 Ming He
Ming He Wenxing Lv
Wenxing Lv Yu Rao
Yu Rao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 22 June 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.685106
This article is part of the Research Topic Aptamer-Based Structural Biology, Computational Modelling, Translational Research & Drug Discovery View all 8 articles
Proteolysis targeting chimeras (PROTAC) represents a new type of small molecule induced protein degradation technology that has emerged in recent years. PROTAC uses bifunctional small molecules to induce ubiquitination of target proteins and utilizes intracellular proteasomes for chemical knockdown. It complements the gene editing and RNA interference for protein knockdown. Compared with small molecule inhibitors, PROTAC has shown great advantages in overcoming tumor resistance, affecting the non-enzymatic function of target proteins, degrading undruggable targets, and providing new rapid and reversible chemical knockout tools. At the same time, its challenges and problems also need to be resolved as a fast-developing newchemical biology technology.
In recent years, targeted protein degradation has attracted much attention from the pharmaceutical industry, among which the focus is undoubtedly the proteolysis targeting chimeras (PROTAC) technology induced by small molecules. PROTAC is a new technology that uses small molecules to induce degradation of target proteins to regulate the protein level. As a new drug development strategy, its model of action is different from that of traditional small molecule inhibitors and it has a huge potentiality in overcoming drug resistance and the traditional “unable to medicine” targets (Toure and Crews, 2016; Lai and Crews, 2017; Ottis and Crews, 2017; Zou et al., 2019; Burslem and Crews, 2020). The ubiquitin-proteasome system in cell (Amm et al., 2014) plays an important role in the process of protein degradation. The system includes two main stages in the degradation of proteins. The first stage is the interaction between ubiquitin and the protein substrate to form a protein substrate-ubiquitin complex. The second stage is the degradation of the protein substrate by the proteasome, followed by the release of ubiquitin. PROTACs mainly play a related role in the first stage in the process of inducing protein degradation. PROTACs induce the target protein and E3 ubiquitin ligase to form a ternary complex by utilizing a bifunctional small molecule, which can simultaneously bind the target protein and E3 ubiquitin ligase. The complex makes the target protein recognized by E3 ubiquitin ligase and then ubiquitinated, finally recognized and degraded by the proteasome in cells (Figure 1).
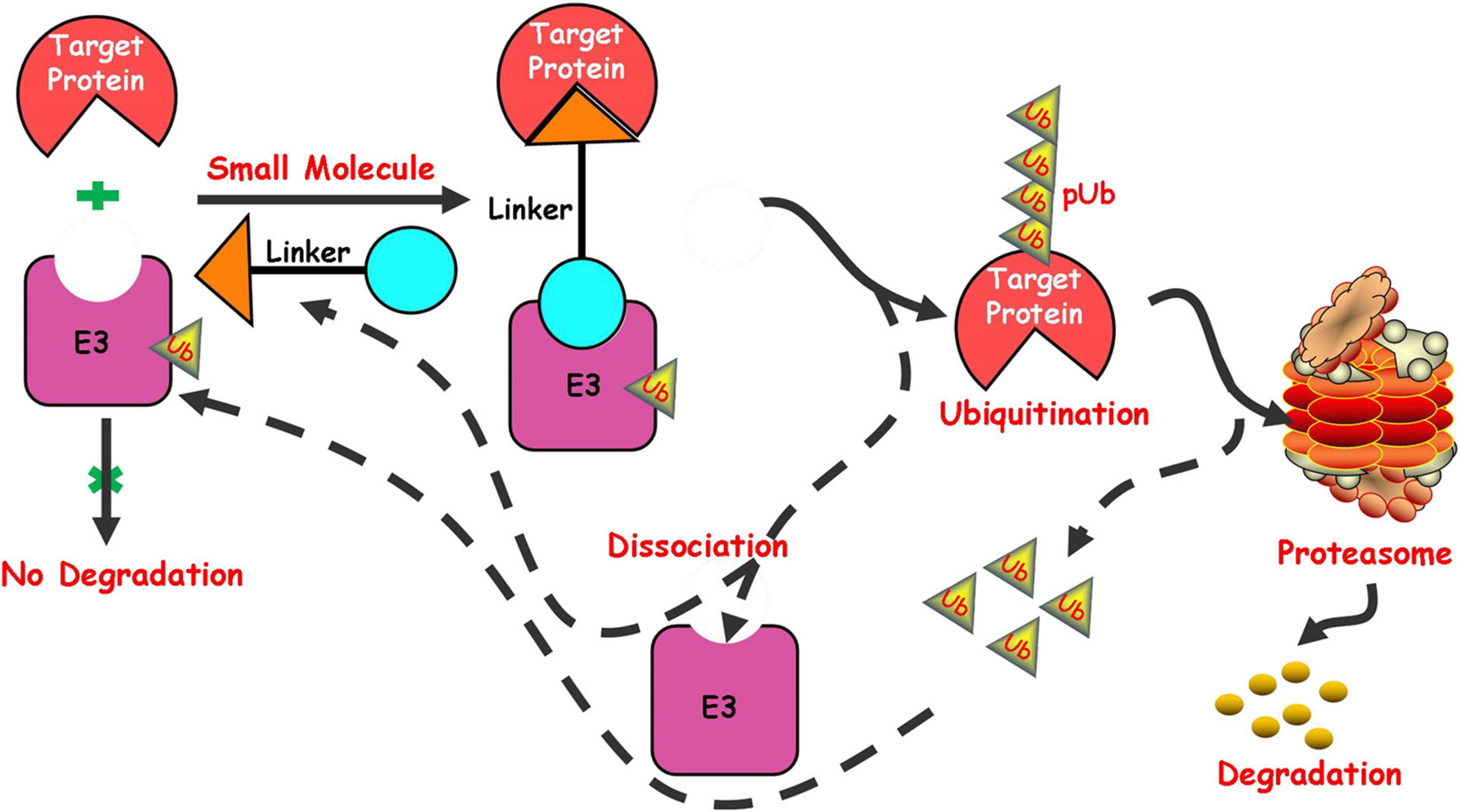
Figure 1. Schematic depiction of the small molecule induced protein degradation.
At present, the regulation of the level of the target protein in cells can be intervened from three levels. Firstly, the regulation of the target protein at the gene level-the gene editing technology (CRISPR-Cas 9) that has emerged in recent years (Ebrahimi and Hashemi, 2020). Although having the advantages of high precision and strong versatility, it cannot dynamically regulate the target protein and is irreversible so that it has a potential genetic compensation effect. Secondly, the regulation of the target protein at the RNA level-RNA interference technology shows great power and charm in inducing the specific target protein degradation to regulate the target protein level. However, this technology is less efficient and not suitable for the study of relatively stable proteins (Agrawal et al., 2003; Mello and Conte, 2004). Finally, the regulation of the target protein at the protein level-the chemical knockdown of the target protein is carried out by PROTAC. This technology has the advantages of high efficiency and reversibility, and can carry out a catalytic cycle process so that PROTAC can play a role at low doses (Burslem and Crews, 2020; Figure 2). Since PROTAC can degrade the entire protein, it can affect the non-enzymatic function of the protein. Unlike traditional small molecule inhibitors that compete with active sites and occupy the pharmacological model of action, PROTAC exerts its effect through the repeated and iterative model of action to induce the target protein degradation. So in the case of target protein mutation or low expression, PROTAC will be better tolerated than traditional small molecule inhibitors. As a new and promising technology, PROTAC shows obvious advantages compared with traditional small molecule, CRISPR and other technologies (Figure 3). Compared with small molecules, PROTAC not only has the advantages of small molecules, but also makes up for its shortcomings in target protein with scaffolding function and catalytic MOA. In addition, compared with CRISPR, monoclonal antibody and siRNA technology, PROTAC has the obvious advantage of oral bioavailability.
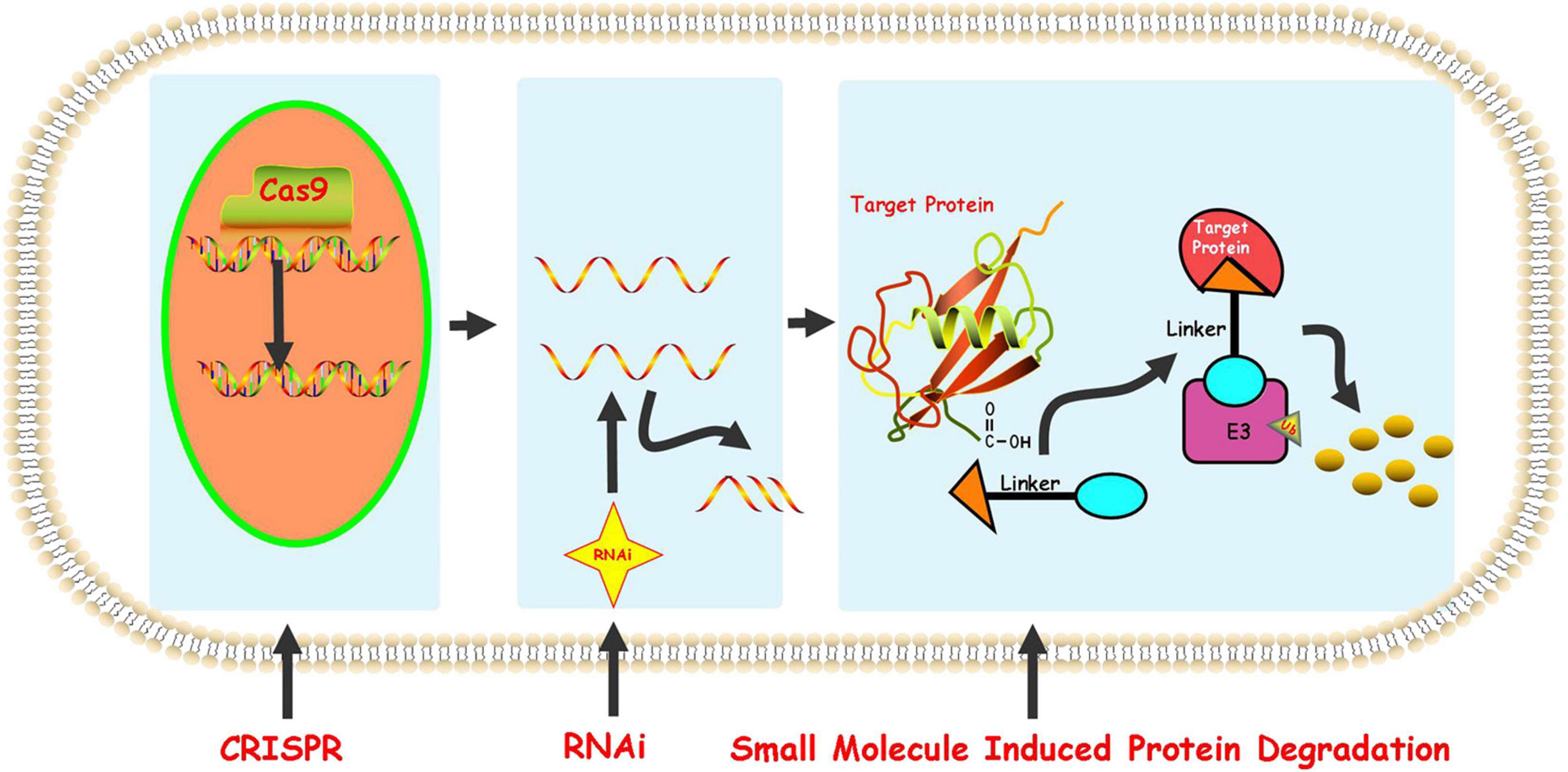
Figure 2. Protein knockdown strategies.
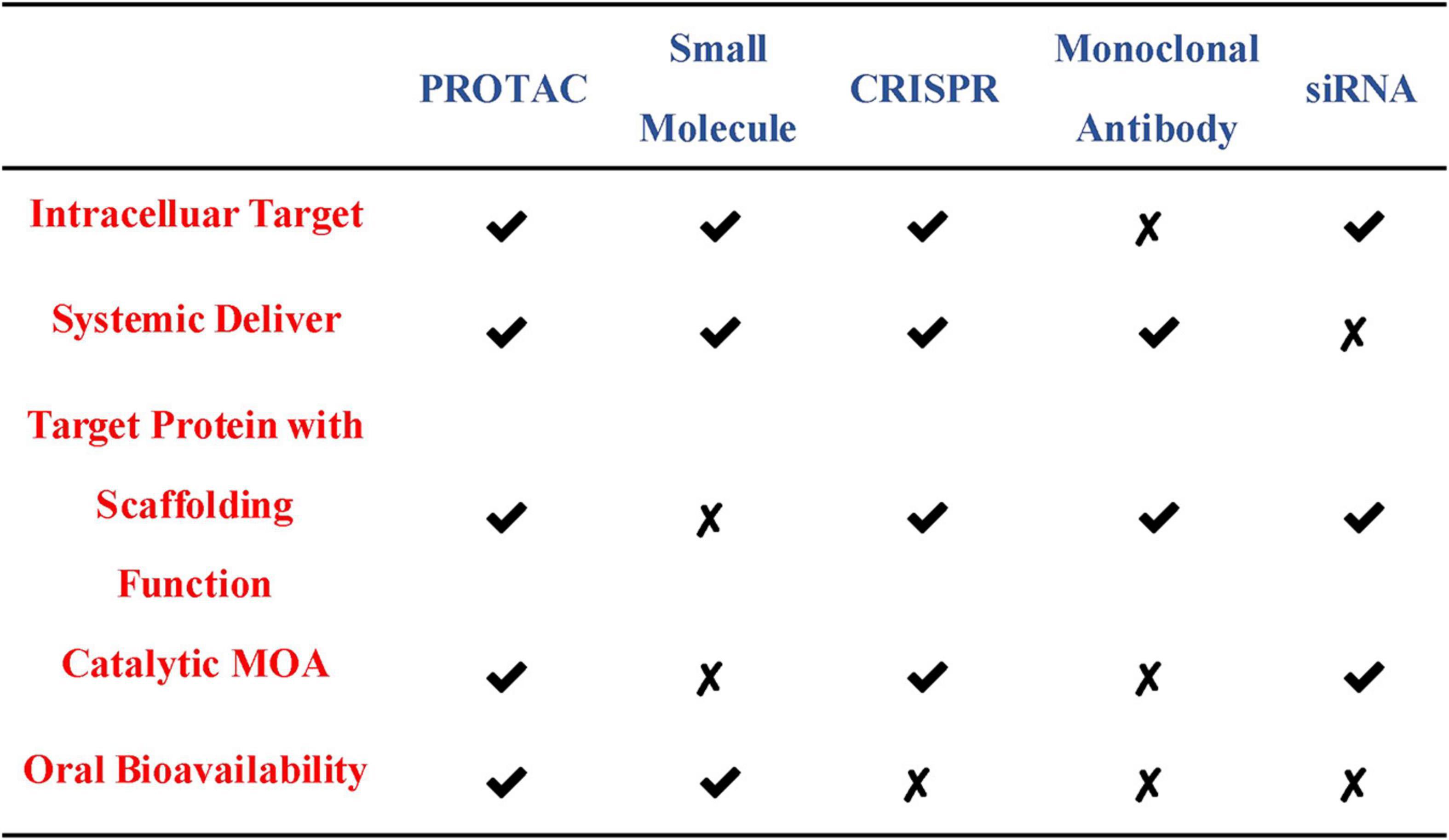
Figure 3. Comparisons of PROTAC with other therapeutic modalities.
The concept of PROTAC was first proposed by Craig Crews in Yale University and Raymond Deshaies in California Institute of Technology in 2001. Their peptide-based Protac-1 (1, Figure 4) can induce methionine aminopeptidase II (MetAP-2) degradation (Sakamoto et al., 2001). Due to the shortcomings of peptide ligand, the degradation efficiency of PROTAC was low and it developed slowly. Until 2008, Crews’s group used MDM2-p53 PPI inhibitor Nutlin-3 (Patton et al., 2006; Tabe et al., 2007; 2, Figure 4) as E3 ligand to design the first PROTAC based on non-polypeptide small molecule (3, Figure 4), and successfully achieved the effective degradation of androgen receptor (Schneekloth et al., 2008). Although the molecule had better transmembrane properties, the degradation efficiency of the androgen receptor protein was relatively average. Since then, a series of new E3 ligase ligands have been reported, which has provided an important research basis for the rapid development of PROTAC.
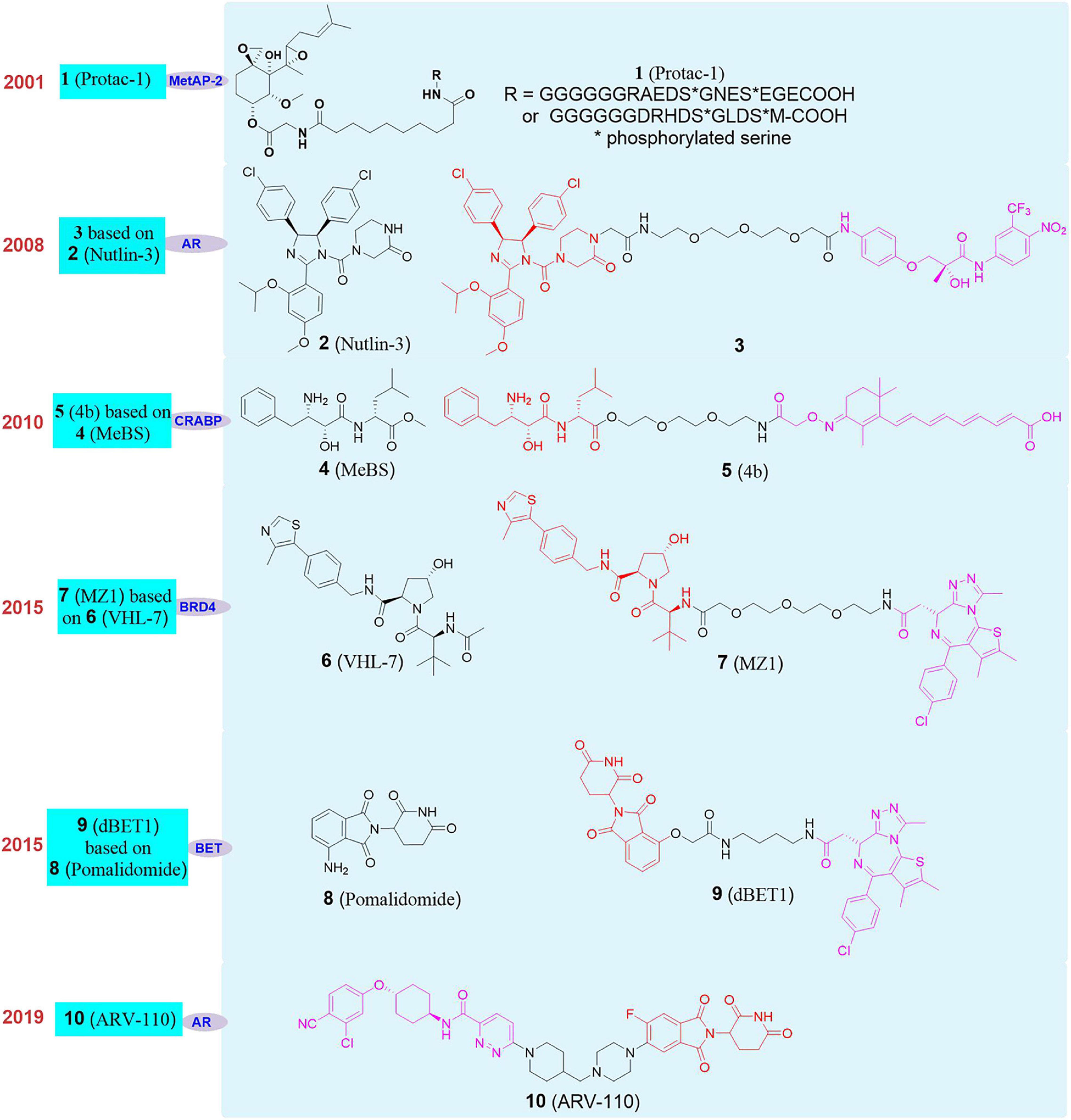
Figure 4. The development and structures of representative E3 ligands and PROTACs.
