 Marisa Cappella
Marisa Cappella Sahar Elouej
Sahar Elouej Maria Grazia Biferi
Maria Grazia Biferi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 13 April 2021
Sec. Stem Cell Research
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.662837
This article is part of the Research Topic Mechanisms of Cellular Differentiation, Organ Development, and Novel Model Systems View all 25 articles
The reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) represents a major advance for the development of human disease models. The emerging of this technique fostered the concept of “disease in a dish,” which consists into the generation of patient-specific models in vitro. Currently, iPSCs are used to study pathological molecular mechanisms caused by genetic mutations and they are considered a reliable model for high-throughput drug screenings. Importantly, precision-medicine approaches to treat monogenic disorders exploit iPSCs potential for the selection and validation of lead candidates. For example, antisense oligonucleotides (ASOs) were tested with promising results in myoblasts or motor neurons differentiated from iPSCs of patients affected by either Duchenne muscular dystrophy or Amyotrophic lateral sclerosis. However, the use of iPSCs needs additional optimization to ensure translational success of the innovative strategies based on gene delivery through adeno associated viral vectors (AAV) for these diseases. Indeed, to establish an efficient transduction of iPSCs with AAV, several aspects should be optimized, including viral vector serotype, viral concentration and timing of transduction. This review will outline the use of iPSCs as a model for the development and testing of gene therapies for neuromuscular and motor neuron disorders. It will then discuss the advantages for the use of this versatile tool for gene therapy, along with the challenges associated with the viral vector transduction of iPSCs.
The development of human induced pluripotent stem cells (iPSCs) (Takahashi et al., 2007) provided unprecedented opportunities to decipher pathophysiological mechanisms of diseases and to test therapeutic approaches in conditions that better translate to humans. This technology allows to obtain an unlimited number of cells from one patient thus representing an ideal model to study in vitro disease’s developmental stages, onset and progression in specific human cells (Park et al., 2008a).
iPSCs are capable of indefinite self-renewal and can differentiate into any cell type under appropriate culture conditions (Takahashi et al., 2007; Yu et al., 2007). iPSCs are generated by reprogramming primary somatic cells, such as dermal fibroblasts or blood cells, using ectopic expression of selected embryonic transcription factors (e.g., Oct4, Sox2, Klf4, and c-Myc) (Takahashi et al., 2007). Over the years, several techniques have been refined to deliver the reprogramming cocktail for iPSCs generation. The first pioneering studies on iPSCs used integrating delivery systems, through retroviral or lentiviral vectors (Takahashi et al., 2007; Yu et al., 2007; Park et al., 2008b). To avoid any incorporation of the foreign genetic material and induction of genomic alterations (Nakagawa et al., 2008; Shao and Wu, 2010), novel delivery systems have been introduced, based on non-integrating vectors (such as the Sendai virus or episomal vectors), self-excising vectors (i.e., Cre-Lox, PiggyBac transposon), and non-viral vectors (i.e., combination of signaling molecules, small bioactive molecules, microRNAs, and other chemicals) (Liu et al., 2020). Interestingly, the delivery of synthetic mRNA expressing the reprogramming factors, was also exploited for the safe generation of iPSCs (Warren et al., 2010). It was also used for iPSCs differentiation (Warren et al., 2012; Mandal and Rossi, 2013; Yoshioka et al., 2013; Goparaju et al., 2017). This technology provides high in vitro transfection efficiency of complex mixtures, with transient expression and absence of genomic integration (Sahin et al., 2014).
iPSCs have the ability to retain the genetic mutation carried by the donor patient together with its genomic background, overcoming the limitations presented by the animal models and leading to a new era of disease modeling and clinical applications (Shi et al., 2017). Moreover, unlike the other unlimited sources of self-renewing cells, the embryonic stem cells (ESCs), which can only be obtained from early-stage blastocysts (4–5 days post fertilization), the iPSCs can be generated from adult patients, eliminating the ethical issues related to the generation of ESCs and leading to the opportunity for studying different stages of the disorders (Romano, 2008; Romito and Cobellis, 2016).
However, genetic background heterogeneity, lack of proper controls, as well as technical challenges in handling and standardizing the culture methods (Doss and Sachinidis, 2019; Volpato and Webber, 2020), contribute to the variability observed in the use of iPSCs as disease model (Hoekstra et al., 2017; Karagiannis et al., 2018; Volpato and Webber, 2020). To deal with genetic background influence on the expression of disease phenotype it is now possible to generate isogenic cell lines, introducing or repairing putative causative mutations through the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9-mediated genomic editing technologies (Ben Jehuda et al., 2018). The use of such controls, when possible, reduces the observed variation in cellular phenotypes caused by the genomic milieu (Soldner and Jaenisch, 2012).
Thanks to the mentioned superior features, iPSCs were exploited to generate in vitro models of severe diseases affecting the neuromuscular system and/or the central nervous system, such as neuromuscular and motor neuron disorders (NMD and MND, respectively). While genetic corrected iPSCs are investigated in the complex field of cell replacement therapies, in which modified cells are reintroduced into patients (Tedesco et al., 2012; Barthélémy and Wein, 2018; Abdul Wahid et al., 2019), the iPSCs platform has already allowed the identification of drug candidates for some of these complex disorders (Ortiz-Vitali and Darabi, 2019; Pasteuning-Vuhman et al., 2020). Recently, the combination of iPSCs and gene targeting approaches is changing the face of modern medicine. In this review, we will thus briefly discuss the successes in the identification of drug candidates for NMD and MND and then we will focus on the efforts toward the validation of gene therapy approaches in iPSCs for muscular dystrophies, amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA). Table 1 summarizes the research efforts in this direction mentioned in this review.
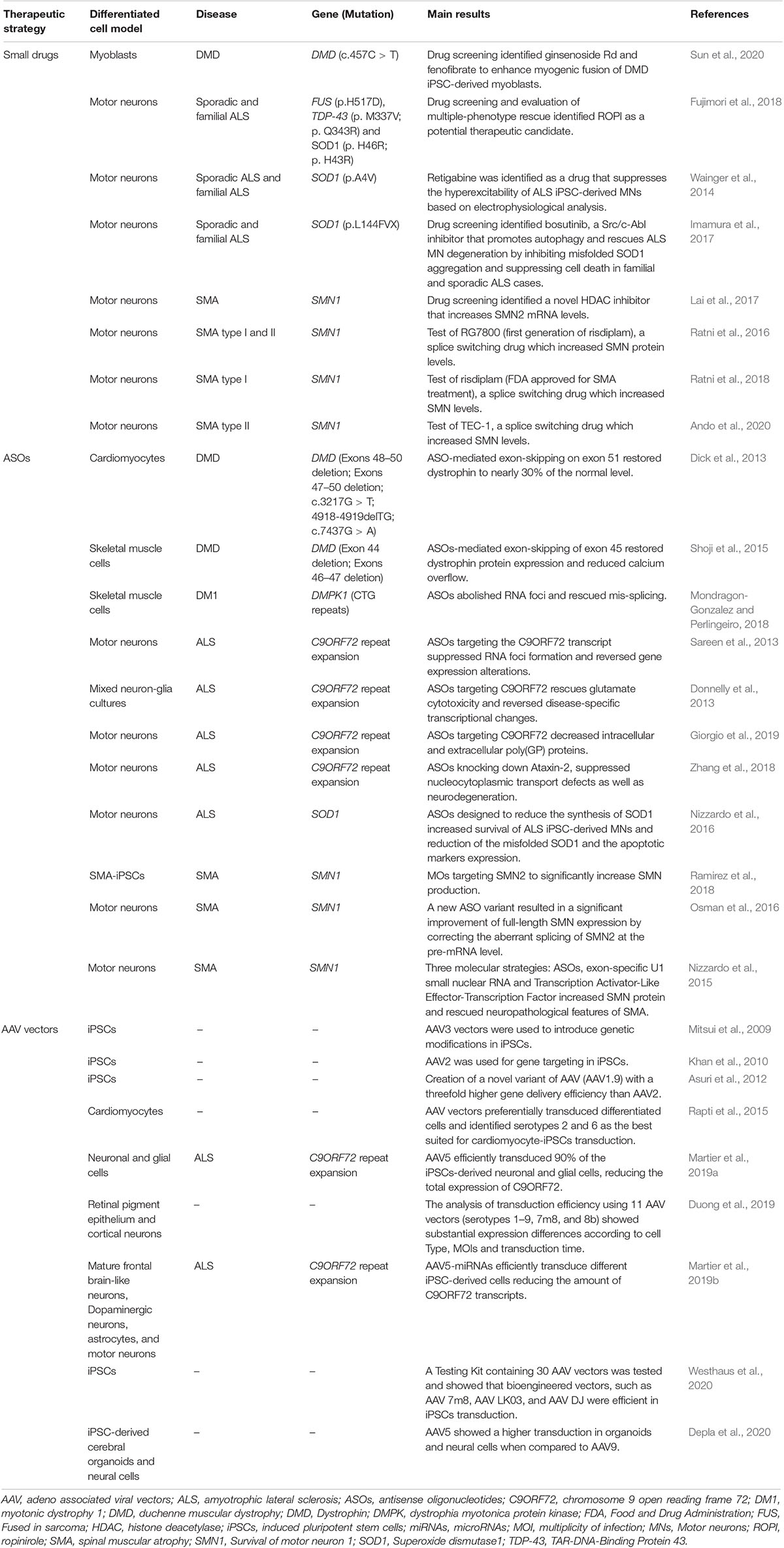
Table 1. Summary of the major findings of the cited articles in which iPSCs were used for therapeutic tests of neuromuscular and motor neuron disorders.
