 Xiaoyan Zhang1,2*
Xiaoyan Zhang1,2*- 1College of Biomedicine and Health, Huazhong Agricultural University, Wuhan, China
- 2College of Life Science and Technology, Huazhong Agricultural University, Wuhan, China
As the central hub in the secretory and endocytic pathways, the Golgi apparatus continually receives the flow of cargos and serves as a major processing station in the cell. Due to its dynamic nature, a sophisticated and constantly remodeling mechanism needs to be set up to maintain the Golgi architecture and function in the non-stop trafficking of proteins and lipids. Abundant evidence has been accumulated that a well-organized Golgi structure is required for its proper functions, especially protein glycosylation. Remarkably, altered glycosylation has been a hallmark of most cancer cells. To understand the causes of Golgi defects in cancer, efforts have been made to characterize Golgi structural proteins under physiological and pathological conditions. This review summarizes the current knowledge of crucial Golgi structural proteins and their connections with tumor progression. We foresee that understanding the Golgi structural and functional defects may help solve the puzzle of whether glycosylation defect is a cause or effect of oncogenesis.
Introduction
Cancer ranks as one of the leading causes of death globally. Governments, research institutes, and pharmaceutical firms spend billions of dollars into the fight against this terrible human disease. Even with the pleasing progress in the tumorigenesis theory, the powerful therapy is still limited due to several real difficulties. The majority of tumors are detected at an advanced stage, leading to unsuccessful targeted treatment. Remarkably, the existence of tumor heterogeneity and cancer stem cells is largely accounted for tumor recurrence and later drug resistance (Barkeer et al., 2018). Despite the significant improvement of traditional methods, such as chemotherapy, radiation therapy, surgery, and the advent of the targeted therapy, only a tiny percentage of patients benefit (Ayyar et al., 2016; Costa et al., 2020). Therefore, the identification of common and reliable targets and the development of related new-targeted therapies are crucial for cancer study and the drug industry (Costa et al., 2020).
The discovery of the difference between normal and tumor cells is the key for targeted therapy. Fortunately, altered glycosylation of cell surface proteins and lipids, and/or extracellular vesicles, are well accepted as a hallmark of cancer cells (Pinho and Reis, 2015; Martins et al., 2021). However, the complexity of the glycosylation process and the underlying regulatory mechanisms make it up-to-date a tough nut to crack for deep investigation. Glycosylation is the most complicated and diverse post-translational modification (PTM), with the attachment of multiple kinds of glycans (carbohydrates) to proteins and lipids. It requires coordinated actions of different glycosyltransferases, glycosidases, nucleotide sugar transporters, and appropriate substrates in an orchestrated manner (Moremen et al., 2012; Hennet and Cabalzar, 2015). Unlike other general types of PTMs, such as phosphorylation and ubiquitination, which occurs in the cytosol or nucleus, the majority of glycosylation processes, except O-GlcNAcylation, happen in the lumen of membranous organelles, the endoplasmic reticulum (ER) and the Golgi apparatus (or the Golgi for simplicity). The limited and narrow space of ER/Golgi lumen may promise the efficacy and accuracy of this complicated modification which function in various crucial cellular events, such as signaling, cell–cell communication, cell-extracellular matrix (ECM) interaction (Moremen et al., 2012). The fact that altered Golgi structure and protein glycosylation pattern have been widely observed in various cancer types prompts researchers to study the mechanism of how Golgi structure regulates protein glycosylation. In this review, we will summarize the recent findings of the correlation between changes of Golgi structural proteins and alterations of glycosylation in tumorigenesis and comment on the enigmatic connection between Golgi morphological alteration and tumor development, aiming to provide insight that may help develop novel cancer therapies by targeting Golgi structural and functional defects.
Glycosylation Is a Common and Crucial Modification
Glycans present in all known living organisms and are as essential as nucleic acids, proteins, lipids, and metabolites (Varki, 2017). In mammals, monosaccharides are linked via a glycosidic bond to form branched or unbranched chains. Such complex glycan linkages are attached to various macromolecules to generate glycoproteins, glycolipids, GPI-anchored proteins, and proteoglycans, which most often cover the cell surface or are secreted as extracellular materials. Except for the single sugar modification O-GlcNAcylation, which refers to the addition of β-N-acetylglucosamine (GlcNAc) onto serine or threonine residues (Ser/Thr) of the targeted protein in the cytosol or nucleus (Bond and Hanover, 2015), oligosaccharides on proteins are primarily divided into two categories, N-linked glycans and O-linked glycans, both of which are commonly found on secretory proteins and the extracellular domains of integral membrane proteins.
About half of all human proteins are glycoproteins, and most of them are N-glycosylated (Apweiler et al., 1999). N-glycans are synthesized as a lipid-linked oligosaccharide (LLO) precursor. When a new protein is synthesized, the 14-sugar chain GlcNAc2Man9Gluc3 of the LLO is transferred by the oligosaccharyltransferase (OST) to the amide group of the asparagine residue in the Asn-X-Ser/Thr motif, where X is any amino acid except proline (Kelleher and Gilmore, 2006). Before the glycoprotein leaves the ER, all three glucose residues and one particular mannose residue are removed. The resulting N-glycans are referred to as the high-mannose subtype and further trimmed in the cis-Golgi. Subsequently, the decoration of GlcNAc on mannose yields sugar branches in the medial-Golgi. Attachment of galactose, sialic acid, and fucose in the trans-Golgi generates complex N-glycans (Stanley et al., 2009). One single protein could possess multiple sugar chains added to different amino acids. Notably, sugar chains could be processed diversely, resulting in hybrid N-glycans which harbor both high-mannose and complex characteristics (Ungar, 2009). Therefore, the precise N-glycan modifications are generated by the accurate removal and addition of sugars in the Golgi, depending on the sequential distribution of the glycosylation enzymes in different cisternae. The same holds true for the nucleotide sugar transporters, which need to be precisely present in the right cisternae (Hirschberg et al., 1998).
Unlike the ER origination of N-glycosylation, O-glycosylation is more diversified and predominantly processed in the Golgi. There are two main types of O-glycans in mammalian cells: the matrix glycosaminoglycan (GAG) chains on proteoglycans and the most common mucin-type glycans (Brockhausen et al., 2009). The repeating disaccharides of unbranched GAG chains are attached to the serine residues on the core proteins of proteoglycans through a common tetrasaccharide linker (xylose–Gal–Gal–glucuronic acid). The disaccharide units contain either N-acetylgalactosamine (GalNAc) or GlcNAc, and a uronic acid, which are extended in the earlier Golgi. The frequent sulfation modification on the disaccharide then occurs in the trans-Golgi (Stanley, 2011). Mucin-type O-glycosylation initiates with the attachment of a GalNAc onto the Ser/Thr residues in a glycoprotein to form GalNAcα1-Ser/Thr, which is also called the tumor-associated Tn antigen (Ju et al., 2014). A family of enzymes known as polypeptide GalNAc-transferases (ppGalNAcTs) catalyzes this reaction. The Tn antigen is usually a precursor and followed by the addition of galactose, GlcNAc, or GalNAc to form core O-glycan structures. The critical step is to transfer galactose by the enzyme termed T-synthase (Core 1 β3-galactosyltransferase, C1GalT1) to form the common core 1 O-glycan (or the T antigen). The T antigen can be further processed into core 2 by core 2 GlcNAc transferases (C2GnTs) to generate GlcNAcβ1–6(Galβ1–3) GalNAcα1-Ser/Thr. The core 1 and/or core 2 O-glycans are ubiquitously expressed in humans. Although O-glycan structures are generally shorter than N-glycans, core 1–4 structures could be further extended to generate diverse glycan chains, such as polyLacNAc, Lewis antigens, and different blood group antigens (Kudelka et al., 2015).
