 Fan Pan
Fan Pan Xin-Rong Lin
Xin-Rong Lin Li-Ping Hao1
Li-Ping Hao1 Rui Wang
Rui Wang- 1Department of Medical Oncology, Affiliated Jinling Hospital, Medical School of Nanjing University, Nanjing, China
- 2Department of Gastroenterology and Hepatology, Affiliated Jinling Hospital, Medical School of Nanjing University, Nanjing, China
Hepatocellular carcinoma (HCC) is the 6th most prevalent cancer and the 4th leading cause of cancer-related death worldwide. Mechanisms explaining the carcinogenesis of HCC are not clear yet. In recent years, rapid development of N6-methyladenosine (m6A) modification provides a fresh approach to disclosing this mystery. As the most prevalent mRNA modification in eukaryotes, m6A modification is capable to post-transcriptionally affect RNA splicing, stability, and translation, thus participating in a variety of biological and pathological processes including cell proliferation, apoptosis, tumor invasion and metastasis. METTL3 has been recognized as a pivotal methyltransferase and essential to the performance of m6A modification. METTL3 can regulate RNA expression in a m6A-dependent manner and contribute to the carcinogenesis, tumor progression, and drug resistance of HCC. In the present review, we are going to make a clear summary of the known roles of METTL3 in HCC, and explicitly narrate the potential mechanisms for these roles.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most prevalent cancer (4.7%) and the fourth leading cause (8.2%) of cancer-related death worldwide, with estimated 841,080 new cases and 781,631 deaths in 2018. The incidence of liver cancer presents obvious geographic heterogeneity, mostly observed in Eastern Asia and Northern Africa (Bray et al., 2018). Generally liver cancer is classified into the primary liver cancer and the secondary liver cancer. HCC, as the most significant subtype of primary liver cancer, comprises almost 75∼85% of the cases (Forner et al., 2012; Sia et al., 2017). The carcinogenesis of HCC is known as a sophisticated multistage process. Multiple risk factors have been validated to be associated with HCC, including hepatitis B virus (HBV) infection, hepatitis C virus (HCV) infection, non-alcoholic fatty liver disease (NAFLD), exposure to aflatoxin B1, alcohol intake, diabetes, and obesity (Singal and El-Serag, 2015). Despite the tremendous efforts devoted to exploring the mechanisms of hepatocarcinogenesis, few progresses have been made. Over the past decades, the rapid development of epigenetics has provided a fresh approach to disclosing the mechanisms of hepatocarcinogenesis, including DNA methylation, histone modification, chromatin remodeling, as well as RNA methylation in which N6-methyladenosine (m6A) modification plays an important role (Schulze et al., 2015; Villanueva et al., 2015; Xu et al., 2017; Liu et al., 2018).
N6-methyladenosine modification, which refers to the insertion of a methyl substituent onto the N-6 position of adenosine, is known as the most prevalent internal messenger RNA (mRNA) modification within eukaryotes (Bokar, 2005). M6A modification has been demonstrated to be capable to post-transcriptionally regulate RNA and affect RNA stability (Huang et al., 2018), splicing (Xiao et al., 2016), and translation (Lin et al., 2016). It has been proposed that m6A modification is involved in various physiological and pathological processes such as cancers (Liu et al., 2018). The conserved enrichment of m6A modification in the coding sequence (CDS) and the 3′ untranslated region (3′UTR) of mRNA has been revealed. A consensus DRACH motif (where D = A, G or U, R = A or G, H = A, C or U) serves as the predominant site of m6A modification (Perry et al., 1975; Csepany et al., 1990; Narayan et al., 1994; Meyer et al., 2012; Linder et al., 2015; Bayoumi and Munir, 2021). In mammalian cells, m6A modification has been found to be dynamically and reversibly regulated by several proteins which have been classified into three groups, including “erasers” with demethylation ability like FTO (Jia et al., 2011) and ALKBH5 (Tang et al., 2018), “readers” such as YTHDF1/2/3 (Li A. et al., 2017) and YTHDC1/2 (Meyer and Jaffrey, 2017; Liu J. et al., 2021) that can recognize and bind to m6A-modified transcripts, and “writers” serving as methyltransferases such as METTL3/14 and WTAP (Bokar et al., 1997; Liu et al., 2014; Figure 1). Typically, as a m6A “writer,” the METTL3-METTL14 complex has been demonstrated to be essential to the performance of m6A modification. In this complex, Methyltransferase-like 3 (METTL3), also known as MT-A70, is believed to be the only catalytic subunit (Liu et al., 2014; Ping et al., 2014).
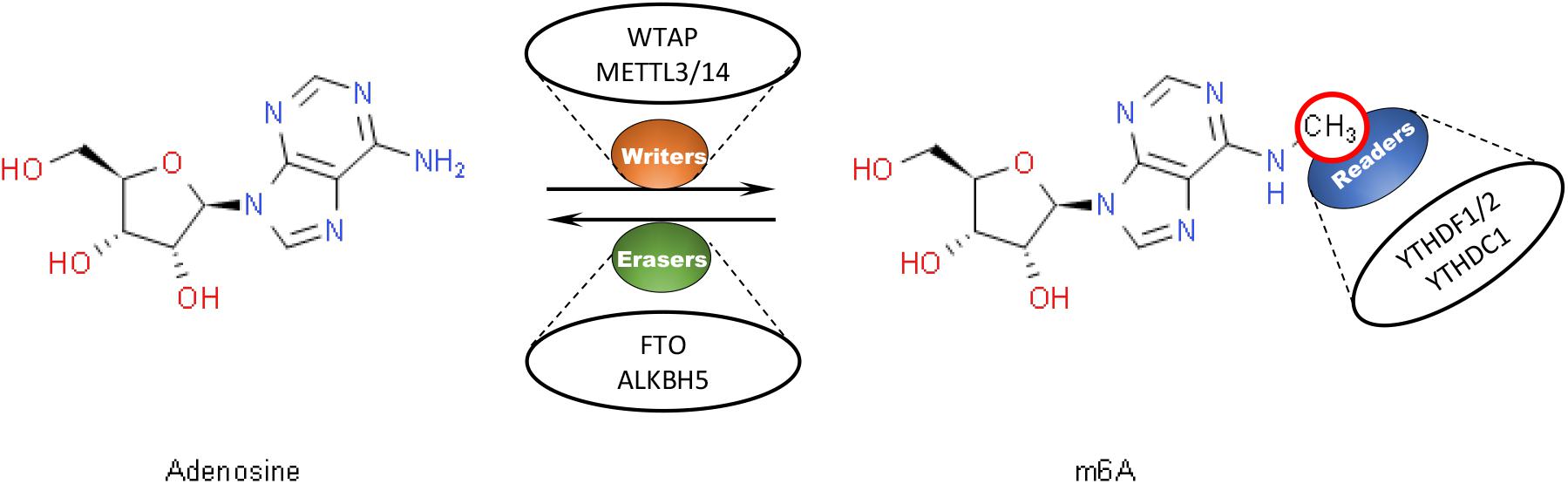
Figure 1. Dynamic and reversible regulation of m6A modification by regulators including “writers,” “readers,” and “erasers.” M6A “writers” serve as methyltransferases, such as WTAP and METTL3/14. M6A “readers” help recognize and bind to m6A-modified transcripts, like YTHDF1/2 and YTHDC1. M6A “erasers” have the ability for demethylation, such as FTO and ALKBH5.