iPSCs are widely exploited in high-throughput drug screenings for genetic disorders. Thus far, the introduction of iPSCs into the drug development pipeline has allowed (i) physiologically improved modeling of disease-relevant phenotypes, (ii) a greater patient stratification, and (iii) discrimination between drug responders and non-responders (Pasteuning-Vuhman et al., 2020). In perspective, this will have an impact on the current limitations of the conventional drug discovery process and consequently improve the success of therapeutic target identification and clinical trial outcomes (Hosoya and Czysz, 2016).
Following their discovery, multiple research efforts focused on the generation of iPSCs for NMD and MND. As example, in 2008 Park and collaborators, established the first iPSCs line from skin fibroblasts from a patient affected by Duchenne muscular dystrophy (DMD), a fatal genetic disorder caused by mutations in the dystrophin (DMD) gene and characterized by progressive muscle wasting (Koenig et al., 1987; Park et al., 2008a; Gao and McNally, 2015). Since then, additional DMD-iPSC lines have been reported by other groups and several differentiation protocols were tested to refine the optimal methods for skeletal muscle and cardiac cell differentiation (reviewed by Danisovic et al., 2018; Piga et al., 2019). These attempts overcame some of the limitations of the commonly used human models of DMD, such as myoblasts obtained from patient biopsies, which are limited in number and phenotypically diverse (Blau et al., 1983; Renault et al., 2000; Sun et al., 2020). In contrast, patient-derived iPSCs allow the generation of large amount of mature skeletal muscle cells (Chal et al., 2016; Caputo et al., 2020) or cardiomyocytes—recapitulating the cardiomyopathy of dystrophic patients (Hashimoto et al., 2016), and can mimic different stages of the disorder (Xia et al., 2018). iPSCs were also converted to neuronal cells to study the impact on the central nervous system in NMD. For example, neuron-iPSCs were generated from patients affected by myotonic dystrophy 1 (DM1) (Du et al., 2013; Xia et al., 2013; Ueki et al., 2017), caused by an expansion of the CTG trinucleotide repeats in the 3′ untranslated region of the dystrophia myotonica protein kinase (DMPK) gene (Brook et al., 1992). Altogether these studies highlight the versatility of iPSCs as model for the thorough study of gene mutations in the main affected tissues (i.e., skeletal and cardiac muscle for DMD) but also in other relevant cell types (such as neurons in DM1), which contribute to the disease manifestations. Furthermore, iPSCs are being exploited for the development of therapies for muscular dystrophies which is usually carried out in mouse models unable to fully recapitulate all the human disease features (Wells, 2018; Ortiz-Vitali and Darabi, 2019; van Putten et al., 2020). Recently, Sun and colleagues developed a platform based on DMD-iPSC–derived myoblasts for drug screening and among 1524 compounds analyzed, they identified 2 promising small molecules with in vivo efficacy (Sun et al., 2020). Further efforts in this direction will likely improve the search for reliable drug candidates and eventually increase the success rate in clinical trials for these severe disorders.
While animal models remain the preferred choice also for modeling and drug testing for MND (Picher-Martel et al., 2016; Dawson et al., 2018; Giorgio et al., 2019), the large genetic variability of these disorders set the ground for the wide use of patient-derived cells. Since 2008, when Eggan’s group (Dimos et al., 2008) used for the first time iPSCs to produce patient-specific motor neurons and glia from skin cells of an 82-year-old female patient diagnosed with ALS—the most common adult onset MND—several groups have designed and validated protocols for spinal motor neurons (MN) (Son et al., 2011; Amoroso et al., 2013; Demestre et al., 2015; Maury et al., 2015; Toli et al., 2015; Sances et al., 2016; Fujimori et al., 2018) and astrocyte differentiation (Madill et al., 2017; Birger et al., 2019; Zhao et al., 2020). The studies performed in ALS-iPSCs with different genetic mutations, facilitated the identification of common pathological features to the various disease forms, such as endoplasmic reticulum stress (Kiskinis et al., 2014; Dafinca et al., 2016), mitochondrial abnormalities (Dafinca et al., 2020; Hor et al., 2020), and impaired excitability (Wainger et al., 2014), but also characteristics related to specific mutations, like protein aggregation or mislocalization (Liu et al., 2015).
Drug screenings using ALS-derived iPSCs additionally allowed the identification of three drugs that are currently explored as therapeutic options in clinical trials.
- The first one, ROPI, a dopamine receptor agonist, was identified from a panel of 1232 Food and Drug Administration (FDA)-approved drugs in a drug screening analysis conducted at Keio University, which examined Fused in sarcoma (FUS)- and TAR-DNA-Binding Protein 43 (TDP-43)-ALS iPSC-derived MN for suppression of ALS-related phenotypes in vitro, such as mislocalization of FUS/TDP43, stress granule formation, MN death/damage, and neurite retraction (Fujimori et al., 2018). This drug is now tested in the ROPALS trial (UMIN000034954 and JMA-IIA00397) as continuation of the Phase I/IIa clinical trial (Morimoto et al., 2019).
- Retigabine (known as an antiepileptic) was identified as a potential suppressor of the hyperexcitability of ALS iPSC-derived MNs based on electrophysiological analysis (Wainger et al., 2014). It is a voltage-gated potassium channel activator (Kv7) able to both block hyperexcitability and improve MN survival in vitro when tested in ALS cases carrying the most common genetic mutations (Wainger et al., 2014). A Phase II Pharmacodynamic Trial of Ezogabine (Retigabine) on neuronal excitability in ALS (NCT02450552) was conducted from 2015 to 2019 showing a decrease of cortical and spinal MN excitability in participants with ALS. These data suggest that such neurophysiological metrics may be used as pharmacodynamic biomarkers in multisite clinical trials (Wainger et al., 2020).
- The third drug is Bosutinib, a proto-oncogene non-receptor protein tyrosine kinase (Src/c-Abl) inhibitor that promoted autophagy and rescued degeneration in iPSC-derived MN, inhibiting misfolded Superoxide Dismutase 1 (SOD1) aggregation and suppressing cell death in genetic and sporadic ALS (Imamura et al., 2017). A new Phase I clinical trial of the drug bosutinib for ALS (UMIN000036295) was initiated in Japan in March 2019.
These examples of drug discovery in iPSCs and their ongoing translation to patients affected by a yet uncurable disease, indicate that this could be a valid paradigm for clinical success in similar diseases, such as SMA. SMA is a MND caused by homozygous mutations in the survival of motor neuron gene (SMN1) leading to infant mortality and motor disabilities in young and adult patients (Lefebvre et al., 1995; Verhaart et al., 2017; Smeriglio et al., 2020). This gene has a paralog called SMN2 that is nearly identical to SMN1, with few nucleotide differences, which result in the exclusion of exon 7 and 90% production of a truncated non-functional survival of motor neuron (SMN) protein (Lefebvre et al., 1995). Several therapeutic strategies have been tested to restore SMN expression (Wirth, 2021). Histone deacetylase (HDAC) inhibitors were tested to induce transcriptional activation of SMN2 and consequent increased production of full length SMN, with successful outcomes in proof-of-concept studies and failure in clinical trials. With the aim to identify compounds with higher efficacy and specificity, Lai and colleagues performed a drug screening in neuron-iPSCs from SMA patients. This study identified novel HDAC inhibitors with therapeutic potential that could be further explored for SMA treatment (Lai et al., 2017). Interestingly, neuron-iPSC from SMA patients were also used to test the efficacy of the recent FDA approved small molecule EvrysdiTM (risdiplam) (Ratni et al., 2016; Ratni et al., 2018; Dhillon, 2020), which forces the inclusion of exon 7 and thus restore SMN protein levels (Poirier et al., 2018). Moreover, the drug called TEC-1 (2-(4,6-dimethylpyrazolo[1,5-a]pyrazin-2-yl)-6-(4-methylpiperazin-1-yl)quinazolin-4(3H)-one) another SMN2 splicing modulator, was recently identified in a screening on SMA patient-derived fibroblasts. The drug’s effects were then confirmed in SMA-MN-iPSCs (Ando et al., 2020).
