 Jun Yu1†
Jun Yu1† Yibo Wu2†
Yibo Wu2† Hong Li3†Hui Zhou2Cong Shen3Tingting Gao4Meng Lin5Xiuliang Dai4Jian Ou3Meiling Liu6Xiaoyan Huang5*
Hong Li3†Hui Zhou2Cong Shen3Tingting Gao4Meng Lin5Xiuliang Dai4Jian Ou3Meiling Liu6Xiaoyan Huang5* Bo Zheng3,6*Fei Sun1*
Bo Zheng3,6*Fei Sun1*- 1Institute of Reproductive Medicine, School of Medicine, Nantong University, Nantong, China
- 2Human Reproductive and Genetic Center, Affiliated Hospital of Jiangnan University, Wuxi, China
- 3State Key Laboratory of Reproductive Medicine, Center for Reproduction and Genetics, Suzhou Municipal Hospital, The Affiliated Suzhou Hospital of Nanjing Medical University, Gusu School, Nanjing Medical University, Suzhou, China
- 4Center of Clinical Reproductive Medicine, The Affiliated Changzhou Maternity and Child Health Care Hospital of Nanjing Medical University, Changzhou, China
- 5State Key Laboratory of Reproductive Medicine, Department of Histology and Embryology, Nanjing Medical University, Nanjing, China
- 6National Health Commission Key Laboratory of Male Reproductive Health, National Research Institute for Family Planning, Beijing, China
Testosterone biosynthesis progressively decreases in aging males primarily as a result of functional changes to Leydig cells. Despite this, the mechanisms underlying steroidogenesis remain largely unclear. Using gene knock-out approaches, we and others have recently identified Bmi1 as an anti-aging gene. Herein, we investigate the role of BMI1 in steroidogenesis using mouse MLTC-1 and primary Leydig cells. We show that BMI1 can positively regulate testosterone production. Mechanistically, in addition to its known role in antioxidant activity, we also report that p38 mitogen-activated protein kinase (MAPK) signaling is activated, and testosterone levels reduced, in BMI1-deficient cells; however, the silencing of the p38 MAPK pathway restores testosterone production. Furthermore, we reveal that BMI1 directly binds to the promoter region of Map3k3, an upstream activator of p38, thereby modulating its chromatin status and repressing its expression. Consequently, this results in the inhibition of the p38 MAPK pathway and the promotion of steroidogenesis. Our study uncovered a novel epigenetic mechanism in steroidogenesis involving BMI1-mediated gene silencing and provides potential therapeutic targets for the treatment of hypogonadism.
Introduction
Reduced serum testosterone, known as hypogonadism, is a clinical condition in aging males resulting from reduced androgen production (Basaria, 2014). Hypogonadism is associated with several metabolic symptoms, such as the loss of muscle mass and bone mineral density, as well as with sexual dysfunction, mood changes, and reduced quality of life (Wu et al., 2010). Based on serum testosterone levels, approximately 20%, 30%, and 50% of men aged 60, 70, and 80 are defined as biochemically hypogonadal, respectively (Salonia et al., 2019). Although exogenous testosterone administration can reverse several hypogonadism-associated symptoms (Mayor, 2016; Snyder et al., 2016), emerging evidence has revealed that it may also increase the risk of cardiovascular disease and cancer, and also impede spermatogenesis (Kolettis et al., 2015; Nguyen et al., 2015; Budoff et al., 2017; Sargis and Davis, 2018). Studies focusing on the mechanisms of testosterone formation can help identify potential targets for use in the treatment of hypogonadism.
In males, testosterone biosynthesis occurs mainly in testicular Leydig cells through the regulation of the hypothalamus–pituitary–gonadal axis (HPG) (Nargund, 2015). Luteinizing hormone (LH) binds to the LH receptor (LHR) and stimulates the production of intercellular cAMP in a G protein-dependent manner. Subsequently, cAMP facilitates cholesterol translocation into the inner mitochondrial membrane. This is aided by steroidogenic acute regulatory protein (StAR), and represents the first rate-limiting step in steroid synthesis. In the mitochondria, cholesterol is converted to pregnenolone by the activity of the cytochrome P450 enzyme CYP11A1. Pregnenolone is subsequently metabolized to testosterone in the smooth endoplasmic reticulum through enzymatic transformation (Miller and Auchus, 2011; Miller, 2017; Zirkin and Papadopoulos, 2018). In both aging humans and rodents, serum testosterone levels diminish as a result of the reduced capacity of Leydig cells to produce testosterone (Wang et al., 2017). Although the mechanism underlying these age-related defects remains uncertain, there is evidence that this is mediated, at least in part, by reactive oxygen species (ROS)-induced oxidative damage to the Leydig cells (Beattie et al., 2015; Wang et al., 2017). For instance, it is well established that age-related oxidative stress inhibits steroidogenesis in adrenal and Leydig cells through the activation of p38 mitogen-activated protein kinase (MAPK), which further transcriptionally represses STAR gene expression (Abidi et al., 2008a, b; Zaidi et al., 2014).
B lymphoma Mo-MLV insertion region 1 (BMI1) is a member of the polycomb group (PcG) of epigenetic silencers first identified as an oncogene and subsequently revealed to be essential for stem cell maintenance (Haupt et al., 1991; van Lohuizen et al., 1991; Park et al., 2004). Studies have shown that Bmi1 gene knock-out (KO) mice display premature aging phenotypes, including severe defects in hematopoietic cell self-renewal, excessive ROS production, and a shortened lifespan (van der Lugt et al., 1994; Molofsky et al., 2003; Park et al., 2003; Liu et al., 2009). More recently, we reported that mice deficient for BMI1 show reduced levels of serum testosterone and steroidogenic enzymes, as well as increased ROS production, leading finally to impaired spermatogenesis in the testes (Dai et al., 2018). We also found that the inhibition of BMI1 significantly reduces testosterone production in mouse MLTC-1 cells and primary Leydig cells, whereas treatment with the antioxidant N-acetylcysteine (NAC) can partially restore steroidogenesis (Gao et al., 2020). The oxidative stress-induced activation of p38 MAPK inhibits steroidogenesis suggests that BMI1 regulates steroidogenesis via the ROS-p38 MAPK pathway. In this study, we found that BMI1 deficiency indeed leads to the activation of the p38 MAPK pathway both in vitro and in vivo. We further found that the activation of the p38 MAPK pathway in BMI1-deficient cells occurs as a direct result of the epigenetic regulation of mitogen-activated protein kinase kinase kinase 3 (Map3k3), an upstream regulator of p38 MAPK, and that this activation was independent of ROS production. These results strongly suggested that BMI1 is a crucial epigenetic mediator of hypogonadism.
Materials and Methods
Mice
Mice were bred and maintained in a temperature- and humidity-controlled room in the Experimental Animal Center of Nanjing Medical University. Mice heterozygous for Bmi1 (C57BL/6J background) were mated to generate wild-type (WT) and Bmi1 KO mice. The mice were genotyped using PCR as previously described (Dai et al., 2018). The animal use protocol was reviewed and approved by the Ethics Committee of Nanjing Medical University.
Cell Culture and Reagents
The mouse MLTC-1 Leydig cell line was purchased from ATCC (Manassas, VA, United States). These cells can synthesize testosterone and retain the full set of steroidogenic enzymes (Mendoza-Villarroel et al., 2014). The cells were maintained in DMEM medium supplemented with 10% fetal bovine serum (FBS) (Thermo Scientific, Waltham, MA, United States) at 37°C in a humidified incubator with 5% CO2. For primary mouse Leydig cell culture, adult male CD-1 mice were euthanized, and the testes dissected and washed in phosphate-buffered saline (PBS). After removing connective tissue, the testes were enzymatically digested in 0.5% (w/v) collagenase IV (Sigma, St. Louis, MO, United States) for 25 min at 37°C. The reaction was stopped with FBS and the supernatant was filtered through a 400-mesh stainless steel filter. Cells were then collected and suspended in DMEM culture medium supplemented with 10% FBS at 37°C with 5% CO2. Testosterone levels were assayed using an ELISA kit (Jiancheng Bioengineering, Nanjing, China) according to the manufacturer’s instructions. PTC-209 and SB203580 were sourced from Selleck (Houston, TX, United States) and NAC was obtained from Sigma.
Plasmids, siRNA, and Transfection
For the construction of plasmids overexpressing WT BMI1 and its nuclear localization signal (NLS2) mutant (BMI1-△NLS2), mouse Bmi1 cDNA was PCR-amplified and then cloned into the pcDNA3.0 vector (Invitrogen, Carlsbad, CA, United States). For the construction of pGL6-Map3k3 plasmids, both full length (−2000 ∼ + 100 relative to the transcriptional start site [TSS]; promoter-1) and partial (−1033 ∼ + 100, promoter-2 and −662 ∼ + 100, promoter-3) Map3k3 promoter sequences were PCR-amplified from mouse genomic DNA and inserted into the pGL6 firefly luciferase reporter vector (Beyotime, Nantong, China). Both the BMI1-△NLS2 and Map3k3 promoter mutant constructs were generated using the ClonExpress Ultra One Step Cloning Kit (Vazyme, Nanjing, China). Bmi1 siRNA (5′-ACAAUGAAAGUUAAAAGUCGU-3′), Map3k3 siRNA (5′-AGUCUAAUGCCUCUUGUUCAU-3′), and negative control (NC) siRNA (5′-ACGUGACACGUUCGGAGAA-3′) were synthesized by GenePharma (Shanghai, China).
The transient transfection of expression plasmids, luciferase reporter plasmids, or siRNAs was performed using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. The transfected plasmids and siRNAs are indicated in the figures and figure legends. NC siRNA and empty vectors (EVs) were used as negative controls. After transfection, cells were utilized for measurements at the times indicated in the figure legends.
Western Blot
Western blot was carried out as previously described (Zheng et al., 2014, 2015). Briefly, cell lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer (Beyotime) containing protease inhibitors (Sigma). Protein was separated by SDS–PAGE and then transferred onto polyvinylidene difluoride membranes. After blocking, the membranes were incubated with the indicated primary antibodies (Supplementary Table 1) overnight at 4°C. The next day, the membranes were washed and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Thermo Scientific) at room temperature (RT) for 2 h. Band signals were detected using an enhanced chemiluminescent substrate (Thermo Scientific) and subsequently analyzed by Image-Pro Plus Software.
Cellular ROS Analysis
Cells were cultured in 96-well plates and treated as indicated in the figure legends. After washing three times with PBS, the cells were incubated with 20 M 2′,7′–dichlorofluorescein diacetate (DCFDA, Genmed Scientifics, Wilmington, DE, United States) for 40 min at 37°C. DCFDA is a fluorogenic dye that allows the measurement of hydroxyl, peroxyl, and other ROS activity within cells. Fluorescence intensity was measured on a fluorescence plate reader at the 488/525 nm excitation/emission wavelengths.
