 Zhi Fang
Zhi Fang Xiang Wang
Xiang Wang Xiaoran Sun
Xiaoran Sun Wenquan Hu
Wenquan Hu Qing R. Miao
Qing R. Miao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 29 April 2021
Sec. Signaling
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.672447
This article is part of the Research TopicMolecular Mechanisms and Signaling in Endothelial Cell Biology and Vascular HeterogeneityView all 27 articles
Endothelial cell (EC), consisting of the innermost cellular layer of all types of vessels, is not only a barrier composer but also performing multiple functions in physiological processes. It actively controls the vascular tone and the extravasation of water, solutes, and macromolecules; modulates circulating immune cells as well as platelet and leukocyte recruitment/adhesion and activation. In addition, EC also tightly keeps coagulation/fibrinolysis balance and plays a major role in angiogenesis. Therefore, endothelial dysfunction contributes to the pathogenesis of many diseases. Growing pieces of evidence suggest that histone protein acetylation, an epigenetic mark, is altered in ECs under different conditions, and the acetylation status change at different lysine sites on histone protein plays a key role in endothelial dysfunction and involved in hyperglycemia, hypertension, inflammatory disease, cancer and so on. In this review, we highlight the importance of histone acetylation in regulating endothelial functions and discuss the roles of histone acetylation across the transcriptional unit of protein-coding genes in ECs under different disease-related pathophysiological processes. Since histone acetylation changes are conserved and reversible, the knowledge of histone acetylation in endothelial function regulation could provide insights to develop epigenetic interventions in preventing or treating endothelial dysfunction-related diseases.
Endothelial cells (ECs), mostly in the inner layer of blood vessels are co-existing with ever-changing environmental conditions, such as fluid shear stress, fluctuations of blood pressure, glucose and cytokines, nutrition availability, as well as pollutant molecules (Potente and Makinen, 2017; Sturtzel, 2017). Sensitive response to rapid changes in both internal and external cues is essential for ECs to maintain cellular homeostasis. Histone post-translational modifications (PTMs) represent crucial epigenetic mechanisms governing the ability of ECs in response to intracellular and extracellular stimuli.
To date, more than 100 different histone modifications have been detected by mass spectrometry or specific antibodies (Choudhary et al., 2014). In all eukaryotes ranging from yeast to humans, DNA is compacted more than 10,000-fold into the nucleus as chromatin. The basic structural subunit of chromatin is the nucleosome, which is comprised of 147 bp of DNA wrapped around a histone octamer core. Each nucleosome core is consisted of two copies of each of the histones: H2A, H2B, H3, and H4 (Tessarz and Kouzarides, 2014). Initially, histones were regarded as merely structural components which act as a repressive barrier to the nuclear processes requiring access to DNA. However, research over the past two decades has illustrated that histone proteins are critical for the organization and function of chromatin. Dynamic histone PTMs, largely occur at the amino-terminal tail of histones, can fundamentally regulate chromatin structure and gene transcription by spatially and temporally coordinating the shifting between condensed/transcriptionally silenced states and accessible/transcriptionally active states (Shahbazian and Grunstein, 2007). Among the diverse histone PTMs, histone acetylation has been recognized as a fundamental process that strongly affects gene expression regulation (Allfrey et al., 1964).
Growing pieces of evidence suggest that histone protein acetylation is altered in ECs under different conditions. The changes of acetylation status at different lysine sites on histone protein strongly affect EC gene expression and thus lead to endothelial dysfunction, which is involved in the pathogenesis of many cardiovascular disease processes. In this review, we will generally introduce histone acetylation and the related writers and erasers and highlight the importance of histone acetylation in regulating endothelial functions and discuss the roles of histone acetylation across the transcriptional unit of protein-coding genes in ECs under different disease-related pathophysiological processes, including vascular tone, inflammation, oxidative stress, angiogenesis, barrier function, thrombosis, and coagulation.
A common form of histone PTMs and, indeed, one of the first discovered is acetylation (Figure 1). Histone acetylation has been linked to active transcription (Phillips, 1963). Mechanically, negatively charged acetyl groups covalently added to specific lysine residues in histone can diminish the electrostatic affinity between histone proteins and DNA, thus disrupts the interaction of these histones with the DNA, leading to chromatin relaxation that enables the activation of gene transcription. Moreover, this modification can serve as docking sites for proteins involved in gene activation, such as transcriptional activators and chromatin remodeling complexes (Marmorstein and Zhou, 2014).
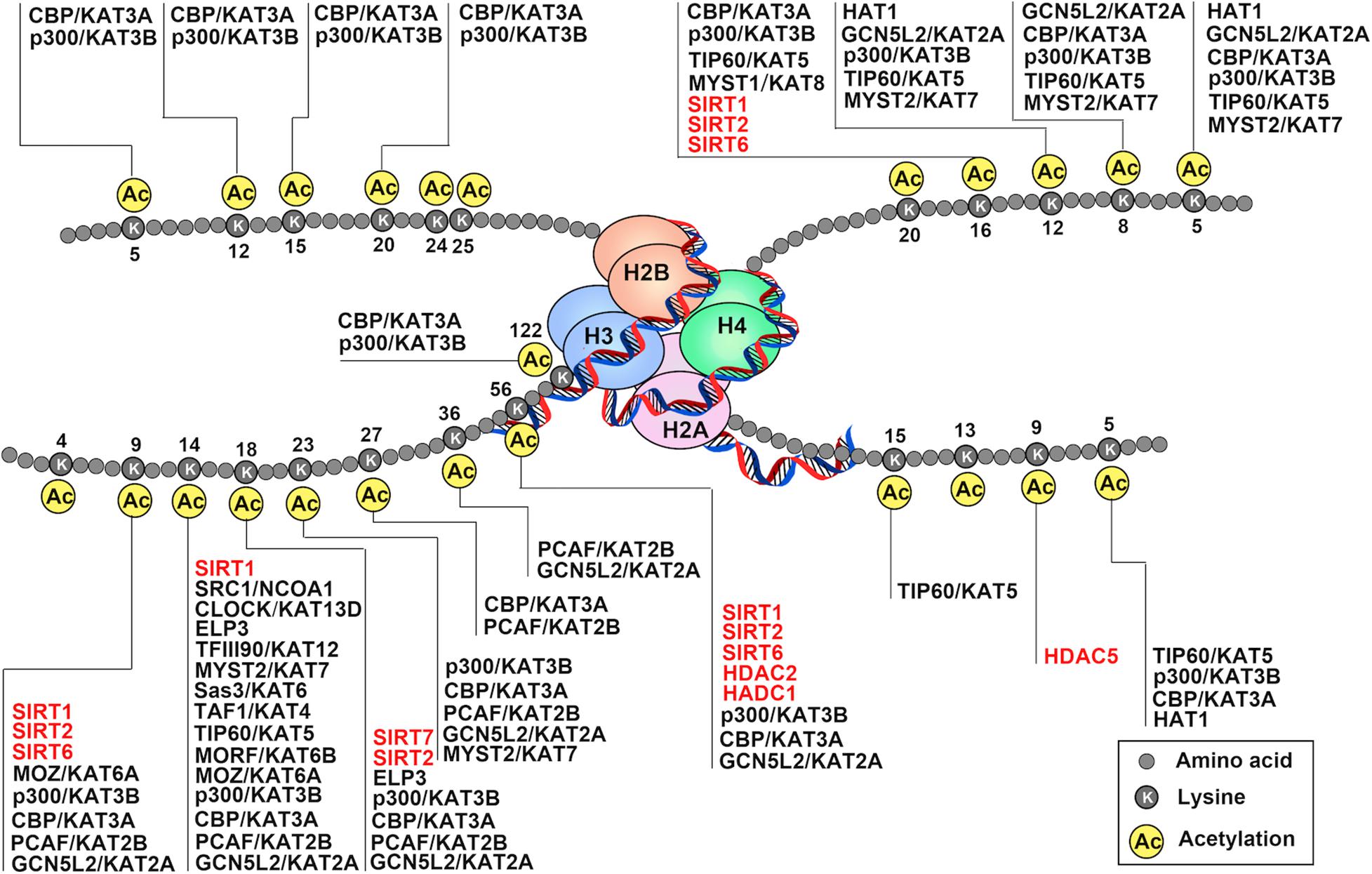
Figure 1. Schematic drawing of histone acetylation. DNA is wound around nucleosomes, which are composed of eight histone molecules with two copies of histones H2A, H2B, H3, and H4. Each histone molecule has a long tail rich in lysine residues (K), which are acetylation modification sites. Gene activation and repression are regulated by acetylation of core histone, and histone acetylation is mediated by coactivators (histone acetyltransferase: HATs) and corepressors (histone deacetylases: HDACs). The addition of acetyl-groups to the tails of histone core proteins leads to a relaxing state of chromatin and includes active transcription. In turn, removel of acetyl-groups from the tails of histone core proteins leads to the adoption of a condensed state of chromatin and transcriptional repression.
Histone acetylation is a highly reversible process operated in a highly regulated manner. A lysine residue becomes acetylated by the action of the histone acetyltransferases (HATs, also referred to as “writers”), which transfer acetyl group from acetyl-coenzyme A (acetyl-CoA) onto the ε-amino group of target lysine residues. To date, 37 mammalian proteins have been suggested to possess endogenous HAT activity, which can be further classified into at least five different subfamilies based on their structural homologies: the CBP/p300, GCN5/PCAF, MYST, the nuclear receptor coactivator family, and basal transcription families (Figures 2A,B; Javaid and Choi, 2017; Sheikh and Akhtar, 2019). However, due to the lack of validated evidence of HAT activities, the latter two subfamilies will not be further discussed in this review. The reverse reaction is catalyzed by histone deacetylases (HDACs, also referred to as “erasers”). In mammals, there are 18 HDAC enzymes that can be grouped into two distinct categories with different catalytic mechanisms: Zn2+-dependent HDACs and NAD+-dependent sirtuin (Sir2 orthologs) deacetylases (Figures 2C,D). On the basis of their domain structures and sequence similarities, they can be further divided into five groups: Class I comprises HDAC1, HDAC2, HDAC3, and HDAC8; the Class IIa includes HDAC4, HDAC5, HDAC7, and HDAC9; class IIb includes HDAC6 and HDAC10; Class III comprise SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7; and the Class IV protein HDAC11 (Seto and Yoshida, 2014; Zhao et al., 2018). Just as histone acetylation is accomplished by “writers” and “erasers,” their actions to govern DNA transcription are mediated by “readers.” According to domain structure, various “readers” can be roughly classified into three categories, including BRD, double PHD finger (DPF), and YEATS domains (Gillette and Hill, 2015; Gong et al., 2016). Emerging evidence has also highlighted the importance of acetylation readers, and some have been implicated in physiological and pathological processes of ECs.
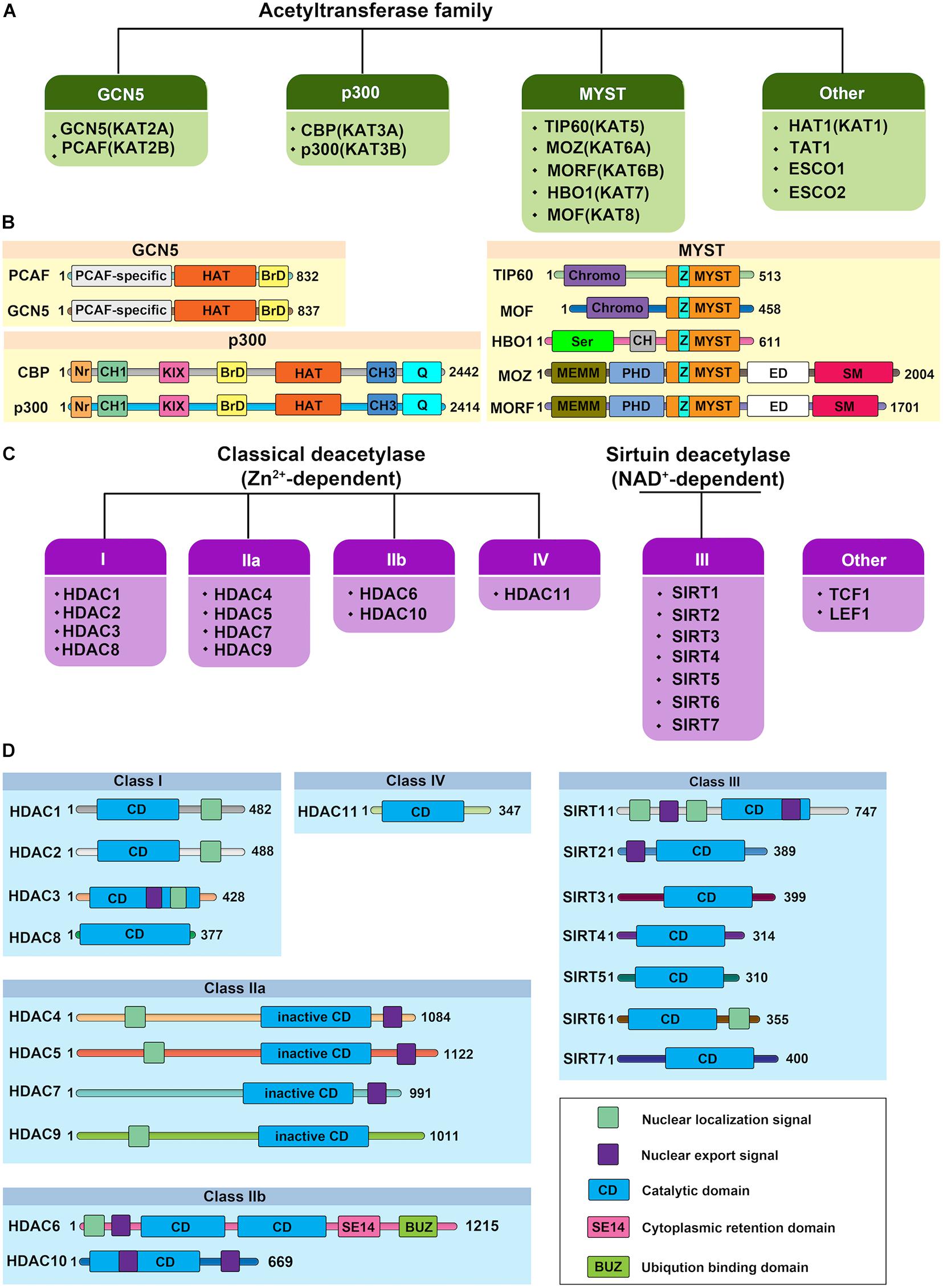
Figure 2. Protein structure of human histone acetylases and deacetylases. (A) The canonical histone acetyltransferases (HATs) are classified into three prominent families: GCN5, p300, and MYST. The other HATs are relatives dissimilar to each other. (B) Protein structure of human histone acetyltransferases. BrD, bromodomain; Nr, nuclear receptor-interacting box; CH, cysteine/histidine-rich module; KIX, phospho-CREB interacting module; Q, glutamine-rich domain; HAT, the lysine acetyltransferase catalytic domain; Chromo, chromodomain; Ser, serine-rich domain; NEMM, N-terminal part of MOZ or MORF; PHD, PHD zinc finger; ED, glutamate/aspartate-rich region; SM, serine/methionine-rich domain; MYST, MYST histone acetyltransferase domain; Z: C2HC zinc finger domain within the MYST domain. Numbers indicate the length of each protein. (C) Histone deacetylases (HDACs) are divided into two categories: the classical Zn2+-dependent HDACs and NAD+-dependent sirtuin deacetylases. Based on their phylogenetic conservation and sequence similarities, the HDACs are further classified into five major families: Class I, Class IIa, Class IIb, Class III, and Class IV. (D) Protein structure of human histone deacetylases. Protein domains of HDAC isoenzymes are presented above. Most HDACs possess a nuclear localization and/or export signal. Numbers indicate the length of each protein.
General control non-derepressible 5 (GCN5, also called KAT2A) was one of the first proteins found to possess HAT activity (Mutlu and Puigserver, 2020). It was also the first histone acetyltransferase confirmed to link histone acetylation to transcriptional activation (Verdin and Ott, 2015). Following its discovery, some other HATs, including PCAF, which contain similar domain structures, have also been identified and consist of the GCN5/PCAF subfamily of HAT proteins. This family of HAT proteins contains PCAF-specific N-terminal domains, a HAT domain of around 160 residues, and a conserved bromodomain within the C-terminal, responsible for recognizing and binding acetyl-lysine recruiting transcriptional machinery to regulate gene expression (Downey, 2020).
GCN5 and PCAF both function as part of unique multi-subunit complexes, where they act as the catalytic HAT domain. For example, GCN5/PCAF is found in the complexes of Spt-Ada-GCN5 acetyltransferase (SAGA) and Ada Two A Containing (ATAC) (Figures 3A,B; Helmlinger and Tora, 2017). While the HAT module within the SAGA complex preferentially acetylates histone H3K9 and H3K14 lysine residues in vivo, GCN5 within the ATAC complex mainly acetylates H4K5, H4K12, and H4K16 (Spedale et al., 2012). Although the exact function of the ATAC complex remains elusive, it has been well demonstrated that following transcriptional activation, GCN5–SAGA mediates H3 acetylation throughout the gene body recruitment of the ATP-dependent chromatin remodeler SWI/SNF to target genes, which initiates a cascade of chromatin remodeling events that promotes gene transcription (Helmlinger et al., 2020).
