 Lauren E. Kueffer
Lauren E. Kueffer Raji E. Joseph
Raji E. Joseph Amy H. Andreotti
Amy H. Andreotti
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 23 June 2021
Sec. Signaling
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.655489
This article is part of the Research Topic Targeting Bruton Tyrosine Kinase View all 9 articles
Since Dr. Ogden Bruton’s 1952 paper describing the first human primary immunodeficiency disease, the peripheral membrane binding signaling protein, aptly named Bruton’s tyrosine kinase (BTK), has been the target of intense study. Dr. Bruton’s description of agammaglobulinemia set the stage for ultimately understanding key signaling steps emanating from the B cell receptor. BTK is a multidomain tyrosine kinase and in the decades since Dr. Bruton’s discovery it has become clear that genetic defects in the regulatory domains or the catalytic domain can lead to immunodeficiency. This finding underscores the intricate regulatory mechanisms within the BTK protein that maintain appropriate levels of signaling both in the resting B cell and during an immune challenge. In recent decades, BTK has become a target for clinical intervention in treating B cell malignancies. The survival reliance of B cell malignancies on B cell receptor signaling has allowed small molecules that target BTK to become essential tools in treating patients with hematological malignancies. The first-in-class Ibrutinib and more selective second-generation inhibitors all target the active site of the multidomain BTK protein. Therapeutic interventions targeting BTK have been successful but are plagued by resistance mutations that render drug treatment ineffective for some patients. This review will examine the molecular mechanisms that drive drug resistance, the long-range conformational effects of active site inhibitors on the BTK regulatory apparatus, and emerging opportunities to allosterically target the BTK kinase to improve therapeutic interventions using combination therapies.
Bruton’s tyrosine kinase (BTK) is a non-receptor tyrosine kinase that belongs to the TEC family. The five members of the TEC family kinases (BTK, ITK, TEC, TXK, and BMX) are expressed in various hematopoietic cell lineages and relay signals downstream of multiple immunological receptors. BTK has been most well studied in B cells in the context of B-cell receptor (BCR) signaling, but it also plays a role in macrophages, mast cells, and dendritic cells downstream of Fc receptors and Toll-like receptors (TLRs) (Weber et al., 2017; Pal Singh et al., 2018). Summarized in Figure 1, antigen binding to the BCR triggers the activation of a trio of tyrosine kinases: LYN, SYK, and BTK (Geahlen, 2009; Stepanek et al., 2013). LYN phosphorylates the immunoreceptor tyrosine-based activation motifs (ITAMs) within the Ig-α/β chains associated with the BCR as well as CD19. The phosphorylation of the ITAMs recruits SYK where it is activated. Activated SYK phosphorylates the B-cell adaptor protein for PI3K (BCAP) and the phosphorylation of BCAP and CD19 by these kinases promote activation of PI3K (Okada et al., 2000). Activated SYK phosphorylates SLP-65 (or BLNK) and activated PI3K phosphorylates PIP2 in the membrane to produce PIP3, which recruits BTK and its substrate phospholipase Cγ2 (PLCγ2) to the BCR complex. BTK association with PIP3 and SLP-65 activates BTK, which phosphorylates and activates PLCγ2 (Li et al., 1997). Activation of PLCγ2 in turn generates the second messengers, inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), to induce a calcium flux and activate protein kinase C (PKC) leading into the MAP kinase pathway (Kurosaki, 2011). These signaling events ultimately lead to the upregulation of specific genes essential for B-cell maturation and proliferation.
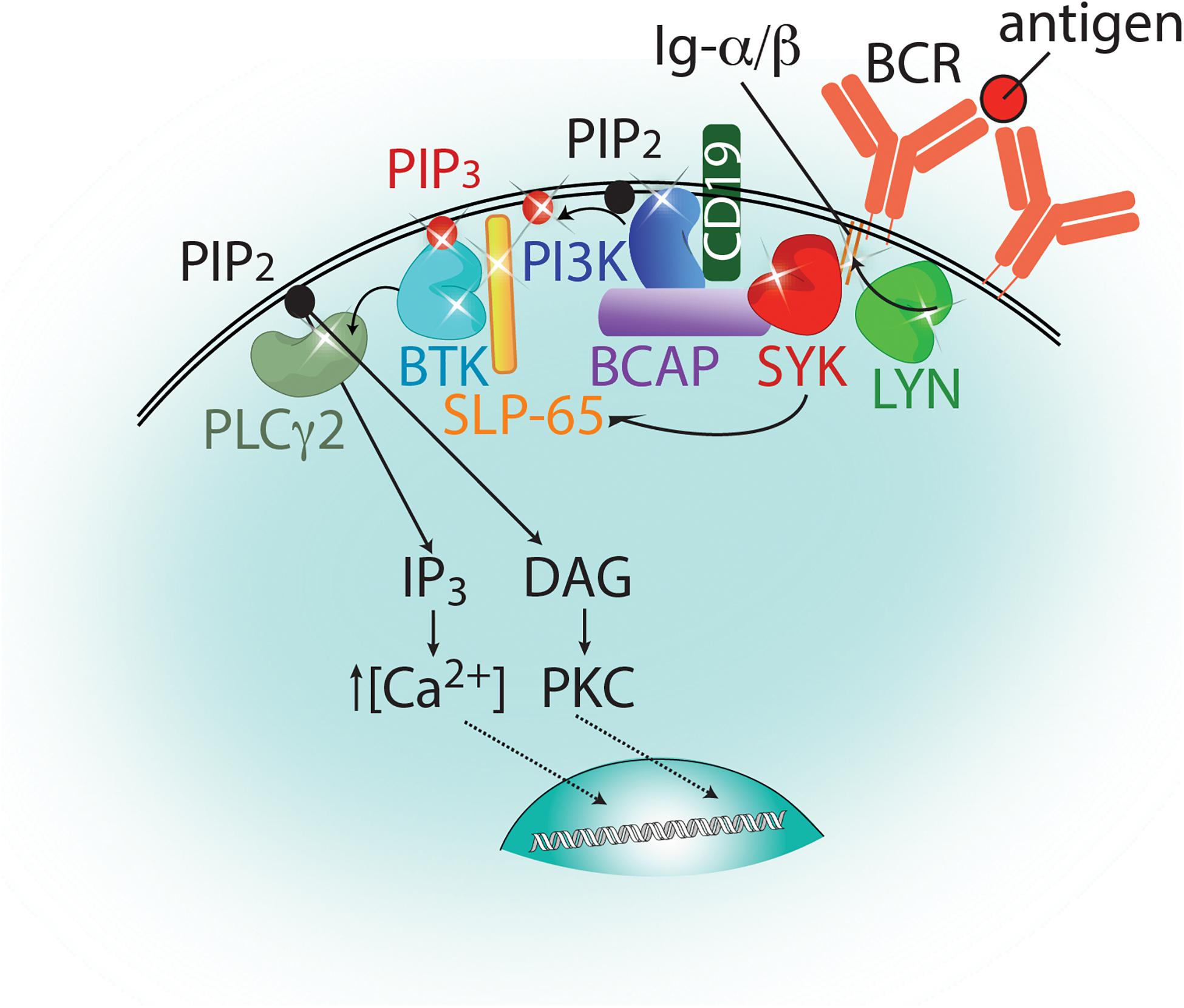
Figure 1. Proximal signaling events emanating from the B cell receptor. Antigen binding to the B cell receptor (BCR, orange) leads to the activation of a trio of kinases: LYN, SYK, and BTK, which are represented as bi-lobed structures colored lime green, red, and cyan, respectively. Phosphorylation events carried out by these kinases are indicated by arrows and sites of phosphorylation are indicated as stars. These include phosphorylation of the ITAMs and CD19 (by LYN), BCAP and SLP-65 (by SYK), and PLCγ2 (by BTK). PI3K, depicted in blue, phosphorylates PIP2 to generate PIP3 at the membrane. PIP3 is indicated as a red circle and PIP2 by a black circle. PLCγ2 hydrolyzes PIP2 at the membrane to produce the secondary messengers IP3 and DAG which will induce a calcium flux into the cell and activate PKC, respectively. Further downstream signaling events that lead to an upregulation of target genes for B cell maturation and proliferation are not shown in this figure for simplicity.
BTK plays an instrumental role in B-cell development as mutations in the BTK gene have been linked to the primary immunodeficiency X-linked agammaglobulinemia (XLA) (Tsukada et al., 1993). XLA patients experience an increased susceptibility to bacterial infections due to an arrest in B-cell development at the pre-B cell stage. Enhanced BTK activity on the other hand is linked to the production of autoantibodies (Kersseboom et al., 2010; Kil et al., 2012; Corneth et al., 2017; Heukels et al., 2019) and this autoimmune phenotype is dependent on the catalytic activity of BTK (Kil et al., 2012). However, the nature of the autoantibodies produced: whether they are natural antibodies derived from B1 cells or immune antibodies is currently unclear (Satterthwaite, 2018). Taken together, it is clear that BTK function must be finely tuned to generate an appropriate immunological response. For this reason, BTK has been studied as a valid target for therapeutic development to tune the BCR signaling cascade. Specifically, BTK is a tissue specific target for inhibition in various B-cell lymphomas including diffuse large B-cell lymphoma (DLBCL) and chronic lymphocytic leukemia (CLL) (Davis et al., 2010; Herishanu et al., 2011); proliferation of these lymphomas strictly depends on the activation of the BTK signaling cascade. Inhibitors targeting BTK for treatment of these lymphomas have shown anti-tumor activity in lymphoma models, and three BTK specific inhibitors (Ibrutinib, Acalabrutinib, and Zanubrutinib) have FDA approval and are being used as a treatment option for patients. The specific role of BTK in B cell lymphomas has been reviewed elsewhere (Pal Singh et al., 2018).
BTK also plays an important role in innate immune signaling pathways in other hematopoietic cell lineages. BTK has been shown to function in antimicrobial responses downstream of TLRs and is involved in Fc receptor signaling (Weber et al., 2017). Given BTK’s role in innate immune signaling pathways, inhibitors of BTK are being investigated in treatment of rheumatoid arthritis (RA) (Di Paolo et al., 2011), and the 2019 novel coronavirus disease (COVID-19) caused by SARS-CoV-2 infection. Administration of Acalabrutinib, a second-generation inhibitor of BTK, improved oxygenation levels in over 70% of patients in a small patient cohort (Roschewski et al., 2020). In other reports, the effect of blocking BTK in the context of thromboinflammation in COVID-19 is considered (Nicolson et al., 2020; Siess et al., 2020). Moreover, as the SARS-CoV-2 virus spreads throughout the population, the number of patients already being treated with a BTK inhibitor that contract COVID-19 has increased. This has resulted in a number of recent reports detailing the effect of BTK inhibition during the course of COVID-19 infection (Chong et al., 2020; Lin et al., 2020; Thibaud et al., 2020; Treon et al., 2020). Currently, two clinical trials are underway to evaluate the efficacy of BTK inhibitors during COVID-19 treatment (NCT numbers: NCT04382586, NCT04346199, ClinicalTrials.gov).
Due to the involvement of BTK in multiple immunological signaling pathways, there have been a host of inhibitors developed with different binding modes all targeting the kinase domain of BTK as a treatment option for B cell lymphomas and other BTK-reliant diseases. Supplementary Table 1 summarizes current BTK inhibitors classified by one of four binding modes: (1) covalent, irreversible, (2) covalent, reversible, (3) non-covalent, reversible, or (4) proteolysis targeting chimeras (PROTACs). Supplementary Figure 1 provides the chemical structures for those inhibitors for which this information has been disclosed. The following sections of this review will focus on inhibitors that have FDA approval, are at Phase 3 clinical trials, or represent a unique approach to targeting BTK. We aim to summarize select clinical data, describe the molecular mechanisms at work in Ibrutinib resistance mutations, and highlight the impact that specific inhibitors have on the conformational ensemble of full-length BTK.
