA has been reported with a more broaden horizon and a more significant depth (Figure 4G). Because of the fast pace of high techniques and their application in research, studies on OA have further emphasized on molecular biology in all aspects. Chen et al. (2017) listed a series mouse model in OA studies such as traditional DMM model, aging mouse model, and available transgenic mouse models. It provided a systemic understandings of the potential pathological mechanisms of OA related to its corresponding signaling pathways, including Wnt/β-catenin, TGF-β, Ihh, FGF, and NF-κB (Zhu et al., 2009; Weng et al., 2012; Shen et al., 2013, 2014; Chang et al., 2019; Kuang et al., 2020). Moreover, Rim et al. (2020) indicated that the shifting from anabolic to catabolic signaling due to chondrocyte dedifferentiation leads to OA initiation and progression. Xu et al. (2021) found that polysaccharide from Angelica sinensis could attenuate OA chondrocyte apoptosis via ERK1/2 inducing autophagy. Current medicines in the treatment of OA focus on reducing pain relief and limited drugs are available in impeding OA initiation and/or progression. Traditional medicines focus on pain relief and anti-inflammation, for example, NSAIDs and vitamin D. With the building-up evidence of signaling pathways in controlling OA pathology, novel drugs targeting specific signaling molecule were developed and have shown a therapeutic potential to some extent. For examples, BMP7 limits progression of OA, FGF-18 targets cartilage of knee OA, TGF-β inhibitor targets subchondral bone remodeling, and resveratrol participates in OA chondrocyte metabolism through upregulation of SIRT1 gene (Kim et al., 2014; Zhang et al., 2019). In addition, other reports indicate that several clinical trials have been conducted to evaluate the efficacy of OA treatment drugs like fisetin, colchicine, GSK3196165, SM04690, MIV-711, Tanezumab, Fasinumab, AmpionTM, and so on. (US National Library of Medicine).
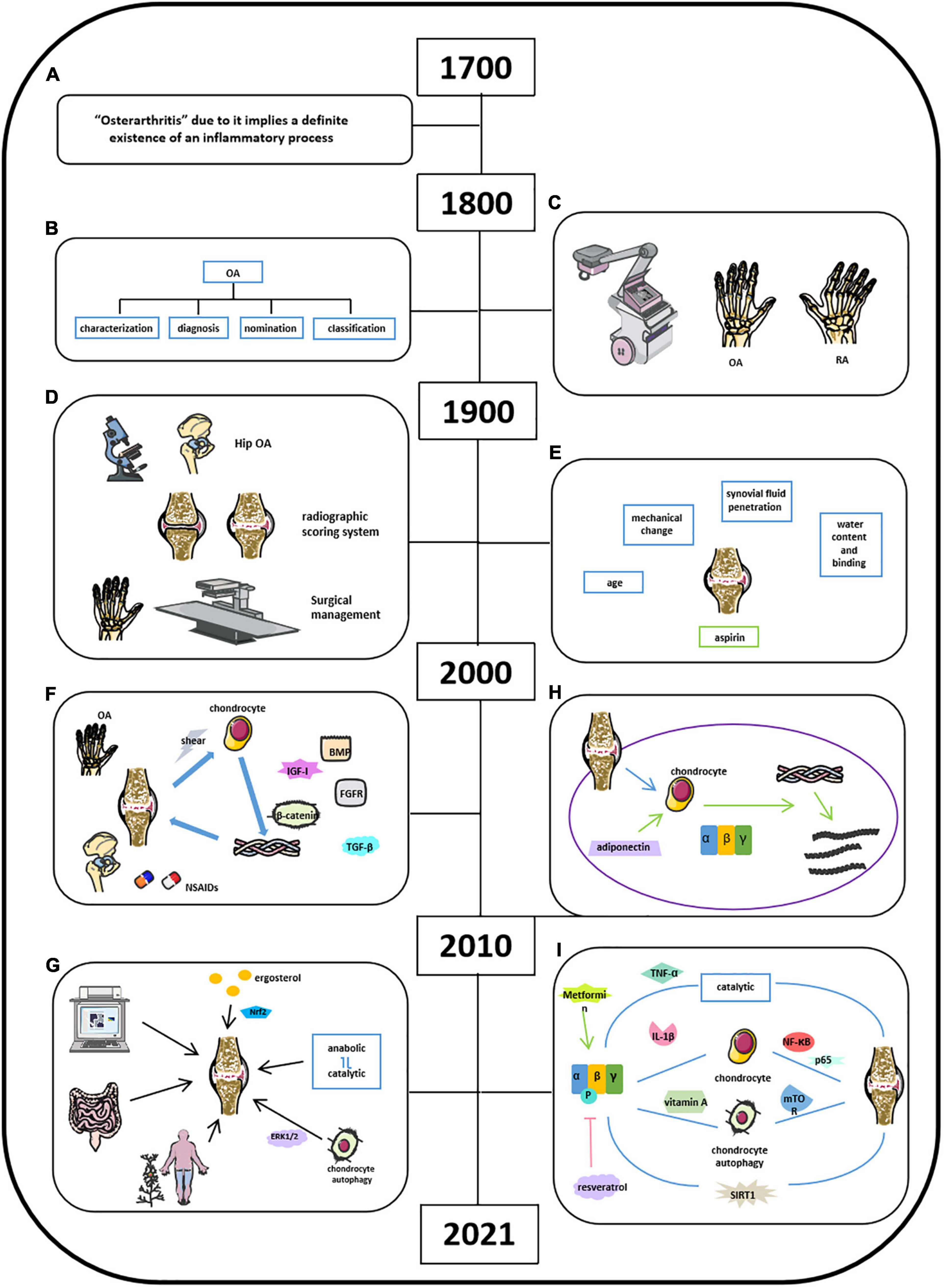
Figure 4. Milestones of discoveries of osteoarthritis and the role of AMPK in cartilage regeneration. (A) Physicians recognize the inflammatory process of OA by 18th century. (B) In 19th century, the knowledge of clinical OA became comprehensive. (C) X-rays applied in clinics helped Goldthwait distinguish OA from rheumatic arthritis (RA). (D) In 1900s, light microscope and electron microscope helped physicians in knee and hip OA treatment. (E) Knowledge of OA related in age, mechanical change, synovial fluid penetration, and water content binging. (F) NSAIDs and some herbal medicines were used to treat OA or relieve OA-related pain; activation of cartilage anabolic factors was associated with the degradation of proteoglycans, collagen type II, and cleaving matrix metalloproteinase. (G) Comprehensive understanding of OA by 2021. (H) Adiponectin mediates matrix degradation in human OA chondrocytes via AMPK and JNK pathways. (I) Molecules involved in OA homeostasis.
