 Yayan Feng1,2†
Yayan Feng1,2† Xiaolei Liu3†
Xiaolei Liu3† Yuqi Liu1Bin Tang3
Yuqi Liu1Bin Tang3 Xue Bai3Chen Li3
Xue Bai3Chen Li3 Xuelin Wang3Yiqun Deng2
Xuelin Wang3Yiqun Deng2 Fei Gao1,4*
Fei Gao1,4* Mingyuan Liu3,5*
Mingyuan Liu3,5*- 1Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen, China
- 2Guangdong Provincial Key Laboratory of Protein Function and Regulation in Agricultural Organisms, College of Life Sciences, South China Agricultural University, Guangzhou, China
- 3Key Laboratory of Zoonosis Research, Ministry of Education, Institute of Zoonosis/College of Veterinary Medicine, Jilin University, Changchun, China
- 4Comparative Pediatrics and Nutrition, Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Frederiksberg, Denmark
- 5Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou, Jiangsu, China
Comparative epigenomics provides new insights on evolutionary biology in relation with complex interactions between species and their environments. In the present study, we focus on deciphering the conservation and divergence of DNA methylomes during Trichinella evolution. Whole-genome bisulfite sequencing and RNA-seq were performed on the two clades of Trichinella species, in addition to whole-genome sequencing. We demonstrate that methylation patterns of sing-copy orthologous genes (SCOs) of the 12 Trichinella species are host-related and can mirror known phylogenetic relationships. Among these SCOs, we identify a panel of genes exhibiting hyper-/hypo-methylated features in gene-bodies or respective promoters that play pivotal roles in transcriptome regulation. These hyper-/hypo-methylated SCOs are also of functional significance across developmental stages, as they are highly enriched species-specific and stage-specific expressed genes both in Ad and ML stages. We further identify a set of parasitism-related functional genes that exhibit host-related differential methylation and expression among those SCOs, including p53-like transcription factor and Cdc37 that are of functional significance for elucidating differential parasitology between the two clades of Trichinella. This comparative epigenome study can help to decipher the environmental effects on differential adaptation and parasitism of the genus Trichinella.
Introduction
Dissecting complex interaction between species and their environments has long been a research hot spot in ecology and evolutionary biology (Duncan et al., 2014). Various factors, such as dietary components, temperature changes, and other external stresses, could modulate the establishment and maintenance of epigenetic modifications, thereby influencing gene expression and phenotype (Feil and Fraga, 2012). As the most studied type of epigenetic modifications found in many taxa, DNA methylation has been confirmed to play a crucial role in transposon silencing, transcriptional regulation, and thus rapid phenotypic variation, which could provide us a deep understanding of how species epigenetically cope with rapidly changing environments (Bender, 2004).
Trichinella spp. are the causative agents of human trichinellosis, a parasitic disease that causes substantial morbidity and mortality in billions of animals and humans worldwide, as well as significant losses to the global food production annually (Wang and Cui, 2001; Gottstein et al., 2009; Jiang et al., 2016). All 12 recognized taxa of Trichinella spp. identified by molecular means and phylogenetic analyses are genetically and biologically delineated into two distinct clades characterized by the presence or absence of an intramuscular collagen capsule (Zarlenga et al., 2006). Thus, one clade is represented by all species and taxa that accordingly encapsulate in host muscle tissue (T. spiralis, T1; T. nativa, T2; T. britovi, T3; T. murrelli, T5; T. nelsoni, T7; T. patagoniensis, T12; and Trichinella genotypes T6, T8, and T9), and the other one does not encapsulate after muscle cell dedifferentiation (T. pseudospiralis, T4; T. papuae, T10; and T. zimbabwensis, T11). Both evolutionary clades share the same life cycles occupying two distinct intracellular niches, intestinal epithelium, and skeletal muscle cells. The muscle larvae (ML) of Trichinella, released by host gastric fluids, invade intestine epithelium and subsequently develop into adult worms (Ad). The newborn larvae (NBL) delivered by female adults migrate to skeletal muscles and invade muscle cells where they develop into ML and survive for years (Gottstein et al., 2009). Despite the similarity in life cycles, the two clades showed substantial differences in parasitological, pathological, and immunological characteristics (Wu et al., 2001, 2002; Boonmars et al., 2005). It has been demonstrated that these differences were driven by post-miocene expansion, colonization, and host switching (Zarlenga et al., 2006). Moreover, the two clades also differed significantly from each other in classes of host species. The encapsulated species can only complete their life cycle in mammals by requiring host body temperature ranging from 37 to 40°C, whereas the non-encapsulated species shows a broader host spectrum, including mammals, birds, and reptiles, as it can complete its life cycle at host body temperature ranging from 25 to 42°C (Pozio, 2005). Previous studies have demonstrated that host body temperature had a significant impact on the epidemiology and parasitology of the parasites using experimental and natural infections of reptiles with all of the encapsulated species and non-encapsulated species, T. pseudospiralis (Pozio, 2005). Similar results were also obtained in other experimental infections of reptiles belonging to other orders (Loricata, Squamata, and Chelonide) (Pozio et al., 2004). However, experimental infections of birds (e.g., hens, ducks, pigeons, crows, and herons) have shown that these vertebrates are suitable hosts only for non-encapsulated species, T. pseudospiralis (Kapel, 2000). Therefore, vertebrate host classes could act as crucial environmental factors to be capable of activating different physiological and immunological mechanisms for parasite development and survival.
Previously, we have characterized DNA methylation profiles in T. spiralis and revealed its functional roles on life cycle transition and transcriptome regulation (Gao et al., 2012). Further, we recently documented substantial epigenome differences between the two representative species T. pseudospiralis and T. spiralis, and confirmed these epigenomic divergences could induce differential interactions at the host-parasite interface (Liu et al., 2021). As a deep coevolutionary association between Trichinella species and vertebrate host classes has been investigated using both nuclear small-subunit rDNA (Zarlenga et al., 2006) and protein-coding gene sets (Korhonen et al., 2016), we speculate that epigenetics in relation with host diversity also play a key role in the evolution and adaptation of the 12 Trichinella species.
Here, we performed whole-genome, methylome, and transcriptome sequencing on the 12 Trichinella species. We observed that methylome divergence of these Trichinella species is host-related, and this epigenomic conservation is a regulatory mechanism of transcription. By dividing SCOs into hyper- or hypo-methylated categories, we showed that these two classes of genes exhibiting functional significance on gene expression, especially for hypo-methylated ones, which are related to active transcription. We further revealed that stage-specific expressed genes are also highly enriched in those genes of the two categories. By comparing DNA methylation patterns between the two clades of Trichinella, we identified a panel of parasitism-related genes exhibiting differential expression under epigenetic regulation, including p53-like transcription factor and Cdc37, etc.
Materials and Methods
Parasite Culture and Collection
Twelve isolates representing all 12 recognized species and genotypes of Trichinella were provided by the International Trichinella Reference Center and preserved by serial passage in BALB/c mice in OIE Collaborating Center on Foodborne Parasites in Asian-Pacific Region. The 12 isolates included the following: T. spiralis (ISS534), T. nativa (ISS70), T. britovi (ISS120), T. pseudospiralis (ISS13), T. murrelli (ISS417), T. nelsoni (ISS37), T. papuae (ISS1980), T. zimbabwensis (ISS1029), and T. patagoniensis (ISS2496) as well as Trichinella genotypes T6 (ISS34), T8 (ISS124), and T9 (ISS409). Muscle larvae (ML), adult worms (Ad), and newborn larvae (NBL) were recovered as described previously (Liu et al., 2011). Briefly, infectious muscle larvae were harvested by 1% pepsin-HCl digestion of minced muscle tissue from rats 35 days post-infection (dpi). Purified adult worms and newborn larvae were collected from intestines of Wistar rats at 30 h or 6 dpi which were inoculated with each Trichinella species with a dose of 8000 larvae per rat. Each sample was snap-frozen in liquid nitrogen and stored at −80°C until nucleic acid isolation. The intestines were then fragmented and further incubated with 0.9% sodium chloride solutions at 37°C for 3 h. Adult worms were harvested by centrifugation. To obtain newborn larvae, adult worms collected at 6 dpi were incubated in Iscove’s Modified Dulbecco’s Medium (IMDM) in 75-cm2 cell culture plates at 37°C. The newborn larvae were harvested every 12 h. The animals were treated according to the guidelines of the National Institutes of Health (publication no. 85-23, revised 1996).
Identification of SCOs in Trichinella spp.