Methyltransferase-like 3 has been observed to be substantially overexpressed and has been viewed as an adverse prognostic factor in HCC patients (Liu G. M. et al., 2020). Overexpression of METTL3 was associated with tumorigenicity and lung metastasis. In vitro experiments suggested that knockdown of METTL3 could lead to reduced capability of HCC cells in proliferation, migration, and colony formation (Chen et al., 2018). However, the mechanisms explaining how METTL3 contributes to HCC are not clear yet. METTL3 has been verified to function as the pivotal unit of the methyltransferase complex of m6A modification (Bokar et al., 1997). Therefore, the roles that METTL3 plays in HCC through m6A modification are worth discussion. In the present review, we are going to make an explicit summary on roles of METTL3 through m6A modification in HCC and the underlying mechanisms. Chances are that uncovering this mystery will provide us with new strategies for treatment of HCC.
Mechanisms Underlying the Carcinogenesis and Progression of HCC
METTL3 Suppresses SOCS2 Dependent on YTHDF2
The YTH domain family member 2 (YTHDF2) is known as a m6A “reader” that is capable to recognize and bind to m6A-modified sequences. YTHDF2 has been found associated with mRNA degradation (Du et al., 2016). Knockdown of YTHDF2 led to increased stability of target mRNAs with extended half-life of those mRNAs. Especially it is the C-terminal domain of YTHDF2 that is responsible for m6A-modified mRNA binding, while the N-terminal domain help execute the decay process (Wang et al., 2014). METTL3 was considered to be involved in YTHDF2 downstream regulation in a m6A-dependent manner. Knockdown of METTL3 led to remarkably reduced binding of YTHDF2 to its targets and thus extended the lifespan of those targets (Chen et al., 2018).
The previously reported YTHDF2 PAR-CLIP-Seq data together with transcriptome-wide m6A profiling data clearly showed the binding between the 3′ end of the suppressor of cytokine signaling 2 (SOCS2) transcript and YTHDF2. Consistently, knockdown of YTHDF2 significantly increased SOCS2 expression, suggesting that SOCS2 probably served as a direct downstream target of YTHDF2 (Dominissini et al., 2012; Liu et al., 2015; Chen et al., 2018). The m6A level of SOCS2 mRNA was strongly reduced after the knockdown of METTL3. SOCS2 expression was dramatically increased after suppressing the effect of METTL3 through the methylation inhibition. However, after mutating the adenosine bases of SOCS2 that abolished m6A modification, METTL3 silencing could not affect the expression of SOCS2. Together, METTL3 was considered to inhibit SOCS2 expression dependent on YTHDF2 through m6A modification (Chen et al., 2018).
Clinically, low expression of SOCS2 was correlated with poor prognosis in HCC patients (Cui et al., 2016). Knockdown of SOCS2 in HCC cells substantially promoted cell proliferation and migration (Chen et al., 2018). The SOCS2, as a member of SOCS family, is a negative regulator of the cytokine-dependent Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway. SOCS2 could inhibit the binding of STAT to its receptors and also target components of the pathways for proteosomal degradation. Enhanced JAK/STAT pathway induced by SOCS2 silencing has been indicated to play a role in cancers. Typically, STAT3 has been verified to contribute significantly to the tumorigenesis, progression, and metastasis process of HCC (Bromberg et al., 1999; Li et al., 2006; Rico-Bautista et al., 2006; Thomas et al., 2015; Xie et al., 2018).
In conclusion, overexpression of METTL3 in HCC probably facilitates the degradation process of SOCS2 dependent on YTHDF2 and reduces its expression, thus leading to aberrant JAK/STAT pathways which is responsible for the proliferation and migration of HCC cells.
METTL3 Inhibits RDM1
RAD52 motif-containing 1 (RDM1) has been verified as a target of METTL3-mediated m6A modification. Incremental expression of METTL3 decreased RDM1 expression, while it increased the m6A level of RDM1 mRNA in HCC cell lines. Overexpression of METTL3 was also correlated with decreased expression of RDM1 in the tissues from HCC patients (Chen S. L. et al., 2020).
RAD52 motif-containing 1 has previously been regarded to be associated with tumorigenesis. Overexpression of RDM1 was observed in several cancers such as the breast cancer and lung adenocarcinoma. In those cancers, RDM1 was regarded as an oncogene that could transcriptionally attenuate p53 expression and increase RAD51 and RAD52 level. This potential regulation of p53/RAD52/RAD51 signaling by increased RDM1 may lead to dysfunctional DNA repair pathways and the suppression of cell cycle arrest and apoptosis, thus promoting the tumor growth (Hermeking and Eick, 1994; Tong et al., 2018; Chen et al., 2019).
In HCC, it was proposed that RDM1 functioned as a tumor suppressor. Clinically low expression of RDM1 was corelated with worse differentiation, higher malignancy, and worse prognosis in HCC patients. Decreased expression of RDM1 has also been validated to improve the proliferation of HCC cells (Chen S. L. et al., 2020). RDM1 was considered to participate in DNA double-strand break (DSB) repair and recombination, which may restrain the process of carcinogenesis in HCC cells (Milne et al., 1995; Hamimes et al., 2006). Decreased expression of RDM1 was noticed to be related to stimulated calcium signaling which contributed to cancer cell survival, together with activated KRAS and RAF pathways which, as upstream of MEK/ERK pathways, enhanced cancer cell growth, survival and metabolism (Chen S. L. et al., 2020) (Asati et al., 2016; Reczek and Chandel, 2018). Overexpression of RDM1 was considered able to activate cell cycle and p53 signaling pathway (Chen S. L. et al., 2020). RDM1 could post-transcriptionally upregulate p53 expression and have a protective effect on wild-type p53, strengthening its stability and elongating its life-time. The process of DNA damage repair was predominantly facilitated by p53, serving as a crucial suppressor in HCC through unions of various DNA-damage-response (DDR) mechanisms (Staib et al., 2003; Williams and Schumacher, 2016).
In brief, RDM1 functions as a tumor suppressor in HCC by inhibiting cancer cell proliferation and promoting DNA damage repair in a p53-dependent manner. However, overexpression of METTL3 in HCC patients is able to decrease the expression of RDM1 in a m6A-dependent manner, thus promoting the survival, proliferation, and stability of HCC cells.
METTL3 Upregulates Snail via YTHDF1
Recently METTL3 has been introduced to participate in the epithelial mesenchymal transition (EMT) process in HCC (Lin et al., 2019). EMT refers to the transformation of epithelial cells into mesenchymal stem cells through specific programs, providing cancer cells with the opportunities for invasion and metastasis (Chaffer et al., 2016). Knockdown of METTL3 strongly suppressed the invasion abilities and EMT process of HCC cells in vitro, with increased expression of E-cadherin and decreased expression of MMP2 and FN (Wong et al., 2018; Lin et al., 2019). A variety of transcription factors have been verified to be related to EMT process such as Snail, Slug, Zeb, and Twist (Puisieux et al., 2014). Typically, Snail (encoded by SNAI1) was suggested to be affected by METTL3 (Xu et al., 2020).
YTHDF1 is known as a m6A “reader” and able to facilitate the translation process of target mRNAs by promoting ribosome loading on those mRNAs. Knockdown of METTL3 could repress this process (Wang et al., 2015; Kapur et al., 2017). Results of m6A RIP-PCR and YTHDF1 RIP-PCR suggested that SNAI1 was the direct target of YTHDF1 on the CDS of SNAI1 during EMT progression. In vitro experiments showed that knockdown of METTL3 could reduce the expression of Snail. In tumor tissues resected from liver cancer patients, increased expression of Snail and malignant behaviors were observed, in line with elevated expression of METTL3 and YTHDF1 (Lin et al., 2019). Together, METTL3 helps increase the expression of Snail through enhanced translation of SNAI1 mediated by YTHDF1.