Although OA has been recognized as low-grade inflammatory disease with elevations in systemic inflammatory markers, such as IL-1, IL-6, IL-10, and TNF, cumulative evidence has indicated that OA is also a metabolic disorder where energy metabolic pathways, including glycolytic and TCA, are upregulated to meet the demand of the ATP for cartilage repair (Zhuo et al., 2012; Zhai, 2019). Also, diabetes could increase ROS and advanced glycosylation end products (AGEs), thus leading to further damage of articular cartilage in OA (Zhai, 2019). Accumulated studies of AMPK and OA were started to be reported since 2010. In detail, Kang et al. (2010) found that adiponectin mediates matrix degradation in human OA chondrocytes as the catabolic effect via AMPK and JNK pathways (Figure 4H). Liu-Bryan et al. (2015) summarized that inflammation disturbed cellular energy balance and increased cell stress via enhancing catabolic activities in degrading articular chondrocytes in OA (Liu-Bryan, 2015; June et al., 2016). Zhou’s group demonstrated that AMPK activity maintains joint homeostasis and OA development by enhancing IL-1β–stimulated catabolic response (Figure 4I; Zhou et al., 2017). Several groups illustrated thaactivationt of AMPK phosphorylation leads to suppressing NF-κB and its downstream molecule p65, which is involved in modulating OA cartilage in response to IL-1β–induced inflammation (Yang et al., 2019; Wang et al., 2020a; Zhu et al., 2020). Kong et al. (2020) suggest that AMPK-mTOR signaling pathway could reduce OA inflammation via activation of chondrocyte autophagy in the presence of active vitamin D. Several investigators at this time also observed the effect of metformin and resveratrol in the treatment of OA. Wei et al. (2018) showed that resveratrol suppresses induction of pAMPK and SIRT1 protein expression in OA rats. Metformin has been demonstrated to prevent cartilage degeneration and reduce pain behavior through activation of AMPK signaling (Feng et al., 2020; Li et al., 2020). Wang’s group concluded that decreased phosphorylation of AMPKα at T172 in chondrocytes aggravated a catabolic metabolism in response to inflammatory cytokines like IL-1β and TNF-α. And they also indicated that AMPK and its downstream molecule SIRT1 could switch chondrocyte metabolic activities from oxidative phosphorylation to glycolysis accompanied by increased production of inflammatory mediators and catabolic factors. Mitochondrial, autophagy, and ER stress may play a catabolic role in OA development through AMPK interacting with multiple signaling pathways (Wang et al., 2020b). Considering the presence of activation of AMPK in OA homeostasis associated with enhanced catabolic response, we suggest that AMPK could provide energy for the inflammatory reactions to OA as shown in metabolic syndrome and diabetes. Although the cumulative evidence has identified that AMPK activities are involved in OA pathogenesis, the exact and clear signaling pathways and molecular mechanisms are needed to be further investigated.
Activated protein kinase is an evolutionarily conserved serine/threonine kinase that is vital for cellular energy metabolism homeostasis (Kahn et al., 2005). AMPK controls cellular energy status when nutrition variation sensed; once activated by low ATP status, it promotes ATP-producing catabolic pathways and shuts down ATP-consuming anabolic pathways to restore cellular energy metabolism homeostasis (Hardie, 2004). AMPK proteins are critical mediators of AMPK signaling activities and participate in extensive cross-talk with other signaling pathways. The cross-talk can occur at different levels by directly or indirectly interactions with AMPKs and three AMPK upstream kinases, which includes LKB1, TGF-β–activated kinase-1 (TAK1) and Ca2+/CaMKKβ. Here we discuss recent progress in our understanding of the cross-talk between AMPK signaling and signaling pathways of NF-κB, mTOR, phosphoinositide 3-kinase (PI3K)–Akt and glycogen synthesis kinase 3 (GSK3) in OA.
NF-κB signaling controls inflammatory responses that develop in OA (Baker et al., 2011; Rigoglou and Papavassiliou, 2013). Zhang et al. (2020) reported that metformin protects chondrocytes against IL-1β–induced injury by regulation of the AMPK/NF-κB signaling pathway. Piao et al. (2020) showed that protectin DX attenuates IL-1β–induced OA inflammation via inhibiting AMPK/NF-κB pathway in chondrocytes and ameliorates OA progression in a rat model. However, whether AMPK directly or indirectly regulates NF-κB is unknown. Activators of AMPK, such as metformin, have been identified to have anti-inflammatory roles, and several studies have proven that AMPK inhibits NF-κB signaling through regulating distinct metabolic pathways including SIRT1 in macrophage, FOXO3 in helper T- cell activation (Lin et al., 2004) and colonic injury and inflammation (Zhou et al., 2009), PGC-1α in aortic smooth muscle and endothelial cells (Alvarez-Guardia et al., 2010), and p53 in aging mice (Salminen and Kaarniranta, 2012). Of interest, AMPK could be a potent regulator of NF-κB function in immune cells, as adiponectin was found to suppress the activation of natural killer cells and IFN-γ secretion via AMPK-mediated inhibition of NF-κB signaling (Kim et al., 2006).
The TOR (target of rapamycin) is a nutrient-sensing signaling pathway that is crucial for the regulation of cell growth and metabolism (Pal et al., 2015). Recent studies demonstrated that mTOR signaling plays a critical role in the development of OA. mTOR signaling pathway was suppressed by IL-1β and promotes chondrocyte autophagy and attenuates the inflammation response in rats with OA (Xue et al., 2017). Upregulation of mTOR in OA cartilage leads to increased chondrocyte apoptosis and reduced chondrocyte autophagy-related genes during OA (Zhang et al., 2015). Furthermore, miR-4262 and miR-27a have been shown to activate the PI3K/AKT/mTOR signaling pathway, indicating that targeting the mTOR signaling cascade by epigenetic regulation regulates chondrocyte viability, autophagy, and apoptosis (Sun et al., 2018; Cai et al., 2019). In addition, bioactive lipids, such as exogenous and endogenous n-3 polyunsaturated fatty acids, were reported to reduce mTORC1 and promote autophagy in chondrocytes (Huang et al., 2014). The AMPK and TOR pathways are interlinked, opposing signaling pathways involved in sensing nutrients and energy variation and regulation of cell growth (Hardie, 2014; Gonzalez et al., 2020). Previous studies have identified that activation of AMPK inhibits the mTORC1 complex by mechanisms of AMPK phosphorylating TSC2 at Thr1271 and Ser1387 (Inoki et al., 2003) and AMPK directly phosphorylating the RAPTOR component of mTORC1 at Ser722 and Ser792 (Gwinn et al., 2008). In OA, regulation of AMPK signaling pathway by mTOR may account for regulating autophagy signaling and the balance of cellular matrix metabolism in articular cartilage (Zhang et al., 2015). However, whether the above mechanism of AMPK switches off the mTOR signaling existing in chondrocytes remains to be determined.