The genomic data resources of the 12 Trichinella species were downloaded from NCBI BioProject database with the following accession codes: PRJNA12603 for T. spiralis, PRJNA451013 for T. pseudospiralis, and PRJNA257433 for 10 other Trichinella species. OrthoMCL (Fischer et al., 2011) was used to cluster protein sequences of the 12 Trichinella species into orthologous groups based on their sequence similarity. As a result, a total of 2708 SCOs among these 12 Trichinella species were identified (Supplementary Data 1).
Whole-Genome Sequencing and Phylogenetic Analysis
High molecular DNA from the 12 Trichinella species were extracted using the phenol-chloroform extraction method from freshly collected muscle larvae. Ultraviolet-Vis spectrophotometry with a NanoDrop 2000 (Thermo Fisher Scientific, CA, United States) was used to determine the quantity and quality of total DNA. Paired-end sequencing libraries with an insert size of 300 bp were conducted on HiSeq 2,500 platform, according to instructions provided by Illumina. FastQC (v0.11.7)1 was used to assess the quality of sequence reads. Adapters and low-quality reads were removed from raw sequencing reads by Trimmomatic (v0.36) (Bolger et al., 2014) with the following parameters: LEADING:3, TRAILING:3, SLIDINGWINDOW:4:15, MINLEN:75. Clean reads from each species were mapped to the respective reference genome with BWA-MEM algorithm (Li and Durbin, 2010). Approximately 86.5% of these clean reads could be uniquely aligned to the respective reference genome (Supplementary Data 2). The mapping results were converted to bam format by SAMtools (v1.6) (Li et al., 2009). The resulting bam files were sorted by coordinate using SortSam.jar embedded in Picard-tools (v1.118). The flow chart in Supplementary Figure 1 describes the strategies we used to identify single nucleotide variant (SNVs) among the 12 Trichinella species. Firstly, we conducted intraspecific single nucleotide polymorphism (SNPs) by Genome Analysis Tool Kit (GATK). Subsequently, we used MUMmer (v3.0) (Kurtz et al., 2004) to examine interspecific SNPs between reference genomes of T. pseudospiralis and other 11 Trichinella species. Lastly, SNPs from uniquely aligned regions with homozygous genotypes (GT = 1/1) constituted the final set of SNVs. In total, we obtained 5,395,250 common SNVs across the 12 Trichinella species. FastTreeMP2 was used to construct a phylogenetic tree of the 12 Trichinella species under GTR+CAT model based on the concatenated common SNVs.
WGBS Library Construction, Sequencing, and Data Analysis
Genomic DNA were extracted using phenol-chloroform extraction method. The quantity and quality of DNA were tested by ultraviolet-Vis spectrophotometry with a NanoDrop 2000 (Thermo Fisher Scientific, CA, United States). Genomic DNA from the three life stages (Ad, ML, and NBL) of all Trichinella species were selected for WGBS library construction as previously described (Jeong et al., 2016). Briefly, approx. two to five micrograms of genomic DNA were first fragmented by Covaris to an average size of 200–300 bp. End-repair, A-tailing, and ligation were conducted as the Illumina PE genomic DNA sample prep kit protocol, except that ligation was performed using methylated cytosine PE adapters provided by Illumina previously (Jeong et al., 2016). Ligation products were purified and bisulfite converted using the EZ DNA Methylation-Gold Kit (ZYMO) according to the manufacturer’s instructions. The libraries were sequenced using Illumina HiSeq 2,500 platform.
After base calling, raw sequencing reads with more than 30% “N”s in over 10% of the reads or smaller than 30 bp were discarded. Clean reads were aligned to the respective reference genome using BSMAP (v2.73) with a maximal four mismatches and gap size up to 2 nucleotides (Xi and Li, 2009). The methylation level of each cytosine was calculated as the number of reads ascertained methylated to the total number of reads for each covered CpG site. Density methylation of any genomic region was measured by the number of methylated CpGs to the total number of CpG sites. To accurately investigate methylation levels in each species, we used CpG sites with a minimal 5× sequencing depth per strand. In total, an average of 33 million reads was generated for each species, among which 84.28% could be uniquely aligned to the respective reference genome after quality control, reaching an average read depth of 19.55× at the covered methylated sites. The mean bisulfite conversion rate of C to T was up to 99.48% (Supplementary Data 2).
RNA-Seq Library Construction, Sequencing, and Data Analysis
Total RNA of the three life stages (Ad, NBL, and ML) of all Trichinella species were extracted using Trizol reagent (Invitrogen, CA, United States) according to the manufacturer’s instructions. Paired-end sequencing libraries with an insert size of 300 bp were conducted on the HiSeq 2,500 platform following Illumina’s protocols. SOAPnuke (Chen et al., 2018) was used for trimming adapters and filtering on length and quality of the raw sequencing reads, with the following parameters: -q 0.5 -d -Q 2 –sanger -l 20. Clean reads were mapped to the respective reference genome using hisat2 (Kim et al., 2015) with default settings. The gene expression levels were quantified using Cufflinks (v2.2.1) (Trapnell et al., 2012) with reads per kilobase million (RPKM). We obtained an average of 41 million raw reads for the three life stages of each species. After quality control and removal of duplication, approximately 66.59% could be uniquely mapped to the respective reference genome. Of total predicted genes in the respective reference genome, the expression levels of 9,060 genes (82.95%) were quantified based on sequenced reads, averagely (Supplementary Data 2).
We adopted the following approaches to identify stage-specific expressed genes for each of these Trichinella species. Firstly, differentially expressed genes between any two developmental stages were identified using DESeq2 (Love et al., 2014). Subsequently, genes exhibiting differential expression both in Ad and NBL stages when comparing with ML stage, whereas differential expression was not observed between Ad and NBL stages, were identified as stage-specifically expressed genes by ML stage (multiple-testing corrected P ≤ 0.05). The same forward approach was applied to identify genes that were specifically expressed by Ad and NBL stages.
Quantifying Evolutionary Changes of DNA Methylation Levels, Gene Expression Levels, and Genomic Sequences
To investigate the evolutionary associations among DNA methylation levels, gene expression levels, and genomic sequences, we examined multiple alignments of the SCOs between T. pseudospiralis and 10 other Trichinella species using mafft (v7.158b) with automatic detection strategy (except for T. spiralis). Then we quantified the epigenetic conservation level using the P-values generated by comparing the common mCGs of orthologous genes between the two species under consideration. In each comparison, the expression level of every gene was regressed to the DNA methylation level of the upstream 2-Kb region of the gene. The predicted interspecies gene expression differences were regressed to the epigenomic differences. Sequence differences were estimated using blastn score differences by comparing differences between the maximum log blastn score of all orthologous promoters (4,000 bp centered at transcriptional start sites) and the blastn score of the orthologous promoter pair under consideration (Xiao et al., 2012).
Classification of SCOs Into Hyper- and Hypo-Methylation Categories
We used two different approaches to distinguish genes into high and low methylation categories following the method described in Sarda et al. (2012). We first used the observed vs. expected proportion of CpG dinucleotides (CpGO/E), which could assess the depletion of CpG dinucleotides for genomic regions of interest. Briefly, this measurement is described as:
where PCpG, PC, and PG represent the frequencies of CpG dinucleotides, C nucleotides, and G nucleotides, respectively. Genes with a CpGO/E value lower than 0.8 were classified as hyper-methylated genes, whereas genes with CpGO/E greater than 1.0 were classified as hypo-methylated genes. In the second approach, we divided genes into high and low categories using fraction methylation levels. If the methylation level of a gene is 0.01 lower than the average methylation level of whole genes, thus the gene could be classified as a low category. Otherwise, the gene could be classified as a high category. The cut-off values of the two approaches were chosen manually, based on density distributions of the respective data. Genes meeting the above two criteria were classified as hyper- or hypo-methylated SCOs. The same forward approach was also applied to promoter regions of the 2708 SCOs.
Statistical Analysis
Hierarchical clustering analysis was performed using “pvclust” package in R (version 3.6.2) based on fractional methylation levels of gene-bodies or promoters of the 2708 SCOs. Differentially methylated SCOs were identified as follows: Firstly, density methylation of genes or promoters of members of SCOs between the two clades of Trichinella species were compared using the Mann-Whitney U test. Subsequently, gene-bodies or promoters with Benjamini-Hochberg corrected P ≤ 0.05 were identified as differentially methylated SCOs. Functional enrichment analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID, v6.8) (Sherman and Lempicki, 2009). Linear regression analysis was conducted using “lm” package in R (version 3.6.2). Expression levels of SCOs between different host classes of Trichinella species were compared using t-test in R (version 3.6.2), and multiple testing-corrected P ≤ 0.05 were reported as differentially expressed genes.
