 Marcelo Santos da Silva
Marcelo Santos da Silva
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 15 April 2021
Sec. Cell Growth and Division
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.669041
This article is part of the Research TopicNuclear Genome Stability: DNA Replication, Telomere Maintenance, and DNA RepairView all 21 articles
For nearly all eukaryotic cells, stochastic DNA double-strand breaks (DSBs) are one of the most deleterious types of DNA lesions. DSB processing and repair can cause sequence deletions, loss of heterozygosity, and chromosome rearrangements resulting in cell death or carcinogenesis. However, trypanosomatids (single-celled eukaryotes parasites) do not seem to follow this premise strictly. Several studies have shown that trypanosomatids depend on DSBs to perform several events of paramount importance during their life cycle. For Trypanosoma brucei, DSBs formation is associated with host immune evasion via antigenic variation. In Trypanosoma cruzi, DSBs play a crucial role in the genetic exchange, a mechanism that is still little explored but appear to be of fundamental importance for generating variability. In Leishmania spp., DSBs are necessary to generate genomic changes by gene copy number variation (CNVs), events that are essential for these organisms to overcome inhospitable conditions. As DSB repair in trypanosomatids is primarily conducted via homologous recombination (HR), most of the events associated with DSBs are HR-dependent. This review will discuss the latest findings on how trypanosomatids balance the benefits and inexorable challenges caused by DSBs.
DNA, the storage center of all genetic information of an organism, is continually assaulted by endogenous and exogenous sources of instability, resulting in a variety of possible injuries. Of these lesions, DNA double-strand breaks (DSBs) are the most threatening. If left unrepaired, DSBs drive genomic instability leading to cell death, and if repaired incorrectly, DSBs can drastically alter the genomic structure, for example, generating chromosomal translocations and rearrangements, both of which contribute to tumorigenesis in metazoans (Kaye et al., 2004; Cannan and Pederson, 2016; Zhao et al., 2020).
In general, endogenous DSBs can arise from metabolic reactions or DNA stressors. For instance, endogenous DSBs can arise during the attempted repair of oxidized DNA bases when they occur simultaneously on opposing strands (Yang et al., 2004; Cannan et al., 2014); or during DNA replication when the replication machinery encounters natural impediments that lead to pausing or blocking of the replication fork (Mirkin and Mirkin, 2007; García-Muse and Aguilera, 2016; da Silva et al., 2019); or during the processing of spontaneous single-stranded DNA breaks (SSBs) generated in the S-phase (Vilenchik and Knudson, 2003; Saleh-Gohari et al., 2005; Elango et al., 2017). Exogenous DSBs are generated predominantly by chemical mutagens or ionizing radiation (Cannan and Pederson, 2016; Carofiglio et al., 2018). Chemical mutagens usually include anticancer chemotherapeutic drugs, such as cross-linking agents (e.g., cisplatin), and radiomimetic compounds (e.g., phleomycin) (Chen and Stubbe, 2005; Wyrobek et al., 2005; Jekimovs et al., 2014). Ionizing radiation (IR) is a source of DSBs, but also SSBs following the production of radiolysis radicals that attack the sugar-phosphate backbone (Ward, 1994; Ma et al., 2012). In short, DSBs are often terminal lesions induced by a wide range of genotoxic conditions that, if unresolved, underpin genomic instability in eukaryotic cells.
DNA double-strand breaks have likely exerted pressure throughout eukaryotic evolution, selecting organisms that had developed a network of pathways and factors capable of efficiently dealing with this lesion (Xu and Price, 2011). The diversity of DNA repair pathways that exist and their conservation across the Eukarya domain support this hypothesis. Among conserved DNA repair pathways able to deal with DSBs are homologous recombination (HR), which requires the presence of a DNA template homologous to the damaged region, and the error-prone non-homologous end joining (NHEJ) pathway, which joins the DNA double-stranded ends in the absence of a homologous sequence (Farlow et al., 2011; Zhao et al., 2020).
Trypanosomatids (supergroup Excavata) have most of their DNA repair pathways conserved. However, notable divergencies exist suggesting a parasite-specific repurposing of the DSBs repair machinery (Glover et al., 2013; Ubeda et al., 2014; Alves et al., 2018; Mehnert et al., 2021). While the HR repair pathway is conserved and functional (McCulloch and Barry, 1999; Glover et al., 2008; Hartley and McCulloch, 2008; Genois et al., 2012; Alves et al., 2018; Marin et al., 2018), canonical NHEJ activities appear absent in trypanosomatids (Burton et al., 2007; Nenarokova et al., 2019). Instead, alternative NHEJ (Alt-NHEJ) pathways (e.g., microhomology-mediated end joining – MMEJ) and single-strand annealing (SSA) predominate to repair chromosomal DSBs in some trypanosomatid species (Glover et al., 2008; Peng et al., 2015; Rose et al., 2020). Several species of trypanosomatids are obligate parasites and can cause human diseases of great medical importance, including Trypanosoma brucei (T. brucei), Trypanosoma cruzi (T. cruzi), and Leishmania spp. These pathogens present a dixenous life cycle, i.e., perform stages of their life cycle in invertebrate and vertebrate hosts (Barratt et al., 2017). To survive and replicate inside their hosts, these organisms must overcome several barriers, including host defense mechanisms and unfavorable environmental conditions (Geiger et al., 2016). Intriguingly, some trypanosomatids can bypass these barriers using recombination events (Beverley et al., 1984; Myler et al., 1984b; Downing et al., 2011; Alves et al., 2018), which in many organisms, can be triggered following a DSB and its subsequent repair (Baudat and Nicolas, 1997; Pâques and Haber, 1999; Pfeiffer et al., 2000; Kuzminov, 2011). Trypanosomatids exploit their DSBs repair pathways and use them to their advantage to survive within a host. Thus, a fine-tuned balance must exist in these organisms to both facilitate the action of pathways DSBs-related and prevent repair machinery from being overwhelmed, which would compromise organism fitness.
In this review, I will discuss this paradoxical effect by which DSBs can act as opportunities for fundamental survival and adaptation mechanisms or as sources of genome instability in trypanosomatids.
Trypanosoma brucei parasites cause debilitating and life-threatening conditions in mammals, including the African trypanosomiasis in humans and nagana in livestock. These infections persist due to the parasite’s ability to undergo antigenic variation. For T. brucei, this involves the stochastic switching of variant surface glycoproteins (VSGs), which hinders recognition and eradication mediated by the host immune system (Barry and McCulloch, 2001; Horn, 2014; Pinger et al., 2017; Ridewood et al., 2017).
Although the precise number of VSG genes that can encode a coat is unknown, around 2500 VSG-encoding genes have been cataloged in the nuclear genomes of T. brucei (Berriman et al., 2005; Cross et al., 2014; Müller et al., 2018). VSGs are located in the subtelomeric regions of the 11 diploid megabase chromosomes and also in the ∼ 100 mini and intermediate chromosomes (Wickstead et al., 2004; Marcello and Barry, 2007). Only one VSG is expressed at one time (i.e., expression is monoallelic) with the active VSG being transcribed from one of ∼15 dedicated telomere-proximal bloodstream expression sites (BESs) distributed among the 11 megabase chromosomes (Hertz-Fowler et al., 2008). Moreover, the activated BES is transcribed exclusively by RNA polymerase I (pol I) from an extranucleolar focus known as Expression Site Body (ESB) (Navarro and Gull, 2001). Recent studies have been demonstrating the strictness of the VSG expression control and how the ESB structure is important for active BES transcription (Kerry et al., 2017; Budzak et al., 2019; Faria et al., 2021).
In general, there are two main mechanisms by which a VSG gene can be switched (Myler et al.,1984a,b). The first one is called transcriptional switching and is characterized by alternating the subtelomeric region containing the ES that is being transcribed (Bernards et al., 1984; Myler et al.,1984a,b). The second involves different types of recombination events to perform VSG gene replacement, i.e., VSG genes can be shuttled from other locations in the chromosomes into an active ES (Myler et al., 1984b; Scholler et al., 1989; McCulloch et al., 1997; Conway et al., 2002; Hall et al., 2013). The VSG switching by recombination events predominates over transcriptional switching because it is possible to have access to the entire VSG repertoire through this mechanism (Robinson et al., 1999; Hovel-Miner et al., 2012). On the other hand, although frequently observed (Bernards et al., 1984; Myler et al., 1984a; Bitter et al., 1998; Rudenko et al., 1998; Barry and McCulloch, 2001), transcriptional switches allow access to only ∼15 VSGs housed in the ES. Recombination is also essential in the segmental gene conversion for generating antigenic diversity, producing a “VSGs mosaic” during chronic infections (Hall et al., 2013; Mugnier et al., 2015).
