 Shan Qiu
Shan Qiu Guixing Jiang1†
Guixing Jiang1† Jun Huang
Jun Huang- 1Department of General Surgery, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2The MOE Key Laboratory of Biosystems Homeostasis and Protection, Zhejiang Provincial Key Laboratory for Cancer Molecular Cell Biology and Innovation Center for Cell Signaling Network, Life Sciences Institute, Zhejiang University, Hangzhou, China
- 3Zhejiang University-University of Edinburgh Institute, Zhejiang University School of Medicine, Zhejiang University, Haining, China
During genome replication, replication forks often encounter obstacles that impede their progression. Arrested forks are unstable structures that can give rise to collapse and rearrange if they are not properly processed and restarted. Replication fork reversal is a critical protective mechanism in higher eukaryotic cells in response to replication stress, in which forks reverse their direction to form a Holliday junction-like structure. The reversed replication forks are protected from nuclease degradation by DNA damage repair proteins, such as BRCA1, BRCA2, and RAD51. Some of these molecules work cooperatively, while others have unique functions. Once the stress is resolved, the replication forks can restart with the help of enzymes, including human RECQ1 helicase, but restart will not be considered here. Here, we review research on the key factors and mechanisms required for the remodeling and protection of stalled replication forks in mammalian cells.
Introduction
Faithful DNA replication during each cell cycle is essential for maintaining genome stability (Jeggo et al., 2016). However, the DNA replication process is frequently challenged by endogenous and exogenous sources of genotoxic stress, including DNA lesions, difficult to replicate sequences, and nucleotide depletion (Mehta and Haber, 2014; Kitao et al., 2018). These challenges, if not properly addressed, would ultimately cause genome instability, a hallmark of tumorigenesis (Jackson and Bartek, 2009; Ou and Schumacher, 2018). Fortunately, organisms have evolved multiple DNA damage repair pathways and DNA damage tolerance (DDT) mechanisms to maintain genome stability (Friedberg, 2005; Huen and Chen, 2010; Branzei and Psakhye, 2016).
DNA damage tolerance refers to the bypassing of DNA lesions and replication restart after the replication fork stalls (Friedberg, 2005). One mode of DDT is replication fork reversal. Proposed in 1976, replication fork reversal was long regarded as a pathological result of fork destabilization, but has now been accepted as a DDT based on recent observations of reversed fork structures in vivo and the identification of molecules involved in fork regression in vitro (Sakaguchi et al., 2009; Bermejo et al., 2011; Neelsen and Lopes, 2015; Berti et al., 2020a). Emerging evidence suggests that replication fork reversal is indispensable for maintaining genome stability in higher eukaryotic cells. For example, it actively slows down replication fork progression via multiple enzymes, such as the recombinase RAD51 and DNA translocase helicase-like transcription factor (HLTF), which provides sufficient time for the DNA repair machinery to become involved and prevent double-strand break (DSB) formation (Poole and Cortez, 2017; Tye et al., 2020). Replication fork reversal also triggers template switching, where the nascent strand is used for error-free DNA synthesis (Zellweger et al., 2015). However, reversal can render replication forks susceptible to nucleolytic attack (Liao et al., 2018; Rickman and Smogorzewska, 2019). Recent studies have explored factors that can protect reversed forks against nuclease processing, like BRCA1, BRCA2, and components of the Fanconi anemia (FA) complex (Rickman and Smogorzewska, 2019; Tye et al., 2020).
This review focuses on the process of replication fork reversal, especially the enzymes, and molecules involved. First, changes in the replication fork structure after damage blockage, and the factors that promote fork regression, are summarized. The review then explores several mechanisms that protect the reversed fork structure. We hope that this review will provide comprehensive insight into replication fork reversal, thereby contributing to future therapies for diseases like cancers.
A Two-Step Mechanism for Replication Fork Reversal
In response to replication perturbation, the DNA fork structure changes depending on the type of damage. If a lesion occurs on the lagging strand, it will likely be bypassed because the semi-discontinuous characteristics of DNA replication allow the lagging strand to leave a single strand DNA (ssDNA) gap to be repaired afterward (McInerney and O’Donnell, 2004). However, if a lesion occurs on the leading strand, the fork structure will be altered. In this case, synthesis of the leading strand is inhibited at the blockage point due to polymerase dissociation (also called fork uncoupling), while the helicase continues to generate ssDNA for hundreds of bases (Atkinson and McGlynn, 2009; Berti et al., 2020a). Thus, stalling the synthesis of the leading strand results in an accumulation of ssDNA; this provides a platform for loading multiple enzymes, thereby promoting fork remodeling (Kolinjivadi et al., 2017).
PCNA Polyubiquitination and Fork Slowing
Proliferation cell nuclear antigen (PCNA) is a highly conserved homotrimer that serves as a DNA clamp and is crucial for DNA replication and associated processes (Boehm et al., 2016; Lee and Park, 2020). It is a critical regulator of DDT, in which PCNA monoubiquitination at lysine 164 (PCNA-Ub) facilitates error-prone translesion DNA synthesis and PCNA polyubiquitination (PCNA-Ubn) promotes error-free damage bypass (Sale, 2013; Branzei and Szakal, 2017). In yeast, PCNA-Ubn is mediated by E3 ubiquitin ligase Rad5, while in mammalian cells it is mediated by the Rad5 orthologs HLTF and SNF2 histone linker PHD RING helicase (SHPRH; Unk et al., 2010). Surprisingly, PCNA-Ubn occurs in Hltf/Shprh double-deficient mouse embryonic fibroblasts (Krijger et al., 2011). Therefore, another E3 ligase must contribute to PCNA-Ubn in mammalian cells. A recent in vitro study found that the HECT-type E3 ligase HECW2 interacted with PCNA and regulated its ubiquitination; its role in DDT needs further study (Krishnamoorthy et al., 2018). Strikingly, a recent study demonstrated that K63-linked, UBC13-dependent PCNA-Ubn is required to slow and reverse replication forks in response to replication stress (Vujanovic et al., 2017).