It was reported that Bestatin methylester (4, Figure 4) can bind to the cellular inhibitor of apoptosis protein 1, and induced cIAP1 to be self-ubiquitinated and degraded by the proteasome. Therefore, cIAP1 ligand was also used as an E3 ligase ligand in the design of PROTAC. Based on these researches, Hashimoto’s group reported the first degrader based on cIAP1 ligand (Itoh et al., 2010; 5, Figure 4) for the degradation of cellular retinol and retinoic acid-binding proteins (CRABP-I and II). Subsequently, Ciulli’s group developed a series of ligands for the E3 ligase VHL (Von Hippel-Lindau), and designed the first degrader MZ1 (Zengerle et al., 2015; 7, Figure 4) targeting degradation of bromodomain-containing protein 4 (BRD4) based on VHl-7 (Galdeano et al., 2014; 6, Figure 4). At the same time, studies have found that CRBN (cereblon) is the target of immunomodulators such as thalidomide, pomalidomide, and lenalidomide. Mechanism studies have shown that combined with CRBN these drugs can recruit such as Ikaros, Aiolos, Casein kinase 1A1 (CK1α) and other substrate proteins, which trigger the ubiquitination of these proteins, then recognized and degraded by the proteasome (Fischer et al., 2014). Then, dBET1 (Winter et al., 2015; 9, Figure 4) as the first degrader based on Pomalidomide (8, Figure 4) as the E3 ligand was successfully used to degrade BET (bromodomain and extra-terminal) protein. Since then, CRBN had gradually become the most widely used E3 ubiquitin ligase in PROTAC. On the basis of the above researches, researchers have designed and synthesized a variety of PROTAC molecules and achieved the degradation of different types of target proteins in recent years. These PROTAC molecules have been used in the treatment of various diseases, including cancer, viral infections, immune disorders, and neurodegenerative diseases (Sun X. Y. et al., 2019; Zhao Q. Y. et al., 2019; Gao et al., 2020a; Sun and Rao, 2020). At present, PROTAC is developing rapidly in various fields, especially in the field of drug development. Arvinas recently announced the latest clinical data of ARV-110 in prostate cancer patients and phase I positive efficacy data of ARV-471 (10, Figure 4) in breast cancer patients. ARV-110 is the first oral degrader and the first PROTAC drug in clinical trial that induced the degradation of androgen receptor (AR) (Neklesa et al., 2019). The data shows that ARV-110 is safe and effective in patients with metastatic castration resistant prostate cancer (mCRPC). ARV-471 is an another oral protein degrader developed by Arvinas based on PROTAC targeting estrogen receptor (ER). Its phase I clinical trial is mainly for adult patients with locally advanced or metastatic ER+/HER2– breast cancer. The results show that ARV-471 has great potential in its safety and tolerability, which is also the milestone in the transformation of PROTAC into a new treatment pattern.
The emergence of PROTAC not only opened a new chapter for new drug development, but also brought unprecedented opportunities to industry and academia. Of course, we also needed to be soberly aware that we are bound to face many new problems and challenges in the process of the rise of any new technology. It was in overcoming all kinds of difficulties that new technologies can be developed and improved, thus broadening the range of applications and influences. In recent years, our group had carried out some exploratory works in the field of small molecule induced protein degradation. The following parts will briefly summarize the main development directions and challenges of PROTAC based on the research work of our group.
In addition to traditional chemotherapy treatments for cancer, kinase inhibitors has developed vigorously in the past two decades. These kinase inhibitors have achieved excellent clinical effects, greatly improving the quality of life and effectively prolonging the survival period of cancer patients. However, although kinase inhibitors are very effective, patients often develop resistance to kinase inhibitors at different times, which causes disease relapse. Therefore, drug resistance caused by targeted tumor therapy is a major problem faced by cancer research.
For example, Bruton’s tyrosine kinase (BTK) covalent inhibitor Ibrutinib (Pan et al., 2007; 11, Figure 5) is used as a treatment drug for mantle cell lymphoma (Wang et al., 2013). But when it is used in clinical, Ibrutinib will induce C481S mutation of BTK and form drug resistance (Chiron et al., 2014; Cheng et al., 2015).
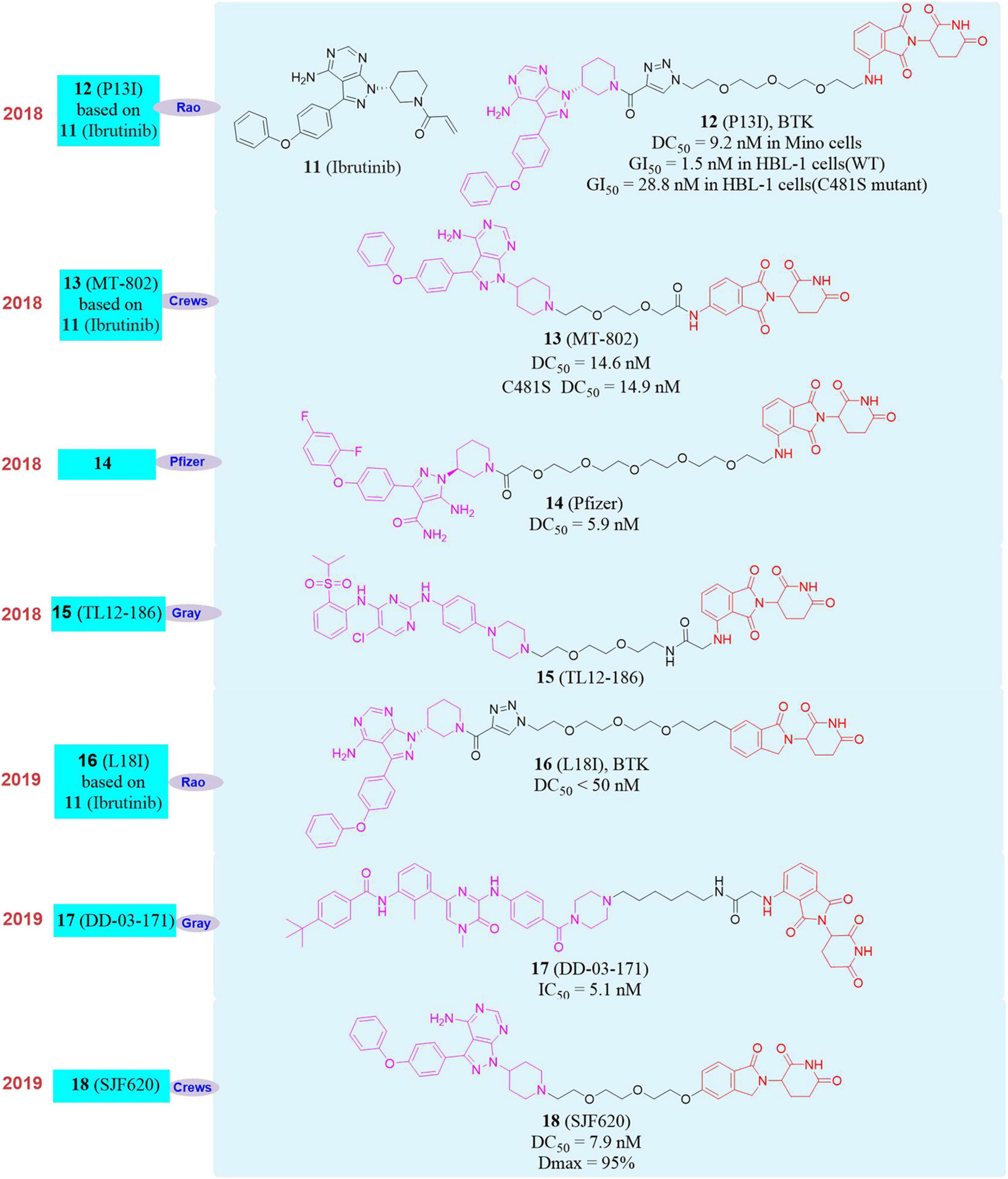
Figure 5. Structures of BTK inhibitor and degraders.
In 2018, our group reported the first new protein degrader P13I (Sun Y. H. et al., 2018; 12, Figure 5), which can be used in a variety of B cell malignancies (mantle cell lymphoma, diffuse large B cell lymphoma, multiple myeloma) with drug concentration less than 10 nM to efficiently degrade BTK protein. The inhibitory activity of P13I on BTK-dependent wild-type human B-cell lymphoma (HBL-1) cells was equivalent to that of Ibrutinib. More importantly, P13I can induce the degradation of C481S mutant BTK protein and overcome the resistance of B-cell malignant tumor BTK kinase to Ibrutinib caused by the C481S mutation. And the selectivity of P13I was better than that of Ibrutinib. Even at high concentrations, it had no degradation or inhibitory activity for the targets that caused serious side effects of Ibrutinib (EGFR, ITK, TEC, etc.), which means that the BTK protein degrader will have better security.
Almost at the same time, Crews’s group reported another BTK degrader MT-802 based on Ibrutinib (Buhimschi et al., 2018; 13, Figure 5). MT-802 can effectively induce the degradation of wild-type BTK protein with DC50 = 14.6 nM as well as the C481S mutant BTK protein with DC50 = 14.9 nM. And MT-802 can reduce phosphorylation of BTK protein in tumor cells isolated from patients with C481S mutant chronic lymphocytic leukemia (CLL), while Ibrutinib cannot. Pfizer also reported the PROTAC molecule 14 (Zorba et al., 2018; Figure 5) targeting the degradation of BTK in 2018. It can efficiently degrade BTK protein after 24 h of treatment of Ramos cells, and the DC50 is 5.9 nM. They found that the molecule can effectively degrade the BTK protein in the rat lung and spleen in vivo activity evaluation, but this research did not report the degradation of the mutant BTK protein by the degrader. In the same year, Nathanael S. Gray’s group reported a multi-target degrader TL12-186 (Huang et al., 2018; 15, Figure 5). They found that TL12-186 can down-regulate the expression levels of 28 kinases through quantitative proteomics research, including BTK.
In 2019, our group constructed a new framework of PROTAC molecule L18I (16, Figure 5) to target the degradation of BTK protein, which can efficiently induce multiple mutation types (C481S/T/A/G/W) BTK protein degradation in transfected HeLa cells at drug concentration less than 50 nM (Sun Y. H. et al., 2019). And more importantly, L18I can cause the decrease of BTK protein level in mutant diffuse large B-cell lymphoma (DLBCL) tumor, thereby effectively inhibiting the growth of the tumor in vivo and overcoming the tumor resistance to Ibrutinib caused by the C481 mutation of BTK. In the same year, Nathanael S. Gray’s group reported the PROTAC molecule DD-03-171 (Dobrovolsky et al., 2019; 17, Figure 5) that induced the degradation of BTK. DD-03-171 had an effect on mantle cell lymphoma (MCL) and showed a stronger anti-proliferation inhibitory effect with IC50 was 5.1 nM in vitro. DD-03-171 was also effective for mouse xenograft models of patient-derived tumor cells. At the same time, Crews’s group reported degrader SJF620 (18, Figure 5) based on MT-802 that can induce the degradation of BTK and had better pharmacokinetic properties. Compared with the MT-802 molecule, SJF620 had a longer half-life, the plasma clearance rate per unit time was lower and the absorption was better (Figueroa et al., 2020).
Cyclin dependent kinase 4/6 (CDK4/6), an important part of the cyclin family, plays a regulatory role in the transition from the G1 phase to the S phase in the cell cycle (Vermeulen et al., 2003). There are mutations or over-activation of CDK4/6 in a variety of cancer tissues (Otto and Sicinski, 2017), so CDK4/6 has always been regarded as an important target for drug development (Niesvizky et al., 2015; Tadesse et al., 2015). Numerous CDK6 inhibitors have been approved for clinical trials, among which Palbociclib (21, Figure 6) has been approved for the treatment of estrogen receptor-positive breast cancer patients (Leonard et al., 2012; Barton et al., 2013). However, because CDK6 point mutations may weaken drug binding affinity and form drug resistance (Yang et al., 2017; Li et al., 2018), the development of traditional small molecule inhibitors of CDK6 is extremely difficult. Therefore, new chemical biological methods to develop a small molecule drug targeting CDK6 has become a practical strategy for the treatment of malignant tumors.
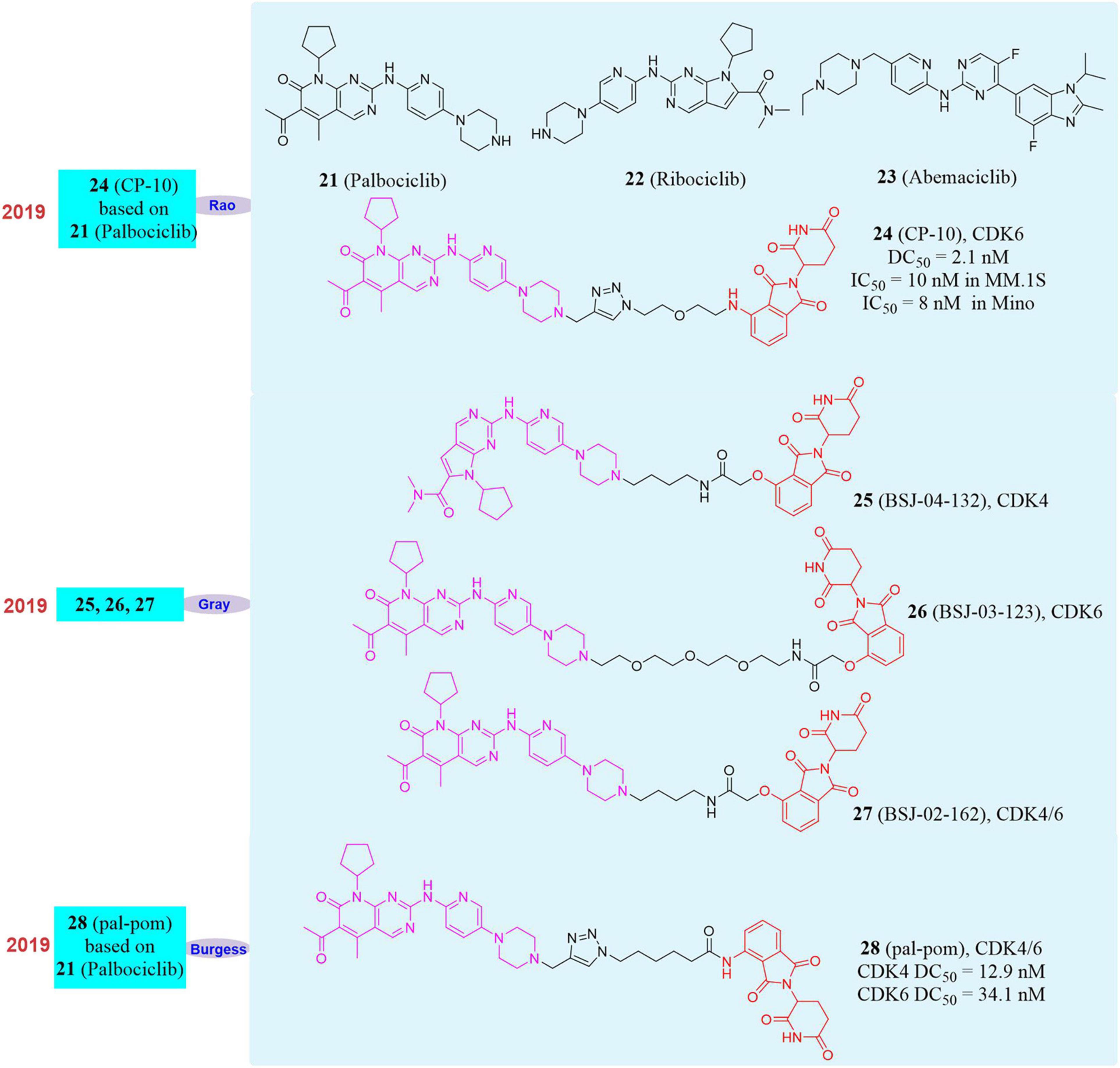
Figure 6. Structures of CDK4/6 inhibitors and degraders.
Until now, three FDA-approved CDK4/6 inhibitors Palbociclib, Ribociclib (22, Figure 6) and Abemaciclib (23, Figure 6) have strong affinity to CDK6, but the binding models are different (Su et al., 2019). Based on these inhibitors, our group developed a series of small molecules degraders. Subsequently, the activity of all small molecules degrading CDK6 in vitro was evaluated. The compound CP-10 (24, Figure 6) was found to have the best CDK6 degradation activity. In human glioblastoma U251 cells, the degradation of CDK6 induced by CP-10 (DC50 = 2.1 nM) was nearly 72% at 10 nM and 89% at 100 nM. The degradation of CDK4 induced by CP-10 was much weaker than that of CDK6 (DC50 was approximately 50–80 times more than that of CDK6). It was found that the degradation effect was dependent on the length of the linker during the evaluation of CDK6 degradation activity. PROTAC with a shorter linker had a higher degradation ability, which mean that these shorter molecules had a better ability to recruit CRBN to CDK6. At the same time, CP-10 can also induce the degradation of CDK6D163G and CDK6S178P mutations. Compared with the wild-type, the degradation of CDK6D163G was slightly weakened, but still remarkable, indicating that although the affinity of Palbociclib in the CP-10 molecule was destroyed due to the mutation of the binding site, it was sufficient to induce CDK6 degradation. These data indicated that small molecules induce CDK4/6 degradation had good application and potential in overcoming Palbociclib resistance. In addition, CP-10 (IC50 = 10 nM) showed a better inhibitory effect than Palbociclib (IC50 = 200 nM) in multiple myeloma cells (MM.1S cells), and the anti-proliferative activity of CP-10 (IC50 = 8 nM) was also better than that of Palbociclib (IC50 = 45 nM) in mantle cell lymphoma cells (Mino cells), and it had comparable activity in leukemia cells. Interestingly, we found that the CDK6 protein was only degraded when PROTAC recruits CRBN, but not when the other three E3 ligase (VHL/MDM2/cIAP) ligands were used.