At least two commonalities between recombination-based VSG switching and DSBs repair by HR strongly support that DSBs are catalysts for switch events (Figure 1A). First, the recombination-based VSG switching can be directly activated by the induction of a DSB in the active BES using the meganuclease I-SceI (Boothroyd et al., 2009; Glover et al., 2013). Second, disruption of the HR pathway through the interruption of some components, such as ATR (Stortz et al., 2017; Black et al., 2020; Marin et al., 2020), Rad51 (McCulloch and Barry, 1999; Proudfoot and McCulloch, 2005), Rad50 (Mehnert et al., 2021), and BRCA2 (Hartley and McCulloch, 2008), impairs the VSG switching by recombination, suggesting that multiple components are shared between these two pathways. In general, these features mirror targeted gene rearrangements in other organisms, such as VAR genes diversity in Plasmodium (Kyes et al., 2007; Claessens et al., 2014), pilin antigenic variation in Neisseria (Cahoon and Seifert, 2011), and V(D)J recombination during the development of B lymphocytes of the vertebrate immune system (Tonegawa, 1983; Brecht et al., 2020).
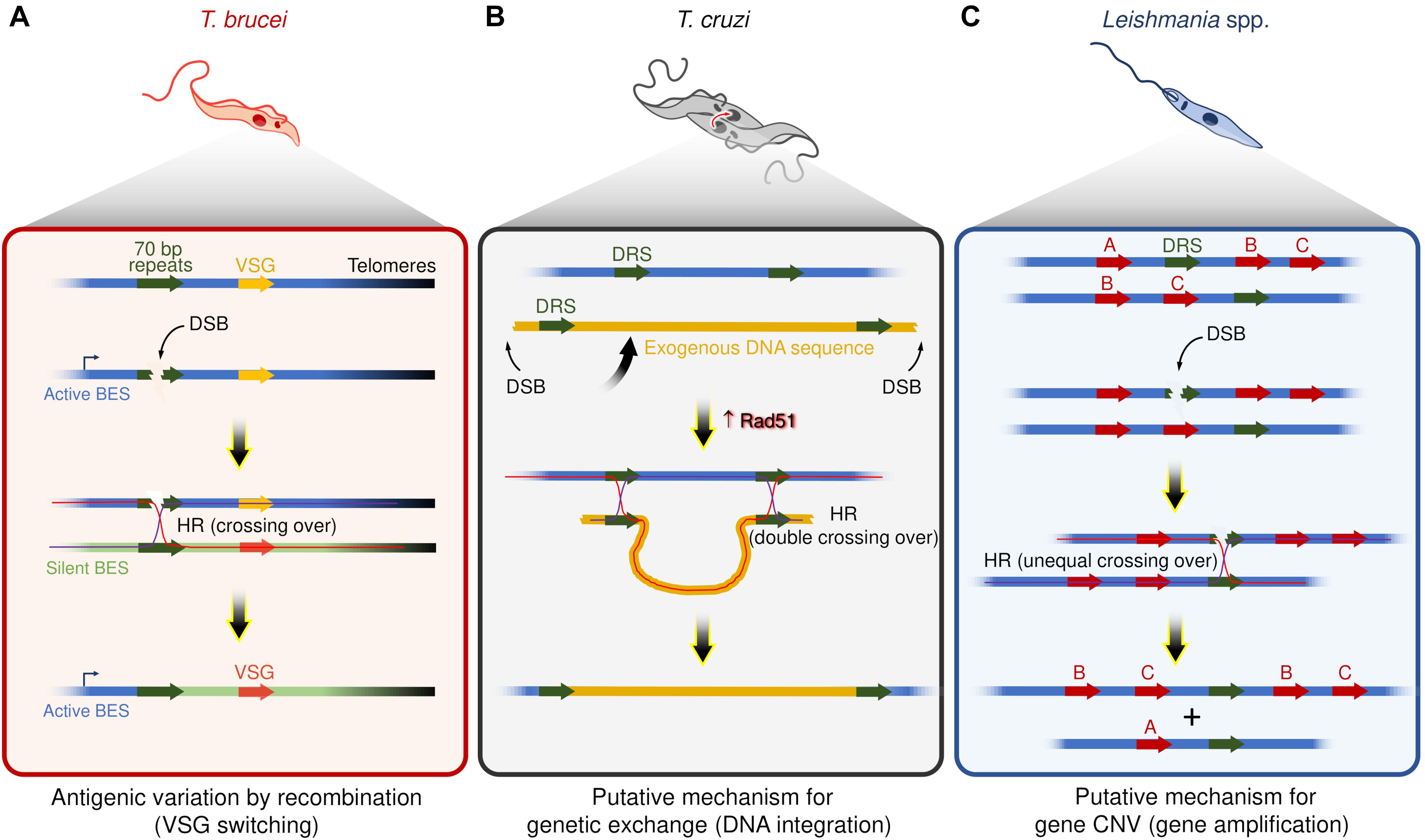
Figure 1. Examples of putative mechanisms dependent on recombination events and DSBs in trypanosomatids. (A) Antigenic variation by recombination events in T. brucei – DSBs within repeat elements (70 bp repeats) are catalysts for VSGs switching. Of note, DSBs occur naturally in active bloodstream expression sites (BES). (B) Genetic exchange in T. cruzi – Hybrid T. cruzi cells have slightly increased Rad51 expression (Alves et al., 2018), which may contribute to driving homologous recombination (HR) between direct repeated sequences (DRS) during the genetic exchange, resulting in the integration of an exogenous DNA. (C) Gene amplification in Leishmania spp. – DSBs nearby or within DRS may trigger HR and lead to gene copy number variation (gene CNV). In the scheme, the genes B and C were amplified.
Briefly, while DSBs may be potentially lethal according to the number, location, and DNA repair capacity of the cell (Marin et al., 2018), this DNA lesion is also a critical factor in the fundamental immune evasion mechanism carried out by T. brucei.
Trypanosoma cruzi is the etiological agent of American trypanosomiasis (also known as Chagas disease), a potentially life-threatening illness afflicting ∼10 million people, predominantly across the Americas (Khare et al., 2016; Browne et al., 2017). Chagas disease encompasses a wide range of clinical manifestations during acute and chronic phases, such as viral−like symptoms (fever, malaise, and lymphadenopathy), arrhythmias, and transient electrocardiogram abnormalities (Morgan et al., 1996; Malik et al., 2015). Most of these symptoms are related to environmental factors and the broad genetic diversity presented by T. cruzi genetic groups (Andrade et al., 2002), of which six discrete typing units (DTUs), TcI to TcVI have been reported (Marcili et al., 2009; Zingales et al., 2012; Brenière et al., 2016).
A pervasive view is that T. cruzi proliferates by binary fission and subsequent clonal expansion (Tibayrenc et al., 1990; Ramírez and Llewellyn, 2014). However, in the last two decades, a growing number of studies support the existence of genetic exchange and possible cryptic sexual cycles among different populations of T. cruzi (Gaunt et al., 2003; Ramírez et al., 2012; Messenger and Miles, 2015; da Silva et al., 2018; Schwabl et al., 2019). For instance, although the evolutive relationships among the different DTUs are largely unclear, at least two DTUs (TcV and TcVI) are hybrids (Machado and Ayala, 2001; Pedroso et al., 2003; Sturm et al., 2003; Lewis et al., 2011; Messenger and Miles, 2015), evidencing that genetic exchange among distinct T. cruzi groups occurs naturally. Intriguingly, naturally occurring hybrid strains of T. cruzi, such as CL Brener (TcVI), show alterations in the expression of core HR factors, displaying high levels of BRCA2 and Rad51 transcripts, indicating that HR repair and DSBs could act as drivers of genetic exchange in these parasites (Alves et al., 2018; Figure 1B).