Ibrutinib (brand name Imbruvica) is the first-in-class covalent irreversible BTK inhibitor that was rationally designed to modify C481 after identifying a chemical scaffold that inhibited BTK kinase activity (Pan et al., 2007; Figure 2A). After demonstrating that Ibrutinib blocks B-cell activation downstream of the BCR in animal models of B-cell malignancy, Ibrutinib moved to randomized human clinical trials (Honigberg et al., 2010). Since its success in clinical trials, Ibrutinib has been approved for use in treatment of CLL, mantle cell lymphoma (MCL), Waldenström’s macroglobulinemia (WM), marginal zone lymphoma (MZL), and chronic graft versus host disease (cGVHD) and is in various stages of clinical trials for the treatment of other immune disorders. At the molecular level, Ibrutinib attaches to C481 within the kinase active site and acts as an ATP competitive inhibitor with an IC50 of 0.5 nM in a cell-free kinase assay (Honigberg et al., 2010). Ibrutinib is also known to bind to and inhibit multiple kinases including ITK, a TEC family kinase important in T-cell signaling (Dubovsky et al., 2013). The off-target binding of Ibrutinib is thought to contribute to undesired side effects namely bleeding events which is linked to platelet dysfunction upon Ibrutinib treatment (Shatzel et al., 2017).
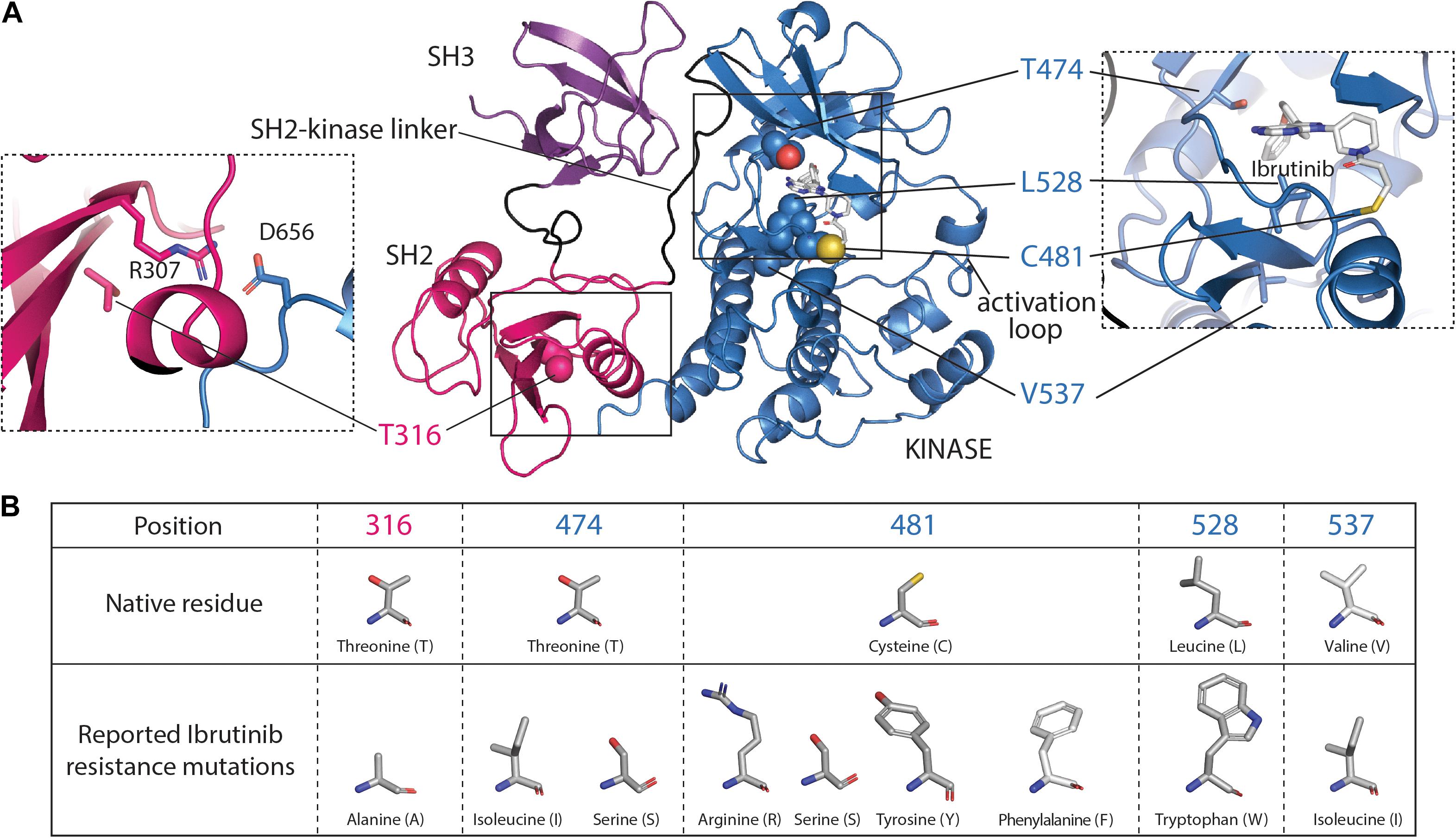
Figure 2. Ibrutinib resistance mutations mapped onto the SH3–SH2-kinase fragment of BTK. (A) The monomeric model of the SH3–SH2-kinase module of BTK is shown (Wang et al., 2015) with the SH3, SH2, and kinase domains colored purple, pink, and blue, respectively. The BTK kinase domain structure bound to Ibrutinib [Protein Data Bank (PDB) code: 5P9J] was used to place Ibrutinib in the SH3–SH2-kinase model. Ibrutinib resistance mutations that have been described at T316, T474, C481, L528, and V537 are shown. The left dashed box highlights the R307/D656 salt bridge in proximity to T316. The right dashed box highlights the Ibrutinib resistance mutations surrounding the active site. The boxed image here was taken from the Ibrutinib-bound kinase domain structure (PDB:5P9J). (B) The table presents structures of native amino acid residues and the corresponding Ibrutinib resistance mutations reported to date.
While Ibrutinib has shown success in the clinic, there is an increasing occurrence of acquired resistance that is driven by either point mutations in the BTK protein itself or gain-of-function mutations in the BTK substrate PLCγ2 (Zhou et al., 2012). A 3-year cohort study found that acquired BTK mutations contributed to CLL progression while Ibrutinib was administered (Quinquenel et al., 2019). The most common Ibrutinib resistance mutation occurs at the site of covalent attachment, BTK C481. The C481S resistance mutation was first identified in the samples of five out of six relapsed CLL patients (Woyach et al., 2014) and has surfaced in other B-cell lymphomas such as WM and MCL supporting the notion that this mutation is a primary acquired resistance mutation that is a consequence of treatment with Ibrutinib (Chiron et al., 2014; Xu et al., 2017). The BTK C481S mutation does not affect the activity of BTK (Joseph et al., 2020) but mutation of this residue renders BTK insensitive to Ibrutinib due to the loss of the covalent bond to Ibrutinib (Cheng et al., 2015; Figure 2). The reversible binding of Ibrutinib by the BTK C481S mutant together with the rapid clearance of Ibrutinib from the plasma (mean half-life of 2–3 h, Advani et al., 2013; Davids and Brown, 2014), would significantly reduce the occupancy of BTK by Ibrutinib, leaving BTK uninhibited and signaling competent. In fact, a recent study showed that BTK occupancy is a critical factor that needs to be considered with the use of BTK covalent inhibitors (Sun et al., 2020; Thompson, 2020). A half dose of inhibitor taken twice daily by patients was found to have higher BTK occupancy and more effective inhibition than a single daily full dose of inhibitor. Sustained presence of a BTK inhibitor in plasma either by increasing inhibitor dosage frequency or by the use of inhibitors with a prolonged plasma half-life will be critical to counter resistance by the BTK C481S mutation.
Other acquired Ibrutinib resistance mutations surrounding the ATP binding site have been uncovered in relapsed patients (Figures 2A,B), including T474I/S, L528W, and V537I (Maddocks et al., 2015; Kanagal-Shamanna et al., 2019). While these kinase domain resistance mutations are thought to destabilize productive Ibrutinib binding, a distinct acquired resistance mutation, T316A, in the regulatory Src homology 2 (SH2) domain of BTK has been described (Sharma et al., 2016; Kadri et al., 2017; Figures 2A,B). The T316A Ibrutinib resistance mutation is unique in that it is the first and only described resistance mutation found outside the kinase domain of BTK (Figure 2A). The T316A mutation does not interfere with Ibrutinib binding but nevertheless confers resistance by permitting downstream phosphorylation signaling events (Sharma et al., 2016). Understanding how these point mutations contribute to Ibrutinib resistance at the molecular level is important as this information can aid inhibitor design to provide treatments for patients who acquire resistance. Recent solution nuclear magnetic resonance (NMR), hydrogen/deuterium exchange mass spectrometry (HDX-MS), and biochemical studies have revealed that the T316A mutation disrupts the autoinhibitory conformation of BTK (described in detail below) thereby increasing the active population of BTK, evading Ibrutinib inhibition (Joseph et al., 2020). There are similar reports of active site inhibitor resistance mutations disrupting autoinhibitory contacts in ABL, another multi-domain kinase (Saleh et al., 2017; Xie et al., 2020) suggesting that this mode of resistance may be a shared mechanism to bypass inhibition.
Second generation covalent irreversible inhibitors have since been developed to increase specificity. There are several covalent irreversible BTK inhibitor candidates that have been developed including the FDA approved Acalabrutinib (brand name Calquence) (Barf et al., 2017) and Zanubrutinib (brand name Brukinsa) (Weaver and Jimeno, 2020), along with Spebrutinib (CC-292) (Evans et al., 2013), Tirabrutinib (Walter et al., 2016), Evobrutinib (Caldwell et al., 2019), and Tolebrutinib (Francesco et al., 2017). All mediate covalent attachment to BTK via C481, the Ibrutinib binding residue. At the biochemical level, kinetic experiments that compared a panel of covalent inhibitors (Ibrutinib, Spebrutinib, Acalabrutinib, and Tirabrutinib) revealed that while Ibrutinib had the most potent IC50 against BTK, it had the lowest selectivity for BTK when tested against a panel of kinases that possess the homologous cysteine binding residue in the ATP binding site (Liclican et al., 2020). The remainder of this section will focus on the second-generation irreversible covalent inhibitors that have either gained FDA approval or are currently in Phase 3 clinical trials.
Acalabrutinib was found to have an improved selectivity profile over other covalent inhibitors, Ibrutinib, and Spebrutinib (Barf et al., 2017). Computational modeling of Acalabrutinib in the ATP binding site of the BTK kinase domain predicts that Acalabrutinib makes more hydrogen bonds with the kinase domain compared to Ibrutinib (Barf et al., 2017). Because of its improved specificity and promising preclinical characterization in an animal model (Harrington et al., 2016), Acalabrutinib moved to clinical trials targeting various B cell lymphomas (Byrd et al., 2015; Wang et al., 2018; Girard et al., 2019). Acalabrutinib is currently FDA approved to use as a treatment for MCL, CLL, and small lymphocytic leukemia (SLL) patients. There is also a preclinical evaluation testing a combination of Acalabrutinib and a PI3K inhibitor in CLL mouse models to target two signaling proteins in the BCR signaling cascade (Niemann et al., 2017). A more extensive review of the preclinical and clinical data for Acalabrutinib has been published (Wu et al., 2016).
Zanubrutinib was rationally designed to improve both specificity for BTK and pharmacokinetic and pharmacodynamic properties such as oral absorption and target occupancy relative to Ibrutinib (Guo et al., 2019). Indeed, Zanubrutinib shows an improved selectivity against common off-targets of Ibrutinib: ITK, TEC, and epidermal growth factor receptor (EGFR) (Guo et al., 2019). Zanubrutinib swiftly moved into clinical trials for a variety of B cell lymphomas (Tam et al., 2019, 2020; Syed, 2020; Weaver and Jimeno, 2020) and has since been FDA approved as a treatment option for MCL patients. Compared to Ibrutinib’s binding mode, Zanubrutinib makes an extra hydrogen bond with M477 (Guo et al., 2019) which could explain its improved selectivity for BTK over other kinases (Kaptein et al., 2018).
