
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 07 April 2021
Sec. Stem Cell Research
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.657456
This article is part of the Research TopicStem Cells and Cardiovascular DiseasesView all 29 articles
 Eric Zhang1
Eric Zhang1 Yanwen Liu1
Yanwen Liu1 Chaoshan Han1
Chaoshan Han1 Chengming Fan1
Chengming Fan1 Lu Wang1
Lu Wang1 Wangping Chen1
Wangping Chen1 Yipeng Du1Dunzheng Han1Baron Arnone1
Yipeng Du1Dunzheng Han1Baron Arnone1 Shiyue Xu1Yuhua Wei1James Mobley2
Shiyue Xu1Yuhua Wei1James Mobley2 Gangjian Qin1*
Gangjian Qin1*Exosomes transport biologically active cargo (e.g., proteins and microRNA) between cells, including many of the paracrine factors that mediate the beneficial effects associated with stem-cell therapy. Stem cell derived exosomes, in particular mesenchymal stem cells (MSCs), have been shown previously to largely replicate the therapeutic activity associated with the cells themselves, which suggests that exosomes may be a useful cell-free alternative for the treatment of cardiovascular disorders. However, the mechanisms that govern how exosomes home to damaged cells and tissues or the uptake and distribution of exosomal cargo are poorly characterized, because techniques for distinguishing between exosomal proteins and proteins in the targeted tissues are lacking. Here, we report the development of an in vivo model that enabled the visualization, tracking, and quantification of proteins from systemically administered MSC exosomes. The model uses bioorthogonal chemistry and cell-selective metabolic labeling to incorporate the non-canonical amino acid azidonorleucine (ANL) into the MSC proteome. ANL incorporation is facilitated via expression of a mutant (L274G) methionyl-tRNA-synthetase (MetRS∗) and subsequent incubation with ANL-supplemented media; after which ANL can be covalently linked to alkyne-conjugated reagents (e.g., dyes and resins) via click chemistry. Our results demonstrate that when the exosomes produced by ANL-treated, MetRS∗-expressing MSCs were systemically administered to mice, the ANL-labeled exosomal proteins could be accurately and reliably identified, isolated, and quantified from a variety of mouse organs, and that myocardial infarction (MI) both increased the abundance of exosomal proteins and redistributed a number of them from the membrane fraction of intact hearts to the cytosol of cells in infarcted hearts. Additionally, we found that Desmoglein-1c is enriched in MSC exosomes and taken up by ischemic myocardium. Collectively, our results indicate that this newly developed bioorthogonal system can provide crucial insights into exosome homing, as well as the uptake and biodistribution of exosomal proteins.
The therapeutic benefits of stem cells are mediated primarily via the secretion of paracrine factors (Bagno et al., 2018), and recent evidence suggests that the exosomes produced by mesenchymal stem cells (MSCs) largely replicate the improvements associated with administration of the cells themselves in large-mammal models of myocardial injury (Charles et al., 2020). Thus, while exosomes could be a useful cell-free alternative to MSC therapy, little is known of how exosomes are directed toward injured tissue, the distribution of the exosomal cargo in target organs and cells, or the physiological mechanisms that are induced by exosome uptake. This information is crucial for identifying the appropriate dose, timing, and route of administration for exosome therapy but remains difficult to decipher, as techniques for tracking exosomes and the exosome cargo are limited. Radiolabeled exosomes have been used to determine the half-life of exosomes in the circulation and to identify some of the major sites of exosome uptake (Morishita et al., 2015), but they cannot track the presence of exosome proteins in target organs and are dependent on half-life of the radiolabel. Thus, methods for accurately and reliably identifying both the source and targets of the exosomal cargo in living organisms will dramatically advance exosome research (Mathieu et al., 2019).
Cell-selective metabolic labeling generates proteins containing non-canonical amino acids with functional groups that can be linked to affinity reagents or fluorescent dyes for subsequent identification, isolation, and imaging. It was previously shown that the mutant E. coli methionyl-tRNA synthetase (MetRSL274G or MetRS∗) selectively appends the azide-bearing non-canonical amino acid azidonorleucine (ANL) to tRNA. Thus, when cells that express MetRS∗ are incubated with ANL, they produce proteins that contain ANL in lieu of endogenous methionine (Ngo et al., 2009) and can be covalently linked to alkyne-tagged reagents (e.g., dyes and resins) via click chemical reactions, such as copper-catalyzed azide-alkyne cycloaddition. These non-canonical amino acids are not produced in vivo and as such are excluded during normal protein synthesis.
For this study, we used non-canonical amino-acid labeling to develop an in vivo model that enables us to visualize, track, and measure the biodistribution of proteins from systemically administered MSC exosomes. MSCs were transfected with a lentiviral vector coding for MetRS∗ and incubated with ANL-supplemented media; then, exosomes were isolated from the MSC-conditioned media and intravenously administered to mice. Our results demonstrate that the ANL-labeled exosomal proteins can be accurately and reliably identified, isolated, and quantified from a variety of mouse organs, and that surgically induced myocardial infarction (MI) both increased the abundance of labeled exosomal proteins and redistributed the localization from the membrane to the cytosol within organs following systemic administration.
Mouse (C57BL/6) bone marrow MSCs were purchased from Cyagen Biosciences (Sunnyvale, CA, United States, Catalog Number: MUBMX-01001), cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and passaged 5–8 times before use. MSC identity was a confirmed via flow-cytometry analyses of MSC surface marker expression and assessments of osteogenic and adipose differentiation potential. Differentiation assays were performed by following the manufacturer’s protocols (Cyagen Biosciences, United States). Briefly, the cells were cultured on 0.1% gelatin-coated six-well plates (2 × 104 cells/cm2) in complete medium until confluent and then treated with osteogenic differentiation medium for 3 weeks or with adipocyte induction medium and maintenance medium for 2–3 weeks; osteogenic and adipose differentiation were analyzed by Alizarin staining and by Oil Red O staining (for lipid droplets), respectively.
Flow cytometry was performed as previously described (Cheng et al., 2019). Briefly, 1 × 106 cells were incubated in 200 μL cold phosphate-buffered saline (PBS) with Fc block (1:100 dilution), washed three times, stained with phycoerythrin (PE)-conjugated antibodies for mouse CD34 (1:50) or CD44 (1:50), or with fluorescein isothiocyanate (FITC)-conjugated antibodies for mouse CD11b (1:50), CD45 (1:50), or Sca-1 (1:50) (ebioscience, Germany). Data were acquired with a BDTM LSR II (BD Biosciences, United States) and analyzed with Cell Quest Software (Becton Dickinson, United Kingdom).
Quantitative reverse transcription PCR (qRT-PCR) was performed as previously described (Tang et al., 2009). Briefly, total MSC RNA was reverse transcribed with random hexamer primer, and 25 ng of the reverse-transcribed product was used for RT-PCR; results with the random primer were consistent with those obtained previously with a primer common to the 3′ untranslated region (UTR) of Dsg1-a,b, and -c. Values were normalized to equivalent assessments of glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA abundance.
For protein extraction, 1 × 107 cells or 100 mg of frozen tissue were homogenized in 1 mL RIPA lysis buffer containing protease inhibitors (Sigma, 4693132001) and phosphatase inhibitors (Sigma, 4906837001). Samples were incubated with agitation for 30 min at 4°C and centrifuged at 13,000 rpm for 10 min at 4°C; then, the protein concentration in the supernatant was determined via bicinchoninic acid (BCA) assay (Pierce). For immunoblotting, proteins in the supernatant were denatured by heating at 95°C for 10 min, separated by SDS-PAGE, and then transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad). The membrane was incubated in 5% nonfat milk blocking buffer (tris-buffered saline [TBS]) for 1 h, incubated with primary antibody in TBS-containing 3% bovine serum albumin (BSA) overnight at 4°C, washed three times with TBS (0.5% Tween 20), incubated with secondary antibody, washed with TBS Tween 20, and then developed with Enhanced Chemiluminescence Detection Reagents (ECL, Thermo Fisher). Protein signals were imaged with a Bio-Rad ChemiDoc System.