Results
Phylogenetic Analysis Across Trichinella spp.
In the present study, we applied a multi-omics strategy to thoroughly decipher the molecular characteristics in response to speciation and adaptation in the genus Trichinella based on their genetic and geographic diversity. Thus, we generated three types of high-resolution sequencing datasets, including whole-genome sequencing for the ML stage, and whole-genome bisulfite sequencing (WGBS) and RNA-sequencing for the three life stages, for the 12 Trichinella species. In total, all of these sequencing reads were well aligned to the respective reference genome after quality control (see section “Materials and Methods” and Supplementary Data 2). Based on the whole-genome sequencing data generated, we obtained high-quality SNV maps/profiles for each of the 12 Trichinella species. A consensus tree was constructed by those common SNVs (N = 5,395,250) across the 12 Trichinella species (see section “Materials and Methods” and Figure 1), which was in agreement with previous phylogenetic analysis using concatenated protein sequence data of other 12 Trichinella species in Korhonen et al. (2016), indicating a similar genomic variation of these Trichinella species in our dataset.
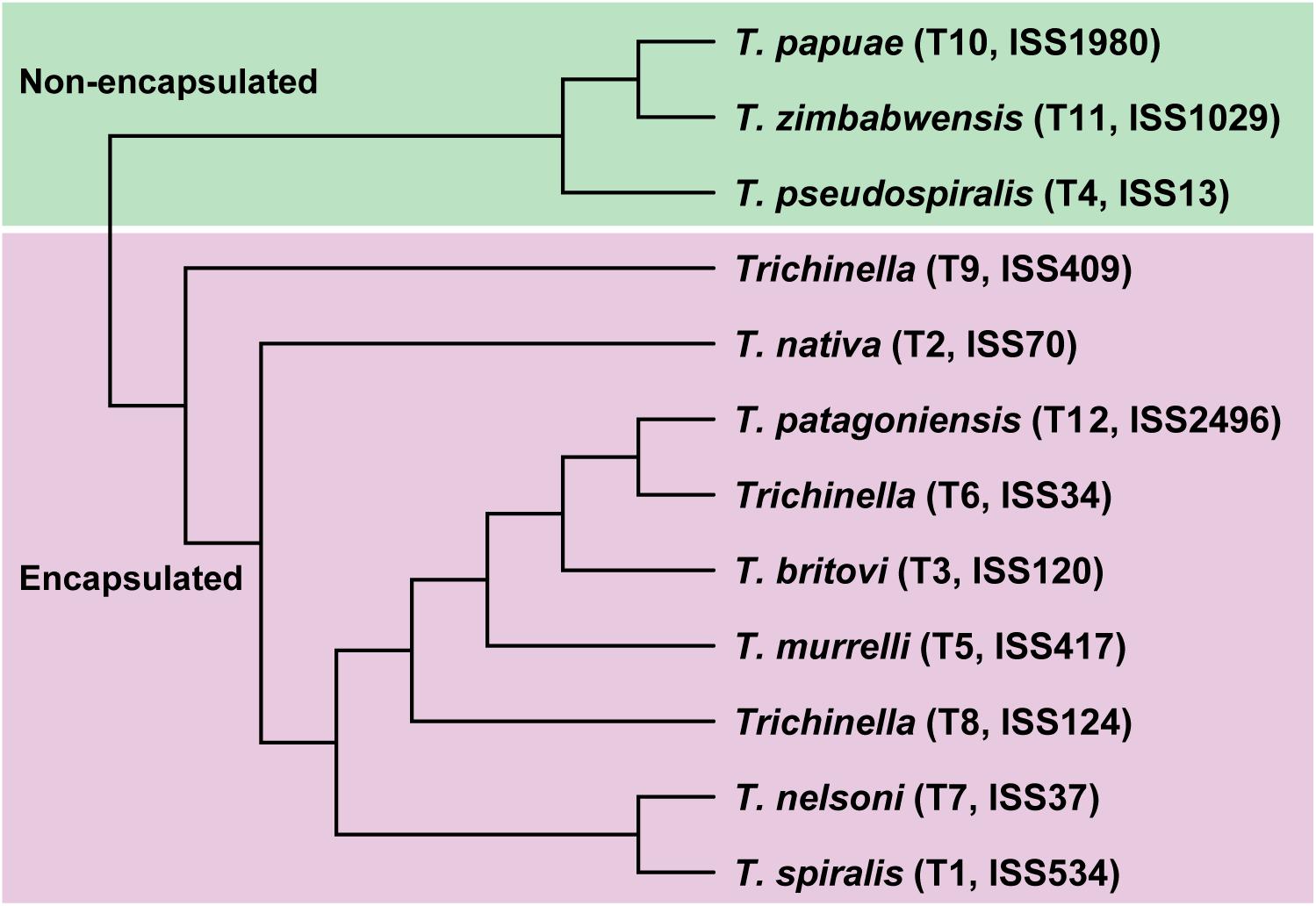
Figure 1. Phylogenetic analysis of the 12 Trichinella species based on common SNVs.
DNA Methylation Patterns Across Trichinella spp.
Reciprocal best BLAST searches confirmed the orthologous genes encoding the machinery for establishing and maintaining DNA methylation in all 12 Trichinella genomes. Phylogenetic analysis further revealed that these three DNMT genes are highly conserved across Trichinella spp. (Supplementary Figure 2). RNA-seq analyses indicated these genes of DNA methylation machinery are functionally expressed in all three life stages (Supplementary Figure 3). Accordingly, our data generated via WGBS revealed a similar distribution pattern of genome-wide methylated cytosines for the 12 Trichinella species. The highest amount of methylated Cs detected in Ad and ML stages were in the CpG sequence context, whereas NBL stage showed higher numbers of mCHH (H represents A, C, or T). Further, the average percentage of methylated CpG was 10.44 and 10.27% in Ad and ML stages, respectively, whereas NBL stage showed rare traces of DNA methylation (0.46%) (Supplementary Data 2). By profiling methylated CpGs around genic regions, a notable trend was observed in the density of methylated CpG and the distance from the transcriptional start site (TSS), with methylated CpG density increasing toward the transcriptional end sites and returned to the same level as 2-Kb upstream at the 2-Kb downstream (Supplementary Figure 4). Further, exons exhibited higher methylation levels in comparison to introns or flanking regions (Supplementary Figure 5). Similar to plants and mammals, DNA methylation levels of repetitive sequences were significantly higher than those in gene-body regions (Supplementary Figure 6).
Host-Related Methylome Variation of SCOs Could Reflect Evolutionary History
We then calculated pairwise sequence differences and methylome variations of conservative sites between these 12 Trichinella species. Upon comparison of sequence divergence and methylation variation of common CpG sites, we showed that interspecies nucleotide substitution rates exhibited a significantly positive correlation with DNA methylation differences (P = 0.0003; Figure 2A).
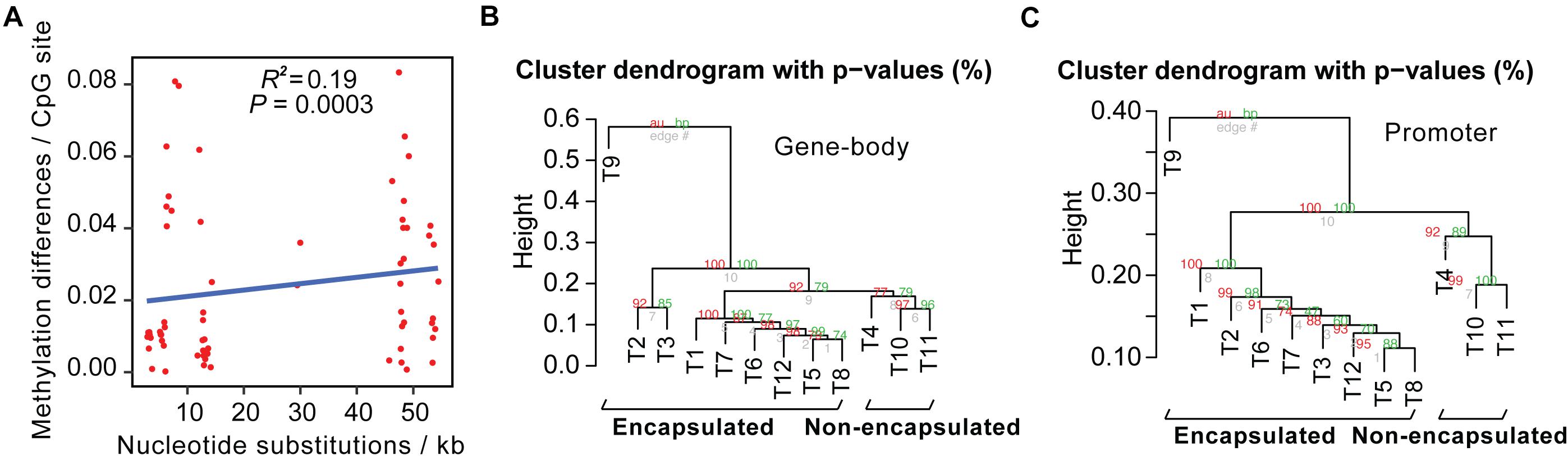
Figure 2. Interspecies methylome variation could reflect evolutionary history across Trichinella spp. (A) Methylation changes correlate with sequence differences. The x-axis represents the number of nucleotide substitutions between the two species per kb. The y-axis represents methylation differences of interspecies conservative methylated CpG sites. (B,C) Hierarchical clustering analysis based on DNA methylation levels of the gene-bodies (B) or promoters (C) of the 2708 SCOs in Ad stage.