Unusually, T. cruzi displays remarkable resistance to ionizing radiation (IR), tolerating radiation exposure levels 50–100 times that of mammalian cells (Yonetani et al., 2005; Regis-da-Silva et al., 2006), an effect attributed to Rad51 directed activities acting to resolve IR-induced DSBs (Regis-da-Silva et al., 2006; Silva et al., 2018; Repolês et al., 2020). Indeed, T. cruzi appears to possess an extreme capacity to repair putative DSBs (Regis-da-Silva et al., 2006). Such capabilities could explain, in part, the ability of T. cruzi to produce hybrid strains. Perhaps unsurprisingly, T. cruzi populations overexpressing Rad51 also accumulate a high percentage of fused-cell hybrids (Alves et al., 2018), where Rad51 both acts to limit the formation/stabilization of fused-cell hybrids and drive HR events during the genetic exchange. T. cruzi hybrid strains appear better adapted to deal with DSBs relative to non-hybrid strains (Regis-da-Silva et al., 2006; Garcia et al., 2016; Cerqueira et al., 2017; Resende et al., 2020). This adaptation is probably related to an efficient HR pathway since Rad51 overexpression or ablation causes significant changes in how T. cruzi deals with DSBs (Regis-da-Silva et al., 2006; Silva et al., 2018).
Furthermore, some studies have been evidencing DSBs as a platform to facilitate other fundamental survival mechanisms, such as increased infectivity (Silva et al., 2018; Repolês et al., 2020), chromosome/gene copy number variation (Reis-Cunha et al., 2015), and variability in multigene families (Chiurillo et al., 2016). The latter is worth highlighting for lead to evasion of host immune response, a strategy like those used by T. brucei through antigenic variation (Myler et al., 1984b; Mugnier et al., 2015). Interestingly, the authors used the meganuclease I-SceI to introduce programmed DSBs into a subtelomeric region of T. cruzi CL Brener (TcVI) and observed that the lesions were predominantly repaired by the Rad51-dependent mechanism: HR (Chiurillo et al., 2016). Whether other non-hybrid T. cruzi strains would repair programmed DSBs by HR is an issue that requires further investigation.
In conclusion, although multiple DSBs are harmful (Regis-da-Silva et al., 2006; Silva et al., 2018; Resende et al., 2020), T. cruzi likely utilizes these lesions to enable an increase in its genome diversity, a feature enhanced by Rad51. However, this raises an intriguing question: what did the naturally high levels of Rad51 expression lead to? The high tolerance to DSBs or the genetic exchange producing fused-cell hybrids? Considering that DSBs can trigger HR-dependent events (Pfeiffer et al., 2000; Li, 2015), and HR plays a crucial role in the genetic exchange (Alves et al., 2018), there will probably never be a satisfactory answer to this question.
Leishmania spp. cause a spectrum of debilitating diseases collectively known as leishmaniasis, which have three main forms: visceral leishmaniasis (also known as kala-azar), which is characterized by the enlargement of the spleen and liver, concomitant with anemia and weight loss; cutaneous leishmaniasis, which causes skin lesions leaving serious disability or stigma; and mucocutaneous leishmaniasis, which destroy the mucous membranes of the nose, mouth, and throat (Nazzaro et al., 2014; Burza et al., 2018). To date, only a few human vaccines are in the clinical trial (Moafi et al., 2019), and parasite resistance to front-line drugs has been documented (Croft and Olliaro, 2011; Perez-Franco et al., 2016; Patino et al., 2019), making leishmaniasis a major global health problem.
Leishmania spp. have remarkably plastic genomes, with genomic alterations such as aneuploidy (Mannaert et al., 2012; Lachaud et al., 2014), and CNVs (Rogers et al., 2011; Bussotti et al., 2018), which seems to be widespread phenomena among the species (Rogers et al., 2011; Lachaud et al., 2014; Negreira et al., 2020). Interspersed throughout the genome of Leishmania spp. are repeated DNA sequence elements, which catalyze DNA rearrangements via the formation of circular and linear sequence amplicons (Beverley, 1991; Ubeda et al., 2014). These amplicons arise in several Leishmania spp. under stress conditions or when parasites are challenged with drugs (Beverley et al., 1984; Downing et al., 2011; Laffitte et al., 2014). As HR factors (e.g., Mre11, Rad50, BRCA2, and Rad51 paralogs) facilitate gene rearrangements (Grondin et al., 1993; Navarro et al., 1994; Genois et al., 2012, 2015; Laffitte et al., 2014; Ubeda et al., 2014), DSBs nearby or within repeat elements could act as initiators of amplicon formation (Figure 1C). Nevertheless, no studies have directly correlated DSBs with the emergence of extrachromosomal DNA elements or DNA rearrangement events in this organism to date. Despite this, DSBs are attractive substrates for this type of adaptive genome re-writing for at least three reasons: first, gene CNVs occur through rearrangements of repeated DNA sequences, a process that relies, at least partially, on HR (Grondin et al., 1993); second, increased expression of Rad51 is observed when DSBs are generated (McKean et al., 2001; Genois et al., 2012); and third, Rad51 inactivation prevents the formation of circular extrachromosomal elements even under drug pressure. However, linear amplicons can still form, suggesting that the production of circular extrachromosomal DNA elements is HR-dependent, whereas linear amplicon likely utilizes an alternative pathway (Ubeda et al., 2014; Genois et al., 2015).
Interestingly, more than half of the predicted extrachromosomal DNA elements in Leishmania spp. are present in wild-type populations in the absence of drug pressure indicating the Leishmania genome is, in fact, undergoing continuous rearrangement (Ubeda et al., 2014; Bussotti et al., 2018). Moreover, these stochastic rearrangements may reflect a strategy by which Leishmania can rapidly adapt to a changing environment (Ubeda et al., 2014). However, if DSBs are continually being generated to trigger these rearrangements is a matter of debate that remains open.
Succinctly, although multiple DSBs can be extremely hazardous for Leishmania spp. (Manna et al., 2010; da Silveira et al., 2013), further studies are necessary to finish the puzzle promoted by these lesions and find out when they can be a benefit or a detriment for this parasite.
Antigenic variation in T. brucei, genetic exchange in T. cruzi, and genomic alterations in Leishmania are examples of some vital processes triggered by DSBs and evidence how fundamental is this type of DNA damage for these organisms. However, some studies have shown that the response to DSBs can be slightly different in each trypanosomatid. For instance, T. brucei apparently fails to trigger a stringent cell cycle checkpoint in response to DSBs and, due to that, DNA breaks may persist during cell division until a template (e.g., sister chromatid) is available (Glover et al., 2019). This finding suggests that MMEJ does not play a major role in T. brucei. In contrast, DSBs generated by CRISPR/Cas9 without a template do not persist in T. cruzi and are repaired by MMEJ (Peng et al., 2015). Curiously, Leishmania donovani predominantly uses SSA instead of MMEJ to repair DSBs introduced by CRISPR/Cas9 (Zhang et al., 2020). These different behaviors in response to DSBs suggest that the cell cycle plays a fundamental role in the trypanosomatids DNA damage response.
In population terms, the cell cycle phase where DSBs are generated is trivial since the predominant phenotype is evidenced by those trypanosomatids that managed to overcome the DNA damage. However, for a single cell, the cell cycle phase in which DSBs are introduced is essential to decide its fate. For instance, DSBs generated outside the S/G2 phases are unlikely to trigger recombination events, mainly due to the absence of a sister chromatid (homologous sequence). This behavior may explain the different and peculiar responses to the DSBs previously mentioned. In this scenario, single-cell analyses (e.g., single-cell transcriptomics) can be a valuable tool to reveal possible cryptic populations capable of dealing with DSBs differently (Briggs et al., 2021). Profiling gene expression of individual cells with single-cell RNA sequencing may detect rare cell types in heterogeneous populations previously challenged with DSBs source agents, such as IR. This approach may contribute to evidence, even more, how relevant are the roles of DSBs in the life cycle of these peculiar organisms.