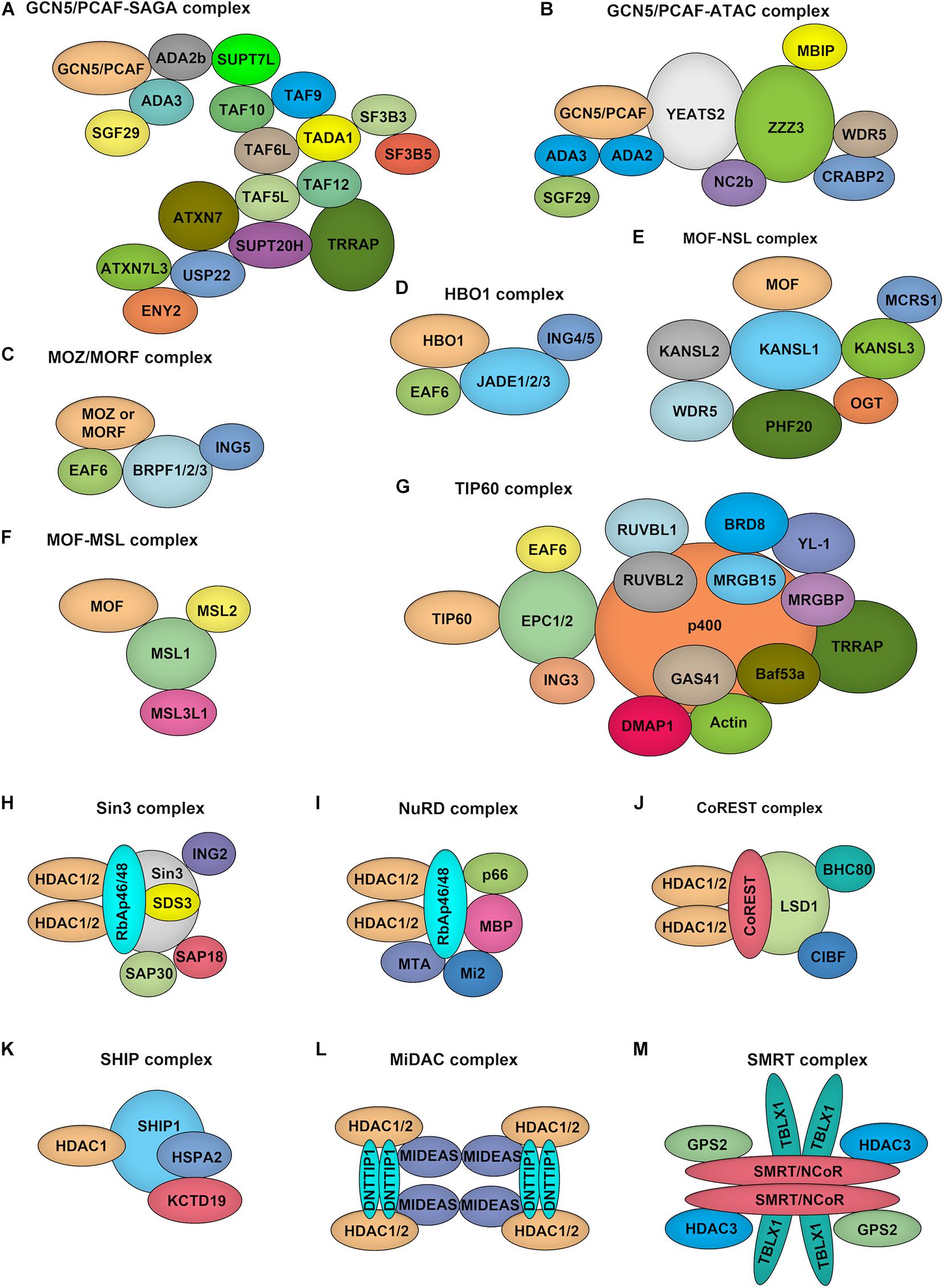
Figure 3. Composition of HATs and HDACs containing multiprotein complexes. (A–G). Depiction of the major histone acetyltransferase complexes: GCN5/PCAF-SAGA (A) and GCN5/PCAF-ATAC (B), MOZ/MORF-ING5 (C), HBO1-JADE (D), MOF-NSL (E), MOF-MSL (F), TIP60 (G), which contains two HATs. (H–M), Overview of class I HDAC containing complexes. Sin3 complex (H): The Sin3 complex is comprised of HDAC1 and HDAC2, delivering enzymatic activity and the structural components Sin3, RbAp46, RbAp48, SAP18, SAP30, and SDS3. NuRD complex (I): RbAp46, RbAp48, HDAC1, and HDAC2 are also found in the NuRD complex, together with the NuRD specific factors MTA, Mi-2, p66, and MBD. CoREST complex (J): The CoREST complex consists of CoREST, CIBF, BHC80, HDAC1, HDAC2 and is targeted to DNA via the REST protein. SHIP complex (K): The SHIP complex was identified recently as a testis specific complex containing SHIP1, HDAC1, HSPA2, and KCTD19. MiDAC complex (L): The mitotic deacetylase complex (MiDAC) is a recently identified histone deacetylase (HDAC) complex using proteomics in cells that are blocked in mitosis by nocodazole. MiDAC contains HDAC1/2, a protein called DNTTIP1 (deoxynucleotidyltransferase terminal-interacting protein 1), and the MIDEAS (mitotic deacetylase-associated SANT domain protein) co-repressor protein. SMRT/NCoR complex (M): The core of the complex consists of SMRT/NCoR bound to TBL1 (transducin beta-like protein 1), GPS2 (G protein pathway suppressor 2), and HDAC3 proteins.
CREB-binding protein [CREB (cyclic-AMP response element-binding protein) binding protein, CBP] and its close paralog EP300 (E1A binding protein p300) consist of another major group of HATs, which are now collectively referred to as CBP/p300 because of extensive structural homology and functional redundancy (Breen and Mapp, 2018). Both proteins are pivotal transcriptional coactivators integrating and coordinating transcriptional activation (Lipinski et al., 2019).
The members of this family are large proteins (with about 2,400 amino acids) that do not have defined molecular complexes but harbor multiple functional domains, including a considerably larger HAT domain of about 500 residues, a bromodomain to capture acetyl-lysine, and some other functional domains including the nuclear receptor-interacting box, cysteine/histidine-rich module, phospho-CREB interacting module, and glutamine-rich domain that can regulate enzyme activity and mediate diverse protein-protein interactions (Figure 2B; Bordoli et al., 2001; He et al., 2021). CBP/p300 facilitates the gene transcription not only through acetylation of histones and transcription factors but also acts as “hubs” that link chromatin-modifying proteins and basal transcriptional machinery (Rando and Chang, 2009). More importantly, recent advances reveal that CBP/p300 can be recruited by enhancers and mediate the acetylation of H3K18 and H3K27, which stimulates the establishment of an active enhancer state and facilitates transcriptional activation (Heintzman et al., 2007; Hnisz et al., 2013; Arner et al., 2015). For example, p300 was found to locate to the tissue-specific enhancers and drive the expression of adjacent genes in the forebrain or limb tissues during mouse embryonic development at a specific time point (Visel et al., 2009). Given that dynamic regulation of enhancers at specific sets of genes involving cellular differentiation, CBP/p300 can orchestrate the transcription of a series of developmentally essential genes in a temporal-specific and cell type-specific manner.
The MYST family of HAT proteins was first reported in 1996 (Reifsnyder et al., 1996), originally named for its founding members in yeast and mammals. The family currently comprises five human HATs: MOZ (MYST3/KAT6a), MORF (MYST4/KAT6b), HBO1 (MYST2/KAT7), MOF (MYST1/KAT8), and TIP60 (HTATIP/KAT5). They are defined together by the presence of a highly conserved 370 residue MYST domain, which consists of an acetyl-CoA-binding motif homologous to the canonical acetyl-CoA binding domain of GCN5/PCAF family and a C2HC zinc finger domain (Thomas and Voss, 2007). Besides the MYST domain, many members contain additional structural features, such as cysteine-rich, zinc-binding domain within the HAT regions and N-terminal chromodomains (Figure 2B; Wiesel-Motiuk and Assaraf, 2020). Based on additional notable domain regions, this family of HATs can be further classified into three subgroups: (1) MOZ and MORF, (2) MOF and TIP60, as well as (3) HBO1 alone. For example, MOZ and MORF contain two PHD-type zinc finger domains, while MOF and TIP60 are characterized by a conserved chromodomain (Santos-Rosa and Caldas, 2005).
The MYST family proteins usually form complexes with other transcriptional cofactors to coordinate transcriptional activation and transcriptional silencing. MOZ and MORF were co-purified from HeLa cells in the ING5 complex, which is consisted of MOZ/MORF, EAF, ING5, and BRPF1/2/3 (Figure 3C; Pillus, 2008). This complex can localize to the promoter regions of target genes to acetylate H3K9 and H3K23, leading to transcriptional activation. MOF occurs in two complexes identified as the MSL (male-specific lethal) complex and the NSL (non-specific lethal) complex (Figures 3E,F; Feller et al., 2012). Either MOF–MSL complex or MOF-NSL complex can trigger H4K16 acetylation at the promoter region to promote transcription, and, most intriguingly, the MOF-NSL complex can be recruited to H3K4 dimethylation positive promoters where it acetylates H4K16. In contrast, the MOF-MSL complex strongly binds to H4K20 monomethylation positive promoters in active gene bodies. TIP60 exists in a multi-subunit protein complex called NuA4 (Figure 3G; Xu et al., 2016), which shows an affinity for multiple histone acetylation sites, including H3K14, H3K27 H3K18, and H3K27. HBO1 functions in the context of a multi-protein complex called HBO1-JADE (Figure 3D), which contains ING4/5, MEAF6, and JADE1/2/3 for H3K14 acetylation throughout the gene body as well as promoters (Saksouk et al., 2009). This complex also shows an affinity for H3K4 dimethylation and trimethylation positive regions.
Among the zinc-dependent HDACs, Class I, II, and IV HDACs share a conserved HDAC domain (Figure 2D). However, they have quite different characteristics. Class I HDACs majorly localize to the nucleus of mammalian cells with robust deacetylase activity. These family proteins usually function as the catalytic core within multi-subunit transcriptional repressor complexes. HDAC1/2 exist in the complexes of NuRD (nucleosome remodeling and deacetylase), SHIP (a testis-specific protein possessing putative DNA binding and chromatin remodeling properties), SIN3 (switch-independent 3), CoREST (corepressor of REST), and MiDAC (mitotic deacetylase) while HDAC3 primarily acts in NCOR1/SMRT repressor protein complex (Figures 3H–M; Kelly and Cowley, 2013; Millard et al., 2017). These complexes play essential roles for Class I HDACs to recruit them to specific target gene regions and activate their enzymatic activity. Due to the absence of a catalytic residue in the HDAC domain, class IIa HDACs, including HDAC4, HDAC5, HDAC7, and HDAC9, possess weak deacetylase activity. However, they still can serve as transcriptional repressors by forming complexes with NCOR1/SMRT (Kelly and Cowley, 2013). Also, the function of class IIa HDACs toward histone proteins is under the control of the shuttling between the cytoplasm and nucleus. Distinguished from other zinc-dependent HDACs, the class IIb family member HDAC6 is a unique member of the HDAC family that not only deacetylates histone but also targets cytoskeleton structure, including α-tubulin and cortactin (Li T. et al., 2018). For the other class IIb HDACs, HDAC10, although it can form a repressor complex with SMRT, has relatively low lysine deacetylase activity but is a robust polyamine deacetylase (Fischer et al., 2002; Hai et al., 2017). As the exclusive class IV HDAC, HDAC11 is the newest member of the HDAC family and shares the catalytic domain both of class I and II HDACs. However, it has been reported that the defatty-acylase activity of HDAC11 is over 10,000 times more than its deacetylase activity (Liu et al., 2020).
Sirtuin deacetylases, which have a conserved nicotinamide adenine dinucleotide (NAD)-binding and catalytic domain, are NAD+-dependent lysine deacetylases or ADP-ribosyltransferases. They were first identified in the yeast, referred to as the class III histone deacetylases (Carafa et al., 2016; Kida and Goligorsky, 2016). To date, seven different SIRT proteins have been identified in mammals. However, growing evidence suggests several members, including SIRT4, SIRT5 have weak deacetylase activity. Instead, extensive studies have clearly shown that SIRT proteins can catalyze the removal of diverse acyl groups from target proteins such as malonyl, succinyl, propionyl, and long-chain fatty acyl (Bosch-Presegue and Vaquero, 2015).
Besides the common deacetylase domain, SIRTs contain quite different N- and C-terminal structural features, which may explain their specified subcellular localization and functions: SIRT1, SIRT2, and SIRT6 can shuttle from nucleus to the cytoplasm; SIRT3, SIRT4, and SIRT5 are majorly mitochondrial proteins; SIRT7 localizes to nuclear and nucleolar compartment (Figure 2B; D’Onofrio et al., 2018; Singh et al., 2018).
Nitric oxide (NO) is a vessel dilator regulating vascular tone (Balligand and Cannon, 1997; Alderton et al., 2001). The production of NO is regulated by the activation of nitric oxide synthase (NOS). Endothelial NOS (eNOS, also called NOS3) is constitutively expressed in vascular ECs (Wilcox et al., 1997) and plays a key role in vascular wall homeostasis and regulation of vascular tone (Balligand and Cannon, 1997; Wilcox et al., 1997; Alderton et al., 2001). Fish et al. (2005) reported that the expression of eNOS is controlled by an EC-specific histone code. Different from non-ECs, the endothelial nucleosomes that encompassed the eNOS core promoter and proximal downstream coding regions are highly enriched in acetylated histones H3K9, H4K12, and di- and tri-methylated H3K4, which are selectively associated with functionally competent RNA polymerase II complexes in ECs (Fish et al., 2005).
Proper expression of eNOS, which is required for maintaining the normal endothelial function, is regulated by histone acetylation under multiple stimuli (Table 1). Laminar shear stress (SS) has an acute stimulation of eNOS mRNA transcription (Dimmeler et al., 1999; Fleming and Busse, 1999; Boo et al., 2002). Chen et al. (2008) reported that shear stress increases p300 histone acetyltransferase activity by 2.5-fold and stimulates acetylation of histones H3 and H4 at the promoter region of the eNOS shear stress response element (SSRE) and extended 3′ toward the eNOS coding region, resulting in the opening of chromatin at the SSRE and rapid transcriptional increase of eNOS. Different from shear stress, Fish et al. (2010) demonstrated that hypoxia causes a rapid decrease in the transcription of the eNOS gene. Hypoxia rapidly evicts histone 2A (H2A.Z) from eNOS proximal promoter, and after longer durations of hypoxia exposure, acetylation of histone H3K9, H3K14, H4K5, H4K8, H4K12, and methylation of histone H3K4 decrease on eNOS proximal promoter (Fish et al., 2010). However, there is a controversy on hypoxia-induced histone acetylation mediating eNOS expression. In a rat model of hypoxia-induced persistent pulmonary hypertension of the newborn (PPHN), eNOS expression in pulmonary vascular ECs exhibits an increase with significantly higher levels of acetylated histone H3 and H4 at the proximal promoter of the eNOS gene (Xu et al., 2010). Postberg et al. (2015) also reported the increased acetylation of histone H3K9 and H2A.Z and eNOS expression after hypoxia. Chao et al. (2018) further reported that early hypoxia exposure (neonatal exposure to hyperoxia) leads to histone modification patterns as H2A.Z and H3K9 hyperacetylation and a substantial increase of the baseline expression of eNOS and STAT3. This histone modification pattern persists and becomes a signature that alters the response of ECs exposed to hypoxia later (Chao et al., 2018). In contrast, eNOS expression is decreased in another PPHN model induced by prenatal ductus arteriosus constriction in lamb. The decreased expression of eNOS is associated with DNA CpG methylation in the gene promoter region accompanied by decreased Sp1 occupancy, H4K12 acetylation, and increased H3K9 trimethylation around Sp1 binding sites (Ke et al., 2018). In human umbilical artery ECs (HUAECs), inhibition of HDAC activity using HDAC inhibitor Trichostatin A (TSA) leads to rapidly increased histone acetylation at the eNOS promoter and increases eNOS mRNA levels (Yang et al., 2012; Postberg et al., 2015). Histone H3K9 acetylation at the eNOS gene transcription start site is directly correlated with eNOS mRNA levels (Postberg et al., 2015). However, HDAC inhibition with TSA decreases eNOS mRNA while paradoxically increasing its promoter activity, suggesting a more complicated mechanism involving HDAC and histone acetylation mediated eNOS mRNA expression (Rossig et al., 2002). The suppression effect of HDAC inhibitor TSA on eNOS expression was further confirmed by Bogaard et al. (2011). Similarly, HDAC inhibitor vorinostat was reported to reduce eNOS expression more than 50% in a mouse model of diabetic nephropathy (Advani et al., 2011), and class I-specific HDAC inhibitor Valproic Acid (VPA) was demonstrated to decrease eNOS expression in HUVECs as well (Murugavel et al., 2018).