Critical Enzymes in Fork Slowing and Reversal
Emerging evidence suggests that active replication fork slowing upon genotoxic stress is linked to replication fork reversal, which is at least partly regulated by SNF2 family chromatin remodelers, including SMARCAL1 (SWI/SNF-related, matrix-associated, actin-dependent, regulator of chromatin, and subfamily A-like 1), ZRANB3 (zinc finger, RAN-binding domain containing 3), and HLTF (Poole and Cortez, 2017; Figure 1). Mutations in SMARCAL1 lead to Schimke immuno-osseous dysplasia (SIOD), while HLTF/ZRANB3-deficient cells are vulnerable to replication stress and contribute to tumorigenesis (Ciccia et al., 2009; Li et al., 2009; Weston et al., 2012; Helmer et al., 2019). Therefore, these helicase-like proteins play critical roles in DDT, and use energy from ATP hydrolysis to remodel chromatin structure (Hargreaves and Crabtree, 2011). They are recruited to the stalled replication forks by interactions with other proteins, like RPA or PCNA, and then bind DNA sequences via substrate-recognition domains. All three of these DNA translocases can catalyze replication fork regression both in vitro and in vivo, and have specific, distinct functions in fork remodeling (Blastyak et al., 2010; Achar et al., 2011; Betous et al., 2012).
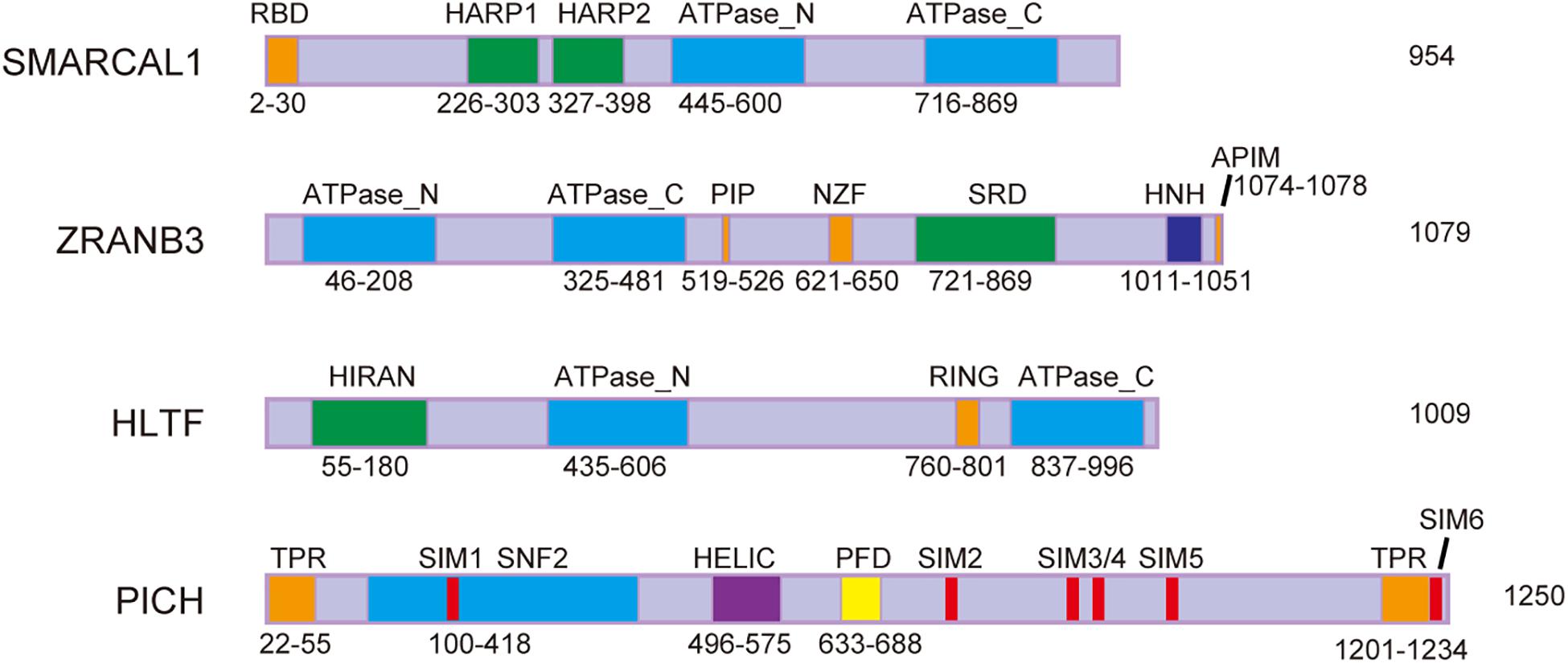
Figure 1. Schematic representation of protein domains of SMARCAL1, ZRANB3, HLTF, and PICH. RBD, RPA-binding domain; HARP, HepA-related protein; PIP, PCNA-interacting protein box; NZF, Npl4 zinc-finger; SRD, substrate recognition domain; HNH, His-Asn-His protein; APIM, AlkB homolog 2 PCNA interacting motif; HIRAN, HIP116 and RAD5 N-terminal; RING, really interesting new gene; TPR, tetratricopeptide repeat; SIM, SUMO-interacting motif; SNF2, sucrose non-fermenting 2; HELIC, helicase superfamily c-terminal domain; and PFD, PICH family domain.