The Lenti-CAG-MetRSL274G–mCherry vector was newly engineered in our lab by using a pLenti.CAG.H2B-Dendra2.W (Addgene #51005) backbone and the MetRSL274G–mCherry coding sequence from pMaRSC (Addgene #89189). Briefly, pLenti.CAG.H2B-Dendra2.W was amplified via high-fidelity PCR with 5′ CAG-WPRE-5 primers (containing a sequence complementary to the WPRE region of the vector sequence with an Asc1-Age1-Mlu1-EcoRV restriction-site overhang) and 3′ CAG-LINK-3 primers (containing a sequence complementary to the Sal1 and upstream region of the vector sequence with an EcoRV-Mlu1-Age1-Asc1 restriction-site overhang). The 7.9-kb PCR product, which included the majority of the pLenti.CAG.H2B-Dendra2.W sequence but lacked the 1.1-kb H2B-Dendra2.W-coding region, was transformed into Top 10 competent cells (Thermo Scientific), where it was circularized via self-assembly cloning (Matsumoto and Itoh, 2011) into a pLenti.CAG.linker (containing multi-cloning sites Sal1-Asc1-Age1-Mlu1-EcoRV) and verified by Mlu1 enzyme digestion. To clone the MetRSL274G–mCherry coding sequence, PCR was performed with 5′ Sal1-L274G-mCherry 5 primers, 3′ Age1-L274G-mCherry 3 primers, and the pMaRSC template (Addgene #89189) for 20 cycles; each cycle consisted of 30 s at 95°C, 30 s at 60°C, and 4 min at 72°C. The 4-kb PCR product, which contained the MetRSL274G-mCherry coding sequence, was subsequently cloned into TA cloning vector PCRTM 2.1 (Thermo Scientific), and positive clones were verified by digestion with Sal1 and Age1; then, the 4-kb MetRSL274G-mCherry fragment was recovered with agarose gel and ligated into the Sal1/Age1-digested pLenti.CAG.linker to produce pLenti.CAG.MetRSL274G-mCherry, which was confirmed by DNA sequencing. The Lenti.CAG.MetRSL274G-mCherry vector was produced by co-transfecting pLenti.CAG.MetRSL274G-mCherry with pMD2.G and psPAX2 into 293FT cells, and then purified and concentrated as previously described (Cheng et al., 2010). All primer sequences are reported in Supplementary Table 1.
For lentivirus transduction, MSCs were seeded at ∼80% confluence, and the lentivirus (MOI = 4) was applied with polybrene (8 μg/mL); transduction efficiency was evaluated 72 h later by monitoring mCherry fluorescence with a fluorescence microscope or via fluorescence-activated cell-sorting (FACS); control assessments were conducted with MSCs that had been transduced with Lenti.CAG.H2B-Dendra2.W. For ANL labeling, cells were washed twice in PBS and then suspended in methionine-, glutamine-, and cystine-depleted DMEM (Gibco, Cat. #21013024) containing 1 mM ANL (IRIS Biotech, Cat. #159610-92-1); the DMEM had been made complete with glutamine and cystine and supplemented with exosome-free FBS (Gibco, Cat. #A270801). Cells were cultured for 72 h before use in subsequent experiments.
MetRS∗-transduced MSCs were incubated in 1 mM ANL for 24 h and lysed in RIPA buffer supplemented with protease inhibitors (Sigma, United States); then, total protein was collected, and the Click-iT reaction was performed with a Click-iT Protein Reaction Buffer Kit (Invitrogen, Waltham, MA, United States) as directed by the manufacturer’s instructions. Briefly, up to 200 μg of ANL-labeled protein was reacted with 100 μL of Click-iT reaction buffer containing alkyne-Cy7 and copper-sulfate catalyst in a rotator for 20 min. Methanol and chloroform were added to remove residual reaction components and precipitate the proteins; then, the precipitated proteins were solubilized in Laemmli buffer and loaded for gel electrophoresis. Gels were immediately imaged with a ChemiDoc MP Imaging System (Bio-Rad, California, United States) under both stain-free mode and Cy7 mode.
For fluorescent non-canonical amino-acid tagging (FUNCAT) of MetRS∗-transduced MSCs in vitro, MSCs were cultured in 1 mM ANL (Jena Bioscience, Jena, Germany) for 24 h, gently rinsed with PBS, fixed in 4% formaldehyde/PBS and permeabilized with 0.5% Triton X-100/PBS; then, the Click-iT reaction was performed with Click-iT reaction cocktails containing Alexa Fluor 488 alkyne (Click-iT Alexa Fluor 488 Protein Synthesis HCS Assay kit, Life Technologies, Waltham, MA, United States) as directed by the manufacturer’s protocol. Cells were stained with Hoechst 33342 prior to imaging. For FUNCAT of MetRS∗ exosomal proteins in vivo, the general methodology was adapted from previous studies (Ngo et al., 2013). Briefly, mice were euthanized 2 h after exosome administration; then, the hearts were explanted, perfused, fixed by incubation in 4% paraformaldehyde at room temperature for 4 h, cryoprotected in 30% sucrose/PBS solution overnight, frozen in OCT compound, cut into 5-μM-thick sections, and stored at −20°C. Frozen sections were incubated at room temperature for 30 min, washed once with PBS, permeabilized in 0.25% Triton/PBS for 35 min, washed in PBS (3 washes, 5 min each), blocked in commercial blocking buffer (Thermo Scientific, TA-125-PBQ) for 7 min, and washed in PBS again (3 washes, 5 min each); then, the ANL-labeled proteins were conjugated to biotin-alkyne by incubating the sections at 4°C overnight in a 1:10,000 dilutio of biotin-alkyne in 2× Catalyst solution from the Click-iT Enrichment kit (Invitrogen, Cat. #C10416). Sections were thoroughly washed in PBS (5 washes, 5 min each) to remove residual biotin, sequentially incubated with primary biotin antibodies (1:1000 dilution) at 4°C overnight and with secondary antibodies at 4°C overnight, washed PBS (three times), incubated with Sudan Black for 30 min, and then washed again in PBS (three times).
MetRS*MSCs were plated at ∼50% confluence in complete DMEM for 24 h, after which they were cultured in methionine-depleted DMEM supplemented with 1 mM ANL for 72 h. Conditioned media was collected from the MSCs, spun at 1,000 G to remove cell debris, passed through a 0.22-micron filter, and ultracentrifuged at 120,000 G for 2 h; then, the supernatant was removed and replaced with cold PBS, and the exosomes were ultracentrifuged at 120,000 G for 2 h. Exosome pellets were resuspended in 10 mL of cold PBS and stored at −80°C until use in subsequent experiments. The size and quantity of freshly isolated exosomes was estimated via nanoparticle-tracking analyses (NanoSight), and morphological assessments were conducted via electron microscopy as previously described (Cheng et al., 2019).
All animal experiments in this report were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Alabama at Birmingham and performed in compliance with the National Institutes of Health (NIH) publication Guide for the Care and Use of Laboratory Animals. Twelve- to 16-week-old C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME, United States) were used for all experiments unless otherwise specified. For surgically induced MI, mice were intubated and connected to a ventilator, and anesthesia was initiated and maintained with inhaled 2% isoflurane USP (FlurisoTM, VetOne). The heart was exposed via left thoracotomy, and MI was induced by permanently ligating the left-anterior descending (LAD) coronary artery, as previously described (Qin et al., 2006); sham-operated animals underwent the identical surgical procedure, except LAD artery ligation was omitted. After chest closure, buprenorphine hydrochloride (0.1 mg/kg, Buprenex, Reckitt Benckiser Pharmaceuticals Inc.) and carprofen (5 mg/kg, Rimadyl, Zoetis) were administered via intraperitoneal injection every 12 h for 3 days. Exosomes (10 × 109 in 200 μL PBS) were delivered via tail-vein injection 24 h after MI or Sham surgery, and mice were euthanized via CO2 inhalation.
Animals were euthanized via CO2 inhalation and perfusion with 10 mL cold PBS; then, the heart, brain, lung, kidney, liver, and spleen were isolated, cut into ∼20-mg sections, rinsed twice in PBS, and flash frozen with liquid nitrogen. Membrane and cytosolic total protein fractions were isolated with a Mem-PER Plus Membrane Protein Extraction kit (Thermo Scientific, Cat. #89842), precipitated via methanol-chloroform extraction, and stored at −80°C. For ANL-labeling and MetRS∗ protein capture, fractions were resolubilized in Click-iT buffer solution and processed as directed by the Click-iT Protein Enrichment Kit protocol (Invitrogen, Cat. #C10416). Captured peptides were stored at −80°C until identification via liquid chromatography mass spectrometry (LC-MS). To collect protein samples from non-administered exosomes, ∼1 × 109 exosomes were suspended in 100 μL PBS and prepared in parallel with organ samples for LC-MS identification.
Dried peptides were reconstituted in 16 μL of 0.1% formic acid (FA); then, 8 μL of each sample was injected onto a 1,260 Infinity nano high-performance liquid chromatography (nHPLC) stack (Agilent Technologies, Santa Clara, CA, United States), and peptides were separated on a 75-micron (internal diameter) by 15-cm pulled tip C-18 column (Jupiter C-18 300 Å, five micron, Phenomenex) that was attached to a Thermo Orbitrap Velos Pro hybrid mass spectrometer equipped with a nano-electrospray source (Thermo Fisher Scientific, Waltham, MA, United States). All data were collected in CID mode, and the nHPLC was configured with binary mobile phases consisting of solvent A (0.1%FA in ddH2O) and solvent B (0.1% FA in 15% ddH2O / 85% ACN) programmed as follows; 10 min at 2% solvent B; 90 min at 5–40% solvent B; 5 min at 70% solvent B; and 10 min at 0% solvent B. After each parent ion scan (300–1,200 m/z; 60 k resolution), fragmentation data (MS2) was collected for the 15 most-intense ions. For data dependent scans, charge state screening and dynamic exclusion were enabled with a repeat count of two, repeat duration of 30 s, and exclusion duration of 90 s.