Next, we detected interspecies methylome divergence of the 12 Trichinella species. A hierarchical clustering analysis based on gene-body methylation (gbM) levels of the 2708 SCOs revealed that these 12 Trichinella species were clustered into two main clades according to host classes in Ad stage (Figure 2B): (i) species that can only complete their life cycle in mammals requiring a host body temperature ranging from 37 to 40°C, including all of the nine encapsulated species; (ii) species that can complete their life cycle in mammals, reptiles, and birds at host body temperature ranging from 25 to 42°C, including the three non-encapsulated larvae. A similar trend was also observed in DNA methylation levels of promoter regions of these SCOs in Ad stage (Figure 2C), suggesting that promoter sequences of these SCOs also exhibited similar conservative features with that observed in gene-bodies under the strength of evolutionary constraints. An almost identical pattern of hierarchical clustering was achieved for the DNA methylation levels of ML stage both in gene-bodies and promoters among these 12 Trichinella species (Supplementary Figure 7). The results of the clustering pattern based on methylation levels are also in concordance with phylogenetic analysis based on common SNVs of the 12 Trichinella species (Figure 1), suggesting that DNA methylation divergence of conservative sites might be associated with the evolutionary history of these Trichinella species. Further study of the correlation between CpG DNA methylation and proximal genetic variations provided strong support for this observation (Supplementary Figure 8).
Interspecies Methylome Variation of SCOs Could Be Predictive of Gene Transcription Changes Across Trichinella spp.
Previously, we have indicated that interspecies epigenomic changes could be predictive of interspecies changes of gene expression, whereas differences of genomic sequences are not well correlated with gene expression changes (Liu et al., 2021). To test whether this trend is pervasive among the genus Trichinella, we compared epigenomic differences to both genomic and transcriptomic changes between reference species T. pseudospiralis and 10 other Trichinella species. Concordant with our previous findings, here, we observed interspecies epigenomic differences exhibited no clear correlations with sequence differences. However, a much higher correlation was observed between epigenomic and transcriptional changes both in the species of encapsulated (Figure 3A and Supplementary Figure 7) and non-encapsulated clade (Figure 3B and Supplementary Figure 9), suggesting that epigenomic variation can provide a better foundation to evaluate the functional divergence of Trichinella species.
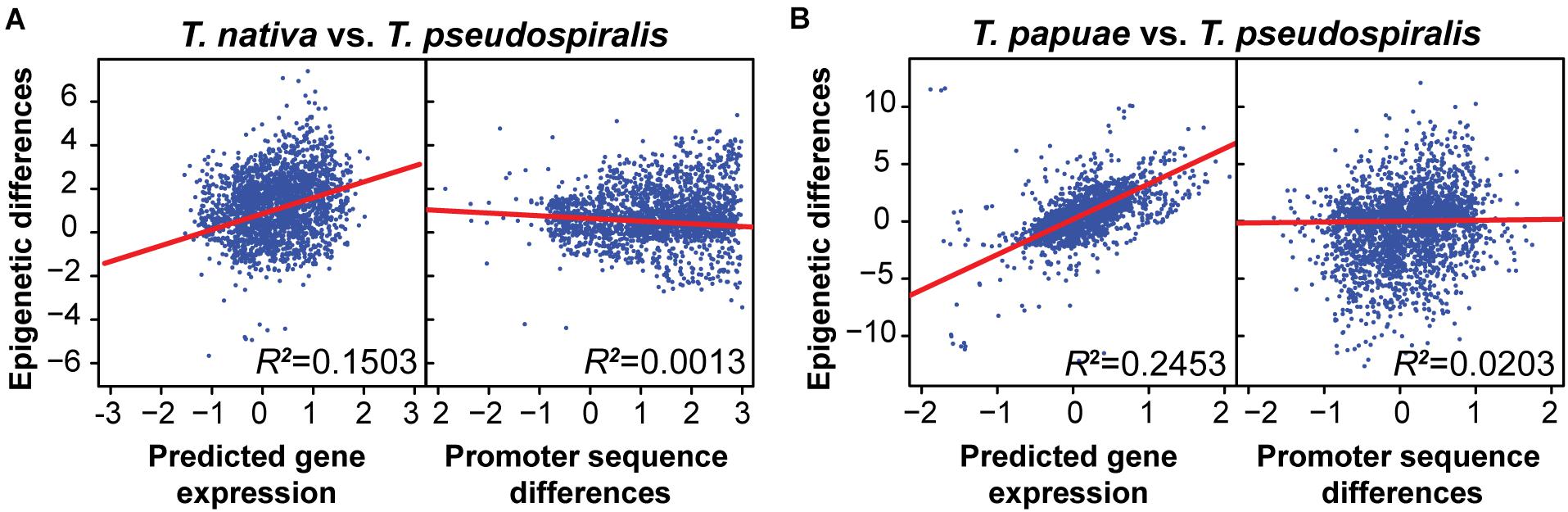
Figure 3. Interspecies methylome conservation is a regulatory mechanism of transcription across Trichinella spp. Correlations among DNA methylation changes, gene expression alterations, and sequence differences in T. nativa (A) and T. papuae (B) when comparing with reference species T. pseudospiralis. Epigenetic differences were computed using the P-values generated by comparing the common methylated CpGs of orthologous genes between the two species under consideration. Predicted gene expression differences of every gene were regressed to the DNA methylation level of the upstream 2-Kb region of the gene. Sequence differences were estimated using blastn score differences by comparing differences between the maximum log blastn score of all orthologous promoters (4,000 bp centered at transcriptional start sites) and the blastn score of the orthologous promoter pair under consideration. R2, square of the sample correlation coefficient.
Gene Expression Patterns of Hyper-/Hypo-Methylated SCOs
To further examine the impact of DNA methylation variation on transcriptome regulation of these SCOs (N = 2708) during Trichinella evolution, we classified these SCOs into high and low categories according to mean methylation levels and CpGO/E values of gene-bodies (section “Materials and Methods”). In total, we identified an average of 774 hyper-methylated (mean = 774 ± 30) and 1,444 hypo-methylated (mean = 1,444 ± 55) genes in Ad stage (Figure 4A). As exemplified by analysis of T. pseudospiralis, functional annotation revealed that many biological processes and pathways bear relevance to Trichinella parasitism, such as regulation of transcription (GO:0006355, P = 6.08E-8), transcription factor activity (GO:0003700, P = 1.15E-5), G-protein coupled receptor activity (GO:0004930, P = 1.5E-4), and collagen trimer (P = 5.6E-4) (Supplementary Data 3). Then we examined the expression patterns of these two categories of genes in comparison to the genome overall. We observed that the hypo-methylated SCOs displayed significantly higher expression levels, whereas hyper-methylated SCOs exhibited similar expression patterns relative to the genome overall in Ad stage (Figure 4B). Next, we adopted a similar approach to analyze the regulatory roles of promoter methylation of these SCOs on mRNA expression levels. Herein, we identified an average of 738 hyper-methylated (mean = 738 ± 40) and 1,459 hypo-methylated (mean = 1,459 ± 47) promoters in Ad stage (Figure 4C). Similarly, a more prominent trend was observed for genes with hypo-methylated promoters to have higher expression levels such that genes with hyper-methylated promoters exhibited extensive expression spectrum similar to the genome overall in Ad stage in T. pseudospiralis (Figure 4D). Such expression patterns were also observed in the SCOs with hyper-/hypo-methylated gene-bodies or promoters of 11 other Trichinella species (data not shown).