SWI/SNF-related, matrix-associated, actin-dependent, regulator of chromatin, and subfamily A-like 1 is an annealing helicase that contains a replication protein A (RPA) binding domain. RPA, a eukaryotic ssDNA-binding protein that regulates various DNA metabolic processes, is required for SMARCAL1 localization to stalled forks (Ciccia et al., 2009; Yuan et al., 2009; Byrne and Oakley, 2019). SMARCAL1 interacts with RPA and catalyzes replication fork regression, which is regulated by the ATM and Rad3-related (ATR) protein kinase (Couch et al., 2013; Bhat and Cortez, 2018). While RPA stimulates SMARCAL1 fork reversal activity when it is bound to a ssDNA gap on the leading template strand, it inhibits SMARCAL1 when bound to a replication fork with a ssDNA gap on the lagging strand (Betous et al., 2013).
Zinc finger, RAN-binding domain containing 3 contains a PCNA-interacting protein box and an AIkB homology 2 PCNA interaction motif (APIM) to bind PCNA, which facilitates its localization to stalled forks (Ciccia et al., 2012; Weston et al., 2012; Yuan et al., 2012). Moreover, its NPL4 zinc-finger motif preferentially interacts with K63-linked polyubiquitinated PCNA and is also required for the localization of ZRANB3 at stalled replication forks (Vujanovic et al., 2017). Because of its homologous sequence, ZRANB3 has functions similar to SMARCAL1, including annealing complementary DNA strands and catalyzing fork reversal. Unlike SMARCAL1, however, RPA inhibits the fork reversal ability of ZRANB3 on the leading-strand gaps substrates (Betous et al., 2013). Moreover, unlike other SNF2 family proteins, ZRANB3 exhibits structure-specific ATP-dependent endonuclease activity and can cleave fork DNA structures (Weston et al., 2012). Exactly how these enzymatic activities work together at stalled replication forks remains unknown.
Similar to SMARCAL1 and ZRANB3, HLTF can catalyze fork reversal via ATP hydrolysis. HLTF binds the leading strand via its N-terminal HIRAN domain to stimulate fork regression (Achar et al., 2015; Kile et al., 2015). In addition, it has been reported that HLTF partly counteracts the activity of the DNA helicase FANCJ at stalled forks to maintain fork remodeling and prevent unlimited replication (Peng et al., 2018). Unlike the other two DNA translocases, no protein interaction motifs have been discovered in HLTF, and how it is recruited to stalled forks requires further investigation. Although a study has demonstrated that RPA and Pax transactivation domain-interacting protein interacts with HLTF, future research should examine their roles in replication stress (MacKay et al., 2009). Since simultaneously depletion of SMARCAL1, ZRANB3, and HLTF did not show an additive effect on reversed fork frequency, these three DNA translocases may function at different stages of a common pathway (Taglialatela et al., 2017; Tian et al., 2021). It is also possible that each translocase works preferentially on specific substrates or genomic regions, which need further investigation (Taglialatela et al., 2017; Tian et al., 2021).
In addition to the SNF2 family proteins, it has been reported that RAD51 is required for replication fork regression. RAD51 is a highly conserved DNA recombinase that facilitates DNA DSB repair in vertebrates by promoting homologous recombination repair (Gachechiladze et al., 2017; Laurini et al., 2020; Sinha et al., 2020). A nascent chromatin capture screening study detected RAD51 on the replication forks (Alabert et al., 2014). Unlike homologous recombination repair, RAD51 has a non-canonical function in fork reversal, since BRCA2-modulated stable RAD51 filaments are not needed in this process (Bhat and Cortez, 2018). Although the mechanisms are not clear, it has been suggested that RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3) may assist RAD51 and DNA translocases in promoting replication fork reversal (Berti et al., 2020b). The loaders and specific role of RAD51 in fork reversal warrant further investigation.
Other enzymes have also been reported to participate in the reversal of replication forks. For example, the branch point translocase FANCM (Fanconi anemia complementation group M) could convert a replication fork from a three-way junction to a four-way junction in an ATP-dependent manner (Gari et al., 2008). Moreover, a study showed that FBH1 (F-box DNA helicase 1) was recruited to the stalled forks and could unwind the lagging strands (Masuda-Ozawa et al., 2013). A more recent study demonstrated that the helicase activity of FBH1 was involved in replication fork regression, which was also dependent on ATP hydrolysis (Fugger et al., 2015). Although many related enzymes and molecules have been discovered, it is not clear whether these proteins work together to promote fork remodeling, or if they work independently in response to different replication obstacles. It will be necessary to explore the interactions among these enzymes in the future.
Although the above enzymes play significant roles in replication fork remodeling, they must be tightly regulated as too little or too much of their activities at stalled forks is deleterious for genomic stability. For example, ATR phosphorylates SMARCAL1 at Ser652 to limit its fork regression activity, thereby preventing replication fork collapse (Couch et al., 2013). Apart from ATR, RAD52 also limits SMARCAL1 activity at stalled forks by counteracting its loading (Malacaria et al., 2019). Moreover, the RPA-like single-strand DNA binding protein RADX antagonizes RAD51 filament formation to prevent inappropriate replication fork reversal (Dungrawala et al., 2017; Schubert et al., 2017; Zhang et al., 2020; Adolph et al., 2021).
The ZATT-TOP2A-PICH Axis and Extensive Replication Fork Reversal
Extrusion of the leading and lagging strands from the template DNA during replication fork reversal, catalyzed by the above enzymes, would cause positive superhelical strain in the newly synthesized sister chromatids (Tian et al., 2021). The resulting superhelical strain prevents further regression of the stalled replication forks and must be dissipated by DNA topoisomerases for reversal to proceed efficiently (Tian et al., 2021). Our recent study found that DNA topoisomerase 2 (mainly TOP2A) can release the superhelical strain in newly synthesized chromatids generated by the DNA translocases SMARCAL1, ZRANB3, and HLTF during limited fork reversal (Tian et al., 2021; Figure 2). Our study also showed that, with replication stress, TOP2A is SUMOylated by the SUMO E3 ligase ZATT, mainly at lysines 1228 and 1240. SUMOylated TOP2A then recruits the SUMO-targeted DNA translocase PICH to stalled replication forks, where PICH branch migrates the Holliday junction structures and drives extensive replication fork reversal (Tian et al., 2021; Figure 2). Based on these findings, we proposed that replication fork reversal has two distinct stages, namely initiation and extension stages (Tian et al., 2021; Figure 2). Like SMARCAL1, ZRANB3, and HLTF, PICH is also a member of the SNF2 family (Figure 1). However, in contrast to SMARCAL1, ZRANB3, and HLTF, PICH possesses branch migration activity but not fork regression activity, indicating that PICH is specifically involved in the extension stage of replication fork reversal.