Immunofluorescence
Cells were harvested, fixed in 4% (w/v) paraformaldehyde (PFA), blocked with 2% bovine serum albumin (BSA) (w/v) (Sigma) for 45 min at RT, and incubated with primary antibodies (Supplementary Table 1) overnight at 4°C according to a previously reported protocol (Zheng et al., 2018; Zhao et al., 2019). Next, the cells were rinsed with PBS and incubated with Alexa Fluor-conjugated secondary antibodies (Thermo Scientific) for 45 min. Nuclei were counterstained with DAPI (Beyotime). Images were acquired using a confocal laser microscope (Zeiss LSM800, Carl Zeiss, Oberkochen, Germany).
RNA Extraction and Quantitative Real-Time Reverse Transcription-PCR
Cellular RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Total RNA was reverse transcribed into cDNA using a PrimeScript Reverse Transcription Kit (Vazyme) according to the instructions of the manufacturer. A total of 1 μL of the synthesized cDNA was mixed with a SYBR green mix (Vazyme) and primers in a final volume of 20 μL and applied to an ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA, United States). Relative gene expression levels were calculated using the 2−ΔΔCt method with 18S rRNA serving as an internal control. The following primers were used: mouse Map3k3, forward 5′-GACTTCAGGACTCGCAGGC-3′ and reverse 5′-TGTTCATCCATGGTGGCGAT-3′; mouse 18sRNA, forward 5′-AAACGGCTACCACATCCAAG-3′ and reverse 5′-CCTCCAATGGATCCTCGTTA-3′.
Luciferase Reporter Assay
Cells were cultured in 12-well plates and transiently transfected with 0.2 μg of the pGL6 luciferase reporter plasmid and Bmi1 effector plasmid. Cells were also cotransfected with an internal control reporter plasmid containing Renilla luciferase (pRL-TK) to normalize the transfection efficiency. Two days after transfection, luciferase activity was detected using the Dual-Luciferase Reporter Gene Assay System (Beyotime).
Chromatin Immunoprecipitation (ChIP)
The ChIP assay was performed using an EZ-ChIP Kit (Millipore, Billerica, MA, United States). Briefly, cells were harvested, washed, and cross-linked using 1% formaldehyde. The cells were then sheared into ∼500-bp fragments using a Branson Sonicator 250. The chromatin DNA–protein complex was reacted with the indicated antibodies (Supplementary Table 1). Approximately 10% of the starting material was used as the input. Immunoprecipitated DNA was analyzed by real-time PCR using the following primers: mouse Map3k3 promoter, forward 5′-ATCGAGCCAAACCTTCCCTG-3′ and reverse 5′-CCACAACCCTCTGCTCAGTT-3′; and the mouse Gapdh gene, forward 5′-AACCCAAACTAACAGTTGTCCCAA-3′ and reverse 5′-ACTCCTTGGAGGCCATGTAGG-3′.
Statistical Analysis
GraphPad Prism software was used for statistical analysis. Data obtained by the Student’s t-test or one-way ANOVA were presented as means ± standard deviation (SD) from at least three independent experiments. p < 0.05, p < 0.01, and p < 0.001 were set as the thresholds for statistical significance.
Results
BMI1 Positively Regulates Testosterone Production in MLTC-1 Cells
In this study, we used the MLTC-1 mouse Leydig cell line for the analysis of the mechanistic and functional roles of BMI1 in steroidogenesis. This line stably and continuously produces androgenic hormones and is a commonly used model to study steroidogenesis (Abdou et al., 2014; Mendoza-Villarroel et al., 2014; Ayoub et al., 2016; Zhang et al., 2018). We first determined the effect of BMI1 on testosterone production by downregulating its expression using a BMI1-specific inhibitor, PTC-209 (Li et al., 2020; Zhu et al., 2020) or Bmi1-siRNA. We observed that both PTC-209 and Bmi1-siRNA treatment led to a substantial reduction in BMI1 levels (Figures 1A,B and Supplementary Figures 1A,B) and inhibited testosterone production in a dose-dependent manner (Figure 1C and Supplementary Figure 1C). In contrast, ectopic BMI1 expression significantly enhanced testosterone production (Figures 1D–F). These results strongly suggested that BMI1 is required for androgen synthesis.
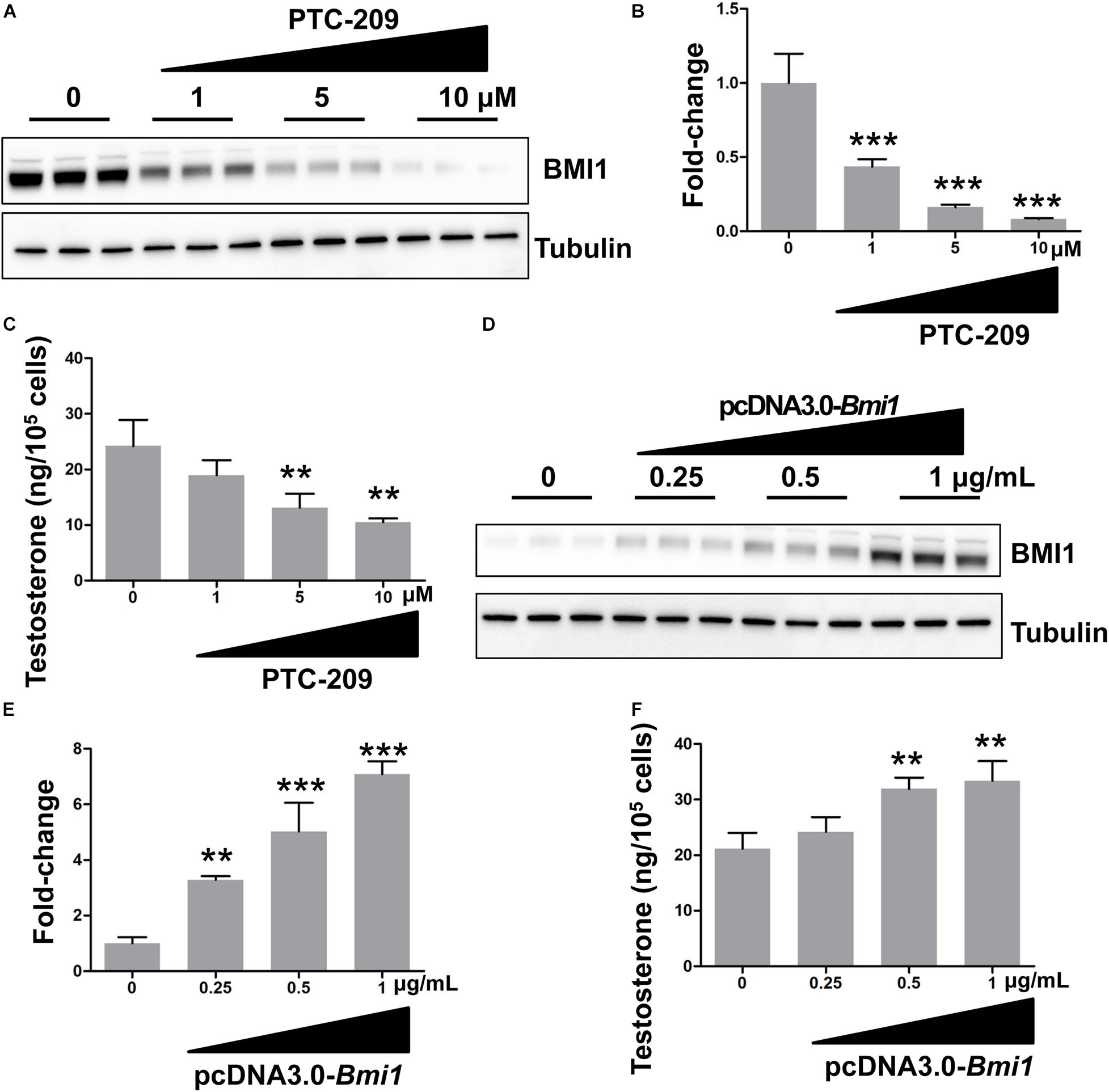
Figure 1. BMI1 promotes steroidogenesis in a dose-dependent manner. (A) MLTC-1 cells were treated with the indicated doses of PTC-209 for 48 h, followed by western blot analysis. (B) Quantification of (A). (C) Assessment of testosterone levels in MLTC-1 cells treated with the indicated doses of PTC-209 after treatment with 1 IU/mL human chorionic gonadotropin (hCG) for 6 h. (D) MLTC-1 cells were transfected with the indicated concentrations of the pcDNA3.0-Bmi1 plasmid for 48 h, followed by western blot analysis. (E) Quantification of (D). (F) The assessment of testosterone levels in MLTC-1 cells transfected with the indicated concentrations of the pcDNA3.0-Bmi1 plasmid after treatment with 1 IU/mL hCG for 6 h. **p < 0.01, ***p < 0.001.
Both Cytoplasmic and Nuclear BMI1 Are Required for Steroidogenesis
In the cytoplasm, BMI1 inhibits ROS generation by regulating mitochondrial homeostasis (Banerjee Mustafi et al., 2016). In line with these observations, we found that BMI1-deficient MLTC-1 cells exhibited increased levels of oxidative stress and DNA double-strand breaks (DSBs) (Supplementary Figures 2A–C) (Gao et al., 2020). When PTC-209-treated MLTC-1 cells were further treated with the oxidant scavenger NAC (Liu et al., 2009; Chagraoui et al., 2011), testosterone production was significantly improved, which suggested that cytoplasmic BMI1 promoted steroidogenesis by maintaining redox homeostasis (Figure 2A). To further clarify the role of cytoplasmic BMI1 in steroidogenesis, we constructed a BMI1-△NLS2 plasmid (Figure 2B), which provided a powerful tool for investigating the role of BMI1 in the cytoplasm independently of its role in the nucleus (Cohen et al., 1996). We found that ectopic BMI1-△NLS2 expression in PTC-209-treated cells also enhanced testosterone production. Altogether, the above data revealed that cytoplasmic BMI1 has an important role in the promotion of steroidogenesis. However, when WT BMI1 was ectopically expressed in PTC-209-treated cells, the testosterone level was markedly higher than that in NAC- and/or BMI1-△NLS2-treated cells (Figure 2A). This indicates that nuclear-localized BMI1 also has a role in testosterone production.