Akt, also named protein kinase B, is a serine/threonine-specific protein kinase and plays a key role in cellular metabolism, proliferation, apoptosis, and migration. Akt is activated through a phosphorylation mechanism dependent on phosphatidylinositol 3-kinase (PI3K) by extracellular factors such as estrogen, serum, and insulin. Akt might play an important role in regulating chondrocyte apoptosis or survival and might be a potential target to prevent OA. Activation of PI3K/AKT signaling in synovial cells and in chondrocytes promoted synovial cell proliferation and the expression of collagenolytic matrix metalloprotease-13 and finally accelerated the hypertrophy and degradation of chondrocytes (Huang et al., 2013, 2019). Thus, it is not surprising that there should be mechanisms by which Akt downregulates AMPK in chondrocytes. Furthermore, recent research reported that treatment with asiatic acid activated AMPK and inhibited PI3K/AKT signaling in vitro in ACLT-induced rat OA model (Liu Z. et al., 2020). In addition, Akt also phosphorylates and inhibits GSK3 to facilitating glycogen synthesis (Cross et al., 1995). Disruption of GSK3 function within the AMPK complex leads to higher AMPK activity and cellular catabolic activities even under anabolic conditions, indicating that GSK3 also acts as a critical sensor for anabolic signaling to inhibit AMPK (Suzuki et al., 2013).
Sedentary at both work and home along with high-sugar and high-fat diet in the modern daily life caused a series of chronic metabolic diseases such as obesity and cancer. Considering the pursuit of higher life quality with longer life expectancy, public awareness of healthy lifestyle, regular diet structure, and fitness endurance are enhanced in every social aspect. OA has been observed for centuries but still has not been solved yet because of its unclear pathogenicity, although its association with age, mechanical loading, and hereditary has been explained to some extent. The clinical treatment of OA evolved from traditional surgical excision and knee joint replacement, anti-inflammation and relieving pain to blocking its initiation and progression, and focusing on prevention from intervening obesity and mechanical loading as well. Nevertheless, researchers are willing to invent therapeutic interventions to prevent or even cure OA. Hence, definitive pathological mechanisms causing OA need to be further clarified. AMPK, as it has been studied for decades, gained significant attention due to its vital role in maintaining energy balance in the body. Although its functional responsibilities in regulating skeletal developmental–related disease have not been well-depicted, its activities retaining chondrocyte balance have been well documented. Besides the well-established structure and function of AMPK at Ser79/Thr172, other allosteric activation sites of AMPK may also need to be identified and investigated. As mentioned previously, the therapeutic effects of metformin and resveratrol (AMPK activators) in the treatment of OA in mice and even in non-human primates have been demonstrated by several research groups (Wei et al., 2018; Feng et al., 2020; Li et al., 2020). Besides, metformin has been suggested as a novel drug to the patients with rheumatoid arthritis because it activates AMPK signaling, leading to the inhibition of mTOR and the differentiation of T cells in vitro and in vivo, thus reducing the invasion of fibroblast-like synovial cells (clinicaltrials.gov). These findings suggest that AMPK signaling molecules may serve as the potential drug targets for the treatment of arthritis. Although more evidence is still needed, promising results from clinical studies using AMPK-mimicking drugs with high efficacy and low toxicity have been used for OA treatment.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This research was supported by the grant from National Natural Science Foundation of China (NSFC) 82030067 to DC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abramson, S. B., and Attur, M. (2009). Developments in the scientific understanding of osteoarthritis. Arthritis Res. Ther. 11:227. doi: 10.1186/ar2655
Aburto, J. M., Villavicencio, F., Basellini, U., Kjaergaard, S., and Vaupel, J. W. (2020). Dynamics of life expectancy and life span equality. Proc. Natl. Acad. Sci. U. S. A. 117, 5250–5259. doi: 10.1073/pnas.1915884117
Alvarez-Guardia, D., Palomer, X., Coll, T., Davidson, M. M., Chan, T. O., Feldman, A. M., et al. (2010). The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc. Res. 87, 449–458. doi: 10.1093/cvr/cvq080
Aulthouse, A. L., Beck, M., Griffey, E., Sanford, J., Arden, K., Machado, M. A., et al. (1989). Expression of the human chondrocyte phenotype in vitro. In. Vitro. Cell. Dev. Biol. 25, 659–668. doi: 10.1007/bf02623638
Baker, R. G., Hayden, M. S., and Ghosh, S. (2011). NF-κB, inflammation, and metabolic disease. Cell Metab. 13, 11–22. doi: 10.1016/j.cmet.2010.12.008
Bandow, K., Kusuyama, J., Kakimoto, K., Ohnishi, T., and Matsuguchi, T. (2015). AMP-activated protein kinase (AMPK) activity negatively regulates chondrogenic differentiation. Bone 74, 125–133. doi: 10.1016/j.bone.2014.12.001
Bohensky, J., Leshinsky, S., Srinivas, V., and Shapiro, I. M. (2010). Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr. Nephrol. 25, 633–642. doi: 10.1007/s00467-009-1310-y
Bougault, C., Priam, S., Houard, X., Pigenet, A., Sudre, L., Lories, R. J., et al. (2014). Protective role of frizzled-related protein B on matrix metalloproteinase induction in mouse chondrocytes. Arthritis Res. Ther. 16:R137.