XCalibur RAW files were collected in profile mode, centroided, and converted to MzXML with ReAdWv.3.5.1; then, the mgf files were created by using MzXML2Search (included in TPP v. 3.5) for all scans. Data was searched via SEQUEST, which was set for two maximum missed cleavages, a precursor mass window of 20 ppm, trypsin digestion, and variable modifications C and M at 57.0293 and 15.9949, respectively. Searches were performed with a specific subset of the UniRef100 database.
The lists of peptide identifications (IDs) generated via SEQUEST (Thermo Fisher Scientific, Waltham, MA, United States) were filtered with Scaffold (Protein Sciences, Portland, Oregon, United States). Scaffold generates and retains only high-confidence IDs while producing normalized spectral counts across all samples, and the spectral counts can subsequently be used for relative quantification. Only peptides with charge states ≥2+, minimum lengths of six amino acids, and nonzero quantities for all six mass tags were analyzed. For large datasets, Scaffold incorporates the two most common methods for statistical validation: false discovery rate (FDR), and protein probability; FDR was set at <1% cutoff, individual peptide probabilities were ≥0.8, protein probabilities were ≥0.99, and at least two peptides were assigned per protein. Relative quantification was performed via spectral counting and spectral count abundances were normalized between samples.
For protein abundance data, the Pearson coefficient was calculated to evaluate correlations in protein expression between samples. Hierarchical clustering of samples and protein profiles was based on the complete linkage method, and differences in protein expression between the MI and Sham groups was evaluated with the Mann Whitney U-test. Differentially expressed proteins (DEPs) were defined as those with nominal p-value < 5% and fold-change >1.5, and were visualized via heatmaps. For proteins that were detected in ≥60% of one group and absent in the other group, we used the method developed by Kojima et al. (2012), with a more stringent cutoff (>60% in only one group); proteins meeting this criteria were defined as “all-or-nothing” hits and included in the list of DEPs.
Murine MSCs were obtained from a commercial vendor, and the characteristic pattern of MSC marker expression (i.e., positive for Sca-1 and CD44; negative for CD11b, CD45, and CD34) was verified via flow-cytometry (Supplementary Figure 1A). Phase-contrast microscopy confirmed that the cells were uniformly spindle-shaped, and robust multipotency was verified by differentiating the cells into adipocytes and osteocytes (Supplementary Figures 1B,C). To facilitate non-canonical amino acid tagging, MSCs were transfected with our newly generated MetRS∗ lentiviral vector (Lenti-CAG-MetRSL274G-mCherry). The vector also coded for mCherry, which enabled stably transfected cells (MetRS*MSCs) to be selected via flow cytometry for mCherry fluorescence, and proteins produced by the MetRS*MSCs were labeled with ANL by incubating the selected cells in 1 μM ANL-supplemented media. Control assessments were conducted with MSCs that had been transfected with an equivalent vector that lacked the MetRSL274G-mCherry cassette (CTLMSCs).
Widespread mCherry expression was observed after just a single transfection (Figure 1A), and there were no apparent morphological differences between MetRS*MSCs and CTLMSCs after 72 h of incubation in the ANL-supplemented media. To confirm that ANL was incorporated into the MetRS*MSC proteome, BONCAT was conducted with lysates from ANL-treated MetRS*MSCs and CTLMSCs: proteins in the lysate were separated on a gel and covalently stained with alkyne-conjugated Cy7. Overall protein levels in the two MSC populations were equivalent; however, the alkyne-Cy7 stain was only observed in proteins from MetRS*MSC lysates, which spanned a wide spectrum of masses (Figure 1B). The results from our BONCAT assessments were also corroborated in intact cells via FUNCAT: the cells were permeabilized, and then click reactions were conducted with a cocktail containing Alexa Fluor 488 alkyne. The fluorescent signal (i.e., ANL-incorporated protein) was highly abundant in the cytosol of MetRS*MSCs but completely absent in CTLMSC cytosol (Figure 1C).
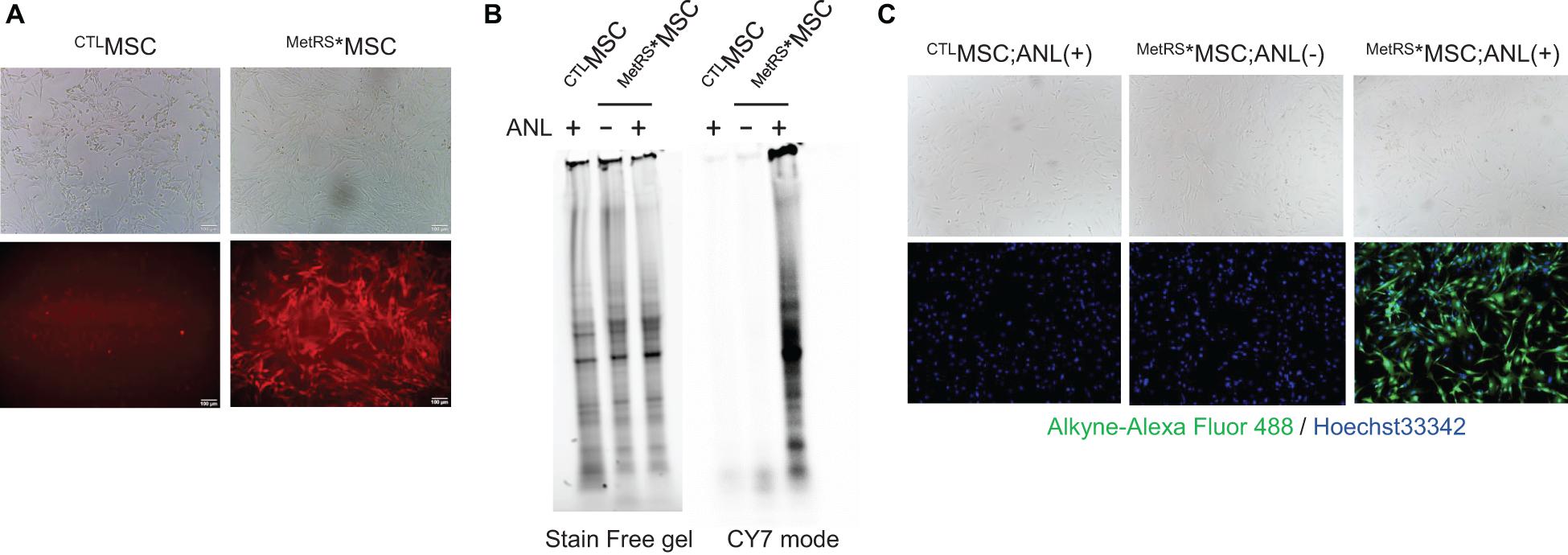
Figure 1. Proteins from ANL-treated MetRS*MSCs can be detected via alkyne-Cy7 click-staining. MSCs were transfected with Lenti-CAG-MetRSL274G–mCherry (MetRS*MSCs) or a control lentiviral vector (CTLMSCs). (A) Morphology of the transfected cells was evaluated via phase contrast microscopy (top), and transfection of MetRS*MSCs was confirmed by visualizing mCherry fluorescence (bottom). (B,C) MetRS*MSCs and CTLMSCs were incubated with ANL (+) or vehicle (–) for 24 h. (B) MSCs were lysed; then, proteins were separated on an SDS-PAGE gel, click-stained with alkyne-Cy7, and imaged in stain-free and Cy7 mode (BONCAT). (C) MSCs were permeabilized, click-stained with alkyne-Alexa Fluor 488, and viewed via phase contrast microscopy (top) or under fluorescence (bottom) (FUNCAT); nuclei were counterstained with Hoechst, and ANL-labeled proteins were identified via alkyne-Alexa Fluor 488 fluorescence.
Assessments via nanoparticle-tracking analysis (Figure 2A) and transmission electron microscopy (Figure 2B) confirmed that treatment with ANL-supplemented media (1 μM for 72 h) did not substantially alter the production, size, and ultrastructure of exosomes produced by MetRS*MSCs. Furthermore, when exosomes were collected from MetRS*MSCs (i.e., MetRS*MSC-Exos) and CTLMSCs (CTLMSC-Exos) that had been grown in media supplemented with or without ANL, BONCAT assessments of their protein cargo indicated that the total amount of protein in all four groups was similar, but alkyne-Cy7 fluorescence was only detected for proteins in exosomes from ANL-treated MetRS*MSC-Exos (Figure 2C). Thus, ANL labeling and subsequent detection was both sensitive and specific for proteins in MetRS*MSC-Exos.