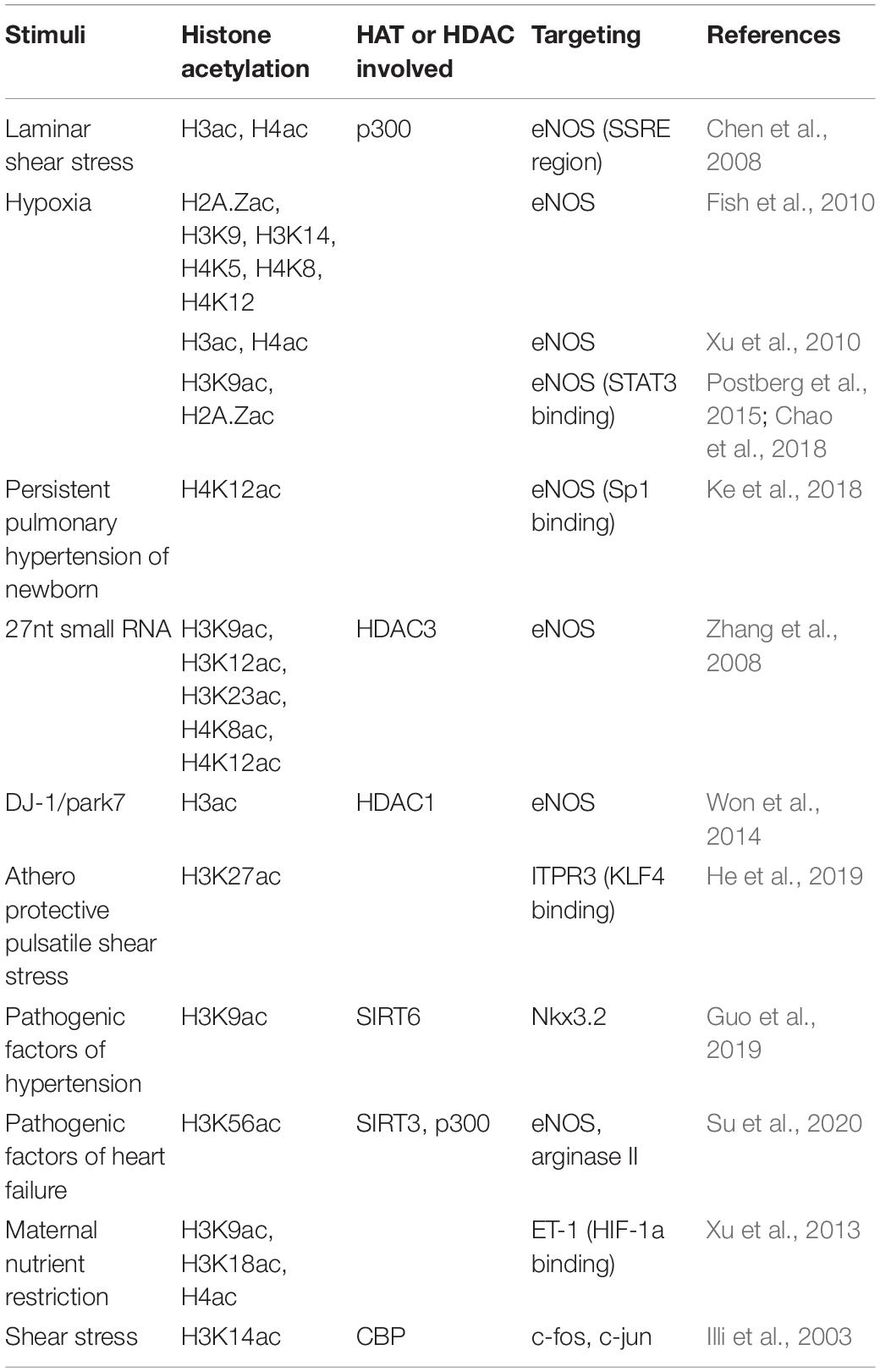
Table 1. Histone acetylation involved in the expression of vascular tone mediators.
The 27nt small RNA, generated from 27nt repeat polymorphism in eNOS intron 4, inhibits eNOS expression. In human aortic ECs (HAECs), the 27 nt small RNA duplex induces hyperacetylation in H3K8, H3K12, H3K23, and H4K9, H4K12 at the 27 nt repeat element and DNA methylation in a region approximately 750nt upstream of the intron 4 repeats of eNOS gene, which is interacted with Non-O (Non-POU Domain Containing Octamer Binding portion) and HDAC3 proteins. The latter deacetylated H3 and H4 at the eNOS core promoter, thereby reduces the expression of eNOS. Treatment with HDAC3 small interfering RNA (siRNA) significantly mitigates the 27 nt small RNA-suppressed expression of eNOS (Zhang et al., 2008). DJ-1/park7, a multifunctional protein, is reported to be implicated in the regulation of vascular contractility and blood pressure by the impairment of NO production through epigenetic inhibition of eNOS expression. HDAC1 recruitment in the eNOS promoter regions is significantly increased in ECs isolated from DJ-1/park7–/– mice compared with control mice. Moreover, the acetylation of H3 histones in the eNOS promoter regions is decreased in DJ-1/park7–/– mice. These DJ-1/park7-dependent epigenetic characteristics in mouse ECs are also appreciated in HUVECs (Won et al., 2014).
Besides by directly binding of acetylated histone proteins on the eNOS promoter region, eNOS expression is also regulated by indirectly histone acetylation-dependent regulation. Atheroprotective pulsatile shear stress induces KLF4-dependent binding of acetylated H3K27 at the ITPR3 promotor region and increases chromatin accessibility, RNA polymerase II recruitment, and ITPR3 transcription. The ITPR3 induction further elevates the expression of eNOS and NO bioavailability (He et al., 2019).
Histone deacetylase SIRT6 in ECs has pleiotropic protective actions, which include promoting endothelium-dependent vasodilatation and vascular NO bioavailability. It induces the expression of GATA-binding protein 5 (GATA5), a novel regulator of blood pressure, through inhibiting NK3 homeobox 2 (Nkx3.2) transcription by deacetylating histone H3K9, thereby regulating GATA5-mediated signaling pathways to prevent endothelial injury (Guo et al., 2019). Histone deacetylase SIRT3 deletion in ECs causes cardiac remodeling and diastolic dysfunction (He et al., 2017), associated closely with vascular tone. Knockout of SIRT3 results in significant increases in p300 expression and histone H3K56 acetylation. P300 inhibitor C646 significantly reduced levels of p300 and H3K56 acetylation and increased eNOS expression (Su et al., 2020). As a product of eNOS, NO also modulates essential endothelial functions and gene expression through histone deacetylation. Mechanistically, NO induces class II HDAC4, and HDAC5 nuclear shuttling via activation of the protein phosphatase 2A (PP2A) (Illi et al., 2008).
Endothelin-1 (ET1), another critical vasoconstrictor secreted by EC, plays a crucial role in vascular tone. ET1-mediated vascular tone increases with age and contributes to the pathogenesis of hypertension (Stauffer et al., 2008). The maternal nutrient restriction increases the acetylation of histone H3K9, H3K18, and H4 and hypoxia-inducible factor-1α (HIF-1α) binding levels at the gene promoter of ET1 in pulmonary vascular ECs (PVEC) of intrauterine growth retardation (IUGR) newborn rats and continues up to 6 weeks after birth. These epigenetic changes result in endothelial dysfunction, and IUGR rats are highly sensitive to hypoxia later in life, causing more significant pulmonary arterial hypertension (PAH) (Xu et al., 2013).
Except for eNOS, shear stress (SS) modulates the expression of several other genes in ECs and regulates vascular tone (Illi et al., 2003; Iring et al., 2019). In HUVECs, SS activates ribosomal S6 kinase-2 and mitogen- and stress-activated kinase-1 protein kinases and promotes the formation of CREB/CBP transcriptional complexes thus increases the histone acetyltransferase activity and acetylates histone H3K14 within the c-fos and c-jun promoters. Consequently, shear stress increases the expression of transcription factors c-fos and c-jun and initiates the transcription of c-fos and c-jun-targeted genes in ECs (Illi et al., 2003).
As an important player in the inflammation process, EC participates in multiple cytokine production and secretion and responds to cytokines stimuli through the mechanism of involving histone acetylation, as summarized in Table 2.
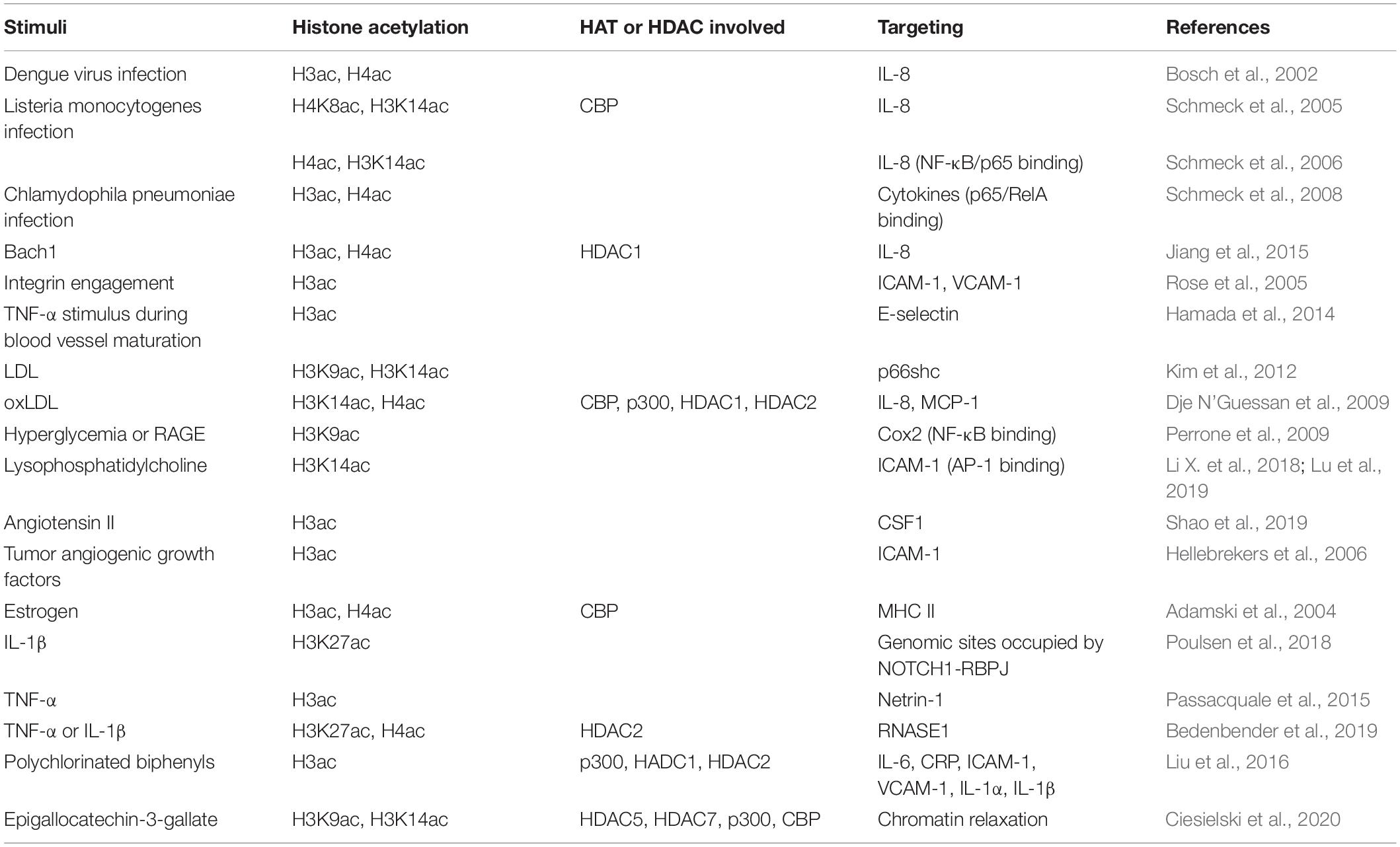
Table 2. Inflammation signals mediated by histone acetylation in ECs.
Histone acetylation mediates cytokine expression and secretion in various infectious diseases. After dengue virus infection, the infected monocytes, ECs, and epithelial cells increase the secretion of interleukin 8 (IL-8) and results in an increased serum level of IL-8 significantly. The induction of IL-8 is associated with the hyperacetylation of histone H3 and H4 at the IL-8 promoter in addition to the transcription activated by NF-kB (Bosch et al., 2002). Intracellular Listeria monocytogenes infection induces time-dependent acetylation of histone H4K8 and H3K14 at the IL-8 promoter in HUVEC, as well as recruitment of the histone acetylase CBP (Schmeck et al., 2005). Inhibition of RhoA, Rac1, and Cdc42 in HUVEC reduces Listeria-promoted recruitment of NF-kB/p65 and RNA polymerase II to the IL-8 promoter, as well as acetylation of histone H4 and H3K14 at the IL-8 gene promoter (Schmeck et al., 2006). Inflammatory activation of the endothelium by Chlamydophila pneumonia infection is involved in the development of chronic vascular lesions. Chlamydophila pneumonia-induced expression of inflammatory genes (IL-6, IL-8, G-CSF, GM-CSF, MIP-1β, and IFN-γ) in ECs is regulated by Rho-GTPase–related acetylation of histone H3 and H4 (Schmeck et al., 2008). Integrin engagement, contributing to antigenotoxic effects of lung endothelium, increases the acetylation of core histone H3 at the promoters of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), similar to the effect of HDAC inhibitor TSA (Rose et al., 2005). However, the effect of HDAC inhibitors on VCAM-1 in ECs is opposite under TNF-α stimulation. Inoue et al. (2006) and Hebbel et al. (2010) reported that HDAC inhibitors TSA and suberoylanilide hydroxamic acid (SAHA) greatly reduced TNF-α-stimulated VCAM-1 expression in ECs. As another case of histone acetylation-mediated cytokine expression in HUVECs, BTB, and CNC homology 1 (Bach1), a basic leucine zipper transcription factor, recruits HDAC1 to the IL-8 promoter and decreases histone H3 and H4 acetylation at the transcription factor 4 (TCF4)-binding site of IL-8 promoter (Jiang et al., 2015). Though histone hyperacetylation is involved in promoting inflammatory cytokine production and secretion, HDAC inhibitors, generally increasing histone acetylation, have a suppressive effect on inflammatory cytokine production induced by lipopolysaccharide (LPS) or TNF-α. A reduction in IL-6 and IL-8 gene expression by HDAC inhibitors, such as butyrate, propionate, and TSA, is observed in HUVECs (Li M. et al., 2018). HDAC inhibitor butyrate also suppresses gene expression and LPS-induced secretion of pro-inflammatory genes CCL2, IL-6, and IL-1β, via enhancing histone H3K9/14 and H2A.Z acetylation and the associated upregulation of oxidative stress-protective genes IRS1, Catalase, SOD2, FOXO3A, and PGC-1α expression in human microvascular ECs (HMEC-1) (Chriett et al., 2019).
Inflammatory response changes during blood vessel maturation and this change has been demonstrated to go through histone H3 acetylation mediated E-selectin expression in response to TNF-α stimulus during blood vessel maturation (Hamada et al., 2014). In many chronic diseases, histone acetylation plays a critical role in endothelial dysfunction associated with inflammation. Hypercholesterolemia induces p66shc-mediated proinflammatory endothelial dysfunction and atheromatous plaque formation by inducing ICAM-1 and reducing thrombomodulin (TM). Mechanistically, low-density lipoprotein (LDL) promotes hypomethylation of two CpG dinucleotides and acetylation of histone H3K9 and H3K14 in the p66shc promoter, thus increasing endothelial p66shc expression (Kim et al., 2012). Similar to LDL, oxidized low-density lipoprotein (oxLDL) also has a proinflammation feature. It induces lectin-like oxidized LDL receptor-1 (LOX-1) and extracellular regulated kinases (ERK1/2)-dependent acetylation of histone H3K14 and H4 binding onto the promoters of IL-8 and monocyte-chemoattractant protein-1 (MCP-1). Consequently, oxLDL increases the expression and secretion of IL-8 and MCP-1 in ECs (Dje N’Guessan et al., 2009). Clinically used statins, which block 3-hydroxy-3-methylglutaryl coenzyme A (HMG Co-A), has a beneficial effect on atherosclerosis depending at least in part on the inhibition of the release of proinflammatory cytokines (Liao and Laufs, 2005). The use of statins alters the histone modification by reducing the recruitment of CBP/p300, NF-kB, and RNA polymerase II but increasing the binding of HDAC1 and HDAC2 at the promoters of IL-8 and MCP-1 genes (Dje N’Guessan et al., 2009). Thioredoxin-interacting protein (TXNIP) plays a causative role in diabetic vascular complications by increasing proinflammatory factor cyclooxygenase (COX)-2 expression in ECs. It induces COX-2 expression by abolishing H3K9 tri-methylation and increasing H3K9 acetylation at the proximal COX-2 promoter bearing the NF-kB-binding site (Perrone et al., 2009). However, HDAC inhibitor TSA decreases LPS-induced COX-2 expression in HUVECs, which is caused by the non-acetylation effect of TSA and may be due to JNK and p38MAPK dephosphorylation induced by TSA activated MKP-1 (Hsu et al., 2011). Lysophosphatidylcholine (LPC) induces mitochondrial reactive oxygen species-dependent site-specific H3K14 acetylation, therefore increases the binding of proinflammatory transcription factor AP-1 in the promoter of ICAM-1 and induces ICAM-1 transcription in HAECs. However, IL-35, an anti-inflammatory cytokine, suppresses LPC-induced monocyte adhesion via ICAM-1 mediated regulation (Li X. et al., 2018). The further RNA-seq and ChIP-seq analysis results demonstrated that LPC induces H3K14 acetylation in the genomic DNA that encodes LPC-induced immunity pathway genes including ICAM-1 (Lu et al., 2019).