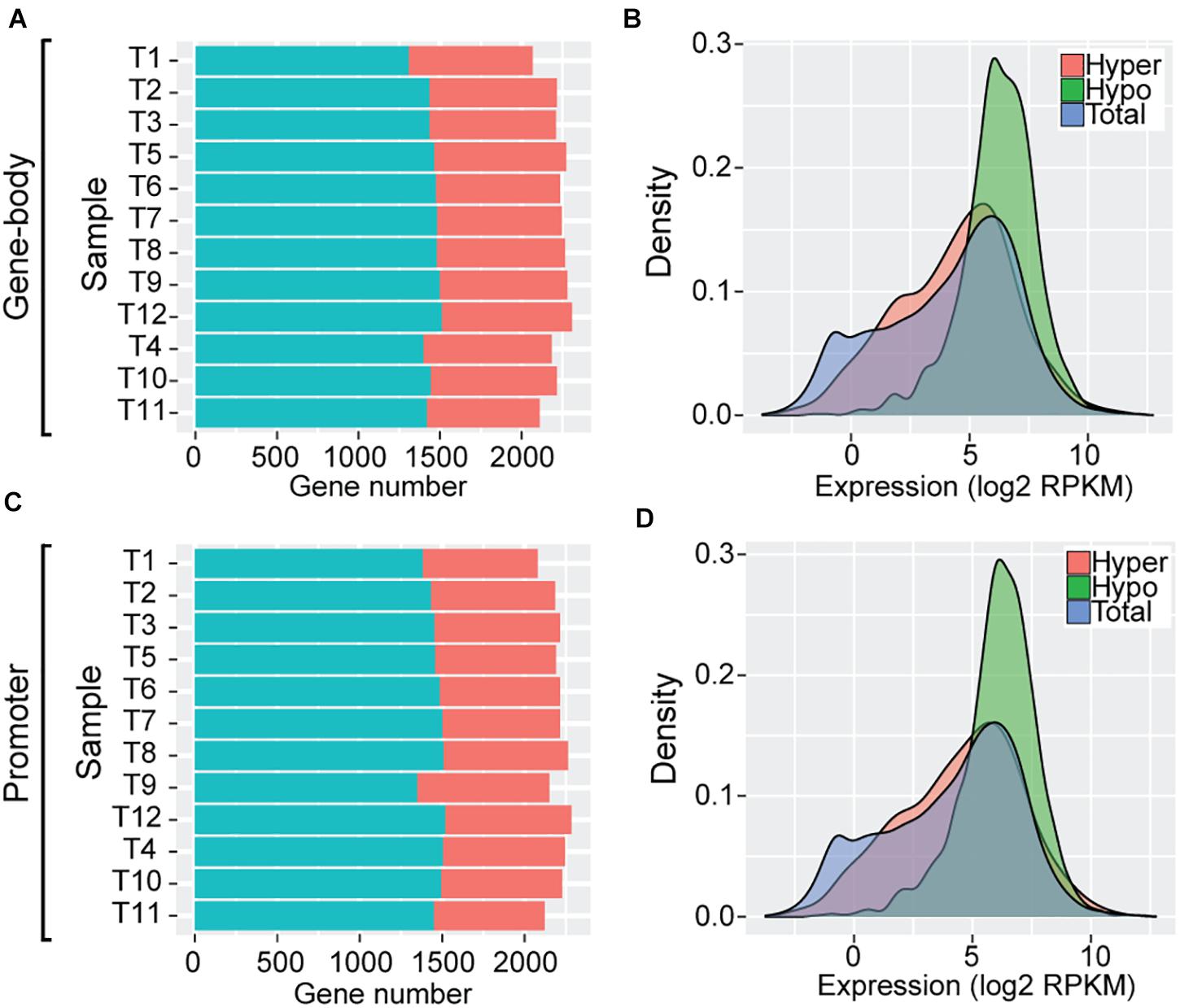
Figure 4. Gene expression patterns of SCOs with hypo/hyper-methylated gene-bodies or promoters of T. pseudospiralis. (A,C) Numbers of hyper-/hypo-methylated SCOs in gene-body (A) and promoter (C) regions. (B,D) Comparison of expression level density of genes exhibiting hyper-/hypo- features in gene-bodies (B) or promoters (D) in Ad stage. “Total” represents the number of total genes across the whole genome and is color-coded in blue. “Hyper” and “Hypo” represent the number of genes with hyper- or hypo- methylated SCOs in gene-bodies or corresponding promoters and are color-coded in red and green, respectively.
Functional Significance of Hyper-/Hypo-Methylated SCOs Across Developmental Stages
We then decipher the functional roles of these hyper-/hypo-methylated SCOs across developmental stages of the 12 Trichinella species. As some proteins involved in parasite development and survival against host physiology in Trichinella are expressed in a stage-specific manner, we focused on characterizing the stage-specific expressed genes in each of the 12 Trichinella species. Based on a stringent criterion (section “Materials and Methods”), an average of 410 Ad stage-specific expressed genes (mean = 410 ± 71) were identified for the 12 Trichinella species (Figure 5A), including casein kinase II subunit alpha, putative kinesin motor domain protein, and putative low-density lipoprotein receptor domain class A, etc. The majority of these genes have been previously reported to play crucial roles at the host-parasite interface, parasite survival, and immune evasion in Trichinella spp. (Nagano et al., 2009). Then we detected an overlapping trend between these stage-specific expressed genes and the aforementioned SCOs showing hyper-/hypo-methylated features in gene-body or promoter regions. In total, we obtained a panel of 390 non-redundant genes (mean = 390 ± 82), accounting for approximately 95.4% of the total stage-specific expressed genes, might be under hyper-/hypo-methylated gene-body or promoter regulation in Ad stage. Accordingly, a similar proportion (mean = 290 ± 67; 95.2%) of ML stage-specific expressed genes were identified, which contains an average of 304 genes (mean = 304 ± 72) across the 12 Trichinella species (Figure 5B). However, these stage-specific expressed genes under hyper-/hypo-methylation regulation are seldomly conserved across the 12 Trichinella species, as only three SCOs (fam2462, fam2831, and fam3343) were identified in Ad stage (Supplementary Data 4), suggesting functional divergence of these stage-specific expressed genes in the genus Trichinella.
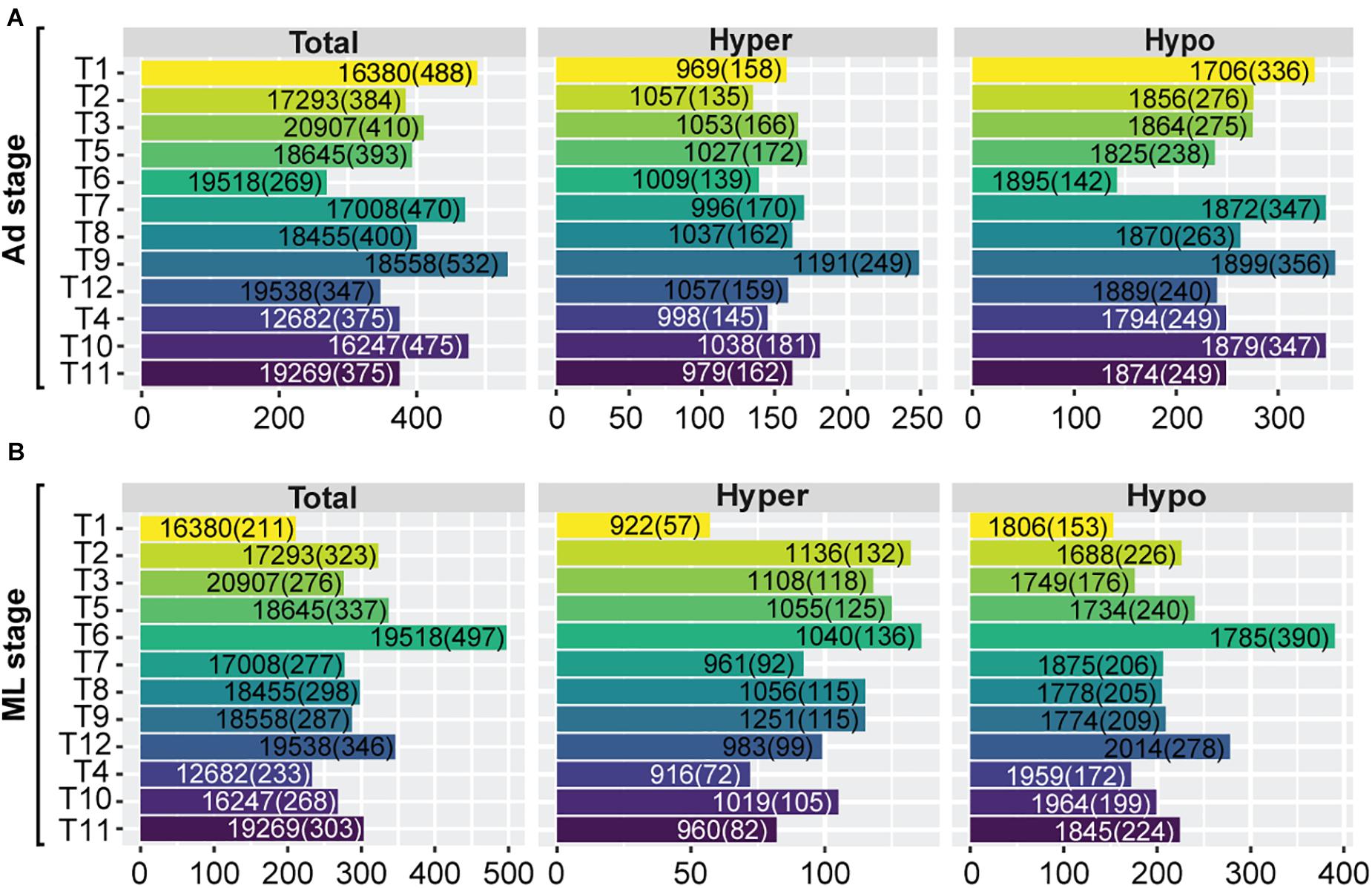
Figure 5. Functional significance of hyper-/hypo-methylated SCOs across developmental stages. Numbers of stage-specific expressed genes of the 12 Trichinella species both in Ad (A) and ML (B) stages. “Total” represents the number of total genes across the whole genome. “Hyper” represents the number of genes with hyper-methylated SCOs in gene-bodies or corresponding promoters. “Hypo” represents the number of genes with hypo-methylated SCOs in gene-bodies or corresponding promoters. Numbers in and outside the brackets represent the number of stage-specific expressed genes and the total number of genes of the respective category, respectively.