Evobrutinib (M2951) and Tolebrutinib (SAR442168, PRN2246) are covalent irreversible BTK inhibitors currently active in Phase 3 clinical trials. Evobrutinib was rationally designed using B43, a moderately potent kinase inhibitor, as a starting structure to pursue structure activity relationship (SAR) drug design and has been evaluated in a Phase 2 clinical trial for multiple sclerosis (MS) (Caldwell et al., 2019; Montalban et al., 2019). This compound has since moved on to a Phase 3 clinical trial targeting the same disease (NCT04338022, ClinicalTrials.gov). At the molecular level, this inhibitor makes two sets of contacts within the kinase active site; a covalent irreversible bond to the sidechain of C481 and a non-covalent interaction with the selectivity pocket surrounding T474, the gatekeeper residue of BTK. The contact Evobrutinib makes in the T474 pocket is thought to impart greater specificity over other BTK inhibitors as a threonine at the gatekeeper position is found in only 20% of human kinases (Lou et al., 2012). Bulky, hydrophobic residues are more often located at this position in other kinases, so the smaller and more polar threonine side chain in BTK creates an additional pocket for ATP binding site inhibitors. Tolebrutinib (SAR442168/PRN2246) is unique from the other covalent irreversible inhibitors mentioned in that it has been designed to cross the blood brain barrier for BTK-dependent disease mechanisms relevant in the nervous system (Francesco et al., 2017). For this reason, Tolebrutinib is currently active in Phase 3 clinical trials for MS (NCT04411641 and NCT04458051, ClinicalTrials.gov).
While Ibrutinib and other covalent, irreversible inhibitors have shown success in the clinic, strategies to modulate residence time with irreversible kinase inhibitors is lacking. To overcome this challenge, design of covalent, reversible kinase inhibitors targeting non-catalytic cysteine residues in the protein has emerged. These covalent, reversible inhibitors usually contain a cyano-acrylamide scaffold that permits elimination of the cysteine residue upon unfolding of the tertiary structure (Serafimova et al., 2012). A covalent, reversible inhibitor targeting C481 in BTK, Rilzabrutinib (PRN1008), has been developed (Hill et al., 2015). In preclinical evaluations, Rilzabrutinib has shown a prolonged BTK occupancy time of over 100 h and also shows great specificity in targeting BTK over other common Ibrutinib off-targets (Hill et al., 2015). Early phase clinical trials indicated that Rilzabrutinib is well-tolerated in healthy volunteers (Smith et al., 2017) and showed a significant response in immune thrombocytopenia patients (Kuter et al., 2020). Rilzabrutinib has since moved to a Phase 3 clinical trial to evaluate its use in treating Immune Thrombocytopenia (NCT04562766, ClinicalTrials.gov). Other covalent, reversible BTK inhibitors are in preclinical evaluations and early stage clinical trials (Herter et al., 2018; Schnute et al., 2019).
Even though Ibrutinib and other covalent BTK inhibitors have been FDA approved for administration to patients, the occurrence of resistance mutations at the cysteine binding residue creates incentive to target BTK with non-covalent inhibitors. A study comparing a panel of covalent and non-covalent BTK inhibitors revealed that the non-covalent inhibitors tested were both more specific toward BTK and were able to target and inhibit Ibrutinib-resistant mutants of BTK (Johnson et al., 2016). There are various non-covalent inhibitors targeting BTK in different clinical stages of development, but our focus will be on the non-covalent inhibitor in Phase 3 clinical trials, Fenebrutinib (GDC-0853). The status of other non-covalent inhibitors is summarized in Supplementary Table 1.
GDC-0853 was rationally designed as a non-covalent BTK inhibitor with greater specificity than existing covalent inhibitors (Crawford et al., 2018). Preclinical characterization of GDC-0853 revealed that BCR signaling events downstream of BTK are inhibited and GDC-0853 has reduced off target effects; no inhibition of ITK or EGFR was observed (Reiff et al., 2018). Furthermore, GDC-0853 has also shown anti-tumor activity in patients harboring the C481S Ibrutinib resistance mutation (Byrd et al., 2019). These findings demonstrate that GDC-0853 could be a treatment option for patients who have relapsed after Ibrutinib and who possess resistance mutations at the covalent cysteine residue. A Phase 1 clinical trial is currently underway to study the safety of GDC-0853 in patients with relapsed CLL or other B-cell lymphomas (NCT01991184, ClinicalTrials.gov). Moreover, GDC-0853 is also being tested as a treatment option for RA, SLE, urticaria (Phase 2 trials), and MS (Phase 3 trial) (NCT02983227, NCT02908100, NCT03137069, NCT04544449, ClinicalTrials.gov).
While small molecule inhibitors developed for BTK have shown success in the clinic, other strategies are being explored to combat resistance. An appealing strategy toward BTK inhibition is targeted degradation using PROTACs. PROTACs are bivalent ligands that are designed to specifically target a protein of interest for degradation via ubiquitination (Sakamoto et al., 2001). Recently, potent PROTACs (MT-802 and DD-03-171) have been developed for BTK. MT-802 is based on the Ibrutinib scaffold, while DD-030-171 is based on the CGI1746 scaffold (Buhimschi et al., 2018; Dobrovolsky et al., 2019). Both chimeric molecules trigger degradation of both wild-type and the C481S BTK in B cell lymphoma cells. However, the potency of PROTACs targeting BTK is context dependent where different cell types and tissues show different levels of BTK degradation even though exposure to the PROTAC is similar across these areas (Zorba et al., 2018). Nevertheless, targeted degradation of BTK could emerge as another tool in the arsenal for treating various immunological diseases. There are emerging PROTACs, including NX-2127 and NX-5948 that target BTK for degradation and show promise in preclinical evaluations (Robbins et al., 2020).
All of the BTK inhibitors described above target the BTK active site situated between the N- and C-lobes of the kinase domain. BTK is a multidomain kinase composed of regulatory regions outside of the carboxy-terminal kinase domain. From the amino- to carboxy terminus is a Pleckstrin homology (PH) domain, a Tec homology (TH) domain, a proline-rich region (PRR), a Src homology 3 (SH3) domain, a SH2 domain, and finally the carboxy terminal kinase domain (Figure 3A). Unlike other families of non-receptor tyrosine kinases, BTK and the other TEC kinases have to date resisted crystallization in their full-length form. Nevertheless, fragment crystal structures and a range of biochemical and biophysical studies are revealing the molecular details of BTK regulation and set the stage to better understand how current BTK inhibitors affect full-length BTK to pave the way to develop allosteric approaches to modulate BTK function in disease.
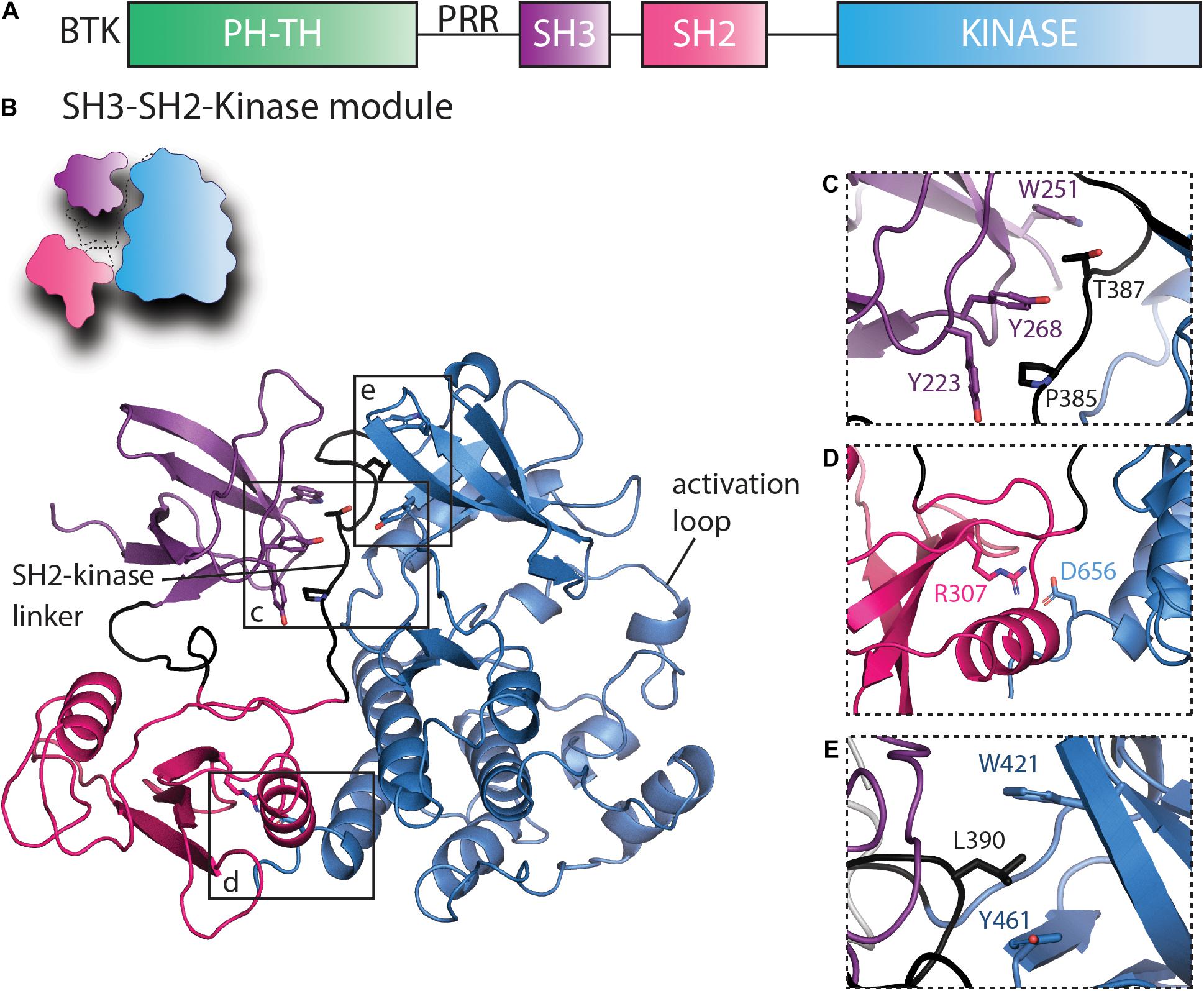
Figure 3. Regulatory interactions within the SH3–SH2-kinase module of BTK that maintain the inactive conformation. (A) Domain schematic of full-length BTK with the PHTH, SH3, SH2, and kinase domains colored green, purple, pink, and blue, respectively. The proline-rich region (PRR) between the PHTH and SH3 domains is labeled. (B) A cartoon of the autoinhibited BTK SH3–SH2-kinase model and the BTK SH3–SH2-kinase structure (Wang et al., 2015) are shown using the same domain colors as in panel (A). The SH2-kinase linker and the activation loop are labeled, and boxes show the regions expanded in panels (C–E). (C) The intramolecular interaction between SH3 domain residues Y223, W251, and Y268 and the SH2-kinase linker residues P385 and T387 stabilize the SH3 domain onto the distal side of the kinase domain (opposite the activation loop face). (D) The salt bridge contact between SH2 R307 and D656 in the kinase domain stabilizes the SH2 domain on the distal side of the kinase domain C-lobe and mimics the SH2/C-terminal tail phosphotyrosine interaction of the SRC kinases (Liu et al., 1993). (E) L390 in the SH2-kinase linker and W421 and Y461 in the kinase domain N-lobe form the hydrophobic stack.