In conclusion, DSB formation poses a conundrum for single-celled organisms like trypanosomatids. On the one hand, DSBs undermine genomic stability compromising parasites fitness and potentially inducing death (Regis-da-Silva et al., 2006; Manna et al., 2010; Marin et al., 2018). On the other, DSBs provide an essential substrate for genome variability and subsequent adaptation to rapidly changing environments, with examples from each parasite harnessing DSBs and its repair to this effect: in T. brucei, DSBs can trigger VSG switching enabling host immune evasion (Boothroyd et al., 2009; Glover et al., 2013); for T. cruzi, DSBs are necessary for HR-dependent events essential for genetic exchange (Gaunt et al., 2003; Alves et al., 2018) and variability in multigene families (Chiurillo et al., 2016); and in the case of Leishmania spp., DSBs can be catalysts for recombination events leading to genomic changes and CNVs, a crucial strategy to overcome hostile environments (Grondin et al., 1993; McKean et al., 2001; Laffitte et al., 2016). Thereby, DSBs represent a “double-edged sword” for trypanosomatids (Figure 2). Now, further studies are required to establish which players (or pathways) wield this heavy blade.
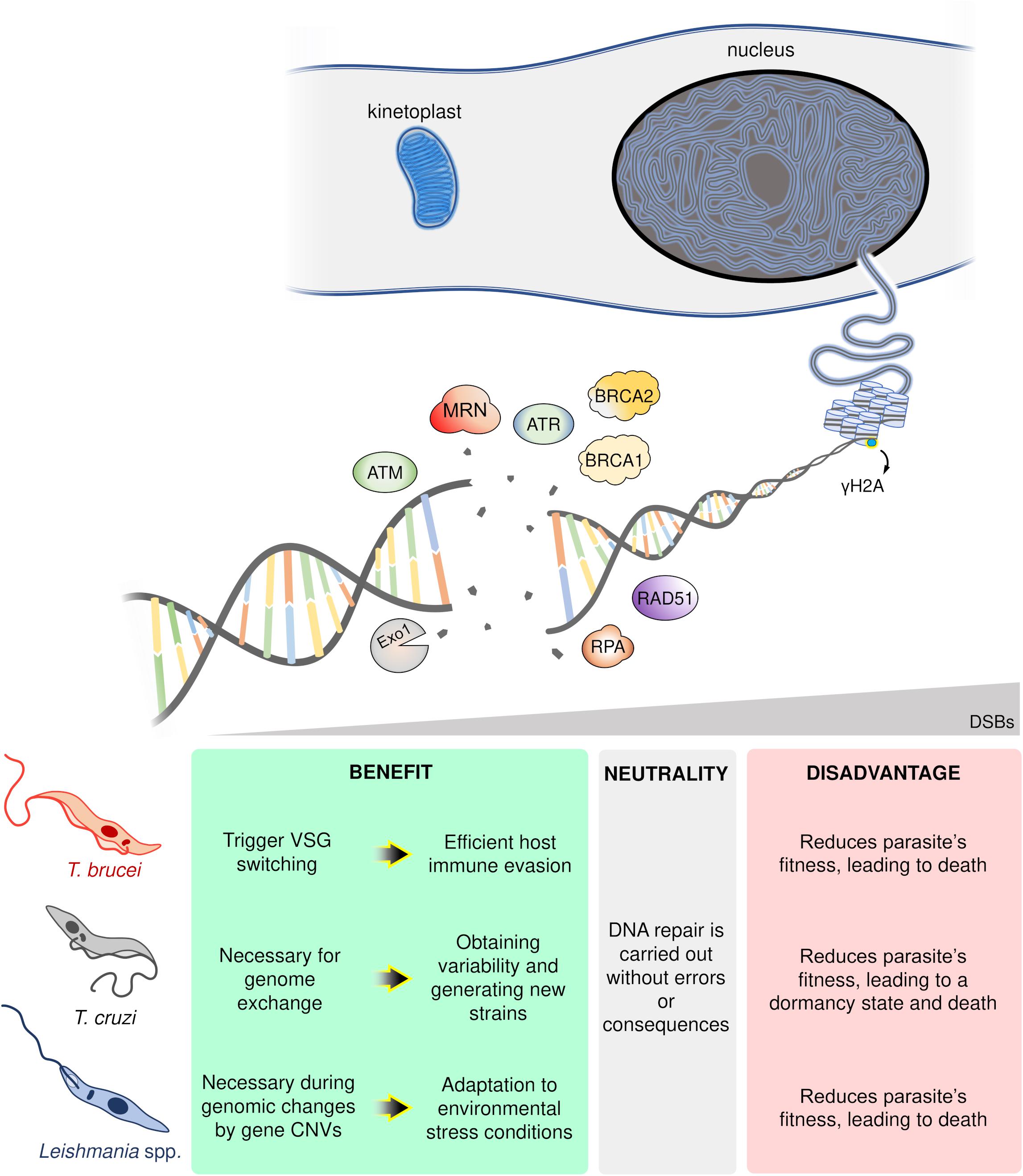
Figure 2. Schematic overview of the possible trypanosomatid cell’s fate in response to DNA double-strand breaks (DSBs). In a hypothetical trypanosomatid, several players act in an orchestrated way in response to DSBs. However, according to the number, location, cell cycle phase, and DNA repair capacity of the cell, these lesions can trigger different consequences: advantages (green box), neutrality (gray box), or disadvantages (red box). ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related; MRN, MRE11-RAD50-NBS1 complex; Exo1, Exonuclease 1; RPA, Replication protein A; BRCA1-2, Breast cancer 1–2; Rad51, Recombinase involved in homologous recombination; γH2A, phosphorylated histone H2A.
MSdS wrote, revised, and approved the submitted version of the manuscript.
This study was supported by the São Paulo Research Foundation (FAPESP) under grants 2019/10753-2, 2020/10277-3, and 2021/02977-8.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author is grateful to Dr. Jennifer A. Black (University of Glasgow, United Kingdom), Dr. Bruno M. Repolês (Umeå University, Sweden), and Dr. Débora Andrade-Silva (Butantan Institute, Brazil) for their critical revision and valuable suggestions to improve the manuscript’s writing.
Alves, C. L., Repolês, B. M., da Silva, M. S., Mendes, I. C., Marin, P. A., Aguiar, P. H. N., et al. (2018). The recombinase Rad51 plays a key role in events of genetic exchange in Trypanosoma cruzi. Sci. Rep. 8:13335. doi: 10.1038/s41598-018-31541-z
Andrade, L. O., Machado, C. R. S., Chiari, E., Pena, S. D. J., and Macedo, A. M. (2002). Trypanosoma cruzi: role of host genetic background in the differential tissue distribution of parasite clonal populations. Exp. Parasitol. 100, 269–275. doi: 10.1016/S0014-4894(02)00024-3
Barratt, J., Kaufer, A., Peters, B., Craig, D., Lawrence, A., Roberts, T., et al. (2017). Isolation of novel trypanosomatid, Zelonia australiensis sp. nov. (Kinetoplastida: Trypanosomatidae) provides support for a Gondwanan origin of dixenous parasitism in the Leishmaniinae. PLoS Negl. Trop. Dis. 11:e0005215. doi: 10.1371/journal.pntd.0005215
Barry, J. D., and McCulloch, R. (2001). Antigenic variation in trypanosomes: enhanced phenotypic variation in a eukaryotic parasite. Adv. Parasitol. 49, 1–70. doi: 10.1016/S0065-308X(01)49037-3
Baudat, F., and Nicolas, A. (1997). Clustering of meiotic double-strand breaks on yeast chromosome III. Proc. Natl. Acad. Sci. U.S.A. 94, 5213–5218. doi: 10.1073/pnas.94.10.5213
Bernards, A., Titia, D. L., Michels, P. A. M., Liu, A. Y. C., Huisman, M. J., and Borst, P. (1984). Two modes of activation of a single surface antigen gene of Trypanosoma brucei. Cell 36, 163–170. doi: 10.1016/0092-8674(84)90085-0
Berriman, M., Ghedin, E., Hertz-Fowler, C., Blandin, G., Renauld, H., Bartholomeu, D. C., et al. (2005). The genome of the African trypanosome Trypanosoma brucei. Science 309, 416–422. doi: 10.1126/science.1112642
Beverley, S. M. (1991). Gene amplification in Leishmania. Annu. Rev. Microbiol. 45, 417–444. doi: 10.1146/annurev.mi.45.100191.002221
Beverley, S. M., Coderre, J. A., Santi, D. V., and Schimke, R. T. (1984). Unstable DNA amplifications in methotrexate resistant Leishmania consist of extrachromosomal circles which relocalize during stabilization. Cell 38, 431–439. doi: 10.1016/0092-8674(84)90498-7
Bitter, W., Gerrits, H., Kieft, R., and Borst, P. (1998). The role of transferrin-receptor variation in the host range of Trypanosoma brucei. Nature 391, 499–502. doi: 10.1038/35166
Black, J. A., Crouch, K., Lemgruber, L., Lapsley, C., Dickens, N., Tosi, L. R. O., et al. (2020). Trypanosoma brucei ATR links DNA damage signaling during antigenic variation with regulation of RNA polymerase I-transcribed surface antigens. Cell Rep. 30, 836–851. doi: 10.1016/j.celrep.2019.12.049
Boothroyd, C. E., Dreesen, O., Leonova, T., Ly, K. I., Figueiredo, L. M., Cross, G. A. M., et al. (2009). A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei. Nature 459, 278–281. doi: 10.1038/nature07982
Brecht, R. M., Liu, C. C., Beilinson, H. A., Khitun, A., Slavoff, S. A., and Schatz, D. G. (2020). Nucleolar localization of RAG1 modulates V(D)J recombination activity. Proc. Natl. Acad. Sci. U.S.A. 117, 4300–4309. doi: 10.1073/pnas.1920021117
Brenière, S. F., Waleckx, E., and Barnabé, C. (2016). Over six thousand Trypanosoma cruzi strains classified into Discrete Typing Units (DTUs): attempt at an Inventory. PLoS Negl. Trop. Dis. 10:e0004792. doi: 10.1371/journal.pntd.0004792
Briggs, E. M., Warren, F. S., Matthews, K. R., McCulloch, R., and Otto, T. D. (2021). Application of single cell transcriptomics to kinetoplastid research. Parasitology 1–51. doi: 10.1017/S003118202100041X
Browne, A. J., Guerra, C. A., Alves, R. V., da Costa, V. M., Wilson, A. L., Pigott, D. M., et al. (2017). The contemporary distribution of Trypanosoma cruzi infection in humans, alternative hosts and vectors. Sci. Data 4:170050. doi: 10.1038/sdata.2017.50
Budzak, J., Kerry, L. E., Aristodemou, A., Hall, B. S., Witmer, K., Kushwaha, M., et al. (2019). Dynamic colocalization of 2 simultaneously active VSG expression sites within a single expression-site body in Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 116, 16561–16570. doi: 10.1073/pnas.1905552116