Aberrant Glycosylation Is a Hallmark of Cancer
Aberrant glycosylation frequently occurs in cancer, playing pivotal roles in cancer progression and metastasis, cell–cell interaction, and epithelial-mesenchyme transition (EMT) (Taniguchi and Kizuka, 2015). Altered glycosylation associated with cancer usually includes the overexpression of Tn and T antigen, and their sialylated counterparts (Sialyl-Tn), sialylated Lewis blood group (SLea and SLex), as well as complex branched N-glycans, including β1,6-GlcNAc branching, bisecting GlcNAc, and core fucose (Taniguchi and Kizuka, 2015; Schneider et al., 2017; Peixoto et al., 2019). These features are most often observed in advanced solid tumors and often correlate with poor survival, indicating that universal mechanisms need to be uncovered. Genetically, protein levels of the responsive glycosylation enzymes are frequently reported dysregulated. For instance, upregulated expression of sialyltransferases seems to be a major cause for SLea/x expression (Kudelka et al., 2015; Bhide and Colley, 2017). Besides, altered expression of the active T-synthase or Cosmc, an ER-localized molecular chaperone required for the stability and activity of T-synthase, can lead to the overexpression of the Tn antigen (Ju et al., 2014). Other than the genetic regulation, the localization changes of glycosylation enzymes could also cause adverse expression of Tn antigen. The activation of the proto-oncogene tyrosine kinase Src could stimulate the COPI trafficking machinery-dependent relocation of GalNAc-transferases (GALNTs) from Golgi to the ER, which leads to increased activity of GALNTs and enhanced cellular Tn level. Therefore, the GALA (GALNT Activation) pathway provided a mechanism of how the membrane trafficking modulates protein glycosylation (Gill et al., 2010; Bard and Chia, 2016). Remarkably, ER-localization of GALNTs in the GALA pathway induces O-Glycosylation of the matrix metalloprotease MMP14 and ER-resident Calnexin, thus drives MMP14 activation and Calnexin/ERp57 cell surface distribution, respectively, both of which promote ECM degradation and tumor development (Nguyen et al., 2017; Ros et al., 2020). Collectively, the GALA pathway shed light on a complete picture of how the signal of oncogene converts to Golgi glycosylation enzyme mis-location, and finally, a vicious circle of tumor development. However, therapeutical options by targeting the featured antigens on cancer cells are very limited due to the complexity of glycans on the cell surface. More information on the underlying mechanisms that regulate glycosylation is deeply needed for future targeting.
Glycan processing certainly has to be tightly supervised. Different from proteins and nucleic acids, glycan structures are not template encoded. Intriguingly, glycosylation enzymes tend to form homomers, or a variety of functionally relevant acting glycosyltransferases form heteromers for sequential modification, providing a regulation mechanism from the enzyme part (Kellokumpu et al., 2016; Khoder-Agha et al., 2019). Remarkably, as the major manufacturing and supply chain, the Golgi has evolved a specific structure to promise proper protein glycosylation.
Changes of Golgi Structural Proteins in Cancer Cells
The Golgi functions as the center in the secretory pathway and receives the uttermost cargos from the ER, modifies and sorts them inside or outside the cell. The cargos, including proteins and lipids, are subjected to extensive Golgi modifications, such as glycosylation, sulfation, phosphorylation, and proteolysis. The Golgi has evolved a particular stacked and ribbon-like structure in mammalian cells to perform its crucial functions. Each polarized stack is usually surrounded with transport vesicles and defined with three separate modules: the cis-Golgi network (CGN); the stacked cis-, medial-, and trans-Golgi cisternae; and the trans-Golgi network (TGN) (Klumperman, 2011; Day et al., 2013). Each cisterna was specialized with distinct enzymes for sequential modification of appropriate substrates.
The multi-compartment stack structure of the Golgi provides an optimal setting for glycosylation. It not only organizes different glycosidases, glycosyltransferases and sugar transporters in an ordered structure and so they can sequentially modify cargo molecules, but also provides each enzyme with an optical microenvironment (e.g., pH, ion, lipid composition, etc.) to maximize its activity (Rivinoja et al., 2012; Wang et al., 2020). Moreover, as the trafficking and processing center of the exocytic and endocytic secretory pathways, the Golgi constantly receives cargo molecules from the ER and endosomes, and thus it needs to maintain its dynamic stack structure to host the Golgi enzymes, and at the same time, allow a constant cargo flow through the Golgi stack at a steady speed. This is not a trivial task. Indeed, there are two seemingly contradictory hypothetical models (and derivatives from each) over the past decades in this field to explain the mechanism of intra-Golgi trafficking, the vesicular transport and the cisternal maturation model. It is possible that both models may not be mutually exclusive and that hybrid models of the two may exist, depending on the cell type and physiological status of the cell. More importantly, both models employ the same set of Golgi building blocks, the Golgi structural proteins, often referred to as the Golgi matrix proteins. Since the first introduction of the concept of “Golgi matrix” (Slusarewicz et al., 1994), several Golgi structural proteins have been identified to be required to maintain Golgi structure and function, including GRASPs and Golgins. Dysregulation of the Golgi structure and Golgi structural proteins has been highly related to glycosylation defects (Hennet and Cabalzar, 2015; Li et al., 2019a). Therefore, it is legitimate to speculate that the dysregulation of Golgi structure may account for the altered glycosylation in tumor progression. The changes of a few critical Golgi structural proteins reported in tumor metastasis are discussed below and depicted in Figure 1.