As suggested by the reported examples, the combination of iPSCs modeling, together with high-throughput drug screening followed by animal tests will likely ensure the identification of effective and safe therapeutic candidates. How this pipeline can be adapted to the development and tests for precision medicine approaches, such as gene therapy, will be discussed in the following paragraphs and is exemplified in Figure 1.
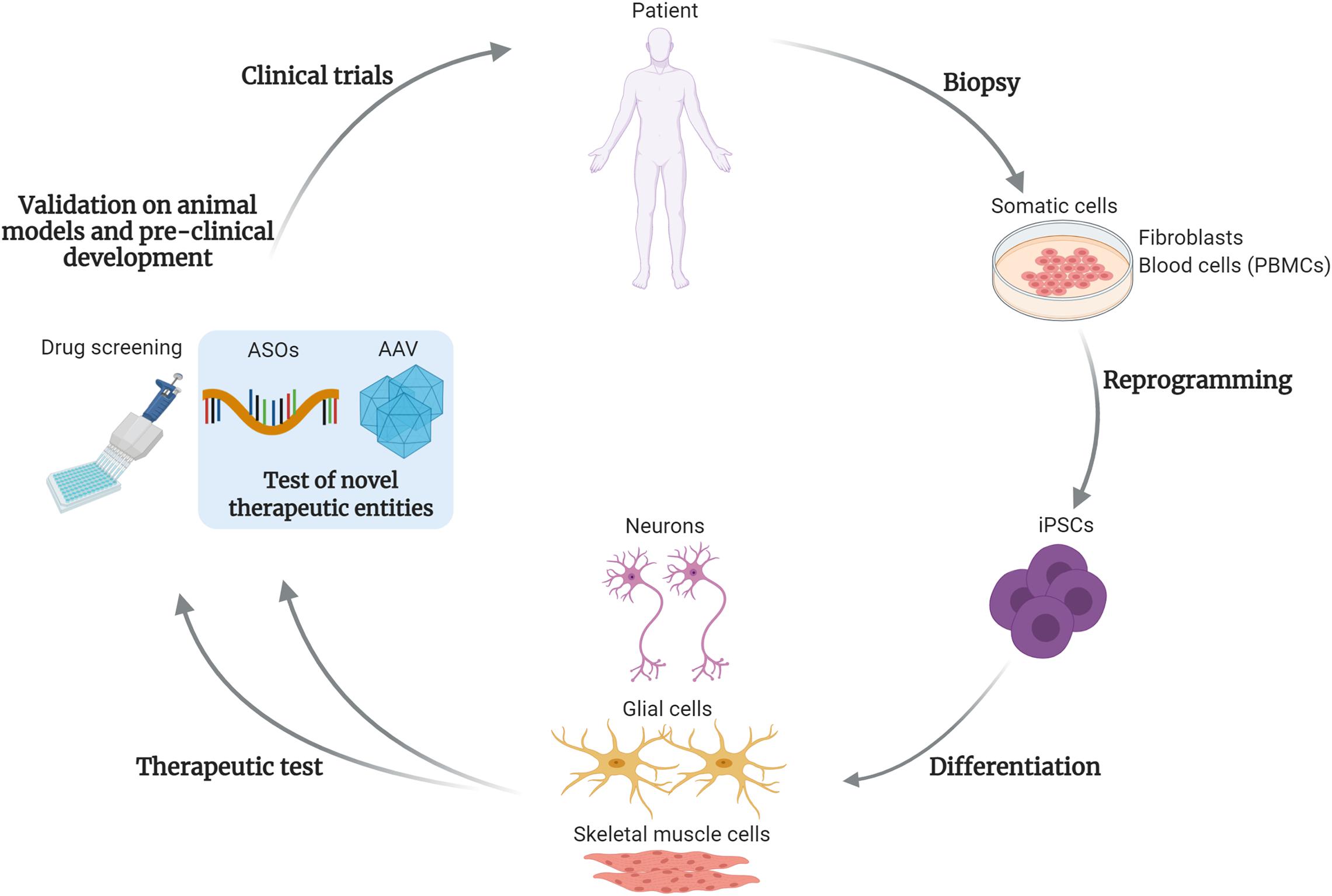
Figure 1. Test and development of gene targeting approaches using iPSCs. This drawing summarizes the steps of development for drugs and gene therapy approaches, using induced pluripotent stem cells (iPSCs). Somatic cells, such as fibroblasts or blood cells (peripheral blood mononuclear cells, PBMCs) are obtained from patient’s biopsies. After reprogramming, the patient-derived iPSCs can be differentiated into disease-relevant cell types, such as skeletal muscle cells, neural or glial cells for neuromuscular or motor neuron disorders. These cells are then subjected to the classical high-throughput drug screening and in perspective will be used to test novel therapeutic entities, based on gene targeting approaches. As example, antisense oligonucleotides (ASOs) or adeno-associated viral vectors (AAV)-based strategies. After validations in animal models and the pre-clinical development process, these novel therapies could enter into clinical trials for patients affected by rare disorders. The use of iPSCs and gene targeting strategies will likely foster the development of personalized medicine approaches. Created with BioRender.com.
Gene targeting approaches are based on the direct correction of the genetic defects (Wang and Gao, 2014; Cappella et al., 2019). For example, antisense oligonucleotides (ASOs) widely tested in pre-clinical and clinical settings, have been approved for SMA (Spinraza®) (Aartsma-Rus, 2017) and DMD (i.e., Exondys 51®) (Stein, 2016) patients, encouraging their use for the treatment of other monogenic disorders.
ASOs are synthetic single-stranded strings of nucleic acids that bind to RNA through standard Watson–Crick base pairing. After binding to the targeted RNA, the antisense drug can modulate the function of the targeted RNA by several mechanisms (Bennett and Swayze, 2010; Crooke et al., 2018), depending on the chemical modifications and the binding position on the target RNA (Wurster and Ludolph, 2018; Talbot and Wood, 2019; Ochoa and Milam, 2020). Briefly, ASOs can promote degradation of the targeted RNA, by mimicking DNA-RNA pairing and activating endogenous nucleases (i.e., RNase H1), or can modulate the processing of the RNA molecule, without inducing its degradation. This can be achieved through several mechanisms, such as by masking RNA splicing sites, as in the examples described below for DMD or SMA (Dick et al., 2013; Shoji et al., 2015; Osman et al., 2016; Ramirez et al., 2018). Other methods of action of ASOs have been previously reviewed (Bennett and Swayze, 2010; Crooke, 2017).
Several strategies, (Miller and Harris, 2016; Schoch and Miller, 2017), are currently investigated to increase ASOs stability, enhance binding affinity to the target RNA, improve tissue distribution and cellular uptake, while decreasing possible adverse effects (Bennett et al., 2017). Here we will focus on the use of iPSCs as model for testing the efficacy of these gene targeting approaches in NMD and MND.
Due to the large size of the DMD gene (Koenig et al., 1987), the restoration of the full-length dystrophin protein is challenging (Gao and McNally, 2015; Duan, 2018). One of the most promising approaches for gene targeting in DMD, is the use of ASOs binding to the pre-mRNA of the DMD gene to restore its reading frame and consequently producing a truncated but yet functional protein.
The ASO-mediated exon-skipping efficacy on exon 51 was tested in cardiomyocytes derived from iPSCs with DMD mutations, restoring dystrophin to nearly 30% of the normal level (Dick et al., 2013). Another similar study tested an ASO forcing exon 45 skipping of the DMD gene in myotubes derived from iPSCs, thus restoring dystrophin expression but also reducing calcium overflow (Shoji et al., 2015). These studies indicate that iPSCs can be used as platforms for therapeutic selection of ASO, based on the gene correction and prevention of skeletal muscle phenotype in DMD. The new frontier for the treatment of DMD patients is the development of mutation-specific ASOs (Schneider and Aartsma-Rus, 2020) and the use of iPSCs will likely speed the path to success of those strategies through the selection of the patient-specific and most efficient candidates.