At the same time, Gray’s group had successively reported different degraders for CDK4, CDK6, and CDK4/6. They used Ribociclib, Palbociclib, and pomalidomide as ligands to achieve the selective degradation of CDK4/6 by changing the type of linker (Brand et al., 2019; Jiang et al., 2019). BSJ-04-132 (25, Figure 6) can selectively induce CDK4 degradation, BSJ-03-123 (26, Figure 6) can selectively induce CDK6 degradation, and BSJ-02-162 (27, Figure 6) can simultaneously induce CDK4/6 degradation. Compared with CDK4/6 inhibitors, these degraders can show stronger protein degradation ability at 100 nM and better anti-tumor cell proliferation activity than inhibitors. In addition, the degrader BSJ-02-162 in the human mantle cell lymphoma cell line (Granta-519) may cause significant degradation of CDK4/6 and at the same time induce tumor cell G1 cell cycle arrest. Burgess’s group also reported the CDK4/6 degrader pal-pom (28, Figure 6) which also used Palbociclib and pomalidomide as ligands (Zhao and Burgess, 2019), and its difference to CP-10 was only on the linker. They found that the compound pal-pom showed better degradation activity to CDK4 than that to CDK6, and its DC50 to CDK4 and CDK6 were 12.9 and 34.1 nM, respectively. Subsequently, the research group tested its anti-tumor cell growth activity and found that the degrader had a poor tumor suppressing activity when the IC50 was at 10–50 μM in MDA-MB-231 cells.
The BCR gene is located on chromosome 22 and the normal BCR gene product is cytosolic phosphoprotein. ABL is a proto-oncogene located on chromosome 9 and the gene product is a non-receptor tyrosine protein kinase, which plays an important role in cell differentiation and cell cycle regulation in normal cells (Talpaz et al., 2006). The chromosomal translocation of t(9; 22) (q34; q11) will lead to the formation of BCR-ABL fusion gene and the gene product is BCR-ABL fusion protein. Its expression leads to the activation of ABL tyrosine kinase, changes of the cell’s tyrosine protein level and capacity of actin binding, which disrupts the normal signal transduction pathway and inhibits the occurrence of apoptosis (Pophali and Patnaik, 2016). Positive (Ph+) acute lymphoblastic leukemia (ALL) and more than 95% of chronic myelogenous leukemia (CML) patients have the BCR-ABL fusion gene. The current new drug for BCR-ABL tyrosine kinase treatment of CML is still ATP competitive inhibitors (Yang and Fu, 2015). Imatinib (29, Figure 7), as the first tyrosine kinase inhibitor (TKI) and the first-generation ABL inhibitor, has achieved significant clinical effects and is used as a first-line treatment for CML patients. Although a model of targeted cancer therapy, Imatinib is ineffective for about 40% patients due to intolerance and drug resistance because of BCR-ABL mutation, especially the T315I mutation, which makes patients form more serious resistance. The advent of the second-generation ABL inhibitors Nilotinib (30, Figure 7), Dasatinib (31, Figure 7) and Bosutinib (32, Figure 7) and the third-generation ABL inhibitors Ponatinib (33, Figure 7) have provided multiple treatment options to patients with mutations. However, the new TKI can not inhibit all resistant mutants and will induce such side effects as vascular disease, which severely limits their clinical use. Ponatinib, as the only drug that targets the BCR-ABL T315I mutation, was temporarily delisted in 2013 due to its serious vascular adverse events, and was later approved for use in the revised indication (Lu et al., 2015). So far, there are no new drugs approved for targeted therapy of T315I mutants, so the design and development of BCR-ABL degrader based on PROTAC is of great significance.
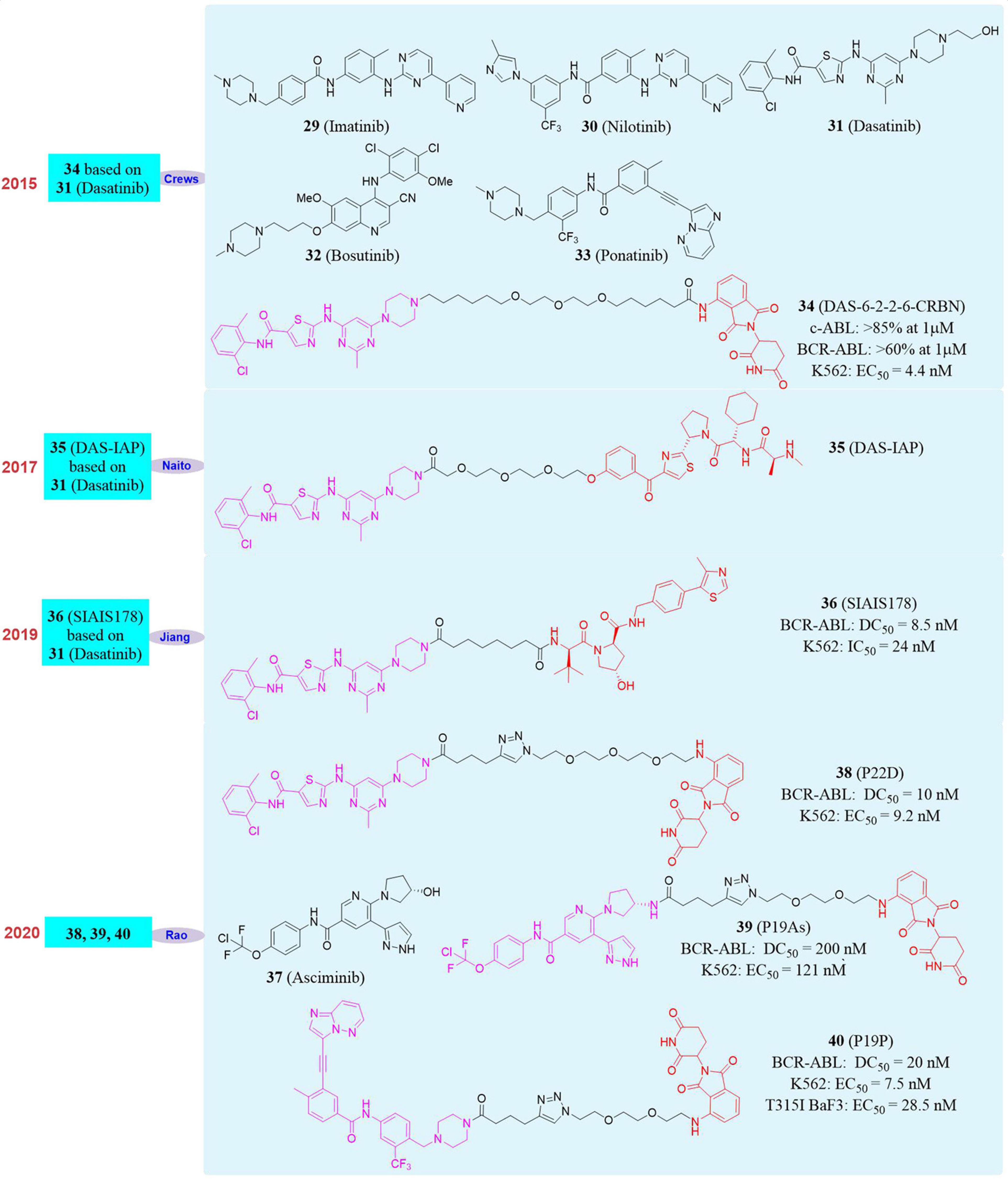
Figure 7. Structures of BCR-ABL inhibitors and degraders.
In 2016, Crews’s group developed the first BCR-ABL degrader (Lai et al., 2016). They constructed the PROTAC molecule DAS-6-2-2-6-CRBN (34, Figure 7) based on CRBN and Dasatinib to induce c-ABL degradation. After evaluation, DAS-6-2-2-6-CRBN derived from Dasatinib caused the degradation of two types of ABL proteins. The degradation effects were c-ABL (>85% at 1 μM) and BCR-ABL (>60% at 1 μM). At the same time, they also found that DAS-6-2-2-6-CRBN had a significant inhibitory effect on the growth of K562 cells, with an EC50 of 4.4 nM. Subsequently, in 2017, Naito’s group reported the second Dasatinib-derived BCR-ABL PROTAC molecule DAS-IAP (35, Figure 7), which had good activity in inhibiting the growth of CML cells and sustaining anti-proliferation effects (Shimokawa et al., 2017). In 2019, Biao Jiang’s group reported the first PROTAC molecule SIAIS178 (36, Figure 7) based on VHL and Dasatinib. The SIAIS178 had good selectivity, and its DC50 to BCR-ABL was 8.5 nM. It showed good anti-proliferative activity on K562 cells with IC50 of 24 nM, and induced in vivo regression of K562 xenograft tumors (Zhao Q. J. et al., 2019). Although the above studies have obtained degraders with obvious degradation effects and excellent cytostatic activity, they had no degradation effect on mutant BCR-ABL. This has become the biggest disadvantage, thus restricting their further use. Therefore, the development of degrader that can induce the degradation of wild-type and mutants BCR-ABL is particularly important.
In 2020, our group used four BCR-ABL inhibitors Imatinib, Dasatinib, Asciminib (37, Figure 7) and Ponatinib as target molecules (Yang Y. Q. et al., 2020). Due to the fact that they had different binding sites and binding models with BCR-ABL, they were used separately to design and synthesize a series of small degraders. Subsequently, the activity of all small degraders to induce BCR-ABL degradation in vitro was evaluated. It was found that the degraders designed with Imatinib did not induce BCR-ABL degradation, and the degraders designed based on the other three BCR-ABL inhibitors can induce BCR-ABL degradation. Especially the compound P22D (38, Figure 7, DC50 = 10 nM) designed based on Dasatinib, the compound P19As (39, Figure 7, DC50 = 200 nM) designed based on Asciminib, the compound P19P (40, Figure 7, DC50 = 20 nM) designed based on Ponatinib performed well, and also showed good cytostatic activity. In K562 cells, its anti-proliferative activity was P22D (EC50 = 9.2 nM) and P19P (EC50 = 7.5 nM), which has comparable cell proliferation activity to that of DAS-6-2-2-6-CRBN (EC50 = 8.8 nM) developed by Crews’s group. Subsequently, our group also tested the degradation effect of these BCR-ABL degraders on mutant BCR-ABL and the anti-proliferation effect on mutant cell lines. It was found that compound P19P has the best degradation effect on T315I mutant BCR-ABL. However, its degradation activity was significantly weaker than that of wild-type BCR-ABL. And P19P had better anti-proliferative activity against T315I mutant BaF3 cell line, with an EC50 of 28.5 nM. In addition to T315I, P19P can also induce the degradation of BCR-ABL with V468F and other mutants (such as E255K and H396R) in transfected HeLa cells. At the same time, it was also proved that these degraders have less side effects on the cardiovascular system and have excellent kinase selectivity.
The BRAF gene is responsible for encoding the RAF kinase protein that transmits cell signals. This protein is part of the RAS-RAF-MEK-ERK signaling pathway (the MAPK/ERK pathway) (Samatar and Poulikakos, 2014). BRAF is one of the most important proto-oncogenes in humans and about 8% of human tumors have BRAF mutations, of which the vast majority are BRAF-V600E mutation and mainly occur in melanoma, colon cancer and thyroid cancer (Holderfield et al., 2014). In normal humans, RAF kinase protein activates the phosphorylation of MEK/ERK, thereby playing a corresponding role in cell proliferation. In this signal pathway, normal human body will have corresponding feedback regulation, so that the phosphorylation of RAF kinase protein can be maintained at a normal level (Karoulia et al., 2017). However, when the RAF protein kinase is mutated, especially the V600E mutation, the feedback effect can only act on the wild-type RAF protein kinase, and there is no feedback effect on the V600E type RAF protein kinase, which leads to the continuous activation of the downstream MEK-ERK signaling pathway. It plays a vital role in tumor growth, proliferation, invasion and metastasis, so BRAF-V600E mutation is one of the effective targets of anti-melanoma and other tumors (Agianian and Gavathiotis, 2018).
In 2011, the first BRAF-V600E targeted inhibitor, Vemurafenib (41, Figure 8), was approved by the FDA for the treatment of patients with BRAF-V600E mutations in advanced melanoma and achieved breakthrough therapeutic effects (Bollag et al., 2012). Vemurafenib is also a typical target drug based on genetic diagnosis. However, it is reported that after 6–12 months of taking Vemurafenib, patients will have varying degrees of drug resistance, which limits the therapeutic effect (Lito et al., 2013). Therefore, the development of new small-molecule drugs targeting BRAF-V600E mutations is particularly important.

Figure 8. Structures of BRAF inhibitors and degraders.
In 2020, Cullgen selected the BRAF-V600E mutation-targeting inhibitor Vemurafenib and the pan-inhibitor BI882370 (42, Figure 8) as the BRAF-V600E ligands (Han et al., 2020). By analyzing the binding model of small molecule inhibitors and proteins, they designed a series of degraders, finding that compound 43 (Figure 8) based on Vemurafenib and compound 44 (Figure 8) based on BI882370 had better activity to degrade BRAF-V600E protein. They also found that compound 43 can induce the degradation BRAF-V600E protein at 12 nM, and the degradation effect was significantly enhanced with the increase of the drug dose; while compound 44 had a degradation effect at 37 nM, and it also showed dose-dependent effect. In order to prove that the degraders had no degrading activity on wild-type BRAF protein, they used compound 43 and compound 44 to perform protein degradation experiments on A549 cells (BRAF-WT), and they found that these two degraders had no degradation activity on the BRAF-WT protein at different concentrations, which proved that the degraders had an excellent selective degradation effect on the BRAF-V600E protein. Finally, in order to prove the anti-tumor effect, they used compounds 43 and 44 to do anti-proliferation experiments on cell lines A375 and HT-29, respectively. The experimental results showed that compound 43 based on Vemurafenib had a worse inhibitory effect on A375 cells than Vemurafenib, but the effect was adverse on the cell line HT-29, with an IC50 of 124 nM. While compound 44 based on BI882370 had the same inhibitory effect on the cell line A375 and HT-29, with IC50 of 46.5 and 51 nM, respectively.
In the same year, Frank Sicheri’s group reported the BRAF-V600E degrader. Based on the screening of E3 ligase binders, linkers and BRAF binders, they obtained the compound P4B (45, Figure 8) with the best degradation effect and the negative control P4BMe (46, Figure 8; Posternak et al., 2020). In subsequent experiments, they found that in A375 cells, the Dmax of P4B to BRAF-V600E was 82%, and the DC50 was 15 nM. At a higher concentration (DC50 = 1000 nM), P4B did not reduce the level of ARAF, but slightly decreased CRAF level (Dmax = 20–45%) and had no effect on the level of RAF family members KSR1 or SRMS. These results were also seen in SK-MEL-28 cells. Next, they tested the anti-proliferative effects of P4B, P4BMe and inhibitor BI882370 on cells. Compared with P4BMe and BI882370, P4B showed excellent anti-proliferative activity. In homozygous A375 cells (V600E), its inhibitory activity was significantly better than that of heterozygous COLO-205 cells (BRAF WT/V600E)/RKO cells (BRAF WT/V600E) and HT-29 cells (BRAF WT/V600E/V600E). Considering that P4B had comparable enzymatic activity to BRAF-WT and BRAF-V600E in vitro (IC50 = 58 nM vs. 12 nM), it showed that P4B had good degradation selectivity to BRAF-V600E in cells. At the same time, they also found that P4B had a certain degradation effect on other BRAF mutants. In the melanoma cell line WM266-4 (V600D), the degrader showed strong degradation activity with DC50 and Dmax of 15 nM and 76%, respectively. At high concentrations, P4B had a weaker proliferation inhibitory effect on the NCI-H1666 cell line with heterozygous BRAF-G466V mutation, and had no effect on the H508 (G596R) and NCI-H1755 cell lines (G469A).
In 2021, Crews’s group reported the degrader SJF-0628 (47, Figure 8) and the negative control SJF-0661 (48, Figure 8) based on Vemurafenib and VHL that target multiple BRAF mutations (Alabi et al., 2021). In their research, they found that the degrader can induce the degradation of BRAF-V600E protein in a variety of cell lines, without inducing the degradation of BRAF-WT protein. In SK-MEL-28 cells, the DC50 to BRAF-V600E was 6.8 nM, and the DCmax was more than 95%. The same phenomenon can be observed in A375 cells and SK-MEL-239 cells. The degrader can completely induce the degradation of BRAF-V600E within 4 h (the drug concentration was 100 nM), its degradation of BRAF-V600E and inhibition of p-ERK lasted up to 72 h. In the wash out experiment, the BRAF level recovered 30% after 24 h. At the same time, they also found that SJF-0628 can inhibit the phosphorylation of MEK and ERK in SK-MEL-28 cells at 10 nM. The degrader SJF-0628 not only can induce the degradation of BRAF-V600E, it can also induce the degradation of a variety of BRAF mutants in a variety of cell lines. In SK-MEL-239 C4 cells, they found that the degrader can induce the degradation of BRAF-p61V600E with DC50 and Dmax of 72 nM and 80%, and had no effect on BRAF-WT and CRAF. In SK-MEL-246 cells, it can induce the degradation of BRAF-G469A, and its DC50 and Dmax were 15 nM and 95%, respectively. In H1666 and CAL-12-T, it can induce the degradation of BRAF-G466V, and its DC50 and Dmax were DC50 of 29 nM, Dmax of 80% and DC50 of 23 nM, Dmax of 90%, respectively. Subsequently, they tested the inhibitory effect of the degrader SJF-0628 on the tumor cells. They found that in SK-MEL-28 cells (BRAF-V600E), the EC50 of Verofenil and the negative control SJF-0661 were 215 ± 1.09 nM and 243 ± 1.09 nM, respectively, while the EC50 of the degrader SJF-0628 was 37 ± 1.2 nM. Although these compounds had comparable binding capacity to BRAF-V600E in vitro (Vemurafenib = 27 nM, SJF-0628 = 39 nM, SJF-0661 = 64 nM), SJF-0628 had significantly stronger anti-tumor activity. In SK-MEL-239 C4 cells (BRAF-WT/BRAF-p61V600E), Vemurafenib and SJF-0661 had weaker growth inhibitory effect compared with SJF-0628, which can reduce about 80% of a cell growth, and its EC50 was 218 nM ± 1.06. In SK-MEL-246 cells (BRAF-G469A), SJF-0628 can also effectively inhibit cell growth, with an EC50 of 45 ± 1.11 nM, and the EC50 of the negative control SJF-0661 was 278 ± 1.07 nM. In H1666 cells (BRAF-G466V), SJF-0628 can induce 65% of cell growth to be inhibited, while the inhibitory effect of Verafenib was less than 50%. These results showed that targeted degradation can be used to overcome acquired resistance to BRAF inhibitor-based therapies.