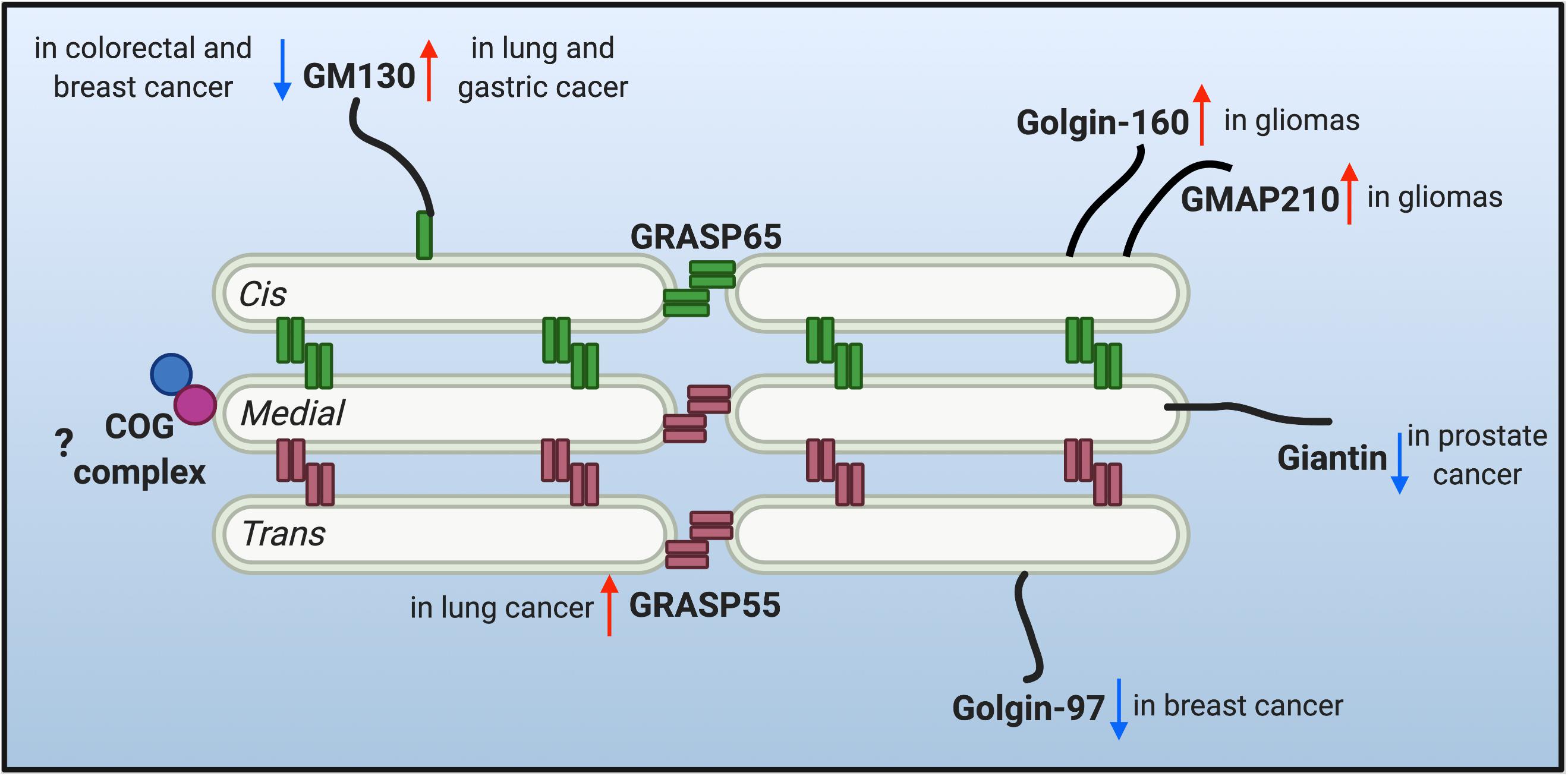
Figure 1. Protein level alteration of the reported Golgi structural proteins in various cancer types. Golgi reassembly-stacking proteins (GRASPs) (GRASP65 and GRASP55) and golgin families of proteins contribute to the structural scaffold that defines the Golgi architecture, and are conceived as the Golgi matrix. Altered protein levels of the Golgi matrix are discovered in different cancer types and may facilitate tumor progression. Upward red arrows indicate upregulated protein level, while downward arrows indicate down-regulated protein level of different matrix proteins in different cancer cells. Few studies are correlating the tethering conserved oligomeric Golgi (COG) complex with tumor metastasis, so a question mark is shown.
GRASP55
Following the establishment of the Golgi matrix hypothesis that Golgi structure formation depends on a network of Golgi structural proteins, Golgi reassembly-stacking protein of 65 kDa (GRASP65) and GRASP55 were identified, characterized, and named for their specific roles in Golgi stack formation (Barr et al., 1997; Shorter et al., 1999). In animal cells, GRASP65 is present in cis-Golgi, while GRASP55 is more distributed in the distal Golgi cisternae. Both GRASPs harbor an N-terminal conserved GRASP domain which can form oligomers and glue the adjacent cisternae together into stacks. GRASPs also contain a less conserved C-terminal serine/proline-rich (SPR) domain, which is highly regulated by PTM, such as phosphorylation and O-GlcNAcylation (Zhang et al., 2018). Phosphorylation of the C-terminus of GRASPs disrupts the oligomerization property of the N-terminal GRASP domain and thus Golgi stack and ribbon in mitosis, facilitating mitotic Golgi fragment formation (Wang et al., 2003; Wang and Seemann, 2011). Dephosphorylation of GRASPs in telophase recovers the compact Golgi morphology in the peripheral area of the nucleus (Tang and Wang, 2013). Therefore, the featured structures of GRASPs endow them with complementary roles in Golgi stack formation and cisternae-specific ribbon linking with the help of Mena (Tang et al., 2016) and DjA1 (Li et al., 2019b) for GRASP65.
With the stacking role of GRASPs in the Golgi, it is not surprising to see that GRASPs knockdown or knockout reduces the abundance and diversity of global N-glycosylation glycan and alters the glycoprotein composition at the cell surface (Xiang et al., 2013; Bekier et al., 2017). The cause of defective glycosylation upon GRASPs depletion can be interpreted in two ways. Firstly, Golgi unstacking enhances vesicle budding, and therefore there is not enough processing time onto the cargos from the glycosylation enzymes (Xiang et al., 2013). Moreover, the cisterna-specific ribbon linking of adjacent stacks is disturbed, and thus the proper Golgi compartmentalization, enzymes localization and glycosylation is violated (Jarvela and Linstedt, 2014). However, it is important to note that there is a discrepancy regarding the role of GRASPs in Golgi cisternal stacking, and recent in vivo studies in mouse showed that lack of both GRASPs did not unstack the cisternal core but disconnect the stacks laterally from each other (Grond et al., 2020). The alternative interpretation for this controversy is that the N-terminus of GRASP55 may still be translated in the conditional double knockout (KO) mice and functional for stacking (Zhang and Wang, 2020). Therefore, a proper GRASPs double KO mouse is still needed to fully understand their roles in Golgi architecture. Most recently, acute double depletion of both GRASPs with a degron system also demonstrates that both GRASPs are dispensable for Golgi stacking but mediates Golgi ribbon linking together with GM130 and Golgin-45 (Zhang and Seemann, 2021). Hence, other Golgi stacking factors need further investigation.
In light of the significant roles of GRASPs in Golgi structure formation and proper protein glycosylation, there were speculations between the dysregulation of GRASPs and tumor-associated glycosylation defects. Still, no effect was made to directly correlate GRASPs expression with tumor progression until Kurie and colleagues recently reported that upregulation of GRASP55 was associated with lung adenocarcinoma (LUAD) progression (Tan et al., 2021). Loss of TP53 increased the expression of GRASP55 by silencing miR-34a, a p53 transcriptional target. Upregulated GRASP55 drives tumor progression by enhancing the secretion of pro-tumorigenic effector proteins, such as IGFBP2 and SPP1. Disruption of GRASP55 and Golgin-45 interaction by a small molecule, GRASPIN, inhibits the secretion and reduces the enhanced metastasis caused by TP53-KO in A549 orthotopic lung tumors (Tan et al., 2021). Nevertheless, no glycosylation alterations are investigated in this report which may need further study. It is necessary as GRASPs depletion reduces cell adhesion and accelerates cell proliferation (Ahat et al., 2019), both highly associated with tumor progression. The success of GRASPIN in inhibiting tumor growth suggests a possibility that GRASP55 or its association with crucial proteins may serve as potential therapeutic targets in cancer (Tan et al., 2021).
GM130
The golgins are a family of predominantly coiled-coil proteins crucial for vesicles tethering to the Golgi (Witkos and Lowe, 2015; Gillingham and Munro, 2016; Muschalik and Munro, 2018). While some golgins are anchored to the Golgi membrane with a signal transmembrane domain at the C-terminus, a majority of them are peripheral membrane proteins that are recruited from the cytoplasm to the cytoplasmic face of the Golgi by association with Rab, Arf, and Arl families small GTPases (Munro, 2011; Kulkarni-Gosavi et al., 2019; Li et al., 2019a). These associations allow the selective localization of a particular golgin to a distinct Golgi sub-compartment (Witkos and Lowe, 2015). Collectively, different golgins localize at specific regions of the Golgi and mediate the tethering of different types of vesicles to specific Golgi subcompartments (Wong and Munro, 2014; Lowe, 2019). For instance, golgins at the cis-Golgi mainly tether vesicles coming from the ER, while those at the trans-Golgi receive vesicles from the endocytic pathway. Golgins located within the Golgi stack are responsible for intra-Golgi vesicle trafficking. The specificity of vesicle traffic by golgins is one of the mechanisms that the Golgi employs for precise protein processing (Gillingham and Munro, 2016).
