 Xue Jiang
Xue Jiang Yihuan Chen1†
Yihuan Chen1† Lingqun Ye
Lingqun Ye Wei Lei
Wei Lei Shijun Hu
Shijun Hu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 17 May 2021
Sec. Stem Cell Research
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.672039
This article is part of the Research TopicStem Cells and Cardiovascular DiseasesView all 29 articles
In the past decades, researchers discovered the contribution of genetic defects to the pathogenesis of primary cardiomyopathy and tried to explain the pathogenesis of these diseases by establishing a variety of disease models. Although human heart tissues and primary cardiomyocytes have advantages in modeling human heart diseases, they are difficult to obtain and culture in vitro. Defects developed in genetically modified animal models are notably different from human diseases at the molecular level. The advent of human induced pluripotent stem cells (hiPSCs) provides an unprecedented opportunity to further investigate the pathogenic mechanisms of inherited cardiomyopathies in vitro using patient-specific hiPSC-derived cardiomyocytes. In this review, we will make a summary of recent advances in in vitro inherited cardiomyopathy modeling using hiPSCs.
Inherited cardiomyopathy is a kind of myocardial disease with a range of different genetic disorders and epigenetic variations (Fatkin and Graham, 2002). Inherited cardiomyopathies mainly include dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), left ventricular non-compaction (LVNC), and arrhythmogenic right ventricular (ARVC) (Figure 1). Among these inherited cardiomyopathies, DCM is characterized by ventricular chamber dilatation and systolic dysfunction. It is usually related to mutations in genes such as BAG3, LMNA, MYH7, TNNT2, TTN, and so on. HCM is a disease with abnormal myocardial thickening. The cause of the disease is often closely related to the mutations of ACTN2, BRAF, CSRP3, LMNA, MYL3, and TNNT2, etc. Thickened myocardium makes it harder for the heart to pump blood. LVNC is characterized by the phenotype that the various tissue layers of the heart are not completely gathered together, resulting in a deep recess in the muscle wall. Patients with LVNC usually have symptoms of heart failure, arrhythmias, and thromboembolism and always accompanied by mutations in genes such as ACTC1, MYBPC3, MYH7, SCN5A, TAZ, TBX20, and so on. ARVC is a heart disease in which the right ventricular muscle is replaced by fat and/or scar tissue. With the aggravation of the degree, the right ventricle (RV) gradually loses its ability to pump blood, and it is mainly caused by mutations in genes such as DSC2, PKP2, and desmoplakin, etc. Patients with ARVC usually have arrhythmias and increased risk of cardiac arrest or death.
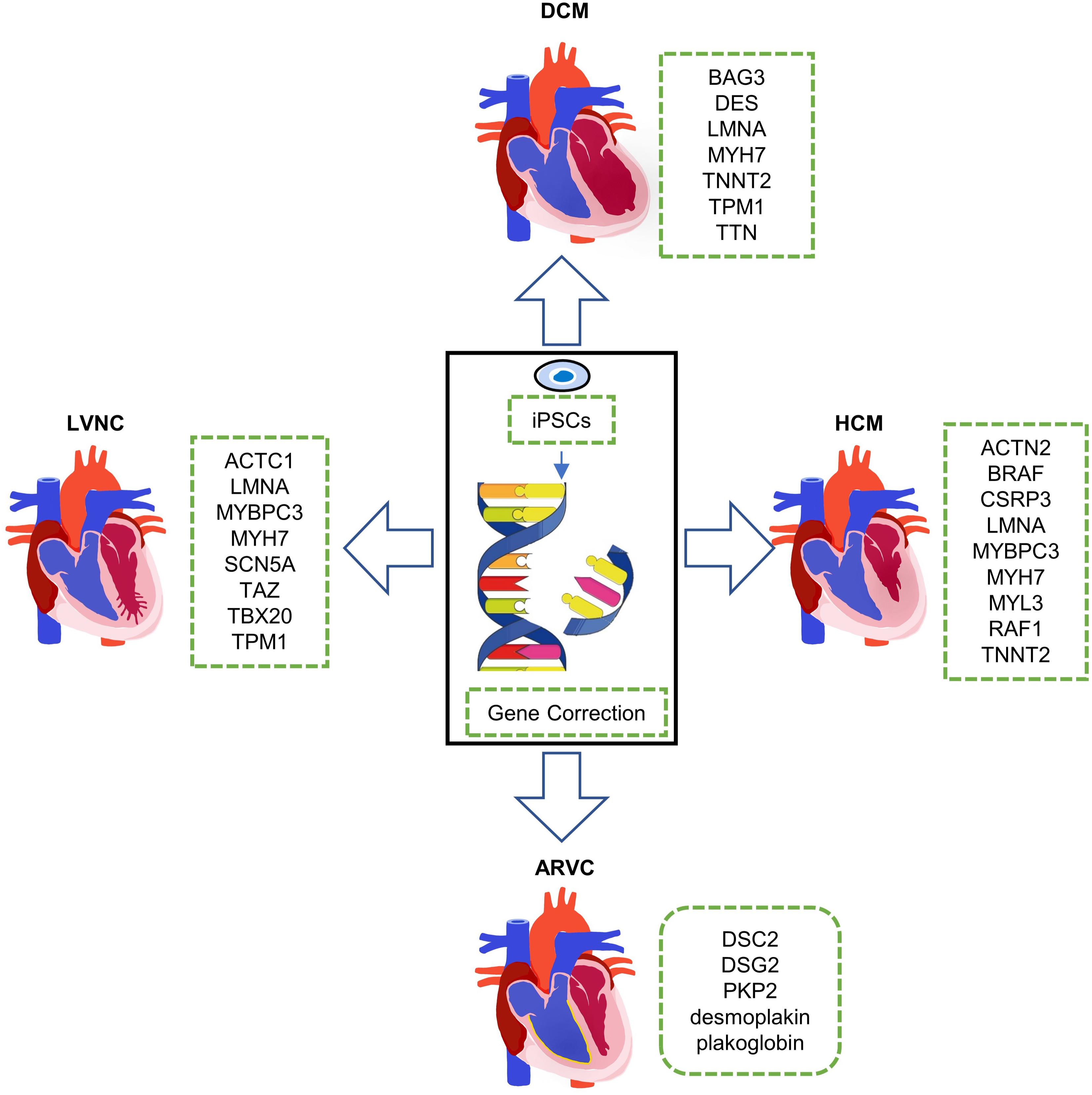
Figure 1. Clinical phenotypes of inherited cardiomyopathies and disease modeling with human induced pluripotent stem cells (hiPSCs). DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; ARVC, arrhythmogenic right ventricular; and LVNC, left ventricular non-compaction.