Angiotensin II (Ang II), a causative factor for hypertension, has been documented to stimulate endothelium-derived colony-stimulating factor (CSF1), which is a key molecule controlling the production, differentiation, and function of macrophage) transcription (da Cunha et al., 2005). Shao et al. (2019) demonstrated that Ang II upregulates both acetylated H3 and tri-methylated H3K4 and downregulates dimethyl H3K9 surrounding the proximal CSF1 promoter via BRG1 interaction with histone methyltransferase SET1A and histone demethylase JMJD1A. In the tumor, another chronic disease, it has been demonstrated that tumor cells could go immune escape through histone acetylation mediated endothelial dysfunction. As a mediator of leukocyte-EC adhesion, ICAM-1 expression in tumor ECs is epigenetically silenced by promoter histone modifications (histone H3 deacetylation and histone H3K4 demethylation) while restored by HDAC inhibitor TSA (Hellebrekers et al., 2006).
Except for the production of cytokines, the responses of ECs to cytokine or inflammatory stimuli also are associated with histone acetylation. Estrogen has immunomodulatory effects and reduces the class II MHC expression, which has been demonstrated to go through attenuating the acetylation of histone H3 and H4 and the association of CBP within the class II MHC promoter (Adamski et al., 2004). Histone H3K27 acetylation is required for proinflammatory cytokine IL-1β-induced rapid recruitment of NF-κB subunit RELA to genomic sites occupied by NOTCH1-RBPJ (Poulsen et al., 2018). Netrin-1, a neuroimmune guidance cue (Boyer and Gupton, 2018), has chemorepellent action of the endothelium against monocyte chemotaxis. TNF-α-induced NF-κB activation upregulates the nuclear isoform of netrin-1 while simultaneously reducing secreted netrin-1 through histone H3 hyperacetylation. Besides, aspirin, a COX inhibitor that is widely used as an antiplatelet agent but exerts anti-inflammatory properties as well (Desborough and Keeling, 2017), also regulates the TNF-α-induced secretion of netrin-1 via epigenetic modification. Mechanistically, aspirin reduces HDAC/HAT ratio and induces higher expression of acetylated histone H3 (Passacquale et al., 2015). Upon long-term inflammation, high amounts of proinflammatory cytokines affect endothelial function by downregulating RNase1, a circulating extracellular endonuclease regulating vascular homeostasis of extracellular RNA and acting as a vessel- and tissue-protective enzyme (Fischer et al., 2007; Cabrera-Fuentes et al., 2015). In HUVECs, TNF-α, or IL-1β stimulation reduces the expression of RNase1 by inducing hypoacetylation of histone H3K27 and histone H4 via accumulation of HDAC2 to the RNASE1 promoter, while class I HDAC specific inhibitor MS275 abolishes the changes (Bedenbender et al., 2019). Lipoxin A4 (LXA4), a primary stop signal of inflammation, exerts potent bioactions by activating a specific G protein-coupled receptor ALX/FPR2. Activation of p300 restores chromatin accessibility and significantly increases ALX/FPR2 mRNA and protein levels in pulmonary artery ECs (PAECs). Changes in the histone acetylation status enhance ALX/FPR2 signaling in response to LXA4 (Simiele et al., 2016).
Environmental pollutants, such as polychlorinated biphenyls (PCBs), are also reported to induce endothelial inflammation via histone acetylation. PCB 126 exposure increases the expression of vascular inflammatory mediators, including IL-6, C-reactive protein (CRP), ICAM-1, VCAM-1, and IL-1α/β in EA.hy926 human ECs (Liu et al., 2016). Anti-inflammatory polyphenol epigallocatechin-3-gallate (EGCG), the main green tea polyphenol, is demonstrated to abolish nuclear translocation of p65, decrease chromatin binding of p65, and p300, and increase chromatin binding of HDAC1/2, therefore deacetylate histone H3 and deactivate the proinflammatory genes (Liu et al., 2016). Besides, EGCG also increases the chromatin marks of H3K9 and H3K14 acetylation as well as H3K4 and H3K9 trimethylation, presenting a broad epigenetic potential in affecting the expression and activity of epigenome modulators including HDAC5 and 7, p300, CBP, Lysine Demethylase 1A (LSD1) or KMT2A (Ciesielski et al., 2020).
Oxidative stress is a complicated factor affecting EC functions. ECs produce both reactive oxygen species (ROS) and anti-oxidative molecules. The proper balance is required to maintain normal endothelial function. Multiple stimuli affect the balance, and excessive ROS causes endothelial dysfunction. Among these processes, histone acetylation plays an important role, as summarized in Table 3. Inactivation of the p66Shc adaptor protein confers EC resistance to oxidative stress. However, high-glucose induces overexpression of p66Shc. It has been reported that p66Shc-deficient mice are protected against age-related and hyperglycemia-induced endothelial dysfunction. SIRT1 inhibits high glucose-induced upregulation of p66Shc via decreasing the acetylation of histone H3 binding onto the p66Shc promoter (Zhou et al., 2011). The p66Shc gene overexpression induced by high-glucose is epigenetically regulated by promoter CpG hypomethylation (Paneni et al., 2012) and histone H3K9 acetylation along with enhanced transcriptional factor p53 binding (Mishra et al., 2019). Besides mediating the endothelial response to oxidative stress, p66Shc is also emerging as a key molecule responsible for ROS generation and vascular damage. Methyltransferase SUV39H1, demethylase JMJD2C, and acetyltransferase SRC-1 are found to be associated with reduced di- and tri-methylation as well as acetylation of H3K9 on the p66Shc promoter in visceral fat arteries (VFA), which results in the elevated expression of p66Shc and ROS-related endothelial dysfunction in VFA (Costantino et al., 2019).
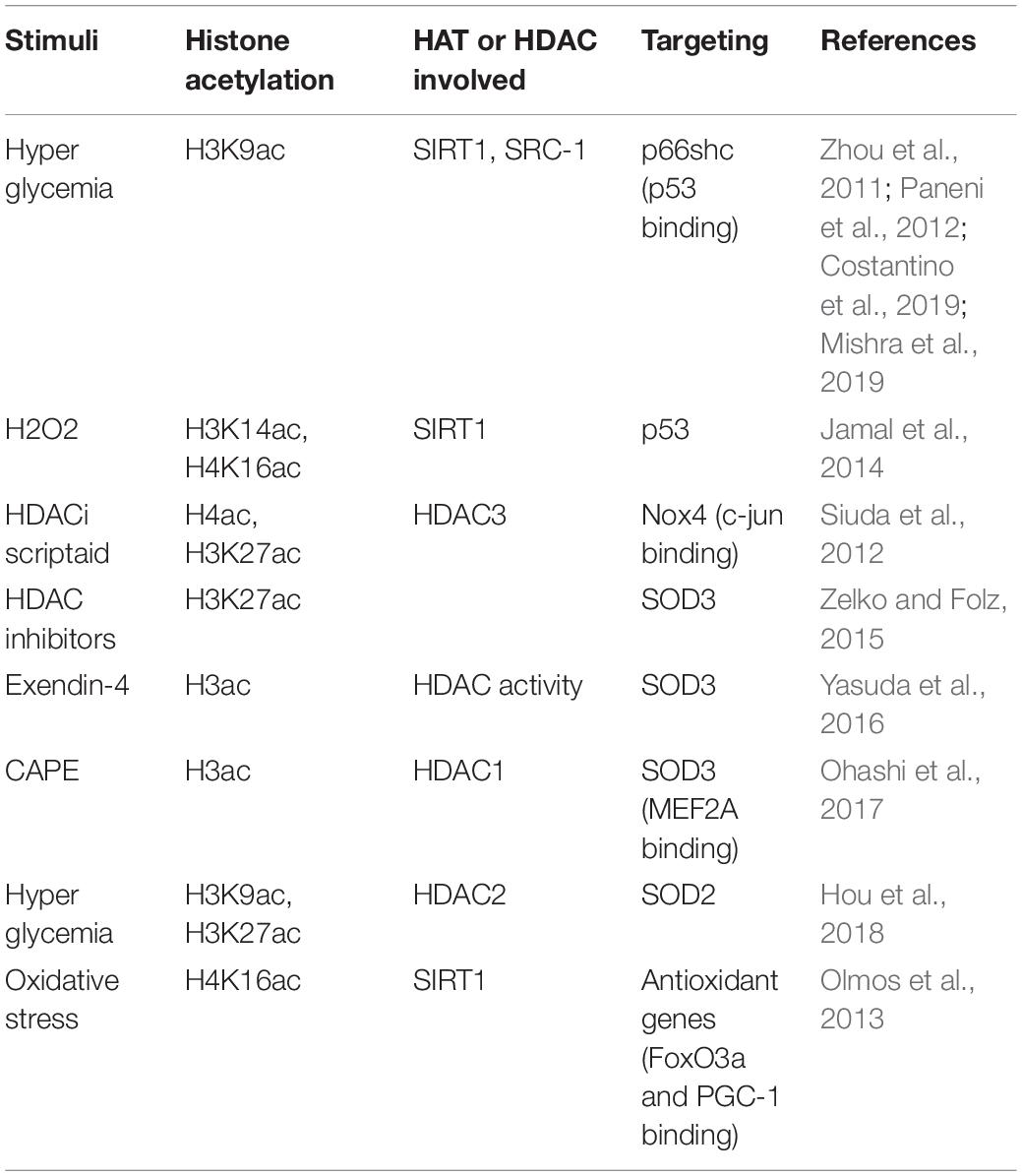
Table 3. Histone acetylation involved in endothelial mediated oxidative stress.
Hydrogen peroxide (H2O2), ROS inducer, upregulates p53 and acetylation of H3K14 and H4K16 due to the decreased activity of SIRT1. Paeonol, a phenolic compound isolated mainly from Moutan cortex, the root bark of the Chinese Peony tree, significantly increases the deacetylase activity of SIRT1 in HUVECs. Pretreatment with paeonol substantially decreases the H2O2–upregulated levels of acetylated H3K14 and H4K16 (Jamal et al., 2014). NADPH oxidase (NOX) promotes ROS production in ECs. In response to hypoxia-reoxygenation (HR), chromatin remodeling protein BRG1 is recruited to the NOX promoter region and induces acetylation of histones H3 and H4 and methylation of histone H3K9 surrounding the NOX promoter (Li Z. et al., 2018). In human EA.hy926 ECs, siRNA-mediated knockdown of HDAC3 leads to a significant downregulation of NOX family member Nox4 expression. Also, HDAC inhibitor Scriptaid leads to the enhanced acetylation of histone H4 and H3K27, and decreases Nox4 transcription by preventing the binding of transcription factor c-Jun and RNA polymerase IIa to the Nox4 promoter due to hyperacetylation-mediated steric inhibition (Siuda et al., 2012).
Extracellular superoxide dismutase (EC-SOD, SOD3) is the major antioxidant enzyme present in the vascular wall. The expression of antioxidant genes in PAECs is regulated by epigenetic mechanisms (Zelko et al., 2011). HDAC inhibitor Scriptaid-mediated augmentation of SOD3 expression is associated with increased histone H3K27 acetylation and H3K4 trimethylation at the promoter of the SOD3 gene (Zelko and Folz, 2015). Exendin-4, a glucagon-like peptide-1 receptor agonist, significantly induces the expression of SOD3 in human retinal microvascular ECs (HRECs) by decreasing HDAC activity and enhancing the status of histone H3 acetylation at the SOD3 proximal promoter and (Yasuda et al., 2016). Moreover, Caffeic acid phenethyl ester (CAPE) increases the transcription of SOD3 through HDAC1-regulated histone acetylation in HRECs (Ohashi et al., 2017). Except for SOD3, SOD2 (Superoxide Dismutase 2, also called MnSOD) is also regulated by histone acetylation. Diabetes-induced HDAC2 upregulation diminishes the acetylation of histone H3K9 and H3K27, therefore, reduces SOD2 expression. The reduced SOD2 expression promotes oxidative stress and contributes to diabetes-related endothelial dysfunction (Hou et al., 2018). SIRT1 activates the expression of antioxidant genes in ECs by deacetylating and increasing the transcription factors FOXO3a and PGC-1α, however reducing H4K16 acetylation and increasing elongating Pol II recruitment occurring at these genes (Olmos et al., 2013).
Angiogenesis is a process of new blood vessel formation, which is the well-characterized function of ECs. As summarized in Table 4, histone acetylation is found to be crucial in regulating angiogenesis.
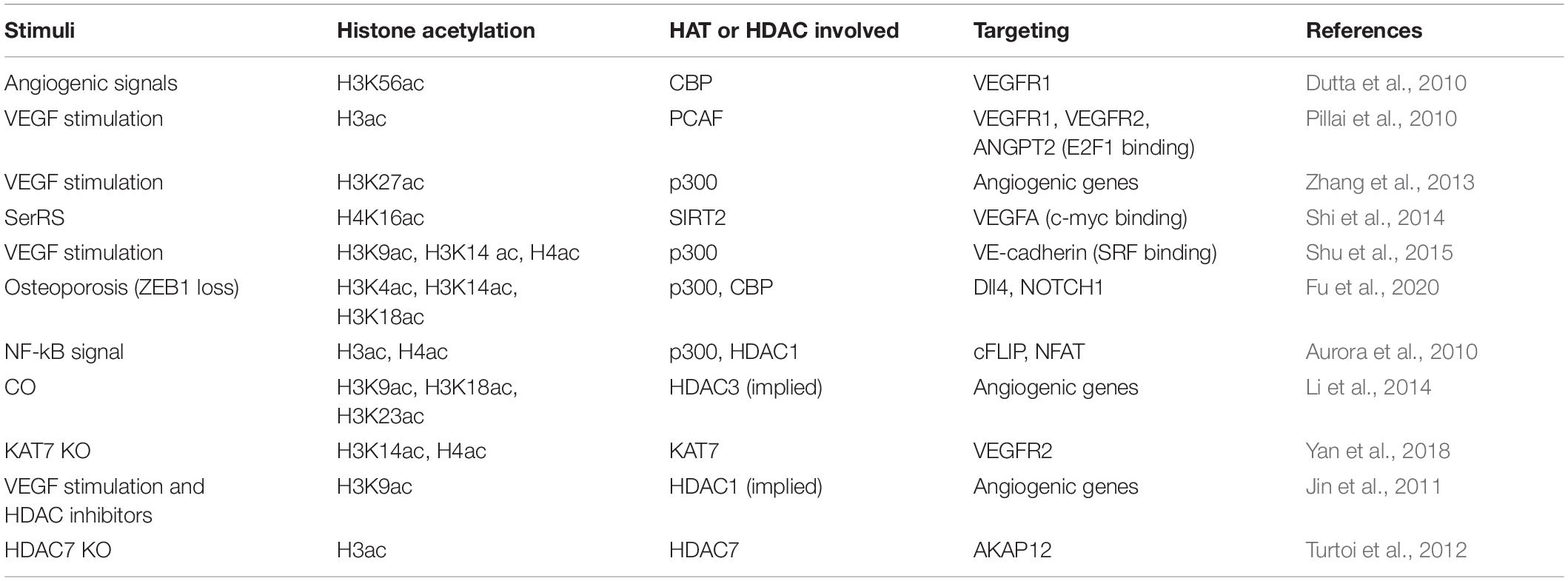
Table 4. Histone acetylation associated with angiogenesis.
In response to angiogenic signals, histone cell cycle regulation defective homolog A (HIRA) is induced in ECs and mediates the incorporation of CBP acetylated H3K56 at the chromatin domain of VEGFR1, which then induces transcription of several angiogenic genes in ECs (Dutta et al., 2010). Upon VEGF stimulation, angiogenic genes VEGFR1, VEGFR2, and angiopoietin 2 (ANGPT2) in HUVECs are induced via increased promotor binding of transcription factor E2F1, which is correlated with the recruitment of p300 and PCAF and acetylation of histones and E2F1 (Pillai et al., 2010). Zhang et al. (2013) also reported that the greatest change of H3K27 acetylation (H3K27ac) is associated tightly with p300 in VEGF-stimulated HUVECs. Dynamic H3K27ac deposition and associated changes in chromatin conformation require p300 activity instead of altered nucleosome occupancy or changes in DNase I hypersensitivity (Zhang et al., 2013). Moreover, human metallothionein 1G (hMT1G) is upregulated upon VEGF stimulation in HAECs. VEGF stimulation leads to dissociation of HDAC1 from the promoter, acetylation of histones, and increased binding of transcription factor E2F1. The upregulation of hMT1G contributes to the proangiogenic functions of VEGF (Joshi et al., 2005). Seryl-tRNA synthetase (SerRS) plays an essential role in regulating vascular development by acetylated histone-mediated regulation of VEGFA expression. In particular, SerRS blocks the binding of c-Myc, the major transcription factor promoting VEGFA, to VEGFA promoter and recruits SIRT2 to erase c-Myc-promoted acetylation of H4K16 so as to intervene c-Myc-dependent VEGFA expression (Shi et al., 2014).