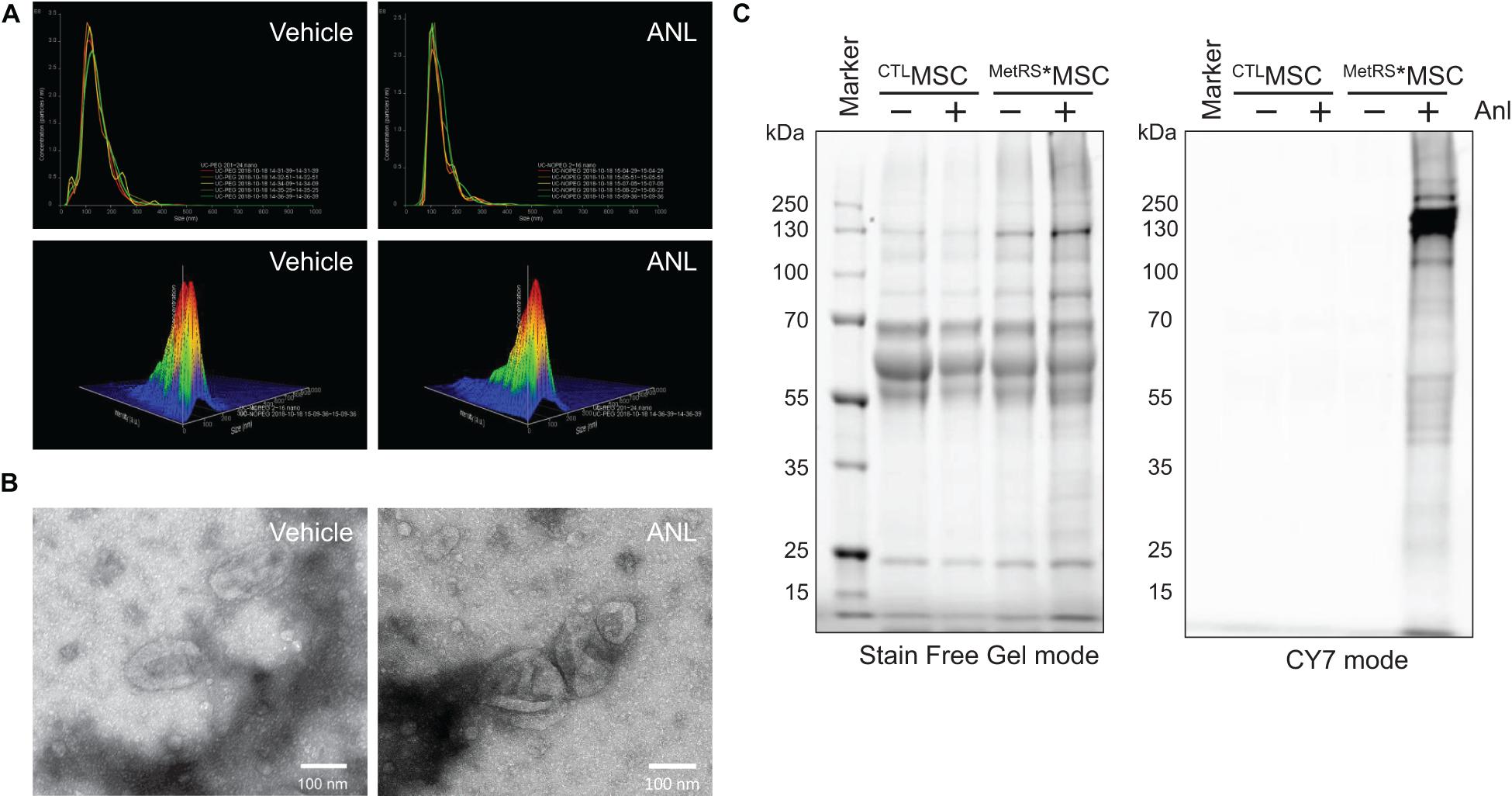
Figure 2. Exosomal proteins from ANL-treated MetRS*MSCs can be detected via alkyne-Cy7 click-staining. (A,B) The MetRS*MSCs were incubated with ANL (+) or vehicle (–) for 24 h; then, exosomes were isolated from culture media and evaluated via (A) nanoparticle-tracking analysis and (B) transmission electron microscopy (TEM). (C) Exosomes were isolated from CTLMSCs and MetRS*MSCs that had been incubated with ANL (+) or vehicle (–) for 24 h; then, the exosomes were lysed, and proteins were separated on an SDS-PAGE gel, click-stained with alkyne-Cy7, and imaged in stain-free and Cy7 mode (BONCAT).
To confirm that our method for detecting ANL-labeled MetRS*MSC-Exo proteins could effectively monitor the trafficking of proteins from systemically administered MetRS*MSC-Exos in vivo, exosomes were isolated from ANL- or vehicle-treated MetRS*MSCs and intravenously delivered into mice (20 μg/mouse); 2 h later, cardiac tissues were harvested, sectioned, and analyzed via FUNCAT. The fluorescent signal was strongly observed in both the vasculature and cardiomyocytes of mice that received the ANL-treated MetRS*MSC-Exos but was completely absent in sections from mice that were administered vehicle-treated MetRS*MSC-Exos (Figure 3).
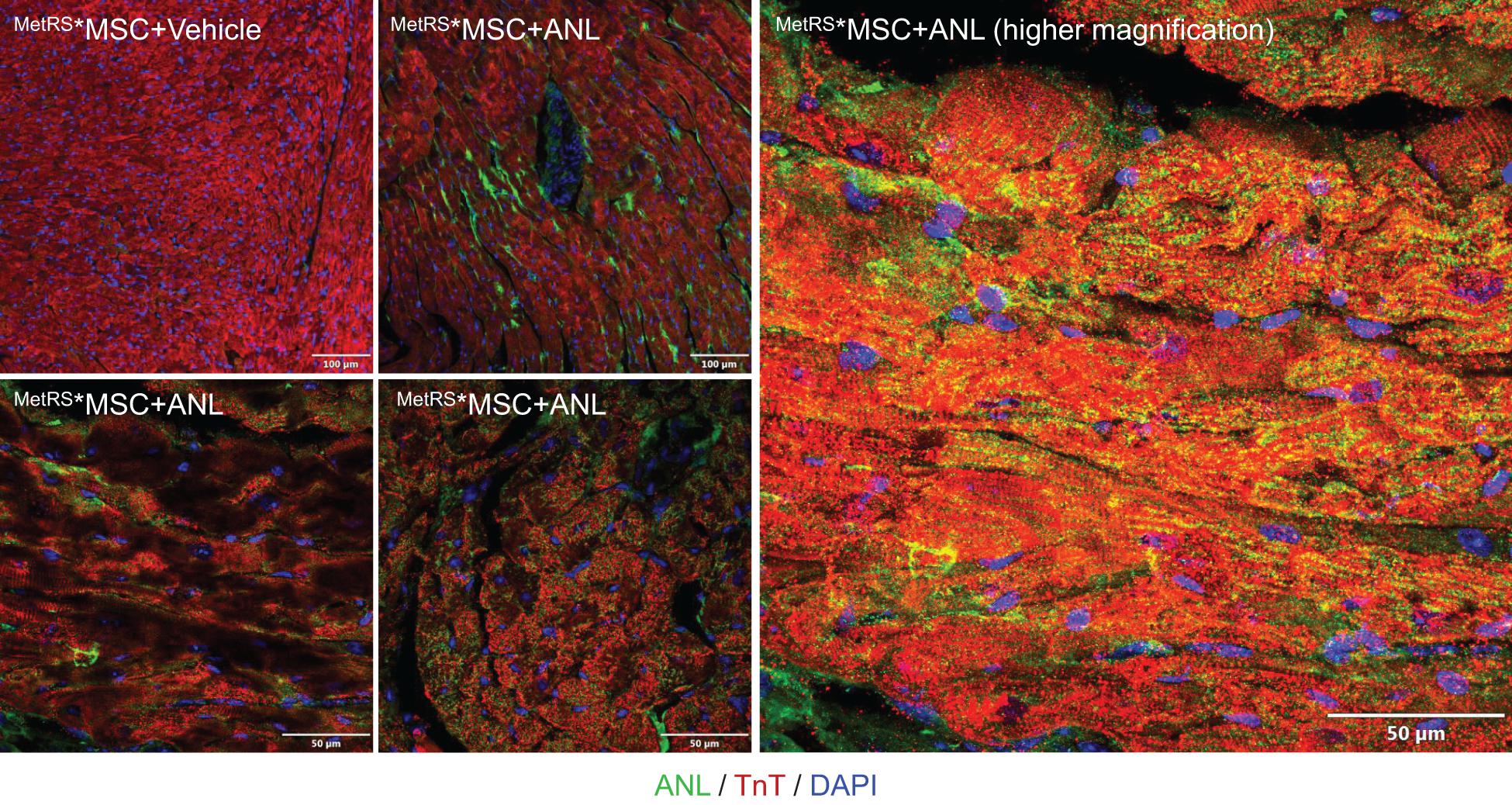
Figure 3. Proteins from systemically administered MetRS*MSC-Exos can be visualized in mouse hearts via click-catalyzed alkyne-Alexa Fluor 488 staining. Exosomes were isolated from the culture media of ANL- or vehicle-treated MetRS*MSCs and injected into the tail vein of mice. Hearts were explanted 2 h later, sectioned, click-stained with reaction cocktails containing Alkyne-conjugated Alexa Fluor 488 (green), and counterstained with anti-Troponin T (red) and Hoechst (blue); regions of overlapping Alexa Fluor 488 and Troponin T fluorescence appear yellow.