ASOs were also proven effective in differentiated myotubes from DM1-iPSCs. A repeat-directed ASO treatment abolished RNA foci accumulation and rescued mis-splicing (Mondragon-Gonzalez and Perlingeiro, 2018) in vitro. These discoveries indicate that once established the proper conversion and differentiation protocols, together with valid disease read-outs, the test of ASOs in iPSCs could be likely applied to a larger spectrum of muscular dystrophies and diseases.
Therapeutic ASOs are currently tested in clinical trials for ALS patients harboring the chromosome 9 open reading frame 72 (C9ORF72) mutations (NCT03626012), SOD1 mutations (NCT03070119, NCT02623699) (recently reviewed by Cappella et al., 2021) or for sporadic ALS patients, with the Ataxin2-ASO (NCT04494256, Becker et al., 2017). Importantly, a splice switching ASO targeted to SMN2 (Spinraza®) was approved for SMA patients in 2016.
To better characterize ASOs ability to rescue disease hallmarks, to dissect pathophysiological mechanisms and to test novel chemistries and molecular technologies, different research groups are studying ASOs in iPSCs for MND. For example, ASOs were proven effective in reducing the accumulation of sense RNA foci or toxic dipeptides in C9ORF72-iPSCs differentiated to neurons or MN (Donnelly et al., 2013; Sareen et al., 2013; Giorgio et al., 2019). More recently, Zhang et al. (2018) demonstrated that nucleocytoplasmic transport deficits and neurodegeneration were alleviated in C9ORF72-MN-iPSCs, after treatment with ASOs directed against the Ataxin 2, an RNA-binding protein. Nizzardo et al. (2016) treated ALS MN-iPSCs with ASOs designed to reduce the synthesis of human SOD1 and observed an increased survival and reduced expression of apoptotic markers in treated cells.
In SMA, iPSCs were used to test novel ASO sequences for their improved capacity of producing the full length SMN protein from splicing modulation of SMN2 and exon 7 inclusion (Osman et al., 2016; Ramirez et al., 2018). They were also used to test novel molecular strategies to restore SMN expression and correct neuropathological feature, namely an U1 small nuclear RNA-mediated splice switching approach and SMN transcription activation, via the Transcription Activator-Like Effector-Transcription Factor (TALE-TF) (Nizzardo et al., 2015). This report suggests that iPSCs could serve for the side-by-side comparison of different gene targeting strategies for monogenic disorders.
The use of adeno-associated viral vectors (AAV) for gene therapy of rare disorders recently became a clinical reality. The approval of Zolgensma® (an AAV-mediated therapy) for the treatment of the most severe form of SMA, endorses the development of similar approaches for NMD and MND. Indeed, several pre-clinical studies report successes of these approaches in disease models (Biferi et al., 2017; Cappella et al., 2019; Crudele and Chamberlain, 2019) and their use in clinical trials (Bowles et al., 2012; Mendell et al., 2015; Mueller et al., 2020).
Some of the challenges associated to the translation of AAV-based therapies from animal models to patients, are linked to (i) the selection of the best AAV serotype for efficient transgene expression, (ii) cell/tissue specificity, as well as (iii) production of high vector titers, and (iv) reduction of immunoreactivity (Colella et al., 2017; Naso et al., 2017). To date, hundreds of natural AAV serotypes, variants and bio-engineered versions have been described (Hester et al., 2009; Choudhury et al., 2016; Deverman et al., 2016; Chan et al., 2017; Hanlon et al., 2019). Beside serotypes, research efforts are also focusing on the combination of the best serotype with the therapeutic and regulatory sequences—such as promoters or enhancers (Colella et al., 2018; Besse et al., 2020; Nieuwenhuis et al., 2020), for efficient, safe and specific transgene expressions (Guilbaud et al., 2019; Hanlon et al., 2019). This will likely contribute to expedite the translational path from bench to clinic. In this context, iPSCs can be used to select the vector with best transduction properties for a specific cell type and/or to test the therapeutic sequences (recombinant transgene, oligonucleotides, antibodies, etc.). These techniques will be further refined to design patient-specific approaches. In perspective, when a therapeutic candidate will be established, iPSCs could be further used for analytical tests of approved gene therapies, such as potency assays.
AAV vectors were initially tested for genetic manipulation of ESCs or iPSCs in vitro, using natural human-derived AAV serotypes (from 1 to 9). After some unsuccessful attempts (Smith-Arica et al., 2003; Jang et al., 2011), some reports showed that natural AAV vector serotypes, such as AAV 2 and 3, were able to target iPSCs, although with limited efficacy (Mitsui et al., 2009; Khan et al., 2010). Through direct evolution, Asuri et al. (2012), derived a novel variant of AAV (AAV1.9) with a threefold higher gene delivery efficiency than AAV2 in iPSCs. These pioneer studies suggested that AAV vectors could be also used for stem cell correction and consequently studies of biological mechanisms in vitro and eventually for therapeutic purposes in cell therapy approaches.
Several studies reported method for AAV-mediated delivery of differentiated iPSCs. For example, Rapti et al. (2015) compared the transduction efficiency of different AAV (serotypes 1, 2, 6, and 9) in cardiomyocyte-iPSCs. Interestingly, they noticed that AAV vectors preferentially transduced differentiated cells and identified in serotypes 2 and 6 the best suited for cardiomyocyte-iPSCs transduction.
For modeling and therapeutic testing of central nervous system cells, AAV serotype 5 expressing the green fluorescent protein (GFP), was proven efficient in iPSCs-derived neuronal and glial cells, resulting in up to 90% of transduction (Martier et al., 2019a). Moreover, Duong et al. examined the level of AAV-GFP expression following the transduction of 11 AAV vectors in iPSCs differentiated into retinal pigment epithelium and cortical neurons (Duong et al., 2019). GFP-expressing cells were examined and compared across doses, time and cell type. They reported that retinal pigmented epithelium had the highest AAV-mediated GFP expression compared to cortical neurons-iPSCs and that AAV7m8 and AAV6 were the best performing, across vector concentrations and cell types. This study suggested that in addition to vector tropisms, cell type significantly affects transgene expression (Duong et al., 2019).
Overall, following optimizations, AAV vectors can be used to efficiently transduce patient-derived cells converted to neural or glial cells, likely facilitating studies for neurological diseases. Indeed, Martier and colleagues investigated the feasibility of a miRNA-based gene therapy to obtain long-term silencing of the repeat-containing transcripts of C9ORF72. Four AAV5 carrying miR candidates were tested in neuron-iPSC, resulting in sufficient transduction and expression of therapeutically relevant levels of the corresponding mature miRNA (Martier et al., 2019b). Two of the tested candidates were then proven efficient in reducing RNA foci accumulation in some brain regions of a disease mouse model (Martier et al., 2019a).
Novel methods are currently developed to select AAV for their fitness in vitro. For example, the group of Lisowski developed an AAV Testing Kit, as novel high-throughput approach based on next-generation sequencing, to study the performance of 30 published AAV variants in vitro, in vivo, and ex vivo. They tested AAV variants in primary cells, immortalized cell lines and iPSCs, showing that iPSCs were most efficiently transduced with bioengineered vectors, such as AAV 7m8, AAV LK03, and AAV DJ (Westhaus et al., 2020). This suggests that further methods for AAV optimization are necessary and will likely improve AAV transduction properties in vitro and in vivo.
Transduction properties of AAV serotypes in the human context have been recently tested in 3D structure iPSC-derived cerebral organoids. The transduction properties of two commonly used AAV serotypes (AAV5 and 9) were compared for transgene expression at the mRNA and protein levels, together with the presence of viral DNA. This study reported a higher transduction of the AAV5 compared to AAV9, in organoids and neural cells (Depla et al., 2020). This work set the ground for the use of iPSCs-derived human organoids as valid system for testing AAV properties and will be likely a valuable platform for holistic characterization of AAV properties in vitro and identification of the best therapeutic candidates.
Gene therapy treatments are revolutionizing the face of modern medicine opening treatment perspectives for patients affected by fatal conditions. Despite the growing success of these approaches, several aspects of gene therapy development need refinement and would benefit of the use of iPSCs. Indeed, together with their most known use, such as disease modeling for high-throughput drug screenings, they can be converted into a reliable platform for testing the novel therapeutic entities. Indeed, after the establishment of proper differentiation protocols and disease readouts, patient-derived models are being utilized to test gene targeting approaches. Here, we have summarized research efforts in testing drugs and gene therapy approaches in iPSCs from patient affected by neuromuscular and motor neuron diseases. We have presented some of the successes in candidate drug identification, such as risdiplam for the treatment of SMA and the research efforts in testing ASOs and AAV-mediated therapies. These studies set the ground for further developments, to select optimized therapeutic molecules and to identify powerful and safe AAV vectors.