Overall, overexpression of METTL3 in HCC is capable to increase the expression of Snail dependent on YTHDF1, which may promote the EMT process and provide HCC cells with opportunities for invasion and metastasis.
METTL3 Promotes Metabolic Reprogramming
Hepatitis B virus X-interacting protein (HBXIP) has been found significantly upregulated in HCC tissues and HCC cell lines (Melegari et al., 1998). Overexpression of HBXIP has been demonstrated to be associated with poor prognosis of HCC patients (Zheng et al., 2019). Several mechanisms have been proposed to account for HBXIP’s oncogenic roles. Among these mechanisms, metabolism reprogramming has been validated to be associated with METTL3 in HCC (Yang N. et al., 2020; Xiu et al., 2021). Liver cancer cells is metabolically characterized by the Warburg effect (or aerobic glycolysis), with enhanced glycolysis and increased level of lactic acid (Koppenol et al., 2011; Vaupel et al., 2019). METTL3, which was positively regulated by HBXIP in HCC, has been verified to be involved in metabolic reprogramming. According to the gene set enrichment analysis, expression of METTL3 was found positively correlated with the expression of genes involved in glycolysis such as glucose transporter member 1 (SLC2A1), hexokinase 2 (HK2), and pyruvate kinase (PKM), while it was negatively correlated with the expression of gluconeogenesis-related genes like glucose-6-phosphatase catalytic subunit (G6PC), pyruvate carboxylase (PC), and Fructose-1,6-bisphosphatase (FBP1) (Lin Y. et al., 2020). It seems that overexpression of METTL3 could promote the glycolysis process and inhibit the gluconeogenesis process. Knockdown of METTL3 was noticed able to repress glycolysis process and activate TCA cycle in HCC cells, with suppressed capability for cell aggression. Further experiments demonstrated that HBXIP’s role in metabolic reprogramming in HCC was dependent on METTL3 (Yang N. et al., 2020).
Hypoxia-Inducible Factor 1α (HIF-1α), associated with the genesis and development of tumors, has been demonstrated to promote the glycolysis process and facilitate the carcinogenesis in HCC (Gwak et al., 2005; Chiavarina et al., 2012; Lin and Wu, 2015). Overexpression of HIF-1α was associated with promoted metabolic reprogramming. METTL3 has been verified to regulate HIF-1α expression in a m6A-dependent manner. Results of MeRIP-qPCR demonstrated that METTL3 could increase the m6A level of HIF-1α mRNA. Knockdown of METTL3 strongly reduced the expression of HIF-1α (Yang N. et al., 2020).
Another strategy that accounted for how METTL3 participated in metabolic reprogramming was proposed. The mammalian target of rapamycin complex 1 (mTORC1) signaling was introduced as a crucial signaling that was involved in cell metabolism. MTORC1 signaling has been demonstrated to promote the glycolysis process (Laplante and Sabatini, 2012; Tian et al., 2019). Expressions of genes that encoded the enzymes of almost every step of glycolysis were found upregulated in response to activation of mTORC1, such as ALDOA, HK1/2, and SLC2A1/GLUT1 (Düvel et al., 2010). It was proposed that METTL3 potentially targeted the mTORC1 to regulate the glycolysis process in HCC. Impaired mTORC1 activity was observed after knockdown of METTL3, with reduced phosphorylation of S6K1 and 4EBP1 which were both substrates of mTORC1 (Burnett et al., 1998; Lin Y. et al., 2020). Additional silencing of METTL3 was unable to further decrease the phosphorylation level of mTORC1 and glycolysis activity in Rapamycin-treated HCC cells, suggesting the regulation of METTL3 on mTORC1. How mTORC1 signaling affected the glycolysis process has been researched. A c-Myc-LDHA axis was proposed to be a downstream target of mTORC1 and contribute to the abnormal glycolysis (Dang et al., 2009; Zhao et al., 2016). In addition, HIF-1α could function as another downstream effector of mTORC1 and participate in this process (Semenza et al., 1994; de la Cruz López et al., 2019).
In summary, overexpression of METTL3 in HCC could owe to the increased expression of HBXIP and be responsible for metabolic reprogramming and the proliferation, migration and invasion of HCC cells. HIF-1α is a potential target of METTL3-mediated m6A modification and is involved in metabolic reprogramming. Enhanced mTORC1 signaling is also closely related to METTL3 overexpression and associated with aberrant glycolysis process.
METTL3 Increases Lipogenesis via Upregulation of LINC00958
In addition to mRNAs, METTL3 has been associated with long non-coding RNAs (lncRNAs) in cancers including HCC (Zuo et al., 2020; Liu G. M. et al., 2021; Qu et al., 2021; Rong et al., 2021). lncRNAs, which comprises about 4–9% of total RNAs, refers to RNAs with limited or no protein-coding potential and possesses transcript sequence of more than 200 nt in length. LncRNAs are believed to execute the function of regulating gene expression and are involved in a variety of biological and pathological processes including cancers. Some lncRNAs have been demonstrated to be involved in carcinogenesis of HCC (Esteller, 2011; Gong et al., 2017; Ji et al., 2019; Ye et al., 2020). Here we take lncRNA LINC00958 as an example and demonstrate the effects on LINC00958 by METTL3, together with the detailed mechanisms explaining how this process affects HCC.
Overexpression of LINC00958 was observed in HCC and correlated with malignant behaviors of HCC cells and poor prognosis in HCC patients. Overexpression of LINC00958 was likely to promote cell growth, proliferation, migration and invasion in HCC cells. Clinically LINC00958 expression was associated with tumor size, tumor differentiation, microvascular invasion, and TNM stage. M6A RIP-qPCR analysis demonstrated that m6A level on LINC00958 was increased appreciably in HCC cells. Knock down of METTL3 reduced the m6A level of LINC00958 which probably decreased the stability of LINC00958 transcript and reduced its expression, suggesting that METTL3 in HCC may positively regulate LINC00958 expression (Liu G. M. et al., 2020; Zuo et al., 2020).
A LINC00958/miR-3619-5p/HDGF axis was proposed. Hepatoma-derived growth factor (HDGF) was regarded as an independent prognostic factor in liver cancer and was significantly upregulated in HCC (Zhou et al., 2010). Aberrant lipogenesis process via HDGF may account for its oncogenic characteristics. HDGF served as a coactivator of the sterol regulatory element binding protein-1 (SREBP-1) to participate in transcriptional activation of lipogenic enzymes associated with fatty acid, triglyceride, and cholesterol synthesis in HCC (Goldstein et al., 2006; Min et al., 2018). HDGF was a direct target of miR-3619-5p. Expression of HDGF has been demonstrated to be negatively regulated by miR-3619-5p (Zuo et al., 2020). However, as a sponge of miR-3619-5p, LINC00958 was regarded to be involved in abnormal lipogenesis process. Competitively binding to miR-3619-5p prevented the interaction between miR-3619-5p and HDGF, resulting in overexpression of HDGF (Peng et al., 2017). With the overexpression of LINC00958, HCC cells exhibited increased cellular levels of cholesterol and triglyceride, suggesting that LINC00958 positively regulated lipogenesis process.
In a word, overexpression of LINC00958 may be ascribed to METTL3-mediated m6A modification. A LINC00958/miR-3619-5P/HDGF axis was proposed to explain how LINC00958 affects lipogenesis and contributes to HCC.