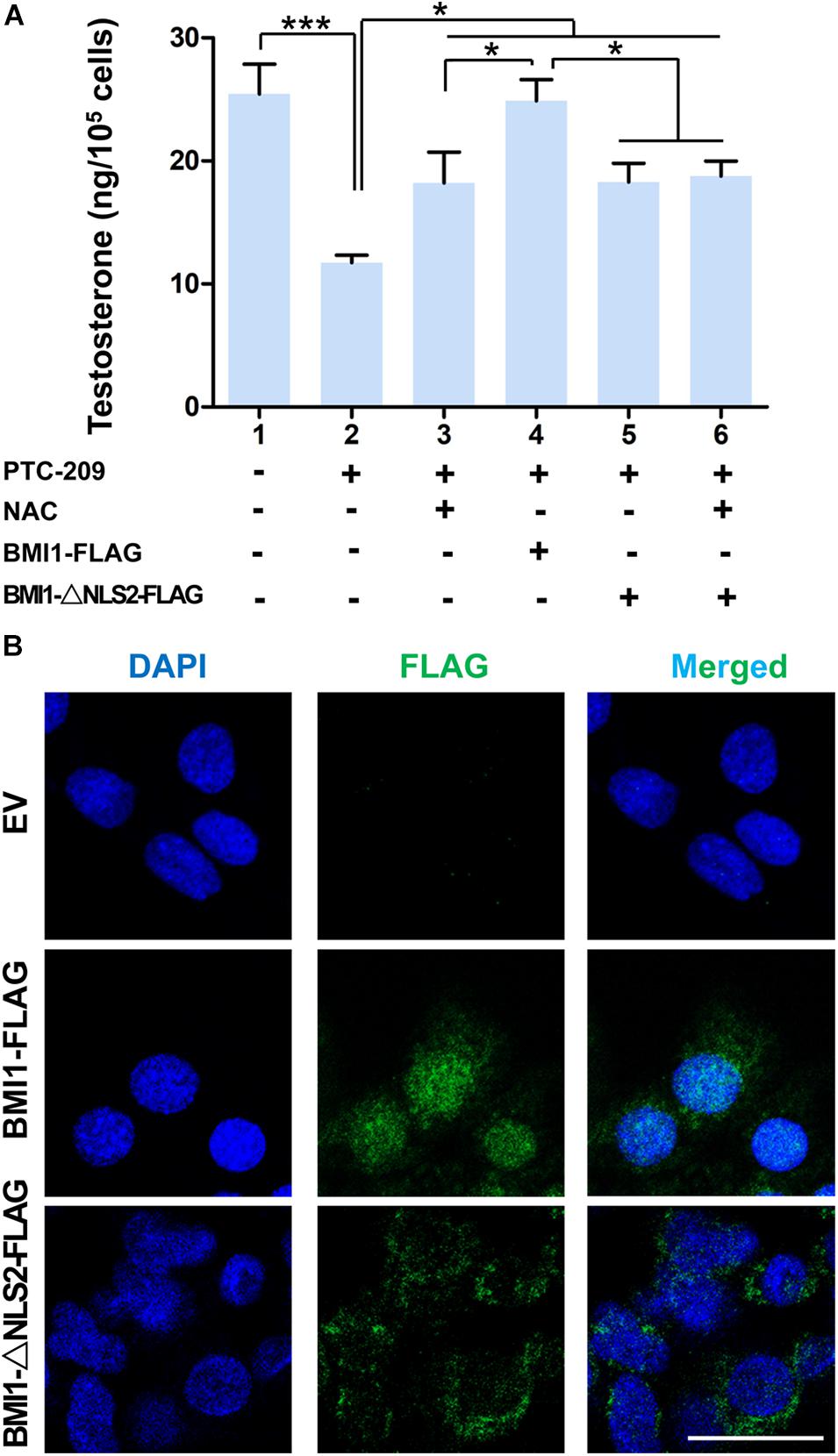
Figure 2. Both cytoplasmic and nuclear BMI1 are required for steroidogenesis. (A) The assessment of testosterone levels in MLTC-1 cells treated as indicated for 48 h. PTC-209, N-acetylcysteine (NAC), pcDNA3.0-Bmi1-flag, and pcDNA3.0-Bmi1-△NLS2-flag were used at the concentrations of 5 μM, 500 μM, 1 μg/mL, and 1 μg/mL, respectively. Testosterone levels were determined by ELISA after treatment with 1 IU/mL human chorionic gonadotropin (hCG) for 6 h. (B) Immunofluorescence staining for FLAG in MLTC-1 cells transfected with the pcDNA3.0-flag empty vector (EV), pcDNA3.0-Bmi1-flag, and pcDNA3.0-Bmi1-△NLS2-flag for 48 h. All constructs were used at the concentration of 1 μg/mL. Scale bar, 20 μm. *p < 0.05, ***p < 0.001.
The Activation of p38 MAPK Is Independent of ROS in BMI1-Deficient Cells
The p38 MAPK pathway is a negative regulator of steroidogenesis through the repression of STAR transcription (Zaidi et al., 2014). Here, we observed that the levels of p38 MAPK and its upstream regulator MAP3K3 were increased in PTC-209-treated MLTC-1 cells, whereas that of StAR was downregulated (Figures 3A,B). This indicated that the p38 MAPK pathway was activated. In contrast, ectopic BMI1 expression reduced the levels of both p38 and MAP3K3, while enhancing that of StAR (Figures 3C,D). However, the p38 MAPK pathway was not affected by the administration of NAC and/or BMI1-△NLS2 to PTC-209-treated cells (Figures 3E,F), whereas ROS production was markedly inhibited (Supplementary Figure 3). Only the ectopic expression of WT BMI1 could reduce p38 MAPK levels in PTC-209-treated cells (Figures 3E,F). To further analyze the expression of p38 MAPK in the differentially treated groups, we examined the expression of phosphorylated (activated) p38 (p-p38) in MLTC-1 cells using immunofluorescence (IF) (Figure 3G). The IF staining results were consistent with those obtained with western blotting (Figures 3E,F). Taken together, these data indicated that the p38 MAPK pathway is not activated by cytoplasmic BMI1-mediated oxidative stress in BMI1-deficient cells, suggesting that p38 MAPK is regulated by nuclear, and not cytoplasmic, BMI1.
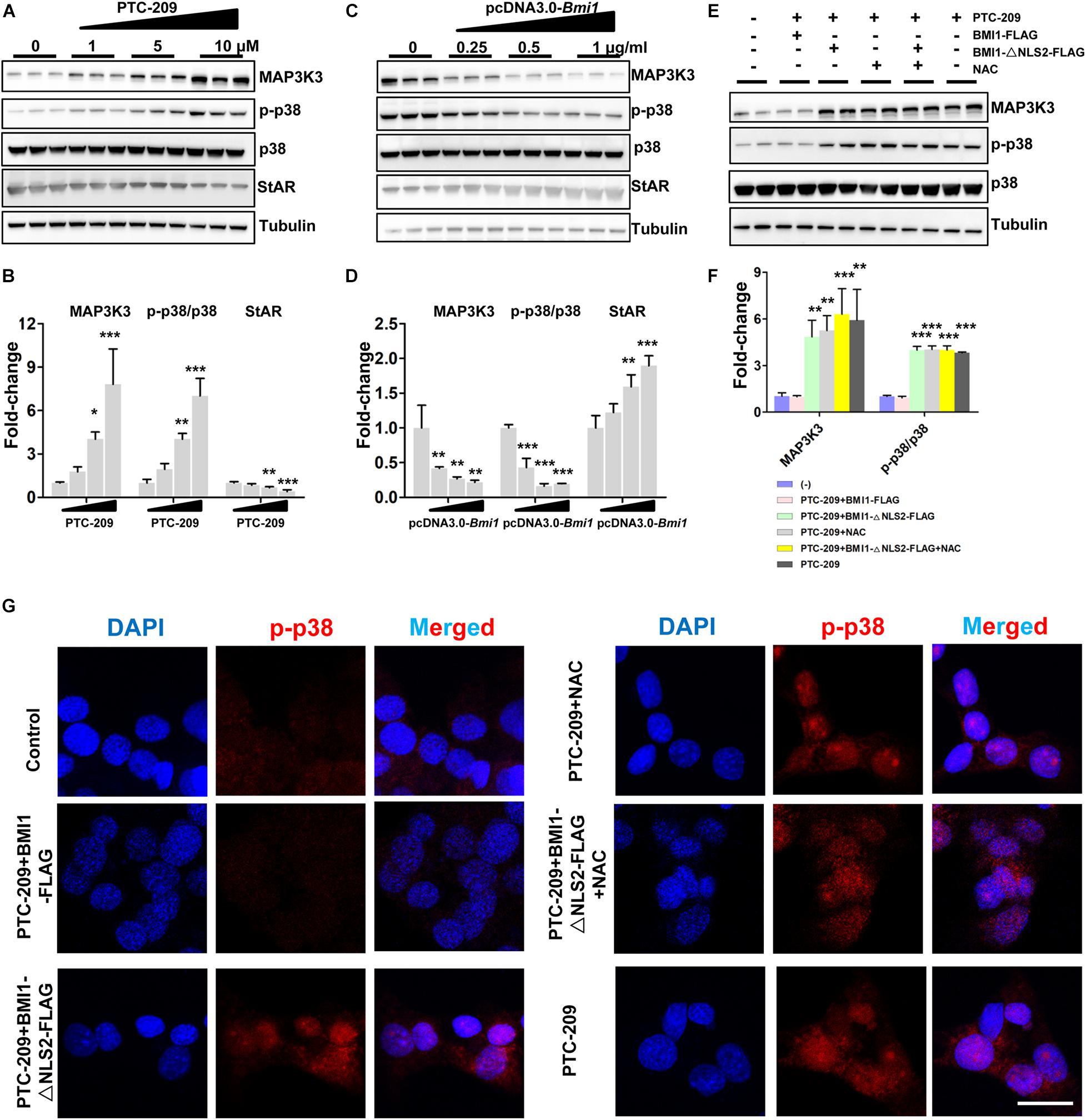
Figure 3. BMI1 inhibits the p38 MAPK pathway. (A) MLTC-1 cells were treated with the indicated concentrations of PTC-209 for 48 h, followed by western blot analysis of p38 MAPK expression. (B) Quantification of (A). (C) MLTC-1 cells were transfected with the indicated concentrations of the pcDNA3.0-Bmi1 plasmid for 48 h, followed by western blot analysis of p38 MAPK expression. (D) Quantification of (C). (E) Western blot analysis of p38 MAPK expression in MLTC-1 cells treated as indicated for 48 h. PTC-209, pcDNA3.0-Bmi1-flag, pcDNA3.0-Bmi1-△NLS2-flag, and N-acetylcysteine (NAC) were used at the concentrations of 5 μM, 1 μg/mL, 1 μg/mL, and 500 μM, respectively. (F) Quantification of (E). (G) Immunofluorescence staining for phospho-p38 in MLTC-1 cells treated as indicated for 48 h. PTC-209, pcDNA3.0-Bmi1-flag, pcDNA3.0-Bmi1-△NLS2-flag, and NAC were used at the concentrations of 5 μM, 1 μg/mL, 1 μg/mL, and 500 μM, respectively. DMSO combined with empty vector (EV) was used as the control. Scale bar, 20 μm. *p < 0.05, **p < 0.01, ***p < 0.001.
