 Yongyu Chen
Yongyu Chen Jing Lei
Jing Lei Song He
Song He
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 06 August 2021
Sec. Epigenomics and Epigenetics
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.692160
This article is part of the Research Topic RNA Modifications and Epitranscriptomics View all 30 articles
Accumulating evidence links m6A modification with immune infiltration. However, the correlation and mechanism by which m6A modification promotes intestinal immune infiltration in inflammatory bowel disease (IBD) is unknown. Here, genomic information from IBD tissues was integrated to evaluate disease-related m6A modification, and the correlation between the m6A modification pattern and the immune microenvironment in the intestinal mucosa was explored. Next, we identified hub genes from the key modules of the m6Acluster and analyzed the correlation among the hub genes, immune infiltration, and therapy. We found that IGF2BP1 and IGF2BP2 expression was decreased in Crohn’s disease (CD) tissues and that IGF2BP2 was decreased in ulcerative colitis (UC) tissues compared with normal tissues (P < 0.05). m6Acluster2, containing higher expressions of IL15, IL16, and IL18, was enriched in M0 macrophage, M1 macrophage, native B cells, memory B cells, and m6Acluster1 with high expression of IL8 and was enriched in resting dendritic and plasma cells (P < 0.05). Furthermore, we reveal that expression of m6A phenotype-related hub genes (i.e., NUP37, SNRPG, H2AFZ) was increased with a high abundance of M1 macrophages, M0 macrophages, and naive B cells in IBD (P < 0.01). Immune checkpoint expression in the genecluster1 with higher expression of hub genes was increased. The anti-TNF therapeutic response of patients in genecluster1 was more significant, and the therapeutic effect of CD was better than that of UC. These findings indicate that m6A modification may affect immune infiltration and therapeutic response in IBD. Assessing the expression of m6A phenotype-related hub genes might guide the choice of IBD drugs and improve the prediction of therapeutic response to anti-TNF therapy.
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic intestinal inflammatory disease (Torres et al., 2017; Ungaro et al., 2017). The precise etiology of IBD remains unclear, but genetic predisposition, disruption of mucosal immune homeostasis, microbial dysbiosis, and environmental factors are involved (Halfvarson et al., 2017; Neurath, 2017). In particular, the immune system is implicated as a major contributor to IBD progression, predominantly through an imbalance between the anti-inflammatory responses of regulatory T cells and M2 macrophages versus the pro-inflammatory responses of M1 macrophages, T helper (Th) 1, Th17, and neutrophils (Canavan et al., 2016; de Souza and Fiocchi, 2016; Zhou et al., 2018, 2019). Current treatments are aimed at relieving inflammation through the use of steroids, biological agents (primarily anti-TNF), and immunosuppressive drugs (Torres et al., 2017; Ungaro et al., 2017). However, approximately 30% of patients using anti-TNF agents do not respond to treatment (Belarif et al., 2019). Thus, the potential mechanism of the progression of IBD needs to be elucidated.
N6-methyladenosine (m6A) modification, as the most prevalent RNA epigenetic modification, occurs transcriptionally (Lan et al., 2019; Huang H. et al., 2020). m6A methylation is catalyzed by the MTase complex referred to as “writers,” including methyltransferase-like 3/14/16 (METTL3/14/16), RNA binding motif protein 15 (RBM15), KIAA1429, and Wilms tumor 1-associated protein (WTAP; Chen X. Y. et al., 2019), whereas its demethylation is mediated by “eraser” proteins (ALKBH5 and FTO) (Huang Y. et al., 2019; Jin et al., 2020). The m6A modification is interpreted by m6A “reader” proteins, including the insulin-like growth factor 2 mRNA-binding protein 1/2/3 (IGF2BP1/2/3), YTH-domain family 1/2/3 (YTHDF1/2/3), and HNRNPA2B1 (Huang H. et al., 2018; Jiang et al., 2021). Accumulating evidence suggests that m6A modification plays an important role in inflammation, various tumors, innate immunity, and immunotherapy through different m6A regulator–related modifications (Chen X. Y. et al., 2019; Wang J. et al., 2019). Most studies focus on various tumors, single regulator, or single immunity cell types, but the influence of m6A modification on disease progression has not been examined in IBD.
Recent studies find that m6A modification is associated with disease-related immune infiltration. For example, METTL3-mediated m6A methylation promotes the activation of dendritic cells (DCs) (Wang H. et al., 2019), and METTL3 leads to an imbalance between Treg cells and native T cells with Treg cells losing the ability to suppress immune and native T cells losing the ability to induce inflammation (Geula et al., 2015; Li et al., 2017). YTHDF1 binds to lysosomal proteases, increases their translational efficiency in DCs, and enhances the tumor infiltrating CD8 + T cell antitumor response (Han et al., 2019). A recent study finds that METTL14 deficiency in T cells promotes spontaneous colitis in mice (Lu et al., 2020). However, the mechanism connecting IBD-related m6A modification and immune cell infiltration has not yet been elucidated. Thus, a comprehensive understanding of the different immune infiltrating characteristics induced by m6A regulators will improve our understanding of immune regulation in IBD.
Herein, the genomic information of IBD samples was integrated to assess the m6A modification patterns. Next, we explored the correlation between the m6A modification pattern and immune infiltration, investigating the functions and related mechanism of m6A modification patterns in the immune microenvironment in IBD. Surprisingly, we reveal that m6A modification patterns are associated with the disease types and infiltration of multiple immune cells. According to differentially expressed genes in different m6A modification patterns, we reveal that m6A phenotype–related hub genes affect disease characteristics and immune cell-infiltrating characteristics in IBD patients and demonstrate that m6A phenotype–related hub genes are connected with disease progression, immune cell infiltration, anti-TNF therapeutic response, and immunotherapy. Assessing the expression of m6A phenotype–related hub genes might guide the choice of IBD drugs and improve the prediction of therapeutic response to anti-TNF therapy.
Public data and full clinical annotation were downloaded from the Gene-Expression Omnibus (GEO) database.1 GSE111889 (Lloyd-Price et al., 2019) (251 samples) was obtained in this study for further analysis (Supplementary Table 1). Data preprocessing was performed as follows: the downloaded data set was the expression profile obtained after data homogenization. First, the probes corresponding to the genes were identified, and no-load probes were removed. When multiple probes corresponded to the same gene, the median number was selected as the expression level of the gene. Four samples with zero expression of all the genes were filtered out, and the final expression matrix of 247 samples was used for subsequent analysis. Then, based on gene expression spectrum data and protein-coding genes from the Ensembl database,2 55,765 target gene expression profile data was obtained. Finally, 27 m6A methylase regulators (i.e., writers, erasers and readers) in three types were used with reference to previous studies (Deng et al., 2018). Among them, 24 m6A methylase regulators were supported by expression profile data (Supplementary Table 2).
Patients with qualitatively different m6A modification patterns were classified by using the R package of ConsensusClusterPlus (gaptools). The optimal k value was chosen by identifying the inflection point of the sum of the squared error (SSE). The decline slows down after k = i, and k = i is selected.
Edger analysis (gaptools) was used to identify differentially expressed genes among m6Acluster patterns, and 4745 genes and gene expression profiles of differentially expressed genes among m6Acluster patterns were obtained. Adjusted P-value < 0.05 was considered to be the criteria of differentially expressed genes.
High-connection genes and modules were identified using the R-package “WGCNA.” Each module was constructed by calculating the soft threshold power β. The WGCNA algorithm was used to construct the co-expression module after setting the soft threshold power value. Adjacency information was converted into topological overlap, and the network connectivity of genes was measured. Based on the dissimilarity of the topological overlap matrix (TOM), a hierarchical clustering function was performed to classify genes with similar expression profiles into modules (the minimum size of gene dendrogram = 25; merged highly similar modules with height = 0.25). The correlation between the clinical traits and the module eigengenes was used to determine the relevant modules. The correlation of gene expression profiles and the module eigengenes was used to quantitatively measure module membership. Gene significance was defined as the absolute value of the correlation between genes and clinical traits. Then, relevant modules of high importance for clinical traits were identified (Supplementary Table 3).
The R package clusterProfiler was used to perform functional enrichment analysis of genes in the key modules. The results of the molecular function (MF), cellular component (CC), and biological functions (BF) in the Gene Ontology (GO) analysis and the enrichment analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) were extracted.
To search for interacting genes, tgenes in the key modules were imported into a search tool (STRING3; score >900) to predict the PPI network. We defined interaction network hub genes as a degree of nodes ≥200 and interaction network non-hub genes as a degree <200 in the PPI (Supplementary Table 4). Genes with significance >0.6 and module membership >0.8 in the modules were defined as key genes (Supplementary Table 5). Finally, hub genes identified in the disease-related module and hub genes in PPI were intersected, and 29 disease-related hub genes were finally obtained (Supplementary Table 6).