In summary, the small molecules induced degradation of BTK, CDK4/6, BCR-ABL and BRAF in cancer cells had a better anti-tumor cell proliferation effect than simply inhibiting, and they also demonstrated promising power in overcoming tumor drug resistance. These research results showed the degradation of protein to overcome drug resistance has good potential and they also increase the potential application of PROTACs in clinical practice.
Traditional small-molecule drugs usually act by inhibiting the enzymatic function of the target and have no effect on the non-enzymatic function, while PROTAC can induce the entire protein degradation, so PROTAC can affect the protein’s enzymatic function and regulate the non-enzymatic function.
Focal adhesion kinase (FAK), also known as protein tyrosine kinase 2 (PTK2), has kinase-dependent enzyme activity functions and kinase-independent backbone functions. These two functions play a vital role in tumors genesis, early embryonic development, and reproduction (Serrels et al., 2007; He et al., 2012; Hartman et al., 2013; Brami-Cherrier et al., 2014; Cromm et al., 2018). Although some FAK small molecule inhibitors have been clinically tested in a variety of malignant tumors, the non-enzymatic functions of FAK still cannot be blocked by reported FAK kinase inhibitors. Because traditional kinase inhibitors can only act on protein kinase domains, drug resistance is likely to occur. Therefore, developing a strategy that can inhibit FAK kinase activity and block its non-kinase activity is an urgent and meaningful need for FAK-related diseases.
In 2018, Crews’s group reported the first PROTAC molecule PROTAC-3 (50, Figure 9) that induced the degradation of FAK (Cromm et al., 2018). At the cellular level, this molecule can induce FAK degradation at nanomolar levels on a variety of cell lines. The PROTAC molecule performed better than inhibitor in the downstream signaling pathways of FAK (p-FAK, p-Paxillian, p-Akt) and in the invasion and migration of human triple-negative breast cancer cell (MDA-MB-231).
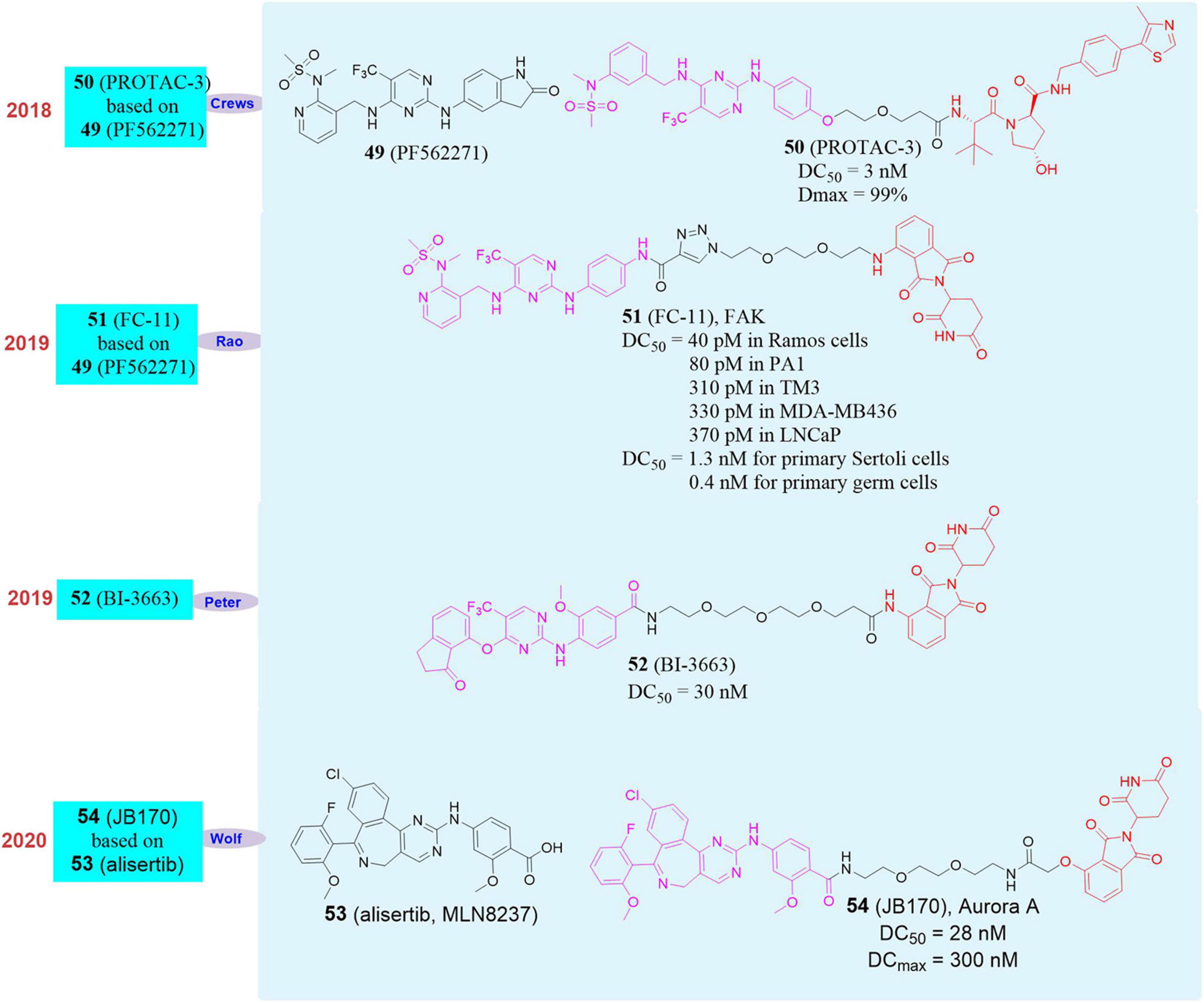
Figure 9. Structures of FAK and Aurora A inhibitors and degraders.
In 2019, our group used PROTAC to successfully obtain an efficient, rapid and reversible PROTAC molecule FC-11 (Gao et al., 2019; 51, Figure 9) that targets FAK and can induce FAK protein degradation in the tested cells. Its FAK protein degradation activity in the tested cells can reach to less than 1 μM and the DC50 can reach to 80 pM when it has been used on PA1 cells for 8 h. In the same year, Peter Ettmayer’s group developed the PROTAC molecule BI-3663 (52, Figure 9) that induced FAK degradation (Popow et al., 2019). BI-3663 efficiently induced FAK degradation with DC50 = 30 nM in 11 human hepatocellular carcinoma cell lines. Although the compound can effectively induce FAK degradation, it basically did not affect the cell proliferation activity of any tested cell line. In 2020, our group conducted the research on the non-enzymatic function of FAK based on FC-11. First of all, under the same action time and concentration, the degradation activity of FC-11 on activated FAK (pFAKtyr397) was much better than that of small molecule inhibitor PF562271 (49, Figure 9). In the mouse model, FC-11 can efficiently induce FAK degradation in the mouse reproductive system, and the FAK in the mouse reproductive system can return to normal levels in 2 weeks after stopping the administration. Compared with the FAK inhibitor PF562271, the sperm motility and motility of mice in the FC-11 administration group were significantly reduced, which in turn led to the decrease of fertility and maldevelopment of embryos in mice (Gao et al., 2020b). In short, FC-11 can induce FAK degradation efficiently and reversibly, thereby affecting the non-enzymatic function of FAK.
The cycle of eukaryotic cells is not only controlled by CDKs, but also affected by other kinases (Nigg, 2001). Aurora A kinase (Aurora kinase A), which is an important subtype in the Aurora kinase family, plays an important role in phosphorylating a variety of proteins during mitosis and its catalytic activity is essential for the entire cell cycle (Marumoto et al., 2005). As scientists studied Aurora kinase, it was discovered that Aurora A kinase activity can be divided into enzymatic activity and non-enzymatic activity (Otto et al., 2009). The inhibition or consumption of Aurora A enzymatic activity may be lethal because it will cause oncogene activation or the loss of tumor suppressor genes. Its non-enzymatic function enables Aurora A to bind to the proto-oncoproteins of the MYC family and protects N-MYC and C-MYC from proteasome degradation, which has nothing to do with the enzymatic activity (Toya et al., 2011; Brockmann et al., 2013; Dauch et al., 2016). Studies have found that Aurora A kinase inhibitors may not eliminate all carcinogenic activities of Aurora A, but whether this effect is related to the non-enzymatic effect of Aurora A kinase is not yet known (Zheng et al., 2016).
In 2020, Elmar Wolf’s group selected the Aurora A kinase inhibitor alisertib (53, MLN8237, Figure 9) and designed a series of degraders that target Aurora A by linking alisertib with CRBN and VHL (Adhikari et al., 2020). They found that the compound JB170 (54, Figure 9) had strong binding ability and degradation activity to Aurora A kinase. The experimental results proved that the DCmax (maximal degradation concentration) and DC50 of JB170 to Aurora A kinase were 300 and 28 nM, respectively. Subsequently, they analyzed the affinity of alisertib (Aurora A EC50 = 18 nM, Aurora B EC50 = 51 nM) and JB170 (Aurora A EC50 = 193 nM, Aurora B EC50 = 1.4 μM), which showed the affinity of JB170 to Aurora A was better than that to Aurora B. After MV4-11 cells were treated with JB170 or alisertib, JB170 reduced Aurora A levels by 73%, which was 57% lower than that of alisertib. Among the 4259 detectable proteins, no other proteins were down-regulated, including Aurora B. At the same time, they also confirmed the selective degradation of Aurora A by JB170 through siRNA.
It is currently known that the enzyme activity of Aurora A is mainly expressed in the G2/M phase of the cell cycle, and its function in the S phase may not be related to its enzymatic activity. Therefore, they analyzed the effect of JB170 and alisertib on the cell cycle of MV4-11 cells. The experimental results showed that almost all cells were blocked in G2/M after 12 h treatment with alisertib. On the contrary, the treatment of JB170 can hardly caused cell accumulation in G2/M phase, but can delay the progress of S phase. RNA sequencing showed that alisertib induced the expression of genes that indicate cell G2/M phase arrest, while JB170 did not activate the same genes. In order to study the effect of JB170-mediated degradation of Aurora A on the survival of cancer cells, they used JB170 (1 μM) to treat MV4-11 cells to measure cell viability. After 72 h, the number of viable cells was 32% of the control level, and similar results were observed in the colony formation test using IMR5 cells. Therefore, they believed that the arresting effect of JB170 in the cell’s S-phase was mainly caused by the effect of Aurora A non-enzymatic activity, which laid a solid foundation for further understanding of the function of Aurora A and the development of drugs for Aurora A kinase.
Therefore, these results indicate that PROTAC can affect the non-enzymatic function of the protein, thus expanding the drug-forming possibility of existing drug targets.
As we all know, only 20–25% of the currently known protein targets are used for drug discovery and related disease treatment, including kinases, GPCRs, nuclear hormone receptors, iron channels, etc. The physiological significance and the relationship with disease of the remaining proteins are still to be explored, so the development of new drug targets is getting more and more attention.
One of the most challenging targets is the signal transducer and activator of transcription 3 (STAT3). STAT3 plays an extremely important role in cell growth, reproduction, apoptosis, metabolism, drug resistance and other processes. It is responsible for transmitting signals from the cell surface receptors to the nucleus. The continuous activation of STAT3 is often associated with the poor prognosis of human cancer, because the activated STAT3 signal can not only promote the growth, survival and metastasis of tumor cells, but also inhibit the anti-tumor immune response, STAT3 is an attractive target for the treatment of human cancer and other diseases (Takeda et al., 1997; Boccaccio et al., 1998; McLemore et al., 2001). Although scientists have been working tirelessly on this target for 20 years, targeting STAT3 is still a huge challenge. Due to the conservative structure and low specificity of STAT3, it is extremely difficult to develop efficient and specific inhibitors. Although some STAT3 inhibitors with anticancer activity have been reported so far, most of the inhibitors have poor activity and lack specificity. At present, most studies on STAT3 inhibitors are based on the SH2 domain on the STAT3 protein. The SH2 domain plays a key role in the dimerization of STAT3. Therefore, drugs targeting the SH2 domain can inhibit the dimerization of STAT3, thus inhibiting its transcriptional activity.
In 2019, Wang Shaomeng’s group developed an SH2 inhibitor SI-109 (55, Figure 10), and successfully screened out the first degrader SD-36 (56, Figure 10) that can target STAT3 by using PROTAC (Bai et al., 2019). This molecule had a high specificity for STAT3 binding and no obvious effect on other members of the STAT family. At the same time, SD-36 still showed a good effect on the mutant SH2, so even if STAT3 had a high mutation rate, it can still be effectively degraded by SD-36. SD-36 also had shown good efficacy data on leukemia model mice and can eliminate tumors lastingly and almost completely.
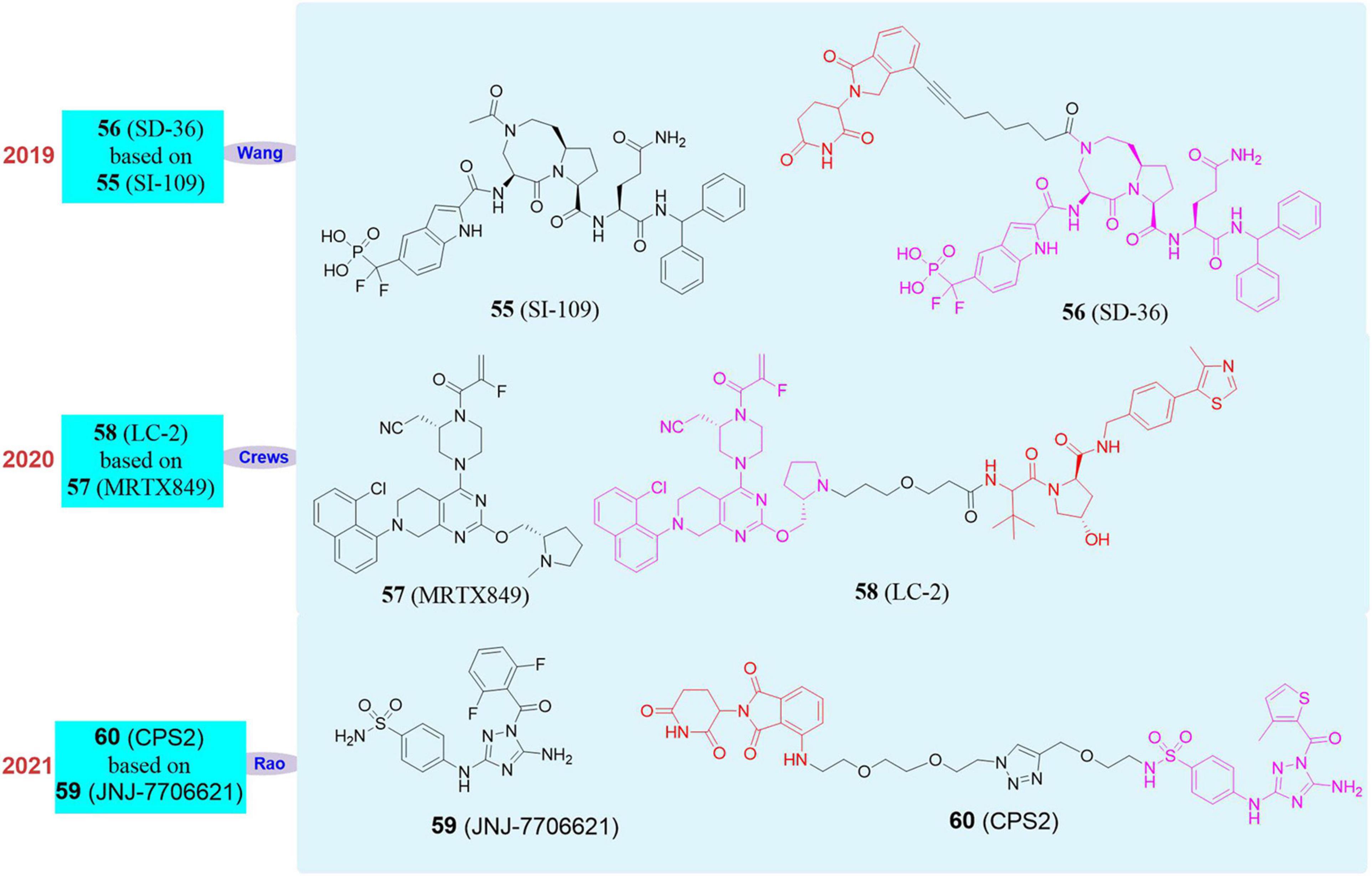
Figure 10. Structures of STAT3, KRASG12C and CDK2 inhibitors and degraders.
Another challenging target is KRAS. KRAS is one of the most frequently mutated oncogenes, which can be activated through a variety of ways and cause tumor development, such as binding to guanosine triphosphate and activated by cell surface receptors. In addition to protein-based activation, KRAS can also be activated due to mutations in key codons. Clinically high-frequency mutations (such as G12A, G12C, G12D, G12S, G12V, G13C, G13D) and some low-frequency mutations can both activate KRAS. The mutated codon interacts with guanosine triphosphate hydrolase, which activates KRAS and ultimately leads to tumorigenesis. KRAS has mutations in a variety of cancers, among which the mutation rate of pancreatic cancer is as high as 90%, that of colon cancer and lung cancer (mostly non-small cell lung cancer) account for 30–50% and 19% respectively, and cholangiocarcinoma accounts for about 26%. Mutations also occur in cancers such as small bowel cancer, skin cancer, bladder cancer and breast cancer (Pylayeva-Gupta et al., 2011; Rojas et al., 2012; Lu et al., 2016; Cagir and Azmi, 2019; Kessler et al., 2019). Since the KRAS protein has no suitable binding pockets for small molecule inhibitors, the development of small molecule drugs targeting KRAS has not made major breakthroughs for a long time. Although the research on covalent inhibitors of KRASG12C mutants has been spotlighted in recent years and there have been many KRASG12C inhibitors in the clinical research stage, the clinical results also show that some patients have already developed drug resistance. However, the PROTAC has unique advantages in such difficult-to-target drug targets.
In 2020, Crews’s group reported the PROTAC molecule LC-2 (58, Figure 10; Bond et al., 2020) based on the KRASG12C inhibitor MRTX849 (57, Figure 10; Hallin et al., 2020). LC-2 can rapidly degrade KRASG12C in different homozygous and heterozygous tumor cells. In NHC-H2030 cells, the DC50 of LC-2 to KRASG12C was 0.59 μM. Since LC-2 was still covalently bound to the target protein, it would affect the catalytic cycle of the PROTAC molecule and may therefore limit its effectiveness. Although there are limitations, the appearance of LC-2 degrader creates new opportunities for targeting KRAS mutants in cancer treatment, which is of great significance. Therefore, it is imperative to develop reversible PROTAC against KRAS mutants.