Burton, P., McBride, D. J., Wilkes, J. M., Barry, J. D., and McCulloch, R. (2007). Ku heterodimer-independent end joining in Trypanosoma brucei cell extracts relies upon sequence microhomology. Eukaryot. Cell 6, 1773–1781. doi: 10.1128/EC.00212-07
Burza, S., Croft, S. L., and Boelaert, M. (2018). Leishmaniasis. Lancet 392, 951–970. doi: 10.1016/S0140-6736(18)31204-2
Bussotti, G., Gouzelou, E., Boité, M. C., Kherachi, I., Harrat, Z., Eddaikra, N., et al. (2018). Leishmania genome dynamics during environmental adaptation reveal strain-specific differences in gene copy number variation, karyotype instability, and telomeric amplification. mBio 9:e01399-18. doi: 10.1128/mBio.01399-18
Cahoon, L. A., and Seifert, H. S. (2011). Focusing homologous recombination: pilin antigenic variation in the pathogenic Neisseria. Mol. Microbiol. 81, 1136–1143. doi: 10.1111/j.1365-2958.2011.07773.x
Cannan, W. J., and Pederson, D. S. (2016). Mechanisms and consequences of double-strand DNA break formation in chromatin. J. Cell. Physiol. 231, 3–14. doi: 10.1002/jcp.25048
Cannan, W. J., Tsang, B. P., Wallace, S. S., and Pederson, D. S. (2014). Nucleosomes suppress the formation of double-strand DNA breaks during attempted base excision repair of clustered oxidative damages. J. Biol. Chem. 289, 19881–19893. doi: 10.1074/jbc.M114.571588
Carofiglio, F., Sleddens-Linkels, E., Wassenaar, E., Inagaki, A., van Cappellen, W. A., Grootegoed, J. A., et al. (2018). Repair of exogenous DNA double-strand breaks promotes chromosome synapsis in SPO11-mutant mouse meiocytes, and is altered in the absence of HORMAD1. DNA Repair (Amst.) 63, 25–38. doi: 10.1016/j.dnarep.2018.01.007
Cerqueira, P. G., Passos-Silva, D. G., Vieira-da-Rocha, J. P., Mendes, I. C., de Oliveira, K. A., Oliveira, C. F. B., et al. (2017). Effect of ionizing radiation exposure on Trypanosoma cruzi ubiquitin-proteasome system. Mol. Biochem. Parasitol. 212, 55–67. doi: 10.1016/j.molbiopara.2017.01.005
Chen, J., and Stubbe, J. A. (2005). Bleomycins: towards better therapeutics. Nat. Rev. Cancer 5, 102–112. doi: 10.1038/nrc1547
Chiurillo, M. A., Moraes Barros, R. R., Souza, R. T., Marini, M. M., Antonio, C. R., Cortez, D. R., et al. (2016). Subtelomeric I-SceI-mediated double-strand breaks are repaired by homologous recombination in Trypanosoma cruzi. Front. Microbiol. 7:2041. doi: 10.3389/fmicb.2016.02041
Claessens, A., Hamilton, W. L., Kekre, M., Otto, T. D., Faizullabhoy, A., Rayner, J. C., et al. (2014). Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of VAR genes during mitosis. PLoS Genet. 10:e1004812. doi: 10.1371/journal.pgen.1004812
Conway, C., Proudfoot, C., Burton, P., Barry, J. D., and McCulloch, R. (2002). Two pathways of homologous recombination in Trypanosoma brucei. Mol. Microbiol. 45, 1687–1700. doi: 10.1046/j.1365-2958.2002.03122.x
Croft, S. L., and Olliaro, P. (2011). Leishmaniasis chemotherapy-challenges and opportunities. Clin. Microbiol. Infect. 17, 1478–1483. doi: 10.1111/j.1469-0691.2011.03630.x
Cross, G. A. M., Kim, H. S., and Wickstead, B. (2014). Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Mol. Biochem. Parasitol. 195, 59–73. doi: 10.1016/j.molbiopara.2014.06.004
da Silva, M. S., Cayres-Silva, G. R., Vitarelli, M. O., Marin, P. A., Hiraiwa, P. M., Araújo, C. B., et al. (2019). Transcription activity contributes to the firing of non-constitutive origins in African trypanosomes helping to maintain robustness in S-phase duration. Sci. Rep. 9:18512. doi: 10.1038/s41598-019-54366-w
da Silva, M. S., Marin, P., Repolês, B., Elias, M., and Machado, C. (2018). Analysis of DNA exchange using thymidine analogs (ADExTA) in Trypanosoma cruzi. Bio Protoc. 8:e3125. doi: 10.21769/bioprotoc.3125
da Silveira, R. D. C. V., da Silva, M. S., Nunes, V. S., Perez, A. M., and Cano, M. I. N. (2013). The natural absence of RPA1N domain did not impair Leishmania amazonensis RPA-1 participation in DNA damage response and telomere protection. Parasitology 140, 547–559. doi: 10.1017/S0031182012002028
Downing, T., Imamura, H., Decuypere, S., Clark, T. G., Coombs, G. H., Cotton, J. A., et al. (2011). Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 21, 2143–2156. doi: 10.1101/gr.123430.111
Elango, R., Sheng, Z., Jackson, J., Decata, J., Ibrahim, Y., Pham, N. T., et al. (2017). Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat. Commun. 8:1790. doi: 10.1038/s41467-017-01987-2
Faria, J., Luzak, V., Müller, L. S. M., Brink, B. G., Hutchinson, S., Glover, L., et al. (2021). Spatial integration of transcription and splicing in a dedicated compartment sustains monogenic antigen expression in African trypanosomes. Nat. Microbiol. 6, 289–300. doi: 10.1038/s41564-020-00833-4
Farlow, A., Meduri, E., and Schlötterer, C. (2011). DNA double-strand break repair and the evolution of intron density. Trends Genet. 27, 1–6. doi: 10.1016/j.tig.2010.10.004
Garcia, J. B. F., da Rocha, J. P. V., Costa-Silva, H. M., Alves, C. L., MacHado, C. R., and Cruz, A. K. (2016). Leishmania major and Trypanosoma cruzi present distinct DNA damage responses. Mol. Biochem. Parasitol. 207, 23–32. doi: 10.1016/j.molbiopara.2016.05.004
García-Muse, T., and Aguilera, A. (2016). Transcription–replication conflicts: how they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 17, 553–563. doi: 10.1038/nrm.2016.88
Gaunt, M. W., Yeo, M., Frame, I. A., Stothard, J. R., Carrasco, H. J., Taylor, M. C., et al. (2003). Mechanism of genetic exchange in American trypanosomes. Nature 421, 936–939. doi: 10.1038/nature01438
Geiger, A., Bossard, G., Sereno, D., Pissarra, J., Lemesre, J. L., Vincendeau, P., et al. (2016). Escaping deleterious immune response in their hosts: lessons from trypanosomatids. Front. Immunol. 7:212. doi: 10.3389/fimmu.2016.00212
Genois, M. M., Mukherjee, A., Ubeda, J. M., Buisson, R., Paquet, E., Roy, G., et al. (2012). Interactions between BRCA2 and RAD51 for promoting homologous recombination in Leishmania infantum. Nucleic Acids Res. 40, 6570–6584. doi: 10.1093/nar/gks306
Genois, M. M., Plourde, M., Éthier, C., Roy, G., Poirier, G. G., Ouellette, M., et al. (2015). Roles of Rad51 paralogs for promoting homologous recombination in Leishmania infantum. Nucleic Acids Res. 43, 2701–2715. doi: 10.1093/nar/gkv118