BMI1 Is Required for the Transcriptional Repression of Map3k3
In the nucleus, as a major member of polycomb repressive complex 1 (PRC1), BMI1 cooperates with E3 ubiquitin-protein ligase RING2 (RING1B) to monoubiquitinate histone H2A at K119, thereby mediating gene silencing (Wang et al., 2004; Morey et al., 2015). Therefore, we sought to determine whether BMI1 directly regulates the transcription of factors in the p38 MAPK pathway. We found that the mRNA expression of Map3k3 was dose-dependently enhanced in PTC-209-treated MLTC-1 cells (Figure 4A), whereas the opposite was observed with ectopic BMI1 expression (Figure 4B). Additionally, bioinformatic analysis of publicly available BMI1 ChIP-seq data for the human MCF-7 cell line (ENCODE1, ENCSR966YYJ) revealed a BMI1-binding peak at the promoter region of MAP3K3 (−359 ∼−742 bp upstream of the TSS) (Supplementary Figure 4), which corresponds to −633 ∼−1033 bp upstream of the TSS of the mouse Map3k3 promoter (Supplementary Figure 5). Accordingly, we cloned both full-length (−2,000 ∼ + 100 bp [promoter-1]) and partial (−1033 ∼ + 100 [promoter-2] and −662 ∼ + 100 [promoter-3]) Map3k3 promoter sequences into the pGL6 firefly luciferase reporter vector (Figure 4C). All three sequences could drive luciferase expression effectively; however, in the absence of the sequence encompassing the BMI1 binding peak (promoter-3), luciferase activity was significantly reduced compared with that for promoter-1 and promoter-2 (Supplementary Figure 6), suggesting that the sequence upstream of position −662 was vital for Map3k3 transcription. Moreover, the inhibition of BMI1 markedly increased luciferase activity, but this effect was least pronounced with promoter-3 (Figure 4D); meanwhile, the overexpression of BMI1 elicited the opposite effect (Figure 4D). The above findings indicated that BMI1 mediates the transcriptional repression of Map3k3 through binding to its core promoter region (−1033 ∼−663 upstream of the TSS). Surprisingly, the putative BMI1 binding sequence in mice showed only 59.7% similarity with that of humans (Supplementary Figure 5), suggesting that BMI1 could bind to the Map3k3 promoter region through a specific motif. Further analysis of the region containing the BMI1 binding peak led to the identification of a 38-bp sequence that was highly conserved among humans, mice, rats, and monkeys (Figure 4E) and contained two functional motifs, namely, 5′-GGGMGGRG-3′ and 5′-TGATTGG-3′ (Figure 4E). To confirm this finding, we generated constructs containing the promoter-1 sequence that lacked the two motifs. A reporter assay revealed that the mutated promoter-1 significantly attenuated the BMI1-mediated transcriptional repression of Map3k3 (Figure 4F). Collectively, these results demonstrated that BMI1 negatively regulates Map3k3 transcription through binding to a set of specific motifs in its promoter region.
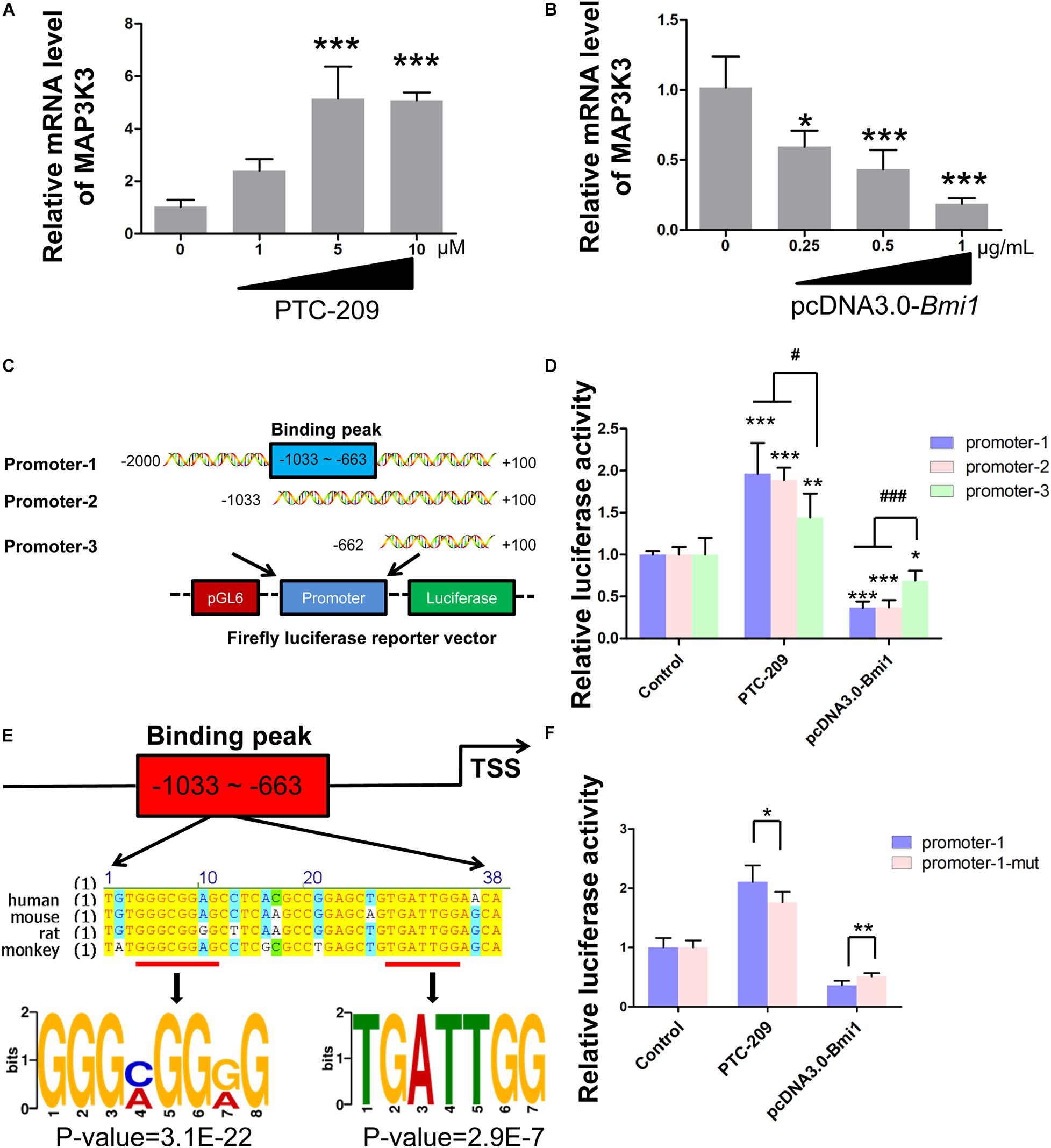
Figure 4. BMI1 inhibits Map3k3 transcription. (A) MLTC-1 cells were treated with the indicated concentrations of PTC-209 for 48 h, followed by quantitative real-time reverse transcription-PCR (RT-qPCR) analysis of Map3k3. (B) MLTC-1 cells were transfected with the indicated concentrations of the pcDNA3.0-Bmi1 plasmid for 48 h, followed by RT-qPCR analysis of Map3k3. (C) Schematic illustration of the firefly luciferase reporter constructs. (D) Luciferase activity assays in MLTC-1 cells transfected with luciferase reporter constructs and treated with PTC-209 (5 μM) or the pcDNA3.0-Bmi1 (1 μg/mL) plasmid for 48 h. DMSO combined with empty vector (EV) was used as the control. *, compared with control; #, compared with promoter-1 and -2. (E) Sequence analysis of the putative BMI1 binding region. (F) Luciferase activity assays in MLTC-1 cells transfected with luciferase reporter constructs containing promoter-1 and mutated promoter-1, PTC-209 (5 μM), or the pcDNA3.0-Bmi1 (1 μg/mL) plasmid for 48 h. DMSO combined with empty vector (EV) was used as the control. *p < 0.05, **p < 0.01, ***p < 0.001; #p < 0.05, ###p < 0.001.
Chromatin Remodeling by BMI1 Is Mediated Through Its Polycomb Function
To obtain further insights into the mechanisms by which BMI1 mediates Map3k3 transcriptional repression, we performed a ChIP assay in MLTC-1 cells using antibodies against BMI1, H2AK119ub, and RING1B. As expected, the BMI1 binding peak sequence was precipitated with BMI1, H2AK119ub, and RING1B (Figures 5A–C). In addition, after BMI1 depletion by PTC-209, significantly less H2AK119ub was precipitated with the region encompassing the BMI1 binding peak (Figure 5D), suggesting that the function of PRC1 was disrupted. Interestingly, bioinformatic analysis further revealed that the putative BMI1 binding region overlapped with the distribution of H3K4me3 (Supplementary Figure 7). In PTC-209-treated cells, the H3K4me3 level was measurably increased at the BMI1 binding peak region (Figure 5E). Taken together, these results demonstrated that BMI1 represses Map3k3 expression through direct binding to its promoter region and modulating chromatin accessibility.
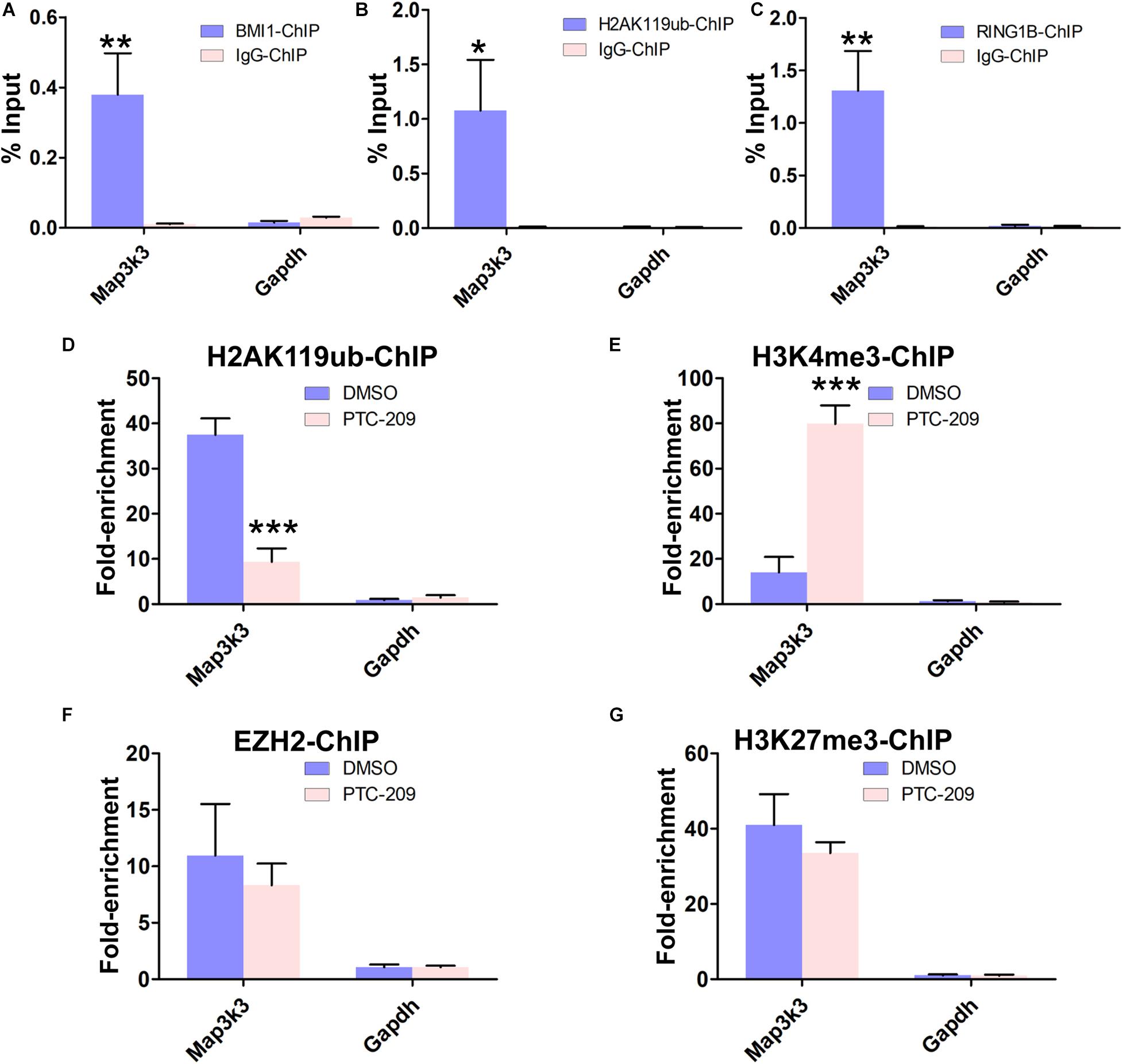
Figure 5. BMI1 inhibition alters the chromatin status at the BMI1 binding peak region in the Map3k3 promoter. (A–C) ChIP-qPCR of BMI1-associated (A), H2AK119ub-associated (B), and RING1B-associated (C) DNA sequences from the putative BMI1-binding region of the Map3k3 promoter in MLTC-1 cells. The Gapdh gene was used as a negative control. (D–G) ChIP-qPCR of H2AK119ub-associated (D), H3K4me3-associated (E), EZH2-associated (F), and H3K27me3-associated (G) DNA sequences in the putative BMI1-binding region of the Map3k3 promoter in DMSO-treated or PTC-209-treated MLTC-1 cells. The y-axis represents enrichment relative to IgG controls. *p < 0.05, **p < 0.01, ***p < 0.001.