Crystallographic studies performed by the Harrison and Kuriyan groups revealed autoinhibitory contacts within the SRC module (Shah et al., 2018) or SH3–SH2-kinase fragment of BTK (Wang et al., 2015). The structure of the SH3–SH2-kinase fragment of BTK shows a compact, SRC-like conformation with the SH3 and SH2 domains docked onto the back side of the kinase domain opposite the activation loop (Figure 3B). The conserved binding groove of the SH3 domain contacts the SH2-kinase linker (Figure 3C) while SH2 domain is docked onto the C-lobe of the kinase domain despite the absence in the TEC family of the phosphorylated tail found in SRC kinases (Figure 3D). Subsequent work applying solution NMR and HDX-MS to full-length BTK further probed these intramolecular interfaces and demonstrated that these interfaces maintain the autoinhibited, inactive conformation of BTK in solution (Joseph et al., 2017). This work also defined a specific salt bridge contact between R307 in the SH2 domain and D656 in the kinase domain (Figure 3D) that serves a role similar to the phosphorylated tail of the SRC kinases by stabilizing the autoinhibitory pose of the SH2 domain.
The regulatory features within the kinase domain have been extensively reviewed elsewhere (Taylor and Kornev, 2011). Key conserved features include the αC helix, which transitions between the αC-out (inactive) and αC-in (active) conformations. A conserved Lys/Glu salt bridge stabilizes the αC-in or active conformation and transition to the αC-out or inactive state is accompanied by loss of the Lys/Glu salt bridge (Taylor et al., 2015). The activation loop also transitions between distinct conformers depending on the activation (and phosphorylation) status of the kinase domain. Phosphorylation of BTK Y551 in the activation loop triggers a conformational shift to favor opening of the activation loop, whereas the inactive state features a compact activation loop folded in toward the active site. This inactive activation loop conformation protects Y551 from phosphorylation and it is interesting to note that loop dynamics differ significantly between BTK and the T cell specific TEC family kinase ITK. The ITK activation loop strongly favors the inactive conformation compared to the BTK activation loop that readily samples the open and Y551 accessible state (Joseph et al., 2013). These findings suggest that the T cell kinase may be under greater regulatory control than BTK in B cells perhaps as a mechanism to limit spurious T cell activation. Additional regulatory features within the kinase domain include the DFG motif, the regulatory spine, and the hydrophobic stack. Briefly, when the DFG motif adopts a DFG-in conformation, the phenylalanine of this motif participates in assembling the regulatory spine which is essential for TEC family kinase activation (Joseph et al., 2010).
The hydrophobic stack is a set of three residues [in BTK W421 and Y461 from the kinase domain N-lobe and L390 from the SH2-kinase linker (Figure 3E)] that when assembled is thought to stabilize the autoinhibited form of the kinase (Von Raußendorf et al., 2017). Thus, contacts between the SH3 domain and the SH2-kinase linker serve to not only protect the SH3 binding groove from engaging with exogenous ligands but also position the side chain of L390 to complete the hydrophobic stack on the back on the kinase domain N-lobe. In the TEC and SRC family kinases, it has been demonstrated that disruption of this hydrophobic stack results in exchange of ADP for ATP promoting full activation (Von Raußendorf et al., 2017).
The domain arrangement in the BTK autoinhibited structure also shields the phosphotyrosine binding pocket of the SH2 domain from interactions with other ligands (Figure 3D). The salt bridge between SH2 and kinase domains ties up R307 preventing that side chain from engaging phosphotyrosine ligands. However, compared to the intramolecular phosphotyrosine/SH2 interaction present in autoinhibited structures of SRC family kinases (Liu et al., 1993), this region of autoinhibited BTK is likely more dynamic and prone to release from the kinase domain. Indeed, crystal structures of both isolated TEC family SH2 domains (Joseph et al., 2012) and the crystal structure of the BTK SH3–SH2-kinase fragment (Wang et al., 2015) show the SH2 domain adopting a domain swapped dimer structure. Whether this mode of dimerization is physiologically relevant is not known. Small angle X-ray scattering (SAXS) data suggested quite early that BTK adopts an extended arrangement of its domains (Márquez et al., 2003), however, the functional state that this extended conformational state reflects is unclear. More recent work has demonstrated that a BTK SH2 specific binding protein abrogates the kinase activity of BTK by blocking a predicted SH2/kinase domain interface required for activation (Duarte et al., 2020; Jeong et al., 2020). The interface between SH2 and kinase domains in TEC family and other tyrosine kinases is known to play a critical role in enhancing kinase activity beyond the isolated kinase domain (Nagar et al., 2003; Joseph et al., 2007; Filippakopoulos et al., 2008; Lamontanara et al., 2014). However, the precise structural features of the interface between the SH2 and kinase domains within active BTK are not yet determined but could eventually provide a route toward targeting BTK to disfavor activating contacts for specific, allosteric inhibition of BTK.
Two distinct autoinhibitory contacts are described in the literature for the BTK PHTH domain. X-ray crystal structures of a tethered BTK PHTH-Kinase construct revealed a docking site for the PHTH domain on the N-lobe of the BTK kinase domain (Figure 4, Pose 1) while solution NMR approaches revealed a different PHTH domain docking site on the C-lobe of the kinase domain (Figure 4, Pose 2). Pose 1 (Figure 4) involves specific contacts between R133 and Y134 in the PHTH domain and the N-lobe of the kinase domain. Importantly, this crystallographically determined structure shows the PHTH domain adopting the “Saraste dimer” (Hyvönen and Saraste, 1997), a structure that has been associated with binding of the BTK PHTH domain to the plasma membrane following production of PIP3 from PIP2 by PI3K (Chung et al., 2019). Molecular dynamics simulations suggest that the BTK PHTH Saraste dimer is stabilized at the membrane by binding multiple PIP3 lipids and mutations that are known to have disease relevance destabilized the dimer interface (Wang et al., 2019). Furthermore, elegant studies using supported lipid bilayers provide evidence that the peripheral site originally identified in the crystal structure of the BTK PH domain bound to IP6 (Wang et al., 2015) stabilizes membrane association (Chung et al., 2019). This requirement for the occupancy of both PIP3 sites for activation of BTK suggests that the PH domain is sensitive to the concentration of PIP3 in the membrane. In a resting cell, due to the broad conformational ensemble of BTK, the PH domain could be sampling the membrane for PIP3 and activation is only triggered when the PIP3 concentration surpasses a certain threshold and both canonical and peripheral sites are occupied, stabilizing dimerization of BTK at the membrane.
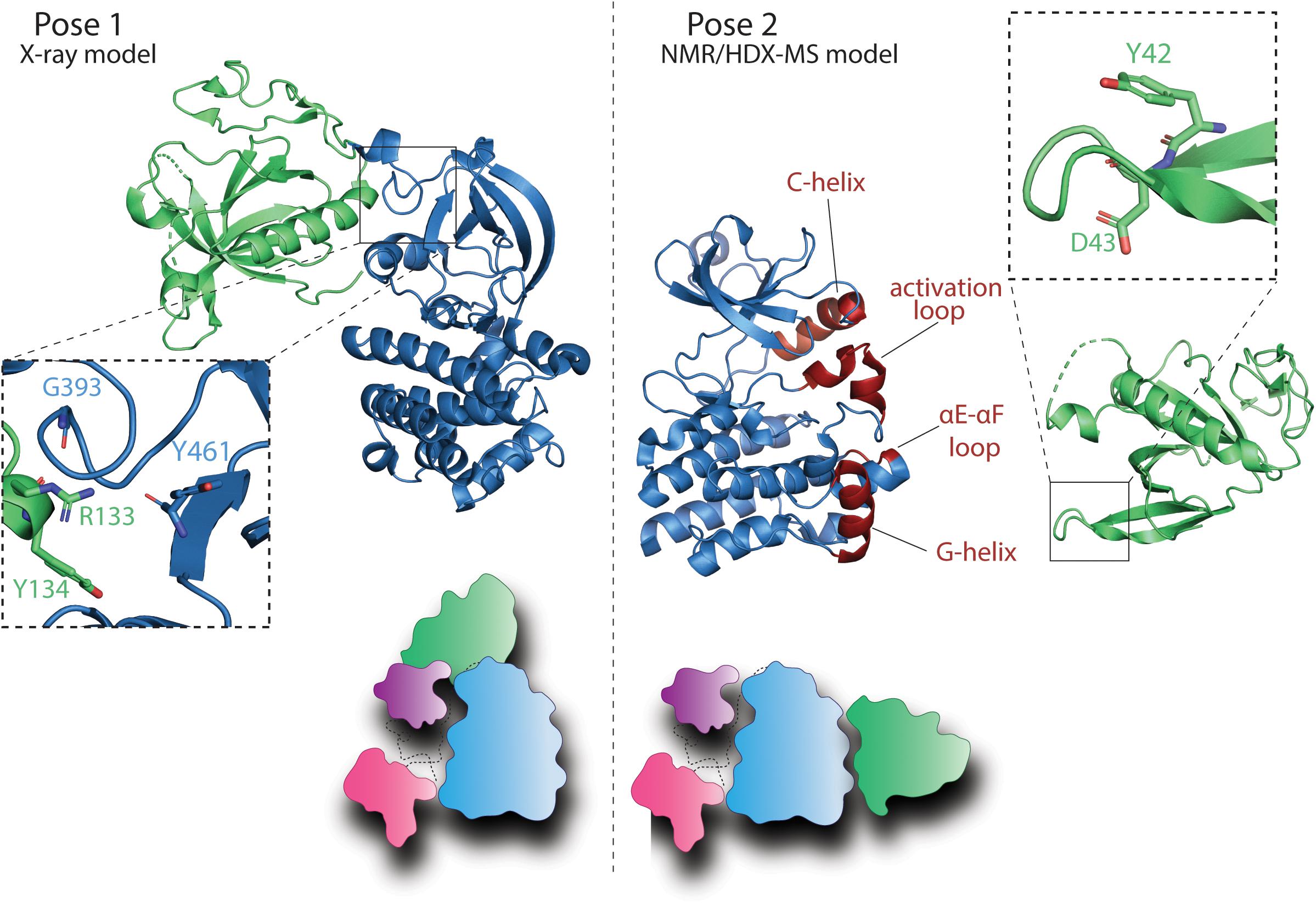
Figure 4. BTK PHTH autoinhibitory interactions. Pose 1: Model derived from the crystal structure of a tethered BTK PHTH-kinase construct (PDB: 4Y93) reveals contacts between the PHTH domain and the kinase domain N-lobe [partially overlapping with the autoinhibitory site of the SH3 domain (Wang et al., 2015)]. Domain colors are as in Figure 3A. The expanded region (dashed box) shows the interaction between R133 and Y134 in the PHTH domain and G393 and Y461 in the kinase domain. The cartoon depiction shows the PHTH/kinase interaction in the context of the autoinhibited SH3–SH2-kinase structure described in Figure 3. Pose 2: Solution NMR, HDX-MS, and biochemical data reveal another autoinhibitory interaction between the PHTH and the activation loop face of the kinase domain (Devkota et al., 2017; Joseph et al., 2017; Amatya et al., 2019). The PHTH domain residues involved in this autoinhibitory pose are Y42 and D43 (dashed box) while the structural elements in the kinase domain reported to mediate this interaction include the αC-helix, the activation loop (A-loop), the αE–αF loop, and the αG-helix (colored in red). The cartoon depiction shows this PHTH/kinase interaction in the context of the autoinhibitory SH3–SH2-kinase structure described in Figure 3.
The BTK PHTH domain in Pose 2 (Figure 4) blocks access of substrate to the kinase domain active site (Amatya et al., 2019) and the residues of the PHTH domain that mediate this autoinhibitory interaction (Y42 and D43) (Devkota et al., 2017; Joseph et al., 2017) are the same residues that mediate the Saraste dimer interface suggesting that this autoinhibitory pose is likely mutually exclusive with the active BTK dimer. The PHTH domain of ITK also blocks the kinase domain active site, impeding access to the activation loop (Devkota et al., 2017). Moreover, phosphatidylinositol binding to the PHTH domain inhibits the interaction between PHTH and kinase domains in solution (Devkota et al., 2017). Interestingly, a similar PH/kinase domain interface has been described for AKT although in this case a small molecule inhibitor seems to promote the domain–domain interaction (Barnett et al., 2005; Calleja et al., 2009; Wu et al., 2010). More recent structural work combining segmental isotopic labeling and NMR spectroscopy has revealed the complexity of the AKT regulatory system (Chu et al., 2018, 2020) and given the domain differences between BTK and AKT it is not yet clear how similar the autoinhibitory role of the shared PH domain will be. Nevertheless, the PH domain of AKT is subject to inhibition by small molecules (Meuillet, 2011; Parikh et al., 2012; Gao et al., 2018; Kang et al., 2018; Yang et al., 2019; Zhang et al., 2020) and therefore provides a template for a similar approach to targeting BTK. Beyond simply targeting the lipid binding function of the BTK PHTH domain (Yoon, 2014), the more mature picture of BTK regulatory interfaces now permits new approaches to target the BTK PHTH/kinase interfaces to allosterically control BTK function.