Mechanisms Accounting for Drug-Resistance of HCC
METTL3 Depletion Contributes to Sorafenib Resistance via FOXO3
Apart from promoting the carcinogenesis and progression of HCC, METTL3 has been related to the resistance of anti-HCC drugs, such as the sorafenib resistance. Clinically, METTL3 silencing was found to noticeably enhance sorafenib resistance in HCC patients (Lin Z. et al., 2020).
Sorafenib has been known as a multi-target oral drug for treatment of tumors. It had dual anti-tumor effects aiming at both tumor cell growth and tumor angiogenesis. Sorafenib functioned as a multi-kinase inhibitor. It was capable to suppress tumor cell proliferation by inhibiting RAF/MEK/ERK pathway, and repress the angiogenesis process through impeding vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR). Sorafenib was the only FDA-approved drug for first-line treatment of advanced HCC and has been validated to prolong the overall survival (OS) of those patients (Cheng et al., 2009; Iyer et al., 2010; Brunetti et al., 2019). However, both primary and acquired resistance to sorafenib have been reported during clinical application.
Hypoxia was observed in HCC resulting from inadequate perfusion and diffusion in tumor tissues, with obvious damaged oxygenation status (Vaupel et al., 2007). Tumor tissues obviously showed higher expression of HIF-1α comparing to adjacent normal tissues. In this situation, reduced expression of METTL3 was noticed in sorafenib-resistant HCC. Catalytic mutant METTL3 did not sensitize METTL3-knockdown HCC cells for sorafenib treatment, suggesting METTL3’s m6A-dependent roles as a methyltransferase. Increased level of autophagosomes and LC3 were also observed in sorafenib-resistant HCC, and could be reversed by overexpression of wild-type METTL3, indicating that autophagy process may be associated with METTL3 and responsible for sorafenib-resistance (Lin Z. et al., 2020). The autophagy process has been considered to participate in multidrug resistance in chemotherapy of cancer (Li Y. J. et al., 2017). FOXO3, as has been elucidated to be associated with autophagy, was introduced to further explain the mechanisms. Knock down of FOXO3 facilitated the transcription of autophagy-related genes and was related to enhanced autophagy. Knock down of METTL3 decreased both mRNA and protein level of FOXO3, which could be reversed by wild-type METTL3 other than catalytic mutant METTL3. RNA m6A-Seq suggested that FOXO3 was probably the direct target of YTHDF1 which is a m6A “reader” and promotes the translation of its targets. The m6A site was located at the 3′UTR region (Wang et al., 2015; Fitzwalter and Thorburn, 2018; Lin Z. et al., 2020). In summary, Knockdown of METTL3 probably reversed the increasing transcription efficiency of FOXO3 mediated by YTHDF1 and brought about decreased FOXO3 expression, thus facilitating transcription of genes related to autophagy including ATG3/5/7/12, ATG16L1, and MAP1LC3B in HCC, ultimately leading to resistance of sorafenib (Lin Z. et al., 2020).
Briefly, under the circumstance of hypoxic microenvironment within tumors, METTL3 depletion has been discovered to substantially contribute to the acquired sorafenib resistance in HCC via FOXO3-mediated autophagy. Thus, METTL3 is a promising target to reverse sorafenib resistance in chemotherapy of HCC patients.
Discussion
In the last decades, m6A modification has been researched to clarify the potential mechanisms accounting for various kinds of cancers (Sun et al., 2019). METTL3, as a critical subunit of the METTL3-METTL14 methyltransferase complex, has been validated to contribute to the process of cancer. The present review focuses on hepatocellular carcinoma, and demonstrates several potential targets of METTL3 and the way METTL3 contributes to HCC in a m6A-dependent manner. METTL3 has been verified to be involved in proliferation, invasion, and metastasis of HCC cells, as well as the glycolysis and lipogenesis processes, promoting the carcinogenesis and progression of HCC (Chen et al., 2018; Chen S. L. et al., 2020; Lin Z. et al., 2020; Xu et al., 2020; Yang N. et al., 2020; Zuo et al., 2020; Table 1). Therefore, METTL3 may function as a potential target of anti-HCC treatment. However, METTL3 silencing has also been noticed to be correlated with sorafenib resistance in chemotherapy of advanced HCC patients. Mechanisms explaining how METTL3 serves as a double-edged sword in HCC deserve discussion. Overexpression of METTL3 was correlated with activation of JAK/STAT and Ras/Raf/ERK pathways, repression of p53 signaling pathway, and enhancement of metabolic reprogramming and the EMT process. Regulation by METTL3 on these signaling pathways and biological processes resulted in carcinogenesis and progression of HCC. However, the sorafenib resistance owed to the enhanced autophagy process which acted as a protective mechanism in cancer and could be induced by METTL3 silencing. Different targets of METTL3 accounted for its distinct influences (Figure 2). Clearly narrating these mechanisms helps support the clinical application of METTL3 in treatment of HCC. Apart from the mechanisms that have already been discussed in this review, some other potential targets of METTL3 may account for the carcinogenesis, progression, and drug resistance process in HCC. More efforts are required to further disclose METTL3’s role in HCC.
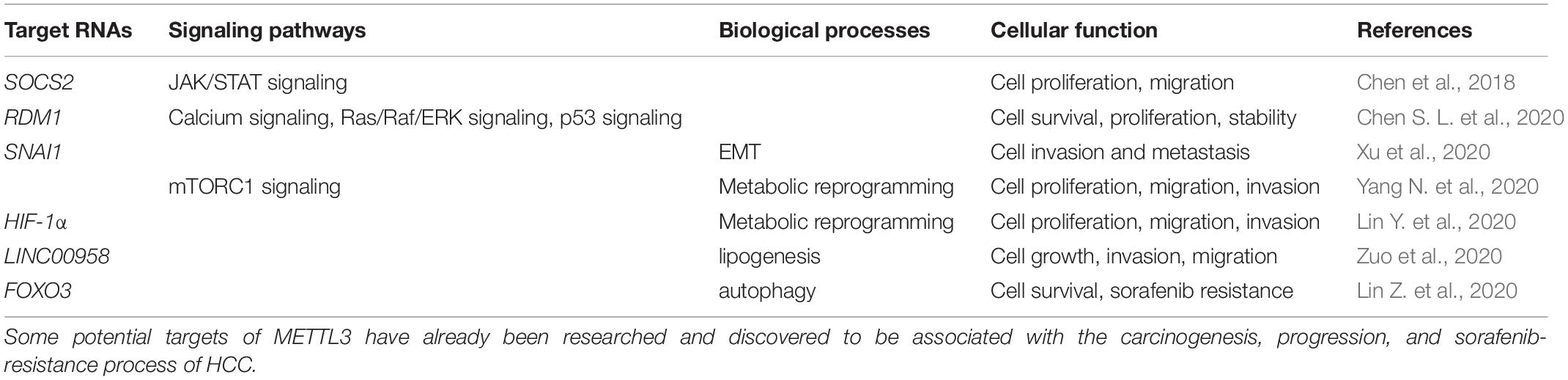
Table 1. Roles of METTL3 in HCC.