The CiberSort algorithm of CiberSort software was performed to calculate the proportion of immune cell infiltration of different types, and a stacking diagram and box plot were drawn for display. The differential expression of genes at the immune examination sites (PD-L1, IDO1, CD86, ICOS, TNFRSF9, and CTLA4) and the response to anti-TNF treatment were analyzed.
Most analyses were performed using R software,4 Prism software,5 and SPSS software6 using two-tailed unpaired Student’s t-test or log-rank test unless otherwise specified. The chi-square test was applied to compare the response rates to therapy. In all analyses, P-values were bilateral, and P < 0.05 was considered statistically significant.
Gene-Expression Omnibus data sets with clinical information and available data (GSE111889, Supplementary Table 1; Lloyd-Price et al., 2019) were enrolled in our study. Based on the previous data (Deng et al., 2018), we identified 24 m6A regulators, including nine “writers,” two “erasers,” and 13 “readers” (Supplementary Table 2). The reversible process of m6A regulator–mediated methylation is summarized in Figure 1A, and the locations of m6A regulators on chromosomes are shown in Figure 1B. To ascertain whether the above m6A regulators influenced the disease progression of IBD, we evaluated the mRNA expression of m6A regulators in UC, CD, and normal tissues and found that expression of “readers” (i.e., IGF2BP1 and IGF2BP2) was dramatically decreased in CD tissues compared with normal tissues (P < 0.01) (Figure 1D). The expression of IGF2BP2 was markedly decreased in UC tissues compared with normal tissues (P < 0.001) (Figure 1C). The expression of IGF2BP1, METTL16, YTHDC2, and KIAA1429 was downregulated in CD tissues compared with UC tissues (P < 0.05) (Supplementary Figure 1A). These results suggest that m6A regulators exhibit high heterogeneity of the expressional alteration landscape among normal, UC, or CD patients, indicating that different m6A regulator expression may affect disease progression in different IBD types.
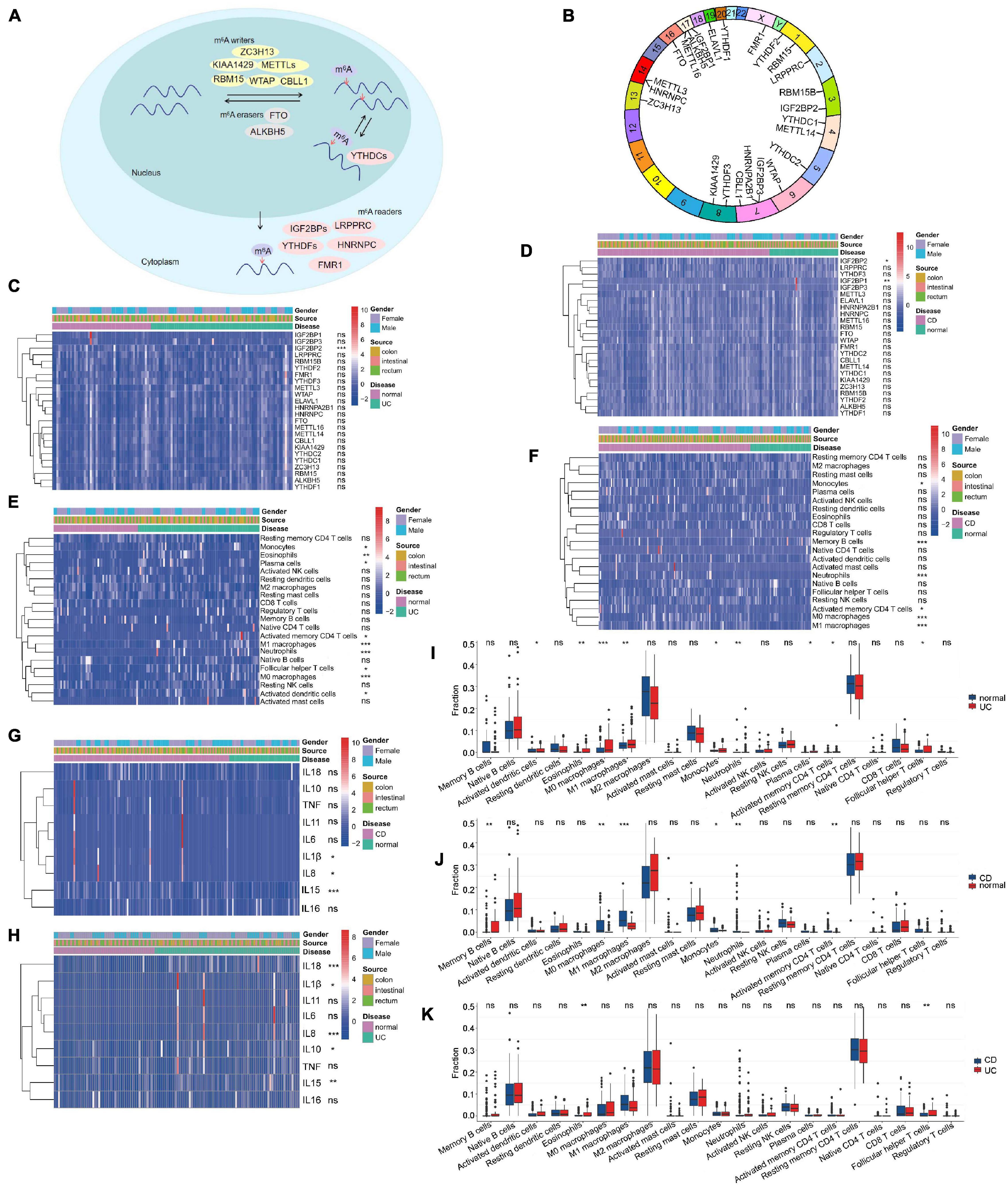
Figure 1. Landscape of m6A regulators and immune microenvironment in inflammatory bowel disease (IBD). (A) Summary of the reversible process of m6A regulator–mediated methylation. (B) The location of m6A regulators on 24 chromosomes using GSE111889 cohort. (C,D) The expression of 24 m6A regulators among ulcerative colitis (UC) panel (C), Crohn’s disease (CD) panel (D), and normal tissues. (E,F,I–K) The difference of immune cell infiltration among UC, CD, and normal tissues. (G,H) The expression of inflammatory factors among normal, UC panel (H), and CD tissues panel (G). The asterisks represented the statistical P-value (*P < 0.05; **P < 0.01; ***P < 0.001).
Additionally, we analyzed immune cell-infiltrating characteristics in the intestinal mucosa in IBD and found that UC was characterized by M1 macrophages (P < 0.001); M0 macrophages (P < 0.001); neutrophils (P < 0.001); eosinophils (P < 0.01) (Figures 1E,I); and the inflammatory factors IL18, IL8, and IL15 (Figure 1H). M1 macrophages, neutrophils, and M0 macrophages were enriched, but memory B cells were decreased in CD tissues (Figures 1F,J; P < 0.001) with the upregulation of IL15, IL8, and IL1β expression (Figure 1G). Compared with CD tissues, eosinophils and follicular helper T cells were more abundant in UC tissues (Figure 1K; P < 0.01). These results indicate that alterations in the mucosal immune microenvironment may be the prominent pathogenic factors of IBD, and local infiltration of M1 macrophages and neutrophils may promote the progression of IBD.
According to the expression of the 24 m6A regulators, m6A modification patterns were classified using the R package of ConsensusClusterPlus (gaptools). The optimal clustering stability (k = 2–10) was determined by the similarity of m6A regulator expression and the proportion of ambiguous clustering measurement, and k = 2 was identified (Figures 2A,B and Supplementary Figure 1B). Based on unsupervised clustering, we eventually identified two distinct modification patterns, including 99 cases in m6Acluster1 and 98 cases in m6Acluster2 (Supplementary Table 7). IBD samples could be completely distinguished based on the expression of these 24 m6A regulators (Figure 2C). To explore the correlation among “writers,” “erasers,” and “readers,” we evaluated pairwise correlations among the expression of 24 m6A regulators in IBD and revealed that positive correlations were more frequent (Figure 2D). We found that remarkable correlations existed among “writers,” “erasers,” and “readers,” and m6A regulators with the same function exhibited significant correlations in expression. IBD with high expression of IGF2BP2 (a “reader” gene) exhibited downregulation of IGF2BP3 (a “reader” gene) although upregulation of IGF2BP2 did not affect expression of other genes except for YTHDF3, LRPPRC, and RBM15B (“writer” genes). The “writer” genes CBLL1, KIAA1429, ZC3H13, and METTL16 presented a common trend in FTO (an “eraser” gene) expression. IBD with upregulation of the “writer” gene KIAA1429 displayed a common trend in ALKBH5 (an “eraser” gene) expression. Additionally, the change of IGF2BPs expression did not affect expression of these “eraser” genes. Therefore, crosstalk among different m6A regulators may be crucial for the generation of m6A modification patterns in individual IBD patients. After unsupervised clustering, we investigated the expression of 24 m6A regulators in different m6A subtypes and disease subtypes (Figure 2E) and found that the expression of m6A methylation regulators was increased in the m6Acluster2 compared with the m6Acluster1 except for IGF2BP3 (P < 0.05). Next, the clinicopathological features between the two subtypes were compared. However, there were no significant differences between m6Acluster1 and m6Acluster2 with respect to patient gender, location, or disease subtypes (P > 0.05).