Glover, L., Alsford, S., and Horn, D. (2013). DNA break site at fragile subtelomeres determines probability and mechanism of antigenic variation in African trypanosomes. PLoS Pathog. 9:e1003260. doi: 10.1371/journal.ppat.1003260
Glover, L., Marques, C. A., Suska, O., and Horn, D. (2019). Persistent DNA damage foci and DNA replication with a broken chromosome in the African trypanosome. mBio 10:e01252-19. doi: 10.1128/mBio.01252-19
Glover, L., McCulloch, R., and Horn, D. (2008). Sequence homology and microhomology dominate chromosomal double-strand break repair in African trypanosomes. Nucleic Acids Res. 36, 2608–2618. doi: 10.1093/nar/gkn104
Grondin, K., Papadopoulon, B., and Ouellette, M. (1993). Homologous recombination between direct repeat sequences yields P-glycoprotein containing amplicons in arsenite resistant Leishmania. Nucleic Acids Res. 21, 1895–1901. doi: 10.1093/nar/21.8.1895
Hall, J. P. J., Wang, H., and David Barry, J. (2013). Mosaic VSGs and the scale of Trypanosoma brucei antigenic variation. PLoS Pathog. 9:e1003502. doi: 10.1371/journal.ppat.1003502
Hartley, C. L., and McCulloch, R. (2008). Trypanosoma brucei BRCA2 acts in antigenic variation and has undergone a recent expansion in BRC repeat number that is important during homologous recombination. Mol. Microbiol. 68, 1237–1251. doi: 10.1111/j.1365-2958.2008.06230.x
Hertz-Fowler, C., Figueiredo, L. M., Quail, M. A., Becker, M., Jackson, A., Bason, N., et al. (2008). Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One 3:e3527. doi: 10.1371/journal.pone.0003527
Horn, D. (2014). Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 195, 123–129. doi: 10.1016/j.molbiopara.2014.05.001
Hovel-Miner, G. A., Boothroyd, C. E., Mugnier, M., Dreesen, O., Cross, G. A. M., and Papavasiliou, F. N. (2012). Telomere length affects the frequency and mechanism of antigenic variation in Trypanosoma brucei. PLoS Pathog. 8:e1002900. doi: 10.1371/journal.ppat.1002900
Jekimovs, C., Bolderson, E., Suraweera, A., Adams, M., O’Byrne, K. J., and Richard, D. J. (2014). Chemotherapeutic compounds targeting the DNA double-strand break repair pathways: the good, the bad, and the promising. Front. Oncol. 4:86. doi: 10.3389/fonc.2014.00086
Kaye, J. A., Melo, J. A., Cheung, S. K., Vaze, M. B., Haber, J. E., and Toczyski, D. P. (2004). DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr. Biol. 14, 2096–2106. doi: 10.1016/j.cub.2004.10.051
Kerry, L. E., Pegg, E. E., Cameron, D. P., Budzak, J., Poortinga, G., Hannan, K. M., et al. (2017). Selective inhibition of RNA polymerase I transcription as a potential approach to treat African trypanosomiasis. PLoS Negl. Trop. Dis. 11:e0005432. doi: 10.1371/journal.pntd.0005432
Khare, S., Nagle, A. S., Biggart, A., Lai, Y. H., Liang, F., Davis, L. C., et al. (2016). Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 537, 229–233. doi: 10.1038/nature19339
Kuzminov, A. (2011). Homologous recombination–experimental systems, analysis, and significance. EcoSal Plus 4. doi: 10.1128/ecosalplus.7.2.6
Kyes, S. A., Kraemer, S. M., and Smith, J. D. (2007). Antigenic variation in Plasmodium falciparum: gene organization and regulation of the var multigene family. Eukaryot. Cell 6, 1511–1520. doi: 10.1128/EC.00173-07
Lachaud, L., Bourgeois, N., Kuk, N., Morelle, C., Crobu, L., Merlin, G., et al. (2014). Constitutive mosaic aneuploidy is a unique genetic feature widespread in the Leishmania genus. Microbes Infect. 16, 61–66. doi: 10.1016/j.micinf.2013.09.005
Laffitte, M. C. N., Genois, M. M., Mukherjee, A., Légaré, D., Masson, J. Y., and Ouellette, M. (2014). Formation of linear amplicons with inverted duplications in Leishmania requires the MRE11 nuclease. PLoS Genet. 10:e1004805. doi: 10.1371/journal.pgen.1004805
Laffitte, M. C. N., Leprohon, P., Hainse, M., Légaré, D., Masson, J. Y., and Ouellette, M. (2016). Chromosomal translocations in the parasite Leishmania by a MRE11/RAD50-independent microhomology-mediated end joining mechanism. PLoS Genet. 12:e1006117. doi: 10.1371/journal.pgen.1006117
Lewis, M. D., Llewellyn, M. S., Yeo, M., Acosta, N., Gaunt, M. W., and Miles, M. A. (2011). Recent, independent and anthropogenic origins of Trypanosoma cruzi hybrids. PLoS Negl. Trop. Dis. 5:e1363. doi: 10.1371/journal.pntd.0001363
Li, B. (2015). DNA double-strand breaks and telomeres play important roles in Trypanosoma brucei antigenic variation. Eukaryot. Cell 14, 196–205. doi: 10.1128/EC.00207-14
Ma, W., Halweg, C. J., Menendez, D., and Resnick, M. A. (2012). Differential effects of poly(ADP-ribose) polymerase inhibition on DNA break repair in human cells are revealed with Epstein-Barr virus. Proc. Natl. Acad. Sci. U.S.A. 109, 6590–6595. doi: 10.1073/pnas.1118078109
Machado, C. A., and Ayala, F. J. (2001). Nucleotide sequences provide evidence of genetic exchange among distantly related lineages of Trypanosoma cruzi. Proc. Natl. Acad. Sci. U.S.A. 98, 7396–7401. doi: 10.1073/pnas.121187198
Malik, L. H., Singh, G. D., and Amsterdam, E. A. (2015). The epidemiology, clinical manifestations, and management of Chagas Heart disease. Clin. Cardiol. 38, 565–569. doi: 10.1002/clc.22421
Manna, S., Chakravarty, R., Manna, B., and Bhattachaiya, A. (2010). Radiation induced alterations in membrane fluidity, microtubular structure, Glycoconjugates and protein in Leishmania donovani. Res. J. Parasitol. 5, 203–213. doi: 10.3923/jp.2006.48.58
Mannaert, A., Downing, T., Imamura, H., and Dujardin, J. C. (2012). Adaptive mechanisms in pathogens: universal aneuploidy in Leishmania. Trends Parasitol. 28, 370–376. doi: 10.1016/j.pt.2012.06.003
Marcello, L., and Barry, J. D. (2007). Analysis of the VSG gene silent archive in Trypanosoma brucei reveals that mosaic gene expression is prominent in antigenic variation and is favored by archive substructure. Genome Res. 17, 1344–1352. doi: 10.1101/gr.6421207
Marcili, A., Lima, L., Cavazzana, M., Junqueira, A. C. V., Veludo, H. H., Maia Da Silva, F., et al. (2009). A new genotype of Trypanosoma cruzi associated with bats evidenced by phylogenetic analyses using SSU rDNA, cytochrome b and histone H2B genes and genotyping based on ITS1 rDNA. Parasitology 136, 641–655. doi: 10.1017/S0031182009005861
Marin, P. A., da Silva, M. S., Pavani, R. S., Machado, C. R., and Elias, M. C. (2018). Recruitment kinetics of the homologous recombination pathway in procyclic forms of Trypanosoma brucei after ionizing radiation treatment. Sci. Rep. 8:5405. doi: 10.1038/s41598-018-23731-6