GM130 is the first identified Golgi matrix protein and the best-characterized golgin. It is mainly present in the cis-Golgi, anchoring adjacent stacks via interaction with GRASP65 at its C-terminus and p115 at its N-terminus (Nakamura et al., 1995; Nakamura et al., 1997; Barr et al., 1998). Depletion of GM130 in culture cells leads to the disconnection of the Golgi ribbon and protein glycosylation defects (Puthenveedu et al., 2006). It has been reported that the GM130–GRASP65 complex directly mediates the localization of the crucial T-synthase (C1GalT1) onto the cis-Golgi (Petrosyan et al., 2012). GM130 is attracting considerable interest in the past decade, and accumulating studies support it as a promising anticancer target (Chang et al., 2012). However, there are discrepancies about the relevance between GM130 protein level and tumor progression. Loss of GM130 expression is frequently observed in colorectal and breast cancer patients. Silencing GM130 reduces Cdc42 activity on the Golgi and down-regulates E-cadherin expression, indicating a loss in cell polarity and epithelial identity (Baschieri et al., 2014; Baschieri and Farhan, 2015). GM130 deletion is also associated with increased migration and invasion of breast cancer cells (Baschieri et al., 2015). In contrast, increased invasion and poor prognosis are associated with high levels of GM130 in lung and gastric tumors (Chang et al., 2012; Zhao et al., 2015). Accordingly, downregulation of GM130 decreases angiogenesis and cancer cell invasion, and suppresses tumorigenesis in the lung cancer mice model (Chang et al., 2012). In gastric cancer cells, GM130 depletion increased the level of E-cadherin, which is an epithelial marker but reduced mesenchymal marker, N-cadherin and vimentin, suppressing cell invasion and tumor formation (Zhao et al., 2015). A recent study of the tumor suppressor PTEN’s role in pre-mRNA splicing may support the connection between the high level of GM130 and tumor progression. PTEN deficiency in cancer cells induces aberrant splicing of GM130, resulting in increased GM130 protein level and dramatic Golgi extension and secretion (Shen et al., 2018). Therefore, GM130 levels should be tightly regulated, and alterations in its expression may have adverse effects. This notion is supported by a study that GM130 is a primary target of the Golgi quality control mechanism, the 26S proteasomes mediated Golgi Apparatus-Related Degradation (GARD) (Eisenberg-Lerner et al., 2020).
Golgin-160 and GMAP210
Golgin-160 and GMAP210 (all called TRIP11) are enriched on the cis-side of the Golgi (Hicks et al., 2006; Cardenas et al., 2009). Golgin-160 has been reported to recruit the dynein microtubule motor to the Golgi, which is crucial for Golgi positioning and structure maintenance (Yadav et al., 2012). By tethering transport vesicles, GMAP210 is engaged in both ER-to-Golgi anterograde and intra-Golgi retrograde trafficking (Roboti et al., 2015). Glial cell line-derived neurotrophic factor (GDNF) triggers glioma cell migration and invasion. Increased expression of golgin-160 and GMAP210 and enlarged Golgi area were observed after GDNF treatment (Tang et al., 2019). Consistently, golgin-160 and GMAP210 depletion reduced the migration and invasion of U251 cells (Tang et al., 2019). In combination, the upregulation of golgin-160 and GMAP210 correlates Gliomas progression, although the detailed mechanisms await further investigation.
Giantin
Giantin, the largest golgin, is mainly distributed at the rims of Golgi cisternae and inserted into the membrane by a C-terminal transmembrane domain (Linstedt et al., 1995). It functions in laterally linking the Golgi cisternae into ribbon structure (Koreishi et al., 2013). There is decreased level of the extracellular hyaluronan and impaired protein glycosylation in giantin mutant embryos, suggesting its role in proper protein glycosylation (Lan et al., 2016). The Golgi targeting of core 2 glycosyltransferase, C2GnT-L and C2GnT-M, is mediated by giantin (Petrosyan et al., 2012; Petrosyan et al., 2014). Interestingly, defective giantin in prostate cancer cells leads to a shift of glycosyltransferases and α-mannosidase IA from giant in to GM130–GRASP65 site, where the full glycosylation processing is prevented, resulting in high mannose N-glycan at the cell surface (Cheng et al., 2020). Therefore, the appearance of cell surface high mannose N-glycans may serve as markers of malignant prostate cancer cells (Bhat et al., 2017; Cheng et al., 2020).
Golgin-97
The trans-Golgi network (TGN) is a major sorting station where newly synthesized proteins and lipids are sent out from the Golgi to different destinations. TGN also serves as the entry point for endocytic cargos in retrograde transport. Golgin-97 acts as a scaffold molecule and is recruited onto the TGN by interacting with Arl1. Depletion of golgin-97 impairs the traffic of Shiga toxin subunit B from early endosomes to the TGN (Lu et al., 2004) and blocks the exit of E-cadherin cargo from the TGN (Lock et al., 2005). Low expression of golgin-97 has been reported to correlate with poor patient survival and increased invasiveness in breast cancer. Mechanism investigation showed that Golgin-97 depletion significantly reduces the IκBα protein levels and activates NF-κB, which can promote cell migration and invasion (Hsu et al., 2018).
Golgi Dispersal as an Indicator of Tumor Progression
The notion that a well-organized Golgi architecture promises its proper function has been widely accepted. Dysregulation of Golgi structural proteins usually correlates with Golgi morphological changes, and Golgi dispersal is frequently observed in various types of cancer cells. Thus, it is simple to speculate that a fragmented or dispersed Golgi morphology may indicate tumor progression. This idea was supported when Golgi phosphoprotein 3 (GOLPH3), the first Golgi resident oncoprotein, was identified (Scott et al., 2009). Compelling evidence demonstrates that tumor cells gain metastatic capacity through a GOLPH3-dependent Golgi membrane dispersal process which enhances vesicular release (Buschman et al., 2015). GOLPH3 is recruited to TGN by binding to phosphatidylinositol-4-phosphate [PI(4)P] in a PITPNC1/RAB1B–containing protein complex dependent manner (Halberg et al., 2016). The PI(4)P-GOLPH3/MYO18A/F-actin module then generates a tensile force that stretches the Golgi membranes and facilitates vesicle budding from the Golgi to the plasma membrane (PM) trafficking (Dippold et al., 2009). Consistently, DNA damage-induced phosphorylation of GOLPH3 by the DNA damage protein kinase (DNA-PK) increases the association between GOLPH3 and MYO18A, leading to enhanced stretching force and thus Golgi dispersal. The altered Golgi morphology and trafficking of cargos to the PM following DNA damage results in cell survival. Therefore, massive Golgi fragmentation has been correlated to tumorigenesis since it is linked to cell survival and resistance to killing by DNA-damaging agents. However, the role of MYO18A/F-actin in Golgi morphology regulation is still controversial, since there is a report that MYO18A does not localize on the Golgi and reduced MYO18A expression does not alter Golgi morphology (Bruun et al., 2017). Hence, further efforts are needed to verify the mechanism of how GOLPH3 modulates Golgi architecture. Other than the morphological effect on the Golgi, recent studies provided evidence that overexpression of GOLPH3 exerts its tumor-promoting activities via enhancing the production of specific growth-inducing glycosphingolipids (GSL). Specifically, GOLPH3 functions as an adaptor between a selectively set of Golgi glycosylation enzymes, especially the GSL biosynthetic pathway enzymes, and COPI coatomer (Eckert et al., 2014; Rizzo et al., 2019). The adaptor role of GOLPH3 mediates the incorporation of GOLPH3 clients into the COPI recycling vesicles, but hinders the clients trafficking to the lysosomes and thus increases the protein levels of glycosylation enzymes (Rizzo et al., 2019). The interaction among GOLPH3 and sialyltransferases also contributes to the oncogenic action of GOLPH3, which efficiently upregulates 2,6-sialylation of β1-integrins and thus enhances integrin-mediated cell migration and signaling pathways (Isaji et al., 2014; Sechi et al., 2020).