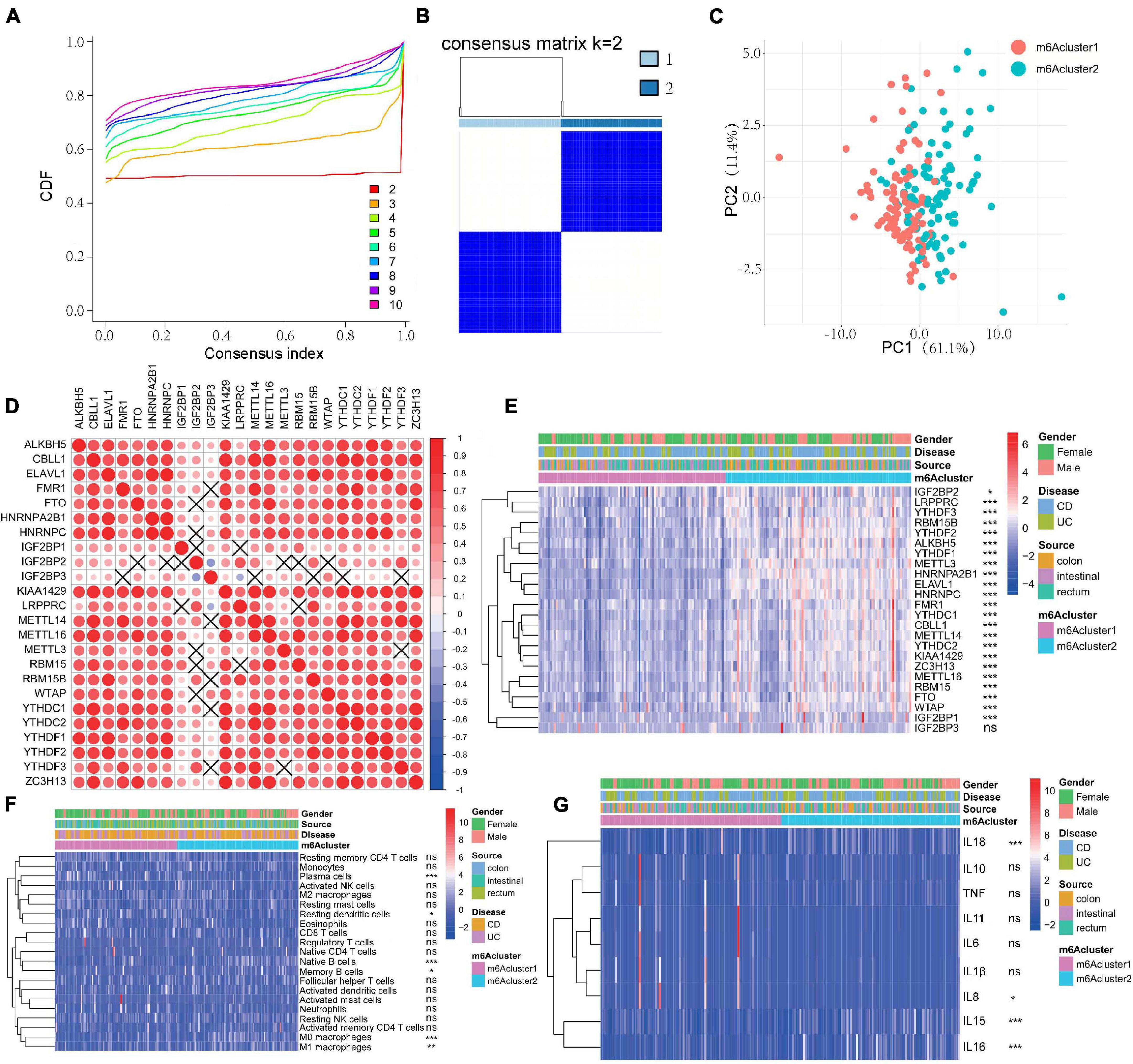
Figure 2. m6A methylation modification patterns are correlated with the clinical characteristics and the mucosal immune microenvironment in IBD. (A–C) Consensus clustering identified two subgroups according to the expression of m6A regulators. (D) Evaluation the pairwise correlations among 24 m6A regulators’ expression. (E) The expression of 24 m6A regulators in different m6A subtypes and disease subtypes. (F,G) The difference of mucosal immune cells infiltration panel (F) and inflammatory factors panel (G) between m6Acluster1 and m6Acluster2. The asterisks represented the statistical P-value (*P < 0.05; **P < 0.01; ***P < 0.001).
We next compared differences in immune cell infiltration between the two m6A modification patterns using CIBERSORT. To our surprise, m6Acluster2 was prominently associated with immune activation (Figure 2F). Subsequent analyses of immune infiltration suggested that m6Acluster2 was significantly abundant in innate immune cell infiltration, including M0 macrophage (P < 0.001), M1 macrophage (P < 0.01), and acquired immune cells (native B cells, memory B cells; P < 0.05). m6Acluster1 was enriched in innate immune cell infiltration, including resting DCs (P < 0.05) and plasma cells (P < 0.001) (Figure 2F). Furthermore, we found that expression of IL15, IL16, and IL18 was upregulated in m6Acluster2 (P < 0.001). However, expression of IL8 in m6Acluster1 was increased (P < 0.05) (Figure 2G). The significant differences in the infiltration of immune cells and inflammatory factors between the two m6Aclusters indicate that m6A methylation modifications may change the mucosal immune microenvironment of IBD.
To explore the differentially expressed genes in the two m6Aclusters, we used the Edger package in GAPTools. Subsequently, 4737 differentially expressed genes were screened out between the two clusters, among which 2274 genes were upregulated and 2463 genes were downregulated. A weighted gene co-expression network was then constructed (Supplementary Figure 1C). After calculating the eigengenes of each module, performing cluster analysis on the modules, and merging the modules close to each other into new modules, we obtained seven modules (Figures 3A,B). The turquoise module was the most correlated with the m6Acluster classification phenotype (Figure 3B and Supplementary Table 3; P = 2e–30). The R package clusterProfiler was used to analyze the function and pathway information of the top 20 enriched genes in the turquoise module. As shown in Figure 3C, genes in the turquoise module were markedly enriched in the biological processes (BP), including ribonucleoprotein complex biogenesis and ribosome biogenesis (Figure 3C; P < 0.05); CC were involved, including the composition of ribosomal subunit, chromosomal region (Figure 3D; P < 0.05); MF-related genes were markedly enriched in structural constituent of ribosome (Figure 3E; P < 0.05); the KEGG pathway was enriched in ribosome and spliceosome (Figure 3F; P < 0.05).