Functional Significance of Hyper-/Hypo-Methylated SCOs Between the Two Clades of Trichinella
Based on the methylome variation suggested by the hierarchical clustering analysis, we then focused our analysis on methylation changes of those hyper-/hypo-methylated SCOs between the encapsulated and non-encapsulated clade. Firstly, we merged the SCOs exhibiting hyper-/hypo-methylated gene-body or promoter features together, thus obtained a panel of 1,076 candidate SCOs in Ad stage across the 12 Trichinella species. By a cut-off of P ≤ 0.05, we identified 139 SCOs that exhibited differentially methylation in gene-body or corresponding promoter regions of these candidates, among which 122 and 17 SCOs exhibited hyper- and hypo-methylation in the encapsulated clade when comparing with the non-encapsulated clade, respectively (Figure 6A). Among these differentially methylated SCOs, 18 of them showed differential expression between the two clades of Trichinella (Figure 6B). Thirteen of them have known functional annotations, such as peptidase activity (GO:0008233), peptidase inhibitor (GO:0030414), and gluconeogenesis (GO:0006094) (Supplementary Data 5). Of note, we observed fam4516, encoding an Hsp90 co-chaperone Cdc37, was an Ad stage-specific expressed SCO with significantly higher methylation in gene-body regions, which results in lower expression levels of fam4516 in the three non-encapsulated species (Figure 6C). Moreover, using similar criteria with Ad stage, we identified a total of 87 SCOs that exhibited differentially methylation in ML stage, among which 54 and 33 SCOs showed hyper- and hypo-methylation, respectively (Figure 6D). Among those SCOs, 21 of them showed differential expression between the two clades of Trichinella (Figure 6E). Functional annotation revealed that majority of these genes involved in biological processes, such as protein kinase activity (GO:0004672), serine-type peptidase activity (GO:0008236), and ATPase activity (GO:0016887) (Supplementary Data 5). Of note, we observed a non-stage-specific expressed SCO fam2698, a p53-like transcription factor involved in the apoptotic process, exhibited higher expression levels in non-encapsulated clade, which might be modulated by hypo-methylation in its corresponding promoter regions when compared to the encapsulated clade (Figure 6F).
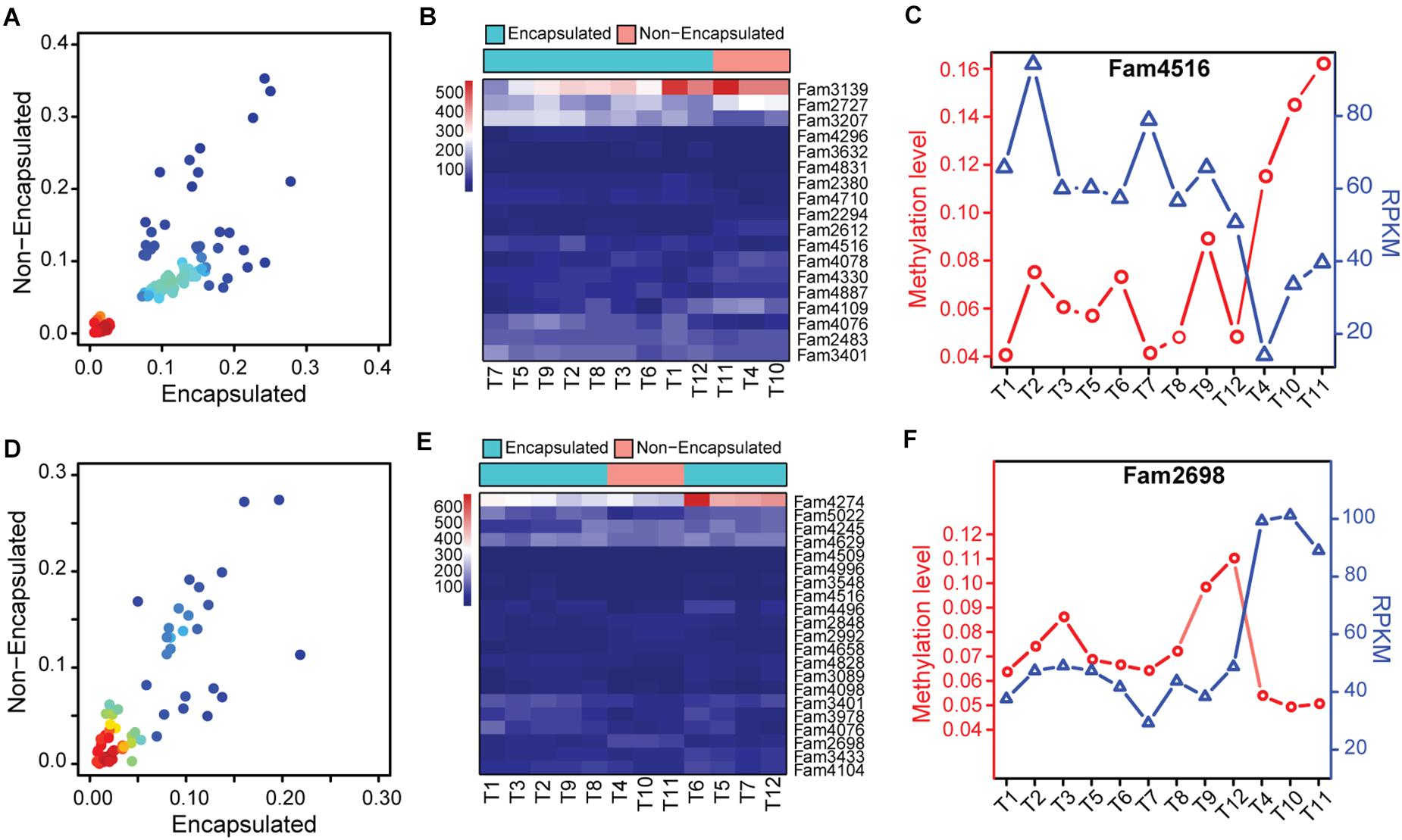
Figure 6. Functional significance of hyper-/hypo-methylated SCOs between the two clades of Trichinella. (A,D) Scatter plot of differentially methylated genes in Ad (A) and ML (D) stages, with density at each point (red for high density, blue for low density). (B,E) Heatmaps depicting the hierarchical clustering of differentially expressed genes that exhibited differential methylation in gene-bodies or corresponding promoter regions in Ad (B) and ML (E) stages between the two clades of Trichinella. (C,F) Methylation and expression levels of fam4516 (C) and fam2698 (F) of the 12 species.