Cyclin-dependent kinases (CDK) are a group of serine/threonine protein kinases with a shorter N-terminus (β-sheet) and a longer C-terminus (α-helix). CDK, who interacts with the corresponding cyclin and cyclin-dependent kinase activated kinase (CAK), plays a significant role in all stages of the cell cycle and participates in the physiological processes of cell growth, differentiation, and proliferation. CDK2 is a member of the CDK family and is widely expressed in mammalian cells. Because its function can be compensated by other CDK family members, CDK2 is not necessary for most normal cells and tissues. However, CDK2 plays an important role in tumorigenesis, cell differentiation, meiosis, and hearing damage repair (Ying et al., 2018). Recent studies have found that CDK2 knockdown can induce differentiation of AML cells (Takada et al., 2017). However, there is no good tool molecule that can successfully knockdown CDK2 selectively because traditional small molecule inhibitors are difficult to achieve selective CDK2 inhibition and they can not eliminate the non-enzymatic function of CDK2 (Tadesse et al., 2019). Gene editing technology has many shortcomings in effective application and clinical use. Although CDK2 is considered a good drug target, it is difficult for existing tools to achieve efficient and selective regulation.
In 2021, our group reported that we used pan-CDK inhibitor JNJ-7706621 (59, Figure 10) and pomalidomide as ligands to design a new type of CDK2 selective degrader 2 (60, Figure 10), which achieved high selectivity and efficient degradation of CDK2 (Wang et al., 2021). CPS2 had good selectivity and wide application range, and we tried to test more than 10 cell lines and found it can effectively induce the degradation of CDK2 at nanomolar concentrations. We also used kinomics, kinase activity test, proteomics, and western blot experiments to have proved that the degradation of CDK2 was the most significant when CPS2 was used to treat cells, while it had no significant effect on other proteins.
At the same time, we also found that under the treatment of CPS2, the differentiation index of AML cells was significantly increased, and the cell morphology was more mature. We conducted multiple sets of rescue experiments to have proved that CPS2 performed differentiation-inducing function by the degradation of CDK2 in cells. In order to fully demonstrate the importance of CPS2 as a selective degrader of CDK2 in the clinical treatment of AML, we collected multiple primary cells from AML patients and added CPS2 for treatment. The results showed that the primary cells can also be significantly differentiated under the treatment of drugs, which fully proved the significance of CPS2 potential in clinical application.
Using PROTAC to establish a protein knockdown animal model can be used as a powerful supplementary method for studying the loss of target gene functional sequence. The traditional genetic method is to establish animal models through genetic modification, such as TALEN or CRISPR-Cas 9. However, these technologies are often difficult to achieve rapid, efficient, and reversible protein degradation, and the long cycle and high cost bring more challenges to research, especially in non-human primates. At the same time, gene knockout models may exist potential gene compensation or gene mutations that led to phenotypic misunderstandings. In addition, the increased possibility of animals activating compensatory pathways may also obscure the phenotype. And right tools are still in urgent need for researches on those indispensable genes during embryonic development. As a new, rapid and efficient method of producing protein knockdown models, PROTAC can be used as an effective supplement to existing genetic tools. The following mainly introduces some PROTAC tool molecules developed by our group.
FKBP12 (FK506 binding protein 12) is a type of protein that specifically binds to macrolide immunosuppressants FK506 and rapamycin, and is widely expressed in mammals. This protein interacts with calcium channel receptor (Nissin receptors) and keeps the calcium channel in a stable closed state. When FKBP12 dissociates from RyRs, RyRs opens and releases calcium ions, thereby realizing the regulation of important functions of the body through the calcium signaling pathway (Andrus and Schreiber, 1993; Holt et al., 1993; Teague and Stocks, 1993; Huse et al., 1999; Wiedeman et al., 1999). One of the important functions of FKBP12 is to participate in the development of the heart and to play an important regulatory role in the phenotypic differentiation of cardiomyocytes, the formation of heart structures and the initiation of heart beats. The absence of FKBP12 in the embryonic heart can cause severe developmental ventricular defects, leading to embryonic death. However, the functional study of whole body knockout FKBP12 in the heart of adult mice is not complete, and there is no research on knockout FKBP12 in large animals.
In 2015, Bradner’s group reported the first PROTAC molecule targeting the degradation of FKBP12. They used CRBN ligand and FKBP12 wild-type inhibitor to design and synthesize two degraders, dFKBP-1 (61, Figure 11) and dFKBP-2 (62, Figure 11; Winter et al., 2015). These two molecules in human myeloid monocytic leukemia cell MV4-11 can well induce FKBP12 degradation at a concentration between 0.01 and 10 μM. However, they only verified that PROTAC can induce the degradation of FKBP12, but did not do further experiments to confirm the physiological function changes caused by FKBP12 degradation. Subsequently, in 2018, Bradner’s group reported a series of PROTAC molecules designed to target the degradation of FKBP12 based on the CRBN ligand thalidomide and FKBP12F36V selective inhibitor AP1867 (Andrade et al., 2007; 63, Figure 11). dTAG-13 (Nabet et al., 2018; 64, Figure 11) showed high selectivity and high efficiency to the degradation of FKBP12F36V. At the same time, they found that the constructed exogenous FKBP12F36V fusion protein FKBP12F36V-BRD4, FKBP12F36V-KRASG12V, FKBP12F36V-EZH2, HDAC1-FKBP12F36V, MYC-FKBP12F36V, and PLK1-FKBP12F36V can also be efficiently degraded by dTAG-13. In addition, in a xenograft mouse model of MV4-11 cells stably expressing the luciferase-FKBP12 fusion protein, dTAG-13 can also successfully induce the FKBP12 protein degradation in tumor cells. This dTAG technology not only reveals the physiological effects of BRD4 and KRASG12V in detail, but also provides a new research strategy for the verification of targets in developing new drugs.
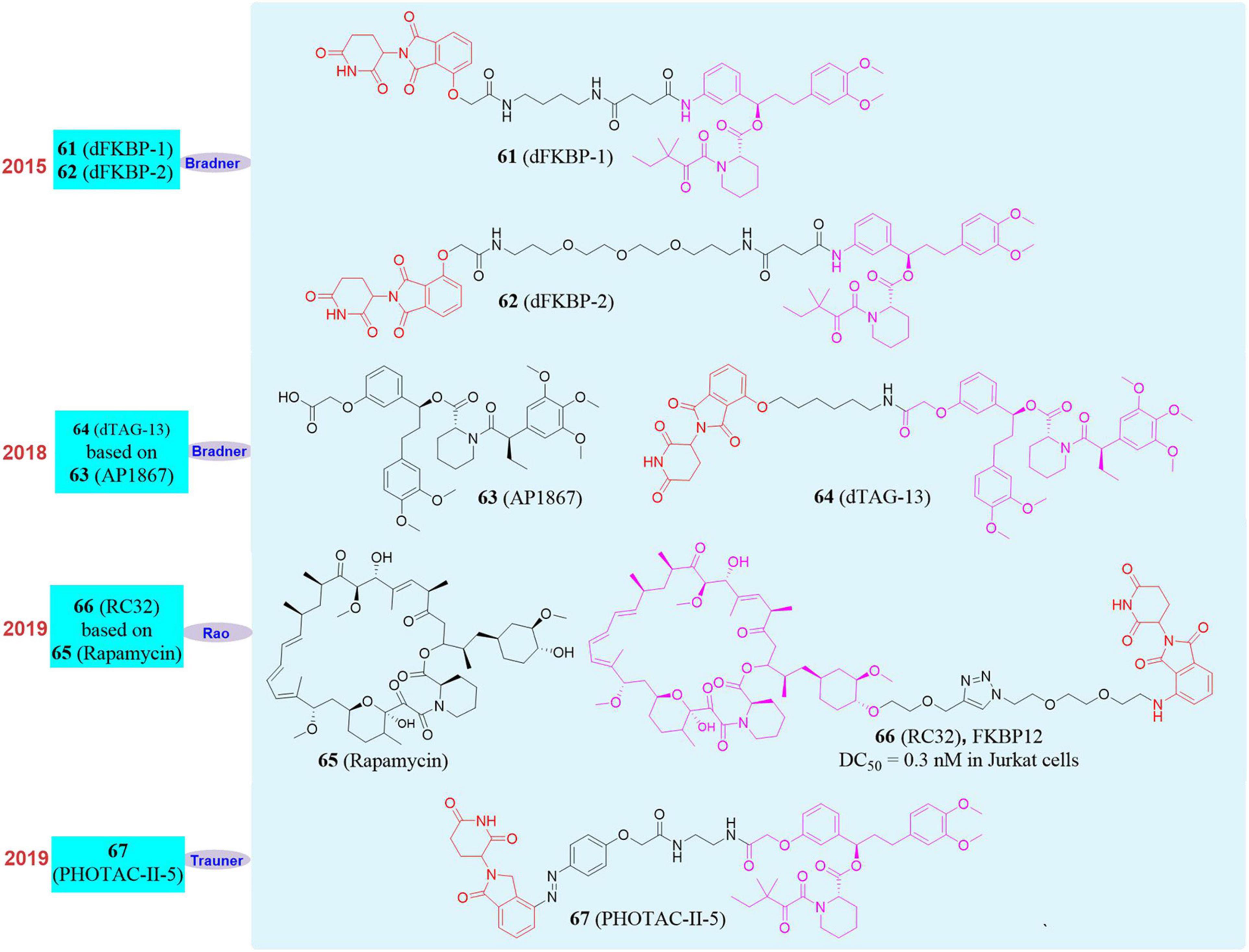
Figure 11. Structures of FKBP12 inhibitors and degraders.
In 2018, our group successfully based on Rapamycin develop a small molecule RC32 (Sun X. Y. et al., 2018, 66, Figure 11; Saxton and Sabatini, 2017, 65, Figure 11) that induces the degradation of FKBP12 protein. The whole body knockdown of the target protein in animals was established, and the whole body knockdown models of mice, rats, pigs and rhesus monkeys were quickly constructed. We also studied and verified the function of FKBP12 protein in mice and rhesus monkeys. Among them, FKBP12 protein in mice and rats, pigs, rhesus monkeys only take 1, 2, 3 days to knock down in the whole body with high efficiency. The conditional knockout of FKBP12 in the brain can be achieved when RC-32 was administered to the cerebral ventricle. If the administration of RC-32 was stopped, the protein in the animal body can gradually recover, which was conducive to the control of animal models and was more accurate for protein function research. Furthermore, this work verified for the first time that PROTACs can maintain high-efficiency protein degradation in the oral route of administration. We also applied this method to the systemic knockdown of other target proteins, such as BTK protein.
In 2020, Trauner’s group developed a series of light-controlled PROTAC for the first time, which was called PHOTOAC. They synthesized and screened the PHOTOAC-II-5 (Reynders et al., 2020; 67, Figure 11) by introducing the light control group azo in the linker of the PROTAC molecule, which showed effective degradation of FKBP12 between the concentration of 10 nM and 3 μM in the human acute lymphocytic leukemia cell line RS4;11 under the irradiation of light with a wavelength of 390 nm. The PHOTAC method precisely regulates the degradation of the target protein through optical control, thus providing a new strategy for photomedicine and photopharmacology. Therefore, the PROTAC is an effective supplement to the current gene knockout methods, and it is an extremely promising technology because of its rapid, efficient, reversible, and controllable realization of systemic protein knockdown in vivo and in vitro.
HDAC6 (Histone deacetylase 6) is the most special histone deacetylase in the HDACs family. It is mainly located in the cytoplasm and the substrates include α-tubulin, HSP90, cortactin, etc. HDAC6 participates in the regulation of misfolded protein degradation, cell morphology and migration. More importantly, the abnormal regulation of HDAC6 is closely related to a variety of diseases, such as neurodegenerative diseases, cancer, and autoimmune diseases (Zhang et al., 2007; Simões-Pires et al., 2013; Krämer et al., 2014; Miyake et al., 2016). Therefore, directly regulating the protein level of HDAC6 is not only of great significance for disease treatment, but also has a profound impact on the biological process of its regulation.
In 2018, Tang’s group reported the first PROTAC molecule 68 (Yang et al., 2018; Figure 12) that induced HDAC6 degradation. This molecule was based on the broad-spectrum HDAC inhibitor Crebinostat (69, Figure 12) and pomalidomide as the E3 ligand. They found that it can selectively induce HDAC6 degradation with a DC50 of 34 nM, but had no effect on the other proteins in the HDACs family.
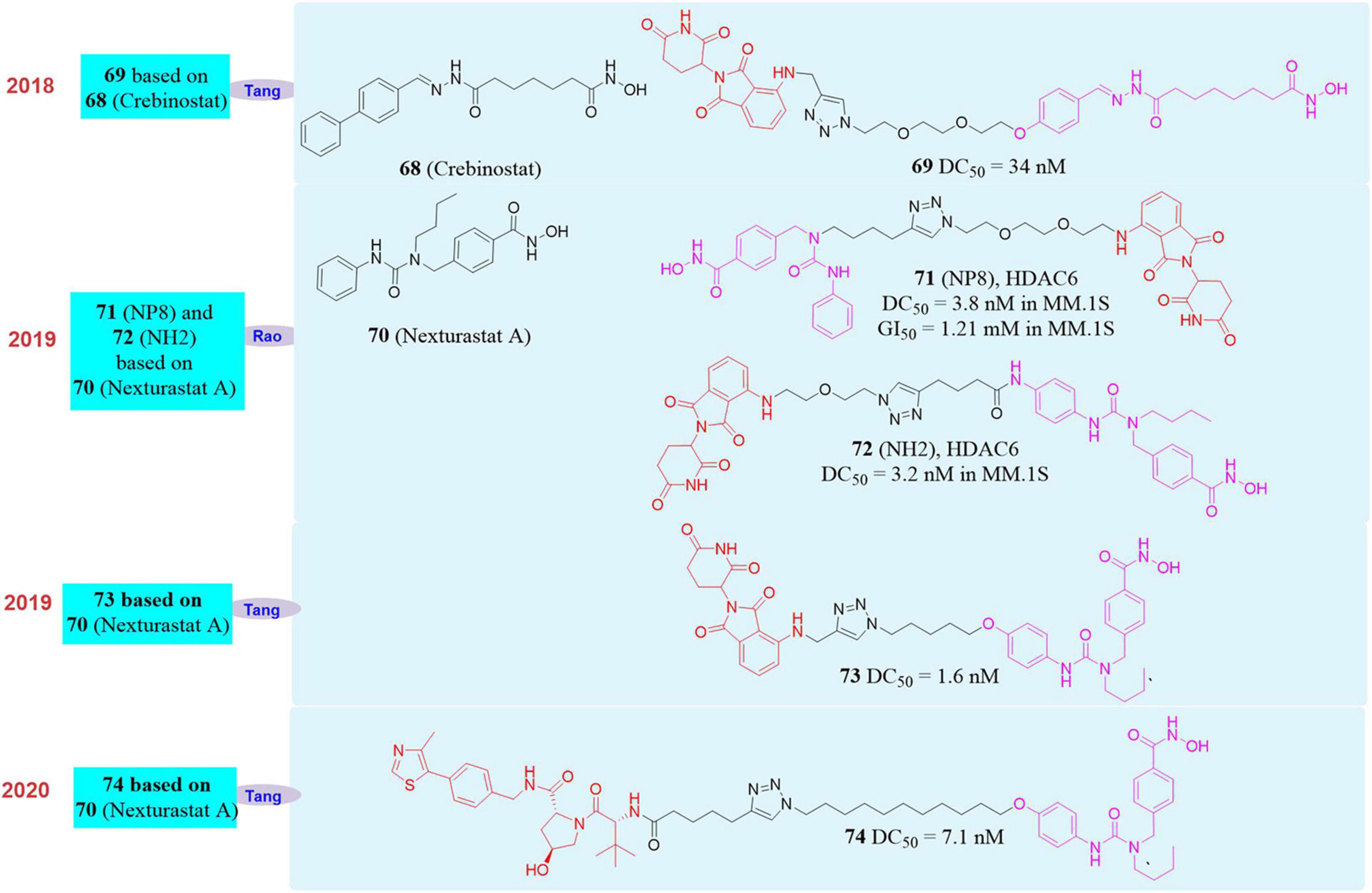
Figure 12. Structures of HDAC6 inhibitors and degraders.
In 2019, our group used PROTAC to construct new HDAC6 protein degrader NP8 (An et al., 2019; 71, Figure 12) and NH2 (Yang et al., 2019; 72, Figure 12). Both NP8 and NH2 can be used in a variety of solid tumor cell lines such as human cervical cancer cells, human lung cancer cells, human glioma cells, human colon cancer cells, and hematoma cell lines (such as acute T cell leukemia cell lines, multiple myeloma cells). They can induce HDAC6 protein degradation in low-dose with high-efficiency, and reversible in those cells, among which the multiple myeloma cell line MM.1S was the most sensitive. In MM.1S cells, the DC50 of NP8 to HDAC6 was only 3.8 nM, and the inhibitory activity of NP8 and HDAC6 selective inhibitor Nexturastat A (Bergman et al., 2012; 70, Figure 12) on MM.1S were equivalent. NP8 was capable of inducing the degradation of the fusion protein of HDAC6 and EGFP, which proved that fluorescent methodology can be used to dynamically monitor the protein degradation process at the cell level, and provided a highly efficient and reversible tool for the research of HDAC6 protein. In the same year, based on Nexturastat A and pomalidomide as the ligands, Tang’s group also reported the HDAC6 protein degrader 73 (Wu et al., 2019; Figure 12), which can effectively induce HDAC6 protein degradation in a variety of tumor cell lines and the DC50 in the multiple myeloma cell line MM.1S was 1.7 nM. Compared with the inhibitor Nexturastat A, 73 had obvious anti-MM.1S cell proliferation function.
In 2020, Tang’s group reported another HDAC6 protein degrader 74 (Yang K. et al., 2020) based on VHL as E3 ligase and Nexturastat A as the protein binder (Figure 12). This molecule also can induce HDAC6 degradation effectively, and the DC50 in MM.1S cell is 7.1 nM. Based on the reports of Tang’s group and our group, it was found that PROTAC molecule based on broad-spectrum HDAC inhibitor such as SAHA can only selectively induce HDAC6 degradation, but did not have degradation activity to other members of HDAC family.
3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase (HMGCR) is the rate-limiting enzyme in the cholesterol synthesis pathway. It consists of a transmembrane domain and a catalytic domain extending into the cytoplasm and locates in the endoplasmic reticulum (ER). Its main physiological role is to catalyze the conversion of HMG-CoA to mevalonate in the cholesterol biosynthesis pathway. Since the production of mevalonate is an irreversible process and inhibiting the activity of HMG-CoA reductase can hinder cholesterol synthesis, so it is one of the most important enzymes in the body’s cholesterol metabolism pathway and also a classic drug target for the treatment of dyslipidemia (Medina and Krauss, 2009; Clendening et al., 2010). In the market, HMGCR inhibitors are mainly statin type drugs. The mechanism is that statins have a HMG-like structure and will compete with the active site of enzymes to prevent the biosynthesis of mevalonate and downstream derivatives (including cholesterol) so as to achieve the effect of reducing plasma cholesterol levels, preventing atherosclerosis and treating cardiovascular diseases. Among all types of statins, atorvastatin (Cohen et al., 1999; Zeng et al., 2012; 75, Figure 13) has shown excellent efficacy in reducing plasma low-density lipoprotein (LDL) cholesterol levels, and has achieved great success in clinical use to prevent and treat heart vascular disease. However, a considerable number of people are intolerant to statins, suffering from such serious side effects as skeletal muscle damage, which may be related to the increase in the compensatory expression of HMGCR in the body through negative feedback regulation after taking statins. Therefore, there is an urgent need for a strategy that can simultaneously eliminate HMGCR activity and abundance in clinical applications.