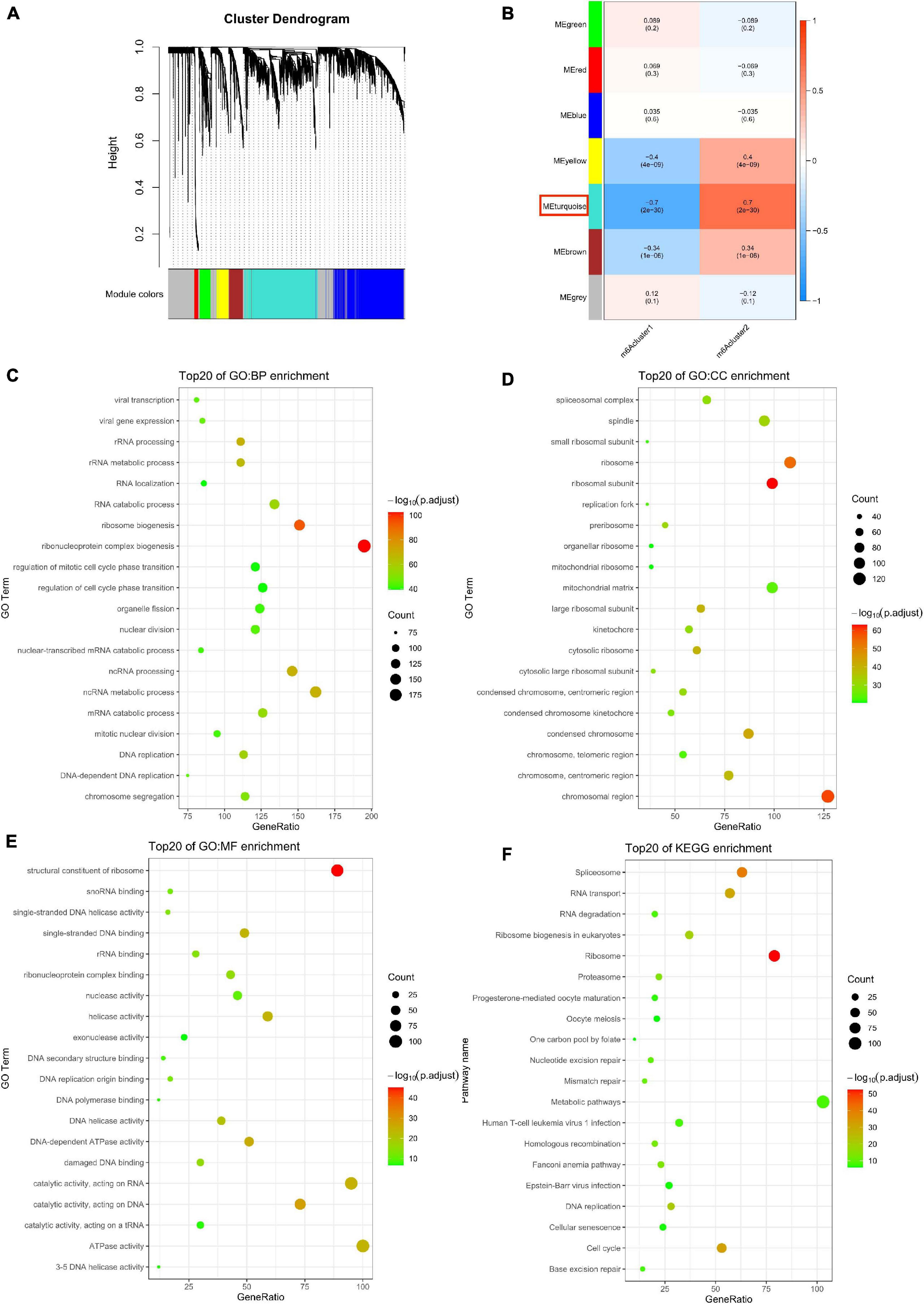
Figure 3. Biological characteristics of key m6A module. (A) Gene dendrogram obtained by average linkage hierarchical clustering. (B) The correlation between the module eigengenes and m6A modification patterns. (C–F) Functional annotation of the genes in the key m6A module by using Gene Ontology (GO) terms of biological processes (BP) panel (C), cellular component (CC) panel (D), molecular function (MF) panel (E), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (F).
To further validate the correlation of m6A phenotype–related hub genes with disease characteristics, we constructed a PPI network to analyze 1463 related genes in the key module (Figure 4A), identifying 112 hub genes (Supplementary Table 4). Genes (gene significance >0.6; module membership >0.8) were identified from the turquoise module as key genes with a total of 321 genes (Figure 4B and Supplementary Table 5). Finally, the hub genes of the interaction network were intersected with the hub genes identified in the m6A-related module, and 29 disease-related hub genes were obtained (Figure 4C and Supplementary Table 6).
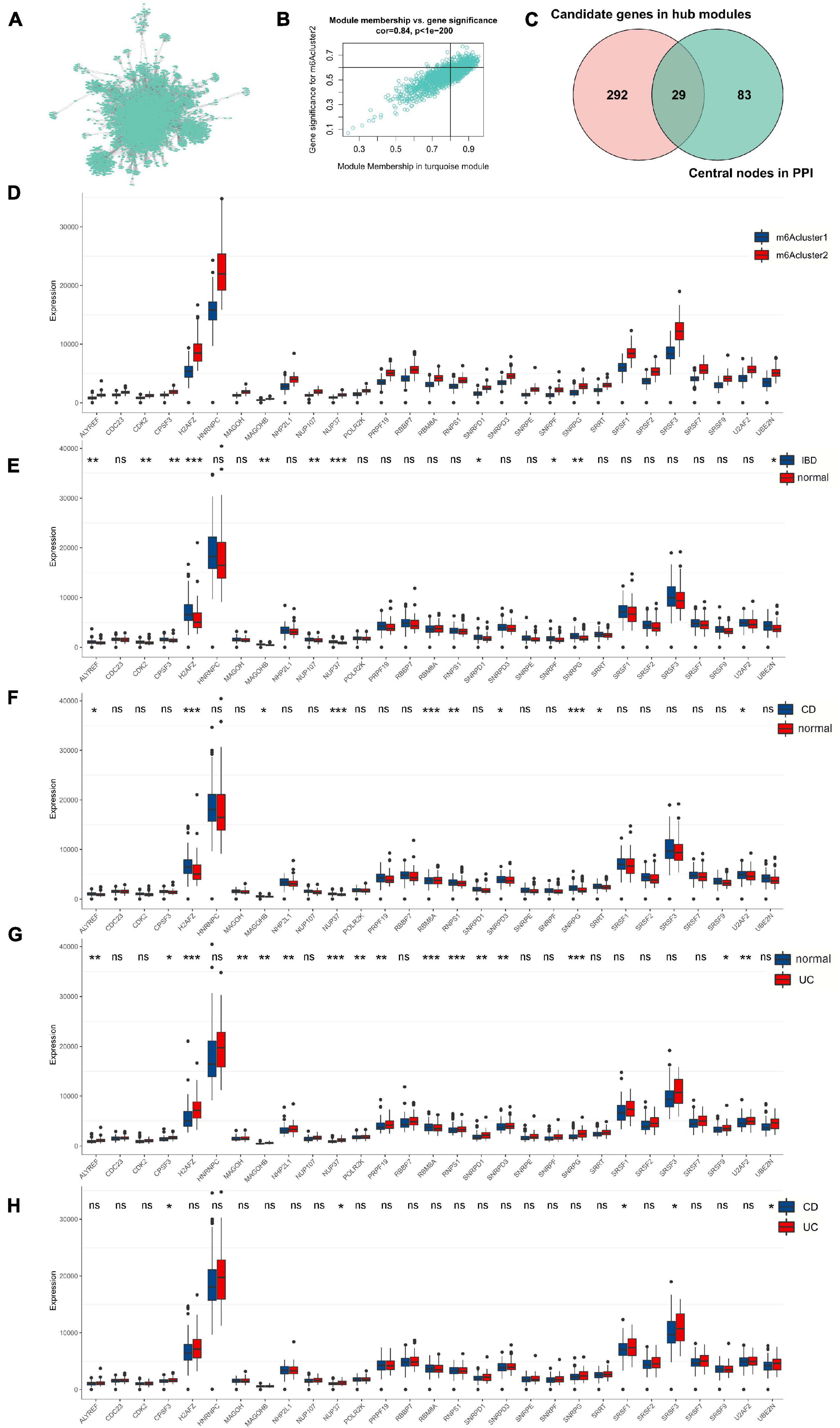
Figure 4. Twenty-nine m6A phenotype-related hub genes affect disease characteristics in IBD. (A) The PPI network of genes in the key module. (B) The significance genes in the turquoise module. (C) The hub genes of the interaction network are intersected with the hub genes identified in the m6A-related module. (D) The 29 genes’ expression in different m6Acluster classification. (E–H) The 29 genes’ expression between normal, UC, and CD tissues. The asterisks represented the statistical P-value (*P < 0.05; **P < 0.01; ***P < 0.001).
We found that 29 disease-related genes were significantly different among the m6Acluster classifications. The expression of 29 genes in m6Acluster2 was higher than that in m6Acluster1 (Figure 4D; P < 0.001). Furthermore, we explored differences in the expression of 29 disease-related genes between normal, CD, and UC tissues and found that the expression of NUP37, SNRPG, H2AFZ, MAGOHB, NUP107, ALYREF, CDK2, F3, SNRPD1, SNRPF, and UBE2N was significantly upregulated in the IBD tissues compared with normal tissues (Figure 4E; P < 0.05) although there was no significant difference in the other 18 genes (Figure 4E; P > 0.05). Compared with normal tissues, NUP37, H2AFZ, SNRPG, RBM8A, RNPS1, SNRPD3, ALYREF, MAGOHB, U2AF2, and SRRT were increased in CD tissues (Figure 4F; P < 0.05). Expression of NUP37, RBM8A, RNPS1, H2AFZ, SNRPG, PRPF19, U2AF2, ALYREF, MAGOHB, NHP2L1, SNRPD1, SNRPD3, MAGOH, POLR2K, CPSF3, and SRSF9 was markedly higher in UC tissues than in normal tissues (Figure 4G; P < 0.05). However, only CPSF3, NUP37, SRSF3, UBE2N, and SRSF1 were upregulated in UC tissues but not in CD tissues (Figure 4H; P < 0.05). These results suggest that 29 m6A phenotype–related hub genes are differentially expressed in different IBD types.