We next sought to determine whether the PRC2 complex can partly compensate for the disrupted PRC1 complex in PTC-209-treated cells. PRC2 is composed of the enhancer of zeste homolog 2 (EZH2), suppressor of zeste 12 (SUZ12), and embryonic ectoderm development (EED) proteins and contributes to the trimethylation of histone H3 at K27 (H3K27me3) (Sauvageau and Sauvageau, 2010). ChIP experiments indicated that PTC-209 treatment did not lead to any noticeable changes in EZH2 and H3K27me3 levels at the BMI1 binding peak-related region (Figures 5F,G).
Knockdown of the P38 MAPK Pathway Restores Steroidogenesis in BMI1-Deficient MLTC-1 Cells
To further test whether the p38 MAPK pathway contributes to the defective steroidogenesis observed in PTC-209-treated cells, we downregulated the p38 MAPK pathway by siRNA-mediated Map3k3 silencing and the application of a widely used pharmacological inhibitor of p38, SB203580 (Li W. et al., 2017; He et al., 2020). Western blotting (Figures 6A–C) and IF (Figure 6D) analyses showed that both Map3k3-siRNA and SB203580 could efficiently inhibit p38 MAPK in PTC-209-treated cells. Meanwhile, western blot results also showed that SB203580 did not affect the levels of BMI1 (Figures 6A,C). Concomitantly, cells treated with PTC-209 and Map3k3-siRNA or SB203580 displayed significantly increased testosterone levels when compared with those in cells treated with PTC-209 alone (Figure 6E). The above data implied that excessively activated p38 signaling serves as a major repressor of BMI1-mediated steroidogenesis.
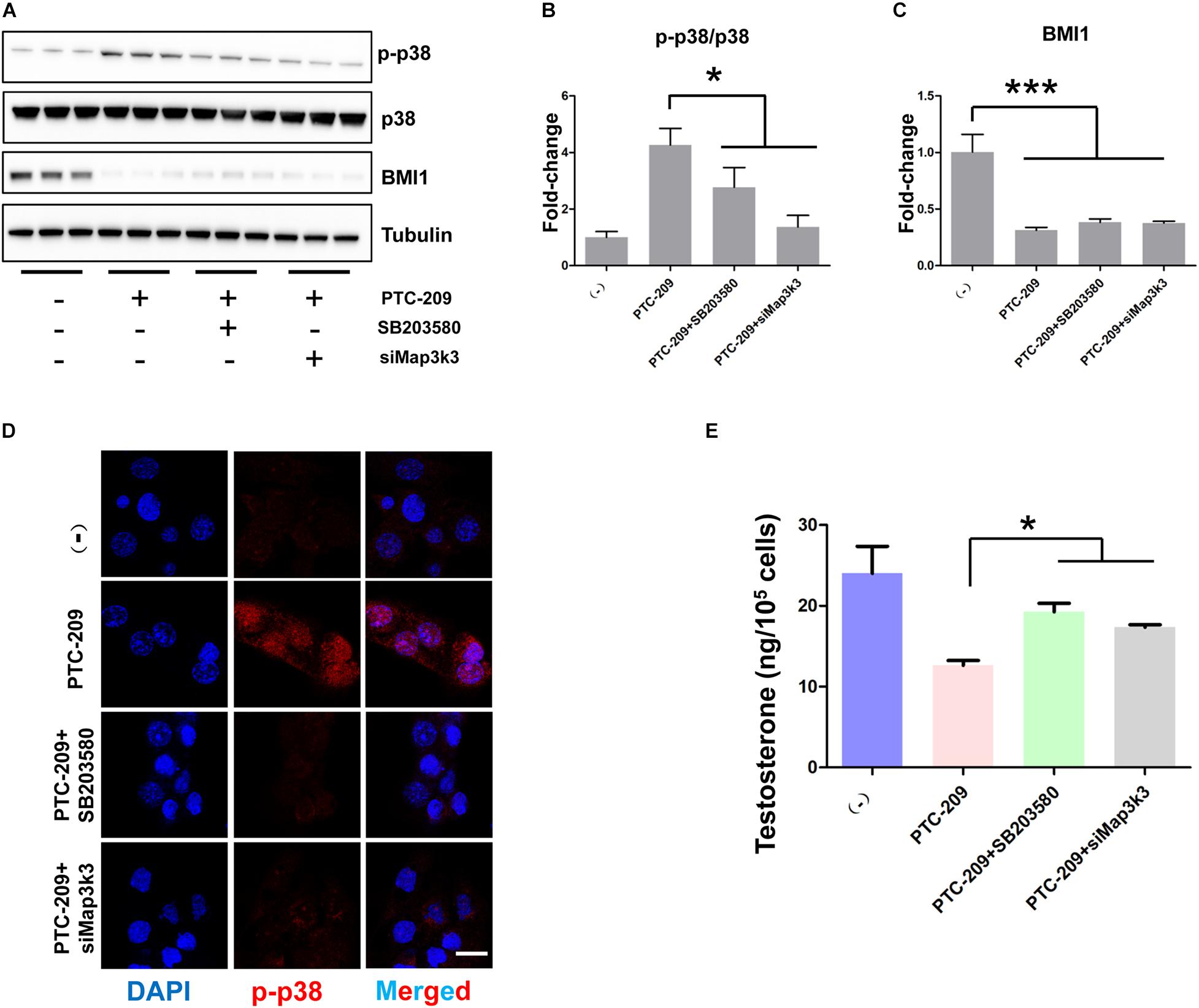
Figure 6. Inhibition of the p38 MAPK pathway rescued disrupted steroidogenesis. (A) Western blot analysis of phospho-p38 (p-p38) and BMI1 in MLTC-1 cells treated with PTC-209 (5 μM), SB203580 (10 μM), or siRNA targeting Map3k3 (50 nM) for 48 h. (B) Quantification of (A). (C) Quantification of (A). (D) Immunofluorescence staining for p-p38 in MLTC-1 cells treated as indicated for 48 h. PTC-209, SB203580, and Map3k3 siRNA were used at the concentrations of 5 μM, 10 μM, and 50 nM, respectively. (E) The assessment of testosterone levels in MLTC-1 cells treated as indicated for 48 h. PTC-209, SB203580, and Map3k3 siRNA were used at the concentrations of 5 μM, 10 μM, and 50 nM, respectively. Testosterone levels were determined by ELISA after treatment with 1 IU/mL human chorionic gonadotropin (hCG) for 6 h. Scale bar, 20 μm. *p < 0.05.
BMI1 Epigenetically Orchestrates Steroidogenesis in Primary Mouse Leydig Cells
Primary mouse Leydig cells were used to further validate our findings in MLTC-1 cells. Consistent with the results obtained with MLTC-1 cells, when SB203580 was added to PTC-209-treated Leydig cells, p38 MAPK expression was inhibited, whereas the levels of testosterone were increased (Figures 7A–D); however, BMI1 levels were not affected (Figures 7A,C). Mechanistically, PTC-209 treatment does-dependently increased the mRNA expression levels of Map3k3 in Leydig cells (Figure 7E). Additionally, a ChIP assay showed that BMI1, H2AK119ub, and RING1B were significantly enriched at the BMI1 binding peak region (Figures 7F–H), while the inhibition of BMI1 by PTC-209 markedly diminished the levels of H2AK119ub, while increasing those of H3K4me3 at the region containing the BMI1 binding peak in the Map3k3 promoter (Figures 7I,J). Finally, we assessed the expression of p38 MAPK in testes from Bmi1-WT and Bmi1-KO mice. Compared with WT mice, the levels of MAP3K3 and p38 were upregulated in the testes of Bmi1-KO mice, whereas those of StAR were downregulated (Supplementary Figure 8). These results suggested that BMI1 also promotes steroidogenesis through the MAP3K3/p38 signaling pathway in vivo. Combined, our in vitro and in vivo findings demonstrated that BMI1 plays an essential role in promoting steroidogenesis by assembling PRC1, modulating chromatin accessibility, and repressing the expression of Map3k3 (Figure 8).