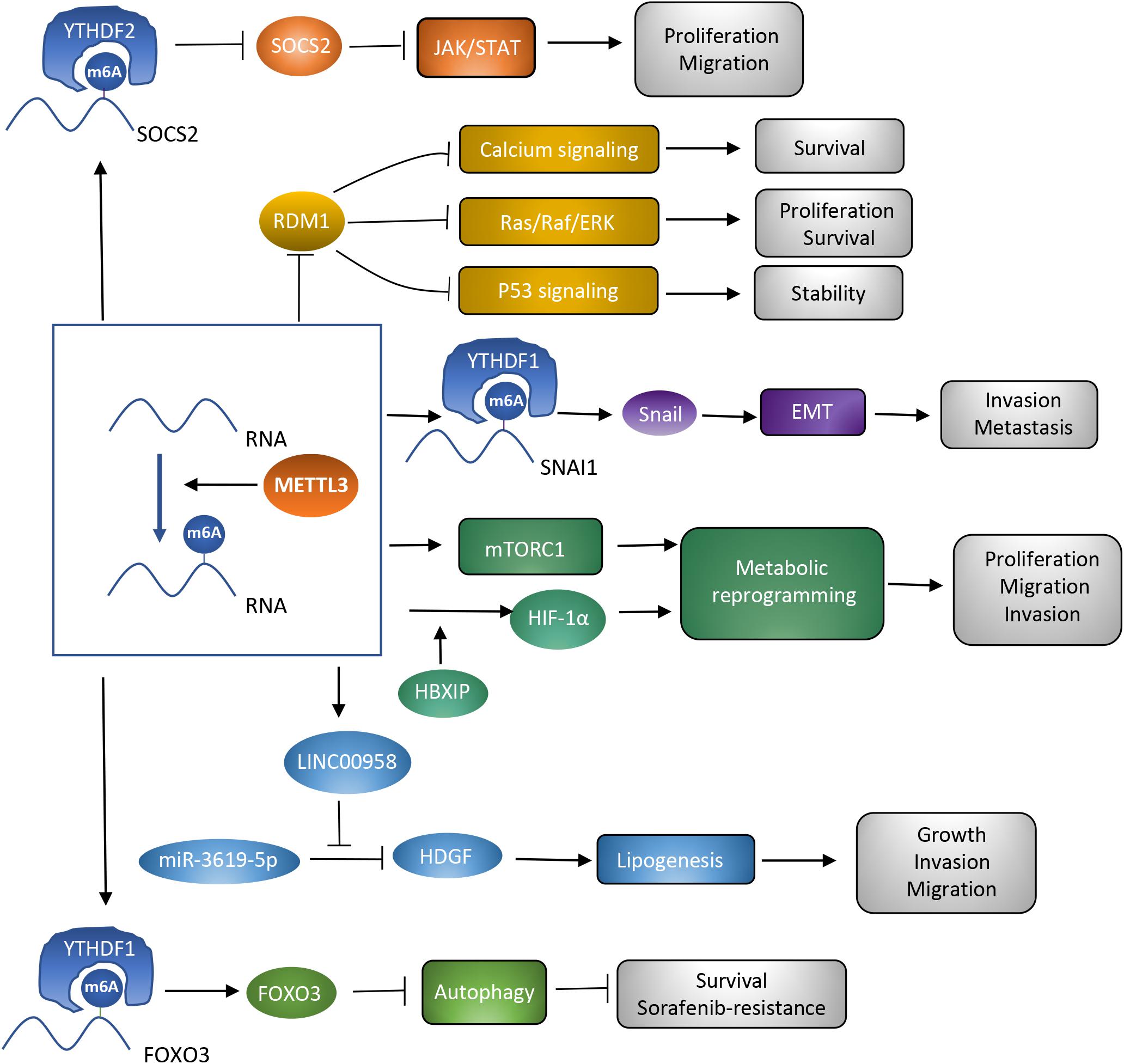
Figure 2. Roles of METTL3 as a methyltransferase in HCC. Different targets of METTL3 are associated with diverse signaling pathways and biological processes, which contribute to the different influences of METTL3 on HCC.
It should be noticed that m6A sequencing has been applied to discuss the interaction between METTL3 and its targets. However, the validity of m6Aseq is still unclear and needs extensive validation. LncRNAs have been associated with m6A modification in some cancers (Zuo et al., 2020; Liu G. M. et al., 2021; Qu et al., 2021; Rong et al., 2021). In the present review, we talked about LINC00958 which was positively correlated with METTL3 and was involved in aberrant lipogenesis in HCC (Zuo et al., 2020). Nevertheless, the detailed mechanisms explaining how METTL3 regulates LINC00958 are lacking. Extensive researches are required to further explain the regulation of METTL3 on lncRNAs. Some researches individually focused on METTL3 and its effects on HCC. Considering that METTL3 and METTL14 collectively form the methyltransferase complex, it is necessary to discuss the interaction between METTL3 and METTL14. In addition to m6A “writers,” some other regulators of m6A modification including YTHDC2, ALKBH5 and FTO have been associated with HCC (Li J. et al., 2019; Chen Y. et al., 2020; Liu J. et al., 2021). It is necessary to disclose their roles and the relevant mechanisms, which could promote their applications in clinical practice of HCC treatment.
Interestingly, opposite regulatory roles of METTL3 and METTL14 were observed in some cancers including HCC (Ma et al., 2017; Chen et al., 2018; Li T. et al., 2019; Yang X. et al., 2020). METTL14 was reported to share almost 56% binding sites with METTL3 (Liu et al., 2014). Functionally, METTL14 was regarded to structurally stabilize METTL3 conformation and help substrate recognition (Wang et al., 2016) Decreased expression of METTL14 was seen in HCC and was correlated with migration, invasion and EMT of HCC cells (Shi et al., 2020). Clinically, HCC patients with lower expression of METTL14 showed poorer prognosis, with lower OS rate. According to a multi-omics analysis, most of the mRNAs, signaling pathways and biological processes were differently regulated after knock down of METTL3 and METTL14, potentially explaining the distinct roles of METTL3 and METTL14 in HCC (Liu X. et al., 2020). More researches are required to clarify the disparate effects of METTL3 and METTL14 on HCC and the relevant mechanisms.
Author Contributions
RW conceptualized the review. FP were the major contributors in writing the manuscript. X-RL and L-PH designed the figures. X-YC and H-JW critically reviewed and edited the manuscript. All the authors read and approved the final manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (Nos. 81772995 and 81472266), the Excellent Youth Foundation of Jiangsu Province, China (No. BK20140032), and Jiangsu Province’s Key Provincial Talents Program (No. ZDRCA2016090).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all the guidance from our colleagues.
References
Asati, V., Mahapatra, D. K., and Bharti, S. K. (2016). PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 109, 314–341. doi: 10.1016/j.ejmech.2016.01.012
Bayoumi, M., and Munir, M. (2021). Evolutionary conservation of the DRACH signatures of potential N6-methyladenosine (m(6)A) sites among influenza A viruses. Sci. Rep. 11:4548. doi: 10.1038/s41598-021-84007-0
Bokar, J. A. (2005). “The biosynthesis and functional roles of methylated nucleosides in eukaryotic mRNA,” in Fine-Tuning of RNA Functions by Modification and Editing, ed. H. Grosjean (Berlin: Springer Berlin Heidelberg), 141–177.
Bokar, J. A., Shambaugh, M. E., Polayes, D., Matera, A. G., and Rottman, F. M. (1997). Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA (New York, NY) 3, 1233–1247.