Discussion
Comparative epigenomics is a powerful tool to understand the conservation and divergence of DNA methylation patterns and regulatory machinery in eukaryotic organisms (Zhong, 2016). For the parasitic nematode Trichinella, which possess a complicated life cycle and undergoing a complex developmental regulation of genes, addressing the epigenetic conservation and variation during Trichinella evolution may help to understand the differential parasitism and adaptation to different host classes. To date, much of the focus in the genus Trichinella has been on the comparison of differential parasitism between the two representative species, T. spiralis and T. pseudospiralis, using traditional molecular biology (Wu et al., 2001; Boonmars et al., 2005). Studies regarding differential regulation of epigenetic mechanisms across all 12 Trichinella species remain largely uncharacterized.
Here we carried out the first comprehensive comparative epigenomic analysis among these Trichinella species and revealed that methylome changes in the genus Trichinella were host-related, which could recapitulate the known phylogenetic relationships arising from the sequence composition. Paralleled patterns between gbM tree and sequence tree were also observed from other four distantly related invertebrate species, including C. intestinalis, N. vectensis, B. mori, and A. mellifera, even though the two trees differ dramatically from the branch length (Sarda et al., 2012). This high interspecies conservation of methylation of orthologous groups implies biological significance, such as assisting acclimatization by modulating gene expression (Takuno and Gaut, 2013; Bewick et al., 2016). That methylation conservation is a regulatory mechanism for transcription is supported by a previous study in which regulatory roles of epigenomic modifications were intensively studied in human, mouse, and pig pluripotent stem cells (Xiao et al., 2012), suggesting that transcriptomes and epigenomes exhibited coevolutionary patterns among those distantly related species.
Relationships between patterns of gbM and transcription regulation in invertebrates have been investigated previously. In insects, genes with high methylation levels tended to be expressed at moderate to high levels. Conversely, a gene with low methylation levels displayed a broad spectrum of mRNA levels (Hunt et al., 2010). In contrast, in C. gigas, a negative correlation was observed between gbM and the mRNA expression levels (Riviere et al., 2013). Thus, associations between gbM and transcription may exhibit species-specific features and heavily depend on the density of methylation (Riviere, 2014). Indeed, here, we observed that in the genus Trichinella, genes heavily methylated exhibited a broad range of expression, whereas genes weakly methylated expressed at high levels. This contradicts the associations observed in other invertebrate species, suggesting more subtle roles and functional outcomes for this epigenetic mark in the genus Trichinella.
Characterization of stage-specific expressed genes that are developmentally regulated in the parasite life-cycle is an essential step toward understanding parasite biology and host-parasite interactions in the genus Trichinella (Liu et al., 2007, 2015; Ren et al., 2019). Yang and colleagues have characterized an NBL stage-specific expressed serine proteinase using monoclonal antibodies and proved its usage as major antigen in the early detection of T. spiralis infection (Yang et al., 2015). However, homologous E/S products may exhibit species-specific antibody responses, such as 53-kDa glycoprotein in mice with infection of five Trichinella species using recombinant antigens, suggesting the need to design species-specific antigens in immunodiagnosis in different host classes (Nagano et al., 2008). This observation is concordant with our findings that there are only three conserved SCOs of stage-specific expressed genes among these Trichinella species. Furthermore, in our recent publication, we revealed that epigenetic mechanism plays a role in regulating the expression of several stage-specific expressed genes, such as TP12446 (Liu et al., 2021), further demonstrating functional significance of these hyper-/hypo-methylated SCOs for the elucidation of parasite development and survival within the host.
Based on these findings, we further assessed the functional significance of SCOs with hyper-/hypo-methylated gene-bodies or promoters on transcriptome variation between the two clades of Trichinella species. We observed fam4516, an Ad stage-specific expressed SCO encoding a kinase-specific co-chaperone Cdc37, was highly expressed in the non-encapsulated clade. Functional roles of Cdc37 have been investigated in rabies virus, which could be proposed as therapeutic targets against rabies by directly affecting the life cycle of rabies virus and stability of viral P protein (Xu et al., 2016). The observed hypermethylation of gene-bodies of Cdc37 in the three species of non-encapsulated clade might provide potential DNA methylation markers preventing parasite invasion and survival. Further, we observed members of fam2698, which was annotated as a p53-like transcription factor, were hypomethylated in promoters with increased expression in ML stage in the non-encapsulated clade. Functions of p53 or p53-like transcription factors have been investigated in Leishmania spp., which has been mediating the apoptosis process of infected host macrophages and thus acting as one of the survival mechanisms of the parasites (Moshrefi et al., 2017). We speculate this higher expression of fam2698 in non-encapsulated clade may be associated with enhanced ability of parasite invasion and attack to adapt to their non-encapsulated phenotype in ML stage.
In the present study, we described the first comprehensively comparative epigenome study in the genus Trichinella. We observed SCOs with hyper-/hypo-methylated features in gene-bodies or promoters exhibited functional roles in gene regulation, especially for stage-specific expressed genes, which showed divergent features among those Trichinella species. By comparing the two clades of Trichinella in epigenomics and transcriptomics, we identified a panel of SCOs with altered methylation and expression, which were important for elucidating differential parasitology and epidemiology between the two clades of Trichinella. Our data provided here could be fully understanding the conservation and divergence of DNA methylation on differential adaptation and parasitism of these Trichinella species, which could lead to the development of new drugs by targeting the DNA methylation process for the treatment of trichinellosis.
Data Availability Statement
Raw sequencing data were deposited in the GEO with the following accession numbers: GSE140382 for RNA-seq data, GSE139486 for WGBS data, and GSE169404 for WGS data.
Ethics Statement
All experimental protocol involving animals have been reviewed and approved by the Ethical Committee of the Jilin University affiliated with the Provincial Animal Health Committee, Jilin Province, China (Ethical Clearance number IZ-2009-08).
Author Contributions
FG and ML conceived the project. YF and FG analyzed and interpreted the high-throughput sequencing data. XL and BT undertook sample collection. XB and XW performed DNA extraction. XL and CL performed RNA extraction experiments. YL constructed high-throughput sequencing libraries, including WGS, WGBS, and RNA-seq. YF performed all of the bioinformatics analyses and was the main contributor in writing the manuscript. FG, ML, and YD critically reviewed the manuscript. All authors read and approved the manuscript for publication.