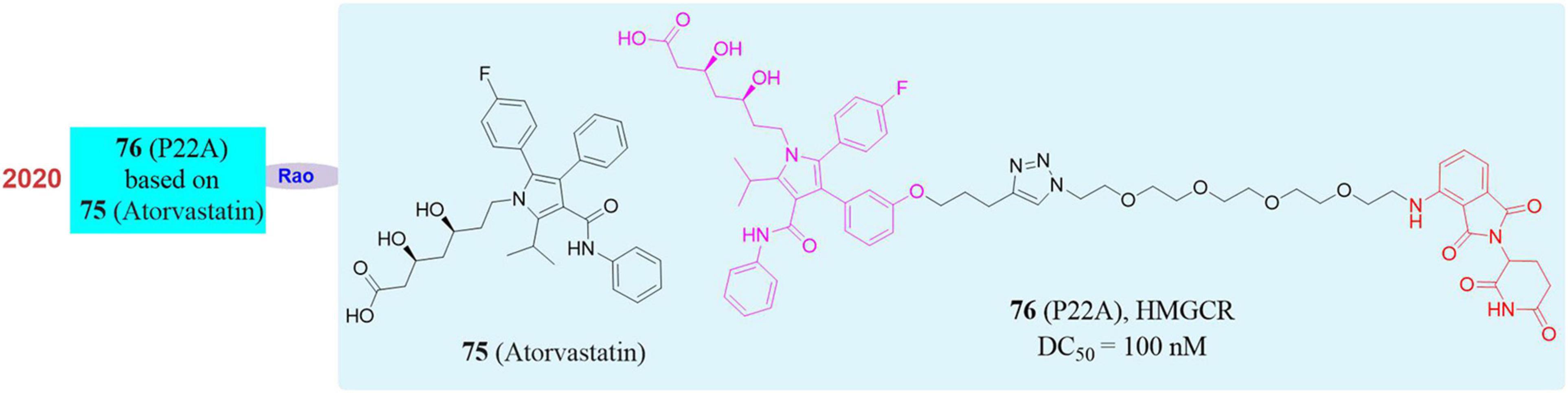
Figure 13. Structures of HMGCR inhibitor and degrader.
In 2020, our group successfully knocked down the HMGCR protein by using PROTAC. A series of small degraders were designed and synthesized based on atorvastatin as the target ligand and Pomalidomide and Lenalidomide as E3 ligands. Subsequently, in vitro degradation activity evaluation, it was found that the compound P22A (Li et al., 2020; 76, Figure 13) induced HMGCR degradation in a dose-dependent manner, and reached up to 70% of the HMGCR protein degradation effect at 1 μM. When higher concentrations of P22A was used, no further degradation was detected. P22A had the best HMGCR degradation activity with a DC50 of 100 nM, which was significantly better than that of other compounds. In contrast, the inhibitor atorvastatin caused a significant upregulation of HMGCR. Besides, proteomics analysis found that the omics responses caused by the inhibitors atorvastatin and compound P22A were also very different. The inhibitors atorvastatin and compound P22A were comparable in inhibiting cholesterol synthesis and in the ability to up-regulate the expression level of LDLR induced by the SREBP pathway. Interestingly, HMGCR is an eight-pass transmembrane protein located on the endoplasmic reticulum. PROTAC had limited degradation ability of such proteins. This work proved the feasibility of using PROTAC to induce endoplasmic reticulum proteins degradation for the first time. In addition, the phenomenon of up-regulation of target proteins also appeared in many other inhibitors. This work showed that PROTAC had greater potential in application in this situation.
Although there are a large number of degraders and tool molecules that can quickly, reversibly and selectively down-regulate various proteins, scientists have little knowledge of the mechanism of degraders in biological systems and their deeper physiological effects. Therefore, the development of degraders with artificial controllability, high temporal and spatial selectivity, and simple control mechanisms is becoming more and more important. Once such degraders are available, scientists can not only learn more deeply in simple biological systems to understand the process and mechanism of protein degradation, it is also possible to explore the role of degraders in complex organisms by controlling the physiological activities. In order to achieve this, the method used must not cause interference to the biological system, and have the characteristics of high efficiency and simple operation (Brieke et al., 2012; Szymanski et al., 2013; Gautier et al., 2014). Therefore, light with high temporal and spatial resolution, which has been widely studied in neurobiology, chemical biology and disease treatment in recent years, appears in the vision of scientists (Madisen et al., 2012; Zhang et al., 2017; Tamura et al., 2019). Its extraordinary characteristics-high temporal and spatial accuracy, non-invasiveness, no interference to biological systems, and qualitative and quantitative control-coincide with the needs of scientists.
At present, two photo molecular biology methods with high temporal and spatial resolution have been widely used to study and control chemical and biological processes. The first method relies on the application of molecular photoelectric switches to perform isomerization between two or more states. This isomerization will lead to changes in molecular properties, which in some cases will be transformed into chemical or biological effects, and it is reversible. However, it is usually not enough to play a role in the major physiological activities of the research system. In this case, the second method, which is to use a photocleavage protecting group (PPG), is usually a better choice (Kessler et al., 2003; Stegmaier et al., 2008). This method can introduce the corresponding PPG into the structure of small molecules or proteins, and provide the corresponding molecule or protein activity through artificial light-induced cleavage of the protecting group (Hansen et al., 2015). This method has such advantages as good release performance and photo-cleavage, large amount of protective group and light source, non-toxicity, etc. However, since the cleavage of protective group is irreversible, this one is also an irreversible light-controlled biological method. On the basis of these studies, more and more scientists are focusing on the study of light-controlled small molecules to induce protein degradation.
In 2019, Pan’s group reported the light-induced degrader pc-PROTAC1 (77, Figure 14) designed based on the BRD4 degrader dBET1 (Xue et al., 2019). They introduced 4,5-dimethoxy-2-nitrobenzyl (DMNB) groups in the linker of dBET1, which has made the newly synthesized degrader pc-PROTAC1 have no physiological activity without light and has shown that it did not induce the degradation of BRD4 protein. However, when irradiated with a 365 nm light source, the degradation of BRD4 protein can be seen clearly when the drug concentration was 300 nM and its maximum degradation (Dmax) reached up to 93% at 1 μM. Although in the absence of light, its affinity for BRD4 protein was significantly weaker than dBET1 (pc-PROTAC1 was 7.6 μM vs. dBET1 was 22 nM) and its inhibitory effect on cells was also significantly worse than dBET1 (pc-PROTAC1 GI50 = 3.1 μM vs. dBET1 GI50 = 0.34 μM), its inhibitory effect on cells was equivalent to dBET1 (pc-PROTAC1 GI50 = 0.4 μM vs. dBET1 GI50 = 0.34 μM) after light treatment. Later, they applied the molecule to zebrafish and found that pc-PROTAC1 can produce the same physiological effects as dBET1 did under light conditions. In order to further verify the rationality and universality of the strategy, they also designed and synthesized the light-controlled degrader pc-PROTAC3 (78, Figure 14) for the BTK protein, and experimentally proved that the molecule can induce the degradation of the BTK protein under light conditions, which proved the practicability of the light-induced degrader release technology.
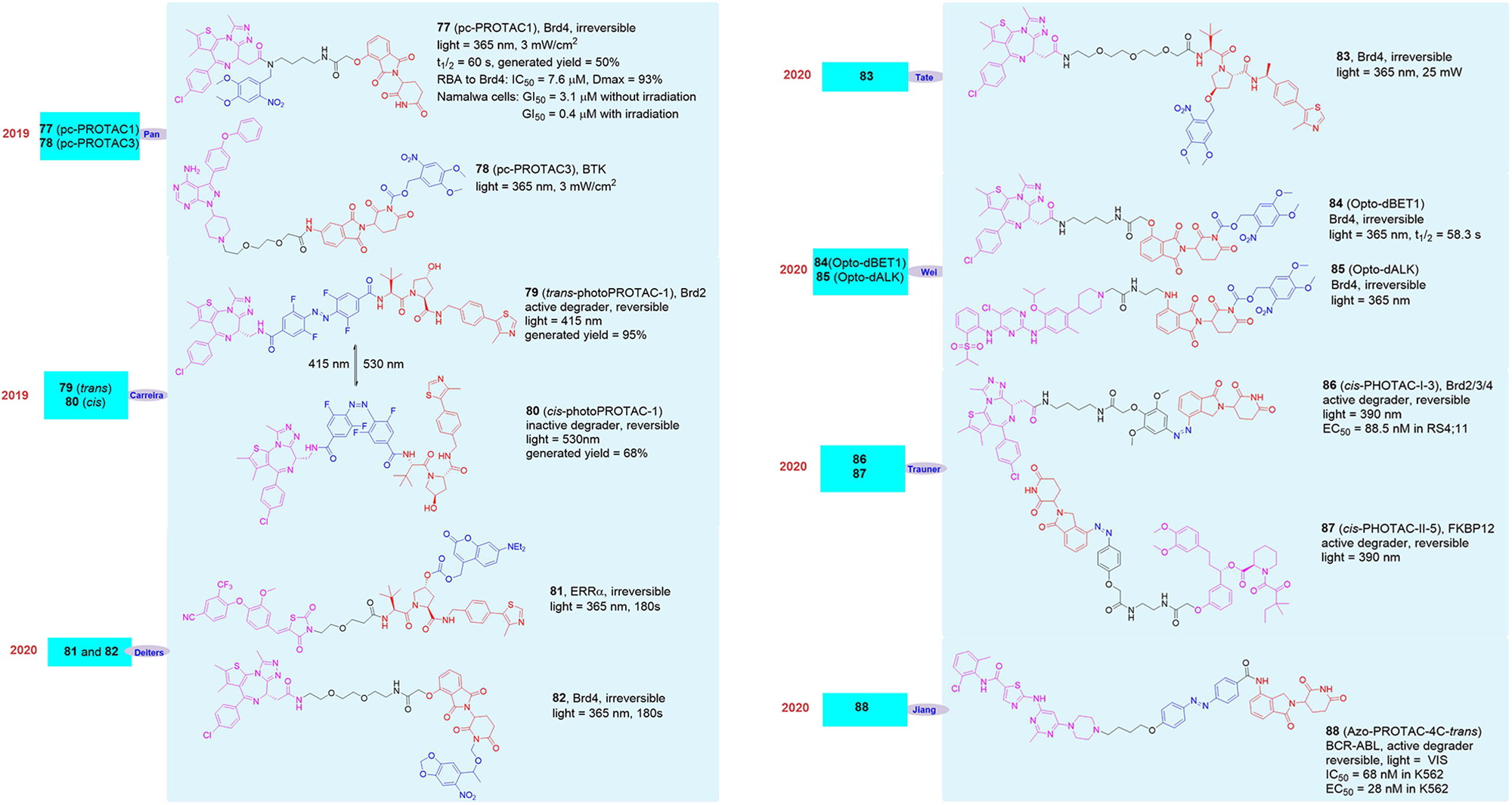
Figure 14. Structures of optical control degraders.
At the same time, Carreira’s group also reported a light-controlled PROTAC based on the BET protein degrader ARV-771 (Pfaff et al., 2019). They analyzed the spatial configuration of ARV-771 and introduced ortho-F4-azobenzene into the linker, which can transform into cis or trans under different light conditions. They synthesized the photo-control degrader photoPROTAC-1 and it can effectively switch between the cis and trans configuration after 530 and 415 nm light irradiation. They found that under 415 nm light conditions photoPROTAC-1 would transform into trans-photoPROTAC-1 (79, Figure 14), in which form can induce BRD2 protein degradation. On the contrary, it will be converted to cis-photoPROTAC-1 (80, Figure 14)under 530 nm light conditions, and this form will not induce BRD2 degradation.
In 2020, Deiters’s group also reported two cases of light-controlled degrader (Naro et al., 2020). The first was a VHL-based degrader 81 (Figure 14) targeting ERRα. They introduced diethylaminocoumarin (DEACM) through carbonate bonds in the degrader. The DEACM group can be activated under 405 nm light conditions, and then leave and peel off the degrader with biological activity, so as to realize the artificial controlled release of the degrader. The second example was the light-controlled degrader 82 (Figure 14) based on the BRD4 protein. They successfully introduced 6-nitropiperidinyloxymethyl (NPOM) into the degrader, of which the NPOM could generate photolysis reaction under 365 nm light condition. However, the activities of these two light-controlled degrader were obvious poor.
In addition to the above examples of light-induced degraders, other research groups have also reported similar molecules in 2020. As reported by Tate’s group, they introduced 4,5-dimethoxy-2-nitrobenzyl (DMNB) group into the VHL structure resulting in the light-controlled degrader 83 (Kounde et al., 2020; Figure 14), which achieved the successful release of the degrader under light conditions. Wei’s group reported that they also modified the CRBN structure with 4,5-dimethoxy-2-nitrobenzyl (DMNB) group, the BRD protein opto-PROTAC opto-dBET1 (84, Figure 14) and the ALK protein opto-PROTAC opto-dALK (85, Figure 14) were successfully obtained, which achieved the successful degradation of Brd protein and ALK protein under light-controlled conditions (Liu et al., 2020). Trauner’s group introduced azo unit into degrader on the benzene ring in CRBN structure, the light-controlled reversible degrader cis-PHOTAC-I-3 (86, Figure 14) and cis-PHOTAC-II-5 (87, Figure 14) were obtained, which realized the light-controlled reversible degradation of BRD protein and FKBP12 protein (Reynders et al., 2020). Jiang’s group reported that the azo unit was introduced into the linker of the degrader, and the BCR-ABL light-controlled degrader Azo-PROTAC-4C-trans (88, Figure 14) was obtained, also it was proved that the light-induced degrader Azo-PROTAC-4C-trans had good biological activity in the cells (Jin et al., 2020).
Analyzing the existing research on light-controlled degraders, we found the following characteristics: (1) Current researchers pay more attention to the mature targets for degraders, such as BRD protein degraders, which had 6 cases and more than half of them, and there are only a few cases for other protein targets (Figure 15A). (2) Among the light-controlled degraders, researchers prefers the structure containing CRBN (Figure 15B). The possible reason is that the synthesis of the degrader containing CRBN is simpler. However, whether the structure of CRBN and VHL in the degrader will affect the efficiency of light-controlled release and degradation activity is currently unknown, so further researches are needed to prove this. (3) The types of light-controlled groups currently are still in limited use (Figure 15C). Its use is mainly concentrated in the range of 365–450 nm, which makes this technology mainly be used in cells and cannot be used in live animals, thus limiting the promotion and application of this technology. Another important related issue that needs to be solved is whether we can find a suitable light control unit in the future to make it possible for fixed-point and timed release in living small animals. (4) The introduction of light-controlling groups is diverse, but in current studies researchers are more inclined to introduce the light control group into CRBN/VHL or linker, but not on the target protein ligand (Figure 15D). It is worth looking forward to whether there will be a breakthrough on this point in the future. (5) Both reversible degraders and irreversible degraders are available, and they have their own characteristics (Figure 15E). However, the currently available methods for reversible degraders are relatively simple, mainly based on azo groups, and there is also the problem of low conversion efficiency.
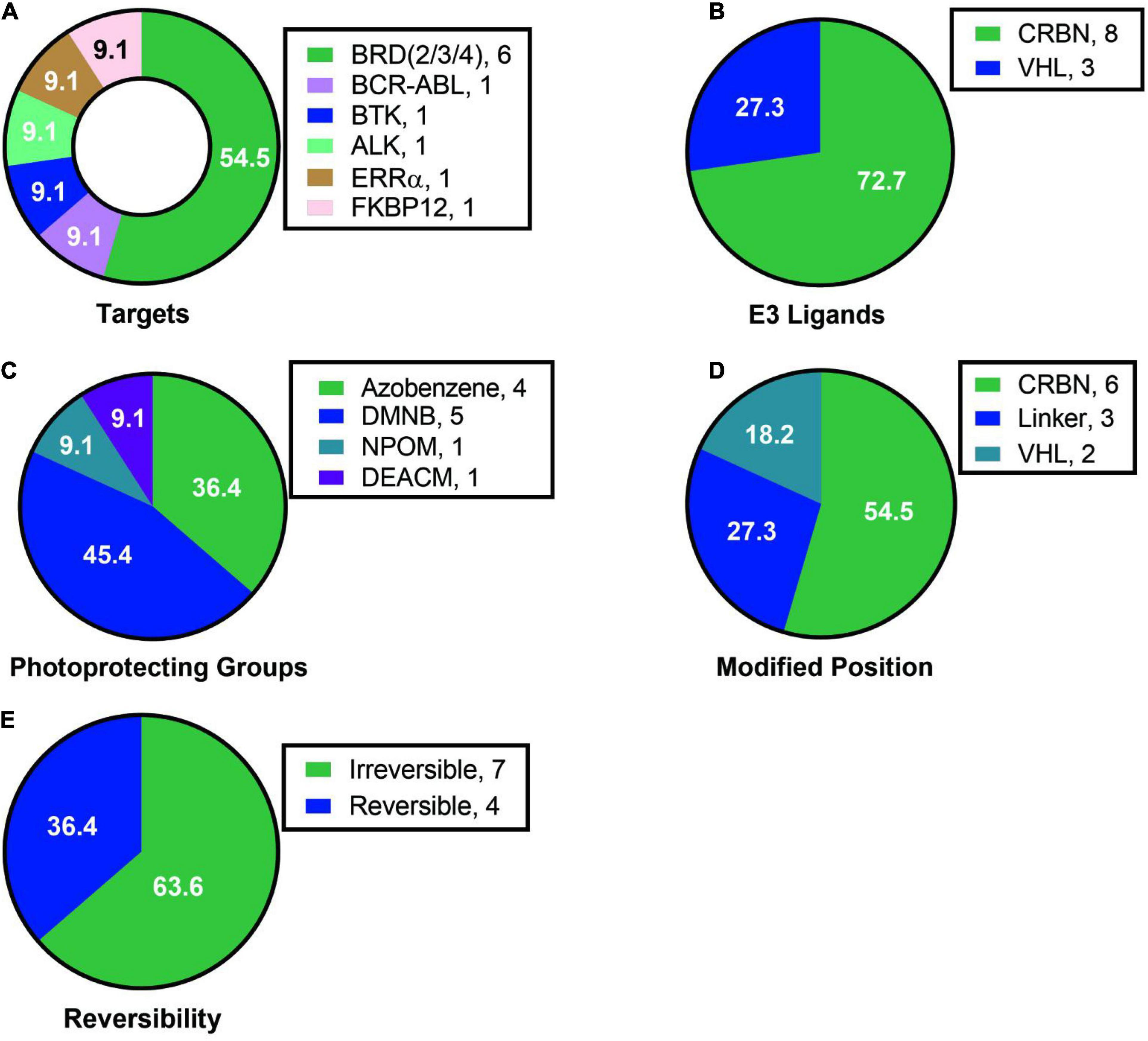
Figure 15. Summary of structure analysis of optical control degraders. (A) Targets used in loptical control degraders. (B) E3 ligands used in loptical control degraders. (C) Photoprotecting groups used in loptical control degraders. (D) Modified position in loptical control degraders. (E) The reversibility in loptical control degraders.