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Bromberg, J. F., Wrzeszczynska, M. H., Devgan, G., Zhao, Y., Pestell, R. G., Albanese, C., et al. (1999). Stat3 as an oncogene. Cell 98, 295–303. doi: 10.1016/s0092-8674(00)81959-5
Brunetti, O., Gnoni, A., Licchetta, A., Longo, V., Calabrese, A., Argentiero, A., et al. (2019). Predictive and Prognostic Factors in HCC Patients Treated with Sorafenib. Medicina (Kaunas, Lithuania) 55, 707. doi: 10.3390/medicina55100707
Burnett, P. E., Barrow, R. K., Cohen, N. A., Snyder, S. H., and Sabatini, D. M. (1998). RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. U.S.A. 95, 1432–1437. doi: 10.1073/pnas.95.4.1432
Chaffer, C. L., San Juan, B. P., Lim, E., and Weinberg, R. A. (2016). EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 35, 645–654. doi: 10.1007/s10555-016-9648-7
Chen, M., Wei, L., Law, C. T., Tsang, F. H., Shen, J., Cheng, C. L., et al. (2018). RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology (Baltimore, Md) 67, 2254–2270. doi: 10.1002/hep.29683
Chen, S. L., Liu, L. L., Wang, C. H., Lu, S. X., Yang, X., He, Y. F., et al. (2020). Loss of RDM1 enhances hepatocellular carcinoma progression via p53 and Ras/Raf/ERK pathways. Mol. Oncol. 14, 373–386. doi: 10.1002/1878-0261.12593
Chen, Y., Sun, Z., and Zhong, T. (2019). RDM1 promotes critical processes in breast cancer tumorigenesis. J. Cell. Mol. Med. 23, 5432–5439. doi: 10.1111/jcmm.14425
Chen, Y., Zhao, Y., Chen, J., Peng, C., Zhang, Y., Tong, R., et al. (2020). ALKBH5 suppresses malignancy of hepatocellular carcinoma via m(6)A-guided epigenetic inhibition of LYPD1. Mol. Cancer 19:123. doi: 10.1186/s12943-020-01239-w
Cheng, A. L., Kang, Y. K., Chen, Z., Tsao, C. J., Qin, S., Kim, J. S., et al. (2009). Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 10, 25–34. doi: 10.1016/s1470-2045(08)70285-7
Chiavarina, B., Martinez-Outschoorn, U. E., Whitaker-Menezes, D., Howell, A., Tanowitz, H. B., Pestell, R. G., et al. (2012). Metabolic reprogramming and two-compartment tumor metabolism: opposing role(s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle (Georgetown, Tex) 11, 3280–3289. doi: 10.4161/cc.21643
Csepany, T., Lin, A., Baldick, C. J. Jr., and Beemon, K. (1990). Sequence specificity of mRNA N6-adenosine methyltransferase. J. Biol. Chem. 265, 20117–20122.
Cui, M., Sun, J., Hou, J., Fang, T., Wang, X., Ge, C., et al. (2016). The suppressor of cytokine signaling 2 (SOCS2) inhibits tumor metastasis in hepatocellular carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 37, 13521–13531. doi: 10.1007/s13277-016-5215-7
Dang, C. V., Le, A., and Gao, P. (2009). MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. Official J. Am. Assoc. Cancer Res. 15, 6479–6483. doi: 10.1158/1078-0432.ccr-09-0889
de la Cruz López, K. G., Toledo Guzmán, M. E., Sánchez, E. O., and García Carrancá, A. (2019). mTORC1 as a regulator of mitochondrial functions and a therapeutic target in cancer. Front. Oncol. 9:1373. doi: 10.3389/fonc.2019.01373
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. doi: 10.1038/nature11112
Du, H., Zhao, Y., He, J., Zhang, Y., Xi, H., Liu, M., et al. (2016). YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 7:12626. doi: 10.1038/ncomms12626
Düvel, K., Yecies, J. L., Menon, S., Raman, P., Lipovsky, A. I., Souza, A. L., et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183. doi: 10.1016/j.molcel.2010.06.022
Esteller, M. (2011). Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861–874. doi: 10.1038/nrg3074
Fitzwalter, B. E., and Thorburn, A. (2018). FOXO3 links autophagy to apoptosis. Autophagy 14, 1467–1468. doi: 10.1080/15548627.2018.1475819
Forner, A., Llovet, J. M., and Bruix, J. (2012). Hepatocellular carcinoma. Lancet (London, England) 379, 1245–1255. doi: 10.1016/s0140-6736(11)61347-0
Goldstein, J. L., DeBose-Boyd, R. A., and Brown, M. S. (2006). Protein sensors for membrane sterols. Cell 124, 35–46. doi: 10.1016/j.cell.2005.12.022
Gong, J., Qi, X., Zhang, Y., Yu, Y., Lin, X., Li, H., et al. (2017). Long noncoding RNA linc00462 promotes hepatocellular carcinoma progression. Biomed. Pharmacother. Biomed. Pharmacother. 93, 40–47. doi: 10.1016/j.biopha.2017.06.004
Gwak, G. Y., Yoon, J. H., Kim, K. M., Lee, H. S., Chung, J. W., and Gores, G. J. (2005). Hypoxia stimulates proliferation of human hepatoma cells through the induction of hexokinase II expression. J. Hepatol. 42, 358–364. doi: 10.1016/j.jhep.2004.11.020
Hamimes, S., Bourgeon, D., Stasiak, A. Z., Stasiak, A., and Van Dyck, E. (2006). Nucleic acid-binding properties of the RRM-containing protein RDM1. Biochem. Biophys. Res. Commun. 344, 87–94. doi: 10.1016/j.bbrc.2006.03.154
Hermeking, H., and Eick, D. (1994). Mediation of c-Myc-induced apoptosis by p53. Science (New York, NY) 265, 2091–2093. doi: 10.1126/science.8091232
Huang, H., Weng, H., Sun, W., Qin, X., Shi, H., Wu, H., et al. (2018). Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295. doi: 10.1038/s41556-018-0045-z
Iyer, R., Fetterly, G., Lugade, A., and Thanavala, Y. (2010). Sorafenib: a clinical and pharmacologic review. Expert Opin. Pharmacother. 11, 1943–1955. doi: 10.1517/14656566.2010.496453
Ji, D., Jiang, C., Zhang, L., Liang, N., Jiang, T., Yang, B., et al. (2019). LncRNA CRNDE promotes hepatocellular carcinoma cell proliferation, invasion, and migration through regulating miR-203/BCAT1 axis. J. Cell. Physiol. 234, 6548–6560. doi: 10.1002/jcp.27396
Jia, G., Fu, Y., Zhao, X., Dai, Q., Zheng, G., Yang, Y., et al. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887. doi: 10.1038/nchembio.687
Kapur, M., Monaghan, C. E., and Ackerman, S. L. (2017). Regulation of mRNA translation in neurons-a matter of life and death. Neuron 96, 616–637. doi: 10.1016/j.neuron.2017.09.057
Koppenol, W. H., Bounds, P. L., and Dang, C. V. (2011). Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337. doi: 10.1038/nrc3038
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Li, A., Chen, Y. S., Ping, X. L., Yang, X., Xiao, W., Yang, Y., et al. (2017). Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 27, 444–447. doi: 10.1038/cr.2017.10
Li, J., Zhu, L., Shi, Y., Liu, J., Lin, L., and Chen, X. (2019). m6A demethylase FTO promotes hepatocellular carcinoma tumorigenesis via mediating PKM2 demethylation. Am. J. Trans. Res. 11, 6084–6092.