To reveal the pathogenesis of these genetic defects, a variety of disease models have been established. Human heart tissues and primary cardiomyocytes are the optimum choice undoubtedly, but they are difficult to obtain and culture in vitro, which hinders their application in cardiovascular research (Savla et al., 2014). Researchers then developed genetically modified animal models, among which mouse model is widely used. These animal models are crucial in progressing knowledge of the pathophysiology of heart disease because the basic principles of cardiac excitation and contraction are relatively conservative in the species. However, although animal models provide some useful results for the pathogenesis of cardiomyopathy, animals and humans are different at the molecular level, such as genetic background, epigenetic inheritance, transcriptional regulation, and protein modification. These, in turn, lead to significant differences between the functional and electrophysiological properties of the animal and human hearts, which limits the interpretation and applicability of these data (Ma et al., 2013). Therefore, the lack of a good source of live human cardiomyocytes in vitro and the failure to establish variant patient-specific disease models have significantly stymied research on inherited cardiomyopathy. Fortunately, human induced pluripotent stem cells (hiPSCs) provided a promising platform for genetic disease modeling.
Human induced pluripotent stem cells can be reprogrammed from somatic cells, which are easily isolated from the skin, urine, blood, and more unexpected samples, by virus-dependent or independent overexpression of a set of reprogramming factors such as Oct3/4, Sox2, Klf4, and c-Myc (Takahashi and Yamanaka, 2006). The hiPSCs can be indefinitely maintained and expanded in the pluripotent state in vitro and theoretically have the ability to generate any adult cell types (Kamp and Lyons, 2009). Through modulating BMP4 and WNT that are critical in heart development, hiPSCs are able to efficiently differentiate into cardiomyocytes (Qyang et al., 2007; Cao et al., 2013). Two major strategies have been developed for hiPSC-based inherited cardiomyopathy modeling: (1) reprogramming the somatic cells from genetic disease patients into hiPSCs and then differentiating them into cardiomyocytes and (2) performing gene editing to introduce pathogenic mutations into hiPSCs of healthy donor and then differentiating them into cardiomyocytes. Three precise genome-editing techniques, including zinc finger nuclease (ZFN; Kim et al., 2010), transcription activator-like effector nuclease (TALEN; Schmid-Burgk et al., 2013), and clustered regularly interspaced short palindromic repeat-associated protein 9 (CRISPR-Cas9; Wu et al., 2014), can be used to correct or introduce mutations in hiPSCs.
In this review, we will discuss the most common inherited cardiomyopathies abovementioned, including their genetic background and the work researchers have done so far using hiPSCs for mechanism research and symptom relief (Figure 1).
Dilated cardiomyopathy is a common form of cardiomyopathy, which is characterized by ventricular chamber dilatation and systolic dysfunction (Maron et al., 2006). DCM contributes to hypertension, sudden cardiac death, progressive heart failure, and eventually leads to heart transplant (Wu et al., 2015). Recent studies have shown that cardiomyocytes from DCM patients often exhibit abnormal sarcomeric structure, which is likely due to pathogenic mutations in sarcomere protein-coding genes such as giant sarcomere protein titin (TTN), cardiac troponin T (TNNT2), tropomyosin 1 (TPM1), and desmin (DES; Fürst et al., 1988; Sehnert et al., 2002; Deacon et al., 2019). In recent years, the pathogenic genes of DCM and their pathological mechanism have been studied by establishing hiPSC-based cell models of DCM (Deacon et al., 2019).
Bearing significant roles in cardiac muscle, single TTN proteins extend from the Z-disk to the M-line, spanning the entire half-sarcomere length, and interact with the thick and thin filaments (Fürst et al., 1988). TTN truncating variants (TTNtv) are a major cause of genetic DCM, accounting for approximately 25% of familial cases (Herman et al., 2012). Hinson and his colleagues (Hinson et al., 2015) found that the TTNtv of DCM patients with TTN mutations in clinical practice are markedly enriched in the A-band. In order to understand the mechanism behind this phenotype, they generated patient-specific hiPSC-derived cardiomyocytes (hiPSC-CMs) with different TTN mutations, including two A-band TTNtvs (p.A22352fs or p.P22582fs) and one missense mutation (p.W976R) within the Z/I junction of TTN protein, as well as mutant isogenic hiPSC-CMs carrying CRISPR/Cas9-mediated TTNtvs in the I- or A-band exons. They observed considerable contractile dysfunction in both hiPSC-CM microtissues with I- or A-band TTNtvs, and attributed it to sarcomere insufficiency and aberrant responses to mechanical and adrenergic stress. However, previous studies revealed a tendency for TTN truncations in DCM to distribute to the A-band, while I-band TTNtvs are better tolerated and have been observed in healthy individuals (Herman et al., 2012; Roberts et al., 2015). Basing on RNA splicing analyses, Hinson and his colleagues further observed extraordinarily high rates of I-band exon incorporation into TTN transcripts in hiPSC-CMs (88% of TTN transcripts), five times more than that in adult left ventricle tissue (18% of TTN transcripts; Hinson et al., 2015). Thus, alternative RNA splicing-mediated elimination of I-band exon in adult cardiomyocytes might be a major mechanism for low incidence of I-band TTNtvs in DCM patients.
Cardiac troponin T, encoded by TNNT2, is another cardiac-specific sarcomere protein that involves in sarcomere assembly and cardiomyocyte contraction. Mutations of TNNT2 could cause abnormal calcium ion dynamics and sarcomeric α-actinin, decreased contractility, and affect β-adrenergic signaling of cardiomyocytes. TNNT2 mutation is also the leading cause of heart failure and sudden death in young athletes (Sehnert et al., 2002). By using patient-specific hiPSC model, Sun et al. (2012) demonstrated that hiPSC-CMs carrying TNNT2 p.R173W mutation could partially recapitulate the disease phenotype of DCM, as indicated in altered regulation of calcium ion (Ca2+), decreased contractility, and abnormal distribution of sarcomeric α-actinin. Interestingly, metoprolol, a β1-selective β-blocker showing beneficial effect on the clinical symptoms of DCM patients, could improve the sarcomeric organization of DCM hiPSC-CMs (Sun et al., 2012). On this basis, Wu et al. found that p.R173W mutation could promote the nuclear localization of TNNT2, and alter the epigenetic regulation of key β-adrenergic signaling genes in DCM hiPSC-CMs (Wu et al., 2015). These studies provide novel targets for the treatment of DCM caused by TNNT2 p.R173W mutation.