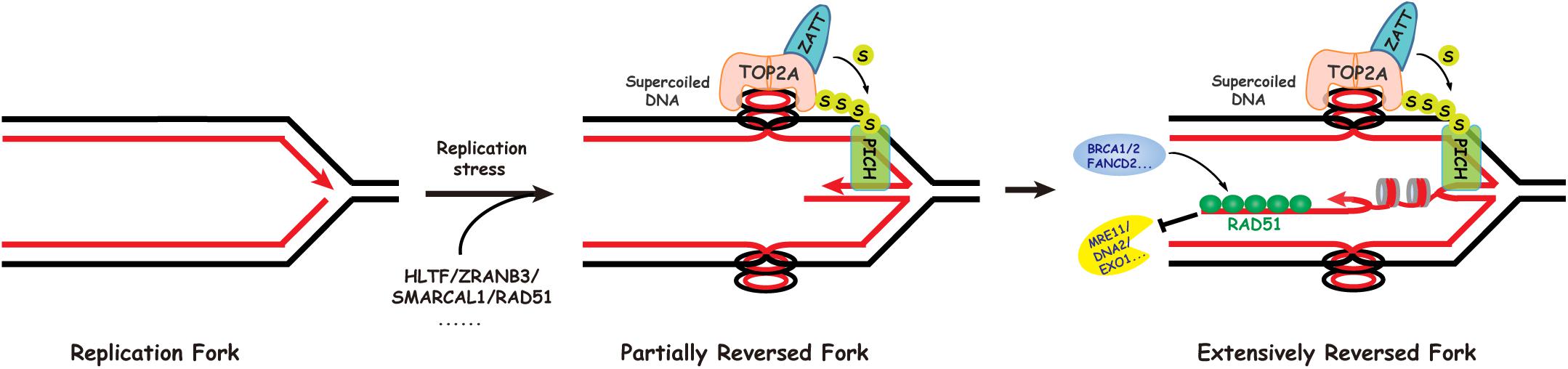
Figure 2. Replication fork reversal occurs via a two-step mechanism. In the first step, SMARCAL1, HLTF, and ZRANB3 cooperate with RAD51 to initiate limited replication fork reversal, generating positive superhelical strain in the newly replicated sister chromatids. The initial fork reversal may be helped by the positive supercoiling ahead of the replication fork created during replication. In the second step, DNA topoisomerase IIalpha (TOP2A) promotes extensive fork reversal, on one hand through resolving the resulting topological barriers, and on the other hand via its role in recruiting the SUMO-targeted DNA translocase PICH to stalled replication forks.
Mechanisms for Replication Fork Maintenance and Stability
Under replication stress, the replication fork reverses to form a four-way Holliday junction structure, as discussed above. However, the nascent strands in this structure resemble a one-ended DNA DSB, which is susceptible to nucleases such as MRE11, EXO1 (exonuclease1), DNA2 (DNA replication helicase/nuclease 2), and MUS81 (Thangavel et al., 2015; Lemacon et al., 2017; Mijic et al., 2017). To prevent excessive degradation at stalled forks, the nucleolytic activity of these enzymes has to be regulated accurately. Recent studies have identified several protective mechanisms that maintain replication fork structure and confer genomic stability.
BRCA1/2 and RAD51
BRCA1/2-mediated stable RAD51 filament formation is required for its protective effect on the regressed arm (Carreira and Kowalczykowski, 2011; Schlacher et al., 2012). Consistent with this, wild-type RAD51, but not its DNA-binding mutant RAD51T131P, stably associates with reversed forks and protects them from Mre11-mediated degradation (Kolinjivadi et al., 2017; Mijic et al., 2017). In addition, inhibition of RAD51 DNA-binding and strand exchange activities by the small molecule B02 destabilizes reversed forks, without causing the fork reversal defects observed upon RAD51 depletion (Taglialatela et al., 2017). Moreover, WRNIP1, a member of the AAA + ATPase family, interacts with the BRCA2/RAD51 complex and participates in the stabilization of RAD51 filaments from degradation by MRE11 (Leuzzi et al., 2016). These findings suggest that RAD51 has both a BRCA1/2-independent fork remodeling function and a BRCA1/2-dependent fork-protecting role. However, it is still unclear exactly how RAD51 protects regressed forks from nuclease-mediated degradation. Physical blocking of nuclease binding, or cooperation with other inhibitory proteins, are putative mechanisms. Furthermore, the RAD51 paralogs also participate in replication fork protection against MRE11 over-resection (Somyajit et al., 2015). Whether RAD51 paralogs dampen nucleases via the same mechanism as the BRCA1/2-RAD51 interaction requires further study.
FA Components
Fanconi anemia is a rare inherited disorder that results from mutations in FA genes, which play key roles in DNA replication and repair (Alter, 2014). The FA core complex is an ubiquitin ligase that detects DNA damage and monoubiquitinates the downstream proteins FANCD2 and FANCI to regulate DNA repair of inter-strand crosslinks (ICL) and homologous recombination repair (Nepal et al., 2017; Liu et al., 2020). In addition to its canonical role in ICL repair, several FA proteins stabilize stalled forks. For example, the FA component FANCD2 prevents MRE11-mediated fork over-processing by stabilizing RAD51 nucleofilaments, similarly to BRCA2 (Schlacher et al., 2012; Kim et al., 2015). Interestingly, a recent study demonstrated that the novel protein BOD1L could also protect stalled forks from genome fragility (Higgs et al., 2015). Being downstream of FANCD2/BRCA2, BOD1L maintained fork stability by inhibiting BLM/FBH1 helicases and stabilizing RAD51 nucleoprotein filaments (Higgs et al., 2015). However, unlike FANCD2, BOD1L suppressed DNA2-mediated degradation rather than MRE11-dependent instability (Higgs et al., 2015). It may seem unintuitive that both FANCD2 and BOD1L stabilize RAD51 at sites of replication damage, but they prevent different types of nucleolytic attack. Future research is required to reveal the precise mechanism underlying RAD51 stabilization.