However, opposite morphological observations were made in studies on the Golgi during epithelial-to-mesenchymal transition (EMT) of lung cancer cells. Rather than causing Golgi dispersal, EMT led to Golgi compaction with improved ribbon linking and cisternal stacking (Tan et al., 2017). In contrast to GOLPH3, depletion of the Golgi scaffolding protein, PAQR11, disperses the Golgi and impairs anterograde vesicle transport to the plasma membrane. Consistently, the high expression level of PAQR11 is correlated with compact Golgi, EMT, and poor prognosis in human tumors (Tan et al., 2017). An alternative explanation is that EMT is not necessary for metastasis or may be relevant to metastasis in limited tumor types. Collectively, it is still mysterious about the relevance between Golgi morphology and tumor status.
We can envision that the Golgi is a highly dynamic structure, actively sensing and reacting to stress stimuli in the surrounding environment to re-establish Golgi homeostasis for adaption. A systematic approach by RNAi screen demonstrated that depletion of approximately 20% of the signaling genes induced diffused, fragmented, or condensed Golgi. These identified Golgi organization regulators affect the general Golgi functions, including protein secretion and glycan biosynthesis. However, there is not a clear correlation between morphological phenotypes and functional perturbations, indicating a complex network of interaction between signaling cascades and Golgi activities (Chia et al., 2012). The fact is the Golgi structure could be stretched or recovered according to the requirement based on physiological conditions. For instance, the Golgi is extensively fragmented in mitosis and returns to compaction in interphase during the cell cycle (Huang and Wang, 2017). It is also experimentally accurate that the Golgi returns quickly back to normal after drug washout, such as nocodazole (Tang et al., 2016) or Brefeldin A washout (Houghton et al., 2009).
Outlook
Defective glycosylation of plasma membrane and extracellular matrix has been identified as a hallmark of tumor metastasis for many years. Multiple sets of antibodies have also been developed to detect tumor-associated glycans, while the benefits are still limited due to the tumor heterogeneity and the complexity of glycans (Costa et al., 2020). Therefore, the fundamental reasons for altered glycosylation in cancer cells await deep investigation. As summarized above, even though the exact mechanisms of how GRASP55, Golgin-160, GMAP210, and Golgin-97 regulates protein glycosylation are still missing in cancer cells, GM130, Giantin, and GOLPH3 were characterized to directly modulate specific localization of glycosylation enzymes in the right cisternae, indicating that Golgi structure or Golgi structural proteins probably functions in providing an ordered space for proper protein glycosylation. Thus, it is reasonable to speculate that altered protein levels of Golgi structural proteins might bring about the disorganized distribution of glycosylation enzymes and resultant cancer-related glycosylation defects. However, the elusive role of other unexplored Golgi structural proteins in protein glycosylation regulation needs future investigation. Moreover, the vesicle transportation machinery, such as the retrograde trafficking tethering complex Conserved Oligomeric Golgi (COG), is involved in regulating the correct localization of glycosylation enzymes in Golgi. Multiple mutations in different COG subunits have been identified as a cause for Congenital Disorders of Glycosylation (CDG) in humans (Laufman et al., 2013), but very limited studies reported the correlation between COG expression and cancer progression. In addition, advanced experimental approaches, such as super-resolution microscopy, need to be employed for a precise view of the exact localization of glycosylation enzymes in cancer cells. Therefore, integrative studies by combining glycobiology, cell biology, and clinical investigation are required to generate a complete picture of cancer-related glycosylation and targeted therapy.
The GALA pathway provided evidence that mis-location of Golgi glycosylation enzymes promotes tumor development, nevertheless, the crucial question “does Golgi alteration facilitates oncogenesis” requires deep investigation. Does the Golgi passively respond to the microenvironment of developing tumor and change its morphology and therefore the glycan structure, or actively maintain and recover the original status? To answer this question, more efforts are needed to define the mechanism of Golgi structure formation, function, and response to cellular stresses. Besides, identifying signaling cascades that regulate cell proliferation, membrane trafficking, glycosylation, and tumor progression may result in major breakthroughs in Golgi physiology and tumor pathology.
Author Contributions
XZ conceived and wrote this manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (XZ, Grant Number: 32070693) and the Fundamental Research Funds for the Central Universities (Program Number: 11041910145).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank the Zhang laboratory members in the Huazhong Agricultural University for helpful discussion and Yanzhuang Wang from University of Michigan for his critical review and insightful comments of the manuscript.
References
Ahat, E., Xiang, Y., Zhang, X., Bekier, M. E. II, and Wang, Y. (2019). GRASP depletion-mediated Golgi destruction decreases cell adhesion and migration via the reduction of alpha5beta1 integrin. Mol. Biol. Cell 30, 766–777. doi: 10.1091/mbc.E18-07-0462
Apweiler, R., Hermjakob, H., and Sharon, N. (1999). On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8. doi: 10.1016/S0304-4165(99)00165-8
Ayyar, B. V., Arora, S., and O’kennedy, R. (2016). Coming-of-age of antibodies in cancer therapeutics. Trends Pharmacol. Sci. 37, 1009–1028. doi: 10.1016/j.tips.2016.09.005
Bard, F., and Chia, J. (2016). Cracking the glycome encoder: signaling, trafficking, and glycosylation. Trends Cell Biol. 26, 379–388. doi: 10.1016/j.tcb.2015.12.004
Barkeer, S., Chugh, S., Batra, S. K., and Ponnusamy, M. P. (2018). Glycosylation of cancer stem cells: function in stemness, tumorigenesis, and metastasis. Neoplasia 20, 813–825. doi: 10.1016/j.neo.2018.06.001
Barr, F. A., Nakamura, N., and Warren, G. (1998). Mapping the interaction between GRASP65 and GM130, components of a protein complex involved in the stacking of Golgi cisternae. EMBO J. 17, 3258–3268. doi: 10.1093/emboj/17.12.3258
Barr, F. A., Puype, M., Vandekerckhove, J., and Warren, G. (1997). GRASP65, a protein involved in the stacking of Golgi cisternae. Cell 91, 253–262. doi: 10.1016/S0092-8674(00)80407-9
Baschieri, F., Confalonieri, S., Bertalot, G., Di Fiore, P. P., Dietmaier, W., Leist, M., et al. (2014). Spatial control of Cdc42 signalling by a GM130-RasGRF complex regulates polarity and tumorigenesis. Nat. Commun. 5:4839. doi: 10.1038/ncomms5839
Baschieri, F., and Farhan, H. (2015). Endomembrane control of cell polarity: relevance to cancer. Small GTPases 6, 104–107. doi: 10.1080/21541248.2015.1018402
Baschieri, F., Uetz-Von Allmen, E., Legler, D. F., and Farhan, H. (2015). Loss of GM130 in breast cancer cells and its effects on cell migration, invasion and polarity. Cell Cycle 14, 1139–1147. doi: 10.1080/15384101.2015.1007771
Bekier, M. E. II, Wang, L., Li, J., Huang, H., Tang, D., Zhang, X., et al. (2017). Knockout of the Golgi stacking proteins GRASP55 and GRASP65 impairs Golgi structure and function. Mol. Biol. Cell 28, 2833–2842. doi: 10.1091/mbc.e17-02-0112
Bhat, G., Hothpet, V. R., Lin, M. F., and Cheng, P. W. (2017). Shifted Golgi targeting of glycosyltransferases and alpha-mannosidase IA from giantin to GM130-GRASP65 results in formation of high mannose N-glycans in aggressive prostate cancer cells. Biochim. Biophys. Acta Gen. Subj. 1861, 2891–2901. doi: 10.1016/j.bbagen.2017.08.006
Bhide, G. P., and Colley, K. J. (2017). Sialylation of N-glycans: mechanism, cellular compartmentalization and function. Histochem. Cell Biol. 147, 149–174. doi: 10.1007/s00418-016-1520-x
Bond, M. R., and Hanover, J. A. (2015). A little sugar goes a long way: the cell biology of O-GlcNAc. J. Cell Biol. 208, 869–880. doi: 10.1083/jcb.201501101
Brockhausen, I., Schachter, H., and Stanley, P. (2009). “O-GalNAc glycans,” in Essentials of Glycobiology, 2nd Edn, eds A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, et al. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press).