Bcl2-associated athanogene 3 (BAG3) encodes an anti-apoptotic co-chaperone protein, which is most highly expressed in skeletal and cardiac muscle and localizing to the sarcomeric Z-disk (Homma et al., 2006). Mutations in BAG3 have been associated with DCM and muscular dystrophy (McDermott-Roe et al., 2019). However, heterozygous BAG3 knockout mice failed to recapitulate the clinical phenotypes of heterozygous BAG3 mutation-related cardiac disease (Homma et al., 2006). Two groups have explored the effects of DCM-related BAG3 deficiency in cardiomyocytes, by using genome-editing approach to create both heterozygous and homozygous mutations in BAG3 gene (Judge et al., 2017; McDermott-Roe et al., 2019). Despite the inconsistent observations at baseline, both groups demonstrated more serious fiber disarray in BAG3-deficient hiPSC-CMs under proteasome inhibition-induced proteotoxic stress. Consistently, nonsense mutations in BAG3 locus have been shown to cause BAG3 haploinsufficiency and disengagement of the BAG3-coordinated cardioprotective chaperone complex in hiPSC-CMs (Judge et al., 2017), which might be the pathologic mechanism of DCM-associated BAG3 mutation. McDermott-Roe et al. demonstrated that stimulating the HSF1 (heat shock factor 1)-driven stress response pathway can reduce fiber disorder mediated by proteasome inhibition in hiPSC-CMs with BAG3 p.R477H mutation (McDermott-Roe et al., 2019). This may help clinical treatment of DCM patients carrying BAG3 mutations.
Some other sacromeric genes have also been involved in DCM. Deacon et al. (2019), through the establishment of hiPSC-CMs containing both sarcomeric gene TPM1 mutation (p.E33K) and the costameric gene vinculin mutation (p.N220fs), demonstrated that composite genetic variants can combine and interact to induce DCM, especially when suffering from other disease-causing stressors. An unfamiliar heterozygous mutation of the desmin (DES), A285V mutation, was recently identified in DCM by Tse and colleagues through whole-exome sequencing (WES; Tse et al., 2013). They found that this mutation could cause three-dimensional structure changes of DES and then induce abnormal DES aggregations in hiPSC-CMs. Moreover, the mutation in β-myosin heavy chain 7 (MYH7) gene, which encodes myosin heavy chain (β-MHC), can also induce DCM. Yang et al. (2018) established an hiPSC-CMs line with MYH7 mutation (p.E848G), and demonstrated that the reduced contractile function of single cells and engineered heart tissues with MYH7 mutation (p.E848G) is due to the damaged interaction between MYH7 and cardiac myosin-binding protein C.
In addition to mutations in sarcomere-related genes, mutations in nuclear membrane proteins can also cause DCM. The lamin A/C gene (LMNA) encodes two major nuclear lamina proteins lamins A and C through alternative splicing. Many LMNA mutations, including p.R225X, p.S143P, and p.K117fs, have been identified in familial DCM patients (Siu et al., 2012; Lee et al., 2019; Shah et al., 2019). The most common features of hiPSC-CMs derived from LMNA mutant DCM patients are arrhythmia, abnormal calcium handling, and increased sensitivity to electric and hypoxia stimulation (Shah et al., 2019). Many reports suggest that LMNA mutation-caused cardiac pathology is related to the activation of the PDGF pathway, and the symptoms caused by LMNA mutations can be moderated by the platelet-derived growth factor reactor β (PDGFRB) inhibitors and statins (Siu et al., 2012; Lee et al., 2019; Shah et al., 2019). Siu et al. (2012) demonstrated that under electrical stimulation, hiPSC-CMs carrying R225X or LMNA frameshift mutation present nuclear bleb formation and micronucleation. In the meanwhile, they found that the pro-apoptotic effects of field electric stimulation could be markedly attenuated by pharmacological blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway with U0126 and selumetinib (AZD6244; Siu et al., 2012).
Consistent with clinical disease manifestations, hiPSC-CMs with DCM mutations often exhibit phenotypes such as altered regulation of calcium ion and abnormal myocardial sarcomere structure. The advantage of modeling diseases through hiPSCs is that they cannot only summarize the disease phenotype but also its pathogenesis, which will greatly promote and deepen our understanding of the underlying mechanisms of DCM.
Hypertrophic cardiomyopathy is a disease of the cardiac sarcomere and tend to link to arrhythmia, electrophysiological abnormalities, and sudden cardiac death (Ramachandra et al., 2021). Practically, about 50% of the cases are caused by mutations either in MYH7, encoding β-myosin heavy chain, or in MYBPC3, encoding cardiac myosin-binding protein C (cMyBP-C; Birket et al., 2015), both of which are sarcomeric protein-encoding genes. Recently, researchers have also used hiPSCs to conduct HCM research (Prajapati et al., 2018; Tang et al., 2019; Ramachandra et al., 2021).
The MYH7 is an important part of myosin and is related to contractile capacity of cells (Fontaine et al., 2021; Li et al., 2021). Studies have found that hiPSC-CMs carrying a missense mutation (p.R663H) in MYH7 displayed the phenotype of cellular enlargement, contractile arrhythmia at the single-cell level, and dysregulation of Ca2+ cycling and elevation in intracellular Ca2+ ([Ca2+]i). The main cause of these defects is the continuous increase in [Ca2+]i, which leads to the activation of calcineurin that further dephosphorylates NFAT3 transcription factors, allowing them to translocate to the nucleus and interact directly with classical transcript factors such as GATA4 and MEF2 (Lan et al., 2013). In hiPSC-CMs carrying p.R442G mutation in MYH7, Han et al. (2014) observed sarcomere disorders, indicating the reduction of contractility in HCM hiPSC-CMs and an abnormal Ca2+ handling. Treatment of verapamil, a calcium channel blocker, successfully alleviated calcium handling abnormalities and arrhythmia in HCM hiPSC-CMs with MYH7 p.R663H or p.R442G mutations (Lan et al., 2013; Han et al., 2014). In addition, some Na+ channel blocker such as lidocaine, mexiletine, and ranolazine, but not drugs targeting K+ channels, are also able to restore normal beat frequency in HCM hiPSC-CMs (Lan et al., 2013). These evidences provide some theoretical basis for the treatment of HCM caused by MYH7 mutation.