It is likely that conformational plasticity is the reason the TEC family kinases have to date resisted crystallization in their full-length form. Indeed, solution NMR data for full-length BTK shows multiple resonances for BTK W395 consistent with the protein adopting multiple distinct conformations that are in slow exchange on the NMR timescale (Joseph et al., 2017). Ultimately, more work is needed to generate a full picture of the BTK activation trajectory, but we can consider the “ensemble” of different conformational states that have been characterized to date. A compact, fully autoinhibited model of BTK can be described based on the crystal structure of the SH3–SH2-kinase fragment and the solution work placing the PHTH domain in Pose 2 across the activation loop of the kinase domain (Figure 5A). In this state, each of the regulatory domains are prevented from binding to other target ligands, the hydrophobic stack is fully assembled favoring ADP bound to the active site, and the activation loop is protected from phosphorylation. NMR and biochemical data (Andreotti et al., 1997; Laederach et al., 2002; Joseph et al., 2017) have previously invoked a role for the PRR in binding the SH3 domain and transiently displacing the SH3 domain and likely the SH2 domain from the fully autoinhibited state (Figure 5B). This transient opening of the BTK autoinhibited structure might favor phospholipid association once PIP3 is present at the membrane (Figure 5C). Once membrane associated, BTK will dimerize via the Saraste dimer and membrane association will be further stabilized by PIP3 binding to both the canonical and peripheral sites on the PH domain (Figure 5D; Chung et al., 2019). Upon membrane dimerization it is likely that further rearrangement of the SH2 domain to create contacts with the kinase domain N-lobe will stabilize the kinase domain in its active state (Figure 5D). Once activated by phosphorylation of Y551 on the activation loop, BTK is poised to phosphorylate its substrate PLCγ2 (Figure 5E). For the related TEC kinase, ITK, a substrate docking site on the C-lobe of the kinase domain has been described (Xie et al., 2013) and it is possible that the BTK/PLCγ2 enzyme/substrate pair shares a similar mechanism to achieve substrate specificity.
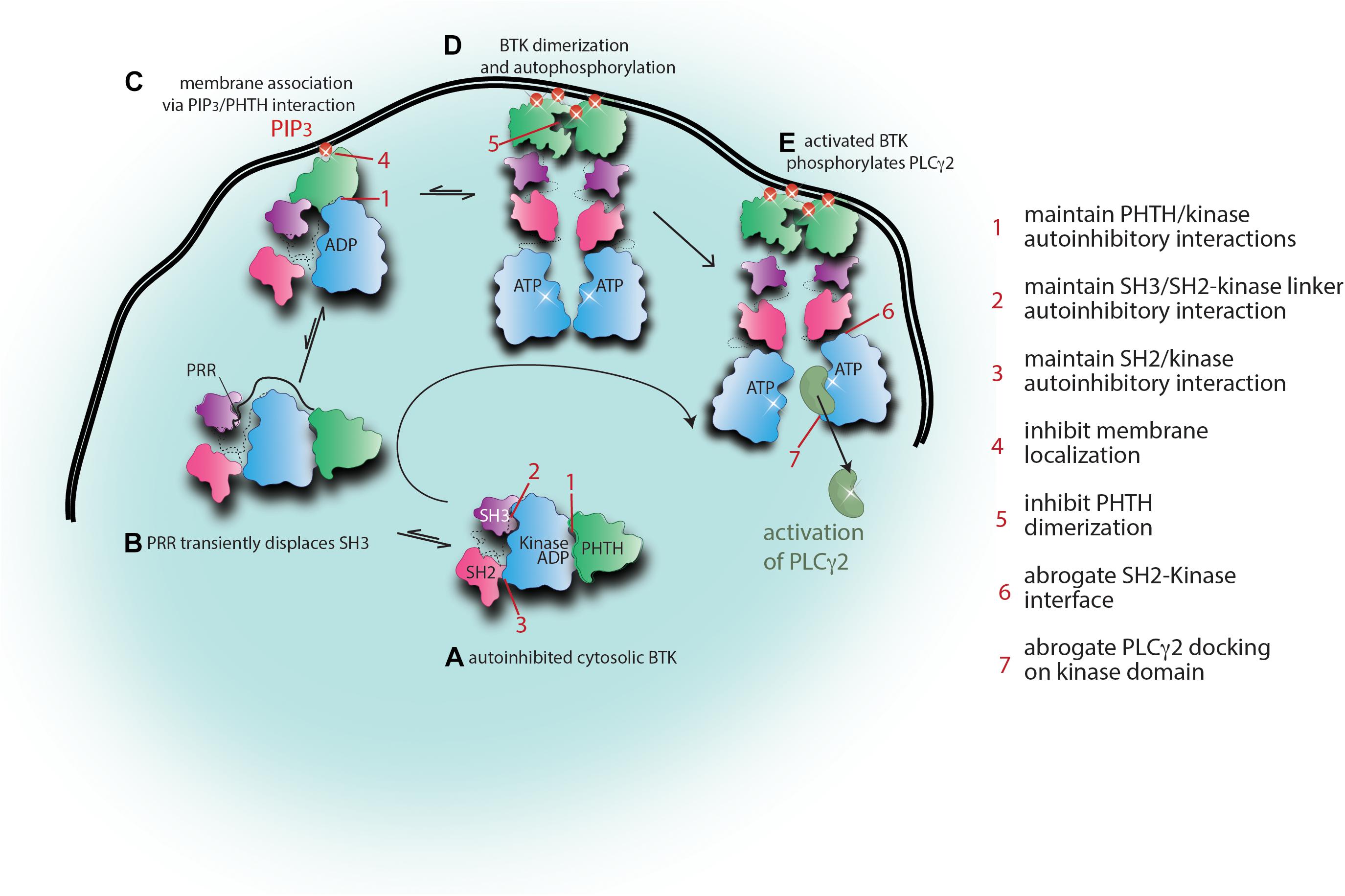
Figure 5. Alternate approaches to targeting BTK. Snapshots of the activation scheme for BTK provide several areas to target for allosteric therapeutic development. Current knowledge of the BTK activation scheme is summarized here with cartoon representations. The domain colors are as in Figures 3, 4. (A) Briefly, autoinhibited cytosolic BTK adopts a compact domain arrangement with the PHTH docked onto the kinase domain blocking the active site (Pose 2) and the SH3 and SH2 domains adopting a SRC-like conformation. (B) Displacement of the SH3 domain from the distal side of the kinase domain by the proline-rich region (PRR) promotes transient opening of the autoinhibited structure which would promote regulatory domain accessibility to exogenous ligands (including PHTH domain sampling of the PIP3 content in the plasma membrane). (C) Initial binding of the PHTH domain to PIP3 might lead to domain rearrangement consistent with the Pose 1 PHTH/kinase interaction. (D) Once PIP3 levels in the membrane surpass the required threshold, BTK engages PIP3 via the canonical and peripheral binding sites in PHTH and BTK dimerization at the membrane is stabilized. Rearrangement of the regulatory domains ensues, the SH2 domain transitions to its activating position on the N-lobe, the hydrophobic stack is disassembled triggering exchange of ADP for ATP in the kinase domain allowing autophosphorylation. (E) Activated BTK phosphorylates its substrate PLCγ2. Seven areas for potential allosteric therapeutic development are listed. These areas encompass stabilizing autoinhibited BTK (points 1, 2, and 3) and/or disfavoring activating or substrate contacts (points 4, 5, 6, and 7).
The emerging model for BTK conformational transitions and interactions in an activated B cell provides a number of possible targets to develop allosteric approaches to BTK inhibition. As already discussed, the drug resistance that is associated with active site inhibitors such as Ibrutinib may be solved by combinations of Ibrutinib and small molecules that act at separate locations on the BTK protein. In particular, resistance mutations remote from the active site such as BTK T316A might be especially vulnerable to combination therapies that include an allosteric inhibitor that compensates for the mutation induced shift toward the active state. Taking a broader view of potential allosteric inhibitors, Figure 5 suggests seven distinct sites that might prove “druggable” in future work. The most compact autoinhibited conformation of BTK may be stabilized by small molecules that target the PHTH/kinase interface (1), the SH3/SH2-kinase linker contact (2), and/or the SH2/kinase interaction that in the native autoinhibited structure is weakly held by a single salt bridge (3). Targeting the membrane bound state of BTK, small molecules that prevent PHTH/PIP3 engagement (4) or PHTH dimerization (5) might prove useful in truncating BTK signaling. Interfering with the activating interaction between the BTK SH2 and kinase domains (6) and/or preventing the PLCγ2 substrate from docking onto the BTK kinase domain (7) might provide yet more avenues toward inhibiting BTK function. As additional structural details and mechanistic insights into BTK regulation continue to emerge the list of potential allosteric target sites will increase.
Due to challenges associated with co-crystallization of drug bound multidomain kinases, the majority of structural work on kinase inhibitor binding tends to be limited to the local effects of the inhibitor on the regulatory features within the kinase domain itself (αC-helix, DFG motif, and activation loop). For example, one study of BTK active site inhibitors found that different small molecule inhibitors differentially sequester the activation loop Y551 and inhibitors that do not sequester this residue exhibit reduced inhibition against Fcε receptor signaling compared to BCR signaling (Bender et al., 2017). Understanding precisely how a given inhibitor affects different BTK mediated signaling pathways is extremely important in a clinical setting and this level of understanding must extend beyond the kinase domain. An increasing volume of work is being published on understanding the molecular level influence of small molecule inhibitors on full-length kinases (Skora et al., 2013; Tong et al., 2017; Chakraborty et al., 2019; Fang et al., 2020). Detailed evaluation of a panel of BTK inhibitors has demonstrated that different active site inhibitors exert a range of distinct dynamic and conformational effects on the remote non-catalytic regulatory domains (Joseph et al., 2020). Ibrutinib proves the most interesting case as covalent binding of Ibrutinib to the BTK active site promotes release of both the SH3 and SH2 domains, as well as the SH2-kinase linker, from their autoinhibitory poses. This is in contrast to the other active site inhibitors; neither CC-292 (also covalent), GDC-0853, nor CGI1764 (both non-covalent) significantly alter the conformational ensemble of the full-length BTK autoinhibitory conformation. The disruption of the BTK autoinhibitory conformation upon Ibrutinib binding makes the BTK regulatory domains available for interaction with upstream and downstream ligands which could promote BTK kinase independent function and/or have dominant negative effects. In fact, in the context of treating B cell lymphomas, PLCγ2 ibrutinib resistant variants are hyper-sensitive to activation regardless of BTK’s kinase activity suggesting that the kinase-independent functions of BTK might be responsible (Wist et al., 2020). Blocking BTK kinase activity alone therefore may not be sufficient in the successful treatment of disease states. Future development of BTK inhibitors and the selection of BTK inhibitor used to treat disease states will need to carefully consider the impact the inhibitor has on the conformation of the full-length protein.