Bruun, K., Beach, J. R., Heissler, S. M., Remmert, K., Sellers, J. R., and Hammer, J. A. (2017). Re-evaluating the roles of myosin 18Aalpha and F-actin in determining Golgi morphology. Cytoskeleton (Hoboken) 74, 205–218. doi: 10.1002/cm.21364
Buschman, M. D., Xing, M., and Field, S. J. (2015). The GOLPH3 pathway regulates Golgi shape and function and is activated by DNA damage. Front. Neurosci. 9:362. doi: 10.3389/fnins.2015.00362
Cardenas, J., Rivero, S., Goud, B., Bornens, M., and Rios, R. M. (2009). Golgi localisation of GMAP210 requires two distinct cis-membrane binding mechanisms. BMC Biol. 7:56. doi: 10.1186/1741-7007-7-56
Chang, S. H., Hong, S. H., Jiang, H. L., Minai-Tehrani, A., Yu, K. N., Lee, J. H., et al. (2012). GOLGA2/GM130, cis-Golgi matrix protein, is a novel target of anticancer gene therapy. Mol. Ther. 20, 2052–2063. doi: 10.1038/mt.2012.125
Cheng, P. W., Davidson, S., and Bhat, G. (2020). Markers of malignant prostate cancer cells: Golgi localization of alpha-mannosidase 1A at GM130-GRASP65 site and appearance of high mannose N-glycans on cell surface. Biochem. Biophys. Res. Commun. 527, 406–410. doi: 10.1016/j.bbrc.2020.03.168
Chia, J., Goh, G., Racine, V., Ng, S., Kumar, P., and Bard, F. (2012). RNAi screening reveals a large signaling network controlling the Golgi apparatus in human cells. Mol. Syst. Biol. 8:629. doi: 10.1038/msb.2012.59
Costa, A. F., Campos, D., Reis, C. A., and Gomes, C. (2020). Targeting glycosylation: a new road for cancer drug discovery. Trends Cancer 6, 757–766. doi: 10.1016/j.trecan.2020.04.002
Day, K. J., Staehelin, L. A., and Glick, B. S. (2013). A three-stage model of Golgi structure and function. Histochem. Cell Biol. 140, 239–249. doi: 10.1007/s00418-013-1128-3
Dippold, H. C., Ng, M. M., Farber-Katz, S. E., Lee, S. K., Kerr, M. L., Peterman, M. C., et al. (2009). GOLPH3 bridges phosphatidylinositol-4- phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 139, 337–351. doi: 10.1016/j.cell.2009.07.052
Eckert, E. S., Reckmann, I., Hellwig, A., Rohling, S., El-Battari, A., Wieland, F. T., et al. (2014). Golgi phosphoprotein 3 triggers signal-mediated incorporation of glycosyltransferases into coatomer-coated (COPI) vesicles. J. Biol. Chem. 289, 31319–31329. doi: 10.1074/jbc.M114.608182
Eisenberg-Lerner, A., Benyair, R., Hizkiahou, N., Nudel, N., Maor, R., Kramer, M. P., et al. (2020). Golgi organization is regulated by proteasomal degradation. Nat. Commun. 11:409. doi: 10.1038/s41467-019-14038-9
Gill, D. J., Chia, J., Senewiratne, J., and Bard, F. (2010). Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J. Cell Biol. 189, 843–858. doi: 10.1083/jcb.201003055
Gillingham, A. K., and Munro, S. (2016). Finding the Golgi: golgin coiled-coil proteins show the way. Trends Cell Biol. 26, 399–408. doi: 10.1016/j.tcb.2016.02.005
Grond, R., Veenendaal, T., Duran, J. M., Raote, I., Van Es, J. H., Corstjens, S., et al. (2020). The function of GORASPs in Golgi apparatus organization in vivo. J. Cell Biol. 219:e202004191. doi: 10.1083/jcb.20200419106242020c
Halberg, N., Sengelaub, C. A., Navrazhina, K., Molina, H., Uryu, K., and Tavazoie, S. F. (2016). PITPNC1 recruits RAB1B to the Golgi network to drive malignant secretion. Cancer Cell 29, 339–353. doi: 10.1016/j.ccell.2016.02.013
Hennet, T., and Cabalzar, J. (2015). Congenital disorders of glycosylation: a concise chart of glycocalyx dysfunction. Trends Biochem. Sci. 40, 377–384. doi: 10.1016/j.tibs.2015.03.002
Hicks, S. W., Horn, T. A., Mccaffery, J. M., Zuckerman, D. M., and Machamer, C. E. (2006). Golgin-160 promotes cell surface expression of the beta-1 adrenergic receptor. Traffic 7, 1666–1677. doi: 10.1111/j.1600-0854.2006.00504.x
Hirschberg, C. B., Robbins, P. W., and Abeijon, C. (1998). Transporters of nucleotide sugars, ATP, and nucleotide sulfate in the endoplasmic reticulum and Golgi apparatus. Annu. Rev. Biochem. 67, 49–69. doi: 10.1146/annurev.biochem.67.1.49
Houghton, F. J., Chew, P. L., Lodeho, S., Goud, B., and Gleeson, P. A. (2009). The localization of the Golgin GCC185 is independent of Rab6A/A’ and Arl1. Cell 138, 787–794. doi: 10.1016/j.cell.2009.05.048
Hsu, R. M., Zhong, C. Y., Wang, C. L., Liao, W. C., Yang, C., Lin, S. Y., et al. (2018). Golgi tethering factor golgin-97 suppresses breast cancer cell invasiveness by modulating NF-kappaB activity. Cell Commun. Signal. 16:19. doi: 10.1186/s12964-018-0230-5
Huang, S., and Wang, Y. (2017). Golgi structure formation, function, and post-translational modifications in mammalian cells. F1000Res 6:2050. doi: 10.12688/f1000research.11900.1
Isaji, T., Im, S., Gu, W., Wang, Y., Hang, Q., Lu, J., et al. (2014). An oncogenic protein Golgi phosphoprotein 3 up-regulates cell migration via sialylation. J. Biol. Chem. 289, 20694–20705. doi: 10.1074/jbc.M113.542688
Jarvela, T., and Linstedt, A. D. (2014). Isoform-specific tethering links the Golgi ribbon to maintain compartmentalization. Mol. Biol. Cell 25, 133–144. doi: 10.1091/mbc.e13-07-0395
Ju, T., Aryal, R. P., Kudelka, M. R., Wang, Y., and Cummings, R. D. (2014). The Cosmc connection to the Tn antigen in cancer. Cancer Biomark. 14, 63–81. doi: 10.3233/CBM-130375
Kelleher, D. J., and Gilmore, R. (2006). An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16, 47R–62R. doi: 10.1093/glycob/cwj066
Kellokumpu, S., Hassinen, A., and Glumoff, T. (2016). Glycosyltransferase complexes in eukaryotes: long-known, prevalent but still unrecognized. Cell Mol. Life Sci. 73, 305–325. doi: 10.1007/s00018-015-2066-0