In short, PROTACs can not only be used as a potential candidate molecule for the treatment of diseases, but also as a tool molecule that can be precisely controlled on time and space scales. The above researches offer ideas for the development of PROTACs in the field of biology, further broaden the practical application of PROTACs, and provide tools for solving more unknown biological problems in the future.
In this review, we have analyzed the related PROTACs that have been reported to illustrate their advantages from different perspectives. Due to the large number of targets and the complex structure of the PROTACs, 51 of them mentioned in this review have been summarized and analyzed. By comparing the types of E3 ligases, the linker types and connection methods, we have shown the results in Figure 16 below. It’s been found that the reported PROTACs were still diverse, but the E3 ligases used were mainly concentrated on CRBN and VHL. The linkers and connection methods were also mainly focused on PEG and alkyl. All of these gradually expose some challenges that currently exist in PROTACs.
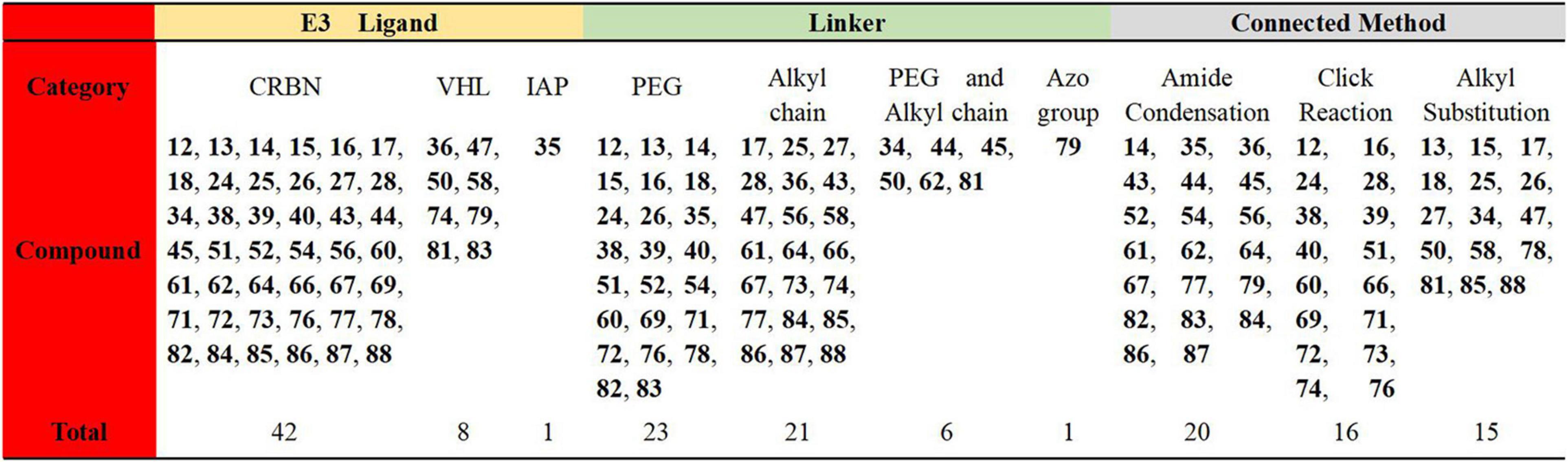
Figure 16. Summary and comparison of the 51 PROTACs mentioned in this review.
The first challenge is about the rational design of PROTACs. For example, the human genome encodes more than 600 E3 ubiquitin ligases, but there are very few E3 ligases (VHL, CRBN, IAPs, and MDM2) currently used in PROTACs design. Therefore, how to expand the E3 ubiquitin ligase to be used in PROTACs is one of the challenges we are facing. In addition, how to expand the types of linkers and the connection methods are questions worth pondering.
The second challenge lies in the evaluation of the biological effects of PROTACs. Firstly, how to quickly and effectively screen the target protein ligand, evaluate the protein degradation activity and biological effects of PROTACs. Secondly, how to understand the relationship between degradation activity, selectivity and possible off-target effects (based on different targets, different cell lines, and different animal models). At last, PROTACs play a role in the catalytic cycle, and traditional methods cannot accurately assess the PK and PD properties of PROTACs; however, there is no mature PK and PD evaluation system. So, how to establish a scientific and credible PK/PD evaluation system is a question worth exploring.
The third challenge is mainly in the application of PROTACs. At present, there is only few cases of degrader reported for undruggable targets, and more cases are needed in the future to support the advantages of PROTACs in undruggable targets. It is reported that certain types of PROTACs can not only play a role through the mechanism of PROTAC, but also induce protein degradation through the mechanism of molecular glue. Therefore, how to distinguish the degradation mechanism (PROTAC or molecular glue or PROTAC and molecular glue) of proteins is also one of the challenges.
These are currently unclear problems that need to be resolved. What’s more, the above problems and challenges involve not only the technical application level, but also the basic fields, so it is necessary to strengthen communications from basic researches and application aspects. It is believed that through the joint efforts of colleagues in academia and industry, these questions can be answered satisfactorily in the near future.
YR designed the project, and reviewed and revised the manuscript. MH and WL summarized the literature, related structures and data. MH wrote the figures and manuscript. WL helped organize the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Nos. 81573277, 81622042, and 81773567), National Major Scientific and Technological Special Project for ‘Significant New Drugs Development’ (Nos. SQ2017ZX095003 and 2018ZX09711001), and ‘National Key R&D Program of China’ (No. 2020YFE0202200).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adhikari, B., Bozilovic, J., Diebold, M., Schwarz, J. D., Hofstetter, J., Schröder, M., et al. (2020). PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat. Chem. Biol. 16, 1179–1188. doi: 10.1038/s41589-020-00652-y
Agianian, B., and Gavathiotis, E. (2018). Current insights of BRAF inhibitors in cancer. J. Med. Chem. 61, 5775–5793. doi: 10.1021/acs.jmedchem.7b01306
Agrawal, N., Dasaradhi, P. V. N., Mohmmed, A., Malhotra, P., Bhatnagar, R. K., and Mukherjee, S. K. (2003). RNA interference: biology, mechanism, and applications. Microbiol. Mol. Biol. Rev. 67, 657–685. doi: 10.1128/mmbr.67.4.657-685.2003
Alabi, S., Jaime-Figueroa, S., Yao, Z., Gao, Y. J., Hines, J., Samarasinghe, K. T. G., et al. (2021). Mutant-selective degradation by BRAF-targeting PROTACs. Nat. Commun. 12, 920–930. doi: 10.1038/s41467-021-21159-7
Amm, I., Sommer, T., and Wolf, D. H. (2014). Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 1843, 182–196. doi: 10.1016/j.bbamcr.2013.06.031
An, Z. X., Lv, W. X., Su, S., Wu, W., and Rao, Y. (2019). Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 10, 606–609. doi: 10.1007/s13238-018-0602-z
Andrade, C. K. Z., Silva, W. A., and Maia, E. R. (2007). Computational approach for the design of AP1867 analogs: aiming at new synthetic routes for potential immunosuppressant agents. J. Biomol. Struct. Dyn. 25, 35–48. doi: 10.1080/07391102.2007.10507153
Andrus, M. B., and Schreiber, S. L. (1993). Structure-based design of an acyclic ligand that bridges FKBP12 and Calcineurin. J. Am. Chem. Soc. 115, 10420–10421. doi: 10.1021/ja00075a092
Bai, L. C., Zhou, H. B., Xu, R. Q., Zhao, Y. J., Chinnaswamy, K., McEachern, D., et al. (2019). A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell 36, 498–511. doi: 10.1016/j.ccell.2019.10.002
Barton, K. L., Misuraca, K., Cordero, F., Dobrikova, E., Min, H. D., Gromeier, M., et al. (2013). PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS One 8:e77639. doi: 10.1371/journal.pone.0077639
Bergman, J. A., Woan, K., Perez-Villarroel, P., Villagra, A., Sotomayor, E. M., and Kozikowski, A. P. (2012). Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J. Med. Chem. 55, 9891–9899. doi: 10.1021/jm301098e
Boccaccio, C., Andò, M., Tamagnone, L., Bardelli, A., Michieli, P., Battistini, C., et al. (1998). Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 391, 285–288. doi: 10.1038/34657
Bollag, G., Tsai, J., Zhang, J. Z., Zhang, C., Ibrahim, P., Nolop, K., et al. (2012). Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 11, 873–886. doi: 10.1038/nrd3847
Bond, M. J., Chu, L., Nalawansha, D. A., Li, K., and Crews, C. M. (2020). Targeted degradation of oncogenic KRASG12C by VHL-recruiting PROTACs. ACS Cent. Sci. 6, 1367–1375. doi: 10.1021/acscentsci.0c00411
Brami-Cherrier, K., Gervasi, N., Arsenieva, D., Walkiewicz, K., Boutterin, M. C., Ortega, A., et al. (2014). FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 33, 356–370. doi: 10.1002/embj.201386399
Brand, M., Jiang, B. S., Bauer, S., Donovan, K. A., Liang, Y. K., Wang, E. S., et al. (2019). Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem. Biol. 26, 300–306. doi: 10.1016/j.chembiol.2018.11.006
Brieke, C., Rohrbach, F., Gottschalk, A., Mayer, G., and Heckel, A. (2012). Light-controlled tools. Angew. Chem. Int. Ed. Engl. 51, 8446–8476. doi: 10.1002/anie.201202134
Brockmann, M., Poon, E., Berry, T., Carstensen, A., Deubzer, H. E., Rycak, L., et al. (2013). Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 24, 75–89. doi: 10.1016/j.ccr.2013.05.005
Buhimschi, A. D., Armstrong, H. A., Toure, M., Figueroa, S. J., Chen, T. L., Lehman, A. M., et al. (2018). Targeting the C481S Ibrutinib-resistance mutation in bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry 57, 3564–3575. doi: 10.1021/acs.biochem.8b00391
Burslem, G. M., and Crews, C. M. (2020). Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 181, 102–114. doi: 10.1016/j.cell.2019.11.031
Cagir, A., and Azmi, A. S. (2019). KRAS(G12C) inhibitors on the horizon. Future Med. Chem. 11, 923–925. doi: 10.4155/fmc-2018-0304
Cheng, S., Guo, A., Lu, P., Ma, J., Coleman, M., and Wang, Y. L. (2015). Functional characterization of BTKC481S mutation that confers ibrutinib resistance: exploration of alternative kinase inhibitors. Leukemia 29, 895–900. doi: 10.1038/leu.2014.263
Chiron, D., Liberto, M. D., Martin, P., Huang, S. G., Sharman, J., Blecua, P., et al. (2014). Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 4, 1022–1035. doi: 10.1158/2159-8290.CD-14-0098
Clendening, J. W., Pandyra, A., Boutros, P. C., Ghamrasni, S. E., Khosravi, F., Trentin, G. A., et al. (2010). Dysregulation of the mevalonate pathway promotes transformation. Proc. Natl. Acad. Sci. U.S.A. 107, 15051–15056. doi: 10.1073/pnas.0910258107
Cohen, D. J., Carrozza, J. P., and Baim, D. S. (1999). Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N. Engl. J. Med. 341, 70–76. doi: 10.1056/NEJM199912093412415
Cromm, P. M., Samarasinghe, K. T. G., Hines, J., and Crews, C. M. (2018). Addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J. Am. Chem. Soc. 140, 17019–17026. doi: 10.1021/jacs.8b08008
Dauch, D., Rudalska, R., Cossa, G., Nault, J. C., Kang, T. W., Wuestefeld, T., et al. (2016). A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 22, 744–753. doi: 10.1038/nm.4107
Dobrovolsky, D., Wang, E. S., Morrow, S., Leahy, C., Faust, T., Nowak, R. P., et al. (2019). Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 133, 952–961. doi: 10.1182/blood-2018-07-862953
Ebrahimi, V., and Hashemi, A. (2020). Challenges of in vitro genome editing with CRISPR/Cas9 and possible solutions: a review. Gene 753:144813. doi: 10.1016/j.gene.2020.144813
Figueroa, S. J., Buhimschi, A. D., Toure, M., Hines, J., and Crews, C. M. (2020). Design, synthesis and biological evaluation of proteolysis targeting chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett. 30:126877. doi: 10.1016/j.bmcl.2019.126877
Fischer, E. S., Böhm, K., Lydeard, J. R., Yang, H. D., Stadler, M. B., Cavadini, S., et al. (2014). Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53. doi: 10.1038/nature13527
Galdeano, C., Gadd, M. S., Soares, P., Scaffidi, S., Molle, I. V., Birced, I., et al. (2014). Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 57, 8657–8663. doi: 10.1021/jm5011258
Gao, H. Y., Sun, X. Y., and Rao, Y. (2020a). PROTAC technology: opportunities and challenges. ACS Med. Chem. Lett. 11, 237–240. doi: 10.1021/acsmedchemlett.9b00597
Gao, H. Y., Zheng, C. W., Du, J., Wu, Y., Sun, Y. H., Han, C. S., et al. (2020b). FAK-targeting PROTAC as a chemical tool for the investigation of non-enzymatic FAK function in mice. Protein Cell 11, 534–539. doi: 10.1007/s13238-020-00732-8
Gao, H. Y., Wu, Y., Sun, Y. H., Yang, Y. Q., Zhou, G. G., and Rao, Y. (2019). Design, synthesis, and evaluation of highly potent FAK-targeting PROTACs. ACS Med. Chem. Lett. 11, 1855–1862. doi: 10.1021/acsmedchemlett.9b00372
Gautier, A., Gauron, C., Volovitch, M., Bensimon, D., Jullien, L., and Vriz, S. (2014). How to control proteins with light in living systems. Nat. Chem. Biol. 10, 533–541. doi: 10.1038/nchembio.1534
Hallin, J., Engstrom, L. D., Hargis, L., Calinisan, A., Aranda, R., Briere, D. M., et al. (2020). The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71. doi: 10.1158/2159-8290.CD-19-1167
Han, X. R., Chen, L. Q., Wei, L., Yu, Q., Yu, W. H., Chen, Y. K., et al. (2020). Discovery of Selective Small Molecule Degraders of BRAF-V600E. J. Med. Chem. 63, 4069–4080. doi: 10.1021/acs.jmedchem.9b02083
Hansen, M. J., Velema, W. A., Lerch, M. M., Szymanski, W., and Feringa, B. L. (2015). Wavelength-selective cleavage of photoprotecting groups: strategies and applications in dynamic systems. Chem. Soc. Rev. 44, 3358–3377. doi: 10.1039/c5cs00118h
Hartman, Z. R., Schaller, M. D., and Agazie, Y. M. (2013). The tyrosine phosphatase SHP2 regulates focal adhesion kinase to promote EGF-induced lamellipodia persistence and cell migration. Mol. Cancer Res. 11, 651–664. doi: 10.1158/1541-7786
He, M. L., Bakken, T., Kassimova, A., Boshoff, C., Philpott, N., and Cannon, M. L. (2012). Focal adhesion kinase is required for KSHV vGPCR signaling. Mol. Carcinog. 51, 339–351. doi: 10.1002/mc.20790
Holderfield, M., Deuker, M. M., McCormick, F., and McMahon, M. (2014). Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 14, 455–467. doi: 10.1038/nrc3760
Holt, D. A., Luengo, J. I., Yamashita, D. S., Oh, H. J., Konialian, A. L., Yen, H. K., et al. (1993). Design, synthesis, and kinetic evaluation of high-affinity FKBP ligands and the X-ray crystal-structures of their complexes with FKBP12. J. Am. Chem. Soc. 115, 9925–9938. doi: 10.1021/ja00075a008
Huang, H. T., Dobrovolsky, D., Paulk, J., Yang, G., Weisberg, E. L., Doctor, Z. M., et al. (2018). A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 25, 88–99. doi: 10.1016/j.chembiol.2017.10.005
Huse, M., Chen, Y. G., Massagué, J., and Kuriyan, J. (1999). Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell 96, 425–436. doi: 10.1016/s0092-8674(00)80555-3
Itoh, Y., Ishikawa, M., Naito, M., and Hashimoto, Y. (2010). Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 132, 5820–5826. doi: 10.1021/ja100691p
Jiang, B. S., Wang, E. S., Donovan, K. A., Liang, Y. K., Fischer, E. S., Zhang, T. H., et al. (2019). Development of dual and selective degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem. Int. Ed. Engl. 58, 6321–6326. doi: 10.1002/anie.201901336
Jin, Y. H., Lu, M. C., Wang, Y., Shan, W. X., Wang, X. Y., You, Q. D., et al. (2020). Azo-PROTAC: novel light-controlled small-molecule tool for protein knockdown. J. Med. Chem. 63, 4644–4654. doi: 10.1021/acs.jmedchem.9b02058
Karoulia, Z., Gavathiotis, E., and Poulikakos, P. I. (2017). New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 17, 676–691. doi: 10.1038/nrc.2017.79
Kessler, D., Gmachl, M., Mantoulidis, A., Martin, L. J., Zoephel, A., Mayer, M., et al. (2019). Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. U.S.A. 341, 70–76. doi: 10.1056/NEJM199912093412415
Kessler, M., Glatthar, R., Giese, B., and Bochet, C. G. (2003). Sequentially photocleavable protecting groups in solid-phase synthesis. Org. Lett. 5, 1179–1181. doi: 10.1021/ol027454g
Kounde, C. S., Shchepinova, M. M., Saunders, C. N., Muelbaier, M., Rackham, M. D., Harling, J. D., et al. (2020). A caged E3 ligase ligand for PROTAC-mediated protein degradation with light. Chem. Commun. 56, 5532–5535. doi: 10.1039/d0cc00523a
Krämer, O. H., Mahboobi, S., and Sellmer, A. (2014). Drugging the HDAC6-HSP90 interplay in malignant cells. Trends Pharmacol. Sci. 35, 501–509. doi: 10.1016/j.tips.2014.08.001
Lai, A. C., and Crews, C. M. (2017). Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114. doi: 10.1038/nrd.2016.211
Lai, A. C., Toure, M., Hellerschmied, D., Salami, J., Jaime-Figueroa, S., Ko, E., et al. (2016). Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 55, 807–810. doi: 10.1002/anie.201507634
Leonard, J. P., LaCasce, A. S., Smith, M. R., Noy, A., Chirieac, L. R., Rodig, S. J., et al. (2012). Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 119, 4597–4607. doi: 10.1182/blood-2011-10-388298
Li, M. X., Yang, Y. Q., Zhao, Q. Y., Wu, Y., Song, L., Yang, H. Y., et al. (2020). Degradation versus inhibition: development of proteolysis-targeting chimeras for overcoming statin-induced compensatory upregulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Med. Chem. 63, 4908–4928. doi: 10.1021/acs.jmedchem.0c00339