Vascular endothelial cadherin (VE-cadherin) is the primary determinant of EC junction integrity during vascular development and angiogenesis. During angiogenesis and neovascularization processes, VEGF induces the binding of myocardin-related transcription factor-A (MRTF-A) to the Serum Response Factor (SRF)-binding site within the VE-cadherin promoter. MRTF-A and p300 synergistically augment VE-cadherin expression by enhancing acetylation of H3K9, H3K14, and H4 at the SRF-binding site within the VE-cadherin promoter (Shu et al., 2015). Osteoporosis-associated downregulation of zinc-finger transcription factor ZEB1 in ECs reduces CBP/p300 mediated acetylation of H3K4, H3K14, and H3K18 within the promoters of Dll4 and Notch1, thereby epigenetically suppressing Notch signaling, a critical pathway that controls bone angiogenesis and osteogenesis (Fu et al., 2020).
NF-kB signal is induced by antiangiogenic molecules and has a dual effect on downstream transcription through histone acetylation regulation. On the one hand, NF-kB acts as an activator of proapoptotic Fas ligand via recruiting p300 and acetylated histones H3 and H4 at its promotor. On the other hand, NF-kB acts as a repressor of antiapoptotic cFLIP [cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein] via reducing the HDAC1 and p300 coordinated histone acetylation and recruitment of transcription factor NFAT critically required for cFLIP transcription (Aurora et al., 2010). Administration of low, safe doses of exogenous carbon monoxide (CO) was demonstrated to enhance EC migration and angiogenesis in the acetylated H3K9, H3K18, and H3K23 dependent manner, nuclear receptor Rev-erbα, a haem-containing transcription factor, responds to CO stimulation, recruits the HDAC/nuclear Receptor Corepressor (N-CoR) complex, and thus suppresses the transcription of genes responsible for EC migration and angiogenesis (Li et al., 2014).
Histone acetyltransferase and deacetylase are potentially involved in regulating angiogenesis through the alteration of histone acetylation. KAT7 participates in regulating VEGFR-2 transcription by promoting the acetylation of H3K14 and H4 and RNA polymerase II binding. Depletion of KAT7 reduces VEGFR2 expression and disrupts angiogenic potential in HUVECs (Yan et al., 2018). HDAC inhibitor VPA results in hyperacetylated H3K9 and greatly enhances the VEGF-promoted spheroid sprouting of HUVECs (Jin et al., 2011). HDAC inhibitor LBH589 induces the acetylation of histone H3 and non-histone protein α-tubulin and induces G2-M cell cycle arrest, inhibits the VEGF-induced expression of ANGPT2, survivin, HIF-1α, and CXCR4 possibly due to histone acetylation modulated upregulation of tumor suppressor genes, such as p53 and VHL (Von Hippel–Lindau) (Qian et al., 2006). Novel HDAC inhibitor N-hydroxy-7-(2-naphthylthio) heptanomide (HNHA) showed a similar effect on histone H3 and non-histone protein α-tubulin to LBH589 (Kim et al., 2009). HDAC inhibitor TSA significantly reduces sVEGFR1 basal expression, suggesting that histone acetylation is involved in activating sVEGFR1 (Choi et al., 2018). The depletion of HDAC7 in HUVEC results in the overexpression of A-kinase anchor protein 12 (AKAP12), which is a tumor/angiogenesis suppressor gene responsible for the inhibition of migration and tube formation. Mechanistically, H3 histone proteins associated with AKAP12 promoter are highly acetylated following the removal of HDAC7, leading to an increase in its mRNA and protein levels (Turtoi et al., 2012). Intriguingly, regulation of gene locus-specific histone acetylation status by the interaction between a lipid signaling mediator LPA and HDAC7 may downregulate the transcription of angiogenic regulator CD36 via protein kinase D pathway, thereby promoting angiogenesis (Ren et al., 2011, 2016).
The endothelial barrier in all organ beds allows the free exchange of water but is restrictive to varying degrees to the transport of solutes (Malik et al., 1989). Functional changes in EC, to a large extent, influence the barrier function. As listed in Table 5, many studies have demonstrated the histone acetylation changes affect the barrier function.
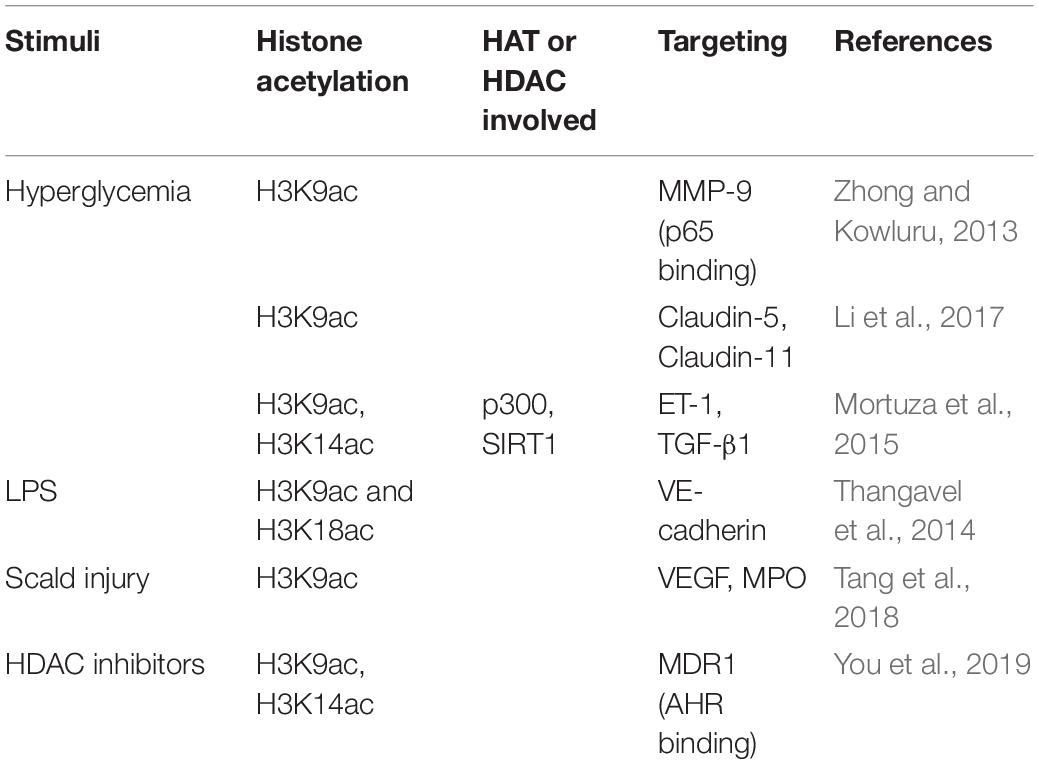
Table 5. EC barrier function mediated by histone acetylation.
High glucose impairs the endothelial barrier through multiple histone acetylation mediated signals. Zhong and Kowluru reported that diabetes activates retinal Matrix metalloproteinases (MMP)-9 via LSD1 mediated decrease of histone H3K9 di-methylation. The decreased H3K9 methylation frees up that lysine 9 for acetylation and facilitates the recruitment of p65 to MMP-9 promoter, thus resulting in MMP-9 activation in retinal ECs (Zhong and Kowluru, 2013). Li et al. (2017) demonstrated high glucose suppresses claudins-5 and -11 gene expression in human cardiac microvascular ECs, possibly due to inhibition of acetylated H3K9 in claudin-5 and claudin-11 gene promoters. Mortuza et al. (2015) revealed that high glucose induces p300 expression, and reduces SIRT1 levels, thus increases the acetylation of H3K9 and H3K14, which is attributed to the upregulation of ET1 and TGFβ1. These changes contribute to high glucose-induced endothelial hyperpermeability (Mortuza et al., 2015). In pulmonary ECs, combinatorial treatment with DNA methyltransferase (DNMT) inhibitor Aza and HDAC inhibitor TSA mitigate the LPS increased endothelial permeability due to the enhanced acetylation of H3K9 and H3K18 on the VE-cadherin promoter (Thangavel et al., 2014). HDAC inhibitor VPA treatment significantly alleviates the microvascular permeability and water content of the lung, lowers VEGF and MPO levels, and promotes the acetylation of H3K9 following scald injury (Tang et al., 2018). In brain ECs, multidrug resistance protein 1 (MDR1, ABCB1, P-glycoprotein) is a critical efflux transporter that extrudes chemicals from the blood–brain barrier and limits neuronal exposure to xenobiotics. HDAC inhibitor SAHA upregulates the expression and activity of the MDR1 transporter in immortalized human brain capillary EC hCMEC/D3 by increasing H3K9/K14 acetylation and facilitating aryl hydrocarbon receptor (AHR) binding at the MDR1 promoter (You et al., 2019).
Several studies revealed the involvement of histone acetylation modification in EC mediated coagulation and thrombosis (Table 6). Tissue-type plasminogen activator (t-PA) plays a key role in the onset of the fibrinolytic process by converting the zymogen plasminogen into the active enzyme plasmin. Stimulated release of t-PA is pivotal for an intravascular fibrinolytic response and protects the circulation from occluding thrombosis. Arts et al. (1995) reported for the first time that t-PA expression could be regulated by histone acetylation. HDAC inhibitors butyrate, TSA, MS275, and VPA were demonstrated to induce a dose-dependent increase of t-PA expression in ECs, which involves histone H3 and H4 acetylation (Arts et al., 1995; Dunoyer-Geindre and Kruithof, 2011; Larsson et al., 2012). VPA induced t-PA expression is associated with increased acetylation of H3K9, H3K18, H3K23, H3K27, H4K8, and H4K16 at the t-PA promoter (Larsson et al., 2012) and dependent on the proximal GC boxes in the t-PA promoter and may involve interactions with transcription factors Sp2, Sp4, and KLF5 (Ulfhammer et al., 2016). The effect of VPA on t-PA expression was then confirmed in vivo (Svennerholm et al., 2014; Larsson et al., 2016). Plasminogen activator inhibitor-1 (PAI-1), the most important serine protease inhibitor of t-PA (Van De Craen et al., 2012), has shown beneficial effects on age-related vascular diseases. PAI-1 is induced in senescent HUVECs and aortas of eld mice. SIRT1 is able to reverse the change by binding to the PAI-1 promoter, resulting in a decreased acetylation of H4K16 at the PAI-1 promoter region (Wan et al., 2014). Clinical study has demonstrated that HDAC inhibitor VPA reduces in exhaustion during repeated and prolonged cumulative stimulated t-PA release and decreases basal PAI-1 levels (Svennerholm et al., 2015), which suggested potentially positive effects of HDAC inhibitors on the expression of t-PA and PAI-1. Von Willebrand factor (vWF), playing an essential role in regulating the balance between blood clotting and bleeding, also is regulated by histone acetylation. Peng et al. (2007) reported that irradiation induces thrombus formation via vWF elevation. The irradiation-caused changes in the association of NF-Y with HDAC1 and PCAF lead to the increased acetylated histone H4 and PCAF recruitment to the vWF promoter, which results in subsequently increased vWF transcription in HUVECs (Peng et al., 2007). Mojiri et al. (2019) reported that, in response to hypoxia, Nuclear Factor IB (NFIB) repressor disassociates from the vWF promoter due to the increased acetylation of the promoter-associated histone H4, and thus increases vWF expression. Tissue factor (TF), another protein involved in coagulation, is demonstrated to lose inducibility during senescence, which occurs following chromatin remodeling at the TF promoter resulting from hypoacetylation of histone H3K9 (Kurz et al., 2014). However, HDAC inhibitors TSA and SAHA were reported to greatly attenuate TF expression induced by TNF-α despite of its effect of promoting histone acetylation (Wang et al., 2007; Hebbel et al., 2010).
Table 6. Histone acetylation associated with thrombosis and coagulation.
Except for vascular tone, inflammation, oxidative stress, angiogenesis, barrier function, and thrombosis and coagulation, histone acetylation is also involved in other EC-associated pathological processes, including endothelial mesenchymal transition (EndMT), senescence, and metabolism. Fu et al. (2009) reported that Notch and TGFβ synergistically upregulate a subset of genes by recruiting Smad3 to both Smad and DNA-binding protein CSL binding sites and cooperatively inducing histone H4 acetylation. Zhang et al. (2016) demonstrated that aging-related transcriptional regulator p49/STRAP alters histone acetylation status and impacts mitochondrial dynamics, thereby reducing mitochondrial function and cardiac performance during mammalian senescence. Zhong et al. found the so-called metabolic memory phenomenon in diabetic retinopathy. This phenomenon is mediated by HDAC activation due to the increased expression of HDAC1, 2, 8 genes and compromised HAT activity, as evidenced by the decreased histone H3 acetylation in the retina and its capillary cells (Zhong and Kowluru, 2010). Pirola et al. (2011) uncovered hyperglycemia-mediated induction of genes and pathways associated with endothelial dysfunction occur through modulation of acetylated H3K9/K14, which is inversely correlated with methyl-CpG content (Pirola et al., 2011). Chen et al. (2010) reported that p300 critically regulates glucose-induced gene expression in ECs via binding to gene promoters and augmenting histone acetylation. In HUVECs, high glucose treatment increases p300 production accompanied by increased binding of p300 to ET-1 and fibronectin promoters and increased transcription of vasoactive factors and extracellular matrix (ECM) proteins (Chen et al., 2010). Tanegashima et al. (2017) demonstrated that fasting-induced production of the ketone body β-hydroxybutyrate (β-OHB) enhances expression of the glucose transporter gene Slc2a1 (Glut1) via HDAC2 mediated H3K9 acetylation at the critical cis-regulatory region of the Slc2a1 gene in brain microvascular ECs.
Histone acetylation has been found to be involved in the pathogenesis of EC dysfunction in the last two decades. The acetylation of histone at different lysine residues alters under various stimuli and affects gene expression in ECs (Figure 4), therefore mediating endothelial functions. In regards to acetylated histone-mediated gene expression, HATs and HDACs are indispensable. The fact that histone acetylation can be reversed makes it an ideal target for drug treatment. With the development of HDAC inhibitors, which could inhibit HDACs, therefore, promoting histone acetylation, modulation of histone acetylation becomes achievable. In the oncology field, HDAC inhibitors have already been tested in clinical trials for treating solid and hematological malignancies (O’Connor et al., 2015; Sekeres et al., 2017; Tambo et al., 2017; Gray et al., 2019; Rodriguez et al., 2020).
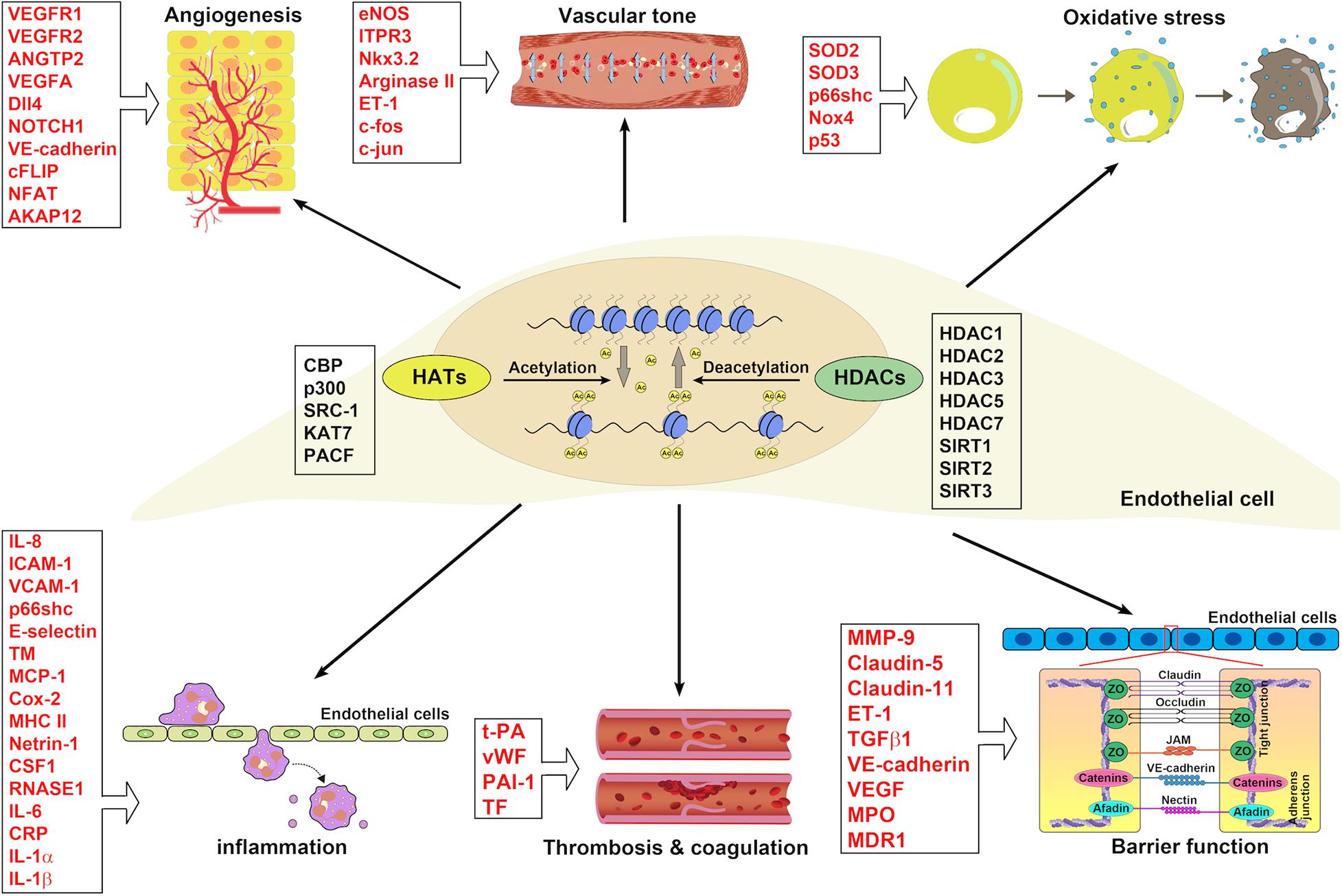
Figure 4. The implications of HATs and HDACs in ECs associated with blood vessel functions. HATs and HDACs have been demonstrated to modulate the histone acetylation of ECs involving blood vessel functions: angiogenesis, vascular tone, oxidative stress, inflammation, thrombosis and coagulation, and barrier function.