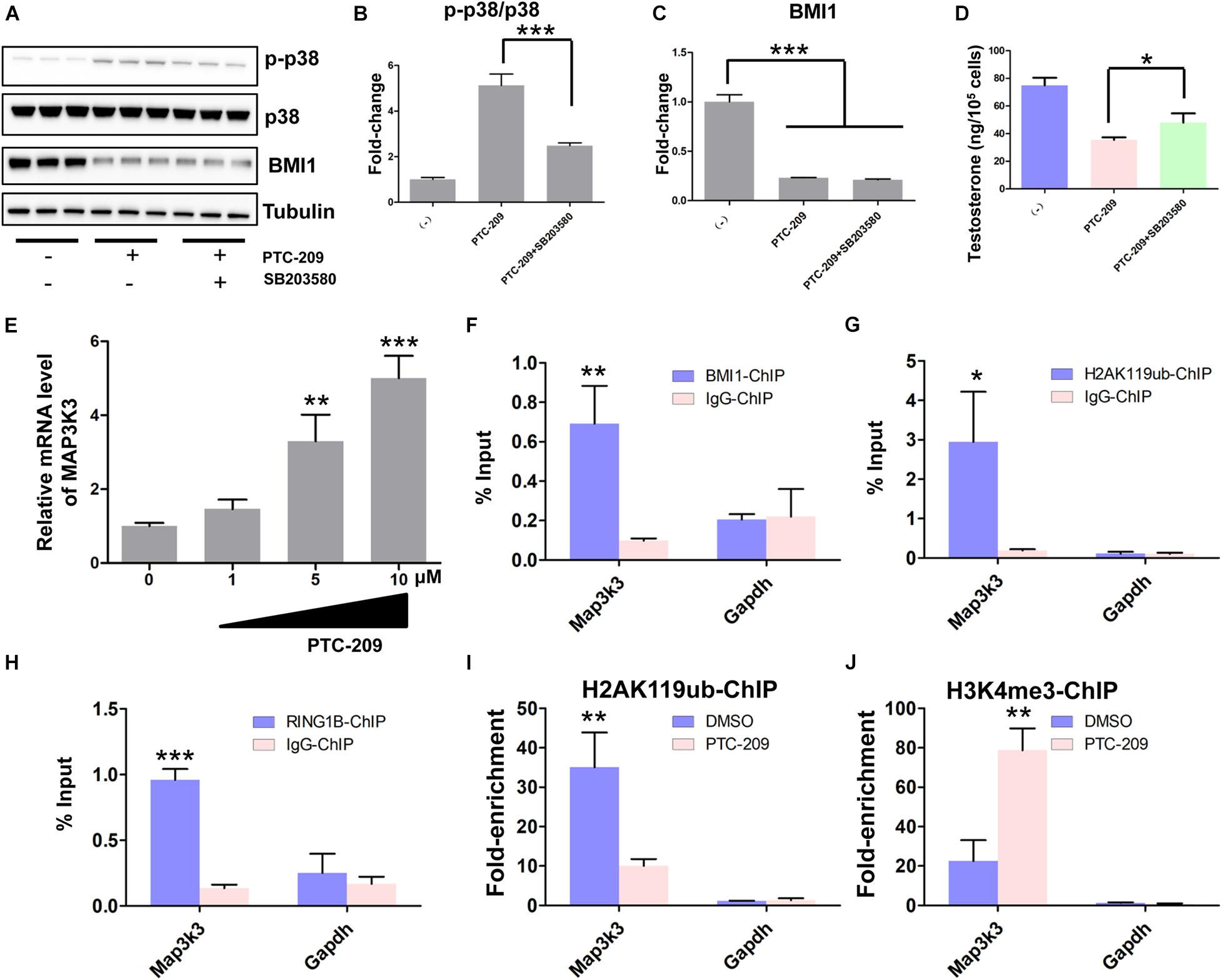
Figure 7. BMI1 epigenetically represses Map3k3 in primary mouse Leydig cells. (A) Western blot analysis of phospho-p38 and BMI1 in Leydig cells treated with PTC-209 (5 μM) or SB203580 (10 μM) for 48 h. (B) Quantification of (A). (C) Quantification of (A). (D) The assessment of testosterone levels in Leydig cells treated with PTC-209 (5 μM) or SB203580 (10 μM) for 48 h. (E) Leydig cells were treated with the indicated concentrations of PTC-209 for 48 h, followed by quantitative real-time reverse transcription-PCR analysis of Map3k3. (F–H) ChIP-qPCR of BMI1-associated (F), H2AK119ub-associated (G), and RING1B-associated (H) DNA sequences in the putative BMI1-binding region of the Map3k3 promoter in Leydig cells. The Gapdh gene was used as a negative control. (I, J) ChIP-qPCR of H2AK119ub- (I) and H3K4me3-associated (J) DNA sequences in the putative BMI1-binding region of the Map3k3 promoter in DMSO-treated or PTC-209-treated Leydig cells. The y-axis represents enrichment relative to IgG controls. *p < 0.05, **p < 0.01, ***p < 0.001.
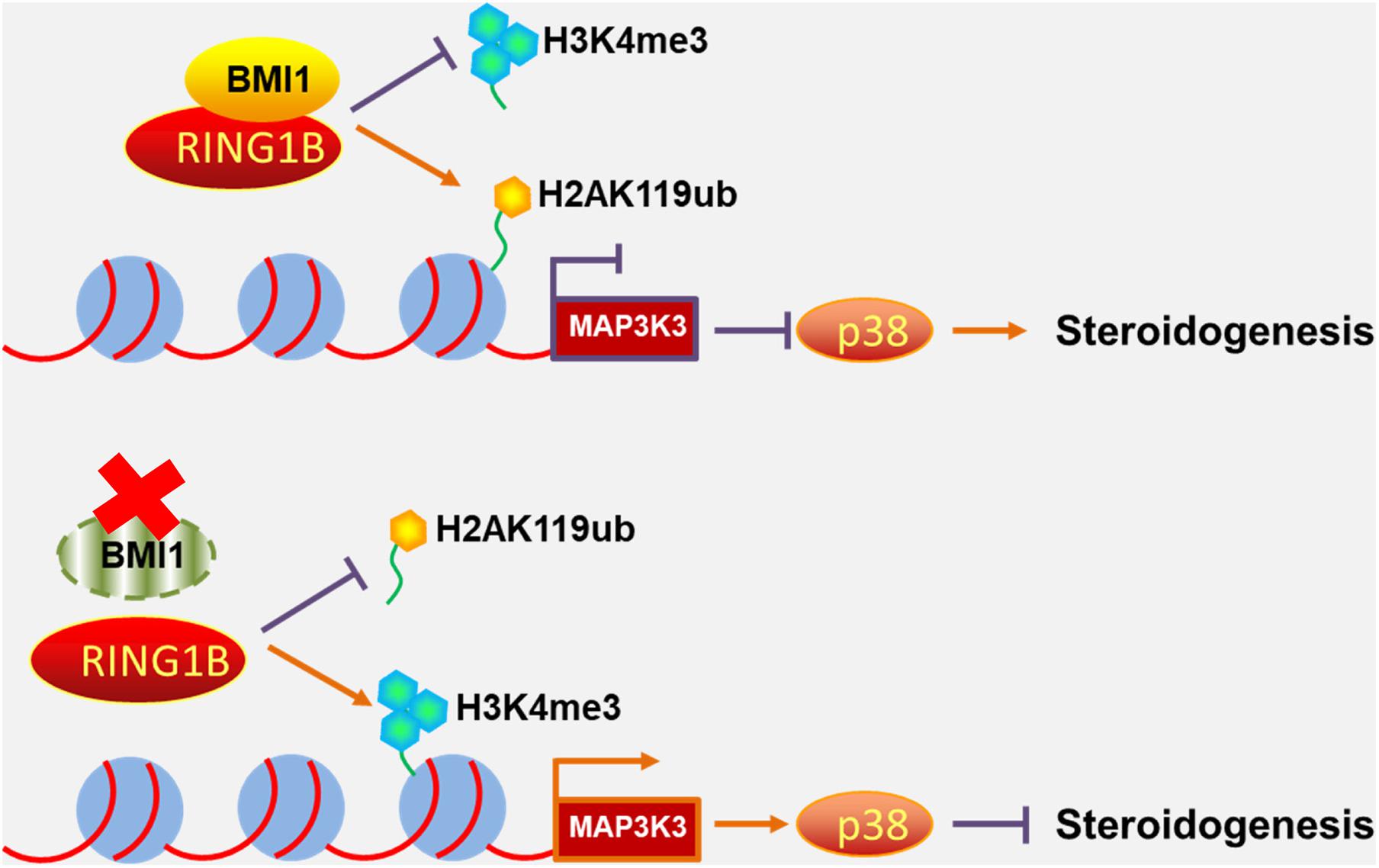
Figure 8. Schematic illustration of the working model for the role of BMI1 in steroidogenesis. In the nucleus, BMI1 is indispensable for PRC1 assembly. Together with RING1B, BMI1 facilitates the monoubiquitination of histone H2A at K119 to repress Map3k3 expression, thereby promoting steroidogenesis. In the absence of BMI1, PRC1 function is disrupted and Map3k3 is transcriptionally activated, thereby blocking steroidogenesis.
Discussion
Serum androgen levels decline in males as they age primarily as a result of functional alterations in the gonad. Aging can be defined as the accumulation of molecular damage in a cell or organism. ROS are known to be key sources of molecular damage and are a hallmark of aging (Lovell and Markesbery, 2007; Sena and Chandel, 2012; Wang and Hekimi, 2015; Kauppila et al., 2017). Several recent studies have implied that a relationship exists between BMI1 and ROS. For instance, it has been reported that BMI1 can prevent excessive ROS generation in neurons by repressing the pro-oxidant function of P53 (Chatoo et al., 2009). The results of a study by Liu et al. (2009) indicated that elevated intracellular ROS levels in Bmi1-KO mice were attributable to impaired mitochondrial function. More recently, Mustafi et al. reported that BMI1 exerts antioxidant activity in CP20 cells. The authors reported that mitochondria-localized BMI1 can stabilize mtRNA and enhance both electron transport chain function and ATP synthesis, and these functions are independent of its known functions in the nucleus (Banerjee Mustafi et al., 2016). Indeed, in this study, we observed that both the antioxidant NAC and cytoplasmic BMI1 could partly restore steroidogenesis in BMI1-deficient cells, which suggested that ROS is an important source of molecular damage during steroidogenesis. Besides these antioxidant functions of cytoplasmic BMI1, we also uncovered a novel epigenetic mechanism in steroidogenesis involving the BMI1-mediated silencing of the p38 MAPK pathway, which promoted steroidogenesis in both mouse MLTC-1 cells and primary Leydig cells. Mechanistically, BMI1 recruited the PRC1 complex to modify the chromatin structure surrounding the Map3k3 promoter, thereby mediating the silencing of the Map3k3 gene. Meanwhile, inhibition of the p38 MAPK pathway could improve steroidogenesis in PTC-209-treated MLTC-1 and Leydig cells. Altogether, our results have identified a role for the BMI1-MAP3K3-p38 MAPK axis in steroidogenesis, thereby providing potential therapeutic targets for the treatment of hypogonadism.