This review has focused on current approaches to BTK inhibition in the clinic, the state-of-the-art knowledge surrounding BTK regulation at the molecular level, and how the FDA approved BTK inhibitor Ibrutinib induces long range structural effects on BTK that might affect drug efficacy in certain contexts. Targeting BTK at allosteric sites could provide better specificity but perhaps more importantly might counteract the conformational influence of resistance mutations or even binding of Ibrutinib and second-generation inhibitors related in structure to Ibrutinib. Targeting both an orthosteric and allosteric site within the same kinase has been achieved for other systems (Eide et al., 2019; To et al., 2019). Furthermore, an explicit conformational connection between an allosteric and active site of a kinase has been shown for phosphoinositide-dependent kinase-1 (PDK1). In that work, HDX-MS data show that binding of ATP destabilized the allosteric PIFtide pocket, making that site more amenable to PIFtide binding (Ghode et al., 2020). In another kinase, cyclin-dependent kinase-2 (CDK2), there is demonstrated positive cooperativity in allosteric inhibitor binding when certain orthosteric inhibitors are present in the ATP site that enhance the allosteric inhibitor’s affinity (Faber et al., 2020). Following these examples, it is intriguing to consider whether the conformational consequences of Ibrutinib binding to BTK might create new allosteric target sites that can be exploited to ultimately gain complete control over BTK function in the catalytic and regulatory domains. As work progresses it will be important to consider how candidate allosteric inhibitors against BTK affect active site structure and dynamics.
Multidomain kinases present many challenges to the full structural characterization needed to elucidate regulatory mechanisms and the effects of inhibitor binding. Multidisciplinary approaches spanning crystallography to solution methods including NMR and HDX-MS to cellular assays are pushing the field forward and as a result we are becoming better equipped to understand and combat disease inducing mutations and drug resistance. It will be exciting to witness the impact of fundamental biophysical characterization on kinase inhibitor development and the use of these inhibitors in a clinical setting.
LK wrote the manuscript and made the figures. AA and RJ edited the manuscript and contributed to some of the writing. All authors contributed to the article and approved the submitted version.
The Roy J. Carver Charitable Trust, Muscatine, Iowa has provided continued research support in the form of an endowed position for the PI. In addition, the National Institute for Allergy and Infectious Diseases at the National Institutes of Health has provided uninterrupted research support for the laboratory since 1999.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as apotential conflict of interest.
We would like to express their gratitude to the Roy J. Carver Charitable Trust, Muscatine, Iowa for continued research support. In addition, we acknowledge the National Institute for Allergy and Infectious Diseases at the National Institutes of Health for support (1R01 AI43957). Finally, we would also like to thank our collaborators John Engen, Thomas Wales, and Leslie Berg for ongoing and significant research contributions and stimulating conversations about BTK and the TEC kinases.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.655489/full#supplementary-material
BCAP, B cell adaptor protein for PI3K; BTK, Bruton’s tyrosine kinase; CDK2, cyclin dependent kinase-2; cGVHD, chronic graft versus host disease; CLL, chronic lymphocytic leukemia; DAG, diacylglycerol; DLBCL, diffuse large B-cell lymphoma; EGFR, epidermal growth factor receptor; HDX-MS, hydrogen/deuterium exchange mass spectrometry; IP3, inositol 1,4,5-trisphosphate; MCL, mantle cell lymphoma; MS, multiple sclerosis; MZL, marginal zone lymphoma; NMR, nuclear magnetic resonance; PDB, Protein Data Bank; PDK1, phosphoinositide-dependent-kinase-1; PH, Pleckstrin homology; PHTH, Pleckstrin homology-Tec homology; PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol (4,5)-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PKC, protein kinase C; PLC γ 2, phospholipase C γ 2; PROTAC, proteolysis targeting chimera; RA, rheumatoid arthritis; SAR, structure activity relationship; SH2, Src homology 2; SH3, Src homology 3; SLE, systemic lupus erythematosus; SLL, small lymphocytic leukemia; SLP-65, SH2 domain-containing leukocyte protein of 65 kDa; SYK, spleen tyrosine kinase; TH, Tec homology; WM, Waldenström’s macroglobulinemia.
Advani, R. H., Buggy, J. J., Sharman, J. P., Smith, S. M., Boyd, T. E., Grant, B., et al. (2013). Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 31, 88–94. doi: 10.1200/JCO.2012.42.7906
Amatya, N., Wales, T. E., Kwon, A., Yeung, W., Joseph, R. E., Bruce Fulton, D., et al. (2019). Lipid-targeting pleckstrin homology domain turns its autoinhibitory face toward the TEC kinases. Proc. Natl. Acad. Sci. U.S.A. 116, 21539–21544. doi: 10.1073/pnas.1907566116
Andreotti, A. H., Bunnell, S. C., Feng, S., Berg, L. J., and Schreiber, S. L. (1997). Regulatory intramolecular association in a tyrosine kinase of the tec family. Nature 385, 93–97. doi: 10.1038/385093a0
Barf, T., Covey, T., Izumi, R., Van De Kar, B., Gulrajani, M., Van Lith, B., et al. (2017). Acalabrutinib (ACP-196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J. Pharmacol. Exp. Ther. 363, 240–252. doi: 10.1124/jpet.117.242909
Barnett, S. F., Defeo-Jones, D., Fu, S., Hancock, P. J., Haskell, K. M., Jones, R. E., et al. (2005). Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem. J. 385, 399–408. doi: 10.1042/BJ20041140
Bender, A. T., Gardberg, A., Pereira, A., Johnson, T., Wu, Y., Grenningloh, R., et al. (2017). Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B-cell receptor signaling. Mol. Pharmacol. 91, 208–219. doi: 10.1124/mol.116.107037
Buhimschi, A. D., Armstrong, H. A., Toure, M., Jaime-Figueroa, S., Chen, T. L., Lehman, A. M., et al. (2018). Targeting the C481S ibrutinib-resistance mutation in bruton’s tyrosine kinase using PROTAC-Mediated degradation. Biochemistry 57, 3564–3575. doi: 10.1021/acs.biochem.8b00391
Byrd, J. C., Harrington, B., O’Brien, S., Jones, J. A., Schuh, A., Devereux, S., et al. (2015). Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 374, 323–332. doi: 10.1056/NEJMoa1509981
Byrd, J. C., Smith, S., Wagner-Johnston, N., Sharman, J., Chen, A. I., Advani, R., et al. (2019). Correction: first-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL (Oncotarget(2018) 9 (13023 -13035)) Oncotarget 10, 3827–3830. doi: 10.18632/oncotarget.24310
Caldwell, R. D., Qiu, H., Askew, B. C., Bender, A. T., Brugger, N., Camps, M., et al. (2019). Discovery of evobrutinib: an oral, potent, and highly selective, covalent bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J. Med. Chem. 62, 7643–7655. doi: 10.1021/acs.jmedchem.9b00794
Calleja, V., Laguerre, M., Parker, P. J., and Larijani, B. (2009). Role of a novel PH-kinase domain interface in PKB/Akt regulation: Structural mechanism for allosteric inhibition. PLoS Biol. 7:1000017. doi: 10.1371/journal.pbio.1000017
Chakraborty, S., Inukai, T., Fang, L., Golkowski, M., and Maly, D. J. (2019). Targeting dynamic ATP-binding site features allows discrimination between highly homologous protein kinases. ACS Chem. Biol. 14, 1249–1259. doi: 10.1021/acschembio.9b00214
Cheng, S., Guo, A., Lu, P., Ma, J., Coleman, M., and Wang, Y. L. (2015). Functional characterization of BTK C481S mutation that confers ibrutinib resistance: exploration of alternative kinase inhibitors. Leukemia 29, 895–900. doi: 10.1038/leu.2014.263
Chiron, D., Di Liberto, M., Martin, P., Huang, X., Sharman, J., Blecua, P., et al. (2014). Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 4, 1022L–1035. doi: 10.1158/2159-8290.CD-14-0098
Chong, E. A., Roeker, L. E., Shadman, M., Davids, M. S., Schuster, S. J., and Mato, A. R. (2020). BTK inhibitors in cancer patients with COVID-19: “The Winner Will be the One Who Controls That Chaos” (Napoleon Bonaparte). Clin. Cancer Res. 26, 3514–3516. doi: 10.1158/1078-0432.CCR-20-1427
Chu, N., Salguero, A. L., Liu, A. Z., Chen, Z., Dempsey, D. R., Ficarro, S. B., et al. (2018). Akt Kinase activation mechanisms revealed using protein semisynthesis. Cell 174, 897–907. doi: 10.1016/j.cell.2018.07.003.Akt
Chu, N., Viennet, T., Bae, H., Salguero, A., Boeszoermenyi, A., Arthanari, H., et al. (2020). The structural determinants of ph domain-mediated regulation of akt revealed by segmental labeling. Elife 9, 1–23. doi: 10.7554/ELIFE.59151
Chung, J. K., Nocka, L. M., Decker, A., Wang, Q., Kadlecek, T. A., Weiss, A., et al. (2019). Switch-like activation of Bruton’s tyrosine kinase by membrane-mediated dimerization. Proc. Natl. Acad. Sci. U.S.A. 166, 10798–10803. doi: 10.1073/pnas.1819309116
Corneth, O. B. J., Verstappen, G. M. P., Paulissen, S. M. J., de Bruijn, M. J. W., Rip, J., Lukkes, M., et al. (2017). Enhanced bruton’s tyrosine kinase activity in peripheral blood B lymphocytes from patients with autoimmune disease. Arthritis Rheumatol. 69, 1313–1324. doi: 10.1002/art.40059
Crawford, J. J., Johnson, A. R., Misner, D. L., Belmont, L. D., Castanedo, G., Choy, R., et al. (2018). Discovery of GDC-0853: a potent, selective, and noncovalent bruton’s tyrosine kinase inhibitor in early clinical development. J. Med. Chem. 61, 2227–2245. doi: 10.1021/acs.jmedchem.7b01712
Davids, M. S., and Brown, J. R. (2014). Ibrutinib: a first in class covalent inhibitor of Bruton’s tyrosine kinase. Future Oncol. 10, 957–967. doi: 10.2217/fon.14.51
Davis, R. E., Ngo, V. N., Lenz, G., Tolar, P., Young, R. M., Romesser, P. B., et al. (2010). Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92. doi: 10.1038/nature08638
Devkota, S., Joseph, R. E., Boyken, S. E., Fulton, D. B., and Andreotti, A. H. (2017). An autoinhibitory role for the pleckstrin homology domain of interleukin-2-inducible tyrosine kinase and its interplay with canonical phospholipid recognition. Biochemistry 56, 2938–2949. doi: 10.1021/acs.biochem.6b01182
Di Paolo, J. A., Huang, T., Balazs, M., Barbosa, J., Barck, K. H., Bravo, B. J., et al. (2011). Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat. Chem. Biol. 7, 41–50. doi: 10.1038/nchembio.481
Dobrovolsky, D., Wang, E. S., Morrow, S., Leahy, C., Faust, T., Nowak, R. P., et al. (2019). Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 133, 952–961. doi: 10.1182/blood-2018-07-862953