Having confirmed the fidelity of click-catalyzed ANL-staining in our in vivo model, we investigated whether ANL-labeled exosome proteins could be isolated from the organs of mice after MetRS*MSC-Exo administration. Proteins were precipitated from whole mouse organs (heart, kidney, spleen, lung, liver, and brain), and then MetRS*MSC-Exo proteins (i.e., proteins containing the ANL-label) were captured from the precipitate with an agarose resin containing alkyne functional groups that, when catalyzed via click reaction, formed covalent links with the ANL label. Because the MetRS*MSC-Exo proteins comprised only a very small fraction of the total amount of protein in each organ, we confirmed the specificity and accuracy of click-catalyzed alkyne-agarose capture by comparing the data for each individual MetRS*MSC-Exo protein to data for the same protein in the corresponding organs from MetRS*MSC-Exo–treated animals when the click catalysis reaction was omitted and from animals that were not treated with MetRS*MSC-Exos.
LC-MS analysis identified 205 MetRS*MSC-Exo proteins that were isolated from the hearts of MetRS*MSC-Exo–treated mice exclusively via click-catalyzed alkyne-agarose capture, compared to 29 proteins that were captured only when the click reaction was omitted, and 48 that were isolated both with and without click catalysis (Figure 4A); the abundance of each individual MetRS*MSC-Exo protein varied widely among organs (Figure 4B). Notably, most (but not all) of the exosome-marker proteins that were present in MetRS*MSC-Exos before administration (Supplementary Table 2) were also isolated from at least one of the organs of MetRS*MSC-Exo–treated mice, but they were not equally distributed across all organs (Figure 4C).
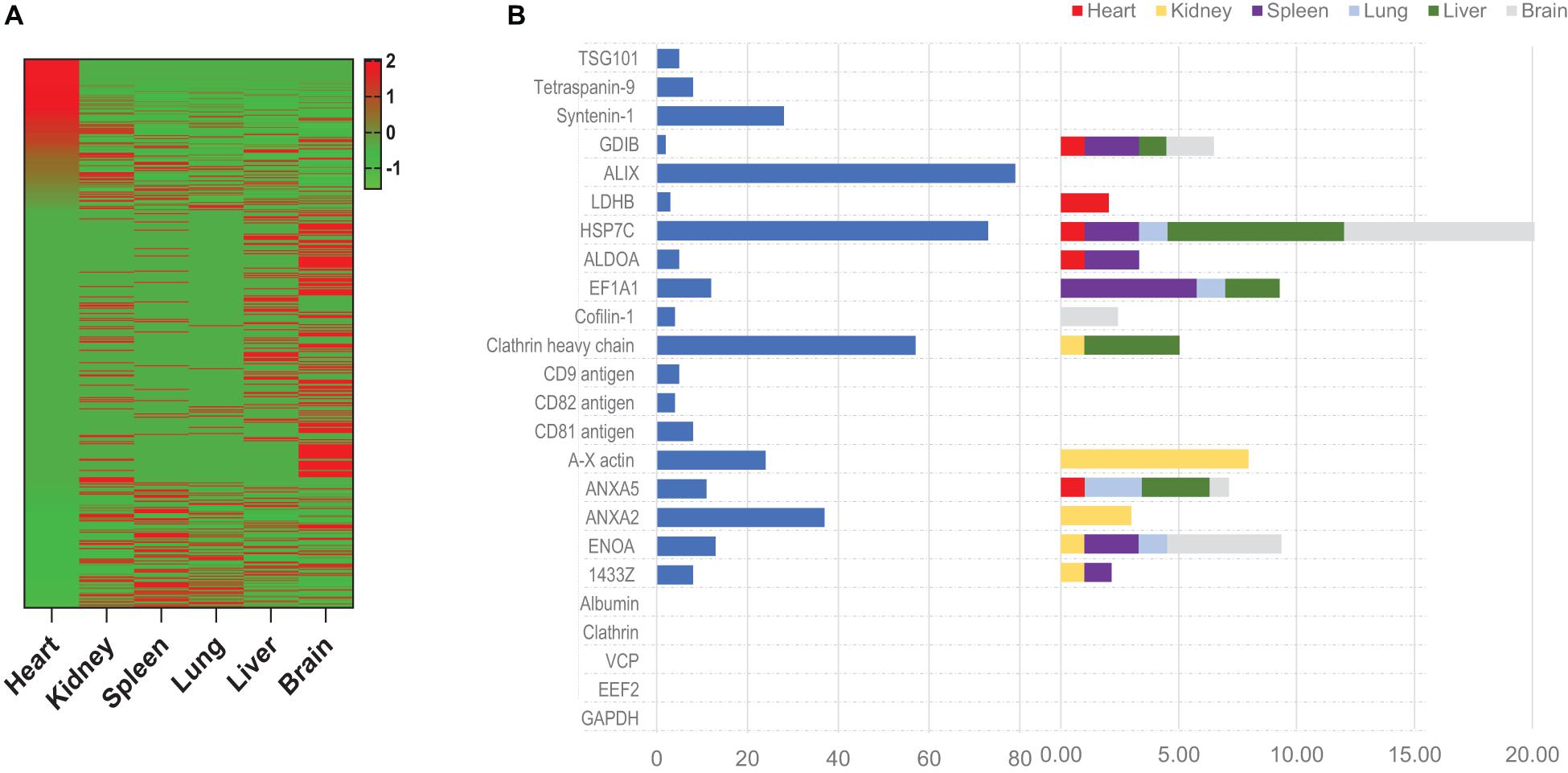
Figure 4. Proteins from systemically administered MetRS*MSC-Exos can be detected, identified, and quantified in mouse organs via click-catalyzed alkyne-agarose capture. MetRS*MSC-Exos were injected into the tail vein of mice. Animals were euthanized 24 h later, and protein extracts were prepared from the heart, kidney, spleen, lung, liver, and brain; then, proteins from the MetRS*MSC-Exos (i.e., those containing the ANL label) were isolated via click-catalyzed alkyne-agarose capture and identified via LC-MS. (A) MetRS*MSC exosomal proteins isolated from the indicated organs (average total spectra) were presented as a heat map. (B) The abundance of canonical positive and negative exosome marker proteins was quantified in MetRS*MSC-Exos before injection (left) and in the indicated organs of mice after MetRS*MSC-Exo injection (right).
To determine whether tissue injury changes the biodistribution of MSC exosomes, ANL-labeled MetRS*MSC-Exos were administered to mice via tail-vein injection 24 h after surgically induced MI (the MI group) or sham surgery (the Sham group). Cardiac tissue was collected 12 h after MetRS*MSC-Exo administration, and MetRS*MSC-Exo proteins were isolated via click-catalyzed alkyne agarose capture. The number of MetRS*MSC-Exo proteins was dramatically greater in hearts from MI than from Sham animals (Figure 5A), and of those that were present in the hearts of both groups, nearly all were more abundant in MI hearts (Figure 5B). These observations are consistent with the MI-induced uptake of MSC exosomes in cardiac cells. Furthermore, when the membrane and cytosol fractions were evaluated separately, a number of MetRS*MSC-Exo proteins that were associated with the membranes of cells in the hearts of Sham animals were found primarily in the cytosol fraction of MI hearts (Figure 6).