Marin, P. A., Obonaga, R., Pavani, R. S., da Silva, M. S., de Araujo, C. B., Lima, A. A., et al. (2020). ATR kinase is a crucial player mediating the DNA damage response in Trypanosoma brucei. Front. Cell Dev. Biol. 8:602956. doi: 10.3389/fcell.2020.602956
McCulloch, R., and Barry, J. D. (1999). A role for RAD51 and homologous recombination in Trypanosoma brucei antigenic variation. Genes Dev. 13, 2875–2888. doi: 10.1101/gad.13.21.2875
McCulloch, R., Rudenko, G., and Borst, P. (1997). Gene conversions mediating antigenic variation in Trypanosoma brucei can occur in variant surface glycoprotein expression sites lacking 70-base-pair repeat sequences. Mol. Cell. Biol. 17, 833–843. doi: 10.1128/MCB.17.2.833
McKean, P. G., Keen, J. K., Smith, D. F., and Benson, F. E. (2001). Identification and characterisation of a RAD51 gene from Leishmania major. Mol. Biochem. Parasitol. 115, 209–216. doi: 10.1016/S0166-6851(01)00288-2
Mehnert, A. K., Prorocic, M., Dujeancourt-Henry, A., Hutchinson, S., McCulloch, R., and Glover, L. (2021). RAD50 promotes DNA repair by homologous recombination and restrains antigenic variation in African trypanosomes. Nucleic Acids Res. 1:gkaa1265. doi: 10.1101/2020.03.17.994905
Messenger, L. A., and Miles, M. A. (2015). Evidence and importance of genetic exchange among field populations of Trypanosoma cruzi. Acta Trop. 151, 150–155. doi: 10.1016/j.actatropica.2015.05.007
Mirkin, E. V., and Mirkin, S. M. (2007). Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 71, 13–35. doi: 10.1128/mmbr.00030-06
Moafi, M., Sherkat, R., Taleban, R., and Rezvan, H. (2019). Leishmania vaccines entered in clinical trials: a review of literature. Int. J. Prev. Med. 10:95. doi: 10.4103/ijpvm.IJPVM_116_18
Morgan, J., Dias, J. C. P., Gontijo, E. D., Bahia-Oliveira, L., Correa-Oliveira, R., Colley, D. G., et al. (1996). Anti-Trypanosoma cruzi antibody isotype profiles in patients with different clinical manifestations of Chagas’ disease. Am. J. Trop. Med. Hyg. 55, 355–359. doi: 10.4269/ajtmh.1996.55.355
Mugnier, M. R., Cross, G. A. M., and Papavasiliou, F. N. (2015). The in vivo dynamics of antigenic variation in Trypanosoma brucei. Science 347, 1470–1473. doi: 10.1126/science.aaa4502
Müller, L. S. M., Cosentino, R. O., Förstner, K. U., Guizetti, J., Wedel, C., Kaplan, N., et al. (2018). Genome organization and DNA accessibility control antigenic variation in trypanosomes. Nature 563, 121–125. doi: 10.1038/s41586-018-0619-8
Myler, P., Nelson, R. G., Agabian, N., and Stuart, K. (1984a). Two mechanisms of expression of a predominant variant antigen gene of Trypanosoma brucei. Nature 309, 282–284. doi: 10.1038/309282a0
Myler, P. J., Allison, J., Agabian, N., and Stuart, K. (1984b). Antigenic variation in African trypanosomes by gene replacement or activation of alternate telomeres. Cell 39, 203–211. doi: 10.1016/0092-8674(84)90206-X
Navarro, M., and Gull, K. (2001). A pol l transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 414, 759–763. doi: 10.1038/414759a
Navarro, M., Liu, J., Muthui, D., Ortiz, G., Segovia, M., and Hamers, R. (1994). Inverted repeat structure and homologous sequences in the LD1 amplicons of Leishmania spp. Mol. Biochem. Parasitol. 68, 69–80. doi: 10.1016/0166-6851(94)00147-2
Nazzaro, G., Rovaris, M., and Veraldi, S. (2014). Leishmaniasis: a disease with many names. JAMA Dermatol. 150, 1202–1204. doi: 10.1001/jamadermatol.2014.1072
Negreira, G., Monsieurs, P., Imamura, H., Maes, I., Kuk, N., Yagoubat, A., et al. (2020). Exploring the evolution and adaptive role of mosaic aneuploidy in a clonal Leishmania donovani population using high throughput single cell genome sequencing. bioRxiv [Preprint]. doi: 10.1101/2020.03.05.976233
Nenarokova, A., Záhonová, K., Krasilnikova, M., Gahura, O., McCulloch, R., Zíková, A., et al. (2019). Causes and effects of loss of classical nonhomologous end joining pathway in parasitic eukaryotes. mBio 10:e01541-19. doi: 10.1128/mbio.01541-19
Pâques, F., and Haber, J. E. (1999). Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63, 349–404. doi: 10.1128/mmbr.63.2.349-404.1999
Patino, L. H., Imamura, H., Cruz-Saavedra, L., Pavia, P., Muskus, C., Méndez, C., et al. (2019). Major changes in chromosomal somy, gene expression and gene dosage driven by SbIII in Leishmania braziliensis and Leishmania panamensis. Sci. Rep. 9:9485. doi: 10.1038/s41598-019-45538-9
Pedroso, A., Cupolillo, E., and Zingales, B. (2003). Evaluation of Trypanosoma cruzi hybrid stocks based on chromosomal size variation. Mol. Biochem. Parasitol. 129, 79–90. doi: 10.1016/S0166-6851(03)00096-3
Peng, D., Kurup, S. P., Yao, P. Y., Minning, T. A., and Tarleton, R. L. (2015). CRISPR-Cas9-mediated single-gene and gene family disruption in Trypanosoma cruzi. mBio 6:e02097-14. doi: 10.1128/mBio.02097-14
Perez-Franco, J. E., Cruz-Barrera, M. L., Robayo, M. L., Lopez, M. C., Daza, C. D., Bedoya, A., et al. (2016). Clinical and parasitological features of patients with American cutaneous leishmaniasis that did not respond to treatment with meglumine antimoniate. PLoS Negl. Trop. Dis. 10:e0004739. doi: 10.1371/journal.pntd.0004739
Pfeiffer, P., Goedecke, W., and Obe, G. (2000). Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis 15, 289–302. doi: 10.1093/mutage/15.4.289
Pinger, J., Chowdhury, S., and Papavasiliou, F. N. (2017). Variant surface glycoprotein density defines an immune evasion threshold for African trypanosomes undergoing antigenic variation. Nat. Commun. 8:828. doi: 10.1038/s41467-017-00959-w
Proudfoot, C., and McCulloch, R. (2005). Distinct roles for two RAD51-related genes in Trypanosoma brucei antigenic variation. Nucleic Acids Res. 33, 6906–6919. doi: 10.1093/nar/gki996
Ramírez, J. D., Guhl, F., Messenger, L. A., Lewis, M. D., Montilla, M., Cucunuba, Z., et al. (2012). Contemporary cryptic sexuality in Trypanosoma cruzi. Mol. Ecol. 21, 4216–4226. doi: 10.1111/j.1365-294X.2012.05699.x
Ramírez, J. D., and Llewellyn, M. S. (2014). Reproductive clonality in protozoan pathogens – truth or artefact? Mol. Ecol. 23, 4195–4202. doi: 10.1111/mec.12872
Regis-da-Silva, C. G., Freitas, J. M., Passos-Silva, D. G., Furtado, C., Augusto-Pinto, L., Pereira, M. T., et al. (2006). Characterization of the Trypanosoma cruzi Rad51 gene and its role in recombination events associated with the parasite resistance to ionizing radiation. Mol. Biochem. Parasitol. 149, 191–200. doi: 10.1016/j.molbiopara.2006.05.012