Duarte, D. P., Lamontanara, A. J., La Sala, G., Jeong, S., Sohn, Y. K., Panjkovich, A., et al. (2020). Btk SH2-kinase interface is critical for allosteric kinase activation and its targeting inhibits B-cell neoplasms. Nat. Commun. 11:16128. doi: 10.1038/s41467-020-16128-5
Dubovsky, J. A., Beckwith, K. A., Natarajan, G., Woyach, J. A., Jaglowski, S., Zhong, Y., et al. (2013). Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 122, 2539–2549. doi: 10.1182/blood-2013-06-507947
Eide, C. A., Zabriskie, M. S., Savage Stevens, S. L., Antelope, O., Vellore, N. A., Than, H., et al. (2019). Combining the allosteric inhibitor asciminib with ponatinib suppresses emergence of and restores efficacy against highly resistant BCR-ABL1 mutants. Cancer Cell 36, 431–443.e5. doi: 10.1016/j.ccell.2019.08.004
Evans, E. K., Tester, R., Aslanian, S., Karp, R., Sheets, M., Labenski, M. T., et al. (2013). Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J. Pharmacol. Exp. Ther. 346, 219–228. doi: 10.1124/jpet.113.203489
Faber, E. B., Tian, D., Burban, D., Levinson, N. M., Hawkinson, J. E., and Georg, G. I. (2020). Cooperativity between Orthosteric Inhibitors and Allosteric Inhibitor 8-Anilino-1-Naphthalene Sulfonic Acid (ANS) in Cyclin-Dependent Kinase 2. ACS Chem. Biol. 15, 1759–1764. doi: 10.1021/acschembio.0c00169
Fang, L., Vilas-Boas, J., Chakraborty, S., Potter, Z. E., Register, A. C., Seeliger, M. A., et al. (2020). How ATP-competitive inhibitors allosterically modulate tyrosine kinases that contain a Src-like regulatory architecture. ACS Chem. Biol. 15, 2005–2016. doi: 10.1021/acschembio.0c00429
Filippakopoulos, P., Kofler, M., Hantschel, O., Gish, G. D., Grebien, F., Salah, E., et al. (2008). Structural coupling of SH2-kinase domains links fes and Abl substrate recognition and kinase activation. Cell 134, 793–803. doi: 10.1016/j.cell.2008.07.047
Francesco, M. R., Wong, M., LaStant, J., Finkle, D., Loewenstein, N., Macsata, R., et al. (2017). PRN2246, a potent and selective blood brain barrier penetrating BTK inhibitor, exhibits efficacy in central nervous system immunity. Mult. Scler. J. 23:P989. doi: 10.1177/1352458517731406
Gao, J., He, X., Ma, Y., Zhao, X., Hou, X., Hao, E., et al. (2018). Chlorogenic acid targeting of the AKT PH domain activates AKT/GSK3β/FOXO1 signaling and improves glucose metabolism. Nutrients 10:1366. doi: 10.3390/nu10101366
Geahlen, R. L. (2009). Syk and pTyr’d: Signaling through the B cell antigen receptor. Biochim. Biophys. Acta 1793, 1115–1127. doi: 10.1016/j.bbamcr.2009.03.004
Ghode, A., Gross, L. Z. F., Tee, W. V., Guarnera, E., Berezovsky, I. N., Biondi, R. M., et al. (2020). Synergistic allostery in multiligand-protein interactions. Biophys. J. 119, 1833–1848. doi: 10.1016/j.bpj.2020.09.019
Girard, J., Reneau, J., Devata, S., Wilcox, R. A., Kaminski, M. S., Mercer, J., et al. (2019). Evaluating acalabrutinib in the treatment of mantle cell lymphoma: design, development, and place in therapy. Onco. Targets. Ther. 12, 8003–8014. doi: 10.2147/OTT.S155778
Guo, Y., Liu, Y., Hu, N., Yu, D., Zhou, C., Shi, G., et al. (2019). Discovery of zanubrutinib (BGB-3111), a novel, potent, and selective covalent inhibitor of bruton’s tyrosine kinase. J. Med. Chem. 62, 7923–7940. doi: 10.1021/acs.jmedchem.9b00687
Harrington, B. K., Gardner, H. L., Izumi, R., Hamdy, A., Rothbaum, W., Coombes, K. R., et al. (2016). Preclinical evaluation of the novel BTK inhibitor acalabrutinib in canine models of B-cell non-hodgkin lymphoma. PLoS One 11:e0159607. doi: 10.1371/journal.pone.0159607
Herishanu, Y., Pérez-Galán, P., Liu, D., Biancotto, A., Pittaluga, S., Vire, B., et al. (2011). The lymph node microenvironment promotes B-cell receptor signaling, NF-κB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 117, 563–574. doi: 10.1182/blood-2010-05-284984
Herter, J. M., Margraf, A., Volmering, S., Correia, B. E., Bradshaw, J. M., Bisconte, A., et al. (2018). PRN473, an inhibitor of Bruton’s tyrosine kinase, inhibits neutrophil recruitment via inhibition of macrophage antigen-1 signalling. Br. J. Pharmacol. 175, 429–439. doi: 10.1111/bph.14090
Heukels, P., van Hulst, J. A. C., van Nimwegen, M., Boorsma, C. E., Melgert, B. N., von der Thusen, J. H., et al. (2019). Enhanced Bruton’s tyrosine kinase in B-cells and autoreactive IgA in patients with idiopathic pulmonary fibrosis. Respir. Res. 20:232. doi: 10.1186/s12931-019-1195-7
Hill, R. J., Bradshaw, J. M., Bisconte, A., Tam, D., Owens, T. D., Brameld, K. A., et al. (2015). THU0068 preclinical characterization of PRN1008, a novel reversible covalent inhibitor of BTK that shows efficacy in a RAT model of collagen-induced arthritis. Ann. Rheum. Dis. 74, 216L–217. doi: 10.1136/annrheumdis-2015-eular.3641
Honigberg, L. A., Smith, A. M., Sirisawad, M., Verner, E., Loury, D., Chang, B., et al. (2010). The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. U.S.A. 107, 13075–13080. doi: 10.1073/pnas.1004594107
Hyvönen, M., and Saraste, M. (1997). Stucture of the PH domain and Btk motif from Bruton’s tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia. EMBO J. 16, 3396–3404. doi: 10.1093/emboj/16.12.3396
Jeong, S., Sohn, Y. K., Choi, Y., Park, J., and Kim, H. S. (2020). A regulatory SH2 domain-targeting protein binder effectively inhibits the activity of Bruton’s tyrosine kinase and its drug-resistant variants. Biochem. Biophys. Res. Commun. 526, 8–13. doi: 10.1016/j.bbrc.2020.03.006
Johnson, A. R., Kohli, P. B., Katewa, A., Gogol, E., Belmont, L. D., Choy, R., et al. (2016). Battling Btk mutants with noncovalent inhibitors that overcome Cys481 and Thr474 mutations. ACS Chem. Biol. 11, 2897–2907. doi: 10.1021/acschembio.6b00480
Joseph, R. E., Amatya, N., Fulton, D. B., Engen, J. R., Wales, T. E., and Andreotti, A. H. (2020). Differential impact of BTK active site inhibitors on the conformational state of full-length BTK. Elife 9, 1–57. doi: 10.7554/eLife.60470
Joseph, R. E., Ginder, N. D., Hoy, J. A., Nix, J. C., Fulton, D. B., Honzatko, R. B., et al. (2012). Structure of the interleukin-2 tyrosine kinase Src homology 2 domain; Comparison between X-ray and NMR-derived structures. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 145–153. doi: 10.1107/S1744309111049761
Joseph, R. E., Kleino, I., Wales, T. E., Xie, Q., Fulton, D. B., Engen, J. R., et al. (2013). Activation loop dynamics determine the different catalytic efficiencies of B cell- and T cell-specific tec kinases. Sci. Signal. 6, 1–13. doi: 10.1126/scisignal.2004298
Joseph, R. E., Min, L., and Andreotti, A. H. (2007). The linker between SH2 and kinase domains positively regulates catalysis of the Tec family kinases. Biochemistry 46, 5455–5462. doi: 10.1021/bi602512e
Joseph, R. E., Wales, T. E., Fulton, D. B., Engen, J. R., and Andreotti, A. H. (2017). Achieving a graded immune response: BTK adopts a range of active/inactive conformations dictated by multiple interdomain contacts. Structure 25, 1481–1494.e4. doi: 10.1016/j.str.2017.07.014
Joseph, R. E., Xie, Q., and Andreotti, A. H. (2010). Identification of an allosteric signaling network within tec family kinases. J. Mol. Biol. 403, 231–242. doi: 10.1016/j.jmb.2010.08.035
Kadri, S., Lee, J., Fitzpatrick, C., Galanina, N., Sukhanova, M., Venkataraman, G., et al. (2017). Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 1, 715–727. doi: 10.1182/bloodadvances.2016003632
Kanagal-Shamanna, R., Jain, P., Patel, K. P., Routbort, M., Bueso-Ramos, C., Alhalouli, T., et al. (2019). Targeted multigene deep sequencing of Bruton tyrosine kinase inhibitor–resistant chronic lymphocytic leukemia with disease progression and Richter transformation. Cancer 125, 559–574. doi: 10.1002/cncr.31831
Kang, Y., Jang, G., Ahn, S., Lee, Y., Shin, S. Y., and Yoon, Y. (2018). Regulation of AKT activity by inhibition of the pleckstrin homology domain-ptdins(3,4,5)P3 interaction using flavonoidss. J. Microbiol. Biotechnol. 28, 1401–1411. doi: 10.4014/jmb.1804.04051
Kaptein, A., de Bruin, G., Emmelot-van Hoek, M., van de Kar, B., de Jong, A., Gulrajani, M., et al. (2018). Potency and selectivity of BTK inhibitors in clinical development for B-cell malignancies. Blood 132:1871. doi: 10.1182/blood-2018-99-109973
Kersseboom, R., Kil, L., Flierman, R., van der Zee, M., Dingjan, G. M., Middendorp, S., et al. (2010). Constitutive activation of Bruton’s tyrosine kinase induces the formation of autoreactive IgM plasma cells. Eur. J. Immunol. 40, 2643–2654. doi: 10.1002/eji.201040521
Kil, L. P., De Bruijn, M. J. W., Van Nimwegen, M., Corneth, O. B. J., Van Hamburg, J. P., Dingjan, G. M., et al. (2012). Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood 119, 3744–3756. doi: 10.1182/blood-2011-12-397919
Kurosaki, T. (2011). Regulation of BCR signaling. Mol. Immunol. 48, 1287–1291. doi: 10.1016/j.molimm.2010.12.007
Kuter, D. J., Efraim, M., Mayer, J., McDonald, V., Bird, R. J., Regenbogen, T., et al. (2020). Oral rilzabrutinib, a bruton tyrosine kinase inhibitor, showed clinically active and durable platelet responses and was well-tolerated in patients with heavily pretreated immune thrombocytopenia. Blood 136, 13–14. doi: 10.1182/blood-2020-134932
Laederach, A., Cradic, K. W., Brazin, K. N., Zamoon, J., Fulton, D. B., Huang, X.-Y., et al. (2002). Competing modes of self-association in the regulatory domains of Bruton’s tyrosine kinase: Intramolecular contact versus asymmetric homodimerization. Protein Sci. 11, 36–57. doi: 10.1110/ps.26702
Lamontanara, A. J., Georgeon, S., Tria, G., Svergun, D. I., and Hantschel, O. (2014). The SH2 domain of Abl kinases regulates kinase autophosphorylation by controlling activation loop accessibility. Nat. Commun. 5:6470. doi: 10.1038/ncomms6470
Li, Z., Wahl, M. I., Eguinoa, A., Stephens, L. R., Hawkins, P. T., and Witte, O. N. (1997). Phosphatidylinositol 3-kinase-γ activates Bruton’s tyrosine kinase in concert with Src family kinases. Proc. Natl. Acad. Sci. U.S.A. 94, 13820–13825. doi: 10.1073/pnas.94.25.13820
Liclican, A., Serafini, L., Xing, W., Czerwieniec, G., Steiner, B., Wang, T., et al. (2020). Biochemical characterization of tirabrutinib and other irreversible inhibitors of Bruton’s tyrosine kinase reveals differences in on - and off - target inhibition. Biochim. Biophys. Acta Gen. Subj. 1864:129531. doi: 10.1016/j.bbagen.2020.129531
Lin, A. Y., Cuttica, M. J., Ison, M. G., and Gordon, L. I. (2020). Ibrutinib for chronic lymphocytic leukemia in the setting of respiratory failure from severe COVID-19 infection: case report and literature review. eJHaem 1, 596–600. doi: 10.1002/jha2.98
Liu, X., Brodeur, S. R., Gish, G., Songyang, Z., Cantley, L. C., Laudano, A. P., et al. (1993). Regulation of c-Src tyrosine kinase activity by the Src SH2 domain. Oncogene 8, 1119–1126.