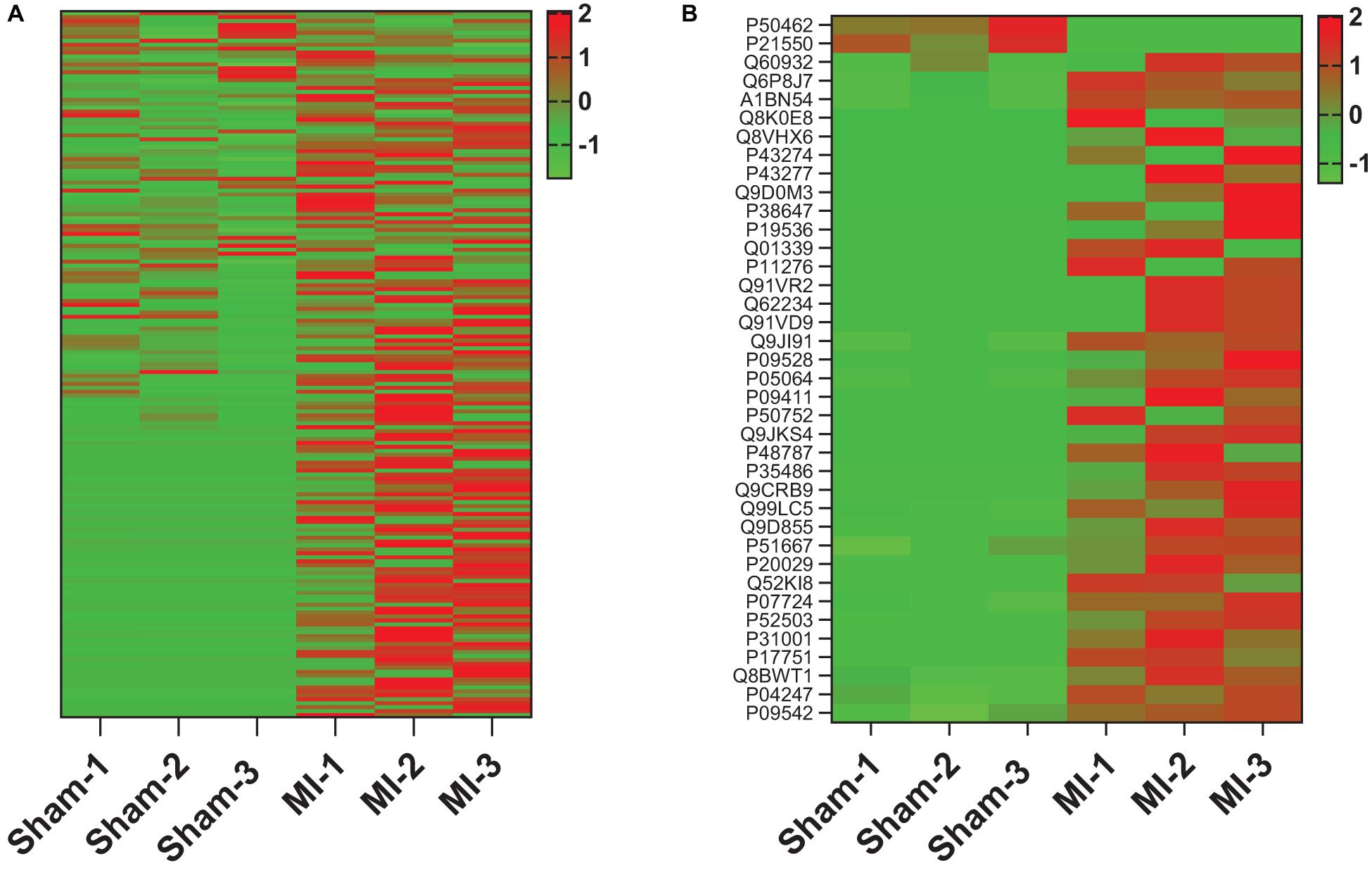
Figure 5. MI increases the abundance of MetRS*MSC exosomal proteins in the heart. MetRS*MSC-Exos were injected into the tail vein of mice 24 h after MI induction or Sham surgery (n = 3 per group). Animals were euthanized 24 h after exosome administration, and protein extracts were prepared from the left ventricle; then, the ANL-labeled MetRS*MSC exosomal proteins were isolated via click-catalyzed alkyne-agarose capture and identified via LC-MS. (A) Heat map of all identified MetRS*MSC exosomal proteins. (B) Heat map of MetRS*MSC exosomal proteins that were differentially expressed in MI and Sham hearts.
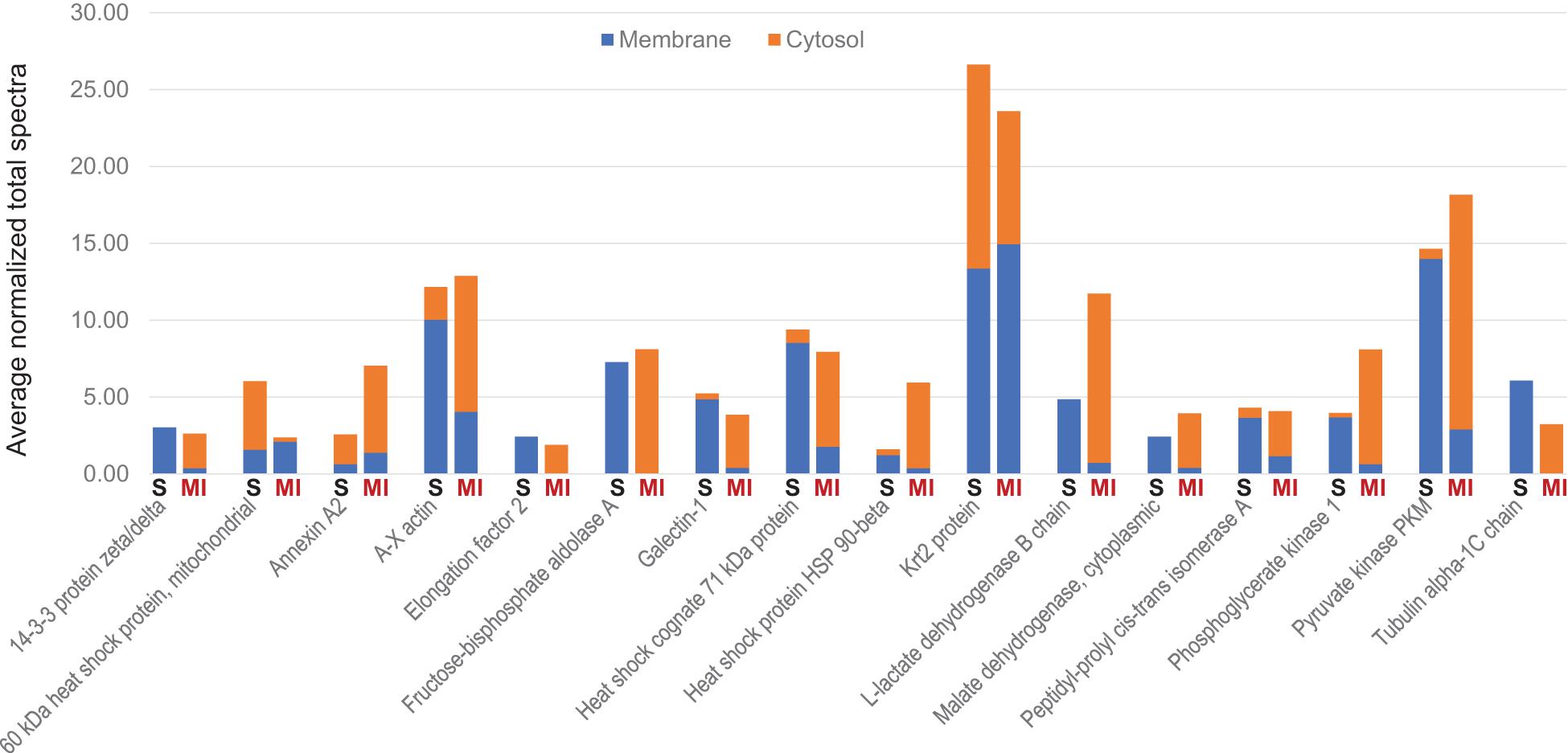
Figure 6. MI alters the subcellular distribution of proteins from systemically administered MetRS*MSC-Exos. MetRS*MSC-Exos were injected into the tail vein of mice 24 h after MI induction or Sham surgery. Animals were euthanized 12 h after exosome administration, and protein extracts from the left ventricle were separated into membrane and cytosolic fractions; then, the ANL-labeled MetRS*MSC exosomal proteins in each fraction were isolated via click-catalyzed alkyne-agarose capture and identified via LC-MS. The abundance of each of the 16 selected canonical exosomal proteins in the membrane and cytosol fractions of hearts from Sham (S) and MI animals was expressed as normalized average spectra from 3 independent samples.
The results from our click-catalyzed alkyne-agarose capture assessments indicated that desmoglein 1 (DSG-1) was more abundant in the heart than in other organs of both MI and Sham animals, and that cardiac DSG-1 levels increased in response to MI; however, to our knowledge, DSG-1 expression had not been previously reported in either MSCs or cardiomyocytes. Thus, we evaluated DSG-1 expression in MSCs via RT-PCR and in both MSCs and MSC exosomes via Western blot: our results confirmed that the DSG-1c isoform was expressed in MSCs (Figure 7A) and enriched in MSC exosomes (Figure 7B). To determine whether we could identify MSC-derived DSG-1 in the infarcted myocardium of MetRS*MSC-Exo–treated mice via FUNCAT, myocardial sections were click-stained with biotin-alkyne, sequentially stained with primary biotin antibodies and fluorescent secondary antibodies, and then counterstained for the presence of DSG-1 and troponin-T (TnT). When visualized via fluorescence, a number of TnT-expressing cells were positive for both the biotin-alkyne and DSG-1 labels (Figure 7C), which confirmed that the cardiomyocytes had taken up MetRS*MSC-Exo–derived DSG-1.