P38 MAPKs, one of the major MAPK subfamilies, are crucial signaling molecules that help transduce extracellular stimuli into cellular responses. The p38 MAPK pathway is activated by MAPK kinases (MAP2Ks or MKKs) such as MKK3 and MKK6; in turn, these MKKs are activated by MAPK kinase kinases (MAP3Ks or MEKKs), including apoptosis signal-regulating kinase 1 (ASK1), thousand-and-one amino acid kinase (TAO), leucine-zipper and sterile-alpha motif kinase (ZAK), transforming growth factor beta-activated kinase 1 (TAK1), and MAP3K3 (Wagner and Nebreda, 2009; Arthur and Ley, 2013). The p38 MAPK pathway is known to act as a ROS sensor. Stimuli such as pathogen attack, ultraviolet (UV) radiation, wounding, and chemotherapeutic agents can induce ROS generation, thereby triggering p38 MAPK pathway activation, which further leads to the modulation of cellular growth and metabolism (Finkel and Holbrook, 2000; Patel et al., 2019). Studies have revealed that ROS-mediated activation of the p38 MAPK pathway impairs steroidogenesis through the transcriptional repression of the STAR gene and the feedback inhibition of a key steroidogenic transcription factor, CREB (Zaidi et al., 2014; Li J. et al., 2017). Besides ROS, substantial evidence has indicated that follicle-stimulating hormone (FSH) can also induce the phosphorylation and activation of p38 MAPK. Moreover, the inhibition of p38 MAPK activity using the p38 MAPK inhibitor SB203580 enhances FSH-induced steroidogenesis in rat granulosa cells (Yu et al., 2005). Intriguingly, in our study, PTC-209-induced ROS production in MLTC-1 cells did not contribute to the activation of the MAP3K3-p38 axis, as evidenced by the fact that treatment with the oxidant scavenger NAC and/or the ectopic expression of BMI1-△NLS2 failed to alleviate the activation of p38 MAPKs. This suggested that MAP3K3 was not an oxidative stress sensor in this situation. Indeed, specific MAP3Ks are sometimes associated with specific stresses. For instance, TAK1 activation is always cytokine receptor-dependent (Sorrentino et al., 2008; Yamashita et al., 2008). Moreover, MAP3K1 mediates p38 activation following UV or peptidoglycan stimulation (Zhuang et al., 2006), while ASK1 activates p38 under conditions of oxidative stress (Dolado et al., 2007).
The identification of histone “codes” has greatly contributed to our understanding of gene regulation networks in almost all stages of development (Jambhekar et al., 2019). Histone modifications must be coordinately regulated for accurate cell fate determination, and dysregulated histone modifications always lead to aging, cancer, or other diseases (Booth and Brunet, 2016; Husmann and Gozani, 2019; Michalak et al., 2019). One histone modification can be repressed or enhanced in the presence or absence of another. For instance, LSD1-mediated demethylation of histone H3K4 is activated upon the removal of histone acetylation (Shi et al., 2005; Lee et al., 2006). Additionally, the transcriptional regulator FOXP3 promotes both H3K4 trimethylation and H4K16 acetylation through displacing the demethylase PLU1 and recruiting the acetyltransferase MOF (Katoh et al., 2011). In the present study, BMI1 knockdown led to a significant decrease in H2AK119ub and an increase in H3K4me3 levels at the promoter region of Map3k3, which derepressed its expression. In mouse embryonic fibroblasts (MEFs), BMI1 deficiency led to the removal of H2AK119ub and the enrichment of H3K4me3 at BMI1 binding regions, which was accompanied by the upregulation of the expression of the corresponding genes. In contrast, gene expressions were not affected when there was a decrease in H2AK119ub but no change in H3K4me3 levels at BMI1 binding regions in BMI1-deficent MEFs (Zhou et al., 2016). Collectively, these findings suggested that H3K4me3 contributes to the derepression of BIM1-regulated genes.
Conclusion
In conclusion, to the best of our knowledge, this study represents the first loss- and gain-of-function analysis of the role of BMI1 in steroidogenesis. Our study uncovered a novel epigenetic mechanism in steroidogenesis involving BMI1-mediated gene silencing and provides potential therapeutic targets for the treatment of hypogonadism.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by The Ethics Committee of Nanjing Medical University.
Author Contributions
YW and HL performed most of the experiments. HZ, CS, TG, MLin, XD, and JO performed some of the experiments. HL and MLiu analyzed the data. BZ, XH, FS and JY initiated the project and designed the experiments. JY and BZ wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2018YFC1003500), National Natural Science Foundation of China (81901532, 81901533, and 81801430), Natural Science Foundation of Jiangsu Province (BK20190188), Open Fund of National Health and Family Planning Key Laboratory of Male Reproductive Health (2019GJP03-1), Suzhou Introduced Project of Clinical Medical Expert Team (SZYJTD201708), Suzhou Key Laboratory of Male Reproduction Research (SZS201718), Scientific Research Project of Wuxi Health Committee (Q201926), and Jiangsu Province “13th Five-Year” Youth Medical Person Program (QNRC2016246).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Smartaoit Co., Ltd. (Seychelles) for providing technological support in our bioinformatic analysis and The Charlesworth Group for language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.665089/full#supplementary-material
Footnotes
References
Abdou, H. S., Bergeron, F., and Tremblay, J. J. (2014). A cell-autonomous molecular cascade initiated by AMP-activated protein kinase represses steroidogenesis. Mol. Cell. Biol. 34, 4257–4271. doi: 10.1128/MCB.00734-14
Abidi, P., Leers-Sucheta, S., Cortez, Y., Han, J., and Azhar, S. (2008a). Evidence that age-related changes in p38 MAP kinase contribute to the decreased steroid production by the adrenocortical cells from old rats. Aging Cell 7, 168–178. doi: 10.1111/j.1474-9726.2007.00364.x
Abidi, P., Zhang, H., Zaidi, S. M., Shen, W. J., Leers-Sucheta, S., Cortez, Y., et al. (2008b). Oxidative stress-induced inhibition of adrenal steroidogenesis requires participation of p38 mitogen-activated protein kinase signaling pathway. J. Endocrinol. 198, 193–207. doi: 10.1677/JOE-07-0570
Arthur, J. S., and Ley, S. C. (2013). Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692. doi: 10.1038/nri3495
Ayoub, M. A., Yvinec, R., Jegot, G., Dias, J. A., Poli, S. M., Poupon, A., et al. (2016). Profiling of FSHR negative allosteric modulators on LH/CGR reveals biased antagonism with implications in steroidogenesis. Mol. Cell. Endocrinol. 436, 10–22. doi: 10.1016/j.mce.2016.07.013
Banerjee Mustafi, S., Aznar, N., Dwivedi, S. K., Chakraborty, P. K., Basak, R., Mukherjee, P., et al. (2016). Mitochondrial BMI1 maintains bioenergetic homeostasis in cells. FASEB J. 30, 4042–4055. doi: 10.1096/fj.201600321R
Beattie, M. C., Adekola, L., Papadopoulos, V., Chen, H., and Zirkin, B. R. (2015). Leydig cell aging and hypogonadism. Exp. Gerontol. 68, 87–91. doi: 10.1016/j.exger.2015.02.014
Booth, L. N., and Brunet, A. (2016). The aging epigenome. Mol. Cell 62, 728–744. doi: 10.1016/j.molcel.2016.05.013
Budoff, M. J., Ellenberg, S. S., Lewis, C. E., Mohler, E. R. III, Wenger, N. K., Bhasin, S., et al. (2017). Testosterone treatment and coronary artery plaque volume in older men with low testosterone. JAMA 317, 708–716. doi: 10.1001/jama.2016.21043
Chagraoui, J., Hebert, J., Girard, S., and Sauvageau, G. (2011). An anticlastogenic function for the polycomb group gene Bmi1. Proc. Natl. Acad. Sci. U.S.A. 108, 5284–5289. doi: 10.1073/pnas.1014263108
Chatoo, W., Abdouh, M., David, J., Champagne, M. P., Ferreira, J., Rodier, F., et al. (2009). The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J. Neurosci. 29, 529–542. doi: 10.1523/JNEUROSCI.5303-08.2009
Cohen, K. J., Hanna, J. S., Prescott, J. E., and Dang, C. V. (1996). Transformation by the Bmi-1 oncoprotein correlates with its subnuclear localization but not its transcriptional suppression activity. Mol. Cell. Biol. 16, 5527–5535. doi: 10.1128/mcb.16.10.5527
Dai, X., Zhang, Q., Yu, Z., Sun, W., Wang, R., and Miao, D. (2018). Bmi1 deficient mice exhibit male infertility. Int. J. Biol. Sci. 14, 358–368. doi: 10.7150/ijbs.23325
Dolado, I., Swat, A., Ajenjo, N., De Vita, G., Cuadrado, A., and Nebreda, A. R. (2007). p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 11, 191–205. doi: 10.1016/j.ccr.2006.12.013
Finkel, T., and Holbrook, N. J. (2000). Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247. doi: 10.1038/35041687
Gao, T., Lin, M., Shao, B., Zhou, Q., Wang, Y., Chen, X., et al. (2020). BMI1 promotes steroidogenesis through maintaining redox homeostasis in mouse MLTC-1 and primary Leydig cells. Cell Cycle 19, 1884–1898. doi: 10.1080/15384101.2020.1779471
Haupt, Y., Alexander, W. S., Barri, G., Klinken, S. P., and Adams, J. M. (1991). Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 65, 753–763. doi: 10.1016/0092-8674(91)90383-a
He, D., Wu, H., Xiang, J., Ruan, X., Peng, P., Ruan, Y., et al. (2020). Gut stem cell aging is driven by mTORC1 via a p38 MAPK-p53 pathway. Nat. Commun. 11:37. doi: 10.1038/s41467-019-13911-x
Husmann, D., and Gozani, O. (2019). Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 26, 880–889. doi: 10.1038/s41594-019-0298-7
Jambhekar, A., Dhall, A., and Shi, Y. (2019). Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 20, 625–641. doi: 10.1038/s41580-019-0151-1
Katoh, H., Qin, Z. S., Liu, R., Wang, L., Li, W., Li, X., et al. (2011). FOXP3 orchestrates H4K16 acetylation and H3K4 trimethylation for activation of multiple genes by recruiting MOF and causing displacement of PLU-1. Mol. Cell 44, 770–784. doi: 10.1016/j.molcel.2011.10.012
Kauppila, T. E. S., Kauppila, J. H. K., and Larsson, N. G. (2017). Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71. doi: 10.1016/j.cmet.2016.09.017