Based on expression of the 29 m6A phenotype-related genes, we classified IBD patients into different genomic subtypes by unsupervised clustering analyses (K-means) and found that the gene clusters could be well distinguished (Figure 5A and Supplementary Figure 1D). We found that there were marked differences among the 29 key disease genes in the gene cluster classification (P < 0.001) with higher gene expression in genecluster1 and lower gene expression in genecluster2 (Figure 5C). As shown in Figure 5B, almost all patients with the m6Acluster2 subtype were classified into genecluster1. However, no significant difference in patient gender, location, or disease subtypes was observed between genecluster1 and genecluster2 (Figures 5B,C; P > 0.05). The expression of all 29 m6A phenotype–related hub genes was significantly higher in genecluster1 than in genecluster2 (Figure 5D; P < 0.001).
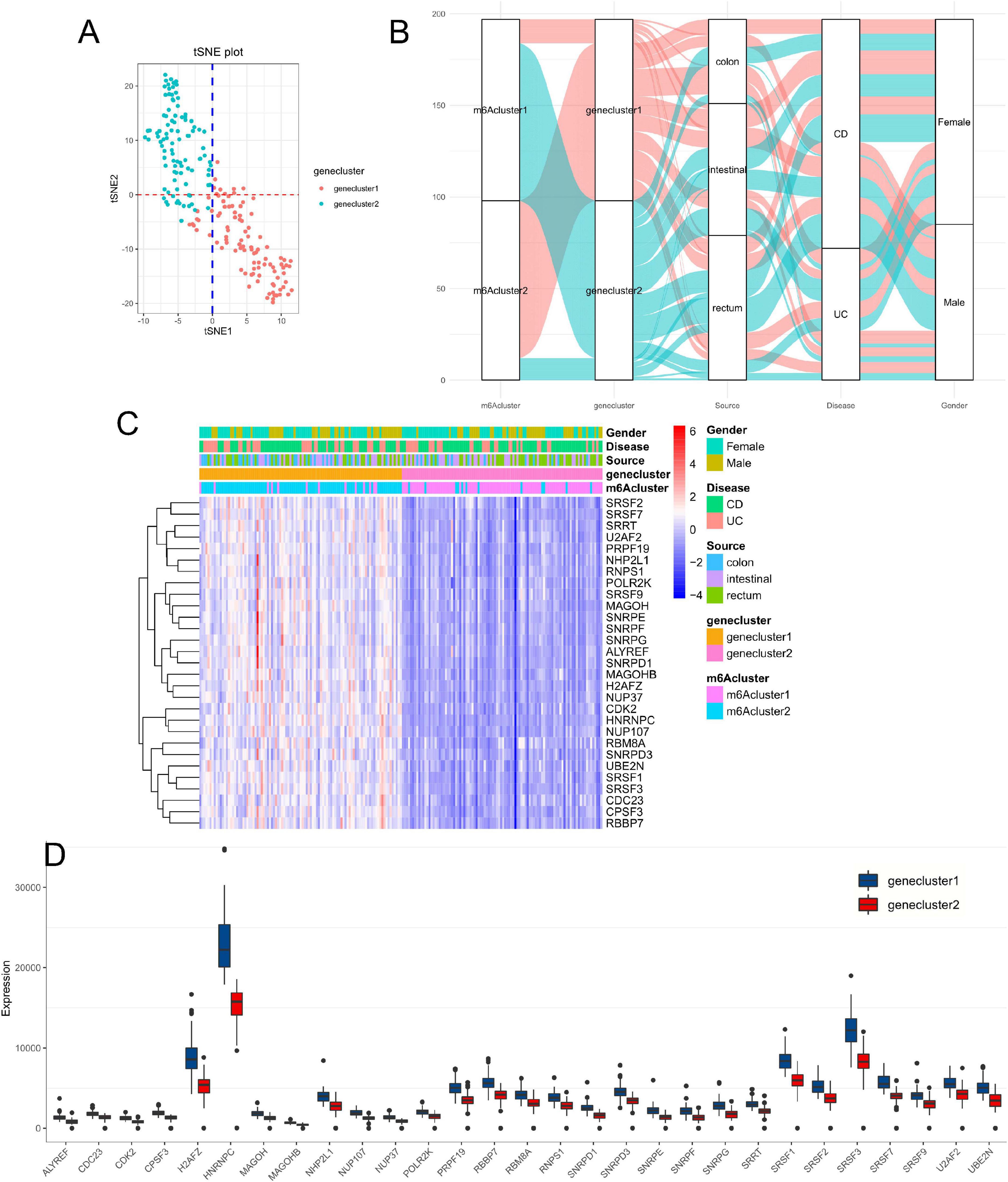
Figure 5. Consensus clustering for m6A phenotype–related hub genes with the characteristics of IBD patients. (A) Clustering the samples with the tSNE method. (B) Alluvial diagram showing the changes of m6Aclusters, disease subtype, location, gender, and gene clusters. (C,D) The expression of 29 hub genes between genecluster1 and genecluster2.
To explore the effect of m6A phenotype–related hub genes on the immune infiltration of IBD, we evaluated immune infiltration between genecluster1 with upregulated m6A phenotype-related hub gene expression and genecluster2 with downregulated m6A phenotype-related hub gene expression. Genecluster1 displayed higher infiltration levels of M1 macrophages, M0 macrophages, naive B cells, CD4 memory-activated T cells, memory B cells, and activated DCs, whereas genecluster2 was more correlated with resting DCs, M2 macrophages, CD4 memory resting T cells, plasma cells, resting mast cells, and eosinophils (Figures 6A,B; P < 0.05).
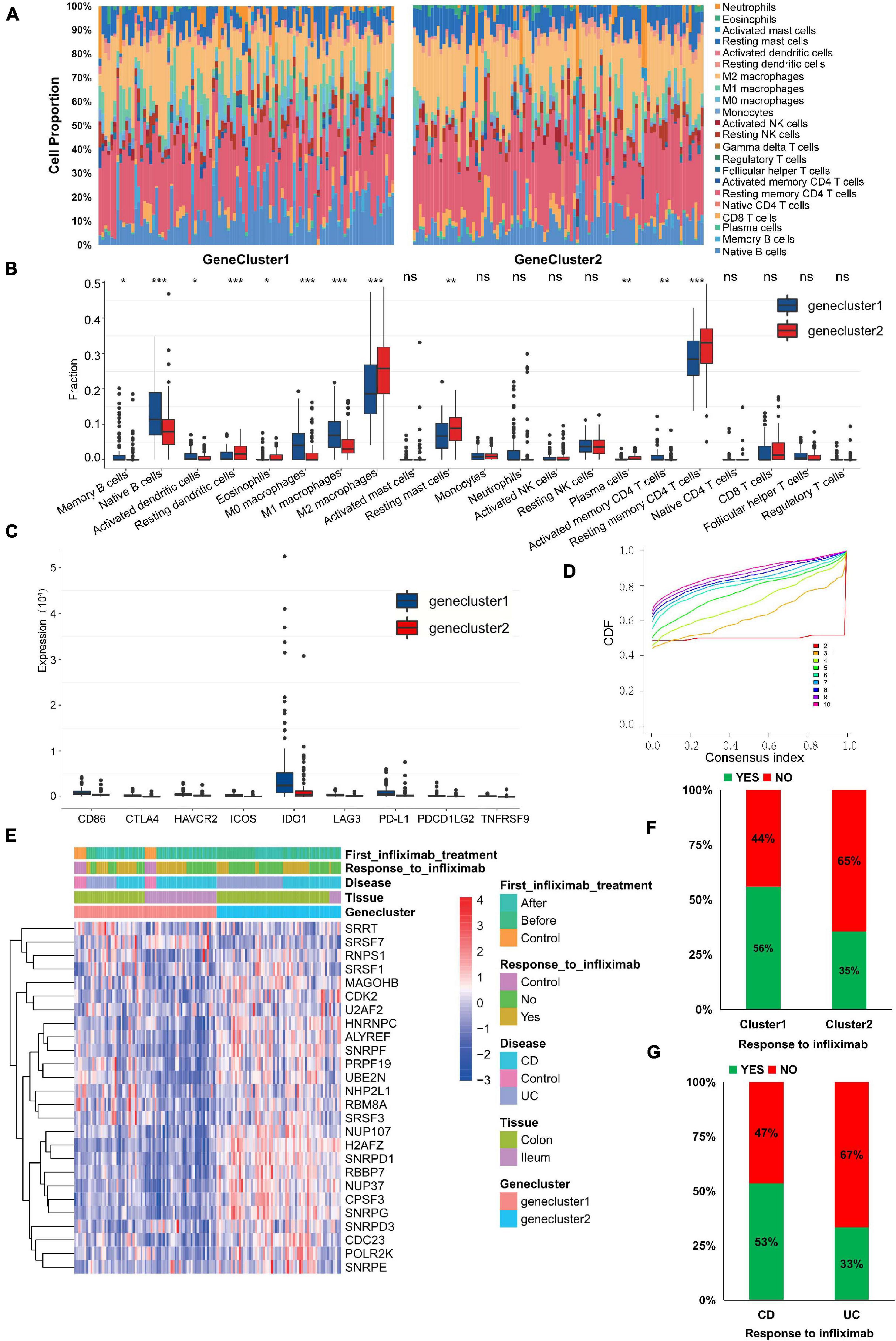
Figure 6. m6A phenotype–related hub genes influence the immune infiltration and therapeutic response. (A,B) The immune cell infiltrating levels in different gene cluster subtypes in the GSE111889 cohort. (C) The expression differences of nine important immune checkpoints among different gene clusters. (D) Consensus clustering cumulative distribution function (CDF) for k = 2–10. (E) The expression of hub genes, anti-TNF therapeutic response, and disease subtype between genecluster1 and genecluster2. (F,G) Infliximab therapeutic response in different gene clusters (F) and disease subtypes (G). The asterisks represented the statistical P-value (*P < 0.05; **P < 0.01; ***P < 0.001).