Reis-Cunha, J. L., Rodrigues-Luiz, G. F., Valdivia, H. O., Baptista, R. P., Mendes, T. A. O., de Morais, G. L., et al. (2015). Chromosomal copy number variation reveals differential levels of genomic plasticity in distinct Trypanosoma cruzi strains. BMC Genomics 16:499. doi: 10.1186/s12864-015-1680-4
Repolês, B. M., Machado, C. R., and Florentino, P. T. V. (2020). DNA lesions and repair in trypanosomatids infection. Genet. Mol. Biol. 43:e20190163. doi: 10.1590/1678-4685-GMB-2019-0163
Resende, B. C., Oliveira, A. C. S., Guañabens, A. C. P., Repolês, B. M., Santana, V., Hiraiwa, P. M., et al. (2020). The influence of recombinational processes to induce dormancy in Trypanosoma cruzi. Front. Cell. Infect. Microbiol 10:5. doi: 10.3389/fcimb.2020.00005
Ridewood, S., Ooi, C. P., Hall, B., Trenaman, A., Wand, N. V., Sioutas, G., et al. (2017). The role of genomic location and flanking 3’UTR in the generation of functional levels of variant surface glycoprotein in Trypanosoma brucei. Mol. Microbiol. 106, 614–634. doi: 10.1111/mmi.13838
Robinson, N. P., Burman, N., Melville, S. E., and Barry, J. D. (1999). Predominance of duplicative VSG gene conversion in antigenic variation in African trypanosomes. Mol. Cell. Biol. 19, 5839–5846. doi: 10.1128/MCB.19.9.5839
Rogers, M. B., Hilley, J. D., Dickens, N. J., Wilkes, J., Bates, P. A., Depledge, D. P., et al. (2011). Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 21, 2129–2142. doi: 10.1101/gr.122945.111
Rose, E., Carvalho, J. L., and Hecht, M. (2020). Mechanisms of DNA repair in Trypanosoma cruzi: what do we know so far? DNA Repair (Amst.) 9:102873. doi: 10.1016/j.dnarep.2020.102873
Rudenko, G., Chaves, I., Dirks-Mulder, A., and Borst, P. (1998). Selection for activation of a new variant surface glycoprotein gene expression site in Trypanosoma brucei can result in deletion of the old one. Mol. Biochem. Parasitol. 95, 97–109. doi: 10.1016/S0166-6851(98)00099-1
Saleh-Gohari, N., Bryant, H. E., Schultz, N., Parker, K. M., Cassel, T. N., and Helleday, T. (2005). Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell. Biol. 25, 7158–7169. doi: 10.1128/mcb.25.16.7158-7169.2005
Scholler, J. K., Myler, P. J., and Stuart, K. D. (1989). A novel telomeric gene conversion in Trypanosoma brucei. Mol. Biochem. Parasitol. 35, 11–19. doi: 10.1016/0166-6851(89)90137-0
Schwabl, P., Imamura, H., van den Broeck, F., Costales, J. A., Maiguashca-Sánchez, J., Miles, M. A., et al. (2019). Meiotic sex in Chagas disease parasite Trypanosoma cruzi. Nat. Commun. 10:3972. doi: 10.1038/s41467-019-11771-z
Silva, D. G. P., da Silva Santos, S., Nardelli, S. C., Mendes, I. C., Freire, A. C. G., Repolês, B. M., et al. (2018). The in vivo and in vitro roles of Trypanosoma cruzi Rad51 in the repair of DNA double strand breaks and oxidative lesions. PLoS Negl. Trop. Dis. 12:e0006875. doi: 10.1371/journal.pntd.0006875
Stortz, J. A., Serafim, T. D., Alsford, S., Wilkes, J., Fernandez-Cortes, F., Hamilton, G., et al. (2017). Genome-wide and protein kinase-focused RNAi screens reveal conserved and novel damage response pathways in Trypanosoma brucei. PLoS Pathog. 13:e1006477. doi: 10.1371/journal.ppat.1006477
Sturm, N. R., Vargas, N. S., Westenberger, S. J., Zingales, B., and Campbell, D. A. (2003). Evidence for multiple hybrid groups in Trypanosoma cruzi. Int. J. Parasitol. 33, 269–279. doi: 10.1016/S0020-7519(02)00264-3
Tibayrenc, M., Kjellberg, F., and Ayala, F. J. (1990). A clonal theory of parasitic protozoa: the population structures of Entamoeba, Giardia, Leishmania, Naegleria, Plasmodium, Trichomonas, and Trypanosoma and their medical and taxonomical consequences. Proc. Natl. Acad. Sci. U.S.A. 87, 2414–2418. doi: 10.1073/pnas.87.7.2414
Tonegawa, S. (1983). Somatic generation of antibody diversity. Nature 302, 575–581. doi: 10.1038/302575a0
Ubeda, J. M., Raymond, F., Mukherjee, A., Plourde, M., Gingras, H., Roy, G., et al. (2014). Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS Biol. 12:e1001868. doi: 10.1371/journal.pbio.1001868
Vilenchik, M. M., and Knudson, A. G. (2003). Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. U.S.A. 100, 12871–12876. doi: 10.1073/pnas.2135498100
Ward, J. F. (1994). The complexity of DNA damage: relevance to biological consequences. Int. J. Radiat. Biol. 66, 427–432. doi: 10.1080/09553009414551401
Wickstead, B., Ersfeld, K., and Gull, K. (2004). The small chromosomes of Trypanosoma brucei involved in antigenic variation are constructed around repetitive palindromes. Genome Res. 14, 1014–1024. doi: 10.1101/gr.2227704
Wyrobek, A. J., Schmid, T. E., and Marchetti, F. (2005). Relative susceptibilities of male germ cells to genetic defects induced by cancer chemotherapies. J. Natl. Cancer Inst. Monogr. 34, 31–35. doi: 10.1093/jncimonographs/lgi001
Xu, Y., and Price, B. D. (2011). Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle 10, 261–267. doi: 10.4161/cc.10.2.14543
Yang, N., Galick, H., and Wallace, S. S. (2004). Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair (Amst.) 3, 1323–1334. doi: 10.1016/j.dnarep.2004.04.014
Yonetani, Y., Hochegger, H., Sonoda, E., Shinya, S., Yoshikawa, H., Takeda, S., et al. (2005). Differential and collaborative actions of Rad51 paralog proteins in cellular response to DNA damage. Nucleic Acids Res. 33, 4544–4552. doi: 10.1093/nar/gki766
Zhang, W. W., Lypaczewski, P., and Matlashewski, G. (2020). Application of CRISPR/Cas9-mediated genome editing in Leishmania. Methods Mol Biol. 2116, 199–224. doi: 10.1007/978-1-0716-0294-2_14
Zhao, F., Kim, W., Kloeber, J. A., and Lou, Z. (2020). DNA end resection and its role in DNA replication and DSB repair choice in mammalian cells. Exp. Mol. Med. 52, 1705–1714. doi: 10.1038/s12276-020-00519-1
Keywords: DNA double-strand breaks, homologous recombination, DNA repair, Trypanosoma brucei, Trypanosoma cruzi, Leishmania spp.
Citation: da Silva MS (2021) DNA Double-Strand Breaks: A Double-Edged Sword for Trypanosomatids. Front. Cell Dev. Biol. 9:669041. doi: 10.3389/fcell.2021.669041
Received: 17 February 2021; Accepted: 29 March 2021;
Published: 15 April 2021.
Edited by:
Andrew Burgess, ANZAC Research Institute, AustraliaReviewed by:
Mathew J. K. Jones, University of Queensland, AustraliaCopyright © 2021 da Silva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo Santos da Silva, bWFyY2Vsby5zYW50b3Mtc2lsdmFAdW5lc3AuYnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.