Li, T., Hu, P. S., Zuo, Z., Lin, J. F., Li, X., Wu, Q. N., et al. (2019). METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol. Cancer 18, 112. doi: 10.1186/s12943-019-1038-7
Li, W. C., Ye, S. L., Sun, R. X., Liu, Y. K., Tang, Z. Y., Kim, Y., et al. (2006). Inhibition of growth and metastasis of human hepatocellular carcinoma by antisense oligonucleotide targeting signal transducer and activator of transcription 3. Clin. Cancer Res. Official J. Am. Assoc. Cancer Res. 12, 7140–7148. doi: 10.1158/1078-0432.ccr-06-0484
Li, Y. J., Lei, Y. H., Yao, N., Wang, C. R., Hu, N., Ye, W. C., et al. (2017). Autophagy and multidrug resistance in cancer. Chinese J. Cancer 36:52. doi: 10.1186/s40880-017-0219-2
Lin, D., and Wu, J. (2015). Hypoxia inducible factor in hepatocellular carcinoma: a therapeutic target. World J. Gastroenterol. 21, 12171–12178. doi: 10.3748/wjg.v21.i42.12171
Lin, S., Choe, J., Du, P., Triboulet, R., and Gregory, R. I. (2016). The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345. doi: 10.1016/j.molcel.2016.03.021
Lin, X., Chai, G., Wu, Y., Li, J., Chen, F., Liu, J., et al. (2019). RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat. Commun. 10:2065. doi: 10.1038/s41467-019-09865-9
Lin, Y., Wei, X., Jian, Z., and Zhang, X. (2020). METTL3 expression is associated with glycolysis metabolism and sensitivity to glycolytic stress in hepatocellular carcinoma. Cancer Med. 9, 2859–2867. doi: 10.1002/cam4.2918
Lin, Z., Niu, Y., Wan, A., Chen, D., Liang, H., Chen, X., et al. (2020). RNA m(6) A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. 39:e103181. doi: 10.15252/embj.2019103181
Linder, B., Grozhik, A. V., Olarerin-George, A. O., Meydan, C., Mason, C. E., and Jaffrey, S. R. (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772. doi: 10.1038/nmeth.3453
Liu, G. M., Zeng, H. D., Zhang, C. Y., and Xu, J. W. (2020). Identification of METTL3 as an adverse prognostic biomarker in hepatocellular carcinoma. Dig. Dis. Sci. 66, 1110–1126.
Liu, G. M., Zeng, H. D., Zhang, C. Y., and Xu, J. W. (2021). Identification of METTL3 as an adverse prognostic biomarker in hepatocellular carcinoma. Dig. Dis. Sci. 66, 1110–1126. doi: 10.1007/s10620-020-06260-z
Liu, H., Flores, M. A., Meng, J., Zhang, L., Zhao, X., Rao, M. K., et al. (2015). MeT-DB: a database of transcriptome methylation in mammalian cells. Nucleic Acids Res. 43, D197–D203. doi: 10.1093/nar/gku1024
Liu, J., Wang, D., Zhou, J., Wang, L., Zhang, N., Zhou, L., et al. (2021). N6-methyladenosine reader YTHDC2 and eraser FTO may determine hepatocellular carcinoma prognoses after transarterial chemoembolization. Arch. Toxicol. doi: 10.1007/s00204-021-03021-3 [Epup ahead of print],
Liu, J., Yue, Y., Han, D., Wang, X., Fu, Y., Zhang, L., et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95. doi: 10.1038/nchembio.1432
Liu, X., Qin, J., Gao, T., Li, C., Chen, X., Zeng, K., et al. (2020). Analysis of METTL3 and METTL14 in hepatocellular carcinoma. Aging 12, 21638–21659. doi: 10.18632/aging.103959
Liu, Z. X., Li, L. M., Sun, H. L., and Liu, S. M. (2018). Link Between m6A modification and cancers. Front. Bioeng. Biotechnol. 6:89. doi: 10.3389/fbioe.2018.00089
Ma, J. Z., Yang, F., Zhou, C. C., Liu, F., Yuan, J. H., Wang, F., et al. (2017). METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology (Baltimore, Md) 65, 529–543. doi: 10.1002/hep.28885
Melegari, M., Scaglioni, P. P., and Wands, J. R. (1998). Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J. Virol. 72, 1737–1743. doi: 10.1128/jvi.72.3.1737-1743.1998
Meyer, K. D., and Jaffrey, S. R. (2017). Rethinking m(6)A readers, writers, and erasers. Annu. Rev. Cell Dev. Biol. 33, 319–342. doi: 10.1146/annurev-cellbio-100616-060758
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3’. UTRs and near stop codons. Cell 149, 1635–1646. doi: 10.1016/j.cell.2012.05.003
Milne, G. T., Ho, T., and Weaver, D. T. (1995). Modulation of Saccharomyces cerevisiae DNA double-strand break repair by SRS2 and RAD51. Genetics 139, 1189–1199.
Min, X., Wen, J., Zhao, L., Wang, K., Li, Q., Huang, G., et al. (2018). Role of hepatoma-derived growth factor in promoting de novo lipogenesis and tumorigenesis in hepatocellular carcinoma. Mol. Oncol. 12, 1480–1497. doi: 10.1002/1878-0261.12357
Narayan, P., Ludwiczak, R. L., Goodwin, E. C., and Rottman, F. M. (1994). Context effects on N6-adenosine methylation sites in prolactin mRNA. Nucleic Acids Res. 22, 419–426. doi: 10.1093/nar/22.3.419
Peng, W. X., Koirala, P., and Mo, Y. Y. (2017). LncRNA-mediated regulation of cell signaling in cancer. Oncogene 36, 5661–5667. doi: 10.1038/onc.2017.184
Perry, R. P., Kelley, D. E., Friderici, K., and Rottman, F. (1975). The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5’ terminus. Cell 4, 387–394. doi: 10.1016/0092-8674(75)90159-2
Ping, X. L., Sun, B. F., Wang, L., Xiao, W., Yang, X., Wang, W. J., et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189. doi: 10.1038/cr.2014.3
Puisieux, A., Brabletz, T., and Caramel, J. (2014). Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 16, 488–494. doi: 10.1038/ncb2976
Qu, T., Mou, Y., Dai, J., Zhang, X., Li, M., Gu, S., et al. (2021). Changes and relationship of N(6)-methyladenosine modification and long non-coding RNAs in oxidative damage induced by cadmium in pancreatic β-cells. Toxicol. Lett. 343, 56–66. doi: 10.1016/j.toxlet.2021.02.014
Reczek, C. R., and Chandel, N. S. R. O. S. (2018). Promotes cancer cell survival through calcium signaling. Cancer Cell 33, 949–951. doi: 10.1016/j.ccell.2018.05.010
Rico-Bautista, E., Flores-Morales, A., and Fernández-Pérez, L. (2006). Suppressor of cytokine signaling (SOCS) 2, a protein with multiple functions. Cytokine Growth Factor Rev. 17, 431–439. doi: 10.1016/j.cytogfr.2006.09.008
Rong, D., Dong, Q., Qu, H., Deng, X., Gao, F., Li, Q., et al. (2021). m(6)A-induced LINC00958 promotes breast cancer tumorigenesis via the miR-378a-3p/YY1 axis. Cell Death Discov. 7:27. doi: 10.1038/s41420-020-00382-z
Schulze, K., Imbeaud, S., Letouzé, E., Alexandrov, L. B., Calderaro, J., Rebouissou, S., et al. (2015). Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 47, 505–511. doi: 10.1038/ng.3252
Semenza, G. L., Roth, P. H., Fang, H. M., and Wang, G. L. (1994). Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 269, 23757–23763.