RecQ Family of DNA Helicases
The RecQ family of DNA helicases, including RECQL1/4/5, WRN (Werner syndrome protein), and BLM (Bloom’s syndrome helicase), have been shown to be important for maintaining genome integrity (Croteau et al., 2014). These proteins are conserved from bacteria to humans, and mutations therein lead to diseases such as Werner syndrome and Bloom syndrome, as well as premature aging, and cancer proneness (Mojumdar, 2020). Since Werner and Bloom syndromes are both characterized by chromosome fragility and increased cancer predisposition, many studies have investigated whether the Bloom syndrome helicase BLM and Werner syndrome helicase WRN play roles in protecting stalled replication forks.
A previous study found that WRN helicase and exonuclease catalytic activities were needed to prevent MUS81-mediated breakage after HU-induced replication fork stalling (Murfuni et al., 2012). However, that study did not reveal how the different enzymatic activities of WRN collaborate at stalled forks. A more recent finding suggested that WRN exonuclease prevented MRE11/EXO1-dependent over-resection at nascent strands, while its helicase ensured the necessary exonucleolytic processing (Iannascoli et al., 2015). A non-enzymatic function of WRN was also reported (Su et al., 2014). The authors found that WRN could limit MRE11 exonuclease activity and prevent excessive degradation on nascent strands, possibly by stabilizing RAD51 (Su et al., 2014).
Bloom’s syndrome helicase, another RecQ helicase, has also been implicated in replication fork protection upon replication stress. It was reported that BLM and FANCD2 co-localized at stalled forks in response to replication fork stalling agents (Pichierri et al., 2004). Moreover, the FA pathway was shown to be essential for BLM phosphorylation and assembly in nuclear foci in response to DNA interstrand crosslinking agents (Pichierri et al., 2004). Surprisingly, a recent study found that BLM helicase activity was also indispensable for FANCM recruitment and function at stalled forks (Ling et al., 2016). Therefore, it is reasonable to hypothesize that BLM and the FA pathway form a positive feedback loop to ensure sufficient protection of the stalled forks.
Other proteins, such as ABRO1, PALB2, and WRNIP, have also been implicated in stalled replication fork protection (Murphy et al., 2014; Liao et al., 2018; Bennett et al., 2020; Berti et al., 2020a). However, it is not clear how these factors interact in this process, or how they function in response to different replication obstacles.
Concluding Remarks
Recent studies have raised many questions about fork remodeling caused by replication stress. Although there are various well-established models of fork reversal and remodeling, some questions remain unanswered. For example, on what basis do cells choose one or several of these mechanisms upon encountering a DNA lesion? How do cells recognize and respond to different DNA lesions? How do factors with similar functions work in non-redundant ways? If helicase and polymerase are dissociated during fork reversal, how is the replisome reloaded onto the replication fork when the fork is restarted? Are other factors vital in the balance between fork reversal and restart? We believe that recent progress in our understanding of fork plasticity under genotoxic stress will spark interest in addressing these questions and clarifying the mechanistic link between fork remodeling and genomic instability. In turn, this should lead to a better understanding of the mechanisms underlying replication and the dynamic relationships among the involved processes, thereby leading to more efficient cancer therapies.
Author Contributions
SQ and GJ wrote the manuscript. JH and LC reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (31730021, 31971220, and 31961160725), National Key Research and Development Program of China (2018YFC2000100), and the China’s Fundamental Research Funds for the Central Universities.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to colleagues whose work could not be cited because of space limitations. We thank all our colleagues in the Huang laboratory for helpful discussions.