Li, Z. Q., Razavi, P., Li, Q., Toy, W. Y., Liu, B., Ping, C., et al. (2018). Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the Hippo pathway. Cancer Cell 34, 893–905. doi: 10.1016/j.ccell.2018.11.006
Lito, P., Rosen, N., and Solit, D. B. (2013). Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 19, 1401–1409. doi: 10.1038/nm.3392
Liu, J., Chen, H., Ma, L. N., He, Z. X., Wang, D., Liu, Y., et al. (2020). Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 6:eaay5154. doi: 10.1126/sciadv.aay5154
Lu, S. Y., Jang, H., Gu, S., Zhang, J., and Nussinov, R. (2016). Drugging Ras GTPase: a comprehensive mechanistic and signaling structural view. Chem. Soc. Rev. 45, 4929–4952. doi: 10.1039/c5cs00911a
Lu, X. Y., Zhang, Z., Ren, X. M., Pan, X. F., Wang, D. P., Zhuang, X. X., et al. (2015). Hybrid pyrimidine alkynyls inhibit the clinically resistance related Bcr-Abl(T315I) mutant. Bioorg. Med. Chem. Lett. 25, 3458–3463. doi: 10.1016/j.bmcl.2015.07.006
Madisen, L., Mao, T. Y., Koch, H., Zhuo, J. M., Berenyi, A., Fujisawa, S., et al. (2012). A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci. 15, 793–802. doi: 10.1038/nn.3078
Marumoto, T., Zhang, D. W., and Saya, H. (2005). Aurora-A - a guardian of poles. Nat. Rev. Cancer 5, 42–50. doi: 10.1038/nrc1526
McLemore, M. L., Grewal, S., Liu, F., Archambault, A., Poursine-Laurent, J., Haug, J., et al. (2001). STAT-3 activation is required for normal G-CSF-dependent proliferation and granulocytic differentiation. Immunity 14, 193–204. doi: 10.1016/s1074-7613(01)00101-7
Medina, M. W., and Krauss, R. M. (2009). The Role of HMGCR alternative splicing in statin efficacy. Trends Cardiovasc. Med. 19, 173–177. doi: 10.1016/j.tcm.2009.10.003
Mello, C. C., and Conte, D. (2004). Revealing the world of RNA interference. Nature 431, 338–342. doi: 10.1038/nature02872
Miyake, Y., Keusch, J. J., Wang, L. L., Saito, M., Hess, D., Wang, X. N., et al. (2016). Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 12, 748–754. doi: 10.1038/nchembio.2140
Nabet, B., Roberts, J. M., Buckley, D. L., Paulk, J., Dastjerdi, S., Yang, A., et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441. doi: 10.1038/s41589-018-0021-8
Naro, Y., Darrah, K., and Deiters, A. (2020). Optical control of small molecule-induced protein degradation. J. Am. Chem. Soc. 142, 2193–2197. doi: 10.1021/jacs.9b12718
Neklesa, T., Snyder, L. B., Willard, R. R., and Vitale, N. (2019). ARV-110: an oral androgen receptor PROTAC degrader for prostate cancer. J. Clin. Oncol. 37:259. doi: 10.1200/JCO.2019.37.7_suppl.259
Niesvizky, R., Badros, A. Z., Costa, L. J., Ely, S. A., Singhal, S. B., Stadtmauer, E. A., et al. (2015). Phase 1/2 study of cyclin-dependent kinase (CDK)4/6 inhibitor palbociclib (PD-0332991) with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Leuk. Lymphoma 56, 320–328. doi: 10.3109/10428194.2015.1030641
Nigg, E. A. (2001). Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2, 21–32. doi: 10.1038/35048096
Ottis, P., and Crews, C. M. (2017). Proteolysis-targeting chimeras: induced protein degradation as a therapeutic strategy. ACS Chem. Biol. 12, 892–898. doi: 10.1021/acschembio.6b01068
Otto, T., Horn, S., Brockmann, M., Eilers, U., Schüttrumpf, L., Popov, N., et al. (2009). Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 15, 67–78. doi: 10.1016/j.ccr.2008.12.005
Otto, T., and Sicinski, P. (2017). Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 93–115. doi: 10.1038/nrc.2016.138
Pan, Z. Y., Scheerens, H., Li, S. J., Schultz, B. E., Sprengeler, P. A., Burrill, L. C., et al. (2007). Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. Chem. Med. Chem. 2, 58–61. doi: 10.1002/cmdc.200600221
Patton, J. T., Mayo, L. D., Singhi, A. D., Gudkov, A. V., Stark, G. R., and Jackson, M. W. (2006). Levels of HdmX expression dictate the sensitivity of normal and transformed cells to nutlin-3. Cancer Res. 66, 3169–3176. doi: 10.1158/0008-5472.CAN-05-3832
Pfaff, P., Samarasinghe, K. T. G., Crews, C. M., and Carreira, E. M. (2019). Reversible spatiotemporal control of induced protein degradation by bistable PhotoPROTACs. ACS Cent. Sci. 5, 1682–1690. doi: 10.1021/acscentsci.9b00713
Pophali, P. A., and Patnaik, M. M. (2016). The role of new tyrosine kinase inhibitors in chronic myeloid leukemia. Cancer J. 22, 40–50. doi: 10.1097/PPO.0000000000000165
Popow, J., Arnhof, H., Bader, G., Berger, H., Ciulli, A., Covini, D., et al. (2019). Highly selective PTK2 proteolysis targeting chimeras to probe Focal Adhesion Kinase scaffolding functions. J. Med. Chem. 62, 2508–2520. doi: 10.1021/acs.jmedchem.8b01826
Posternak, G., Tang, X. J., Maisonneuve, P., Jin, T., Lavoie, H., Daou, S., et al. (2020). Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat. Chem. Biol. 16, 1170–1178. doi: 10.1038/s41589-020-0609-7
Pylayeva-Gupta, Y., Grabocka, E., and Bar-Sagi, D. (2011). RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774. doi: 10.1038/nrc3106
Reynders, M., Matsuura, B. S., Bérouti, M., Simoneschi, D., Marzio, A., Pagano, M., et al. (2020). PHOTACs enable optical control of protein degradation. Sci. Adv. 6:eaay5064. doi: 10.1126/sciadv.aay5064
Rojas, A. M., Fuentes, G., Rausell, A., and Valencia, A. (2012). The Ras protein superfamily: evolutionary tree and role of conserved amino acids. J. Cell Biol. 196, 189–201. doi: 10.1083/jcb.201103008
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U.S.A. 98, 8554–8559. doi: 10.1073/pnas.141230798
Samatar, A. A., and Poulikakos, P. I. (2014). Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov. 13, 928–942. doi: 10.1038/nrd4281
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. doi: 10.1016/j.cell.2017.03.035
Schneekloth, A. R., Pucheault, M., Tae, H. S., and Crews, C. M. (2008). Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg. Med. Chem. Lett. 18, 5904–5908. doi: 10.1016/j.bmcl.2008.07.114
Serrels, B., Serrels, A., Brunton, V. G., Holt, M., McLean, G. W., Gray, C. H., et al. (2007). Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 9, 1046–1056. doi: 10.1038/ncb1626
Shimokawa, K., Shibata, N., Sameshima, T., Miyamoto, N., Ujikawa, O., Nara, H., et al. (2017). Targeting the allosteric site of oncoprotein BCR-ABL as an alternative strategy for effective target protein degradation. ACS Med. Chem. Lett. 8, 1042–1047. doi: 10.1021/acsmedchemlett.7b00247
Simões-Pires, C., Zwick, V., Nurisso, A., Schenker, E., Carrupt, P., and Cuendet, M. (2013). HDAC6 as a target for neurodegenerative diseases: what makes it different from the other HDACs? Mol. Neurodegener. 8:7. doi: 10.1186/1750-1326-8-7
Stegmaier, P., Alonso, J. M., and Campo, A. (2008). Photoresponsive surfaces with two independent wavelength-selective functional levels. Langmuir 24, 11872–11879. doi: 10.1021/la802052u
Su, S., Yang, Z. M., Gao, H. Y., Yang, H. Y., Zhu, S. B., An, Z. X., et al. (2019). Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J. Med. Chem. 62, 7575–7582. doi: 10.1021/acs.jmedchem.9b00871
Sun, X. Y., Gao, H. Y., Yang, Y. Q., He, M., Wu, Y., Song, Y. G., et al. (2019). PROTACs: great opportunities for academia and industry. Signal Transduct. Target Ther. 4, 64–96. doi: 10.1038/s41392-019-0101-6
Sun, X. Y., and Rao, Y. (2020). PROTACs as potential therapeutic agents for cancer drug resistance. Biochemistry 59, 240–249. doi: 10.1021/acs.biochem.9b00848
Sun, X. Y., Wang, J., Yao, X., Zheng, W., Mao, Y., Lan, T. L., et al. (2018). A chemical approach for global protein knockdown from mice to non-human primates. Cell Discov. 5:10. doi: 10.1038/s41421-018-0079-1
Sun, Y. H., Ding, N., Song, Y. Q., Yang, Z. M., Liu, W. L., Zhu, J., et al. (2019). Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia 33, 2105–2110. doi: 10.1038/s41375-019-0440-x
Sun, Y. H., Zhao, X. W., Ding, N., Gao, H. Y., Wu, Y., Yang, Y. Q., et al. (2018). PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 28, 779–781. doi: 10.1038/s41422-018-0055-1
Szymanski, W., Beierle, J. M., Kistemaker, H. A. V., Velema, W. A., and Feringa, B. L. (2013). Reversible photocontrol of biological systems by the incorporation of molecular photoswitches. Chem. Rev. 113, 6114–6178. doi: 10.1021/cr300179f
Tabe, Y., Sebasigari, D., Rudelius, M., Pittaluga, S., and Raffel, M. (2007). The MDM2 antagonist nutlin-3 is lethal to mantle cell lymphoma with wild type p53. Blood 110:1382. doi: 10.1182/blood.V110.11.1382.1382
Tadesse, S., Caldon, E. C., Tilley, W., and Wang, S. D. (2019). Cyclin-dependent kinase 2 inhibitors in cancer therapy: an update. J. Med. Chem. 62, 4233–4251. doi: 10.1021/acs.jmedchem.8b01469
Tadesse, S., Yu, M. F., Kumarasiri, M., Le, B. T., and Wang, S. D. (2015). Targeting CDK6 in cancer: state of the art and new insights. Cell Cycle 14, 3220–3230. doi: 10.1080/15384101.2015.1084445
Takada, M., Zhang, W. G., Suzuki, A., Kuroda, T. S., Yu, Z. L., Inuzuka, H., et al. (2017). FBW7 loss promotes chromosomal instability and tumorigenesis via cyclin E1/CDK2-mediated phosphorylation of CENP-A. Cancer Res. 77, 4881–4893. doi: 10.1158/0008-5472.CAN-17-1240
Takeda, K., Noguchi, K., Shi, W., Tanaka, T., Matsumoto, M., Yoshida, N., et al. (1997). Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. U.S.A. 94, 3801–3804. doi: 10.1073/pnas.94.8.3801
Talpaz, M., Shah, N. P., Kantarjian, H., Donato, N., Nicoll, J., Paquette, R., et al. (2006). Dasatinib in imatinib-resistant philadelphia chromosome-positive leukemias. N. Engl. J. Med. 354, 2531–2541. doi: 10.1056/NEJMoa055229
Tamura, R., Balabanova, A., Frakes, S. A., Bargmann, A., Grimm, J. A., Koch, T. H., et al. (2019). Photoactivatable prodrug of doxazolidine targeting exosomes. J. Med. Chem. 62, 1959–1970. doi: 10.1021/acs.jmedchem.8b01508
Teague, S. J., and Stocks, M. J. (1993). The affinity of the excised binding domain of FK-506 for the immunophilin FKBP12. Bioorg. Med. Chem. Lett. 3, 1947–1950. doi: 10.1016/S0960-894X(01)80992-6
Toure, M., and Crews, C. M. (2016). Small-molecule PROTACS: new approaches to protein degradation. Angew. Chem. Int. Ed. Engl. 55, 1966–1973. doi: 10.1002/anie.201507978
Toya, M., Terasawa, M., Nagata, K., Iida, Y., and Sugimoto, A. (2011). A kinase-independent role for Aurora A in the assembly of mitotic spindle microtubules in Caenorhabditis elegans embryos. Nat. Cell Biol. 13, 708–714. doi: 10.1038/ncb2242
Vermeulen, K., Bockstaele, D. R. V., and Berneman, Z. N. (2003). The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 36, 131–149. doi: 10.1046/j.1365-2184.2003.00266.x
Wang, L. G., Shao, X. J., Zhong, T. B., Wu, Y., Xu, A. X., Sun, X. Y., et al. (2021). Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nat. Chem. Biol. 17, 567–575. doi: 10.1038/s41589-021-00742-5
Wang, M. L., Rule, S., Martin, P., Goy, A., Auer, R., Kahl, B. S., et al. (2013). Targeting BTK with Ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 369, 507–516. doi: 10.1056/NEJMoa1306220
Wiedeman, P. E., Fesik, S. W., Petros, A. M., Nettesheim, D. G., Mollison, K. W., Lane, B. C., et al. (1999). Retention of immunosuppressant activity in an ascomycin analogue lacking a hydrogen-bonding interaction with FKBP12. J. Med. Chem. 42, 4456–4461. doi: 10.1021/jm980252z
Winter, G. E., Buckley, D. L., Paulk, J., Roberts, J. M., Souza, A., Dhe-Paganon, S., et al. (2015). Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. doi: 10.1126/science.aab1433
Wu, H., Yang, K., Zhang, Z. R., Leisten, E. D., Li, Z. Y., and Xie, H. B. (2019). Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J. Med. Chem. 62, 7042–7057. doi: 10.1021/acs.jmedchem.9b00516
Xue, G., Wang, K., Zhou, D. L., Zhong, H. B., and Pan, Z. Y. (2019). Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc. 141, 18370–18374. doi: 10.1021/jacs.9b06422
Yang, C., Li, Z., Bhatt, T., Dickler, M., Giri, D., Scaltriti, M., et al. (2017). Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36, 2255–2264. doi: 10.1038/onc.2016.379
Yang, H. Y., Lv, W. X., He, M., Deng, H. T., Li, H. T., Wu, W., et al. (2019). Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. 55, 14848–14851. doi: 10.1039/c9cc08509b
Yang, K., and Fu, L. W. (2015). Mechanisms of resistance to BCR-ABL TKIs and the therapeutic strategies: a review. Crit. Rev. Oncol. Hematol. 93, 277–292. doi: 10.1016/j.critrevonc.2014.11.001
Yang, K., Song, Y. L., Xie, H. B., Wu, H., Wu, Y. T., Leisten, E. D., et al. (2018). Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 28, 2493–2497. doi: 10.1016/j.bmcl.2018.05.057
Yang, K., Wu, H., Zhang, Z. R., Leisten, E. D., Nie, X. Q., Liu, B. K., et al. (2020). Development of selective histone deacetylase 6(HDAC6) degraders recruiting Von Hippel-Lindau (VHL) E3 ubiquitin ligase. ACS Med. Chem. Lett. 11, 575–581. doi: 10.1021/acsmedchemlett.0c00046
Yang, Y. Q., Gao, H. Y., Sun, X. Y., Sun, Y. H., Qiu, Y. P., Weng, Q. J., et al. (2020). A global PROTAC toolbox for degrading BCR-ABL overcomes drug-resistant mutants and adverse effects. J. Med. Chem. 63, 8567–8583. doi: 10.1021/acs.jmedchem.0c00967
Ying, M. D., Shao, X. J., Jing, H., Liu, Y. J., Qi, X. T., Cao, L., et al. (2018). Ubiquitin-dependent degradation of CDK2 drives the therapeutic diferentiation of AML by targeting PRDX2. Blood 131, 2698–2711. doi: 10.1182/blood-2017-10-813139
Zeng, W. J., Xiong, C. M., Zhao, L., Shan, G. L., Liu, Z. H., Xue, F., et al. (2012). Atorvastatin in Pulmonary Arterial Hypertension (APATH) study. Eur. Respir. J. 40, 67–74. doi: 10.1183/09031936.00149011
Zengerle, M., Chan, K. H., and Ciulli, A. (2015). Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 10, 1770–1777. doi: 10.1021/acschembio.5b00216
Zhang, H. Y., Aaonbangkhen, C., Tarasovetc, E. V., Ballister, E., Chenoweth, D. M., and Lampson, M. A. (2017). Optogenetic control of kinetochore function. Nat. Chem. Biol. 13, 1096–1101. doi: 10.1038/nchembio.2456
Zhang, X. H., Yuan, Z. G., Zhang, Y. T., Yong, S., Salas-Burgos, A., Koomen, J., et al. (2007). HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell. 27, 197–213. doi: 10.1016/j.molcel.2007.05.033
Zhao, B. S., and Burgess, K. (2019). PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem. Commun. 55, 2704–2707. doi: 10.1039/c9cc00163h
Zhao, Q. J., Ren, C. W., Liu, L. Y., Chen, J. J., Shao, Y. B., Sun, N., et al. (2019). Discovery of SIAIS178 as an effective BCR-ABL degrader by recruiting Von Hippel-Lindau (VHL) E3 ubiquitin ligase. J. Med. Chem. 62, 9281–9298. doi: 10.1021/acs.jmedchem.9b01264
Zhao, Q. Y., Lan, T. L., Su, S., and Rao, Y. (2019). Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem. Commun. 55, 369–372. doi: 10.1039/c8cc07813k
Zheng, F. M., Yue, C. F., Li, G. H., He, B., Cheng, W., Wang, X., et al. (2016). Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat. Commun. 7:10180. doi: 10.1038/ncomms10180
Zorba, A., Nguyen, C., Xu, Y. R., Starr, J., Borzilleri, K., Smith, J., et al. (2018). Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. U.SA. 115, E7285–E7292. doi: 10.1073/pnas.1803662115
Keywords: PROTAC, protein degradation, E3 ubiquitin ligase, proteasome, small molecule inhibitor
Citation: He M, Lv W and Rao Y (2021) Opportunities and Challenges of Small Molecule Induced Targeted Protein Degradation. Front. Cell Dev. Biol. 9:685106. doi: 10.3389/fcell.2021.685106
Received: 24 March 2021; Accepted: 24 May 2021;
Published: 22 June 2021.
Edited by:
Baoting Zhang, The Chinese University of Hong Kong, ChinaReviewed by:
Qingping Dou, Wayne State University, United StatesCopyright © 2021 He, Lv and Rao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Rao, eXJhb0B0c2luZ2h1YS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.