Brown, J. A., Bugescu, R., Mayer, T. A., Gata-Garcia, A., Kurt, G., Woodworth, H. L., et al. (2017). Loss of Action via Neurotensin-Leptin Receptor Neurons Disrupts Leptin and Ghrelin-Mediated Control of Energy Balance. Endocrinology 158, 1271–1288. doi: 10.1210/en.2017-00122
Bruckner, P., Horler, I., Mendler, M., Houze, Y., Winterhalter, K. H., Eich-Bender, S. G., et al. (1989). Induction and prevention of chondrocyte hypertrophy in culture. J. Cell Biol. 109, 2537–2545. doi: 10.1083/jcb.109.5.2537
Cai, C., Min, S., Yan, B., Liu, W., Yang, X., Li, L., et al. (2019). MiR-27a promotes the autophagy and apoptosis of IL-1beta treated-articular chondrocytes in osteoarthritis through PI3K/AKT/mTOR signaling. Aging 11, 6371–6384. doi: 10.18632/aging.102194
Chang, S. H., Mori, D., Kobayashi, H., Mori, Y., Nakamoto, H., Okada, K., et al. (2019). Excessive mechanical loading promotes osteoarthritis through the gremlin-1-NF-κB pathway. Nat. Commun. 10:1442.
Chao, P. H., Roy, R., Mauck, R. L., Liu, W., Valhmu, W. B., and Hung, C. T. (2000). Chondrocyte translocation response to direct current electric fields. J. Biomech. Eng. 122, 261–267. doi: 10.1115/1.429661
Chen, D., Shen, J., Zhao, W., Wang, T., Han, L., Hamilton, J. L., et al. (2017). Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res. 5:16044.
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789.
Dai, B., Zhu, F., Chen, Y., Zhou, R., Wang, Z., Xie, Y., et al. (2017). ASIC1a Promotes Acid-Induced Autophagy in Rat Articular Chondrocytes through the AMPK/FoxO3a Pathway. Int. J. Mol. Sci. 18:2125. doi: 10.3390/ijms18102125
Dietrich, M. O., Antunes, C., Geliang, G., Liu, Z. W., Borok, E., Nie, Y., et al. (2010). Agrp neurons mediate Sirt1’s action on the melanocortin system and energy balance: roles for Sirt1 in neuronal firing and synaptic plasticity. J. Neurosci. 30, 11815–11825. doi: 10.1523/jneurosci.2234-10.2010
Dreier, R. (2010). Hypertrophic differentiation of chondrocytes in osteoarthritis: the developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 12:216. doi: 10.1186/ar3117
Eichelberger, L., Brower, T. D., and Roma, M. (1951). Histochemical characterization of inorganic constituents, connective tissue and the chondroitin sulfate of extracellular and intracellular compartments of hyaline cartilages. Am. J. Physiol. 166, 328–329. doi: 10.1152/ajplegacy.1951.166.2.328
Feng, X., Pan, J., Li, J., Zeng, C., Qi, W., Shao, Y., et al. (2020). Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR. Aging 12, 1087–1103. doi: 10.18632/aging.102635
Garcia, D., and Shaw, R. J. (2017). AMPK: mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 66, 789–800. doi: 10.1016/j.molcel.2017.05.032
Ge, Y., Zhou, S., Li, Y., Wang, Z., Chen, S., Xia, T., et al. (2019). Estrogen prevents articular cartilage destruction in a mouse model of AMPK deficiency via ERK-mTOR pathway. Ann. Transl. Med. 7:336. doi: 10.21037/atm.2019.06.77
Gerber, M., and Corpet, D. (1999). Energy balance and cancers. Eur. J. Cancer Prev. 8, 77–89. doi: 10.1097/00008469-199904000-00002
Gervis, W. H. (1949). Excision of the trapezium for osteoarthritis of the trapezio-metacarpal joint. J. Bone Joint Surg. Br. 31B, 537–539. doi: 10.1302/0301-620x.31b4.537
Godman, G. C., and Porter, K. R. (1960). Chondrogenesis, studied with the electron microscope. J. Biophys. Biochem. Cytol. 8, 719–760. doi: 10.1083/jcb.8.3.719
Gonzalez, A., Hall, M. N., Lin, S. C., and Hardie, D. G. (2020). AMPK and TOR: the Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 31, 472–492. doi: 10.1016/j.cmet.2020.01.015
Grande, D. A., Pitman, M. I., Peterson, L., Menche, D., and Klein, M. (1989). The repair of experimentally produced defects in rabbit articular cartilage by autologous chondrocyte transplantation. J. Orthop. Res. 7, 208–218. doi: 10.1002/jor.1100070208
Green, W. T. Jr. (1977). Articular cartilage repair. Behavior of rabbit chondrocytes during tissue culture and subsequent allografting. Clin. Orthop. Relat. Res. 124, 237–250.
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. doi: 10.1016/j.molcel.2008.03.003
Hall, K. D., Sacks, G., Chandramohan, D., Chow, C. C., Wang, Y. C., Gortmaker, S. L., et al. (2011). Quantification of the effect of energy imbalance on bodyweight. Lancet 378, 826–837. doi: 10.1016/s0140-6736(11)60812-x
Halliday, D., Robert, R., and Walker, J. (2011). Fundamentals Of Physics, 9th Edn. New York: John Wiley & Sons, Inc, 485.
Hardcastle, S. A., Dieppe, P., Gregson, C. L., Davey Smith, G., and Tobias, J. H. (2015). Osteoarthritis and bone mineral density: are strong bones bad for joints? Bonekey Rep. 4:624.
Hardie, D. G. (2004). The AMP-activated protein kinase pathway–new players upstream and downstream. J. Cell Sci. 117, 5479–5487. doi: 10.1242/jcs.01540
Hardie, D. G. (2014). AMP-activated protein kinase: maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 34, 31–55. doi: 10.1146/annurev-nutr-071812-161148
Hardie, D. G., Salt, P. I., Hawley, S. A., and Davies, S. P. (1999). AMP-activated protein kinase: an ultrasensitive system for monitoring cellular energy charge. Biochem. J. 338, 717–722. doi: 10.1042/0264-6021:3380717
He, F., Jin, J. Q., Qin, Q. Q., Zheng, Y. Q., Li, T. T., Zhang, Y., et al. (2018). Resistin Regulates Fatty Acid Beta Oxidation by Suppressing Expression of Peroxisome Proliferator Activator Receptor Gamma-Coactivator 1alpha (PGC-1alpha). Cell. Physiol. Biochem. 46, 2165–2172. doi: 10.1159/000489546
Herzig, S., and Shaw, R. J. (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135. doi: 10.1038/nrm.2017.95
Hill, J. O., and Commerford, R. (1996). Physical activity, fat balance, and energy balance. Int. J. Sport Nutr. 6, 80–92. doi: 10.1123/ijsn.6.2.80
Hill, J. O., and Saris, W. H. M. (2003). “Energy expenditure and physical activity,” in Handbook of Obesity, 2nd Edn, eds G. Bray and C. Bouchard (New York, NY: Marcel Dekker, Inc), 631–654.