References
Achar, Y. J., Balogh, D., and Haracska, L. (2011). Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc. Natl. Acad. Sci. U.S.A. 108, 14073–14078. doi: 10.1073/pnas.1101951108
Achar, Y. J., Balogh, D., Neculai, D., Juhasz, S., Morocz, M., Gali, H., et al. (2015). Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res. 43, 10277–10291. doi: 10.1093/nar/gkv896
Adolph, M. B., Mohamed, T. M., Balakrishnan, S., Xue, C., Morati, F., Modesti, M., et al. (2021). RADX controls RAD51 filament dynamics to regulate replication fork stability. Mol. Cell 81, 1074 e1075–1083 e1075. doi: 10.1016/j.molcel.2020.12.036
Alabert, C., Bukowski-Wills, J. C., Lee, S. B., Kustatscher, G., Nakamura, K., de Lima Alves, F., et al. (2014). Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 16, 281–293. doi: 10.1038/ncb2918
Alter, B. P. (2014). Fanconi anemia and the development of leukemia. Best Practice Res. Clin. Haematol. 27, 214–221. doi: 10.1016/j.beha.2014.10.002
Atkinson, J., and McGlynn, P. (2009). Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 37, 3475–3492. doi: 10.1093/nar/gkp244
Bennett, L. G., Wilkie, A. M., Antonopoulou, E., Ceppi, I., Sanchez, A., Vernon, E. G., et al. (2020). MRNIP is a replication fork protection factor. Sci. Adv. 6:eaba5974. doi: 10.1126/sciadv.aba5974
Bermejo, R., Capra, T., Jossen, R., Colosio, A., Frattini, C., Carotenuto, W., et al. (2011). The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell 146, 233–246. doi: 10.1016/j.cell.2011.06.033
Berti, M., Cortez, D., and Lopes, M. (2020a). The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 21, 633–651. doi: 10.1038/s41580-020-0257-5
Berti, M., Teloni, F., Mijic, S., Ursich, S., Fuchs, J., Palumbieri, M. D., et al. (2020b). Sequential role of RAD51 paralog complexes in replication fork remodeling and restart. Nat. Commun. 11:3531. doi: 10.1038/s41467-020-17324-z
Betous, R., Couch, F. B., Mason, A. C., Eichman, B. F., Manosas, M., and Cortez, D. (2013). Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 3, 1958–1969. doi: 10.1016/j.celrep.2013.05.002
Betous, R., Mason, A. C., Rambo, R. P., Bansbach, C. E., Badu-Nkansah, A., Sirbu, B. M., et al. (2012). SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 26, 151–162. doi: 10.1101/gad.178459.111
Bhat, K. P., and Cortez, D. (2018). RPA and RAD51: fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 25, 446–453. doi: 10.1038/s41594-018-0075-z
Blastyak, A., Hajdu, I., Unk, I., and Haracska, L. (2010). Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol. Cell. Biol. 30, 684–693. doi: 10.1128/MCB.00863-09
Boehm, E. M., Gildenberg, M. S., and Washington, M. T. (2016). The many roles of PCNA in Eukaryotic DNA replication. Enzymes 39, 231–254. doi: 10.1016/bs.enz.2016.03.003
Branzei, D., and Psakhye, I. (2016). DNA damage tolerance. Curr. Opin. Cell. Biol. 40, 137–144. doi: 10.1016/j.ceb.2016.03.015
Branzei, D., and Szakal, B. (2017). Building up and breaking down: mechanisms controlling recombination during replication. Crit. Rev. Biochem. Mol. Biol. 52, 381–394. doi: 10.1080/10409238.2017.1304355
Byrne, B. M., and Oakley, G. G. (2019). Replication protein A, the laxative that keeps DNA regular: the importance of RPA phosphorylation in maintaining genome stability. Semin. Cell. Dev. Biol. 86, 112–120. doi: 10.1016/j.semcdb.2018.04.005
Carreira, A., and Kowalczykowski, S. C. (2011). Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc. Natl. Acad. Sci. U.S.A. 108, 10448–10453. doi: 10.1073/pnas.1106971108
Ciccia, A., Bredemeyer, A. L., Sowa, M. E., Terret, M. E., Jallepalli, P. V., Harper, J. W., et al. (2009). The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 23, 2415–2425. doi: 10.1101/gad.1832309
Ciccia, A., Nimonkar, A. V., Hu, Y., Hajdu, I., Achar, Y. J., Izhar, L., et al. (2012). Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell. 47, 396–409. doi: 10.1016/j.molcel.2012.05.024
Couch, F. B., Bansbach, C. E., Driscoll, R., Luzwick, J. W., Glick, G. G., Betous, R., et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 27, 1610–1623. doi: 10.1101/gad.214080.113
Croteau, D. L., Popuri, V., Opresko, P. L., and Bohr, V. A. (2014). Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 83, 519–552. doi: 10.1146/annurev-biochem-060713-035428
Dungrawala, H., Bhat, K. P., Le Meur, R., Chazin, W. J., Ding, X., Sharan, S. K., et al. (2017). RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol. Cell. 67:e375. doi: 10.1016/j.molcel.2017.06.023
Friedberg, E. C. (2005). Suffering in silence: the tolerance of DNA damage. Nat. Rev. Mol. Cell. Biol. 6, 943–953. doi: 10.1038/nrm1781
Fugger, K., Mistrik, M., Neelsen, K. J., Yao, Q., Zellweger, R., Kousholt, A. N., et al. (2015). FBH1 catalyzes regression of stalled replication forks. Cell Rep. 10, 1749–1757. doi: 10.1016/j.celrep.2015.02.028
Gachechiladze, M., Skarda, J., Soltermann, A., and Joerger, M. (2017). RAD51 as a potential surrogate marker for DNA repair capacity in solid malignancies. Int. J. Cancer 141, 1286–1294. doi: 10.1002/ijc.30764
Gari, K., Decaillet, C., Delannoy, M., Wu, L., and Constantinou, A. (2008). Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl. Acad. Sci. U.S.A. 105, 16107–16112. doi: 10.1073/pnas.0804777105
Hargreaves, D. C., and Crabtree, G. R. (2011). ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420. doi: 10.1038/cr.2011.32