The application of HDAC inhibitors in EC dysfunction-related disease is promising but still in its infantile stage. Many challenges are awaiting to be conquered. The side effects of HDAC inhibitors on the acetylation of non-histone proteins and other non-acetylation effects may limit their application. For example, the non-selective HDAC inhibitor vorinostat, inhibiting the activities of class I and class II HDAC enzymes, potentially increases the lysine acetylation status of more than 1,700 non-histone proteins (Choudhary et al., 2009). HDAC inhibitor LBH589 and HNHA have an acetylation effect on non-histone protein α-tubulin (Qian et al., 2006; Kim et al., 2009). HDAC inhibitor TSA could acetylate non-histone protein p53 (Zeng et al., 2003) and has ubiquitination effect on histone acetyltransferase p300 (Hakami et al., 2016). HDAC inhibitor VPA could induce ERK1/2 phosphorylation independent of the HDAC inhibition effect (Michaelis et al., 2006). In ECs, except for the histone deacetylation effect, HDACs were also reported to deacetylate non-histone proteins. HDAC1 was demonstrated to deacetylate p53 and transactivate the p21 under laminar flow (Zeng et al., 2003). SIRT1 activates the expression of antioxidant genes in ECs by deacetylating and increasing the activity of the transcription factors FOXO3a and PGC-1α (Olmos et al., 2013). Furthermore, many HDACs form corepressor complexes to silence gene activity, which again amplifies the potential non-specific effects that HDAC inhibitors can have within the cell (Krishna et al., 2015). Therefore, there is a critical need for further investigation to elucidate the roles of HDACs and HATs in regulating the acetylation of non-histone proteins in ECs.
Not all the HATs and HDACs, which are potentially involved in endothelial dysfunction-related histone acetylation, have been elucidated. The systemic study using individual HATs and HDACs endothelial-specific knockout mice should be essential for filling up our current knowledge gaps. Same HAT or HDAC may regulate different genes that have beneficial or detrimental EC functions in various physiological and pathological settings. HDAC1 has an anti-inflammatory effect via histone acetylation mediated IL-8 expression (Jiang et al., 2015) and pro-oxidative stress through SOD3 as well (Ohashi et al., 2017). Though HDAC2 could decrease pro-inflammatory gene expression (Liu et al., 2016), it also inhibits anti-oxidative stress gene SOD3 expression (Hou et al., 2018). It could be another challenge in how to target HDAC mediated histone acetylation for modulating endothelial functions precisely. In addition, histone acetylation and methylation always work together. The complexity of histone posttranslational modification in ECs needs our endeavor to be revealed in the future.
QM provided the outline of the manuscript. ZF, XW, and XS searched the literature for the article and wrote the manuscript. WH coordinated and revised the content and prepared the figures. ZF prepared the tables. QM supervised the work and completed the final editing. All authors discussed its content and edited the submitted manuscript.
This work was supported in part by start-up funds from NYU Langone Hospital-Long Island, NIH R01HL141733 to QM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adamski, J., Ma, Z., Nozell, S., and Benveniste, E. N. (2004). 17beta-Estradiol inhibits class II major histocompatibility complex (MHC) expression: influence on histone modifications and cbp recruitment to the class II MHC promoter. Mol. Endocrinol. 18, 1963–1974. doi: 10.1210/me.2004-0098
Advani, A., Huang, Q., Thai, K., Advani, S. L., White, K. E., Kelly, D. J., et al. (2011). Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am. J. Pathol. 178, 2205–2214. doi: 10.1016/j.ajpath.2011.01.044
Alderton, W. K., Cooper, C. E., and Knowles, R. G. (2001). Nitric oxide synthases: structure, function and inhibition. Biochem. J. 357(Pt 3), 593–615. doi: 10.1042/0264-6021:3570593
Allfrey, V. G., Faulkner, R., and Mirsky, A. E. (1964). Acetylation and methylation of histones and their possible role in the regulation of Rna synthesis. Proc. Natl. Acad. Sci. U.S.A. 51, 786–794. doi: 10.1073/pnas.51.5.786
Arner, E., Daub, C. O., Vitting-Seerup, K., Andersson, R., Lilje, B., Drablos, F., et al. (2015). Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014. doi: 10.1126/science.1259418
Arts, J., Lansink, M., Grimbergen, J., Toet, K. H., and Kooistra, T. (1995). Stimulation of tissue-type plasminogen activator gene expression by sodium butyrate and trichostatin A in human endothelial cells involves histone acetylation. Biochem. J. 310(Pt 1), 171–176. doi: 10.1042/bj3100171
Aurora, A. B., Biyashev, D., Mirochnik, Y., Zaichuk, T. A., Sanchez-Martinez, C., Renault, M. A., et al. (2010). NF-kappaB balances vascular regression and angiogenesis via chromatin remodeling and NFAT displacement. Blood 116, 475–484. doi: 10.1182/blood-2009-07-232132
Balligand, J. L., and Cannon, P. J. (1997). Nitric oxide synthases and cardiac muscle. Autocrine and paracrine influences. Arterioscler. Thromb. Vasc. Biol. 17, 1846–1858. doi: 10.1161/01.atv.17.10.1846
Bedenbender, K., Scheller, N., Fischer, S., Leiting, S., Preissner, K. T., Schmeck, B. T., et al. (2019). Inflammation-mediated deacetylation of the ribonuclease 1 promoter via histone deacetylase 2 in endothelial cells. FASEB J. 33, 9017–9029. doi: 10.1096/fj.201900451R
Bogaard, H. J., Mizuno, S., Hussaini, A. A., Toldo, S., Abbate, A., Kraskauskas, D., et al. (2011). Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am. J. Respir. Crit. Care Med. 183, 1402–1410. doi: 10.1164/rccm.201007-1106OC
Boo, Y. C., Hwang, J., Sykes, M., Michell, B. J., Kemp, B. E., Lum, H., et al. (2002). Shear stress stimulates phosphorylation of eNOS at Ser(635) by a protein kinase A-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 283, H1819–H1828. doi: 10.1152/ajpheart.00214.2002
Bordoli, L., Husser, S., Luthi, U., Netsch, M., Osmani, H., and Eckner, R. (2001). Functional analysis of the p300 acetyltransferase domain: the PHD finger of p300 but not of CBP is dispensable for enzymatic activity. Nucleic Acids Res. 29, 4462–4471. doi: 10.1093/nar/29.21.4462
Bosch, I., Xhaja, K., Estevez, L., Raines, G., Melichar, H., Warke, R. V., et al. (2002). Increased production of interleukin-8 in primary human monocytes and in human epithelial and endothelial cell lines after dengue virus challenge. J. Virol. 76, 5588–5597. doi: 10.1128/jvi.76.11.5588-5597.2002
Bosch-Presegue, L., and Vaquero, A. (2015). Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 282, 1745–1767. doi: 10.1111/febs.13053
Boyer, N. P., and Gupton, S. L. (2018). Revisiting Netrin-1: one who guides (Axons). Front. Cell Neurosci. 12:221. doi: 10.3389/fncel.2018.00221
Breen, M. E., and Mapp, A. K. (2018). Modulating the masters: chemical tools to dissect CBP and p300 function. Curr. Opin. Chem. Biol. 45, 195–203. doi: 10.1016/j.cbpa.2018.06.005
Cabrera-Fuentes, H. A., Niemann, B., Grieshaber, P., Wollbrueck, M., Gehron, J., Preissner, K. T., et al. (2015). RNase1 as a potential mediator of remote ischaemic preconditioning for cardioprotectiondagger. Eur. J. Cardiothorac. Surg. 48, 732–737. doi: 10.1093/ejcts/ezu519
Carafa, V., Rotili, D., Forgione, M., Cuomo, F., Serretiello, E., Hailu, G. S., et al. (2016). Sirtuin functions and modulation: from chemistry to the clinic. Clin. Epigenetics 8:61. doi: 10.1186/s13148-016-0224-3
Chao, C. M., van den Bruck, R., Lork, S., Merkle, J., Krampen, L., Weil, P. P., et al. (2018). Neonatal exposure to hyperoxia leads to persistent disturbances in pulmonary histone signatures associated with NOS3 and STAT3 in a mouse model. Clin. Epigenetics 10:37. doi: 10.1186/s13148-018-0469-0
Chen, S., Feng, B., George, B., Chakrabarti, R., Chen, M., and Chakrabarti, S. (2010). Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 298, E127–E137. doi: 10.1152/ajpendo.00432.2009
Chen, W., Bacanamwo, M., and Harrison, D. G. (2008). Activation of p300 histone acetyltransferase activity is an early endothelial response to laminar shear stress and is essential for stimulation of endothelial nitric-oxide synthase mRNA transcription. J. Biol. Chem. 283, 16293–16298. doi: 10.1074/jbc.M801803200
Choi, S., Uehara, H., Wu, Y., Das, S., Zhang, X., Archer, B., et al. (2018). RNA activating-double stranded RNA targeting flt-1 promoter inhibits endothelial cell proliferation through soluble FLT-1 upregulation. PLoS One 13:e0193590. doi: 10.1371/journal.pone.0193590
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. doi: 10.1126/science.1175371
Choudhary, C., Weinert, B. T., Nishida, Y., Verdin, E., and Mann, M. (2014). The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550. doi: 10.1038/nrm3841
Chriett, S., Dabek, A., Wojtala, M., Vidal, H., Balcerczyk, A., and Pirola, L. (2019). Prominent action of butyrate over beta-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 9:742. doi: 10.1038/s41598-018-36941-9
Ciesielski, O., Biesiekierska, M., and Balcerczyk, A. (2020). Epigallocatechin-3-gallate (EGCG) alters histone acetylation and methylation and impacts chromatin architecture profile in human endothelial cells. Molecules 25:2326. doi: 10.3390/molecules25102326
Costantino, S., Paneni, F., Virdis, A., Hussain, S., Mohammed, S. A., Capretti, G., et al. (2019). Interplay among H3K9-editing enzymes SUV39H1, JMJD2C and SRC-1 drives p66Shc transcription and vascular oxidative stress in obesity. Eur. Heart J. 40, 383–391. doi: 10.1093/eurheartj/ehx615
da Cunha, V., Tham, D. M., Martin-McNulty, B., Deng, G., Ho, J. J., Wilson, D. W., et al. (2005). Enalapril attenuates angiotensin II-induced atherosclerosis and vascular inflammation. Atherosclerosis 178, 9–17. doi: 10.1016/j.atherosclerosis.2004.08.023
Desborough, M. J. R., and Keeling, D. M. (2017). The aspirin story - from willow to wonder drug. Br. J. Haematol. 177, 674–683. doi: 10.1111/bjh.14520
Dimmeler, S., Fleming, I., Fisslthaler, B., Hermann, C., Busse, R., and Zeiher, A. M. (1999). Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605. doi: 10.1038/21224
Dje N’Guessan, P., Riediger, F., Vardarova, K., Scharf, S., Eitel, J., et al. (2009). Statins control oxidized LDL-mediated histone modifications and gene expression in cultured human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 29, 380–386. doi: 10.1161/ATVBAHA.108.178319
D’Onofrio, N., Servillo, L., and Balestrieri, M. L. (2018). SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid Redox Signal. 28, 711–732. doi: 10.1089/ars.2017.7178
Downey, M. (2020). Non-histone protein acetylation by the evolutionarily conserved GCN5 and PCAF acetyltransferases. Biochim. Biophys. Acta Gene Regul. Mech. 1864:194608. doi: 10.1016/j.bbagrm.2020.194608
Dunoyer-Geindre, S., and Kruithof, E. K. (2011). Epigenetic control of tissue-type plasminogen activator synthesis in human endothelial cells. Cardiovasc. Res. 90, 457–463. doi: 10.1093/cvr/cvr028
Dutta, D., Ray, S., Home, P., Saha, B., Wang, S., Sheibani, N., et al. (2010). Regulation of angiogenesis by histone chaperone HIRA-mediated incorporation of lysine 56-acetylated histone H3.3 at chromatin domains of endothelial genes. J. Biol. Chem. 285, 41567–41577. doi: 10.1074/jbc.M110.190025
Feller, C., Prestel, M., Hartmann, H., Straub, T., Soding, J., and Becker, P. B. (2012). The MOF-containing NSL complex associates globally with housekeeping genes, but activates only a defined subset. Nucleic Acids Res. 40, 1509–1522. doi: 10.1093/nar/gkr869
Fischer, D. D., Cai, R., Bhatia, U., Asselbergs, F. A., Song, C., Terry, R., et al. (2002). Isolation and characterization of a novel class II histone deacetylase. HDAC10. J. Biol. Chem. 277, 6656–6666. doi: 10.1074/jbc.M108055200
Fischer, S., Gerriets, T., Wessels, C., Walberer, M., Kostin, S., Stolz, E., et al. (2007). Extracellular RNA mediates endothelial-cell permeability via vascular endothelial growth factor. Blood 110, 2457–2465. doi: 10.1182/blood-2006-08-040691
Fish, J. E., Matouk, C. C., Rachlis, A., Lin, S., Tai, S. C., D’Abreo, C., et al. (2005). The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J. Biol. Chem. 280, 24824–24838. doi: 10.1074/jbc.M502115200
Fish, J. E., Yan, M. S., Matouk, C. C., St Bernard, R., Ho, J. J., Gavryushova, A., et al. (2010). Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J. Biol. Chem. 285, 810–826. doi: 10.1074/jbc.M109.067868
Fleming, I., and Busse, R. (1999). Signal transduction of eNOS activation. Cardiovasc. Res. 43, 532–541. doi: 10.1016/s0008-6363(99)00094-2
Fu, R., Lv, W. C., Xu, Y., Gong, M. Y., Chen, X. J., Jiang, N., et al. (2020). Endothelial ZEB1 promotes angiogenesis-dependent bone formation and reverses osteoporosis. Nat. Commun. 11:460. doi: 10.1038/s41467-019-14076-3
Fu, Y., Chang, A., Chang, L., Niessen, K., Eapen, S., Setiadi, A., et al. (2009). Differential regulation of transforming growth factor beta signaling pathways by Notch in human endothelial cells. J. Biol. Chem. 284, 19452–19462. doi: 10.1074/jbc.M109.011833
Gillette, T. G., and Hill, J. A. (2015). Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circ. Res. 116, 1245–1253. doi: 10.1161/CIRCRESAHA.116.303630
Gong, F., Chiu, L. Y., and Miller, K. M. (2016). Acetylation reader proteins: linking acetylation signaling to genome maintenance and cancer. PLoS Genet. 12:e1006272. doi: 10.1371/journal.pgen.1006272
Gray, J. E., Saltos, A., Tanvetyanon, T., Haura, E. B., Creelan, B., Antonia, S. J., et al. (2019). Phase I/ib study of pembrolizumab plus vorinostat in advanced/metastatic non-small cell lung cancer. Clin. Cancer Res. 25, 6623–6632. doi: 10.1158/1078-0432.CCR-19-1305
Guo, J., Wang, Z., Wu, J., Liu, M., Li, M., Sun, Y., et al. (2019). Endothelial SIRT6 Is vital to prevent hypertension and associated cardiorenal injury through targeting Nkx3.2-GATA5 signaling. Circ. Res. 124, 1448–1461. doi: 10.1161/CIRCRESAHA.118.314032
Hai, Y., Shinsky, S. A., Porter, N. J., and Christianson, D. W. (2017). Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun. 8:15368. doi: 10.1038/ncomms15368
Hakami, N. Y., Dusting, G. J., and Peshavariya, H. M. (2016). Trichostatin A, a histone deacetylase inhibitor suppresses NADPH Oxidase 4-Derived redox signalling and angiogenesis. J. Cell Mol. Med. 20, 1932–1944. doi: 10.1111/jcmm.12885
Hamada, K., Osaka, M., and Yoshida, M. (2014). Cell density impacts epigenetic regulation of cytokine-induced E-selectin gene expression in vascular endothelium. PLoS One 9:e90502. doi: 10.1371/journal.pone.0090502