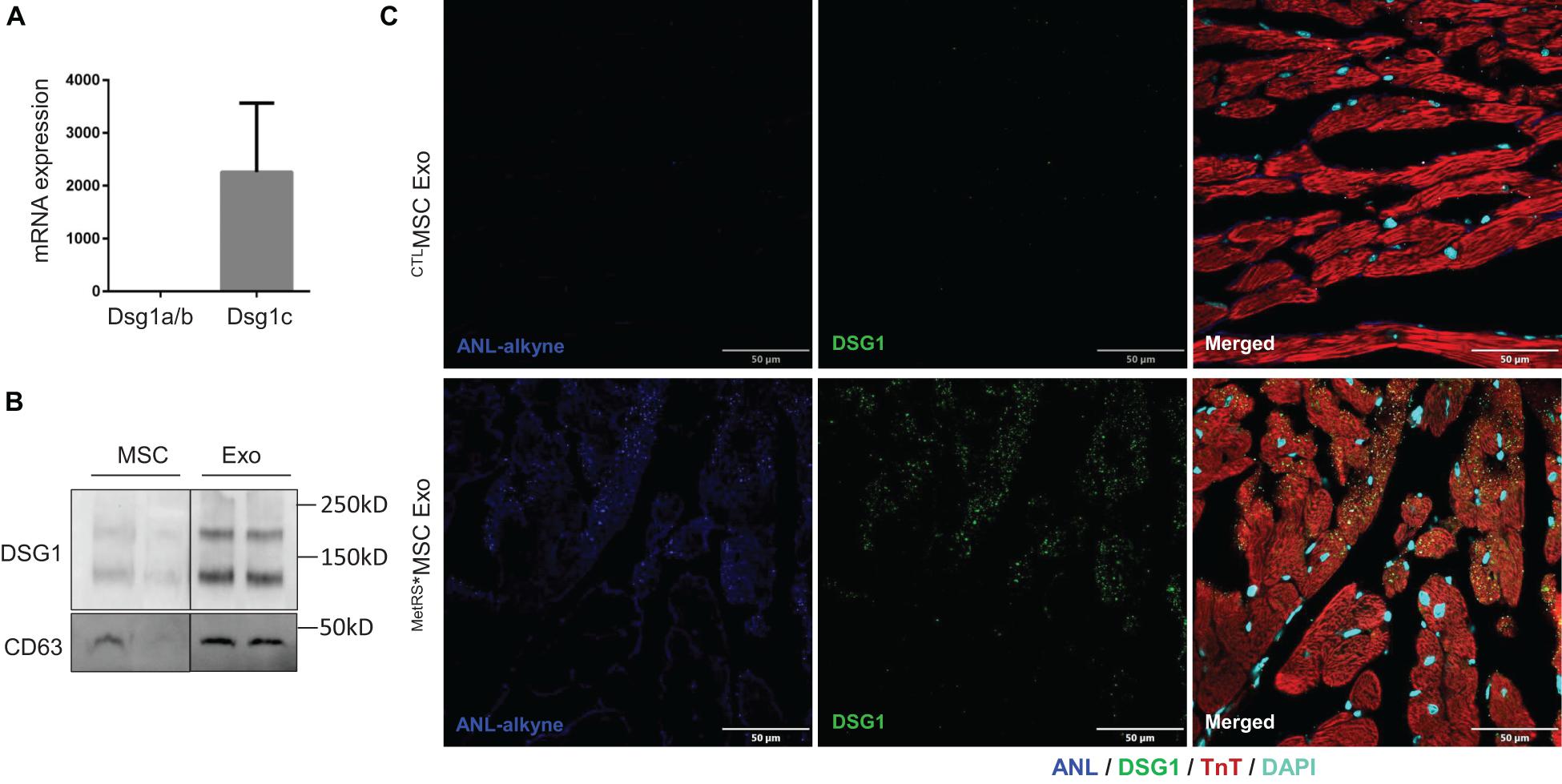
Figure 7. MSC exosomes contain DSG1 and deliver DSG1 to the cardiomyocytes of infarcted hearts. (A) mRNA levels of Dsg1 isoforms a/b and c were evaluated in MSCs via qRT-PCR and normalized to GAPDH mRNA levels. (B) The abundance of DSG1 protein in MSCs and MSC exosomes was evaluated via Western blot. (C) MetRS*MSC-Exos and CTLMSC-Exos were injected into the tail vein of mice 3 days after surgically induced MI. Mice were euthanized 4 h after exosome administration, and heart-tissue sections were click-stained to identify the ANL-labeled MetRS*MSC exosomal proteins and immunofluorescently stained for the presence of DSG1 and troponin T (TnT) protein; nuclei were counterstained with DAPI.
Because the benefits of stem-cell therapy for myocardial repair are primarily mediated via paracrine mechanisms, the factors produced by these cells, many of which are transported by exosomes, could serve as a potent, cell-free alternative for the treatment of patients with cardiovascular disease (Liu et al., 2018). Results from a number of preclinical studies indicate that MSCs and the exosomes produced by them are equally beneficial (Xiao et al., 2018); however, the mechanisms that direct the homing of exosomes to injured tissues, as well as the uptake and biodistribution of their cargo, remain largely uncharacterized, because techniques for distinguishing between exosomal proteins and proteins from the targeted tissues are either unavailable or inefficient. Here, we demonstrate that when MSCs are transfected with a vector coding for MetRS∗ and cultured in ANL-supplemented medium, the proteins produced by the transfected MSCs are selectively labeled with ANL, which can be covalently linked (via click chemistry) to alkyne-tagged reagents for subsequent visualization, identification, and isolation. Thus, when exosomes produced by the MetRS*MSCs were systemically administered to mice, the protein components of their cargo could be accurately and reliably tracked, visualized, identified, and quantified in tissues from a variety of organs, including both intact and infarcted hearts. Notably, as bioorthogonal chemistry can occur in living systems without disrupting native biochemical processes (Prescher et al., 2004; Prescher and Bertozzi, 2005; Sletten and Bertozzi, 2011; Ngo et al., 2013; Han et al., 2020a), this approach can be applied to other cell populations and even extended to wholly in vivo investigations by generating MetRS∗-transgenic mice (Alvarez-Castelao et al., 2017; Han et al., 2020a).
The overall pattern of MetRS*MSC-Exo protein distribution differed substantially between organs; thus, the mechanisms that govern exosome homing and the processing of exosomal proteins after uptake are likely cell- and tissue-type specific. Furthermore, our observation that a number of canonical exosome marker proteins (e.g., TSG101, Syntenin-1, ALIX, CD9, and CD81) were not present in any of the organs analyzed suggests that they are not suitable for tracking exosome uptake; whether their absence is attributable to rapid degradation in the targeted tissues or another process will be investigated in future studies. We also observed that surgically induced MI led to an increase in the abundance of exosomal proteins in cardiac tissue, which is consistent with the therapeutic mechanism of systemically delivered exosomes (Xiong et al., 2021) but has not been explicitly demonstrated previously. MI was also associated with the redistribution of numerous exosomal proteins from the membrane fraction in intact hearts to the cytosol of cells in infarcted hearts. This observation was not anticipated but likely evolves from increases in membrane permeability, because the loss of cardiomyocyte membrane integrity is a hallmark of MI, and some proposed treatment strategies target membrane dysfunction (Han et al., 2020b). Our results also indicated that the desmosome protein DSG-1c was expressed in MSCs, enriched in MSC exosomes, and taken up by cardiomyocytes in response to MI and MetRS*MSC-Exo administration. DSG-1 expression has not been previously reported in cardiomyocytes; however, cardiomyocytes express DSG-2, which is critical for cardiac integrity and electric conduction. Notably, human DSG-2 mutations are associated with cardiac arrhythmia (Schinner et al., 2020), and genetic deletion of DSG-2 leads to arrhythmogenic cardiomyopathy and heart failure in mice (Pilichou et al., 2009); thus, whether the exosome-mediated transfer of DSG-1 to cardiomyocytes contributes to the benefit of MSC therapy warrants continued investigation, particularly since MSC exosomes appear to reduce the arrhythmogenicity of human cardiomyocytes (Sattayaprasert et al., 2020).
A limitation of this study is that N-terminal processing by methionyl aminopeptidases results in cleavage of the first residue during protein maturation, preventing identification of these proteins. Methionine is the first amino acid translated during protein synthesis in eukaryotes and the average methionine content of mammalian proteins is ∼2.13% (Dieterich et al., 2006). Nevertheless, N-terminal posttranslational processing and acetylation occurs in ∼30% of all mammalian proteins (Meinnel et al., 2005) and only ∼1.02% of the open reading frames of the human database (predicted to be <25 residues and incomplete sequences) do not contain a single methionine residue (Dieterich et al., 2006).