In parallel to iPSCs development, research efforts are currently focused on the generation of even more advanced disease models. Indeed, despite iPSCs represent a reliable model for the understanding of pathological mechanisms and therapeutic development, they do not fully recapitulate the complexity of a tissue, with its architecture and interactions (Costamagna et al., 2019). In this direction, 3D culture methods are being implemented for NMD and MND, for example with the generation of artificial skeletal muscle for DMD (Maffioletti et al., 2018) or spinal cord organoids for SMA, which were used for drug test (Hor et al., 2018). Interestingly, the group of Pasça, has recently reported the generation of iPSC-derived 3D culture, in which cerebral cortex or hindbrain/spinal cord organoids were assembled with skeletal muscle spheroids (Andersen et al., 2020). These so-called 3D cortico-motor assembloids hold promise for the development of effective therapeutics for NMD and MND.
In conclusion, the advances in novel technologies, such as production of mature organoids, will endorse the development of efficient personalized medicine approaches.
MC and SE: writing of the manuscript draft. MB: conceptualization, writing, and review. All authors contributed to the article and approved the submitted version.
MC was supported by the ANR grant no. ANR-19-CE18-0014-01. MB and SE were supported by the Association Institut de Myologie (AIM)
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Association Française contre les Myopathies (AFM), the Association Institut de Myologie (AIM), the Sorbonne Université, the Institut National de la Santé et de la Recherche Médicale (INSERM). We also thank Piera Smeriglio for critical reading of the manuscript.
Aartsma-Rus, A. (2017). FDA approval of nusinersen for spinal muscular atrophy makes 2016 the year of splice modulating oligonucleotides. Nucleic Acid Ther. 27, 67–69. doi: 10.1089/nat.2017.0665
Abdul Wahid, S. F., Law, Z. K., Ismail, N. A., and Lai, N. M. (2019). Cell-based therapies for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst. Rev. 12:CD011742.
Amoroso, M. W., Croft, G. F., Williams, D. J., O’Keeffe, S., Carrasco, M. A., Davis, A. R., et al. (2013). Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J. Neurosci. 33, 574–586. doi: 10.1523/jneurosci.0906-12.2013
Andersen, J., Revah, O., Miura, Y., Thom, N., Amin, N. D., Kelley, K. W., et al. (2020). Generation of functional human 3D cortico-motor assembloids. Cell 183, 1913–1929.e26.
Ando, S., Suzuki, S., Okubo, S., Ohuchi, K., Takahashi, K., Nakamura, S., et al. (2020). Discovery of a CNS penetrant small molecule SMN2 splicing modulator with improved tolerability for spinal muscular atrophy. Sci. Rep. 10:17472.
Asuri, P., Bartel, M. A., Vazin, T., Jang, J.-H., Wong, T. B., and Schaffer, D. V. (2012). Directed evolution of adeno-associated virus for enhanced gene delivery and gene targeting in human pluripotent stem cells. Mol. Ther. 20, 329–338. doi: 10.1038/mt.2011.255
Barthélémy, F., and Wein, N. (2018). Personalized gene and cell therapy for Duchenne muscular dystrophy. Neuromuscul. Disord. 28, 803–824. doi: 10.1016/j.nmd.2018.06.009
Becker, L. A., Huang, B., Bieri, G., Ma, R., Knowles, D. A., Jafar-Nejad, P., et al. (2017). Therapeutic reduction of ataxin 2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371. doi: 10.1038/nature22038
Ben Jehuda, R., Shemer, Y., and Binah, O. (2018). Genome editing in induced pluripotent stem cells using CRISPR/Cas9. Stem Cell Rev. Rep. 14, 323–336. doi: 10.1007/s12015-018-9811-3
Bennett, C. F., and Swayze, E. E. (2010). RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 50, 259–293. doi: 10.1146/annurev.pharmtox.010909.105654
Bennett, C. F., Baker, B. F., Pham, N., Swayze, E., and Geary, R. S. (2017). Pharmacology of antisense drugs. Annu. Rev. Pharmacol. Toxicol. 57, 81–105.
Besse, A., Astord, S., Marais, T., Roda, M., Giroux, B., Lejeune, F.-X., et al. (2020). AAV9-mediated expression of SMN restricted to neurons does not rescue the spinal muscular atrophy phenotype in mice. Mol. Ther. 28, 1887–1901. doi: 10.1016/j.ymthe.2020.05.011
Biferi, M. G., Cohen-Tannoudji, M., Cappelletto, A., Giroux, B., Roda, M., Astord, S., et al. (2017). A new AAV10-U7-mediated gene therapy prolongs survival and restores function in an ALS mouse model. Mol. Ther. 25, 2038–2052. doi: 10.1016/j.ymthe.2017.05.017
Birger, A., Ben-Dor, I., Ottolenghi, M., Turetsky, T., Gil, Y., Sweetat, S., et al. (2019). Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 50, 274–289. doi: 10.1016/j.ebiom.2019.11.026
Blau, H. M., Webster, C., and Pavlath, G. K. (1983). Defective myoblasts identified in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U.S.A. 80, 4856–4860. doi: 10.1073/pnas.80.15.4856
Bowles, D. E., McPhee, S. W., Li, C., Gray, S. J., Samulski, J. J., Camp, A. S., et al. (2012). Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 20, 443–455.
Brook, J. D., McCurrach, M. E., Harley, H. G., Buckler, A. J., Church, D., Aburatani, H., et al. (1992). Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68, 799–808. doi: 10.1016/0092-8674(92)90154-5
Cappella, M., Ciotti, C., Cohen-Tannoudji, M., and Biferi, M. G. (2019). Gene therapy for ALS—a perspective. Int. J. Mol. Sci. 20:4388. doi: 10.3390/ijms20184388
Cappella, M., Pradat, P.-F., Querin, G., and Biferi, M. G. (2021). Beyond the traditional clinical trials for amyotrophic lateral sclerosis and the future impact of gene therapy. J. Neuromuscul. Dis. 8, 25–38. doi: 10.3233/jnd-200531
Caputo, L., Granados, A., Lenzi, J., Rosa, A., Ait-Si-Ali, S., Puri, P. L., et al. (2020). Acute conversion of patient-derived Duchenne muscular dystrophy iPSC into myotubes reveals constitutive and inducible over-activation of TGFβ-dependent pro-fibrotic signaling. Skelet. Muscle 10:13.
Chal, J., Al Tanoury, Z., Hestin, M., Gobert, B., Aivio, S., Hick, A., et al. (2016). Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc. 11, 1833–1850. doi: 10.1038/nprot.2016.110
Chan, K. Y., Jang, M. J., Yoo, B. B., Greenbaum, A., Ravi, N., Wu, W.-L., et al. (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 20, 1172–1179. doi: 10.1038/nn.4593
Choudhury, S. R., Fitzpatrick, Z., Harris, A. F., Maitland, S. A., Ferreira, J. S., Zhang, Y., et al. (2016). In vivo selection yields AAV-B1 capsid for central nervous system and muscle gene therapy. Mol. Ther. 24, 1247–1257. doi: 10.1038/mt.2016.84
Colella, P., Ronzitti, G., and Mingozzi, F. (2017). Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 8, 87–104. doi: 10.1016/j.omtm.2017.11.007
Colella, P., Sellier, P., Costa Verdera, H., Puzzo, F., van Wittenberghe, L., Guerchet, N., et al. (2018). AAV gene transfer with tandem promoter design prevents anti-transgene immunity and provides persistent efficacy in neonate pompe mice. Mol. Ther. Methods Clin. Dev. 12, 85–101. doi: 10.1016/j.omtm.2018.11.002
Costamagna, G., Andreoli, L., Corti, S., and Faravelli, I. (2019). iPSCs-based neural 3D systems: a multidimensional approach for disease modeling and drug discovery. Cells 8:1438. doi: 10.3390/cells8111438
Crooke, S. T. (2017). Molecular mechanisms of antisense oligonucleotides. Nucleic Acid Ther. 27, 70–77. doi: 10.1089/nat.2016.0656
Crooke, S. T., Witztum, J. L., Bennett, C. F., and Baker, B. F. (2018). RNA-targeted therapeutics. Cell Metabol. 27, 714–739. doi: 10.1016/j.cmet.2018.03.004
Crudele, J. M., and Chamberlain, J. S. (2019). AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet. 28, R102–R107.