He, M., Huang, T. S., Li, S., Hong, H. C., Chen, Z., Martin, M., et al. (2019). Atheroprotective Flow Upregulates ITPR3 (Inositol 1,4,5-Trisphosphate Receptor 3) in vascular endothelium via KLF4 (Kruppel-Like Factor 4)-Mediated histone modifications. Arterioscler. Thromb. Vasc. Biol. 39, 902–914. doi: 10.1161/ATVBAHA.118.312301
He, X., Zeng, H., Chen, S. T., Roman, R. J., Aschner, J. L., Didion, S., et al. (2017). Endothelial specific SIRT3 deletion impairs glycolysis and angiogenesis and causes diastolic dysfunction. J. Mol. Cell Cardiol. 112, 104–113. doi: 10.1016/j.yjmcc.2017.09.007
He, Z. X., Wei, B. F., Zhang, X., Gong, Y. P., Ma, L. Y., and Zhao, W. (2021). Current development of CBP/p300 inhibitors in the last decade. Eur. J. Med. Chem. 209:112861. doi: 10.1016/j.ejmech.2020.112861
Hebbel, R. P., Vercellotti, G. M., Pace, B. S., Solovey, A. N., Kollander, R., Abanonu, C. F., et al. (2010). The HDAC inhibitors trichostatin A and suberoylanilide hydroxamic acid exhibit multiple modalities of benefit for the vascular pathobiology of sickle transgenic mice. Blood 115, 2483–2490. doi: 10.1182/blood-2009-02-204990
Heintzman, N. D., Stuart, R. K., Hon, G., Fu, Y., Ching, C. W., Hawkins, R. D., et al. (2007). Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318. doi: 10.1038/ng1966
Hellebrekers, D. M., Castermans, K., Vire, E., Dings, R. P., Hoebers, N. T., Mayo, K. H., et al. (2006). Epigenetic regulation of tumor endothelial cell anergy: silencing of intercellular adhesion molecule-1 by histone modifications. Cancer Res. 66, 10770–10777. doi: 10.1158/0008-5472.CAN-06-1609
Helmlinger, D., Papai, G., Devys, D., and Tora, L. (2020). What do the structures of GCN5-containing complexes teach us about their function? Biochim. Biophys. Acta Gene Regul. Mech. 1864:194614. doi: 10.1016/j.bbagrm.2020.194614
Helmlinger, D., and Tora, L. (2017). Sharing the SAGA. Trends Biochem. Sci. 42, 850–861. doi: 10.1016/j.tibs.2017.09.001
Hnisz, D., Abraham, B. J., Lee, T. I., Lau, A., Saint-Andre, V., Sigova, A. A., et al. (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. doi: 10.1016/j.cell.2013.09.053
Hou, Q., Hu, K., Liu, X., Quan, J., and Liu, Z. (2018). HADC regulates the diabetic vascular endothelial dysfunction by targetting MnSOD. Biosci. Rep. 38:BSR20181042. doi: 10.1042/BSR20181042
Hsu, Y. F., Sheu, J. R., Lin, C. H., Chen, W. C., Hsiao, G., Ou, G., et al. (2011). MAPK phosphatase-1 contributes to trichostatin A inhibition of cyclooxygenase-2 expression in human umbilical vascular endothelial cells exposed to lipopolysaccharide. Biochim. Biophys. Acta 1810, 1160–1169. doi: 10.1016/j.bbagen.2011.08.015
Illi, B., Dello Russo, C., Colussi, C., Rosati, J., Pallaoro, M., Spallotta, F., et al. (2008). Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circ. Res. 102, 51–58. doi: 10.1161/CIRCRESAHA.107.157305
Illi, B., Nanni, S., Scopece, A., Farsetti, A., Biglioli, P., Capogrossi, M. C., et al. (2003). Shear stress-mediated chromatin remodeling provides molecular basis for flow-dependent regulation of gene expression. Circ. Res. 93, 155–161. doi: 10.1161/01.RES.0000080933.82105.29
Inoue, K., Kobayashi, M., Yano, K., Miura, M., Izumi, A., Mataki, C., et al. (2006). Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule-1 expression. Arterioscler. Thromb. Vasc. Biol. 26, 2652–2659. doi: 10.1161/01.ATV.0000247247.89787.e7
Iring, A., Jin, Y. J., Albarran-Juarez, J., Siragusa, M., Wang, S., Dancs, P. T., et al. (2019). Shear stress-induced endothelial adrenomedullin signaling regulates vascular tone and blood pressure. J. Clin. Invest. 129, 2775–2791. doi: 10.1172/JCI123825
Jamal, J., Mustafa, M. R., and Wong, P. F. (2014). Paeonol protects against premature senescence in endothelial cells by modulating Sirtuin 1 pathway. J. Ethnopharmacol. 154, 428–436. doi: 10.1016/j.jep.2014.04.025
Javaid, N., and Choi, S. (2017). Acetylation- and methylation-related epigenetic proteins in the context of their targets. Genes 8:196. doi: 10.3390/genes8080196
Jiang, L., Yin, M., Wei, X., Liu, J., Wang, X., Niu, C., et al. (2015). Bach1 represses wnt/beta-catenin signaling and angiogenesis. Circ. Res. 117, 364–375. doi: 10.1161/CIRCRESAHA.115.306829
Jin, G., Bausch, D., Knightly, T., Liu, Z., Li, Y., Liu, B., et al. (2011). Histone deacetylase inhibitors enhance endothelial cell sprouting angiogenesis in vitro. Surgery 150, 429–435. doi: 10.1016/j.surg.2011.07.001
Joshi, B., Ordonez-Ercan, D., Dasgupta, P., and Chellappan, S. (2005). Induction of human metallothionein 1G promoter by VEGF and heavy metals: differential involvement of E2F and metal transcription factors. Oncogene 24, 2204–2217. doi: 10.1038/sj.onc.1208206
Ke, X., Johnson, H., Jing, X., Michalkiewicz, T., Huang, Y. W., Lane, R. H., et al. (2018). Persistent pulmonary hypertension alters the epigenetic characteristics of endothelial nitric oxide synthase gene in pulmonary artery endothelial cells in a fetal lamb model. Physiol. Genomics 50, 828–836. doi: 10.1152/physiolgenomics.00047.2018
Kelly, R. D., and Cowley, S. M. (2013). The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem. Soc. Trans. 41, 741–749. doi: 10.1042/BST20130010
Kida, Y., and Goligorsky, M. S. (2016). Sirtuins, cell senescence, and vascular aging. Can. J. Cardiol. 32, 634–641. doi: 10.1016/j.cjca.2015.11.022
Kim, J. H., Kim, J. H., Oh, M., Yu, Y. S., Kim, K. W., and Kwon, H. J. (2009). N-hydroxy-7-(2-naphthylthio) heptanomide inhibits retinal and choroidal angiogenesis. Mol. Pharm. 6, 513–519. doi: 10.1021/mp800178b
Kim, Y. R., Kim, C. S., Naqvi, A., Kumar, A., Kumar, S., Hoffman, T. A., et al. (2012). Epigenetic upregulation of p66shc mediates low-density lipoprotein cholesterol-induced endothelial cell dysfunction. Am. J. Physiol. Heart Circ. Physiol. 303, H189–H196. doi: 10.1152/ajpheart.01218.2011
Krishna, S. M., Trollope, A. F., and Golledge, J. (2015). The relevance of epigenetics to occlusive cerebral and peripheral arterial disease. Clin. Sci. 128, 537–558. doi: 10.1042/CS20140491
Kurz, D. J., Payeli, S., Greutert, H., Briand Schumacher, S., Luscher, T. F., and Tanner, F. C. (2014). Epigenetic regulation of tissue factor inducibility in endothelial cell senescence. Mech. Ageing Dev. 140, 1–9. doi: 10.1016/j.mad.2014.07.002
Larsson, P., Alwis, I., Niego, B., Sashindranath, M., Fogelstrand, P., Wu, M. C., et al. (2016). Valproic acid selectively increases vascular endothelial tissue-type plasminogen activator production and reduces thrombus formation in the mouse. J. Thromb. Haemost. 14, 2496–2508. doi: 10.1111/jth.13527
Larsson, P., Ulfhammer, E., Magnusson, M., Bergh, N., Lunke, S., El-Osta, A., et al. (2012). Role of histone acetylation in the stimulatory effect of valproic acid on vascular endothelial tissue-type plasminogen activator expression. PLoS One 7:e31573. doi: 10.1371/journal.pone.0031573
Li, B., Li, Y., Liu, K., Wang, X., Qi, J., Wang, B., et al. (2017). High glucose decreases claudins-5 and -11 in cardiac microvascular endothelial cells: antagonistic effects of tongxinluo. Endocr. Res. 42, 15–21. doi: 10.3109/07435800.2016.1163723
Li, M., Gallo, D., Csizmadia, E., Otterbein, L. E., and Wegiel, B. (2014). Carbon monoxide induces chromatin remodelling to facilitate endothelial cell migration. Thromb. Haemost. 111, 951–959. doi: 10.1160/TH13-09-0748
Li, M., van Esch, B., Henricks, P. A. J., Folkerts, G., and Garssen, J. (2018). The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor alpha-stimulated endothelial cells via activation of GPR41/43 and inhibition of HDACs. Front. Pharmacol. 9:533. doi: 10.3389/fphar.2018.00533
Li, T., Zhang, C., Hassan, S., Liu, X., Song, F., Chen, K., et al. (2018). Histone deacetylase 6 in cancer. J. Hematol. Oncol. 11:111. doi: 10.1186/s13045-018-0654-9
Li, X., Shao, Y., Sha, X., Fang, P., Kuo, Y. M., Andrews, A. J., et al. (2018). IL-35 (Interleukin-35) suppresses endothelial cell activation by inhibiting mitochondrial reactive oxygen species-mediated site-specific acetylation of H3K14 (Histone 3 Lysine 14). Arterioscler. Thromb. Vasc. Biol. 38, 599–609. doi: 10.1161/ATVBAHA.117.310626
Li, Z., Zhang, X., Liu, S., Zeng, S., Yu, L., Yang, G., et al. (2018). BRG1 regulates NOX gene transcription in endothelial cells and contributes to cardiac ischemia-reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 3477–3486. doi: 10.1016/j.bbadis.2018.08.002
Liao, J. K., and Laufs, U. (2005). Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 45, 89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748
Lipinski, M., Del Blanco, B., and Barco, A. (2019). CBP/p300 in brain development and plasticity: disentangling the KAT’s cradle. Curr. Opin. Neurobiol. 59, 1–8. doi: 10.1016/j.conb.2019.01.023
Liu, D., Perkins, J. T., and Hennig, B. (2016). EGCG prevents PCB-126-induced endothelial cell inflammation via epigenetic modifications of NF-kappaB target genes in human endothelial cells. J. Nutr. Biochem. 28, 164–170. doi: 10.1016/j.jnutbio.2015.10.003
Liu, S. S., Wu, F., Jin, Y. M., Chang, W. Q., and Xu, T. M. (2020). HDAC11: a rising star in epigenetics. Biomed. Pharmacother. 131:110607. doi: 10.1016/j.biopha.2020.110607
Lu, Y., Sun, Y., Drummer, C. T., Nanayakkara, G. K., Shao, Y., Saaoud, F., et al. (2019). Increased acetylation of H3K14 in the genomic regions that encode trained immunity enzymes in lysophosphatidylcholine-activated human aortic endothelial cells - Novel qualification markers for chronic disease risk factors and conditional DAMPs. Redox Biol. 24:101221. doi: 10.1016/j.redox.2019.101221
Malik, A. B., Lynch, J. J., and Cooper, J. A. (1989). Endothelial barrier function. J. Invest. Dermatol. 93(2 Suppl.), 62S–67S. doi: 10.1111/1523-1747.ep12581072
Marmorstein, R., and Zhou, M. M. (2014). Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 6:a018762. doi: 10.1101/cshperspect.a018762
Michaelis, M., Suhan, T., Michaelis, U. R., Beek, K., Rothweiler, F., Tausch, L., et al. (2006). Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ. 13, 446–453. doi: 10.1038/sj.cdd.4401759
Millard, C. J., Watson, P. J., Fairall, L., and Schwabe, J. W. R. (2017). Targeting class i histone deacetylases in a “Complex” environment. Trends Pharmacol. Sci. 38, 363–377. doi: 10.1016/j.tips.2016.12.006
Mishra, M., Duraisamy, A. J., Bhattacharjee, S., and Kowluru, R. A. (2019). Adaptor protein p66Shc: a link between cytosolic and mitochondrial dysfunction in the development of diabetic retinopathy. Antioxid Redox Signal 30, 1621–1634. doi: 10.1089/ars.2018.7542
Mojiri, A., Alavi, P., Lorenzana Carrillo, M. A., Nakhaei-Nejad, M., Sergi, C. M., Thebaud, B., et al. (2019). Endothelial cells of different organs exhibit heterogeneity in von Willebrand factor expression in response to hypoxia. Atherosclerosis 282, 1–10. doi: 10.1016/j.atherosclerosis.2019.01.002
Mortuza, R., Feng, B., and Chakrabarti, S. (2015). SIRT1 reduction causes renal and retinal injury in diabetes through endothelin 1 and transforming growth factor beta1. J. Cell Mol. Med. 19, 1857–1867. doi: 10.1111/jcmm.12557
Murugavel, S., Bugyei-Twum, A., Matkar, P. N., Al-Mubarak, H., Chen, H. H., Adam, M., et al. (2018). Valproic acid induces endothelial-to-mesenchymal transition-like phenotypic switching. Front. Pharmacol. 9:737. doi: 10.3389/fphar.2018.00737
Mutlu, B., and Puigserver, P. (2020). GCN5 acetyltransferase in cellular energetic and metabolic processes. Biochim. Biophys. Acta Gene Regul. Mech. 1864:194626. doi: 10.1016/j.bbagrm.2020.194626
O’Connor, O. A., Horwitz, S., Masszi, T., Van Hoof, A., Brown, P., Doorduijn, J., et al. (2015). Belinostat in patients with relapsed or refractory Peripheral T-Cell lymphoma: results of the pivotal phase II BELIEF (CLN-19) study. J. Clin. Oncol. 33, 2492–2499. doi: 10.1200/JCO.2014.59.2782
Ohashi, A., Yasuda, H., Kamiya, T., Hara, H., and Adachi, T. (2017). CAPE increases the expression of SOD3 through epigenetics in human retinal endothelial cells. J. Clin. Biochem. Nutr. 61, 6–13. doi: 10.3164/jcbn.16-109
Olmos, Y., Sanchez-Gomez, F. J., Wild, B., Garcia-Quintans, N., Cabezudo, S., Lamas, S., et al. (2013). SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1alpha complex. Antioxid Redox Signal 19, 1507–1521. doi: 10.1089/ars.2012.4713
Paneni, F., Mocharla, P., Akhmedov, A., Costantino, S., Osto, E., Volpe, M., et al. (2012). Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ. Res. 111, 278–289. doi: 10.1161/CIRCRESAHA.112.266593
Passacquale, G., Phinikaridou, A., Warboys, C., Cooper, M., Lavin, B., Alfieri, A., et al. (2015). Aspirin-induced histone acetylation in endothelial cells enhances synthesis of the secreted isoform of netrin-1 thus inhibiting monocyte vascular infiltration. Br. J. Pharmacol. 172, 3548–3564. doi: 10.1111/bph.13144
Peng, Y., Stewart, D., Li, W., Hawkins, M., Kulak, S., Ballermann, B., et al. (2007). Irradiation modulates association of NF-Y with histone-modifying cofactors PCAF and HDAC. Oncogene 26, 7576–7583. doi: 10.1038/sj.onc.1210565
Perrone, L., Devi, T. S., Hosoya, K., Terasaki, T., and Singh, L. P. (2009). Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell Physiol. 221, 262–272. doi: 10.1002/jcp.21852
Phillips, D. M. (1963). The presence of acetyl groups of histones. Biochem. J. 87, 258–263. doi: 10.1042/bj0870258
Pillai, S., Kovacs, M., and Chellappan, S. (2010). Regulation of vascular endothelial growth factor receptors by Rb and E2F1: role of acetylation. Cancer Res. 70, 4931–4940. doi: 10.1158/0008-5472.CAN-10-0501
Pillus, L. (2008). MYSTs mark chromatin for chromosomal functions. Curr. Opin. Cell Biol. 20, 326–333. doi: 10.1016/j.ceb.2008.04.009
Pirola, L., Balcerczyk, A., Tothill, R. W., Haviv, I., Kaspi, A., Lunke, S., et al. (2011). Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res. 21, 1601–1615. doi: 10.1101/gr.116095.110
Postberg, J., Kanders, M., Forcob, S., Willems, R., Orth, V., Hensel, K. O., et al. (2015). CpG signalling, H2A.Z/H3 acetylation and microRNA-mediated deferred self-attenuation orchestrate foetal NOS3 expression. Clin. Epigenetics 7:9. doi: 10.1186/s13148-014-0042-4
Potente, M., and Makinen, T. (2017). Vascular heterogeneity and specialization in development and disease. Nat. Rev. Mol. Cell Biol. 18, 477–494. doi: 10.1038/nrm.2017.36
Poulsen, L. C., Edelmann, R. J., Kruger, S., Dieguez-Hurtado, R., Shah, A., Stav-Noraas, T. E., et al. (2018). Inhibition of endothelial NOTCH1 signaling attenuates inflammation by reducing cytokine-mediated histone acetylation at inflammatory enhancers. Arterioscler. Thromb. Vasc. Biol. 38, 854–869. doi: 10.1161/ATVBAHA.117.310388
Qian, D. Z., Kato, Y., Shabbeer, S., Wei, Y., Verheul, H. M., Salumbides, B., et al. (2006). Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin. Cancer Res. 12, 634–642. doi: 10.1158/1078-0432.CCR-05-1132
Rando, O. J., and Chang, H. Y. (2009). Genome-wide views of chromatin structure. Annu. Rev. Biochem. 78, 245–271. doi: 10.1146/annurev.biochem.78.071107.134639