The mutation of MYBPC3, encoding a thick filament contractile protein that regulates sarcomere organization and cardiac contractility, is another important cause of HCM, which in turn causes damage to the contractile ability of cardiomyocytes and ultimately leads to HCM (Jin et al., 2020). Birket et al. (2015) found that the amount of MyBPC3 protein was reduced by nearly half in MYBPC3-mutant (c.2373dupG) hiPSC-CMs when compared with the normal level. Due to the haploinsufficiency, the maximum Ca2+-activated force markedly decreased. Premature termination codon (PTC) is another common mutation form of MYBPC3 that causes HCM. Seeger et al. found that PTC mutation led to nonsense-mediated decay (NMD) and ubiquitin–proteasome system (UPS), leading to haploinsufficiency of healthy alleles and ultimately to HCM. Inhibition of the NMD pathway in HCM hiPSC-CMs carrying MYBPC3 (p.R943X) mutation resulted in the restoration of molecular phenotypes and calcium-processing kinetics (Seeger et al., 2019). These studies have shown that when dealing with HCM caused by mutations in the MYBPC3 gene, targeting the NMD pathway might be effective.
In addition to sarcomeric genes, mutations in some genes related to cell signaling pathways can also cause HCM. The BRAF protein is a RAF family serine/threonine protein kinase involved in RAS/MAPK signaling. Researchers have found that its mutations can cause pro-fibrotic phenotype, enhanced susceptibility to arrhythmias, aberrant MEK1/2 signaling and sarcomere disorders, and eventually lead to HCM (Cashman et al., 2016; Josowitz et al., 2016; Jaffré et al., 2019). Josowitz et al. used hiPSC-CMs to construct disease models carrying BRAF p.T599R or p.Q257R mutations. They sorted out SIRPα+/CD90– and SIRPα–/CD90+ cardiomyocytes so as to examine hiPSC-derived cell type-specific phenotypes and cellular interactions underpinning HCM. They found that BRAF-mutant SIRPα+/CD90– cardiomyocytes displayed the phenotype of HCM, and BRAF-mutant SIRPα–/CD90+ cells exhibited a pro-fibrotic phenotype and partially modulated cardiomyocyte hypertrophy through transforming growth factor (TGF) paracrine signaling. These symptoms can be relieved by inhibition of TGFβ or RAS/MAPK signaling (Josowitz et al., 2016). Furthermore, hiPSCs from a patient with cardio-facio-cutaneous syndrome were also used to develop the 3D human-engineered cardiac tissue model of BRAF p.T599R mutation-induced HCM. This model reflects an enhanced susceptibility to arrhythmias (Cashman et al., 2016). The RAF1 p.S257L mutation also demonstrated potential to cause cell-autonomous hypertrophy and myofibrillar disarray in hiPSC-CMs, through activation of ERK5 and MEK1/2 signaling, respectively, (Jaffré et al., 2019).
Moreover, researchers have also established disease models through hiPSCs to study HCM caused by mutations in genes such as SCO cytochrome oxidase-deficient homolog 2 (SCO2), CSRP3, which encodes muscle LIM protein, and ACTN2, encoding α-actinin 2 (Hallas et al., 2018; Li et al., 2019; Prondzynski et al., 2019), which are also some common genetic mutations that cause HCM. These mutations can, respectively, result in reduced mitochondrial oxidative ATP production capacity, multinucleation, disorganized sarcomeric ultrastructure, and higher myofilament Ca2+ sensitivity (Hallas et al., 2018; Li et al., 2019; Prondzynski et al., 2019).
Diastolic dysfunction (DD) is commonly diagnosed in HCM patients, and is a major cause of morbidity and mortality among HCM patients. Wu et al. (2019) generated four hiPSC-CM models of DD that carry different mutations in sarcomeric proteins, including MYH7 (p.R663H), MYBPC3 (p.V321M), MYBPC3 (p.V219L), and TNNT2 (p.R92W). These human hiPSC-CM models recapitulated the disease phenotype of DD in HCM, as evidenced by shortened diastolic sarcomere length, decreased maximum relaxation rate, and prolonged relaxation duration. Both diastolic Ca2+ overload and enhanced myofilament Ca2+ sensitivity are involved in impaired diastolic function in HCM hiPSC-CMs, which can be alleviated by treatment with partial Ca2+ or late Na+ current inhibitors (Wu et al., 2019).
On the one hand, the above research shows that hiPSC-CMs with HCM pathogenic genes often have the characteristics of cellular enlargement, contractile arrhythmia, and disorganized sarcomeric, which are consistent with their arrhythmia and electrophysiological abnormalities in diseased organs. On the other, these studies point out the direction for further exploring the possible pathogenesis of HCM and provide a theoretical basis for the rational formulation of clinical protocols. The hiPSC-CM-based HCM studies have shown that abnormal calcium homeostasis is an important factor leading to inherited HCM. The development of drugs that inhibit Ca2+ and Na+ channels may provide a direction for HCM treatment.
Left ventricular non-compaction is caused by an arrest of normal endomyocardial morphogenesis. Patients with LVNC often have symptoms of heart failure, arrhythmias, and thromboembolism. LVNC is mainly related to mutations in genes encoding taffazin (TAZ), lamin A/C (LMNA), sodium channel type Vα (SCN5A), MYH7, MYBPC3, α-cardiac actin 1 (ACTC1), and α-tropomyosin (TPM1). Among them, researchers have established hiPSC-CMs models of ACTC1, TPM1, and TBX20 genes with mutations to study the pathological mechanisms of LVNC.
Among them, ACTC1 and TPM1 are genes encoding sarcomere proteins. To elucidate the pathological role, hiPSCs from a patient carrying a E99K-ACTC1 (α-cardiac actin 1) mutation has been developed and studied (Smith et al., 2018). Smith et al., through the study of two different training methods, 3D and 2D, found that E99K-ACTC1 hiPSC-CMs have obviously arrhythmogenesis in both kinds of models, that is because of the abnormal Ca2+ handling, such as prolonged Ca2+ transients (Smith et al., 2018). Takasaki et al. found that hiPSC-CMs carrying a p.R178H mutation in TPM1 exhibited obvious pathological changes, such as disturbed sarcomere structure and impaired calcium handling in cardiomyocytes. Microarray analysis showed that TPM1 mutations lead to down-regulated expression of many genes involved in heart development and beneficial regulation cellular processes, particularly the calcium signaling pathway (Takasaki et al., 2018).