Hill, J. O., Wyatt, H. R., and Peters, J. C. (2012). Energy balance and obesity. Circulation 126, 126–132.
Hill, J. O., Wyatt, H. R., and Peters, J. C. (2013). The importance of energy balance. Eur. Endocrinol. 9, 111–115.
Huang, C. Y., Lin, H. J., Chen, H. S., Cheng, S. Y., Hsu, H. C., and Tang, C. H. (2013). Thrombin promotes matrix metalloproteinase-13 expression through the PKCdelta c-Src/EGFR/PI3K/Akt/AP-1 signaling pathway in human chondrocytes. Mediators Inflamm. 2013:326041.
Huang, J., Zhao, L., Fan, Y., Liao, L., Ma, P. X., Xiao, G., et al. (2019). The microRNAs miR-204 and miR-211 maintain joint homeostasis and protect against osteoarthritis progression. Nat. Commun. 10:2876.
Huang, M. J., Wang, L., Jin, D. D., Zhang, Z. M., Chen, T. Y., Jia, C. H., et al. (2014). Enhancement of the synthesis of n-3 PUFAs in fat-1 transgenic mice inhibits mTORC1 signalling and delays surgically induced osteoarthritis in comparison with wild-type mice. Ann. Rheum. Dis. 73, 1719–1727. doi: 10.1136/annrheumdis-2013-203231
Hu, S., Zhang, C., Ni, L., Huang, C., Chen, D., and Shi, K., et al. (2020). Stabilization of HIF-1alpha alleviates osteoarthritis via enhancing mitophagy. Cell Death Dis. 11:481.
Ichinose, S., Muneta, T., Koga, H., Segawa, Y., Tagami, M., Tsuji, K., et al. (2010). Morphological differences during in vitro chondrogenesis of bone marrow-, synovium-MSCs, and chondrocytes. Lab. Invest. 90, 210–221. doi: 10.1038/labinvest.2009.125
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/s0092-8674(03)00929-2
June, R. K., Liu-Bryan, R., Long, F., and Griffin, T. M. (2016). Emerging role of metabolic signaling in synovial joint remodeling and osteoarthritis.. J. Orthop. Res. 34, 2048–2058. doi: 10.1002/jor.23420
Kahn, B. B., Alquier, T., Carling, D., and Hardie, D. G. (2005). AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25. doi: 10.1016/j.cmet.2004.12.003
Kang, E. H., Lee, Y. J., Kim, T. K., Chang, C. B., Chung, J. H., Shin, K., et al. (2010). Adiponectin is a potential catabolic mediator in osteoarthritis cartilage. Arthritis Res. Ther. 12:R231.
Kawamura, S., Wakitani, S., Kimura, T., Maeda, A., Caplan, A. I., Shino, K., et al. (1998). Articular cartilage repair. Rabbit experiments with a collagen gel-biomatrix and chondrocytes cultured in it. Acta Orthop. Scand. 69, 56–62. doi: 10.3109/17453679809002358
Kim, K. Y., Kim, J. K., Han, S. H., Lim, J. S., Kim, K. I., Cho, D. H., et al. (2006). Adiponectin is a negative regulator of NK cell cytotoxicity. J. Immunol. 176, 5958–5964. doi: 10.4049/jimmunol.176.10.5958
Kim, H. J., Braun, H. J., and Dragoo, J. L. (2014). The effect of resveratrol on normal and osteoarthritic chondrocyte metabolism. Bone Joint Res. 3, 51–59. doi: 10.1302/2046-3758.33.2000226
Kim, K. H., Park, T. S., Cho, B. W., and Kim, T. M. (2020). Nanoparticles from Equine Fetal Bone Marrow-Derived Cells Enhance the Survival of Injured Chondrocytes. Animals 10:1723 doi: 10.3390/ani10101723
Kishimoto, H., Akagi, M., Zushi, S., Teramura, T., Onodera, Y., Sawamura, T., et al. (2010). Induction of hypertrophic chondrocyte-like phenotypes by oxidized LDL in cultured bovine articular chondrocytes through increase in oxidative stress. Osteoarthritis Cartilage 18, 1284–1290. doi: 10.1016/j.joca.2010.05.021
Kloppenburg, M., and Berenbaum, F. (2020). Osteoarthritis year in review 2019: epidemiology and therapy. Osteoarthritis Cartilage 28, 242–248. doi: 10.1016/j.joca.2020.01.002
Keith, N. F. (2013). “Energy Balance and Body Weight Regulation,” in Metabolic Regulation: A Human Perspective, 3rd Edn, Chap. 11, ed. N. F. Keith (New York: John Wiley & Sons), 329–349.
Kong, C., Wang, C., Shi, Y., Yan, L., Xu, J., and Qi, W. (2020). Active vitamin D activates chondrocyte autophagy to reduce osteoarthritis via mediating the AMPK-mTOR signaling pathway. Biochem. Cell Biol. 98, 434–442. doi: 10.1139/bcb-2019-0333
Kuang, L., Wu, J., Su, N., Qi, H., Chen, H., Zhou, S., et al. (2020). FGFR3 deficiency enhances CXCL12-dependent chemotaxis of macrophages via upregulating CXCR7 and aggravates joint destruction in mice. Ann. Rheum. Dis. 79, 112–122. doi: 10.1136/annrheumdis-2019-215696
Lam, Y. Y., and Ravussin, E. (2016). Analysis of energy metabolism in humans: a review of methodologies. Mol. Metab. 5, 1057–1071. doi: 10.1016/j.molmet.2016.09.005
Lee, T. J., Jang, J., Kang, S., Jin, M., Shin, H., Kim, D. W., et al. (2013). Enhancement of osteogenic and chondrogenic differentiation of human embryonic stem cells by mesodermal lineage induction with BMP-4 and FGF2 treatment. Biochem. Biophys. Res. Commun. 430, 793–797. doi: 10.1016/j.bbrc.2012.11.067
Lewis, C. A., Pratt, R. M., Pennypacker, J. P., and Hassell, J. R. (1978). Inhibition of limb chondrogenesis in vitro by vitamin A: alterations in cell surface characteristics. Dev. Biol. 64, 31–47. doi: 10.1016/0012-1606(78)90058-1
Li, J., and Dong, S. (2016). The Signaling Pathways Involved in Chondrocyte Differentiation and Hypertrophic Differentiation. Stem Cells Int. 2016:2470351.