Helmer, R. A., Kaur, G., Smith, L. A., and Chilton, B. S. (2019). Helicase-like transcription factor (Hltf) gene-deletion promotes oxidative phosphorylation (OXPHOS) in colorectal tumors of AOM/DSS-treated mice. PLoS One 14:e0221751. doi: 10.1371/journal.pone.0221751
Higgs, M. R., Reynolds, J. J., Winczura, A., Blackford, A. N., Borel, V., Miller, E. S., et al. (2015). BOD1L is required to suppress deleterious resection of stressed replication forks. Mol. Cell. 59, 462–477. doi: 10.1016/j.molcel.2015.06.007
Huen, M. S., and Chen, J. (2010). Assembly of checkpoint and repair machineries at DNA damage sites. Trends Biochem. Sci. 35, 101–108. doi: 10.1016/j.tibs.2009.09.001
Iannascoli, C., Palermo, V., Murfuni, I., Franchitto, A., and Pichierri, P. (2015). The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 43, 9788–9803. doi: 10.1093/nar/gkv836
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Jeggo, P. A., Pearl, L. H., and Carr, A. M. (2016). DNA repair, genome stability and cancer: a historical perspective. Nat. Rev. Cancer 16, 35–42. doi: 10.1038/nrc.2015.4
Kile, A. C., Chavez, D. A., Bacal, J., Eldirany, S., Korzhnev, D. M., Bezsonova, I., et al. (2015). HLTF’s Ancient HIRAN domain binds 3’ DNA ends to drive replication fork reversal. Mol. Cell 58, 1090–1100. doi: 10.1016/j.molcel.2015.05.013
Kim, T. M., Son, M. Y., Dodds, S., Hu, L., Luo, G., and Hasty, P. (2015). RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res. 43, 893–903. doi: 10.1093/nar/gku1334
Kitao, H., Iimori, M., Kataoka, Y., Wakasa, T., Tokunaga, E., Saeki, H., et al. (2018). DNA replication stress and cancer chemotherapy. Cancer Sci. 109, 264–271. doi: 10.1111/cas.13455
Kolinjivadi, A. M., Sannino, V., De Antoni, A., Zadorozhny, K., Kilkenny, M., Techer, H., et al. (2017). Smarcal1-mediated fork reversal triggers Mre11-Dependent degradation of nascent DNA in the absence of Brca2 and Stable Rad51 nucleofilaments. Mol. Cell. 67:e867. doi: 10.1016/j.molcel.2017.07.001
Krijger, P. H., Lee, K. Y., Wit, N., van den Berk, P. C., Wu, X., Roest, H. P., et al. (2011). HLTF and SHPRH are not essential for PCNA polyubiquitination, survival and somatic hypermutation: existence of an alternative E3 ligase. DNA Repair (Amst) 10, 438–444. doi: 10.1016/j.dnarep.2010.12.008
Krishnamoorthy, V., Khanna, R., and Parnaik, V. K. (2018). E3 ubiquitin ligase HECW2 targets PCNA and lamin B1. Biochim. Biophys. Acta Mol. Cell. Res. 1865, 1088–1104. doi: 10.1016/j.bbamcr.2018.05.008
Laurini, E., Marson, D., Fermeglia, A., Aulic, S., Fermeglia, M., and Pricl, S. (2020). Role of Rad51 and DNA repair in cancer: a molecular perspective. Pharmacol. Ther. 208:107492. doi: 10.1016/j.pharmthera.2020.107492
Lee, K. Y., and Park, S. H. (2020). Eukaryotic clamp loaders and unloaders in the maintenance of genome stability. Exp. Mol. Med. 52, 1948–1958. doi: 10.1038/s12276-020-00533-3
Lemacon, D., Jackson, J., Quinet, A., Brickner, J. R., Li, S., Yazinski, S., et al. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 8:860. doi: 10.1038/s41467-017-01180-5
Leuzzi, G., Marabitti, V., Pichierri, P., and Franchitto, A. (2016). WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J. 35, 1437–1451. doi: 10.15252/embj.201593265
Li, Y., Bolderson, E., Kumar, R., Muniandy, P. A., Xue, Y., Richard, D. J., et al. (2009). HSSB1 and hSSB2 form similar multiprotein complexes that participate in DNA damage response. J. Biol. Chem. 284, 23525–23531. doi: 10.1074/jbc.C109.039586
Liao, H., Ji, F., Helleday, T., and Ying, S. (2018). Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 19:e46263. doi: 10.15252/embr.201846263
Ling, C., Huang, J., Yan, Z., Li, Y., Ohzeki, M., Ishiai, M., et al. (2016). Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov. 2:16047. doi: 10.1038/celldisc.2016.47
Liu, W., Palovcak, A., Li, F., Zafar, A., Yuan, F., and Zhang, Y. (2020). Fanconi anemia pathway as a prospective target for cancer intervention. Cell. Biosci. 10:39. doi: 10.1186/s13578-020-00401-7
MacKay, C., Toth, R., and Rouse, J. (2009). Biochemical characterisation of the SWI/SNF family member HLTF. Biochem. Biophys. Res. Commun. 390, 187–191. doi: 10.1016/j.bbrc.2009.08.151
Malacaria, E., Pugliese, G. M., Honda, M., Marabitti, V., Aiello, F. A., Spies, M., et al. (2019). Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat. Commun. 10:1412. doi: 10.1038/s41467-019-09196-9
Masuda-Ozawa, T., Hoang, T., Seo, Y. S., Chen, L. F., and Spies, M. (2013). Single-molecule sorting reveals how ubiquitylation affects substrate recognition and activities of FBH1 helicase. Nucleic Acids Res. 41, 3576–3587. doi: 10.1093/nar/gkt056
McInerney, P., and O’Donnell, M. (2004). Functional uncoupling of twin polymerases: mechanism of polymerase dissociation from a lagging-strand block. J. Biol. Chem. 279, 21543–21551. doi: 10.1074/jbc.M401649200
Mehta, A., and Haber, J. E. (2014). Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect Biol. 6:a016428. doi: 10.1101/cshperspect.a016428
Mijic, S., Zellweger, R., Chappidi, N., Berti, M., Jacobs, K., Mutreja, K., et al. (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 8:859. doi: 10.1038/s41467-017-01164-5
Mojumdar, A. (2020). Mutations in conserved functional domains of human RecQ helicases are associated with diseases and cancer: a review. Biophys. Chem. 265:106433. doi: 10.1016/j.bpc.2020.106433
Murfuni, I., De Santis, A., Federico, M., Bignami, M., Pichierri, P., and Franchitto, A. (2012). Perturbed replication induced genome wide or at common fragile sites is differently managed in the absence of WRN. Carcinogenesis 33, 1655–1663. doi: 10.1093/carcin/bgs206