In addition, researchers have also studied many other mutations associated with LVNC by using patient-specific hiPSC-CMs. LVNC hiPSC-CMs carrying p.T262M mutation and stop-gain mutation p.Y317∗ in TBX20 (T-box transcription factor 20) possess a defect of proliferative capacity due to abnormal activation of TGF-β signaling. Compared with mild DCM hiPSC-CMs, LVNC hiPSC-CMs have a significantly higher expression of TGF-β signaling pathway-related genes, while the expression of PRDM16, the repressor of TGF-β signaling, was significantly down-regulated in all LVNC hiPSC-CMs. These data indicate that the abnormal activation of TGF-β signaling by TBX20 mutation is associated with the proliferation defect of LVNC hiPSC-CMs (Kodo et al., 2016). These studies provide more possibilities for the follow-up investigators’ work and lay the foundation for the clinical treatment of LVNC.
Arrhythmogenic right ventricular is an inherited primary heart muscle disorder characterized by fibrofatty infiltration of the myocardium and cardiomyocyte loss basically in the RV that may lead to sudden cardiac death (Chen et al., 2020; Jahng et al., 2021). More than 50% of the ARVC patients had mutations in the desmosome genes including plakophilin 2 (PKP2), desmoglein 2 (DSG2), desmoplakin, and plakoglobin (Lombardi and Marian, 2010).
Among them, the most common mutations occur in the PKP2 gene (Calkins and Marcus, 2008), which is important for desmosomal protein localization, lipid formation, β-catenin activity, and the activity of Wnt signaling pathway. For this reason, in recent years, researchers have begun to use hiPSCs to construct disease models of ARVC caused by PKP2 mutations. In the beginning, Ma and his colleagues investigated the role of 1841T > C mutation of PKP2 gene in hiPSC-CMs. The PKP2 mutant hiPSC-CMs exhibit reduced localization of desmosomal proteins on the cell surface, enlarged cardiomyocytes, and a more lipid formation. They also found that after exposure to adipogenic differentiation medium for 2 weeks, the lipid content of ARVC-hiPSC-derived cardiomyocytes was significantly higher than that in the control group (Ma et al., 2013). In hiPSC-CMs with the c.2484C > T mutation of PKP2, Kim and his colleagues manifested abnormal nuclear translocation of plakoglobin and decreased β-catenin activity in cardiogenic conditions (Kim et al., 2013). However, these hiPSC-CMs cannot reproduce the pathological phenotypes of ARVC in standard cardiogenic conditions only if adjusting the cultivation conditions to mature them (Kim et al., 2013). Khudiakov et al. (2015) using hiPSC-CMs with two mutations (c.23321del.T, p.K859R) in the PKP2 gene, found that the PKP2 mutations could decrease the activity of Wnt signaling pathway during adipogenic and cardiomyogenic differentiation. To improve this hiPSC-CMs model, Wen et al. developed a method to induce metabolic maturation of hiPSC-CMs. Moreover, they found that coactivation of normal peroxisome proliferator-activated receptor (PPAR)-α and abnormal PPAR-γ pathways could stimulate the pathological signatures such as excessive CM adipogenesis, CM apoptosis, Na+ channel downregulation, and intracellular calcium deficiency of ARVC in hiPSC-CMs carrying two mutations (c.2484C > T, c.2013delC) in PKP2 (Wen et al., 2015). The abovementioned studies indicate that induction of adult-like metabolic energetics is essential to model a cardiac disease with patient-specific hiPSCs.
Besides, Padrón-Barthe et al. demonstrated the genetic mechanism of ARVC5, the most aggressive ARVC subtype, by using the hiPSC-CM model. ARVC5 is a devastating disease that is caused by a point mutation in TMEM43, a transmembrane protein that is located in the nuclear membrane. Their research demonstrated that the hiPSC-CMs with p.S358L mutation in TMEM43 had an increased contraction duration, time to peak, and relaxation time in mutants, and these contraction abnormalities could be relieved by inhibition of glucogen synthase kinase 3 β (Padrón-Barthe et al., 2019). In addition to mutations in desmosomal and nuclear membrane proteins that can cause ARVC, abnormalities in ion channels can also cause ARVC. Cardiomyocytes derived from DSG2 mutant hiPSCs displayed multiple ion channel dysfunction, cellular electrophysiological abnormalities, and increased sensitivity to adrenergic stimulation, which may underlie ARVC patients’ arrhythmias (El-Battrawy et al., 2018). In the above studies, it was found that hiPSC-CMs derived from patient’s somatic cells showed various phenotypes such as enlarged cardiomyocytes, increased lipid formation, and abnormal cell contraction, which are related to the fibrofatty infiltration of the myocardium in ARVC patients. Therefore, patient-specific hiPSC-CMs have been shown to be beneficial in studying the pathogenesis of ARVC.
In addition to the abovementioned cardiomyopathies, researchers have also used iPSCs to study restrictive cardiomyopathy (RCM), a rare heart disease, which is usually caused by increased myocardial stiffness, which results in limited ventricular filling (Galea et al., 2020). Brodehl et al. constructed a disease model with p.Y122H mutation of DSE by using iPSCs and found that the mutant desmin with a severe filament assembly defect supports the pathogenicity of the RCM. By establishing a disease model of this mutation, they first classified the p.Y122H mutation of DSE as a novel pathogenic mutation (Brodehl et al., 2019).
As one of the increasingly serious problems of the 21st century, cardiomyopathy still has many difficulties waiting for us to overcome, for example, the pathogenesis of many genetic diseases has not yet been clarified, and the corresponding treatment options have not been studied. As we described above, hiPSC technology is a powerful means that can be used (Figure 2). Through this technology, researchers have established various disease models of cardiomyopathy, such as HCM, DCM, ARVC, and LVNC, and studied the mechanisms of pathogenesis caused by mutations of different genes. Therefore, hiPSC-based cardiovascular disease models provide more possibilities for the follow-up investigators’ work and will greatly promote and deepen our understanding of the underlying mechanisms of cardiomyopathy. In addition to these 2D disease models, researchers have also constructed 3D organoid models to simulate real human organs and relieve the limitations of spatial factors on disease models (Giacomelli et al., 2020). In addition, iPSC-CMs is also widely used in drug screening (Ito et al., 2020; Li et al., 2020), and iPSC-CM and its derivatives are also used to treat heart failure (Nakamura and Murry, 2019). These all lay the foundation for the clinical treatment of cardiac disease.

Figure 2. The whole process of constructing hiPSC-derived cardiomyocyte (hiPSC-CM) disease model from patients’ somatic cells and the application of these disease models.