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. A., et al. (2020). Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell Reports 14, 892–908. doi: 10.1016/j.stemcr.2020.03.023
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
Danisovic, L., Culenova, M., and Csobonyeiova, M. (2018). Induced pluripotent stem cells for Duchenne muscular Dystrophy modeling and therapy. Cells 7:253. doi: 10.3390/cells7120253
Dawson, T. M., Golde, T. E., and Tourenne, C. L. (2018). Animal models of neurodegenerative diseases. Nat. Neurosci. 21, 1370–1379.
Demestre, M., Orth, M., Föhr, K. J., Achberger, K., Ludolph, A. C., Liebau, S., et al. (2015). Formation and characterisation of neuromuscular junctions between hiPSC derived motoneurons and myotubes. Stem Cell Res. 15, 328–336. doi: 10.1016/j.scr.2015.07.005
Depla, J. A., Sogorb-Gonzalez, M., Mulder, L. A., Heine, V. M., Konstantinova, P., van Deventer, S. J., et al. (2020). Cerebral organoids: a human model for AAV capsid selection and therapeutic transgene efficacy in the brain. Mol. Ther. Methods Clin. Dev. 18, 167–175. doi: 10.1016/j.omtm.2020.05.028
Deverman, B. E., Pravdo, P. L., Simpson, B. P., Kumar, S. R., Chan, K. Y., Banerjee, A., et al. (2016). Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 34, 204–209. doi: 10.1038/nbt.3440
Dick, E., Kalra, S., Anderson, D., George, V., Ritso, M., Laval, S. H., et al. (2013). Exon skipping and gene transfer restore dystrophin expression in human induced pluripotent stem cells-cardiomyocytes harboring DMD mutations. Stem Cells Dev. 22, 2714–2724. doi: 10.1089/scd.2013.0135
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218–1221. doi: 10.1126/science.1158799
Donnelly, C. J., Zhang, P.-W., Pham, J. T., Haeusler, A. R., Heusler, A. R., Mistry, N. A., et al. (2013). RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80, 415–428. doi: 10.1016/j.neuron.2013.10.015
Doss, M. X., and Sachinidis, A. (2019). Current challenges of iPSC-based disease modeling and therapeutic implications. Cells 8:403. doi: 10.3390/cells8050403
Du, J., Campau, E., Soragni, E., Jespersen, C., and Gottesfeld, J. M. (2013). Length-dependent CTG⋅CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 22, 5276–5287.
Duan, D. (2018). Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol. Ther. 26, 2337–2356. doi: 10.1016/j.ymthe.2018.07.011
Duong, T. T., Lim, J., Vasireddy, V., Papp, T., Nguyen, H., Leo, L., et al. (2019). Comparative AAV-eGFP transgene expression using vector serotypes 1–9, 7m8, and 8b in human pluripotent stem cells, RPEs, and human and rat cortical neurons (Hindawi). Stem Cells Int. 2019:7281912.
Fujimori, K., Ishikawa, M., Otomo, A., Atsuta, N., Nakamura, R., Akiyama, T., et al. (2018). Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 24, 1579–1589. doi: 10.1038/s41591-018-0140-5
Gao, Q., and McNally, E. M. (2015). The dystrophin complex: structure, function and implications for therapy. Compr. Physiol. 5, 1223–1239. doi: 10.1002/cphy.c140048
Gendron, T. F., Chew, J., Stankowski, J. N., Hayes, L. R., Zhang, Y.-J., Prudencio, M., et al. (2017). Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med. 9:eaai7866. doi: 10.1126/scitranslmed.aai7866
Giorgio, F. D., Maduro, C., Fisher, E. M. C., and Acevedo-Arozena, A. (2019). Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Model. Mech. 12:dmm037424. doi: 10.1242/dmm.037424
Goparaju, S. K., Kohda, K., Ibata, K., Soma, A., Nakatake, Y., Akiyama, T., et al. (2017). Rapid differentiation of human pluripotent stem cells into functional neurons by mRNAs encoding transcription factors. Sci. Rep. 7:42367.
Guilbaud, M., Devaux, M., Couzinié, C., Le Duff, J., Toromanoff, A., Vandamme, C., et al. (2019). Five years of successful inducible transgene expression following locoregional adeno-associated virus delivery in nonhuman primates with no detectable immunity. Hum. Gene Ther. 30, 802–813. doi: 10.1089/hum.2018.234
Hanlon, K. S., Meltzer, J. C., Buzhdygan, T., Cheng, M. J., Sena-Esteves, M., Bennett, R. E., et al. (2019). Selection of an efficient AAV vector for robust CNS transgene expression. Mol. Ther. Methods Clin. Dev. 15, 320–332. doi: 10.1016/j.omtm.2019.10.007
Hashimoto, A., Naito, A. T., Lee, J.-K., Kitazume-Taneike, R., Ito, M., Yamaguchi, T., et al. (2016). Generation of induced pluripotent stem cells from patients with Duchenne muscular dystrophy and their induction to cardiomyocytes. Int. Heart J. 57, 112–117. doi: 10.1536/ihj.15-376
Hester, M., Foust, K., Kaspar, R., and Kaspar, B. (2009). AAV as a gene transfer vector for the treatment of neurological disorders: novel treatment thoughts for ALS. Curr. Gene Ther. 9, 428–433. doi: 10.2174/156652309789753383
Hoekstra, S. D., Stringer, S., Heine, V. M., and Posthuma, D. (2017). Genetically-informed patient selection for iPSC studies of complex diseases may aid in reducing cellular heterogeneity. Front. Cell. Neurosci. 11:164. doi: 10.3389/fncel.2017.00164
Hor, J. H., Soh, E. S.-Y., Tan, L. Y., Lim, V. J. W., Santosa, M. M., and Winanto, et al. (2018). Cell cycle inhibitors protect motor neurons in an organoid model of spinal muscular atrophy. Cell Death Dis. 9:1100.
Hor, J.-H., Santosa, M. M., Lim, V. J. W., Ho, B. X., Taylor, A., Khong, Z. J., et al. (2020). ALS motor neurons exhibit hallmark metabolic defects that are rescued by SIRT3 activation. Cell Death Differ. doi: 10.1038/s41418-020-00664-0 [Epub ahead of print].
Hosoya, M., and Czysz, K. (2016). Translational prospects and challenges in human induced pluripotent stem cell research in drug discovery. Cells 5:46. doi: 10.3390/cells5040046
Imamura, K., Izumi, Y., Watanabe, A., Tsukita, K., Woltjen, K., Yamamoto, T., et al. (2017). The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 9:eaaf3962.
Jang, J.-H., Koerber, J. T., Kim, J.-S., Asuri, P., Vazin, T., Bartel, M., et al. (2011). An evolved adeno-associated viral variant enhances gene delivery and gene targeting in neural stem cells. Mol. Ther. 19, 667–675. doi: 10.1038/mt.2010.287
Karagiannis, P., Takahashi, K., Saito, M., Yoshida, Y., Okita, K., Watanabe, A., et al. (2018). Induced pluripotent stem cells and their use in human models of disease and development. Physiol. Rev. 99, 79–114.
Khan, I. F., Hirata, R. K., Wang, P.-R., Li, Y., Kho, J., Nelson, A., et al. (2010). Engineering of human pluripotent stem cells by AAV-mediated gene targeting. Mol. Ther. 18, 1192–1199. doi: 10.1038/mt.2010.55
Kiskinis, E., Sandoe, J., Williams, L. A., Boulting, G. L., Moccia, R., Wainger, B. J., et al. (2014). Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795. doi: 10.1016/j.stem.2014.03.004
Koenig, M., Hoffman, E. P., Bertelson, C. J., Monaco, A. P., Feener, C., and Kunkel, L. M. (1987). Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50, 509–517. doi: 10.1016/0092-8674(87)90504-6
Lai, J.-I., Leman, L. J., Ku, S., Vickers, C. J., Olsen, C. A., Montero, A., et al. (2017). Cyclic tetrapeptide HDAC inhibitors as potential therapeutics for spinal muscular atrophy: screening with iPSC-derived neuronal cells. Bioorg. Med. Chem. Lett. 27, 3289–3293. doi: 10.1016/j.bmcl.2017.06.027
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165.
Liu, G., David, B. T., Trawczynski, M., and Fessler, R. G. (2020). Advances in pluripotent stem cells: history, mechanisms, technologies, and applications. Stem Cell Rev. Rep. 16, 3–32. doi: 10.1007/s12015-019-09935-x
Liu, X., Chen, J., Liu, W., Li, X., Chen, Q., Liu, T., et al. (2015). The fused in sarcoma protein forms cytoplasmic aggregates in motor neurons derived from integration-free induced pluripotent stem cells generated from a patient with familial amyotrophic lateral sclerosis carrying the FUS-P525L mutation. Neurogenetics 16, 223–231. doi: 10.1007/s10048-015-0448-y
Madill, M., McDonagh, K., Ma, J., Vajda, A., McLoughlin, P., O’Brien, T., et al. (2017). Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 10:22.