Expression differences of nine important immune checkpoints supported by expression data among different gene clusters are shown in Figure 6C. We found that the expression of all immune checkpoints in genecluster1 was significantly higher than that in genecluster2 (P < 0.01), indicating that m6A phenotype–related hub genes might affect immunotherapy. Current treatments are biological agents anti-TNF (primarily infliximab) for refractory and severe forms of IBD. Thus, we further verified the 29 m6A phenotype–related hub genes in GSE16879 (Arijs et al., 2009), which was available data and clinical treatment information. However, the expression of only 26 hub genes was supported by expression data in the GSE16879 data set. Based on the expression of 26 hub genes, we classified patients using the R package of ConsensusClusterPlus (gaptools), and k = 2 was identified (Figure 6D and Supplementary Figure 1E). Significant differences in infliximab therapy existed between the two gene clusters (Figure 6E). The expression of hub genes (H2AFZ, NUP37, SNRPD1, CPSF3, RBBP7, and SNRPG) in genecluster2 was significantly higher than in genecluster1, whereas expression of SRSF7 and SRRT in genecluster2 were lower than in genecluster1 (Figure 6E; P < 0.05). The response to infliximab in genecluster1 was better than that in genecluster2 (Figure 6F), and the response to infliximab in CD was better than that in UC (Figure 6G), suggesting that m6A-related hub gene expression may affect the therapeutic effect of infliximab. These results indicate that IBD with high expression of some m6A phenotype–related hub genes may represent those characterized by resistance to infliximab. Assessing the expression of m6A phenotype–related hub genes might guide the choice of IBD drugs and predict the response to anti-TNF therapy.
Currently, as most studies on disease-related m6A modification are limited to various tumors, single regulators, or single immune cell types, the effects of multiple m6A regulators on the overall immune infiltration characteristics and on IBD progression are poorly understood. Clarifying the role of m6A modification in the immune cell infiltration of IBD will improve our understanding of disease progression and disease-related local immune infiltration and strengthen more effective therapeutic strategies.
N6-methyladenosine regulators are involved in inflammatory development. However, only one study reports an association between m6A regulators and disease progression in IBD, and it reports that METTL14 (a “writer” gene) deficiency in T cells promotes spontaneous colitis in mice (Lu et al., 2020). IGF2BPs, a new family of m6A “readers,” are composed of two RNA recognition motif domains and four K homology domains (Bell et al., 2013) and promote the processes of various tumors (Huang X. et al., 2018; Huang S. et al., 2019; Hanniford et al., 2020). MYC, ACTIN, and LIN28B are reported as targets of IGF2BPs (Nielsen et al., 1999). Here, consistent with a previous study (Chen et al., 2021), we find that not only remarkable correlations exist among “writers,” “erasers,” and “readers,” but also that m6A regulators with the same function display significant correlations in expression. We find that expression of “readers” (i.e., IGF2BP1 and IGF2BP2) was downregulated in CD tissues compared with normal tissues, and expression of the IGF2BP2 was markedly decreased in UC tissues. The m6A regulators among normal, UC, and CD patients exhibit high heterogeneity of the expressional alteration landscape. However, the exact molecular mechanisms by which m6A regulators modulate IBD (UC and CD) development remain elusive.
As a reversible RNA methylation, m6A is added by “writers” and removed by “erasers,” indicating that they may represent opposite roles (Lan et al., 2019). However, it is interesting to find that m6A writers and erasers were positively correlated in our study. The “writer” genes CBLL1, KIAA1429, ZC3H13, and METTL16 presented a common trend in FTO (an “eraser” gene) expression. The “writer” gene KIAA1429 displayed a common trend in ALKBH5 (an “eraser” gene) expression in IBD. Previous studies find that m6A writers that increase the RNA m6A level are oncogenes (Lan et al., 2019; Qian et al., 2019; Wang X. K. et al., 2021), and FTO and ALKBH5 that decrease the RNA m6A level are also oncogenes (Shen et al., 2020; Su et al., 2020). The targets of “erasers” and “writers” that are reported in different studies are different, suggesting different mechanisms and other regulatory factors exist in disease development other than the “writer” and “eraser.” Additionally, the m6A “readers” exert post-transcriptional functions, which may partly account for the seemingly contradictory roles between the “eraser” and “writer.” In the future, the effect of m6A “writers” and “erasers” on the same RNA simultaneously should be examined to explore whether other regulatory factors are involved in m6A modification.
More recently, m6A regulators are shown to play crucial roles in disease-related immune cell infiltration. ALKBH5 inhibits antiviral innate responses by erasing their m6A modification (Zheng et al., 2017). METTL3, potentially serving as an anti-inflammatory target, methylates STAT1 mRNA and drives M1 macrophage polarization (Liu et al., 2019; Wang J. et al., 2019). LPS-induced inflammatory responses are regulated by the m6A reader YTHDF2 in macrophages (Yu et al., 2019). FTO knockdown induces a decrease of PPAR-γ and STAT1 transcripts through accelerating mRNA degradation mediated by YTHDF and inhibiting M1/M2 activation (Gu et al., 2020). It is reported that the immune cells contributing to IBD progression are predominantly M1 macrophages, neutrophils, and Th cells (de Souza and Fiocchi, 2016; Zhou et al., 2018, 2019). The m6A “writer” protein METTL3 has important roles in reprograming naive T cells for proliferation and differentiation (Li et al., 2017). Deficiency of METTL14, another m6A “writer,” causes induction of native T cells into induced Treg cells (Lu et al., 2020). In this study, we find that m6Acluster2 with a higher expression of m6A regulators was significantly abundant in innate immune cell infiltration, including M0 macrophage, M1 macrophage, and acquired immune cells (native B cells and memory B cells), and m6Acluster1 with a lower expression of m6A regulators was enriched in innate immune cell infiltration, including resting DCs and acquired immune cells (plasma cells), suggesting that m6A methylation modification may upregulate innate immune cells associated with inflammation and inhibit the activation of acquired immune B cells during IBD disease progression. In addition, neutrophils and activated memory CD4 T cells were more highly expressed in CD and UC tissues than in normal tissues, but no significant differences in the T cell types or neutrophils exist between the two m6A modification patterns, indicating that m6A regulators may influence inflammation development primarily by recruiting macrophages. METTL3 deficiency increases the secretion of IL8 and elevates the recruitment of neutrophils (He et al., 2021). In our study, IL8 expression in m6Acluster1 was increased, and expression of IL15, IL16, and IL18 was upregulated in m6Acluster2, suggesting that m6A methylation modification may change mucosal inflammatory factors in IBD. Nevertheless, our data on m6A regulators are confined to bioinformatics analysis, and more research is needed to explore the exact molecular mechanisms by which these disease-related m6A regulators induce immune cell infiltration in IBD.