Li, J., Zhang, B., Liu, W. X., Lu, K., Pan, H., Wang, T., et al. (2020). Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann. Rheum. Dis. 79, 635–645. doi: 10.1136/annrheumdis-2019-216713
Li, T. F., Gao, L., Sheu, T. J., Sampson, E. R., Flick, L. M., Konttinen, Y. T., et al. (2010). Aberrant hypertrophy in Smad3-deficient chondrocytes is rescued by restoring TAK1-ATF-2 signaling: a potential clinical implication for osteoarthritis. Arthritis Rheum. 62, 2359–2369. doi: 10.1002/art.27537
Lin, L., Hron, J. D., and Peng, S. L. (2004). Regulation of NF-kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity 21, 203–213. doi: 10.1016/j.immuni.2004.06.016
Lim, C. T., Kola, B., and Korbonits, M. (2010). AMPK as a mediator of hormonal signalling. J. Mol. Endocrinol. 44, 87–97. doi: 10.1677/jme-09-0063
Liu, N., Fu, D., Yang, J., Liu, P., Song, X., Wang, X., et al. (2020). Asiatic acid attenuates hypertrophic and fibrotic differentiation of articular chondrocytes via AMPK/PI3K/AKT signaling pathway. Arthritis Res. Ther. 22:112.
Liu-Bryan, R. (2015). Inflammation and intracellular metabolism: new targets in OA. Osteoarthritis Cartilage 23, 1835–1842. doi: 10.1016/j.joca.2014.12.016
Liu, Z., Zhang, H., Wang, H., Wei, L., and Niu, L. (2020). Magnolol Alleviates IL-1beta-Induced Dysfunction of Chondrocytes Through Repression of SIRT1/AMPK/PGC-1alpha Signaling Pathway. J. Interferon Cytokine Res. 40, 145–151. doi: 10.1089/jir.2019.0139
Long, L., Soeken, K., and Ernst, E. (2001). Herbal medicines for the treatment of osteoarthritis: a systematic review. Rheumatology 40, 779–793. doi: 10.1093/rheumatology/40.7.779
Lopez, M., Nogueiras, R., Tena-Sempere, M., and Dieguez, C. (2016). Hypothalamic AMPK: a canonical regulator of whole-body energy balance. Nat. Rev. Endocrinol. 12, 421–432. doi: 10.1038/nrendo.2016.67
Ma, C. H., Chiua, Y. C., Wu, C. H. I, Jou, M., Tu, Y. K., Hung, C. H., et al. (2018). Homocysteine causes dysfunction of chondrocytes and oxidative stress through repression of SIRT1/AMPK pathway: a possible link between hyperhomocysteinemia and osteoarthritis. Redox Biol. 15, 504–512. doi: 10.1016/j.redox.2018.01.010
MacLean, H. E., Guo, J., Knight, M. C., Zhang, P., Cobrinik, D., and Kronenberg, H. M. (2004). The cyclin-dependent kinase inhibitor p57(Kip2) mediates proliferative actions of PTHrP in chondrocytes. J. Clin. Invest. 113, 1334–1343. doi: 10.1172/jci200421252
Manore, M. M., Larson-Meyer, D. E., Lindsay, A. R., Hongu, N., and Houtkooper, L. (2017). Dynamic Energy Balance: an Integrated Framework for Discussing Diet and Physical Activity in Obesity Prevention-Is it More than Eating Less and Exercising More? Nutrients 9:905. doi: 10.3390/nu9080905
Mauck, R. L., Soltz, M. A., Wang, C. C., Wong, D. D., Chao, P. H., Valhmu, W. B., et al. (2000). Functional tissue engineering of articular cartilage through dynamic loading of chondrocyte-seeded agarose gels. J. Biomech. Eng. 122, 252–260. doi: 10.1115/1.429656
Mbalilaki, J. A., Masesa, Z., Stromme, S. B., Hostmark, A. T., Sundquist, J., Wandell, P., et al. (2010). Daily energy expenditure and cardiovascular risk in Masai, rural and urban Bantu Tanzanians. Br. J. Sports Med. 44, 121–126. doi: 10.1136/bjsm.2007.044966
Moyes, K. M., Drackley, J. K., Morin, D. E., Rodriguez-Zas, S. L., Everts, R. E., Lewin, H. A., et al. (2010). Mammary gene expression profiles during an intramammary challenge reveal potential mechanisms linking negative energy balance with impaired immune response. Physiol. Genom. 41, 161–170. doi: 10.1152/physiolgenomics.00197.2009
Murphy, K. G., and Bloom, S. R. (2006). Gut hormones and the regulation of energy homeostasis. Nature 444, 854–859. doi: 10.1038/nature05484
Nerucci, F., Fioravanti, A., Cicero, M. R., Collodel, G., and Marcolongo, R. (2000). Effects of chondroitin sulfate and interleukin-1beta on human chondrocyte cultures exposed to pressurization: a biochemical and morphological study. Osteoarthritis Cartilage 8, 279–287. doi: 10.1053/joca.1999.0302
Ng, M., Fleming, T., Robinson, M., Thomson, B., Graetz, N., Margono, C., et al. (2014). Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384, 766–781.
Okada, T. S. (1959). Regeneration of cartilaginous matrix from the dissociated chondrocytes in vitro. Experientia 15, 147–149. doi: 10.1007/bf02165532
Pal, B., Endisha, H., Zhang, Y., and Kapoor, M. (2015). mTOR: a potential therapeutic target in osteoarthritis? Drugs R D 15, 27–36.
Peter, W. F., Dekker, J., Tilbury, C., Tordoir, R. L., Verdegaal, S. H., and Onstenk, R. (2015). The association between comorbidities and pain, physical function and quality of life following hip and knee arthroplasty. Rheumatol. Int. 35, 1233–1241. doi: 10.1007/s00296-015-3211-7
Petursson, F., Husa, M., June, R., Lotz, M., Terkeltaub, R., and Liu-Bryan, R. (2013). Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res. Ther. 15:R77.
Piao, S., Du, W., Wei, Y., Yang, Y., Feng, X., and Bai, L. (2020). Protectin DX attenuates IL-1beta-induced inflammation via the AMPK/NF-kappaB pathway in chondrocytes and ameliorates osteoarthritis progression in a rat model. Int. Immunopharmacol. 78:106043. doi: 10.1016/j.intimp.2019.106043
Radomska, D. M., Osiecka, A., and Malejczyk, J. (1989). Natural cell-mediated cytotoxicity against syngeneic rat chondrocytes originating from different types of cartilage. Immunol. Cell Biol. 67, 209–213. doi: 10.1038/icb.1989.32
Rim, Y. A., Nam, Y., and Ju, J. H. (2020). The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 21:2358. doi: 10.3390/ijms21072358
Rigoglou, S., and Papavassiliou, A. G. (2013). The NF-kappaB signalling pathway in osteoarthritis. Int. J. Biochem. Cell Biol. 45, 2580–2584.