Khoder-Agha, F., Harrus, D., Brysbaert, G., Lensink, M. F., Harduin-Lepers, A., Glumoff, T., et al. (2019). Assembly of B4GALT1/ST6GAL1 heteromers in the Golgi membranes involves lateral interactions via highly charged surface domains. J. Biol. Chem. 294, 14383–14393. doi: 10.1074/jbc.RA119.009539
Klumperman, J. (2011). Architecture of the mammalian Golgi. Cold Spring Harb. Perspect. Biol. 3:a005181, doi: 10.1101/cshperspect.a005181
Koreishi, M., Gniadek, T. J., Yu, S., Masuda, J., Honjo, Y., and Satoh, A. (2013). The golgin tether giantin regulates the secretory pathway by controlling stack organization within Golgi apparatus. PLoS One 8:e59821. doi: 10.1371/journal.pone.0059821
Kudelka, M. R., Ju, T., Heimburg-Molinaro, J., and Cummings, R. D. (2015). Simple sugars to complex disease–mucin-type O-glycans in cancer. Adv. Cancer Res. 126, 53–135. doi: 10.1016/bs.acr.2014.11.002
Kulkarni-Gosavi, P., Makhoul, C., and Gleeson, P. A. (2019). Form and function of the Golgi apparatus: scaffolds, cytoskeleton and signalling. FEBS Lett. 593, 2289–2305. doi: 10.1002/1873-3468.13567
Lan, Y., Zhang, N., Liu, H., Xu, J., and Jiang, R. (2016). Golgb1 regulates protein glycosylation and is crucial for mammalian palate development. Development 143, 2344–2355. doi: 10.1242/dev.134577
Laufman, O., Freeze, H. H., Hong, W., and Lev, S. (2013). Deficiency of the Cog8 subunit in normal and CDG-derived cells impairs the assembly of the COG and Golgi SNARE complexes. Traffic 14, 1065–1077. doi: 10.1111/tra.12093
Li, J., Ahat, E., and Wang, Y. (2019a). Golgi structure and function in health, stress, and diseases. Results Probl. Cell Differ. 67, 441–485. doi: 10.1007/978-3-030-23173-6_19
Li, J., Tang, D., Ireland, S. C., and Wang, Y. (2019b). DjA1 maintains Golgi integrity via interaction with GRASP65. Mol. Biol. Cell 30, 478–490. doi: 10.1091/mbc.E18-10-0613
Linstedt, A. D., Foguet, M., Renz, M., Seelig, H. P., Glick, B. S., and Hauri, H. P. (1995). A C-terminally-anchored Golgi protein is inserted into the endoplasmic reticulum and then transported to the Golgi apparatus. Proc. Natl. Acad. Sci. U. S. A. 92, 5102–5105. doi: 10.1073/pnas.92.11.5102
Lock, J. G., Hammond, L. A., Houghton, F., Gleeson, P. A., and Stow, J. L. (2005). E-cadherin transport from the trans-Golgi network in tubulovesicular carriers is selectively regulated by golgin-97. Traffic 6, 1142–1156. doi: 10.1111/j.1600-0854.2005.00349.x
Lowe, M. (2019). The physiological functions of the golgin vesicle tethering proteins. Front. Cell Dev. Biol. 7:94. doi: 10.3389/fcell.2019.00094
Lu, L., Tai, G., and Hong, W. (2004). Autoantigen Golgin-97, an effector of Arl1 GTPase, participates in traffic from the endosome to the trans-golgi network. Mol. Biol. Cell 15, 4426–4443. doi: 10.1091/mbc.e03-12-0872
Martins, A. M., Ramos, C. C., Freitas, D., and Reis, C. A. (2021). Glycosylation of cancer extracellular vesicles: capture strategies, functional roles and potential clinical applications. Cells 10:109. doi: 10.3390/cells10010109
Moremen, K. W., Tiemeyer, M., and Nairn, A. V. (2012). Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462. doi: 10.1038/nrm3383
Munro, S. (2011). The golgin coiled-coil proteins of the Golgi apparatus. Cold Spring Harb. Perspect. Biol. 3:a005256. doi: 10.1101/cshperspect.a005256
Muschalik, N., and Munro, S. (2018). Golgins. Curr. Biol. 28, R374–R376. doi: 10.1016/j.cub.2018.01.006
Nakamura, N., Lowe, M., Levine, T. P., Rabouille, C., and Warren, G. (1997). The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell 89, 445–455. doi: 10.1016/S0092-8674(00)80225-1
Nakamura, N., Rabouille, C., Watson, R., Nilsson, T., Hui, N., Slusarewicz, P., et al. (1995). Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 131, 1715–1726. doi: 10.1083/jcb.131.6.1715
Nguyen, A. T., Chia, J., Ros, M., Hui, K. M., Saltel, F., and Bard, F. (2017). Organelle specific O-glycosylation drives MMP14 activation, tumor growth, and metastasis. Cancer Cell 32:e636. doi: 10.1016/j.ccell.2017.10.001
Peixoto, A., Relvas-Santos, M., Azevedo, R., Santos, L. L., and Ferreira, J. A. (2019). Protein glycosylation and tumor microenvironment alterations driving cancer hallmarks. Front. Oncol. 9:380. doi: 10.3389/fonc.2019.00380
Petrosyan, A., Ali, M. F., and Cheng, P. W. (2012). Glycosyltransferase-specific Golgi-targeting mechanisms. J. Biol. Chem. 287, 37621–37627. doi: 10.1074/jbc.C112.403006
Petrosyan, A., Holzapfel, M. S., Muirhead, D. E., and Cheng, P. W. (2014). Restoration of compact Golgi morphology in advanced prostate cancer enhances susceptibility to galectin-1-induced apoptosis by modifying mucin O-glycan synthesis. Mol. Cancer Res. 12, 1704–1716. doi: 10.1158/1541-7786.MCR-14-0291-T
Pinho, S. S., and Reis, C. A. (2015). Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer 15, 540–555. doi: 10.1038/nrc3982
Puthenveedu, M. A., Bachert, C., Puri, S., Lanni, F., and Linstedt, A. D. (2006). GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat. Cell Biol. 8, 238–248. doi: 10.1038/ncb1366
Rivinoja, A., Pujol, F. M., Hassinen, A., and Kellokumpu, S. (2012). Golgi pH, its regulation and roles in human disease. Ann. Med. 44, 542–554. doi: 10.3109/07853890.2011.579150
Rizzo, R., Russo, D., Kurokawa, K., Sahu, P., Lombardi, B., Supino, D., et al. (2019). Retrograde transport of Golgi enzymes by GOLPH3 across maturing Cisternae regulates glycan assembly on sphingolipids and cell growth. bioRxiv [Preprint]. doi: 10.1101/870477
Roboti, P., Sato, K., and Lowe, M. (2015). The golgin GMAP-210 is required for efficient membrane trafficking in the early secretory pathway. J. Cell Sci. 128, 1595–1606. doi: 10.1242/jcs.166710