However, at present, hiPSC-CMs generally have the problem of immaturity. Mature and immature cells have huge differences in their cell morphology, number of mitochondria, sarcomere structure, calcium transient, and electrophysiology, etc. In addition, the maturation of cardiomyocytes will bring more detailed subtypes, such as ventricular myocardium, atrial myocardium, and sinoatrial node cells, etc. All of these have a significant impact on our research on specific diseases. If the subtypes of differentiated and mature cardiomyocytes can be obtained, we can make more detailed and precise studies on the physiological processes of diseases. Besides, the cellular and molecular crosstalk between cardiac cell lineages is difficult to model by using only hiPSC-derived cardiomyocytes in a two-dimensional model. Therefore, we should use hiPSCs to differentiate a variety of cardiac cells, such as endothelial cells, and construct a three-dimensional model of the cardiac cells to better simulate the overall changes in the heart and the interaction between cells when the disease occurs. In summary, hiPSC technology will not only completely alter our understanding of diseases but also will eventually change the future of medical practice and make personalized medicine possible.
XJ, YC, and XL mainly wrote the manuscript. LY, MY, and ZS revised the manuscript. WL and SH wrote and revised the manuscript, as well as provided the funding. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key R&D Program of China (2017YFA0103700), the National Natural Science Foundation of China (91949111, 81770257, 81970223, 82000263, and 82003756), the Natural Science Foundation of Jiangsu Province (BK20201409 and BK20200880), Top Six Talents in Jiangsu Province (SWYY-081), Jiangsu Province’s Key Discipline/Laboratory of Medicine (XK201118), National Center for International Research (2017B01012), and Introduction Project of Clinical Medicine Expert Team for Suzhou (SZYJTD201704).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Birket, M. J., Ribeiro, M. C., Kosmidis, G., Ward, D., Leitoguinho, A. R., van de Pol, V., et al. (2015). Contractile Defect Caused by Mutation in MYBPC3 Revealed under Conditions Optimized for Human PSC-Cardiomyocyte Function. Cell Rep 13, 733–745. Epub 2015/10/23. doi: 10.1016/j.celrep.2015.09.025,
Brodehl, A., Pour Hakimi, S., Stanasiuk, C., Ratnavadivel, S., Hendig, D., Gaertner, A., et al. (2019). DESRestrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 10:918. doi: 10.3390/genes10110918
Calkins, H., and Marcus, F. (2008). Arrhythmogenic right ventricular cardiomyopathy/dysplasia: an update. Curr. Cardiol. Rep. 10, 367–375. doi: 10.1007/s11886-008-0059-4
Cao, N., Liang, H., Huang, J., Wang, J., Chen, Y., Chen, Z., et al. (2013). Highly efficient induction and long-term maintenance of multipotent cardiovascular progenitors from human pluripotent stem cells under defined conditions. Cell Res. 23, 1119–1132. doi: 10.1038/cr.2013.102
Cashman, T., Josowitz, R., Johnson, B., Gelb, B., and Costa, K. (2016). Human Engineered Cardiac Tissues Created Using Induced Pluripotent Stem Cells Reveal Functional Characteristics of BRAF-Mediated Hypertrophic Cardiomyopathy. PloS One 11:e0146697. doi: 10.1371/journal.pone.0146697
Chen, P., Xiao, Y., Wang, Y., Zheng, Z., Chen, L., Yang, X., et al. (2020). OBSCNIntracellular calcium current disorder and disease phenotype in mutant iPSC-based cardiomyocytes in arrhythmogenic right ventricular cardiomyopathy. Theranostics 10, 11215–11229. doi: 10.7150/thno.45172
Deacon, D., Happe, C., Chen, C., Tedeschi, N., Manso, A., Li, T., et al. (2019). Combinatorial interactions of genetic variants in human cardiomyopathy. Nat. Biomed. Eng. 3, 147–157. doi: 10.1038/s41551-019-0348-9
El-Battrawy, I., Zhao, Z., Lan, H., Cyganek, L., Tombers, C., Li, X., et al. (2018). Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. Europace 20, f46–f56. doi: 10.1093/europace/euy042
Fatkin, D., and Graham, R. (2002). Molecular mechanisms of inherited cardiomyopathies. Physiol. Rev. 82, 945–980. doi: 10.1152/physrev.00012.2002
Fontaine, V., Duboscq-Bidot, L., Jouve, C., Hamlin, M., Curjol, A., Briand, V., et al. (2021). Generation of iPSC line from MYH7 R403L mutation carrier with severe hypertrophic cardiomyopathy and isogenic CRISPR/Cas9 corrected control. Stem Cell Res. 52:102245. doi: 10.1016/j.scr.2021.102245
Fürst, D., Osborn, M., Nave, R., and Weber, K. (1988). The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J. Cell. Biol. 106, 1563–1572. doi: 10.1083/jcb.106.5.1563
Galea, N., Polizzi, G., Gatti, M., Cundari, G., Figuera, M., and Faletti, R. (2020). Cardiovascular magnetic resonance (CMR) in restrictive cardiomyopathies. Radiol. Med. 125, 1072–1086. doi: 10.1007/s11547-020-01287-8
Giacomelli, E., Meraviglia, V., Campostrini, G., Cochrane, A., Cao, X., van Helden, R., et al. (2020). Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 26, 862-879.e11.