In conclusion, when exosomes were collected from MetRS∗MSCs that had been incubated with ANL and then systemically administered to mice, the ANL-labeled exosomal proteins could be accurately and reliably identified, isolated, and quantified from a variety of mouse organs, including both intact and infarcted hearts, via ANL-alkyne click chemistry. Thus, this bioorthogonal technique can provide crucial insights into the mechanisms that govern exosome homing and the uptake of exosomal proteins.
The datasets presented in this study can be found in online repositories. The name of the repository and accession numbers can be found below: European Bioinformatics Institute (EBI) PRoteomics IDEntifications Database (PRIDE) Archive, https://www.ebi.ac.uk/pride/archive/, PXD024482, PXD024483, PXD024484, and PXD024485.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Alabama at Birmingham.
EZ and GQ conceptualized the study, interpreted the data, and wrote the manuscript. EZ, YL, CH, CF, LW, WC, YD, DH, SX, YW, and JM performed the experiments and analyzed the data. BA and JM made intellectual contributions and assisted in data interpretation. All authors contributed to the article and approved the submitted version.
This work was supported by the National Institute of Health (R01 Grant Nos. HL113541, HL130052, HL131110, and HL138990 to GQ), American Diabetes Association (Grant No. 1-15-BS-148 to GQ), and American Heart Association (Grant Nos. 19TPA34910227 to GQ, 18POST34070088 to SX, and 18PRE34080358 to EZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank W. Kevin Meisner, Ph.D. ELS, for editorial assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.657456/full#supplementary-material
Alvarez-Castelao, B., Schanzenbacher, C. T., Hanus, C., Glock, C., Tom Dieck, S., Dorrbaum, A. R., et al. (2017). Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nat. Biotechnol. 35, 1196–1201. doi: 10.1038/nbt.4016
Bagno, L., Hatzistergos, K. E., Balkan, W., and Hare, J. M. (2018). Mesenchymal stem cell-based therapy for cardiovascular disease: progress and challenges. Mol. Ther. 26, 1610–1623. doi: 10.1016/j.ymthe.2018.05.009
Charles, C. J., Li, R. R., Yeung, T., Mazlan, S. M. I., Lai, R. C., De Kleijn, D. P. V., et al. (2020). Systemic mesenchymal stem cell-derived exosomes reduce myocardial infarct size: characterization with MRI in a porcine model. Front. Cardiovasc. Med. 7:601990. doi: 10.3389/fcvm.2020.601990
Cheng, M., Yang, J., Zhao, X., Zhang, E., Zeng, Q., Yu, Y., et al. (2019). Circulating myocardial microRNAs from infarcted hearts are carried in exosomes and mobilise bone marrow progenitor cells. Nat. Commun. 10:959.
Cheng, M., Zhou, J., Wu, M., Boriboun, C., Thorne, T., Liu, T., et al. (2010). CXCR4-mediated bone marrow progenitor cell maintenance and mobilization are modulated by c-kit activity. Circ. Res. 107, 1083–1093. doi: 10.1161/circresaha.110.220970
Dieterich, D. C., Link, A. J., Graumann, J., Tirrell, D. A., and Schuman, E. M. (2006). Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. U.S.A. 103, 9482–9487. doi: 10.1073/pnas.0601637103
Han, D., Yang, J., Zhang, E., Liu, Y., Boriboun, C., Qiao, A., et al. (2020a). Analysis of mesenchymal stem cell proteomes in situ in the ischemic heart. Theranostics 10, 11324–11338. doi: 10.7150/thno.47893
Han, X., Zhao, Z. A., Yan, S., Lei, W., Wu, H., Lu, X. A., et al. (2020b). CXADR-like membrane protein protects against heart injury by preventing excessive pyroptosis after myocardial infarction. J. Cell. Mol. Med. 24, 13775–13788. doi: 10.1111/jcmm.15955
Kojima, K., Bowersock, G. J., Kojima, C., Klug, C. A., Grizzle, W. E., and Mobley, J. A. (2012). Validation of a robust proteomic analysis carried out on formalin-fixed paraffin-embedded tissues of the pancreas obtained from mouse and human. Proteomics 12, 3393–3402. doi: 10.1002/pmic.201100663
Liu, B., Lee, B. W., Nakanishi, K., Villasante, A., Williamson, R., Metz, J., et al. (2018). Cardiac recovery via extended cell-free delivery of extracellular vesicles secreted by cardiomyocytes derived from induced pluripotent stem cells. Nat. Biomed. Eng. 2, 293–303. doi: 10.1038/s41551-018-0229-7
Mathieu, M., Martin-Jaular, L., Lavieu, G., and Thery, C. (2019). Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 21, 9–17. doi: 10.1038/s41556-018-0250-9
Matsumoto, A., and Itoh, T. Q. (2011). Self-assembly cloning: a rapid construction method for recombinant molecules from multiple fragments. Biotechniques 51, 55–56.
Meinnel, T., Peynot, P., and Giglione, C. (2005). Processed N-termini of mature proteins in higher eukaryotes and their major contribution to dynamic proteomics. Biochimie 87, 701–712. doi: 10.1016/j.biochi.2005.03.011
Morishita, M., Takahashi, Y., Nishikawa, M., Sano, K., Kato, K., Yamashita, T., et al. (2015). Quantitative analysis of tissue distribution of the B16BL6-derived exosomes using a streptavidin-lactadherin fusion protein and iodine-125-labeled biotin derivative after intravenous injection in mice. J. Pharm. Sci. 104, 705–713. doi: 10.1002/jps.24251
Ngo, J. T., Champion, J. A., Mahdavi, A., Tanrikulu, I. C., Beatty, K. E., Connor, R. E., et al. (2009). Cell-selective metabolic labeling of proteins. Nat. Chem. Biol. 5, 715–717.
Ngo, J. T., Schuman, E. M., and Tirrell, D. A. (2013). Mutant methionyl-tRNA synthetase from bacteria enables site-selective N-terminal labeling of proteins expressed in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 110, 4992–4997. doi: 10.1073/pnas.1216375110
Pilichou, K., Remme, C. A., Basso, C., Campian, M. E., Rizzo, S., Barnett, P., et al. (2009). Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 206, 1787–1802. doi: 10.1084/jem.20090641
Prescher, J. A., and Bertozzi, C. R. (2005). Chemistry in living systems. Nat. Chem. Biol. 1, 13–21.
Prescher, J. A., Dube, D. H., and Bertozzi, C. R. (2004). Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877. doi: 10.1038/nature02791
Qin, G., Ii, M., Silver, M., Wecker, A., Bord, E., Ma, H., et al. (2006). Functional disruption of alpha4 integrin mobilizes bone marrow-derived endothelial progenitors and augments ischemic neovascularization. J. Exp. Med. 203, 153–163. doi: 10.1084/jem.20050459
Sattayaprasert, P., Vasireddi, S. K., Bektik, E., Jeon, O., Hajjiri, M., Mackall, J. A., et al. (2020). Human cardiac mesenchymal stem cells remodel in disease and can regulate arrhythmia substrates. Circ. Arrhythm. Electrophysiol. 13:e008740.
Schinner, C., Erber, B. M., Yeruva, S., Schlipp, A., Rotzer, V., Kempf, E., et al. (2020). Stabilization of desmoglein-2 binding rescues arrhythmia in arrhythmogenic cardiomyopathy. JCI Insight 5:e130141.
Sletten, E. M., and Bertozzi, C. R. (2011). From mechanism to mouse: a tale of two bioorthogonal reactions. Acc. Chem. Res. 44, 666–676. doi: 10.1021/ar200148z
Tang, Y. L., Zhu, W., Cheng, M., Chen, L., Zhang, J., Sun, T., et al. (2009). Hypoxic preconditioning enhances the benefit of cardiac progenitor cell therapy for treatment of myocardial infarction by inducing CXCR4 expression. Circ. Res. 104, 1209–1216. doi: 10.1161/circresaha.109.197723
Xiao, C., Wang, K., Xu, Y., Hu, H., Zhang, N., Wang, Y., et al. (2018). Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of mir-125b. Circ. Res. 123, 564–578.
Keywords: exosomes, extracellular vesicles, mesenchymal stem cells, bioorthogonal labeling, click reaction, cardiac ischemia
Citation: Zhang E, Liu Y, Han C, Fan C, Wang L, Chen W, Du Y, Han D, Arnone B, Xu S, Wei Y, Mobley J and Qin G (2021) Visualization and Identification of Bioorthogonally Labeled Exosome Proteins Following Systemic Administration in Mice. Front. Cell Dev. Biol. 9:657456. doi: 10.3389/fcell.2021.657456
Received: 22 January 2021; Accepted: 12 March 2021;
Published: 07 April 2021.
Edited by:
Shijun Hu, Soochow University, ChinaReviewed by:
Raj Kishore, Temple University, United StatesCopyright © 2021 Zhang, Liu, Han, Fan, Wang, Chen, Du, Han, Arnone, Xu, Wei, Mobley and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gangjian Qin, Z3FpbkB1YWIuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.