Ros, M., Nguyen, A. T., Chia, J., Le Tran, S., Le Guezennec, X., Mcdowall, R., et al. (2020). ER-resident oxidoreductases are glycosylated and trafficked to the cell surface to promote matrix degradation by tumour cells. Nat. Cell Biol. 22, 1371–1381. doi: 10.1038/s41556-020-00590-w
Schneider, M., Al-Shareffi, E., and Haltiwanger, R. S. (2017). Biological functions of fucose in mammals. Glycobiology 27, 601–618. doi: 10.1093/glycob/cwx034
Scott, K. L., Kabbarah, O., Liang, M. C., Ivanova, E., Anagnostou, V., Wu, J., et al. (2009). GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 459, 1085–1090. doi: 10.1038/nature08109
Sechi, S., Frappaolo, A., Karimpour-Ghahnavieh, A., Piergentili, R., and Giansanti, M. G. (2020). Oncogenic roles of GOLPH3 in the physiopathology of cancer. Int. J. Mol. Sci. 21:933. doi: 10.3390/ijms21030933
Shen, S. M., Ji, Y., Zhang, C., Dong, S. S., Yang, S., Xiong, Z., et al. (2018). Nuclear PTEN safeguards pre-mRNA splicing to link Golgi apparatus for its tumor suppressive role. Nat. Commun. 9:2392. doi: 10.1038/s41467-018-04760-1
Shorter, J., Watson, R., Giannakou, M. E., Clarke, M., Warren, G., and Barr, F. A. (1999). GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 18, 4949–4960. doi: 10.1093/emboj/18.18.4949
Slusarewicz, P., Nilsson, T., Hui, N., Watson, R., and Warren, G. (1994). Isolation of a matrix that binds medial Golgi enzymes. J. Cell Biol. 124, 405–413. doi: 10.1083/jcb.124.4.405
Stanley, P. (2011). Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 3:a005199, doi: 10.1101/cshperspect.a005199
Stanley, P., Schachter, H., and Taniguchi, N. (2009). “N-Glycans,” in Essentials of Glycobiology, eds A. Nd, R. D. Varki, J. D. Cummings, H. H. Esko, P. Freeze, C. R. Stanley, et al. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press).
Tan, X., Banerjee, P., Guo, H. F., Ireland, S., Pankova, D., Ahn, Y. H., et al. (2017). Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J. Clin. Invest. 127, 117–131. doi: 10.1172/JCI88736
Tan, X., Shi, L., Banerjee, P., Liu, X., Guo, H. F., Yu, J., et al. (2021). A protumorigenic secretory pathway activated by p53 deficiency in lung adenocarcinoma. J. Clin. Invest. 131:e137186. doi: 10.1172/JCI137186
Tang, C. X., Luan, L., Zhang, L., Wang, Y., Liu, X. F., Wang, J., et al. (2019). Golgin-160 and GMAP210 play an important role in U251 cells migration and invasion initiated by GDNF. PLoS One 14:e0211501. doi: 10.1371/journal.pone.0211501
Tang, D., and Wang, Y. (2013). Cell cycle regulation of Golgi membrane dynamics. Trends Cell Biol. 23, 296–304. doi: 10.1016/j.tcb.2013.01.008
Tang, D., Zhang, X., Huang, S., Yuan, H., Li, J., and Wang, Y. (2016). Mena-GRASP65 interaction couples actin polymerization to Golgi ribbon linking. Mol. Biol. Cell 27, 137–152. doi: 10.1091/mbc.E15-09-0650
Taniguchi, N., and Kizuka, Y. (2015). Glycans and cancer: role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv. Cancer Res. 126, 11–51. doi: 10.1016/bs.acr.2014.11.001
Ungar, D. (2009). Golgi linked protein glycosylation and associated diseases. Semin. Cell Dev. Biol. 20, 762–769. doi: 10.1016/j.semcdb.2009.03.004
Wang, H. Y., Bharti, D., and Levental, I. (2020). Membrane heterogeneity beyond the plasma membrane. Front. Cell Dev. Biol. 8:580814. doi: 10.3389/fcell.2020.580814
Wang, Y., and Seemann, J. (2011). Golgi biogenesis. Cold Spring Harb. Perspect. Biol. 3:a005330. doi: 10.1101/cshperspect.a005330
Wang, Y., Seemann, J., Pypaert, M., Shorter, J., and Warren, G. (2003). A direct role for GRASP65 as a mitotically regulated Golgi stacking factor. EMBO J. 22, 3279–3290. doi: 10.1093/emboj/cdg317
Witkos, T. M., and Lowe, M. (2015). The golgin family of coiled-coil tethering proteins. Front. Cell Dev. Biol. 3:86. doi: 10.3389/fcell.2015.00086
Wong, M., and Munro, S. (2014). Membrane trafficking. The specificity of vesicle traffic to the Golgi is encoded in the golgin coiled-coil proteins. Science 346:1256898. doi: 10.1126/science.1256898
Xiang, Y., Zhang, X., Nix, D. B., Katoh, T., Aoki, K., Tiemeyer, M., et al. (2013). Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat. Commun. 4:1659. doi: 10.1038/ncomms3310
Yadav, S., Puthenveedu, M. A., and Linstedt, A. D. (2012). Golgin160 recruits the dynein motor to position the Golgi apparatus. Dev. Cell 23, 153–165. doi: 10.1016/j.devcel.2012.05.023
Zhang, X., Wang, L., Lak, B., Li, J., Jokitalo, E., and Wang, Y. (2018). GRASP55 senses glucose deprivation through O-GlcNAcylation to promote autophagosome-lysosome fusion. Dev. Cell 45, 245–261.e6. doi: 10.1016/j.devcel.2018.03.023
Zhang, X., and Wang, Y. (2020). Nonredundant roles of GRASP55 and GRASP65 in the Golgi apparatus and beyond. Trends Biochem. Sci. 45, 1065–1079. doi: 10.1016/j.tibs.2020.08.001
Zhang, Y., and Seemann, J. (2021). Rapid degradation of GRASP55 and GRASP65 reveals their immediate impact on the Golgi structure. J. Cell Biol. 220:e202007052. doi: 10.1083/jcb.202007052
Keywords: glycosylation, cancer, Golgi fragmentation, Golgi matrix, GM130, GRASP55, COG complex
Citation: Zhang X (2021) Alterations of Golgi Structural Proteins and Glycosylation Defects in Cancer. Front. Cell Dev. Biol. 9:665289. doi: 10.3389/fcell.2021.665289
Received: 07 February 2021; Accepted: 19 April 2021;
Published: 12 May 2021.
Edited by:
Celso A. Reis, University of Porto, PortugalReviewed by:
Frederic A. Bard, Institute of Molecular and Cell Biology (A∗STAR), SingaporeMartin Lowe, The University of Manchester, United Kingdom
Copyright © 2021 Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Zhang, eGlhb3lhbnpAbWFpbC5oemF1LmVkdS5jbg==