Reifsnyder, C., Lowell, J., Clarke, A., and Pillus, L. (1996). Yeast SAS silencing genes and human genes associated with AML and HIV-1 Tat interactions are homologous with acetyltransferases. Nat. Genet. 14, 42–49. doi: 10.1038/ng0996-42
Ren, B., Best, B., Ramakrishnan, D. P., Walcott, B. P., Storz, P., and Silverstein, R. L. (2016). LPA/PKD-1-FoxO1 signaling axis mediates endothelial cell CD36 transcriptional repression and proangiogenic and proarteriogenic reprogramming. Arterioscler. Thromb. Vasc. Biol. 36, 1197–1208. doi: 10.1161/ATVBAHA.116.307421
Ren, B., Hale, J., Srikanthan, S., and Silverstein, R. L. (2011). Lysophosphatidic acid suppresses endothelial cell CD36 expression and promotes angiogenesis via a PKD-1-dependent signaling pathway. Blood 117, 6036–6045. doi: 10.1182/blood-2010-12-326017
Rodriguez, C. P., Wu, Q. V., Voutsinas, J., Fromm, J. R., Jiang, X., Pillarisetty, V. G., et al. (2020). A Phase II trial of pembrolizumab and vorinostat in recurrent metastatic head and neck squamous cell carcinomas and salivary gland cancer. Clin. Cancer Res. 26, 837–845. doi: 10.1158/1078-0432.CCR-19-2214
Rose, J. L., Huang, H., Wray, S. F., and Hoyt, D. G. (2005). Integrin engagement increases histone H3 acetylation and reduces histone H1 association with DNA in murine lung endothelial cells. Mol. Pharmacol. 68, 439–446. doi: 10.1124/mol.104.010876
Rossig, L., Li, H., Fisslthaler, B., Urbich, C., Fleming, I., Forstermann, U., et al. (2002). Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ. Res. 91, 837–844. doi: 10.1161/01.res.0000037983.07158.b1
Saksouk, N., Avvakumov, N., Champagne, K. S., Hung, T., Doyon, Y., Cayrou, C., et al. (2009). HBO1 HAT complexes target chromatin throughout gene coding regions via multiple PHD finger interactions with histone H3 tail. Mol. Cell 33, 257–265. doi: 10.1016/j.molcel.2009.01.007
Santos-Rosa, H., and Caldas, C. (2005). Chromatin modifier enzymes, the histone code and cancer. Eur. J. Cancer 41, 2381–2402. doi: 10.1016/j.ejca.2005.08.010
Schmeck, B., Beermann, W., N’Guessan, P. D., Hocke, A. C., Opitz, B., Eitel, J., et al. (2008). Simvastatin reduces Chlamydophila pneumoniae-mediated histone modifications and gene expression in cultured human endothelial cells. Circ. Res. 102, 888–895. doi: 10.1161/CIRCRESAHA.107.161307
Schmeck, B., Beermann, W., van Laak, V., Opitz, B., Hocke, A. C., Meixenberger, K., et al. (2006). Listeria monocytogenes induced Rac1-dependent signal transduction in endothelial cells. Biochem. Pharmacol. 72, 1367–1374. doi: 10.1016/j.bcp.2006.06.033
Schmeck, B., Beermann, W., van Laak, V., Zahlten, J., Opitz, B., Witzenrath, M., et al. (2005). Intracellular bacteria differentially regulated endothelial cytokine release by MAPK-dependent histone modification. J. Immunol. 175, 2843–2850. doi: 10.4049/jimmunol.175.5.2843
Sekeres, M. A., Othus, M., List, A. F., Odenike, O., Stone, R. M., Gore, S. D., et al. (2017). Randomized phase II study of azacitidine alone or in combination with lenalidomide or with vorinostat in higher-risk myelodysplastic syndromes and chronic myelomonocytic leukemia: north american intergroup study SWOG S1117. J. Clin. Oncol. 35, 2745–2753. doi: 10.1200/JCO.2015.66.2510
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6:a018713. doi: 10.1101/cshperspect.a018713
Shahbazian, M. D., and Grunstein, M. (2007). Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100. doi: 10.1146/annurev.biochem.76.052705.162114
Shao, J., Weng, X., Zhuo, L., Yu, L., Li, Z., Shen, K., et al. (2019). Angiotensin II induced CSF1 transcription is mediated by a crosstalk between different epigenetic factors in vascular endothelial cells. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 1–11. doi: 10.1016/j.bbagrm.2018.10.001
Sheikh, B. N., and Akhtar, A. (2019). The many lives of KATs - detectors, integrators and modulators of the cellular environment. Nat. Rev. Genet. 20, 7–23. doi: 10.1038/s41576-018-0072-4
Shi, Y., Xu, X., Zhang, Q., Fu, G., Mo, Z., Wang, G. S., et al. (2014). tRNA synthetase counteracts c-Myc to develop functional vasculature. eLife 3:e02349. doi: 10.7554/eLife.02349
Shu, X. Z., Zhang, L. N., Zhang, R., Zhang, C. J., He, H. P., Zhou, H., et al. (2015). Histone acetyltransferase p300 promotes MRTF-A-mediates transactivation of VE-cadherin gene in human umbilical vein endothelial cells. Gene 563, 17–23. doi: 10.1016/j.gene.2015.02.076
Simiele, F., Recchiuti, A., Patruno, S., Plebani, R., Pierdomenico, A. M., Codagnone, M., et al. (2016). Epigenetic regulation of the formyl peptide receptor 2 gene. Biochim. Biophys. Acta 1859, 1252–1258. doi: 10.1016/j.bbagrm.2016.07.007
Singh, C. K., Chhabra, G., Ndiaye, M. A., Garcia-Peterson, L. M., Mack, N. J., and Ahmad, N. (2018). The role of sirtuins in antioxidant and redox signaling. Antioxid Redox Signal 28, 643–661. doi: 10.1089/ars.2017.7290
Siuda, D., Zechner, U., El Hajj, N., Prawitt, D., Langer, D., Xia, N., et al. (2012). Transcriptional regulation of Nox4 by histone deacetylases in human endothelial cells. Basic Res. Cardiol. 107:283. doi: 10.1007/s00395-012-0283-3
Spedale, G., Timmers, H. T., and Pijnappel, W. W. (2012). ATAC-king the complexity of SAGA during evolution. Genes Dev. 26, 527–541. doi: 10.1101/gad.184705.111
Stauffer, B. L., Westby, C. M., and DeSouza, C. A. (2008). Endothelin-1, aging and hypertension. Curr. Opin. Cardiol. 23, 350–355. doi: 10.1097/HCO.0b013e328302f3c6
Sturtzel, C. (2017). Endothelial cells. Adv. Exp. Med. Biol. 1003, 71–91. doi: 10.1007/978-3-319-57613-8_4
Su, H., Zeng, H., He, X., Zhu, S. H., and Chen, J. X. (2020). Histone acetyltransferase p300 inhibitor improves coronary flow reserve in SIRT3 (Sirtuin 3) knockout mice. J. Am. Heart Assoc. 9:e017176. doi: 10.1161/JAHA.120.017176
Svennerholm, K., Bergh, N., Larsson, P., Jern, S., Johansson, G., Biber, B., et al. (2014). Histone deacetylase inhibitor treatment increases coronary t-PA release in a porcine ischemia model. PLoS One 9:e97260. doi: 10.1371/journal.pone.0097260
Svennerholm, K., Haney, M., Biber, B., Ulfhammer, E., Saluveer, O., Larsson, P., et al. (2015). Histone deacetylase inhibition enhances tissue plasminogen activator release capacity in atherosclerotic man. PLoS One 10:e0121196. doi: 10.1371/journal.pone.0121196
Tambo, Y., Hosomi, Y., Sakai, H., Nogami, N., Atagi, S., Sasaki, Y., et al. (2017). Phase I/II study of docetaxel combined with resminostat, an oral hydroxamic acid HDAC inhibitor, for advanced non-small cell lung cancer in patients previously treated with platinum-based chemotherapy. Invest. New Drugs 35, 217–226. doi: 10.1007/s10637-017-0435-2
Tanegashima, K., Sato-Miyata, Y., Funakoshi, M., Nishito, Y., Aigaki, T., and Hara, T. (2017). Epigenetic regulation of the glucose transporter gene Slc2a1 by beta-hydroxybutyrate underlies preferential glucose supply to the brain of fasted mice. Genes Cells 22, 71–83. doi: 10.1111/gtc.12456
Tang, F. B., Dai, Y. L., Zhou, G. Y., Zhang, W. H., Wang, H. B., Li, Y. G., et al. (2018). Valproic acid treatment inhibits vasopermeability and improves survival in rats with lethal scald injury. J. Burn Care Res. 39, 209–217. doi: 10.1097/BCR.0000000000000568
Tessarz, P., and Kouzarides, T. (2014). Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 15, 703–708. doi: 10.1038/nrm3890
Thangavel, J., Malik, A. B., Elias, H. K., Rajasingh, S., Simpson, A. D., Sundivakkam, P. K., et al. (2014). Combinatorial therapy with acetylation and methylation modifiers attenuates lung vascular hyperpermeability in endotoxemia-induced mouse inflammatory lung injury. Am. J. Pathol. 184, 2237–2249. doi: 10.1016/j.ajpath.2014.05.008
Thomas, T., and Voss, A. K. (2007). The diverse biological roles of MYST histone acetyltransferase family proteins. Cell Cycle 6, 696–704. doi: 10.4161/cc.6.6.4013
Turtoi, A., Mottet, D., Matheus, N., Dumont, B., Peixoto, P., Hennequiere, V., et al. (2012). The angiogenesis suppressor gene AKAP12 is under the epigenetic control of HDAC7 in endothelial cells. Angiogenesis 15, 543–554. doi: 10.1007/s10456-012-9279-8
Ulfhammer, E., Larsson, P., Magnusson, M., Karlsson, L., Bergh, N., and Jern, S. (2016). Dependence of proximal GC boxes and binding transcription factors in the regulation of basal and valproic acid-induced expression of t-PA. Int. J. Vasc. Med. 2016:7928681. doi: 10.1155/2016/7928681
Van De Craen, B., Declerck, P. J., and Gils, A. (2012). The biochemistry, physiology and pathological roles of PAI-1 and the requirements for PAI-1 inhibition in vivo. Thromb. Res. 130, 576–585. doi: 10.1016/j.thromres.2012.06.023
Verdin, E., and Ott, M. (2015). 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 16, 258–264. doi: 10.1038/nrm3931
Visel, A., Blow, M. J., Li, Z., Zhang, T., Akiyama, J. A., Holt, A., et al. (2009). ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858. doi: 10.1038/nature07730
Wan, Y. Z., Gao, P., Zhou, S., Zhang, Z. Q., Hao, D. L., Lian, L. S., et al. (2014). SIRT1-mediated epigenetic downregulation of plasminogen activator inhibitor-1 prevents vascular endothelial replicative senescence. Aging Cell 13, 890–899. doi: 10.1111/acel.12247
Wang, J., Mahmud, S. A., Bitterman, P. B., Huo, Y., and Slungaard, A. (2007). Histone deacetylase inhibitors suppress TF-kappaB-dependent agonist-driven tissue factor expression in endothelial cells and monocytes. J. Biol. Chem. 282, 28408–28418. doi: 10.1074/jbc.M703586200
Wiesel-Motiuk, N., and Assaraf, Y. G. (2020). The key roles of the lysine acetyltransferases KAT6A and KAT6B in physiology and pathology. Drug Resist. Updat. 53:100729. doi: 10.1016/j.drup.2020.100729
Wilcox, J. N., Subramanian, R. R., Sundell, C. L., Tracey, W. R., Pollock, J. S., Harrison, D. G., et al. (1997). Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler. Thromb. Vasc. Biol. 17, 2479–2488. doi: 10.1161/01.atv.17.11.2479
Won, K. J., Jung, S. H., Jung, S. H., Lee, K. P., Lee, H. M., Lee, D. Y., et al. (2014). DJ-1/park7 modulates vasorelaxation and blood pressure via epigenetic modification of endothelial nitric oxide synthase. Cardiovasc. Res. 101, 473–481. doi: 10.1093/cvr/cvt274
Xu, P., Li, C., Chen, Z., Jiang, S., Fan, S., Wang, J., et al. (2016). The NuA4 core complex acetylates nucleosomal histone H4 through a double recognition mechanism. Mol. Cell 63, 965–975. doi: 10.1016/j.molcel.2016.07.024
Xu, X. F., Lv, Y., Gu, W. Z., Tang, L. L., Wei, J. K., Zhang, L. Y., et al. (2013). Epigenetics of hypoxic pulmonary arterial hypertension following intrauterine growth retardation rat: epigenetics in PAH following IUGR. Respir. Res. 14:20. doi: 10.1186/1465-9921-14-20
Xu, X. F., Ma, X. L., Shen, Z., Wu, X. L., Cheng, F., and Du, L. Z. (2010). Epigenetic regulation of the endothelial nitric oxide synthase gene in persistent pulmonary hypertension of the newborn rat. J. Hypertens 28, 2227–2235. doi: 10.1097/HJH.0b013e32833e08f1
Yan, M. S., Turgeon, P. J., Man, H. J., Dubinsky, M. K., Ho, J. J. D., El-Rass, S., et al. (2018). Histone acetyltransferase 7 (KAT7)-dependent intragenic histone acetylation regulates endothelial cell gene regulation. J. Biol. Chem. 293, 4381–4402. doi: 10.1074/jbc.RA117.001383
Yang, D., Xie, P., and Liu, Z. (2012). Ischemia/reperfusion-induced MKP-3 impairs endothelial NO formation via inactivation of ERK1/2 pathway. PLoS One 7:e42076. doi: 10.1371/journal.pone.0042076
Yasuda, H., Ohashi, A., Nishida, S., Kamiya, T., Suwa, T., Hara, H., et al. (2016). Exendin-4 induces extracellular-superoxide dismutase through histone H3 acetylation in human retinal endothelial cells. J. Clin. Biochem. Nutr. 59, 174–181. doi: 10.3164/jcbn.16-26
You, D., Wen, X., Gorczyca, L., Morris, A., Richardson, J. R., and Aleksunes, L. M. (2019). Increased MDR1 transporter expression in human brain endothelial cells through enhanced histone acetylation and activation of aryl hydrocarbon receptor signaling. Mol. Neurobiol. 56, 6986–7002. doi: 10.1007/s12035-019-1565-7
Zelko, I. N., and Folz, R. J. (2015). Regulation of oxidative stress in pulmonary artery endothelium. modulation of extracellular superoxide dismutase and NOX4 expression using histone deacetylase Class I inhibitors. Am. J. Respir. Cell Mol. Biol. 53, 513–524. doi: 10.1165/rcmb.2014-0260OC
Zelko, I. N., Stepp, M. W., Vorst, A. L., and Folz, R. J. (2011). Histone acetylation regulates the cell-specific and interferon-gamma-inducible expression of extracellular superoxide dismutase in human pulmonary arteries. Am. J. Respir. Cell Mol. Biol. 45, 953–961. doi: 10.1165/rcmb.2011-0012OC
Zeng, L., Zhang, Y., Chien, S., Liu, X., and Shyy, J. Y. (2003). The role of p53 deacetylation in p21Waf1 regulation by laminar flow. J. Biol. Chem. 278, 24594–24599. doi: 10.1074/jbc.M301955200
Zhang, B., Day, D. S., Ho, J. W., Song, L., Cao, J., Christodoulou, D., et al. (2013). A dynamic H3K27ac signature identifies VEGFA-stimulated endothelial enhancers and requires EP300 activity. Genome Res. 23, 917–927. doi: 10.1101/gr.149674.112
Zhang, M. X., Zhang, C., Shen, Y. H., Wang, J., Li, X. N., Chen, L., et al. (2008). Effect of 27nt small RNA on endothelial nitric-oxide synthase expression. Mol. Biol. Cell 19, 3997–4005. doi: 10.1091/mbc.E07-11-1186
Zhang, X., Williams, E. D., Azhar, G., Rogers, S. C., and Wei, J. Y. (2016). Does p49/STRAP, a SRF-binding protein (SRFBP1), modulate cardiac mitochondrial function in aging? Exp. Gerontol 82, 150–159. doi: 10.1016/j.exger.2016.06.008
Zhao, S., Zhang, X., and Li, H. (2018). Beyond histone acetylation-writing and erasing histone acylations. Curr. Opin. Struct. Biol. 53, 169–177. doi: 10.1016/j.sbi.2018.10.001
Zhong, Q., and Kowluru, R. A. (2010). Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J. Cell Biochem. 110, 1306–1313. doi: 10.1002/jcb.22644
Zhong, Q., and Kowluru, R. A. (2013). Regulation of matrix metalloproteinase-9 by epigenetic modifications and the development of diabetic retinopathy. Diabetes 62, 2559–2568. doi: 10.2337/db12-1141
Keywords: epigenetic regulation, histone acetylation, acetyltransferase, deacetylase, endothelial dysfunction
Citation: Fang Z, Wang X, Sun X, Hu W and Miao QR (2021) The Role of Histone Protein Acetylation in Regulating Endothelial Function. Front. Cell Dev. Biol. 9:672447. doi: 10.3389/fcell.2021.672447
Received: 25 February 2021; Accepted: 06 April 2021;
Published: 29 April 2021.
Edited by:
Xiaofeng Yang, Temple University, United StatesReviewed by:
Bin Ren, University of Alabama at Birmingham, United StatesCopyright © 2021 Fang, Wang, Sun, Hu and Miao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenquan Hu, d2VucXVhbi5odUBueXVsYW5nb25lLm9yZw==; Qing R. Miao, cWluZy5taWFvQG55dWxhbmdvbmUub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.