Robinson, D., Halperin, N., and Nevo, Z. (1990). Regenerating hyaline cartilage in articular defects of old chickens using implants of embryonal chick chondrocytes embedded in a new natural delivery substance. Calcif. Tissue Int. 46, 246–253. doi: 10.1007/bf02555003
Robson, H., Siebler, T., Stevens, D. A., Shalet, S. M., and Williams, G. R. (2000). Thyroid hormone acts directly on growth plate chondrocytes to promote hypertrophic differentiation and inhibit clonal expansion and cell proliferation. Endocrinology 141, 3887–3897. doi: 10.1210/endo.141.10.7733
Rosier, R. N., O’Keefe, R. J. I, Crabb, D., and Puzas, J. E. (1989). Transforming growth factor beta: an autocrine regulator of chondrocytes. Connect. Tissue Res. 20, 295–301. doi: 10.3109/03008208909023900
Ross, R., Soni, S., and Houle, S. A. (2020). Negative Energy Balance Induced by Exercise or Diet: effects on Visceral Adipose Tissue and Liver Fat. Nutrients 12:891. doi: 10.3390/nu12040891
Salminen, A., and Kaarniranta, K. (2012). AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 11, 230–241. doi: 10.1016/j.arr.2011.12.005
Searls, R. L., Hilfer, S. R., and Mirow, S. M. (1972). An ultrastructural study of early chondrogenesis in the chick wing bud. Dev. Biol. 28, 123–137. doi: 10.1016/0012-1606(72)90131-5
Seed, S. M., Dunican, K. C., and Lynch, A. M. (2009). Osteoarthritis: a review of treatment options. Geriatrics 64, 20–29.
Sharifi, N., and Gharravi, A. M. (2019). Shear bioreactors stimulating chondrocyte regeneration, a systematic review. Inflamm. Regen. 39:16.
Shen, J., Li, J., Wang, B., Jin, H., Wang, M., Zhang, Y., et al. (2013). Deletion of the type II TGF-β receptor gene in articular chondrocytes leads to progressive OA-like phenotype in mice. Arthritis Rheum. 65, 3107–3119. doi: 10.1002/art.38122
Srinivas, V., Bohensky, J., and Shapiro, I. M. (2009). Autophagy: a new phase in the maturation of growth plate chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells Tissues Organs. 189, 88–92. doi: 10.1159/000151428
Steinberg, G. R., and Carling, D. (2019). AMP-activated protein kinase: the current landscape for drug development. Nat. Rev. Drug Discov. 18, 527–551. doi: 10.1038/s41573-019-0019-2
Steinberg, G. R., and Kemp, B. E. (2009). AMPK in Health and Disease. Physiol. Rev. 89, 1025–1078. doi: 10.1152/physrev.00011.2008
Strassburg, S., Anker, S. D., Castaneda, T. R., Burget, L., Perez-Tilve, D., Pfluger, P. T., et al. (2008). Long-term effects of ghrelin and ghrelin receptor agonists on energy balance in rats. Am. J. Physiol. Endocrinol. Metab. 295, E78–E84.
Studer, R. K., Levicoff, E., Georgescu, H., Miller, L., Jaffurs, D., and Evans, C. H. (2000). Nitric oxide inhibits chondrocyte response to IGF-I: inhibition of IGF-IRbeta tyrosine phosphorylation. Am. J. Physiol. Cell. Physiol. 279, C961–C969.
Suri, P., Morgenroth, D. C., and Hunter, D. J. (2012). Epidemiology of osteoarthritis and associated comorbidities. PM R 4, S10–S19.
Sun, W., Li, Y., and Wei, S. (2018). miR-4262 regulates chondrocyte viability, apoptosis, autophagy by targeting SIRT1 and activating PI3K/AKT/mTOR signaling pathway in rats with osteoarthritis. Exp. Ther. Med. 15, 1119–1128.
Suzuki, T., Bridges, D., Nakada, D., Skiniotis, G., Morrison, S. J., Lin, J. D., et al. (2013). Inhibition of AMPK catabolic action by GSK3. Mol. Cell 50, 407–419. doi: 10.1016/j.molcel.2013.03.022
Takada, K., Hirose, J., Senba, K., Yamabe, S., Oike, Y., Gotoh, T., et al. (2011). Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int. J. Exp. Pathol. 92, 232–242. doi: 10.1111/j.1365-2613.2010.00758.x
Takada, K., Hirose, J., Yamabe, S., Uehara, Y., and Mizuta, H. (2013). Endoplasmic reticulum stress mediates nitric oxide-induced chondrocyte apoptosis. Biomed. Rep. 1, 315–319. doi: 10.3892/br.2013.52
Terkeltaub, R., Yang, B., Lotz, M., and Liu-Bryan, R. (2011). Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1beta and tumor necrosis factor alpha. Arthritis Rheum. 63, 1928–1937. doi: 10.1002/art.30333
van Baak, M. A. (1999). Physical activity and energy balance. Public Health Nutr. 2, 335–339. doi: 10.1017/s1368980099000452
Vasan, N. S., and Lash, J. W. (1975). Chondrocyte metabolism as affected by vitamin A. Calcif. Tissue Res. 19, 99–107. doi: 10.1007/bf02563995
Wakitani, S., Kimura, T., Hirooka, A., Ochi, T., Yoneda, M., Yasui, N., et al. (1989). Repair of rabbit articular surfaces with allograft chondrocytes embedded in collagen gel. J. Bone Joint Surg. Br. 71, 74–80. doi: 10.1302/0301-620x.71b1.2915011
Wallace, I. J., Worthington, S., Felson, D. T., Jurmain, R. D., Wren, K. T., Maijanen, H., et al. (2017). Knee osteoarthritis has doubled in prevalence since the mid-20th century. Proc. Natl. Acad. Sci. U. S. A. 114, 9332–9336. doi: 10.1073/pnas.1703856114
Wang, J., Fang, L., Ye, L., Ma, S., Huang, H., Lan, X., et al. (2020a). miR-137 targets the inhibition of TCF4 to reverse the progression of osteoarthritis through the AMPK/NF-kappaB signaling pathway. Biosci. Rep. 40:BSR20200466.