Hallas, T., Eisen, B., Shemer, Y., Ben Jehuda, R., Mekies, L., Naor, S., et al. (2018). Investigating the cardiac pathology of SCO2-mediated hypertrophic cardiomyopathy using patients induced pluripotent stem cell-derived cardiomyocytes. J. cell. Mol. Med. 22, 913–925. doi: 10.1111/jcmm.13392
Han, L., Li, Y., Tchao, J., Kaplan, A. D., Lin, B., Li, Y., et al. (2014). Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 104, 258–269Epub 2014/09/12. doi: 10.1093/cvr/cvu205
Herman, D., Lam, L., Taylor, M., Wang, L., Teekakirikul, P., Christodoulou, D., et al. (2012). Truncations of titin causing dilated cardiomyopathy. New Eng. J. Med. 366, 619–628. doi: 10.1056/NEJMoa1110186
Hinson, J., Chopra, A., Nafissi, N., Polacheck, W., Benson, C., Swist, S., et al. (2015). HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986. doi: 10.1126/science.aaa5458
Homma, S., Iwasaki, M., Shelton, G., Engvall, E., Reed, J., and Takayama, S. (2006). BAG3 deficiency results in fulminant myopathy and early lethality. Am. J. Pathol. 169, 761–773. doi: 10.2353/ajpath.2006.060250
Ito, M., Nomura, S., Morita, H., and Komuro, I. (2020). Trends and Limitations in the Assessment of the Contractile Properties of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes From Patients With Dilated Cardiomyopathy. Front. Cardiovasc. Med. 7:154. doi: 10.3389/fcvm.2020.00154
Jaffré, F., Miller, C., Schänzer, A., Evans, T., Roberts, A., Hahn, A., et al. (2019). Inducible Pluripotent Stem Cell-Derived Cardiomyocytes Reveal Aberrant Extracellular Regulated Kinase 5 and Mitogen-Activated Protein Kinase Kinase 1/2 Signaling Concomitantly Promote Hypertrophic Cardiomyopathy in RAF1-Associated Noonan Syndrome. Circulation 140, 207–224. doi: 10.1161/circulationaha.118.037227
Jahng, J., Black, K., Liu, L., Bae, H., Perez, M., Ashley, E., et al. (2021). Generation of three induced pluripotent stem cell lines, SCVIi003-A, SCVIi004-A, SCVIi005-A, from patients with ARVD/C caused by heterozygous mutations in the PKP2 gene. Stem Cell Res. 53:102284. doi: 10.1016/j.scr.2021.102284
Jin, J., Lu, L., Chen, J., Wang, K., Han, J., Xue, S., et al. (2020). Generation of an induced pluripotential stem cell (iPSC) line from a patient with hypertrophic cardiomyopathy carrying myosin binding protein C (MYBPC3) c.3369-3370 insC mutation. New Eng. J Med. 50:102144. doi: 10.1016/j.scr.2020.102144
Josowitz, R., Mulero-Navarro, S., Rodriguez, N. A., Falce, C., Cohen, N., Ullian, E. M., et al. (2016). Autonomous and Non-autonomous Defects Underlie Hypertrophic Cardiomyopathy in BRAF-Mutant hiPSC-Derived Cardiomyocytes. Stem Cell Reports 7, 355–369. Epub 2016/08/30. doi: 10.1016/j.stemcr.2016.07.018,
Judge, L., Perez-Bermejo, J., Truong, A., Ribeiro, A., Yoo, J., Jensen, C., et al. (2017). A BAG3 chaperone complex maintains cardiomyocyte function during proteotoxic stress. JCI Insight 2:e94623. doi: 10.1172/jci.insight.94623
Kamp, T. J., and Lyons, G. E. (2009). On the road to iPS cell cardiovascular applications. Circ. Res. 105, 617–619. Epub 2009/10/03. doi: 10.1161/CIRCRESAHA.109.205740,
Khudiakov, A., Kostina, D., Kostareva, A., Tomilin, A., and Malashicheva, A. (2015). The effect of plakophilin-2 gene mutations on activity of the canonical wnt signaling pathway. Tsitologiia 57, 868–875.
Kim, C., Wong, J., Wen, J., Wang, S., Wang, C., Spiering, S., et al. (2013). Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494, 105–110. doi: 10.1038/nature11799
Kim, J., Lee, H., and Carroll, D. (2010). Genome editing with modularly assembled zinc-finger nucleases. Nat. Methods 7:91. doi: 10.1038/nmeth0210-91a
Kodo, K., Ong, S., Jahanbani, F., Termglinchan, V., Hirono, K., InanlooRahatloo, K., et al. (2016). iPSC-derived cardiomyocytes reveal abnormal TGF-β signalling in left ventricular non-compaction cardiomyopathy. Nat. Cell. Biol. 18, 1031–1042. doi: 10.1038/ncb3411
Lan, F., Lee, A. S., Liang, P., Sanchez-Freire, V., Nguyen, P. K., Wang, L., et al. (2013). Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 12, 101–113.
Lee, J., Termglinchan, V., Diecke, S., Itzhaki, I., Lam, C., Garg, P., et al. (2019). Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 572, 335–340. doi: 10.1038/s41586-019-1406-x
Li, H., Huang, M., Wang, J., and Chen, D. A. (2020). method for establishing the electrophysiological model of alcoholic cardiomyopathy. Zhong Nan Da Xue Xue Bao Yi Xue Ban 45, 386–394. doi: 10.11817/j.issn.1672-7347.2020.190086
Li, X., Fu, W., Guo, G., Liu, M., Du, W., Zhao, J., et al. (2021). A heterozygous MYH7 (c. 2156G > A) mutant human induced pluripotent stem cell line (ZZUNEUi020-A) generated from a patient with hypertrophic cardiomyopathy. Stem Cell Res. 51:102158. doi: 10.1016/j.scr.2021.102158
Li, X., Lu, W., Li, Y., Wu, F., Bai, R., Ma, S., et al. (2019). MLP-deficient human pluripotent stem cell derived cardiomyocytes develop hypertrophic cardiomyopathy and heart failure phenotypes due to abnormal calcium handling. Cell. death Dis. 10:610. doi: 10.1038/s41419-019-1826-4
Lombardi, R., and Marian, A. (2010). Arrhythmogenic right ventricular cardiomyopathy is a disease of cardiac stem cells. Curr. opinion Cardiol. 25, 222–228. doi: 10.1097/HCO.0b013e3283376daf
Ma, D., Wei, H., Lu, J., Ho, S., Zhang, G., Sun, X., et al. (2013). Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 34, 1122–1133. doi: 10.1093/eurheartj/ehs226
Maron, B., Towbin, J., Thiene, G., Antzelevitch, C., Corrado, D., Arnett, D., et al. (2006). Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 113, 1807–1816. doi: 10.1161/circulationaha.106.174287
McDermott-Roe, C., Lv, W., Maximova, T., Wada, S., Bukowy, J., Marquez, M., et al. (2019). Investigation of a dilated cardiomyopathy-associated variant in BAG3 using genome-edited iPSC-derived cardiomyocytes. JCI Insight 4:e128799. doi: 10.1172/jci.insight.128799