Funding
This study was supported by the National Natural Science Foundation of China (31872467 and 31520103916), the Guangdong Innovative and Entrepreneurial Research Team Program (No. 2014ZT05S123), the Program for JLU Science and Technology Innovative Research Team (2017TD-32), the Agricultural Science and Technology Innovation Program, and the Elite Young Scientists Program of CAAS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank J. Ruan, P. Cui, and Q. Lin for help guidance on bioinformatic analyses.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.681839/full#supplementary-material
Footnotes
- ^ https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ http://www.microbesonline.org/fasttree/
References
Bender, J. (2004). DNA methylation and epigenetics. Annu. Rev. Plant Biol. 55, 41–68. doi: 10.1146/annurev.arplant.55.031903.141641
Bewick, A. J., Ji, L., Niederhuth, C. E., Willing, E.-M., Hofmeister, B. T., Shi, X., et al. (2016). On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. U.S.A. 113, 9111–9116. doi: 10.1073/pnas.1604666113
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boonmars, T., Wu, Z., Nagano, I., and Takahashi, Y. (2005). Trichinella pseudospiralis infection is characterized by more continuous and diffuse myopathy than T. spiralis infection. Parasitol. Res. 97, 13–20. doi: 10.1007/s00436-005-1359-x
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6. doi: 10.1093/gigascience/gix120
Duncan, E. J., Gluckman, P. D., and Dearden, P. K. (2014). Epigenetics, plasticity, and evolution: how do we link epigenetic change to phenotype? J. Exp. Zool. B Mol. Dev. Evol. 322, 208–220. doi: 10.1002/jez.b.22571
Feil, R., and Fraga, M. F. (2012). Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 13, 97–109. doi: 10.1038/nrg3142
Fischer, S., Brunk, B. P., Chen, F., Gao, X., Harb, O. S., Iodice, J. B., et al. (2011). Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinformatics 6, 11–19. doi: 10.1002/0471250953.bi0612s35
Gao, F., Liu, X., Wu, X. P., Wang, X. L., Gong, D., Lu, H., et al. (2012). Differential DNA methylation in discrete developmental stages of the parasitic nematode Trichinella spiralis. Genome Biol. 13:R100. doi: 10.1186/gb-2012-13-10-r100
Gottstein, B., Pozio, E., and Nockler, K. (2009). Epidemiology, diagnosis, treatment, and control of trichinellosis. Clin. Microbiol. Rev. 22, 127–145. doi: 10.1128/CMR.00026-08
Hunt, B. G., Brisson, J. A., Yi, S. V., and Goodisman, M. A. (2010). Functional conservation of DNA methylation in the pea aphid and the honeybee. Genome Biol. Evol. 2, 719–728. doi: 10.1093/gbe/evq057
Jeong, Y. H., Lu, H., Park, C. H., Li, M., Luo, H., Kim, J. J., et al. (2016). Stochastic anomaly of methylome but persistent SRY hypermethylation in disorder of sex development in canine somatic cell nuclear transfer. Sci. Rep. 6:31088. doi: 10.1038/srep31088
Jiang, P., Zhang, X., Wang, L. A., Han, L. H., Yang, M., Duan, J. Y., et al. (2016). Survey of Trichinella infection from domestic pigs in the historical endemic areas of henan province, central China. Parasitol. Res. 115, 4707–4709. doi: 10.1007/s00436-016-5240-x
Kapel, C. M. (2000). Host diversity and biological characteristics of the Trichinella genotypes and their effect on transmission. Vet. Parasitol. 93, 263–278. doi: 10.1016/s0304-4017(00)00345-9
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Korhonen, P. K., Pozio, E., La Rosa, G., Chang, B. C., Koehler, A. V., Hoberg, E. P., et al. (2016). Phylogenomic and biogeographic reconstruction of the Trichinella complex. Nat. Commun. 7:10513. doi: 10.1038/ncomms10513
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liu, M., Wang, X., Fu, B., Li, C., Wu, X., Le Rhun, D., et al. (2007). Identification of stage-specifically expressed genes of Trichinella spiralis by suppression subtractive hybridization. Parasitology 134, 1443–1455. doi: 10.1017/S0031182007002855
Liu, R., Cui, J., Liu, X., Jiang, P., Sun, G., Zhang, X., et al. (2015). Comparative proteomic analysis of surface proteins of Trichinella spiralis muscle larvae and intestinal infective larvae. Acta Trop. 150, 79–86. doi: 10.1016/j.actatropica.2015.07.002
Liu, X., Feng, Y., Bai, X., Wang, X., Qin, R., Tang, B., et al. (2021). Comparative multi-omics analyses reveal differential expression of key genes relevant for parasitism between non-encapsulated and encapsulated Trichinella. Commun. Biol. 4:134. doi: 10.1038/s42003-021-01650-z
Liu, X., Song, Y., Lu, H., Tang, B., Piao, X., Hou, N., et al. (2011). Transcriptome of small regulatory RNAs in the development of the zoonotic parasite Trichinella spiralis. PLoS One 6:e26448. doi: 10.1371/journal.pone.0026448
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Moshrefi, M., Spotin, A., Kafil, H. S., Mahami-Oskouei, M., Baradaran, B., Ahmadpour, E., et al. (2017). Tumor suppressor p53 induces apoptosis of host lymphocytes experimentally infected by Leishmania major, by activation of Bax and caspase-3: a possible survival mechanism for the parasite. Parasitol. Res. 116, 2159–2166. doi: 10.1007/s00436-017-5517-8
Nagano, I., Wu, Z., and Takahashi, Y. (2008). Species-specific antibody responses to the recombinant 53-kilodalton excretory and secretory proteins in mice infected with Trichinella spp. Clin. Vaccine Immunol. 15, 468–473. doi: 10.1128/CVI.00467-07
Nagano, I., Wu, Z., and Takahashi, Y. (2009). Functional genes and proteins of Trichinella spp. Parasitol. Res. 104, 197–207. doi: 10.1007/s00436-008-1248-1
Pozio, E. (2005). The broad spectrum of Trichinella hosts: from cold- to warm-blooded animals. Vet. Parasitol. 132, 3–11. doi: 10.1016/j.vetpar.2005.05.024
Pozio, E., Marucci, G., Casulli, A., Sacchi, L., Mukaratirwa, S., Foggin, C., et al. (2004). Trichinella papuae and Trichinella zimbabwensis induce infection in experimentally infected varans, caimans, pythons and turtles. Parasitology 128, 333–342. doi: 10.1017/S0031182003004542
Ren, H. N., Liu, R. D., Song, Y. Y., Zhuo, T. X., Guo, K. X., Zhang, Y., et al. (2019). Label-free quantitative proteomic analysis of molting-related proteins of Trichinella spiralis intestinal infective larvae. Vet. Res. 50:70. doi: 10.1186/s13567-019-0689-0
Riviere, G. (2014). Epigenetic features in the oyster Crassostrea gigas suggestive of functionally relevant promoter DNA methylation in invertebrates. Front. Physiol. 5:129. doi: 10.3389/fphys.2014.00129
Riviere, G., Wu, G. C., Fellous, A., Goux, D., Sourdaine, P., and Favrel, P. (2013). DNA methylation is crucial for the early development in the Oyster C. gigas. Mar. Biotechnol. 15, 739–753. doi: 10.1007/s10126-013-9523-2
Sarda, S., Zeng, J., Hunt, B. G., and Yi, S. V. (2012). The evolution of invertebrate gene body methylation. Mol. Biol. Evol. 29, 1907–1916. doi: 10.1093/molbev/mss062
Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Takuno, S., and Gaut, B. S. (2013). Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc. Natl. Acad. Sci. U.S.A. 110, 1797–1802. doi: 10.1073/pnas.1215380110
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Wang, Z. Q., and Cui, J. (2001). The epidemiology of human trichinellosis in China during 1964-1999. Parasite 8(2 Suppl), S63–S66. doi: 10.1051/parasite/200108s2063
Wu, Z., Matsuo, A., Nakada, T., Nagano, I., and Takahashi, Y. (2001). Different response of satellite cells in the kinetics of myogenic regulatory factors and ultrastructural pathology after Trichinella spiralis and T. pseudospiralis infection. Parasitology 123, 85–94. doi: 10.1017/S0031182001007958
Wu, Z., Nagano, I., Nakada, T., and Takahashi, Y. (2002). Expression of excretory and secretory protein genes of Trichinella at muscle stage differs before and after cyst formation. Parasitol. Int. 51, 155–161.
Xi, Y., and Li, W. (2009). BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10:232. doi: 10.1186/1471-2105-10-232
Xiao, S., Xie, D., Cao, X., Yu, P., Xing, X., Chen, C. C., et al. (2012). Comparative epigenomic annotation of regulatory DNA. Cell 149, 1381–1392. doi: 10.1016/j.cell.2012.04.029
Xu, Y., Liu, F., Liu, J., Wang, D., Yan, Y., Ji, S., et al. (2016). The co-chaperone Cdc37 regulates the rabies virus phosphoprotein stability by targeting to Hsp90AA1 machinery. Sci. Rep. 6:27123. doi: 10.1038/srep27123
Yang, Y., Lacour, S. A., Laine-Prade, V., Versille, N., Grasset-Chevillot, A., Feng, S., et al. (2015). Trichinella spiralis newborn larvae: characterization of a stage specific serine proteinase expression, NBL1, using monoclonal antibodies. Parasitology 142, 783–790. doi: 10.1017/S0031182014001851
Zarlenga, D. S., Rosenthal, B. M., La Rosa, G., Pozio, E., and Hoberg, E. P. (2006). Post-miocene expansion, colonization, and host switching drove speciation among extant nematodes of the archaic genus Trichinella. Proc. Natl. Acad. Sci. U.S.A. 103, 7354–7359. doi: 10.1073/pnas.0602466103
Keywords: comparative epigenomics, host-related methylomes, hyper-/hypo-methylated SCOs, differential parasitism, Trichinella
Citation: Feng Y, Liu X, Liu Y, Tang B, Bai X, Li C, Wang X, Deng Y, Gao F and Liu M (2021) Comparative Epigenomics Reveals Host Diversity of the Trichinella Epigenomes and Their Effects on Differential Parasitism. Front. Cell Dev. Biol. 9:681839. doi: 10.3389/fcell.2021.681839
Received: 17 March 2021; Accepted: 27 April 2021;
Published: 11 June 2021.
Edited by:
Qingfeng Zhang, Tongji University, ChinaReviewed by:
Zhong Quan Wang, Zhengzhou University, ChinaXiao Wang, Technical University of Denmark, Denmark
Nian-Zhang Zhang, Chinese Academy of Agricultural Sciences, China
Copyright © 2021 Feng, Liu, Liu, Tang, Bai, Li, Wang, Deng, Gao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Gao, Zmx5czgyOEBnbWFpbC5jb20=; Mingyuan Liu, bGl1bXkzNkAxNjMuY29t
†These authors have contributed equally to this work