Maffioletti, S. M., Sarcar, S., Henderson, A. B. H., Mannhardt, I., Pinton, L., Moyle, L. A., et al. (2018). Three-dimensional human iPSC-derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Rep. 23, 899–908. doi: 10.1016/j.celrep.2018.03.091
Mandal, P. K., and Rossi, D. J. (2013). Reprogramming human fibroblasts to pluripotency using modified mRNA. Nat. Protoc. 8, 568–582. doi: 10.1038/nprot.2013.019
Martier, R., Liefhebber, J. M., García-Osta, A., Miniarikova, J., Cuadrado-Tejedor, M., Espelosin, M., et al. (2019a). Targeting RNA-mediated toxicity in C9orf72 ALS and/or FTD by RNAi-based gene therapy. Mol. Ther. Nucleic Acids 16, 26–37. doi: 10.1016/j.omtn.2019.02.001
Martier, R., Liefhebber, J. M., Miniarikova, J., van der Zon, T., Snapper, J., and Kolder, I. (2019b). Artificial microRNAs targeting C9orf72 can reduce accumulation of intra-nuclear transcripts in ALS and FTD patients. Mol. Ther. Nucleic Acids 14, 593–608. doi: 10.1016/j.omtn.2019.01.010
Maury, Y., Côme, J., Piskorowski, R. A., Salah-Mohellibi, N., Chevaleyre, V., Peschanski, M., et al. (2015). Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 33, 89–96. doi: 10.1038/nbt.3049
Mendell, J. R., Sahenk, Z., Malik, V., Gomez, A. M., Flanigan, K. M., Lowes, L. P., et al. (2015). A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 23, 192–201. doi: 10.1038/mt.2014.200
Miller, C. M., and Harris, E. N. (2016). Antisense oligonucleotides: treatment strategies and cellular internalization. RNA Dis. 3:e1393.
Mitsui, K., Suzuki, K., Aizawa, E., Kawase, E., Suemori, H., Nakatsuji, N., et al. (2009). Gene targeting in human pluripotent stem cells with adeno-associated virus vectors. Biochem. Biophys. Res. Commun. 388, 711–717. doi: 10.1016/j.bbrc.2009.08.075
Mondragon-Gonzalez, R., and Perlingeiro, R. C. R. (2018). Recapitulating muscle disease phenotypes with myotonic dystrophy 1 induced pluripotent stem cells: a tool for disease modeling and drug discovery. Dis. Model Mech. 11:dmm034728. doi: 10.1242/dmm.034728
Morimoto, S., Takahashi, S., Fukushima, K., Saya, H., Suzuki, N., Aoki, M., et al. (2019). Ropinirole hydrochloride remedy for amyotrophic lateral sclerosis – protocol for a randomized, double-blind, placebo-controlled, single-center, and open-label continuation phase I/IIa clinical trial (ROPALS trial). Regen. Ther. 11, 143–166. doi: 10.1016/j.reth.2019.07.002
Mueller, C., Berry, J. D., McKenna-Yasek, D. M., Gernoux, G., Owegi, M. A., Pothier, L. M., et al. (2020). SOD1 suppression with adeno-associated virus and microRNA in familial ALS. N. Engl. J. Med. 383, 151–158.
Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., Ichisaka, T., Aoi, T., et al. (2008). Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 26, 101–106. doi: 10.1038/nbt1374
Naso, M. F., Tomkowicz, B., Perry, W. L., and Strohl, W. R. (2017). Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs 31, 317–334.
Nieuwenhuis, B., Haenzi, B., Hilton, S., Carnicer-Lombarte, A., Hobo, B., Verhaagen, J., et al. (2020). Optimization of adeno-associated viral vector-mediated transduction of the corticospinal tract: comparison of four promoters. Gene Ther. 28, 56–74. doi: 10.1038/s41434-020-0169-1
Nizzardo, M., Simone, C., Dametti, S., Salani, S., Ulzi, G., Pagliarani, S., et al. (2015). Spinal muscular atrophy phenotype is ameliorated in human motor neurons by SMN increase via different novel RNA therapeutic approaches. Sci. Rep. 5:11746.
Nizzardo, M., Simone, C., Rizzo, F., Ulzi, G., Ramirez, A., Rizzuti, M., et al. (2016). Morpholino-mediated SOD1 reduction ameliorates an amyotrophic lateral sclerosis disease phenotype. Sci. Rep. 6:21301.
Ochoa, S., and Milam, V. T. (2020). Modified nucleic acids: expanding the capabilities of functional oligonucleotides. Molecules 25:4659. doi: 10.3390/molecules25204659
Ortiz-Vitali, J. L., and Darabi, R. (2019). iPSCs as a platform for disease modeling, drug screening, and personalized therapy in muscular dystrophies. Cells 8:20. doi: 10.3390/cells8010020
Osman, E. Y., Washington, C. W., Kaifer, K. A., Mazzasette, C., Patitucci, T. N., Florea, K. M., et al. (2016). Optimization of morpholino antisense oligonucleotides targeting the intronic repressor element1 in spinal muscular atrophy. Mol. Ther. 24, 1592–1601. doi: 10.1038/mt.2016.145
Park, I.-H., Arora, N., Huo, H., Maherali, N., Ahfeldt, T., Shimamura, A., et al. (2008a). Disease-specific induced pluripotent stem (iPS) cells. Cell 134, 877–886.
Park, I.-H., Zhao, R., West, J. A., Yabuuchi, A., Huo, H., Ince, T. A., et al. (2008b). Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146. doi: 10.1038/nature06534
Pasteuning-Vuhman, S., de Jongh, R., Timmers, A., and Pasterkamp, R. J. (2020). Towards advanced iPSC-based drug development for neurodegenerative disease. Trends Mol. Med. 27, 263–279. doi: 10.1016/j.molmed.2020.09.013
Picher-Martel, V., Valdmanis, P. N., Gould, P. V., Julien, J.-P., and Dupré, N. (2016). From animal models to human disease: a genetic approach for personalized medicine in ALS. Acta Neuropathol. Commun. 4:70.
Piga, D., Salani, S., Magri, F., Brusa, R., Mauri, E., Comi, G. P., et al. (2019). Human induced pluripotent stem cell models for the study and treatment of Duchenne and Becker muscular dystrophies. Ther. Adv. Neurol. Disord. 12:1756286419833478. doi: 10.1177/1756286419833478
Poirier, A., Weetall, M., Heinig, K., Bucheli, F., Schoenlein, K., Alsenz, J., et al. (2018). Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol. Res. Perspect. 6:e00447. doi: 10.1002/prp2.447
Ramirez, A., Crisafulli, S. G., Rizzuti, M., Bresolin, N., Comi, G. P., Corti, S., et al. (2018). Investigation of new morpholino oligomers to increase survival motor neuron protein levels in spinal muscular atrophy. Int. J. Mol. Sci. 19:167. doi: 10.3390/ijms19010167
Rapti, K., Stillitano, F., Karakikes, I., Nonnenmacher, M., Weber, T., Hulot, J.-S., et al. (2015). Effectiveness of gene delivery systems for pluripotent and differentiated cells. Mol. Ther. Methods Clin. Dev. 2:14067. doi: 10.1038/mtm.2014.67
Ratni, H., Ebeling, M., Baird, J., Bendels, S., Bylund, J., Chen, K. S., et al. (2018). Discovery of risdiplam, a selective survival of motor neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J. Med. Chem. 61, 6501–6517.
Ratni, H., Karp, G. M., Weetall, M., Naryshkin, N. A., Paushkin, S. V., Chen, K. S., et al. (2016). Specific correction of alternative survival motor neuron 2 splicing by small molecules: discovery of a potential novel medicine to treat spinal muscular atrophy. J. Med. Chem. 59, 6086–6100.
Renault, V., Piron-Hamelin, G., Forestier, C., DiDonna, S., Decary, S., Hentati, F., et al. (2000). Skeletal muscle regeneration and the mitotic clock. Exp. Gerontol. 35, 711–719. doi: 10.1016/s0531-5565(00)00151-0
Romano, G. (2008). Artificial reprogramming of human somatic cells to generate pluripotent stem cells: a possible alternative to the controversial use of human embryonic stem cells. Drug News Perspect. 21, 440–445.
Romito, A., and Cobellis, G. (2016). Pluripotent stem cells: current understanding and future directions. Stem Cells Int. 2016:9451492.