Kolettis, P. N., Purcell, M. L., Parker, W., Poston, T., and Nangia, A. K. (2015). Medical testosterone: an iatrogenic cause of male infertility and a growing problem. Urology 85, 1068–1073. doi: 10.1016/j.urology.2014.12.052
Lee, M. G., Wynder, C., Bochar, D. A., Hakimi, M. A., Cooch, N., and Shiekhattar, R. (2006). Functional interplay between histone demethylase and deacetylase enzymes. Mol. Cell. Biol. 26, 6395–6402. doi: 10.1128/MCB.00723-06
Li, B., Chen, Y., Wang, F., Guo, J., Fu, W., Li, M., et al. (2020). Bmi1 drives hepatocarcinogenesis by repressing the TGFbeta2/SMAD signalling axis. Oncogene 39, 1063–1079. doi: 10.1038/s41388-019-1043-8
Li, J., Zhou, Q., Ma, Z., Wang, M., Shen, W. J., Azhar, S., et al. (2017). Feedback inhibition of CREB signaling by p38 MAPK contributes to the negative regulation of steroidogenesis. Reprod. Biol. Endocrinol. 15:19. doi: 10.1186/s12958-017-0239-4
Li, W., Zhu, J., Dou, J., She, H., Tao, K., Xu, H., et al. (2017). Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat. Commun. 8:1763. doi: 10.1038/s41467-017-01609-x
Liu, J., Cao, L., Chen, J., Song, S., Lee, I. H., Quijano, C., et al. (2009). Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 459, 387–392. doi: 10.1038/nature08040
Lovell, M. A., and Markesbery, W. R. (2007). Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 35, 7497–7504. doi: 10.1093/nar/gkm821
Mayor, S. (2016). Testosterone may improve sexual function and mood in older men with low levels. BMJ 352:i989. doi: 10.1136/bmj.i989
Mendoza-Villarroel, R. E., Robert, N. M., Martin, L. J., Brousseau, C., and Tremblay, J. J. (2014). The nuclear receptor NR2F2 activates star expression and steroidogenesis in mouse MA-10 and MLTC-1 Leydig cells. Biol. Reprod. 91:26. doi: 10.1095/biolreprod.113.115790
Michalak, E. M., Burr, M. L., Bannister, A. J., and Dawson, M. A. (2019). The roles of DNA, RNA and histone methylation in ageing and cancer. Nat. Rev. Mol. Cell Biol. 20, 573–589. doi: 10.1038/s41580-019-0143-1
Miller, W. L. (2017). Steroidogenesis: unanswered questions. Trends Endocrinol. Metab. 28, 771–793. doi: 10.1016/j.tem.2017.09.002
Miller, W. L., and Auchus, R. J. (2011). The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151. doi: 10.1210/er.2010-0013
Molofsky, A. V., Pardal, R., Iwashita, T., Park, I. K., Clarke, M. F., and Morrison, S. J. (2003). Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 425, 962–967. doi: 10.1038/nature02060
Morey, L., Santanach, A., Blanco, E., Aloia, L., Nora, E. P., Bruneau, B. G., et al. (2015). Polycomb regulates mesoderm cell fate-specification in embryonic stem cells through activation and repression mechanisms. Cell Stem Cell 17, 300–315. doi: 10.1016/j.stem.2015.08.009
Nargund, V. H. (2015). Effects of psychological stress on male fertility. Nat. Rev. Urol. 12, 373–382. doi: 10.1038/nrurol.2015.112
Nguyen, C. P., Hirsch, M. S., Moeny, D., Kaul, S., Mohamoud, M., and Joffe, H. V. (2015). Testosterone and “age-related hypogonadism”–FDA concerns. N. Engl. J. Med. 373, 689–691. doi: 10.1056/NEJMp1506632
Park, I. K., Morrison, S. J., and Clarke, M. F. (2004). Bmi1, stem cells, and senescence regulation. J. Clin. Invest. 113, 175–179. doi: 10.1172/JCI20800
Park, I. K., Qian, D., Kiel, M., Becker, M. W., Pihalja, M., Weissman, I. L., et al. (2003). Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423, 302–305. doi: 10.1038/nature01587
Patel, P. H., Penalva, C., Kardorff, M., Roca, M., Pavlovic, B., Thiel, A., et al. (2019). Damage sensing by a Nox-Ask1-MKK3-p38 signaling pathway mediates regeneration in the adult Drosophila midgut. Nat. Commun. 10:4365. doi: 10.1038/s41467-019-12336-w
Salonia, A., Rastrelli, G., Hackett, G., Seminara, S. B., Huhtaniemi, I. T., Rey, R. A., et al. (2019). Paediatric and adult-onset male hypogonadism. Nat. Rev. Dis. Primers 5:38. doi: 10.1038/s41572-019-0087-y
Sargis, R. M., and Davis, A. M. (2018). Evaluation and treatment of male hypogonadism. JAMA 319, 1375–1376. doi: 10.1001/jama.2018.3182
Sauvageau, M., and Sauvageau, G. (2010). Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 7, 299–313. doi: 10.1016/j.stem.2010.08.002
Sena, L. A., and Chandel, N. S. (2012). Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167. doi: 10.1016/j.molcel.2012.09.025
Shi, Y. J., Matson, C., Lan, F., Iwase, S., Baba, T., and Shi, Y. (2005). Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 19, 857–864. doi: 10.1016/j.molcel.2005.08.027
Snyder, P. J., Bhasin, S., Cunningham, G. R., Matsumoto, A. M., Stephens-Shields, A. J., Cauley, J. A., et al. (2016). Effects of testosterone treatment in older men. N. Engl. J. Med. 374, 611–624. doi: 10.1056/NEJMoa1506119
Sorrentino, A., Thakur, N., Grimsby, S., Marcusson, A., von Bulow, V., Schuster, N., et al. (2008). The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 10, 1199–1207. doi: 10.1038/ncb1780
van der Lugt, N. M., Domen, J., Linders, K., van Roon, M., Robanus-Maandag, E., te Riele, H., et al. (1994). Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 8, 757–769. doi: 10.1101/gad.8.7.757
van Lohuizen, M., Verbeek, S., Scheijen, B., Wientjens, E., van der Gulden, H., and Berns, A. (1991). Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 65, 737–752. doi: 10.1016/0092-8674(91)90382-9
Wagner, E. F., and Nebreda, A. R. (2009). Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 9, 537–549. doi: 10.1038/nrc2694
Wang, H., Wang, L., Erdjument-Bromage, H., Vidal, M., Tempst, P., Jones, R. S., et al. (2004). Role of histone H2A ubiquitination in polycomb silencing. Nature 431, 873–878. doi: 10.1038/nature02985
Wang, Y., Chen, F., Ye, L., Zirkin, B., and Chen, H. (2017). Steroidogenesis in Leydig cells: effects of aging and environmental factors. Reproduction 154, R111–R122. doi: 10.1530/REP-17-0064
Wang, Y., and Hekimi, S. (2015). Mitochondrial dysfunction and longevity in animals: untangling the knot. Science 350, 1204–1207. doi: 10.1126/science.aac4357
Wu, F. C., Tajar, A., Beynon, J. M., Pye, S. R., Silman, A. J., Finn, J. D., et al. (2010). Identification of late-onset hypogonadism in middle-aged and elderly men. N. Engl. J. Med. 363, 123–135. doi: 10.1056/NEJMoa0911101
Yamashita, M., Fatyol, K., Jin, C., Wang, X., Liu, Z., and Zhang, Y. E. (2008). TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 31, 918–924. doi: 10.1016/j.molcel.2008.09.002
Yu, F. Q., Han, C. S., Yang, W., Jin, X., Hu, Z. Y., and Liu, Y. X. (2005). Activation of the p38 MAPK pathway by follicle-stimulating hormone regulates steroidogenesis in granulosa cells differentially. J. Endocrinol. 186, 85–96. doi: 10.1677/joe.1.05955
Zaidi, S. K., Shen, W. J., Bittner, S., Bittner, A., McLean, M. P., Han, J., et al. (2014). p38 MAPK regulates steroidogenesis through transcriptional repression of STAR gene. J. Mol. Endocrinol. 53, 1–16. doi: 10.1530/JME-13-0287
Zhang, X., Guan, T., Yang, B., Chi, Z., Wang, Z. Y., and Gu, H. F. (2018). A novel role for zinc transporter 8 in the facilitation of zinc accumulation and regulation of testosterone synthesis in Leydig cells of human and mouse testicles. Metabolism 88, 40–50. doi: 10.1016/j.metabol.2018.09.002
Zhao, D., Shen, C., Gao, T., Li, H., Guo, Y., Li, F., et al. (2019). Myotubularin related protein 7 is essential for the spermatogonial stem cell homeostasis via PI3K/AKT signaling. Cell Cycle 18, 2800–2813. doi: 10.1080/15384101.2019.1661174
Zheng, B., Yu, J., Guo, Y., Gao, T., Shen, C., Zhang, X., et al. (2018). Cellular nucleic acid-binding protein is vital to testis development and spermatogenesis in mice. Reproduction 156, 59–69. doi: 10.1530/REP-17-0666
Zheng, B., Zhao, D., Zhang, P., Shen, C., Guo, Y., Zhou, T., et al. (2015). Quantitative proteomics reveals the essential roles of stromal interaction molecule 1 (STIM1) in the testicular cord formation in mouse testis. Mol. Cell. Proteomics 14, 2682–2691. doi: 10.1074/mcp.M115.049569
Zheng, B., Zhou, Q., Guo, Y., Shao, B., Zhou, T., Wang, L., et al. (2014). Establishment of a proteomic profile associated with gonocyte and spermatogonial stem cell maturation and differentiation in neonatal mice. Proteomics 14, 274–285. doi: 10.1002/pmic.201300395
Zhou, Y., Wang, L., Vaseghi, H. R., Liu, Z., Lu, R., Alimohamadi, S., et al. (2016). Bmi1 is a key epigenetic barrier to direct cardiac reprogramming. Cell Stem Cell 18, 382–395. doi: 10.1016/j.stem.2016.02.003
Zhu, S., Zhao, D., Li, C., Li, Q., Jiang, W., Liu, Q., et al. (2020). BMI1 is directly regulated by androgen receptor to promote castration-resistance in prostate cancer. Oncogene 39, 17–29. doi: 10.1038/s41388-019-0966-4
Zhuang, Z. H., Zhou, Y., Yu, M. C., Silverman, N., and Ge, B. X. (2006). Regulation of Drosophila p38 activation by specific MAP2 kinase and MAP3 kinase in response to different stimuli. Cell Signal. 18, 441–448. doi: 10.1016/j.cellsig.2005.05.013
Keywords: testosterone, Bmi1, p38 mitogen-activated protein kinase (MAPK) signaling, epigenetic mechanism, steroidogenesis
Citation: Yu J, Wu Y, Li H, Zhou H, Shen C, Gao T, Lin M, Dai X, Ou J, Liu M, Huang X, Zheng B and Sun F (2021) BMI1 Drives Steroidogenesis Through Epigenetically Repressing the p38 MAPK Pathway. Front. Cell Dev. Biol. 9:665089. doi: 10.3389/fcell.2021.665089
Received: 07 February 2021; Accepted: 22 March 2021;
Published: 13 April 2021.
Edited by:
Joaquim S.L. Vong, The Chinese University of Hong Kong, ChinaReviewed by:
Qingyu Huang, Institute of Urban Environment (CAS), ChinaZhihong Yang, Université de Fribourg, Switzerland
Copyright © 2021 Yu, Wu, Li, Zhou, Shen, Gao, Lin, Dai, Ou, Liu, Huang, Zheng and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Zheng, bWFuc25vb3B5QDE2My5jb20=; Fei Sun, c3VuZmVpQG50dS5lZHUuY24=; Xiaoyan Huang, YmJoeHlAbmptdS5lZHUuY24=
†These authors have contributed equally to this work