Murphy, A. K., Fitzgerald, M., Ro, T., Kim, J. H., Rabinowitsch, A. I., Chowdhury, D., et al. (2014). Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J. Cell Biol. 206, 493–507. doi: 10.1083/jcb.201404111
Neelsen, K. J., and Lopes, M. (2015). Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell. Biol. 16, 207–220. doi: 10.1038/nrm3935
Nepal, M., Che, R., Zhang, J., Ma, C., and Fei, P. (2017). Fanconi anemia signaling and cancer. Trends Cancer 3, 840–856. doi: 10.1016/j.trecan.2017.10.005
Ou, H. L., and Schumacher, B. (2018). DNA damage responses and p53 in the aging process. Blood 131, 488–495. doi: 10.1182/blood-2017-07-746396
Peng, M., Cong, K., Panzarino, N. J., Nayak, S., Calvo, J., Deng, B., et al. (2018). Opposing roles of FANCJ and HLTF protect forks and restrain replication during stress. Cell Rep. 24, 3251–3261. doi: 10.1016/j.celrep.2018.08.065
Pichierri, P., Franchitto, A., and Rosselli, F. (2004). BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 23, 3154–3163. doi: 10.1038/sj.emboj.7600277
Poole, L. A., and Cortez, D. (2017). Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 52, 696–714. doi: 10.1080/10409238.2017.1380597
Rickman, K., and Smogorzewska, A. (2019). Advances in understanding DNA processing and protection at stalled replication forks. J. Cell. Biol. 218, 1096–1107. doi: 10.1083/jcb.201809012
Sakaguchi, K., Ishibashi, T., Uchiyama, Y., and Iwabata, K. (2009). The multi-replication protein A (RPA) system–a new perspective. FEBS J. 276, 943–963. doi: 10.1111/j.1742-4658.2008.06841.x
Sale, J. E. (2013). Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect Biol. 5:a012708. doi: 10.1101/cshperspect.a012708
Schlacher, K., Wu, H., and Jasin, M. (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. doi: 10.1016/j.ccr.2012.05.015
Schubert, L., Ho, T., Hoffmann, S., Haahr, P., Guerillon, C., and Mailand, N. (2017). RADX interacts with single-stranded DNA to promote replication fork stability. EMBO Rep. 18, 1991–2003. doi: 10.15252/embr.201744877
Sinha, A., Saleh, A., Endersby, R., Yuan, S. H., Chokshi, C. R., Brown, K. R., et al. (2020). RAD51-Mediated DNA homologous recombination is independent of PTEN mutational status. Cancers (Basel) 12:3178. doi: 10.3390/cancers12113178
Somyajit, K., Saxena, S., Babu, S., Mishra, A., and Nagaraju, G. (2015). Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res. 43, 9835–9855. doi: 10.1093/nar/gkv880
Su, F., Mukherjee, S., Yang, Y., Mori, E., Bhattacharya, S., Kobayashi, J., et al. (2014). Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep. 9, 1387–1401. doi: 10.1016/j.celrep.2014.10.025
Taglialatela, A., Alvarez, S., Leuzzi, G., Sannino, V., Ranjha, L., Huang, J. W., et al. (2017). Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell 68:e418. doi: 10.1016/j.molcel.2017.09.036
Thangavel, S., Berti, M., Levikova, M., Pinto, C., Gomathinayagam, S., Vujanovic, M., et al. (2015). DNA2 drives processing and restart of reversed replication forks in human cells. J. Cell Biol. 208, 545–562. doi: 10.1083/jcb.201406100
Tian, T., Bu, M., Chen, X., Ding, L., Yang, Y., Han, J., et al. (2021). The ZATT-TOP2A-PICH Axis Drives Extensive Replication Fork Reversal to Promote Genome Stability. Mol. Cell. 81, 198 e196–211 e196. doi: 10.1016/j.molcel.2020.11.007
Tye, S., Ronson, G. E., and Morris, J. R. (2020). A fork in the road: where homologous recombination and stalled replication fork protection part ways. Semin. Cell. Dev. Biol. [Online ahead of print] S1084–S9521. doi: 10.1016/j.semcdb.2020.07.004
Unk, I., Hajdu, I., Blastyak, A., and Haracska, L. (2010). Role of yeast Rad5 and its human orthologs, HLTF and SHPRH in DNA damage tolerance. DNA Repair (Amst) 9, 257–267. doi: 10.1016/j.dnarep.2009.12.013
Vujanovic, M., Krietsch, J., Raso, M. C., Terraneo, N., Zellweger, R., Schmid, J. A., et al. (2017). Replication fork slowing and reversal upon DNA damage require PCNA Polyubiquitination and ZRANB3 DNA translocase activity. Mol. Cell. 67, 882–890e885. doi: 10.1016/j.molcel.2017.08.010
Weston, R., Peeters, H., and Ahel, D. (2012). ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev. 26, 1558–1572. doi: 10.1101/gad.193516.112
Yuan, J., Ghosal, G., and Chen, J. (2009). The annealing helicase HARP protects stalled replication forks. Genes Dev. 23, 2394–2399. doi: 10.1101/gad.1836409
Yuan, J., Ghosal, G., and Chen, J. (2012). The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol. Cell 47, 410–421. doi: 10.1016/j.molcel.2012.05.025
Zellweger, R., Dalcher, D., Mutreja, K., Berti, M., Schmid, J. A., Herrador, R., et al. (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell. Biol. 208, 563–579. doi: 10.1083/jcb.201406099
Keywords: replication stress, replication fork stalling, genome instability, replication fork reversal, DNA translocase
Citation: Qiu S, Jiang G, Cao L and Huang J (2021) Replication Fork Reversal and Protection. Front. Cell Dev. Biol. 9:670392. doi: 10.3389/fcell.2021.670392
Received: 21 February 2021; Accepted: 19 April 2021;
Published: 10 May 2021.
Edited by:
Huiqiang Lou, China Agricultural University, ChinaReviewed by:
Ian Grainge, The University of Newcastle, AustraliaPietro Pichierri, National Institute of Health (ISS), Italy
Copyright © 2021 Qiu, Jiang, Cao and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liping Cao, Y2FvbGlwaW5nemp1QHpqdS5lZHUuY24=; Jun Huang, amh1YW5nQHpqdS5lZHUuY24=
†These authors have contributed equally to this study