Sahin, U., Karikó, K., and Türeci, Ö (2014). mRNA-based therapeutics — developing a new class of drugs. Nat. Rev. Drug Discov. 13, 759–780. doi: 10.1038/nrd4278
Sances, S., Bruijn, L. I., Chandran, S., Eggan, K., Ho, R., Klim, J. R., et al. (2016). Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 19, 542–553.
Sareen, D., O’Rourke, J. G., Meera, P., Muhammad, A. K. M. G., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5:208ra149. doi: 10.1126/scitranslmed.3007529
Schneider, A.-F. E., and Aartsma-Rus, A. (2020). Developments in reading frame restoring therapy approaches for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 21, 343–359. doi: 10.1080/14712598.2021.1832462
Schoch, K. M., and Miller, T. M. (2017). Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron 94, 1056–1070. doi: 10.1016/j.neuron.2017.04.010
Shao, L., and Wu, W.-S. (2010). Gene-delivery systems for iPS cell generation. Expert Opin. Biol. Ther. 10, 231–242. doi: 10.1517/14712590903455989
Shi, Y., Inoue, H., Wu, J. C., and Yamanaka, S. (2017). Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 16, 115–130. doi: 10.1038/nrd.2016.245
Shoji, E., Sakurai, H., Nishino, T., Nakahata, T., Heike, T., Awaya, T., et al. (2015). Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep. 5:12831.
Smeriglio, P., Langard, P., Querin, G., and Biferi, M. G. (2020). The identification of novel biomarkers is required to improve adult SMA patient stratification, diagnosis and treatment. J. Pers. Med. 10:75. doi: 10.3390/jpm10030075
Smith-Arica, J. R., Thomson, A. J., Ansell, R., Chiorini, J., Davidson, B., and McWhir, J. (2003). Infection efficiency of human and mouse embryonic stem cells using adenoviral and adeno-associated viral vectors. Cloning Stem Cells 5, 51–62. doi: 10.1089/153623003321512166
Soldner, F., and Jaenisch, R. (2012). iPSC disease modeling. Science 338, 1155–1156. doi: 10.1126/science.1227682
Son, E. Y., Ichida, J. K., Wainger, B. J., Toma, J. S., Rafuse, V. F., Woolf, C. J., et al. (2011). Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9, 205–218. doi: 10.1016/j.stem.2011.07.014
Stein, C. A. (2016). Eteplirsen approved for Duchenne muscular dystrophy: the FDA faces a difficult choice. Mol. Ther. 24, 1884–1885. doi: 10.1038/mt.2016.188
Sun, C., Choi, I. Y., Gonzalez, Y. I. R., Andersen, P., Talbot, C. C., and Iyer, S. R. (2020). Duchenne muscular dystrophy hiPSC–derived myoblast drug screen identifies compounds that ameliorate disease in mdx mice. JCI Insight 5:e134287.
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Talbot, K., and Wood, M. J. A. (2019). Wrangling RNA: antisense oligonucleotides for neurological disorders. Sci. Transl. Med. 11:eaay2069. doi: 10.1126/scitranslmed.aay2069
Tedesco, F. S., Gerli, M. F. M., Perani, L., Benedetti, S., Ungaro, F., Cassano, M., et al. (2012). Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci. Transl. Med. 4:140ra89. doi: 10.1126/scitranslmed.3003541
Toli, D., Buttigieg, D., Blanchard, S., Lemonnier, T., Lamotte d’Incamps, B., Bellouze, S., et al. (2015). Modeling amyotrophic lateral sclerosis in pure human iPSc-derived motor neurons isolated by a novel FACS double selection technique. Neurobiol. Dis. 82, 269–280. doi: 10.1016/j.nbd.2015.06.011
Ueki, J., Nakamori, M., Nakamura, M., Nishikawa, M., Yoshida, Y., Tanaka, A., et al. (2017). Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci. Rep. 7:42522.
van Putten, M., Lloyd, E. M., de Greef, J. C., Raz, V., Willmann, R., and Grounds, M. D. (2020). Mouse models for muscular dystrophies: an overview. Dis. Model. Mech. 13:dmm043562. doi: 10.1242/dmm.043562
Verhaart, I. E. C., Robertson, A., Wilson, I. J., Aartsma-Rus, A., Cameron, S., Jones, C. C., et al. (2017). Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy – a literature review. Orphanet J. Rare Dis. 12:124.
Volpato, V., and Webber, C. (2020). Addressing variability in iPSC-derived models of human disease: guidelines to promote reproducibility. Dis. Model. Mech. 13:dmm042317. doi: 10.1242/dmm.042317
Wainger, B. J., Kiskinis, E., Mellin, C., Wiskow, O., Han, S. S. W., Sandoe, J., et al. (2014). Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11. doi: 10.1016/j.celrep.2014.03.019
Wainger, B. J., Macklin, E. A., Vucic, S., McIlduff, C. E., Paganoni, S., Maragakis, N. J., et al. (2020). Effect of ezogabine on cortical and spinal motor neuron excitability in amyotrophic lateral sclerosis: a randomized clinical trial. JAMA Neurol. 78, 186–196.
Wang, D., and Gao, G. (2014). STATE-of-the-art human gene therapy: part II. Gene therapy strategies and applications. Discov. Med. 18, 151–161.
Warren, L., Manos, P. D., Ahfeldt, T., Loh, Y.-H., Li, H., Lau, F., et al. (2010). Highly efficient reprogramming to pluripotency and directed differentiation of human cells using synthetic modified mRNA. Cell Stem Cell 7, 618–630. doi: 10.1016/j.stem.2010.08.012
Warren, L., Ni, Y., Wang, J., and Guo, X. (2012). Feeder-free derivation of human induced pluripotent stem cells with messenger RNA. Sci. Rep. 2:657.
Wells, D. J. (2018). Tracking progress: an update on animal models for Duchenne muscular dystrophy. Dis. Model Mech. 11:dmm035774. doi: 10.1242/dmm.035774
Westhaus, A., Cabanes-Creus, M., Rybicki, A., Baltazar, G., Navarro, R. G., Zhu, E., et al. (2020). High-throughput in vitro, ex vivo, and in vivo screen of adeno-associated virus vectors based on physical and functional transduction. Hum. Gene Ther. 31, 575–589. doi: 10.1089/hum.2019.264
Wirth, B. (2021). Spinal muscular atrophy: in the challenge lies a solution. Trends Neurosci.44, 306–322. doi: 10.1016/j.tins.2020.11.009
Wurster, C. D., and Ludolph, A. C. (2018). Antisense oligonucleotides in neurological disorders. Ther. Adv. Neurol. Disord. 11:1756286418776932. doi: 10.1177/1756286418776932
Xia, G., Santostefano, K. E., Goodwin, M., Liu, J., Subramony, S. H., Swanson, M. S., et al. (2013). Generation of neural cells from DM1 induced pluripotent stem cells as cellular model for the study of central nervous system neuropathogenesis. Cell. Reprogram. 15, 166–177. doi: 10.1089/cell.2012.0086
Xia, G., Terada, N., and Ashizawa, T. (2018). Human iPSC models to study orphan diseases: muscular dystrophies. Curr. Stem Cell Rep. 4, 299–309. doi: 10.1007/s40778-018-0145-5
Yoshioka, N., Gros, E., Li, H.-R., Kumar, S., Deacon, D. C., Maron, C., et al. (2013). Efficient generation of human iPS cells by a synthetic self-replicative RNA. Cell Stem Cell 13, 246–254. doi: 10.1016/j.stem.2013.06.001
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920.
Zhang, K., Daigle, J. G., Cunningham, K. M., Coyne, A. N., Ruan, K., Grima, J. C., et al. (2018). Stress granule assembly disrupts nucleocytoplasmic transport. Cell 173, 958–971.e17.
Keywords: IPSCs, gene therapy, AAV, NMD, MND, ASOs
Citation: Cappella M, Elouej S and Biferi MG (2021) The Potential of Induced Pluripotent Stem Cells to Test Gene Therapy Approaches for Neuromuscular and Motor Neuron Disorders. Front. Cell Dev. Biol. 9:662837. doi: 10.3389/fcell.2021.662837
Received: 01 February 2021; Accepted: 22 March 2021;
Published: 13 April 2021.
Edited by:
Benedetta Artegiani, Princess Maxima Center for Pediatric Oncology, NetherlandsReviewed by:
Francesco Roselli, University of Ulm, GermanyCopyright © 2021 Cappella, Elouej and Biferi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Grazia Biferi, bWcuYmlmZXJpQGluc3RpdHV0LW15b2xvZ2llLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.