Next, we identified 29 disease-related hub genes from the key module, which was the most correlated with the m6Acluster phenotype, and found that expression of NUP37, SNRPG, H2AFZ, and ALYREF was significantly upregulated in the IBD (CD and UC) tissues compared with normal tissues. NUP37, a component of the nuclear pore complex, is both a significantly mutated gene and a tumor-destructive gene in various carcinomas (Chen J. et al., 2019; Huang L. et al., 2020). SNRPG belongs to the small nuclear ribonucleoprotein peptide family, which might participate in tumor chemotherapeutic resistance (Lan et al., 2020). H2AFZ is significantly upregulated in hepatocellular carcinoma and is related to poor prognosis (Tang et al., 2020). ALYREF activates the Wnt/β-catenin pathway to promote cell proliferation (Wang J. et al., 2021). However, there are few reports on the association of these genes with IBD or other inflammatory diseases. Our study suggests that these hub genes may be downstream targets of m6A regulators and can be expected to trigger new therapeutic strategies for IBD. However, further experiments are needed to verify their roles in IBD development.
Furthermore, we classified patients into different genomic subtypes according to expression of the 29 obtained m6A phenotype–related hub genes. Genecluster1, with higher expression of m6A phenotype-related hub genes, exhibited higher infiltration levels of M1 macrophages, M0 macrophages, naive B cells, CD4 memory-activated T cells, memory B cells, and activated DCs, whereas genecluster2 was more correlated with resting DCs, M2 macrophages, CD4 memory resting T cells, plasma cells, resting mast cells, and eosinophils. Despite these findings, these hub genes could, at least in part, explain how m6A regulators stimulate the inflammation-associated immune cell infiltration in IBD.
N6-methyladenosine regulators are closely associated with immunotherapy (Yang et al., 2019; Li et al., 2020). For example, inhibiting FTO sensitizes melanoma cells to anti-PD-1 and interferon gamma (IFNγ) treatment (Yang et al., 2019). Knockout of YTHDF1 enhances the therapeutic efficacy of PD-L1 checkpoint blockade in mice (Han et al., 2019). Downregulation of ALKBH5 correlates with a positive response to PD-1 blockade in melanoma patients (Li et al., 2020). In this study, we found that expression levels of all immune checkpoints in genecluster1 with a higher expression of m6A phenotype-related hub genes was significantly increased, suggesting that m6A phenotype–related hub genes might influence immunotherapy. Furthermore, as current treatments are mostly the biological agent anti-TNF (primarily infliximab) for refractory and severe forms of IBD (Koelink et al., 2020), we verified the m6A phenotype–related hub genes in GSE16879 and found that the therapeutic effect of cluster1 with the lower expression of hub genes, including H2AFZ, NUP37, SNRPD1, CPSF3, and RBBP7, was better than that of cluster2. Furthermore, the therapeutic effect in CD was better than that in UC, indicating that IBD with high expression of some m6A phenotype-related hub genes (H2AFZ, NUP37, SNRPD1, CPSF3, and RBBP7) may represent those characterized by resistance to infliximab. Our findings provide new possibilities for enhancing the efficacy of anti-TNF therapy for IBD. However, our study is on bioinformatics and is retrospective in nature, and the predictive value of gene signatures should be further verified.
We identified for the first time the alterations and correlations among m6A modulators, m6A-related immune infiltration, m6A phenotype–related hub genes, and therapeutic response in IBD. Assessing the expression of m6A phenotype–related hub genes may guide the choice of IBD drugs and improve the prediction of therapeutic response to anti-TNF therapy.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The IBD patient data in our study were downloaded from the publicly available data sets in which informed consent were complete.
SH and YC: study design and drafting manuscript. YC and JL: data analysis and statistical analysis. All authors contributed to the article and approved the submitted version.
This work was supported by grants from China Postdoctoral Science Foundation (No. 2020M680145).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.692160/full#supplementary-material
Supplementary Figure 1 | Landscape of m6A regulators in IBD subtypes and consensus clustering and gene co-expression network in different IBD cohort. (A) The expression of 24 m6A regulators between UC and CD tissues. (B) Consensus clustering CDF for k = 2–10 based on m6A regulators’ expression in the GSE111889 cohort. (C) A weighted gene co-expression network is constructed for candidate gene sets. (D) Clustering CDF for k = 2–10 based on the expression of hub genes in the GSE111889 cohort. (E) Consensus clustering CDF for k = 2–10 based on hub genes expression in the GSE16879 cohort. The asterisks represented the statistical P-value (∗P < 0.05).
Arijs, I., De Hertogh, G., Lemaire, K., Quintens, R., Van Lommel, L., Van Steen, K., et al. (2009). Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PLoS One 4:e7984. doi: 10.1371/journal.pone.0007984
Belarif, L., Danger, R., Kermarrec, L., Nerriere-Daguin, V., Pengam, S., Durand, T., et al. (2019). IL-7 receptor influences anti-TNF responsiveness and T cell gut homing in inflammatory bowel disease. J. Clin. Invest. 129, 1910–1925. doi: 10.1172/JCI121668
Bell, J. L., Wachter, K., Muhleck, B., Pazaitis, N., Kohn, M., Lederer, M., et al. (2013). Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell. Mol. Life Sci. 70, 2657–2675. doi: 10.1007/s00018-012-1186-z
Canavan, J. B., Scotta, C., Vossenkamper, A., Goldberg, R., Elder, M. J., Shoval, I., et al. (2016). Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut 65, 584–594. doi: 10.1136/gutjnl-2014-306919
Chen, H., Yao, J., Bao, R., Dong, Y., Zhang, T., Du, Y., et al. (2021). Cross-talk of four types of RNA modification writers defines tumor microenvironment and pharmacogenomic landscape in colorectal cancer. Mol. Cancer 20:29. doi: 10.1186/s12943-021-01322-w
Chen, J., Wo, D., Ma, E., Yan, H., Peng, J., Zhu, W., et al. (2019). Deletion of low-density lipoprotein-related receptor 5 inhibits liver cancer cell proliferation via destabilizing Nucleoporin 37. Cell Commun. Signal. 17, 174. doi: 10.1186/s12964-019-0495-3
Chen, X. Y., Zhang, J., and Zhu, J. S. (2019). The role of m(6)A RNA methylation in human cancer. Mol. Cancer 18:103. doi: 10.1186/s12943-019-1033-z
de Souza, H. S., and Fiocchi, C. (2016). Immunopathogenesis of IBD: current state of the art. Nat. Rev. Gastroenterol. Hepatol. 13, 13–27. doi: 10.1038/nrgastro.2015.186
Deng, X., Su, R., Weng, H., Huang, H., Li, Z., and Chen, J. (2018). RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 28, 507–517. doi: 10.1038/s41422-018-0034-6
Geula, S., Moshitch-Moshkovitz, S., Dominissini, D., Mansour, A. A., Kol, N., Salmon-Divon, M., et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006. doi: 10.1126/science.1261417
Gu, X., Zhang, Y., Li, D., Cai, H., Cai, L., and Xu, Q. (2020). N6-methyladenosine demethylase FTO promotes M1 and M2 macrophage activation. Cell. Signal. 69:109553. doi: 10.1016/j.cellsig.2020.109553
Halfvarson, J., Brislawn, C. J., Lamendella, R., Vazquez-Baeza, Y., Walters, W. A., Bramer, L. M., et al. (2017). Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2:17004. doi: 10.1038/nmicrobiol.2017.4
Han, D., Liu, J., Chen, C., Dong, L., Liu, Y., Chang, R., et al. (2019). Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 566, 270–274. doi: 10.1038/s41586-019-0916-x