Nakamura, K., and Murry, C. (2019). Function Follows Form–A Review of Cardiac Cell Therapy. Circ. J. 83, 2399–2412. doi: 10.1253/circj.CJ-19-0567
Padrón-Barthe, L., Villalba-Orero, M., Gómez-Salinero, J., Domínguez, F., Román, M., Larrasa-Alonso, J., et al. (2019). Severe Cardiac Dysfunction and Death Caused by Arrhythmogenic Right Ventricular Cardiomyopathy Type 5 Are Improved by Inhibition of Glycogen Synthase Kinase-3β. Circulation 140, 1188–1204. doi: 10.1161/circulationaha.119.040366
Prajapati, C., Ojala, M., and Aalto-Setala, K. (2018). Divergent effects of adrenaline in human induced pluripotent stem cell-derived cardiomyocytes obtained from hypertrophic cardiomyopathy. Dis Model Mech 11:dmm032896. doi: 10.1242/dmm.032896
Prondzynski, M., Lemoine, M., Zech, A., Horváth, A., Di Mauro, V., Koivumäki, J., et al. (2019). Disease modeling of a mutation in α-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med 11:e11115. doi: 10.15252/emmm.201911115
Qyang, Y., Martin-Puig, S., Chiravuri, M., Chen, S., Xu, H., Bu, L., et al. (2007). The renewal and differentiation of Isl1+ cardiovascular progenitors are controlled by a Wnt/beta-catenin pathway. Cell Stem Cell 1, 165–179. doi: 10.1016/j.stem.2007.05.018
Ramachandra, C., Kp, M., Chua, J., Hernandez-Resendiz, S., Liehn, E., Gan, L., et al. (2021). Inhibiting cardiac myeloperoxidase alleviates the relaxation defect in hypertrophic cardiomyocytes. Cardiovasc. Res. 2021. cvab077 doi: 10.1093/cvr/cvab077
Roberts, A., Ware, J., Herman, D., Schafer, S., Baksi, J., Bick, A., et al. (2015). Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 7:270ra6. doi: 10.1126/scitranslmed.3010134
Savla, J., Nelson, B., Perry, C., and Adler, E. (2014). Induced pluripotent stem cells for the study of cardiovascular disease. J. Am. Coll. Cardiol. 64, 512–519. doi: 10.1016/j.jacc.2014.05.038
Schmid-Burgk, J., Schmidt, T., Kaiser, V., Höning, K., and Hornung, V. A. (2013). ligation-independent cloning technique for high-throughput assembly of transcription activator–like effector genes. Nat. Biotechnol. 31, 76–81. doi: 10.1038/nbt.2460
Seeger, T., Shrestha, R., Lam, C., Chen, C., McKeithan, W., Lau, E., et al. (2019). A Premature Termination Codon Mutation in MYBPC3 Causes Hypertrophic Cardiomyopathy via Chronic Activation of Nonsense-Mediated Decay. Circulation 139, 799–811. doi: 10.1161/circulationaha.118.034624
Sehnert, A., Huq, A., Weinstein, B., Walker, C., Fishman, M., and Stainier, D. (2002). Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat. Gene. 31, 106–110. doi: 10.1038/ng875
Shah, D., Virtanen, L., Prajapati, C., Kiamehr, M., Gullmets, J., West, G., et al. (2019). Modeling of LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cells. Cells 8:594. doi: 10.3390/cells8060594
Siu, C., Lee, Y., Ho, J., Lai, W., Chan, Y., Ng, K., et al. (2012). Modeling of lamin A/C mutation premature cardiac aging using patient−specific induced pluripotent stem cells. Aging 4, 803–822. doi: 10.18632/aging.100503
Smith, J., Owen, T., Bhagwan, J., Mosqueira, D., Scott, E., Mannhardt, I., et al. (2018). Isogenic Pairs of hiPSC-CMs with Hypertrophic Cardiomyopathy/LVNC-Associated ACTC1 E99K Mutation Unveil Differential Functional Deficits. Stem. Cell. Rep. 11, 1226–1243. doi: 10.1016/j.stemcr.2018.10.006
Sun, N., Yazawa, M., Liu, J., Han, L., Sanchez-Freire, V., Abilez, O., et al. (2012). Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 4:130ra47. doi: 10.1126/scitranslmed.3003552
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Takasaki, A., Hirono, K., Hata, Y., Wang, C., Takeda, M., Yamashita, J., et al. (2018). Sarcomere gene variants act as a genetic trigger underlying the development of left ventricular noncompaction. Pediatric. Res. 84, 733–742. doi: 10.1038/s41390-018-0162-1
Tang, L., Yao, F., Wang, H., Wang, X., Shen, J., Dai, B., et al. (2019). Inhibition of TRPC1 prevents cardiac hypertrophy via NF-κB signaling pathway in human pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 126, 143–154. doi: 10.1016/j.yjmcc.2018.10.020
Tse, H. F., Ho, J. C., Choi, S. W., Lee, Y. K., Butler, A. W., Ng, K. M., et al. (2013). Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum. Mol. Genet. 22, 1395–1403.
Wen, J., Wei, C., Shah, K., Wong, J., Wang, C., and Chen, H. (2015). Maturation-Based Model of Arrhythmogenic Right Ventricular Dysplasia Using Patient-Specific Induced Pluripotent Stem Cells. Circ. J. 79, 1402–1408. doi: 10.1253/circj.CJ-15-0363
Wu, H., Lee, J., Vincent, L., Wang, Q., Gu, M., Lan, F., et al. (2015). Epigenetic Regulation of Phosphodiesterases 2A and 3A Underlies Compromised β-Adrenergic Signaling in an iPSC Model of Dilated Cardiomyopathy. Cell Stem Cell 17, 89–100. doi: 10.1016/j.stem.2015.04.020
Wu, H., Yang, H., Rhee, J., Zhang, J., Lam, C., Sallam, K., et al. (2019). Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur. Heart J. 40, 3685–3695. doi: 10.1093/eurheartj/ehz326
Wu, X., Scott, D., Kriz, A., Chiu, A., Hsu, P., Dadon, D., et al. (2014). Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 32, 670–676. doi: 10.1038/nbt.2889
Keywords: induced pluripotent stem cell, inherited cardiomyopathy, disease modeling, heart disease, pathogenic mechanism
Citation: Jiang X, Chen Y, Liu X, Ye L, Yu M, Shen Z, Lei W and Hu S (2021) Uncovering Inherited Cardiomyopathy With Human Induced Pluripotent Stem Cells. Front. Cell Dev. Biol. 9:672039. doi: 10.3389/fcell.2021.672039
Received: 25 February 2021; Accepted: 06 April 2021;
Published: 17 May 2021.
Edited by:
Wei Jiang, Wuhan University, ChinaReviewed by:
Sang-Ging Ong, University of Illinois at Chicago, United StatesCopyright © 2021 Jiang, Chen, Liu, Ye, Yu, Shen, Lei and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shijun Hu, c2hpanVuaHVAc3VkYS5lZHUuY24=; Wei Lei, bGVpd2VpQHN1ZGEuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.