Shi, Y., Zhuang, Y., Zhang, J., Chen, M., and Wu, S. (2020). METTL14 inhibits hepatocellular carcinoma metastasis through regulating EGFR/PI3K/AKT signaling pathway in an m6A-dependent manner. Cancer Manag. Res. 12, 13173–13184. doi: 10.2147/cmar.s286275
Sia, D., Villanueva, A., Friedman, S. L., and Llovet, J. M. (2017). Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 152, 745–761. doi: 10.1053/j.gastro.2016.11.048
Singal, A. G., and El-Serag, H. B. (2015). Hepatocellular carcinoma from epidemiology to prevention: translating knowledge into practice. Clin. Gastroenterol. Hepatol. Official Clin. Practice J. Am. Gastroenterol. Assoc. 13, 2140–2151. doi: 10.1016/j.cgh.2015.08.014
Staib, F., Hussain, S. P., Hofseth, L. J., Wang, X. W., and Harris, C. C. (2003). TP53 and liver carcinogenesis. Hum. Mutation 21, 201–216. doi: 10.1002/humu.10176
Sun, T., Wu, R., and Ming, L. (2019). The role of m6A RNA methylation in cancer. Biomed. Pharmacother. Biomed. Pharmacother. 112:108613. doi: 10.1016/j.biopha.2019.108613
Tang, C., Klukovich, R., Peng, H., Wang, Z., Yu, T., Zhang, Y., et al. (2018). ALKBH5-dependent m6A demethylation controls splicing and stability of long 3’-UTR mRNAs in male germ cells. Proc. Natl. Acad. Sci. U.S.A. 115, E325–E333. doi: 10.1073/pnas.1717794115
Thomas, S. J., Snowden, J. A., Zeidler, M. P., and Danson, S. J. (2015). The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 113, 365–371. doi: 10.1038/bjc.2015.233
Tian, T., Li, X., and Zhang, J. (2019). mTOR signaling in cancer and mTOR inhibitors in solid tumor targeting therapy. Int. J. Mol. Sci. 20:755. doi: 10.3390/ijms20030755
Tong, L., Liu, J., Yan, W., Cao, W., Shen, S., Li, K., et al. (2018). RDM1 plays an oncogenic role in human lung adenocarcinoma cells. Sci. Rep. 8:11525. doi: 10.1038/s41598-018-30071-y
Vaupel, P., Höckel, M., and Mayer, A. (2007). Detection and characterization of tumor hypoxia using pO2 histography. Antiox. Redox Signal. 9, 1221–1235. doi: 10.1089/ars.2007.1628
Vaupel, P., Schmidberger, H., and Mayer, A. (2019). The Warburg effect: essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiation Biol. 95, 912–919. doi: 10.1080/09553002.2019.1589653
Villanueva, A., Portela, A., Sayols, S., Battiston, C., Hoshida, Y., Méndez-González, J., et al. (2015). DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology (Baltimore, Md) 61, 1945–1956. doi: 10.1002/hep.27732
Wang, P., Doxtader, K. A., and Nam, Y. (2016). Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 63, 306–317. doi: 10.1016/j.molcel.2016.05.041
Wang, X., Lu, Z., Gomez, A., Hon, G. C., Yue, Y., Han, D., et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. doi: 10.1038/nature12730
Wang, X., Zhao, B. S., Roundtree, I. A., Lu, Z., Han, D., Ma, H., et al. (2015). N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399. doi: 10.1016/j.cell.2015.05.014
Williams, A. B., and Schumacher, B. (2016). p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 6:a026070. doi: 10.1101/cshperspect.a026070
Wong, S. H. M., Fang, C. M., Chuah, L. H., Leong, C. O., and Ngai, S. C. (2018). E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 121, 11–22. doi: 10.1016/j.critrevonc.2017.11.010
Xiao, W., Adhikari, S., Dahal, U., Chen, Y. S., Hao, Y. J., Sun, B. F., et al. (2016). Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 61, 507–519. doi: 10.1016/j.molcel.2016.01.012
Xie, Y., Li, J., and Zhang, C. (2018). STAT3 promotes the proliferation and migration of hepatocellular carcinoma cells by regulating AKT2. Oncol. Lett. 15, 3333–3338. doi: 10.3892/ol.2017.7681
Xiu, M., Zeng, X., Shan, R., Wen, W., Li, J., and Wan, R. (2021). The oncogenic role of HBXIP. Biomed. Pharmacother. Biomed. Pharmacother. 133:111045. doi: 10.1016/j.biopha.2020.111045
Xu, H., Wang, H., Zhao, W., Fu, S., Li, Y., Ni, W., et al. (2020). SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating Snail mRNA homeostasis in hepatocellular carcinoma. Theranostics 10, 5671–5686. doi: 10.7150/thno.42539
Xu, R. H., Wei, W., Krawczyk, M., Wang, W., Luo, H., Flagg, K., et al. (2017). Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat. Materials 16, 1155–1161. doi: 10.1038/nmat4997
Yang, N., Wang, T., Li, Q., Han, F., Wang, Z., Zhu, R., et al. (2020). HBXIP drives metabolic reprogramming in hepatocellular carcinoma cells via METTL3-mediated m6A modification of HIF-1α. J. Cell. Physiol. 236, 3863–3880. doi: 10.1002/jcp.30128
Yang, X., Zhang, S., He, C., Xue, P., Zhang, L., He, Z., et al. (2020). METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol. Cancer 19:46. doi: 10.1186/s12943-020-1146-4
Ye, Y., Guo, J., Xiao, P., Ning, J., Zhang, R., Liu, P., et al. (2020). Macrophages-induced long noncoding RNA H19 up-regulation triggers and activates the miR-193b/MAPK1 axis and promotes cell aggressiveness in hepatocellular carcinoma. Cancer Lett. 469, 310–322. doi: 10.1016/j.canlet.2019.11.001
Zhao, X., Jiang, P., Deng, X., Li, Z., Tian, F., Guo, F., et al. (2016). Inhibition of mTORC1 signaling sensitizes hepatocellular carcinoma cells to glycolytic stress. Am. J. Cancer Res. 6, 2289–2298.
Zheng, S., Wu, H., Wang, F., Lv, J., Lu, J., Fang, Q., et al. (2019). The oncoprotein HBXIP facilitates metastasis of hepatocellular carcinoma cells by activation of MMP15 expression. Cancer Manag. Res. 11, 4529–4540. doi: 10.2147/cmar.s198783
Zhou, Y., Zhou, N., Fang, W., and Huo, J. (2010). Overexpressed HDGF as an independent prognostic factor is involved in poor prognosis in Chinese patients with liver cancer. Diagnostic Pathol. 5:58. doi: 10.1186/1746-1596-5-58
Keywords: RNA modification, METTL3, m6A, Hepatocellular carcinoma, drug-resistance
Citation: Pan F, Lin X-R, Hao L-P, Chu X-Y, Wan H-J and Wang R (2021) The Role of RNA Methyltransferase METTL3 in Hepatocellular Carcinoma: Results and Perspectives. Front. Cell Dev. Biol. 9:674919. doi: 10.3389/fcell.2021.674919
Received: 02 March 2021; Accepted: 19 April 2021;
Published: 11 May 2021.
Edited by:
Filippo Cernilogar, Ludwig Maximilian University of Munich, GermanyReviewed by:
Isaia Barbieri, University of Cambridge, United KingdomLuca Pandolfini, Italian Institute of Technology (IIT), Italy
Copyright © 2021 Pan, Lin, Hao, Chu, Wan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hai-Jun Wan, d2FuaGFpanVuNzkwNjIwQHNpbmEuY29t; Rui Wang, d2FuZ3J1aTIxOEAxNjMuY29t