Wang, J., Li, J., Song, D., Ni, J., Ding, M., Huang, J., et al. (2020b). AMPK: implications in osteoarthritis and therapeutic targets. Am. J. Transl. Res. 12, 7670–7681.
Wei, Y., Jia, J., Jin, X., Tong, W., and Tian, H. (2018). Resveratrol ameliorates inflammatory damage and protects against osteoarthritis in a rat model of osteoarthritis. Mol. Med. Rep. 17, 1493–1498.
Weng, T., Yi, L., Huang, J., Luo, F., Wen, X., Du, X., et al. (2012). Genetic inhibition of FGFR1 in cartilage attenuates articular cartilage degeneration in adult mice. Arthritis Rheum. 64, 3982–3992. doi: 10.1002/art.34645
Wilkins, R. J., Browning, J. A., and Urban, J. P. (2000). Chondrocyte regulation by mechanical load. Biorheology 37, 67–74.
Xing, W., Godwin, C., Pourteymoor, S., and Mohan, S. (2019). Conditional disruption of the osterix gene in chondrocytes during early postnatal growth impairs secondary ossification in the mouse tibial epiphysis. Bone Res. 7:24.
Xu, C., Ni, S., Zhuang, C., Li, C., Zhao, G., Jiang, S., et al. (2021). Polysaccharide from Angelica sinensis attenuates SNP-induced apoptosis in osteoarthritis chondrocytes by inducing autophagy via the ERK1/2 pathway. Arthritis Res. Ther. 23:47.
Xue, J. F., Shi, Z. M., Zou, J., and Li, X. L. (2017). Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed. Pharmacother. 89, 1252–1261. doi: 10.1016/j.biopha.2017.01.130
Yamabe, S., Hirose, J., Uehara, Y., Okada, T., Okamoto, N., Oka, K., et al. (2013). Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. FEBS J. 280, 1617–1629. doi: 10.1111/febs.12170
Yamaoka, H., Nishizawa, S., Asawa, Y., Fujihara, Y., Ogasawara, T., Yamaoka, K., et al. (2010). Involvement of fibroblast growth factor 18 in dedifferentiation of cultured human chondrocytes. Cell Prolif. 43, 67–76. doi: 10.1111/j.1365-2184.2009.00655.x
Yang, Y., Wang, Y., Kong, Y., Zhang, X., Zhang, H., Gang, Y., et al. (2019). Mechanical stress protects against osteoarthritis via regulation of the AMPK/NF-kappaB signaling pathway. J. Cell. Physiol. 234, 9156–9167. doi: 10.1002/jcp.27592
Yu, R. G., Zhang, J. Y., Liu, Z. T., Zhuo, Y. G., Wang, H. Y., and Ye, J., et al. (2021). Text mining-based drug discovery in osteoarthritis. J. Healthc Eng. 2021:6674744.
Zhang, W., Robertson, W. B., Zhao, J., Chen, W., and Xu, J. (2019). Emerging Trend in the Pharmacotherapy of Osteoarthritis. Front. Endocrinol. 10:431. doi: 10.3389/fendo.2019.00431
Zhao, X., Li, Y., Lin, X., Wang, J., Zhao, X., Xie, J., et al. (2018). Ozone induces autophagy in rat chondrocytes stimulated with IL-1beta through the AMPK/mTOR signaling pathway. J. Pain Res. 11, 3003–3017. doi: 10.2147/jpr.s183594
Zheng, Z., Xiang, S., Wang, Y., Dong, Y., Li, Z., Xiang, Y., et al. (2020). NR4A1 promotes TNFalphainduced chondrocyte death and migration injury via activating the AMPK/Drp1/mitochondrial fission pathway. Int. J. Mol. Med. 45, 151–161.
Zhou, S., Lu, W., Chen, L., Ge, Q., Chen, D., Xu, Z., et al. (2017). AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci. Rep. 7:43245.
Zhu, H., Yan, H., Ma, J., Zhang, H., Zhang, J., Hu, Z., et al. (2020). CCAL1 enhances osteoarthritis through the NF-kappaB/AMPK signaling pathway. FEBS Open Bio. 10, 2553–2563. doi: 10.1002/2211-5463.12989
Zurlo, F., Larson, K., Bogardus, C., and Ravussin, E. (1990). Skeletal muscle metabolism is a major determinant of resting energy expenditure. J. Clin. Invest. 86, 1423–1427. doi: 10.1172/jci114857
Zhang, M., Liu, Y., Huan, Z., Wang, Y., and Xu, J. (2020). Metformin protects chondrocytes against IL-1beta induced injury by regulation of the AMPK/NF-kappa B signaling pathway. Pharmazie 75, 632–636.
Zhang, Y., Vasheghani, F., Li, Y. H., Blati, M., Simeone, K., Fahmi, H., et al. (2015). Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 74, 1432–1440. doi: 10.1136/annrheumdis-2013-204599
Zhai, G. (2019). Alteration of Metabolic Pathways in Osteoarthritis. Metabolites 9:11. doi: 10.3390/metabo9010011
Zhuo, Q., Yang, W., Chen, J., and Wang, Y. (2012). Metabolic syndrome meets osteoarthritis. Nat. Rev. Rheumatol. 8, 729–737. doi: 10.1038/nrrheum.2012.135
Zhu, M., Tang, D., Wu, Q., Hao, S., Chen, M., Xie, C., et al. (2009). Activation of β-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult β-catenin conditional activation mice. J. Bone Miner. Res. 24, 12–21. doi: 10.1359/jbmr.080901
Keywords: AMPK, energy balance, chondrocyte, osteoarthritis, signaling interaction
Citation: Yi D, Yu H, Lu K, Ruan C, Ding C, Tong L, Zhao X and Chen D (2021) AMPK Signaling in Energy Control, Cartilage Biology, and Osteoarthritis. Front. Cell Dev. Biol. 9:696602. doi: 10.3389/fcell.2021.696602
Received: 17 April 2021; Accepted: 24 May 2021;
Published: 22 June 2021.
Edited by:
Guanghua Lei, Central South University, ChinaCopyright © 2021 Yi, Yu, Lu, Ruan, Ding, Tong, Zhao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Di Chen, ZGkuY2hlbkBzaWF0LmFjLmNu; Xiaoli Zhao, emhhby54bEBzaWF0LmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.