Hanniford, D., Ulloa-Morales, A., Karz, A., Berzoti-Coelho, M. G., Moubarak, R. S., Sanchez-Sendra, B., et al. (2020). Epigenetic silencing of CDR1as drives IGF2BP3-mediated melanoma invasion and metastasis. Cancer Cell 37, 55–70.e15. doi: 10.1016/j.ccell.2019.12.007
He, J., Zhou, M., Yin, J., Wan, J., Chu, J., Jia, J., et al. (2021). METTL3 restrains papillary thyroid cancer progression via m(6)A/c-Rel/IL-8-mediated neutrophil infiltration. Mol. Ther. 29, 1821–1837. doi: 10.1016/j.ymthe.2021.01.019
Huang, H., Weng, H., and Chen, J. (2020). The biogenesis and precise control of RNA m(6)A methylation. Trends Genet. 36, 44–52. doi: 10.1016/j.tig.2019.10.011
Huang, H., Weng, H., Sun, W., Qin, X., Shi, H., Wu, H., et al. (2018). Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295. doi: 10.1038/s41556-018-0045-z
Huang, L., Wang, T., Wang, F., Hu, X., Zhan, G., Jin, X., et al. (2020). NUP37 silencing induces inhibition of cell proliferation, G1 phase cell cycle arrest and apoptosis in non-small cell lung cancer cells. Pathol. Res. Pract. 216:152836. doi: 10.1016/j.prp.2020.152836
Huang, S., Wu, Z., Cheng, Y., Wei, W., and Hao, L. (2019). Insulin-like growth factor 2 mRNA binding protein 2 promotes aerobic glycolysis and cell proliferation in pancreatic ductal adenocarcinoma via stabilizing GLUT1 mRNA. Acta Biochim. Biophys. Sin. 51, 743–752. doi: 10.1093/abbs/gmz048
Huang, X., Zhang, H., Guo, X., Zhu, Z., Cai, H., and Kong, X. (2018). Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in cancer. J. Hematol. Oncol. 11:88. doi: 10.1186/s13045-018-0628-y
Huang, Y., Su, R., Sheng, Y., Dong, L., Dong, Z., Xu, H., et al. (2019). Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 35, 677–691.e610. doi: 10.1016/j.ccell.2019.03.006
Jiang, X., Liu, B., Nie, Z., Duan, L., Xiong, Q., Jin, Z., et al. (2021). The role of m6A modification in the biological functions and diseases. Signal. Transduct. Target Ther. 6:74. doi: 10.1038/s41392-020-00450-x
Jin, D., Guo, J., Wu, Y., Yang, L., Wang, X., Du, J., et al. (2020). m(6)A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2-mediated YAP activity in NSCLC. Mol. Cancer 19:40. doi: 10.1186/s12943-020-01161-1
Koelink, P. J., Bloemendaal, F. M., Li, B., Westera, L., Vogels, E. W. M., van Roest, M., et al. (2020). Anti-TNF therapy in IBD exerts its therapeutic effect through macrophage IL-10 signalling. Gut 69, 1053–1063. doi: 10.1136/gutjnl-2019-318264
Lan, Q., Liu, P. Y., Haase, J., Bell, J. L., Huttelmaier, S., and Liu, T. (2019). The critical role of RNA m(6)A methylation in cancer. Cancer Res. 79, 1285–1292. doi: 10.1158/0008-5472.CAN-18-2965
Lan, Y., Lou, J., Hu, J., Yu, Z., Lyu, W., and Zhang, B. (2020). Downregulation of SNRPG induces cell cycle arrest and sensitizes human glioblastoma cells to temozolomide by targeting Myc through a p53-dependent signaling pathway. Cancer Biol. Med. 17, 112–131. doi: 10.20892/j.issn.2095-3941.2019.0164
Li, H. B., Tong, J., Zhu, S., Batista, P. J., Duffy, E. E., Zhao, J., et al. (2017). m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 548, 338–342. doi: 10.1038/nature23450
Li, N., Kang, Y., Wang, L., Huff, S., Tang, R., Hui, H., et al. (2020). ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc. Natl. Acad. Sci. U.S.A. 117, 20159–20170. doi: 10.1073/pnas.1918986117
Liu, Y., Liu, Z., Tang, H., Shen, Y., Gong, Z., Xie, N., et al. (2019). The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation of STAT1 mRNA. Am. J. Physiol. Cell Physiol. 317, C762–C775. doi: 10.1152/ajpcell.00212.2019
Lloyd-Price, J., Arze, C., Ananthakrishnan, A. N., Schirmer, M., Avila-Pacheco, J., Poon, T. W., et al. (2019). Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. doi: 10.1038/s41586-019-1237-9
Lu, T. X., Zheng, Z., Zhang, L., Sun, H. L., Bissonnette, M., Huang, H., et al. (2020). A new model of spontaneous colitis in mice induced by deletion of an RNA m(6)A methyltransferase component METTL14 in T cells. Cell. Mol. Gastroenterol. Hepatol. 10, 747–761. doi: 10.1016/j.jcmgh.2020.07.001
Neurath, M. (2017). Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 14:688. doi: 10.1038/nrgastro.2017.138
Nielsen, J., Christiansen, J., Lykke-Andersen, J., Johnsen, A. H., Wewer, U. M., and Nielsen, F. C. (1999). A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol. Cell. Biol. 19, 1262–1270. doi: 10.1128/MCB.19.2.1262
Qian, J. Y., Gao, J., Sun, X., Cao, M. D., Shi, L., Xia, T. S., et al. (2019). KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene 38, 6123–6141. doi: 10.1038/s41388-019-0861-z
Shen, C., Sheng, Y., Zhu, A. C., Robinson, S., Jiang, X., Dong, L., et al. (2020). RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-renewal in acute myeloid leukemia. Cell Stem Cell 27, 64–80.e69. doi: 10.1016/j.stem.2020.04.009
Su, R., Dong, L., Li, Y., Gao, M., Han, L., Wunderlich, M., et al. (2020). Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell 38, 79–96.e11. doi: 10.1016/j.ccell.2020.04.017
Tang, S., Huang, X., Wang, X., Zhou, X., Huang, H., Qin, L., et al. (2020). Vital and distinct roles of H2A.Z isoforms in hepatocellular carcinoma. Onco Targets Ther. 13, 4319–4337. doi: 10.2147/OTT.S243823
Torres, J., Mehandru, S., Colombel, J. F., and Peyrin-Biroulet, L. (2017). Crohn’s disease. Lancet 389, 1741–1755. doi: 10.1016/S0140-6736(16)31711-1
Ungaro, R., Mehandru, S., Allen, P. B., Peyrin-Biroulet, L., and Colombel, J. F. (2017). Ulcerative colitis. Lancet 389, 1756–1770. doi: 10.1016/S0140-6736(16)32126-2
Wang, H., Hu, X., Huang, M., Liu, J., Gu, Y., Ma, L., et al. (2019). Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat. Commun. 10:1898. doi: 10.1038/s41467-019-09903-6
Wang, J., Li, Y., Xu, B., Dong, J., Zhao, H., Zhao, D., et al. (2021). ALYREF drives cancer cell proliferation through an ALYREF-MYC positive feedback loop in glioblastoma. Onco Targets Ther. 14, 145–155. doi: 10.2147/OTT.S286408
Wang, J., Yan, S., Lu, H., Wang, S., and Xu, D. (2019). METTL3 attenuates LPS-induced inflammatory response in macrophages via NF-kappaB signaling pathway. Mediators Inflamm. 2019:3120391. doi: 10.1155/2019/3120391
Wang, X. K., Zhang, Y. W., Wang, C. M., Li, B., Zhang, T. Z., Zhou, W. J., et al. (2021). METTL16 promotes cell proliferation by up-regulating cyclin D1 expression in gastric cancer. J. Cell. Mol. Med. 25, 6602–6617. doi: 10.1111/jcmm.16664
Yang, S., Wei, J., Cui, Y. H., Park, G., Shah, P., Deng, Y., et al. (2019). m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 10:2782. doi: 10.1038/s41467-019-10669-0
Yu, R., Li, Q., Feng, Z., Cai, L., and Xu, Q. (2019). m6A reader YTHDF2 regulates LPS-induced inflammatory response. Int. J. Mol. Sci. 20:1323. doi: 10.3390/ijms20061323
Zheng, Q., Hou, J., Zhou, Y., Li, Z., and Cao, X. (2017). The RNA helicase DDX46 inhibits innate immunity by entrapping m(6)A-demethylated antiviral transcripts in the nucleus. Nat. Immunol. 18, 1094–1103. doi: 10.1038/ni.3830
Zhou, G., Yu, L., Fang, L., Yang, W., Yu, T., Miao, Y., et al. (2018). CD177(+) neutrophils as functionally activated neutrophils negatively regulate IBD. Gut 67, 1052–1063. doi: 10.1136/gutjnl-2016-313535
Keywords: m6A modification, IBD, immune infiltration, immunotherapy, anti-TNF response
Citation: Chen Y, Lei J and He S (2021) m6A Modification Mediates Mucosal Immune Microenvironment and Therapeutic Response in Inflammatory Bowel Disease. Front. Cell Dev. Biol. 9:692160. doi: 10.3389/fcell.2021.692160
Received: 07 April 2021; Accepted: 12 July 2021;
Published: 06 August 2021.
Edited by:
Jia Meng, Xi’an Jiaotong-Liverpool University, ChinaCopyright © 2021 Chen, Lei and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongyu Chen, NjE3NTg1MjE4QHFxLmNvbQ==; Song He, aGVkb2N0b3I2NUBjcW11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.