Lou, Y., Owens, T. D., Kuglstatter, A., Kondru, R. K., and Goldstein, D. M. (2012). Bruton’s tyrosine kinase inhibitors: approaches to potent and selective inhibition, preclinical and clinical evaluation for inflammatory diseases and B cell malignancies. J. Med. Chem. 55, 4539–4550. doi: 10.1021/jm300035p
Maddocks, K. J., Ruppert, A. S., Lozanski, G., Heerema, N. A., Zhao, W., Abruzzo, L., et al. (2015). Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 1, 80–87. doi: 10.1001/jamaoncol.2014.218
Márquez, J. A., Smith, C. I. E., Petoukhov, M. V., Surdo, P., Lo Mattsson, P. T., Knekt, M., et al. (2003). Conformation of full-length Bruton tyrosine kinase (Btk) from synchrotron X-ray solution scattering. EMBO J. 22, 4616–4624. doi: 10.1093/emboj/cdg448
Meuillet, E. J. (2011). Novel inhibitors of AKT: assessment of a different approach targeting the pleckstrin homology domain. Curr. Med. Chem. 18, 2727–2742. doi: 10.2174/092986711796011292
Montalban, X., Arnold, D. L., Weber, M. S., Staikov, I., Piasecka-Stryczynska, K., Willmer, J., et al. (2019). Placebo-controlled trial of an Oral BTK inhibitor in multiple sclerosis. N. Engl. J. Med. 380, 2406–2417. doi: 10.1056/nejmoa1901981
Nagar, B., Hantschel, O., Young, M. A., Scheffzek, K., Veach, D., Bornmann, W., et al. (2003). Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 112, 859–871. doi: 10.1016/S0092-8674(03)00194-6
Nicolson, P. L. R., Welsh, J. D., Chauhan, A., Thomas, M. R., Kahn, M. L., and Watson, S. P. (2020). A rationale for blocking thromboinflammation in COVID-19 with Btk inhibitors. Platelets 31, 685–690. doi: 10.1080/09537104.2020.1775189
Niemann, C. U., Mora-Jensen, H. I., Dadashian, E. L., Krantz, F., Covey, T., Chen, S.-S., et al. (2017). Combined BTK and PI3Kδ Inhibition with Acalabrutinib and ACP-319 Improves Survival and Tumor Control in CLL Mouse Model. Clin. Cancer Res. 23, 5814L–5823. doi: 10.1158/1078-0432.CCR-17-0650
Okada, T., Maeda, A., Iwamatsu, A., Gotoh, K., and Kurosaki, T. (2000). BCAP: the tyrosine kinase substrate that connects B cell receptor to phosphoinositide 3-kinase activation. Immunity 13, 817–827. doi: 10.1016/s1074-7613(00)00079-0
Pal Singh, S., Dammeijer, F., and Hendriks, R. W. (2018). Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 17, 1–23. doi: 10.1186/s12943-018-0779-z
Pan, Z., Scheerens, H., Li, S. J., Schultz, B. E., Sprengeler, P. A., Burrill, L. C., et al. (2007). Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2, 58–61. doi: 10.1002/cmdc.200600221
Parikh, C., Janakiraman, V., Wu, W.-I., Foo, C. K., Kljavin, N. M., Chaudhuri, S., et al. (2012). Disruption of PH–kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc. Natl. Acad. Sci. U.S.A. 109, 19368L–19373. doi: 10.1073/pnas.1204384109
Quinquenel, A., Fornecker, L. M., Letestu, R., Ysebaert, L., Fleury, C., Lazarian, G., et al. (2019). Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: A FILO group study. Blood 134, 641–644. doi: 10.1182/blood.2019000854
Reiff, S. D., Muhowski, E. M., Guinn, D., Lehman, A., Fabian, C. A., Cheney, C., et al. (2018). Noncovalent inhibition of C481S Bruton tyrosine kinase by GDC-0853: A new treatment strategy for ibrutinib-resistant CLL. Blood 132, 1039–1049. doi: 10.1182/blood-2017-10-809020
Robbins, D. W., Kelly, A., Tan, M., McIntosh, J., Wu, J., Konst, Z., et al. (2020). Nx-2127, a Degrader of BTK and IMiD Neosubstrates, for the Treatment of B-Cell Malignancies. Blood 136:34. doi: 10.1182/blood-2020-141461
Roschewski, M., Lionakis, M. S., Sharman, J. P., Roswarski, J., Goy, A., Monticelli, M. A., et al. (2020). Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci. Immunol. 5:110. doi: 10.1126/SCIIMMUNOL.ABD0110
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 98, 8554–8559. doi: 10.1073/pnas.141230798
Saleh, T., Rossi, P., and Kalodimos, C. G. (2017). Atomic view of the energy landscape in the allosteric regulation of Abl kinase. Nat. Struct. Mol. Biol. 24, 893–901. doi: 10.1038/nsmb.3470
Satterthwaite, A. B. (2018). Bruton’s tyrosine kinase, a component of b cell signaling pathways, has multiple roles in the pathogenesis of lupus. Front. Immunol. 8:1986. doi: 10.3389/fimmu.2017.01986
Schnute, M. E., Benoit, S. E., Buchler, I. P., Caspers, N., Grapperhaus, M. L., Han, S., et al. (2019). Aminopyrazole carboxamide bruton’s tyrosine kinase inhibitors. irreversible to reversible covalent reactive group tuning. ACS Med. Chem. Lett. 10, 80–85. doi: 10.1021/acsmedchemlett.8b00461
Serafimova, I. M., Pufall, M. A., Krishnan, S., Duda, K., Cohen, M. S., Maglathlin, R. L., et al. (2012). Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 8, 471–476. doi: 10.1038/nchembio.925
Shah, N. H., Amacher, J. F., Nocka, L. M., and Kuriyan, J. (2018). The Src module: an ancient scaffold in the evolution of cytoplasmic tyrosine kinases. Crit. Rev. Biochem. Mol. Biol. 53, 535–563. doi: 10.1080/10409238.2018.1495173
Sharma, S., Galanina, N., Guo, A., Lee, J., Kadri, S., Van Slambrouck, C., et al. (2016). Identification of a structurally novel BTK mutation that drives ibrutinib resistance in CLL. Oncotarget 7, 68833–68841. doi: 10.18632/ONCOTARGET.11932
Shatzel, J. J., Olson, S. R., Tao, D. L., McCarty, O. J. T., Danilov, A. V., and DeLoughery, T. G. (2017). Ibrutinib-associated bleeding: pathogenesis, management and risk reduction strategies. J. Thromb. Haemost. 15, 835–847. doi: 10.1111/jth.13651
Siess, W., Hundelshausen, P., and Von Lorenz, R. (2020). Selective inhibition of thromboinflammation in COVID-19 by Btk inhibitors. Platelets 31, 989–992. doi: 10.1080/09537104.2020.1809647
Skora, L., Mestan, J., Fabbro, D., Jahnke, W., and Grzesiek, S. (2013). NMR reveals the allosteric opening and closing of Abelson tyrosine kinase by ATP-site and myristoyl pocket inhibitors. Proc. Natl. Acad. Sci. U.S.A. 110, E4437L–E4445. doi: 10.1073/pnas.1314712110
Smith, P. F., Krishnarajah, J., Nunn, P. A., Hill, R. J., Karr, D., Tam, D., et al. (2017). A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br. J. Clin. Pharmacol. 83, 2367–2376. doi: 10.1111/bcp.13351
Stepanek, O., Draber, P., Drobek, A., Horejsi, V., and Brdicka, T. (2013). Nonredundant roles of src-family kinases and syk in the initiation of B-cell antigen receptor signaling. J. Immunol. 190, 1807L–1818. doi: 10.4049/jimmunol.1202401
Sun, C., Nierman, P., Kendall, E. K., Cheung, J., Gulrajani, M., Herman, S. E. M., et al. (2020). Clinical and biological implications of target occupancy in CLL treated with the BTK inhibitor acalabrutinib. Blood 136, 93–105. doi: 10.1182/blood.2019003715
Tam, C. S., Opat, S., D’Sa, S., Jurczak, W., Lee, H.-P., Cull, G., et al. (2020). A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood 136, 2038–2050. doi: 10.1182/blood.2020006844
Tam, C. S., Trotman, J., Opat, S., Burger, J. A., Cull, G., Gottlieb, D., et al. (2019). Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood 134, 851–859. doi: 10.1182/blood.2019001160
Taylor, S. S., and Kornev, A. P. (2011). Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77. doi: 10.1016/j.tibs.2010.09.006
Taylor, S. S., Shaw, A. S., Kannan, N., and Alexandr, K. (2015). Integration of Signaling in the Kinome: architecture and regulation of the aC Helix. Biochim Biophys Acta. 1854, 1567–1574. doi: 10.1016/j.bbapap.2015.04.007.Integration
Thibaud, S., Tremblay, D., Bhalla, S., Zimmerman, B., Sigel, K., and Gabrilove, J. (2020). Protective role of Bruton tyrosine kinase inhibitors in patients with chronic lymphocytic leukaemia and COVID-19. Br. J. Haematol. 190, e73–e76. doi: 10.1111/bjh.16863
Thompson, P. A. (2020). Occupy BTK: the key to controlling CLL. Blood 136, 4–6. doi: 10.1182/blood.2020005877
To, C., Jang, J., Chen, T., Park, E., Mushajiang, M., De Clercq, D. J. H., et al. (2019). Single and dual targeting of mutant egfr with an allosteric inhibitor. Cancer Discov. 9, 926–943. doi: 10.1158/2159-8290.CD-18-0903
Tong, M., Pelton, J. G., Gill, M. L., Zhang, W., Picart, F., and Seeliger, M. A. (2017). Survey of solution dynamics in Src kinase reveals allosteric cross talk between the ligand binding and regulatory sites. Nat. Commun. 8:2160. doi: 10.1038/s41467-017-02240-6
Treon, S. P., Castillo, J. J., Skarbnik, A. P., Soumerai, J. D., Ghobrial, I. M., Guerrera, M. L., et al. (2020). The BTK inhibitor ibrutinib may protect against pulmonary injury in COVID-19–infected patients. Blood 135, 1912–1915. doi: 10.1182/blood.2020006288
Tsukada, S., Saffran, D. C., Rawlings, D. J., Parolini, O., Allen, R. C., Klisak, I., et al. (1993). Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 72, 279–290. doi: 10.1016/0092-8674(93)90667-f
Von Raußendorf, F., De Ruiter, A., and Leonard, T. A. (2017). A switch in nucleotide affinity governs activation of the Src and Tec family kinases. Sci. Rep. 7, 1–14. doi: 10.1038/s41598-017-17703-5
Walter, H. S., Rule, S. A., Dyer, M. J. S., Karlin, L., Jones, C., Cazin, B., et al. (2016). A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood 127, 411–419. doi: 10.1182/blood-2015-08-664086
Wang, M., Rule, S., Zinzani, P. L., Goy, A., Casasnovas, O., Smith, S. D., et al. (2018). Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet 391, 659–667. doi: 10.1016/S0140-6736(17)33108-2
Wang, Q., Pechersky, Y., Sagawa, S., Pan, A. C., and Shaw, D. E. (2019). Structural mechanism for Bruton’s tyrosine kinase activation at the cell membrane. Proc. Natl. Acad. Sci. U.S.A. 116, 9390–9399. doi: 10.1073/pnas.1819301116
Wang, Q., Vogan, E. M., Nocka, L. M., Rosen, C. E., Zorn, J. A., Harrison, S. C., et al. (2015). Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. Elife 2015, 1–31. doi: 10.7554/eLife.06074
Weaver, A. N., and Jimeno, A. (2020). Zanubrutinib: a new BTK inhibitor for treatment of relapsed/refractory mantle cell lymphoma. Drugs Today (Barc). 56, 531–539. doi: 10.1358/dot.2020.56.8.3158047
Weber, A. N. R., Bittner, Z., Liu, X., Dang, T. M., Radsak, M. P., and Brunner, C. (2017). Bruton’s tyrosine kinase: An emerging key player in innate immunity. Front. Immunol. 8:1454. doi: 10.3389/fimmu.2017.01454
Wist, M., Meier, L., Gutman, O., Haas, J., Endres, S., Zhou, Y., et al. (2020). Noncatalytic Bruton’s tyrosine kinase activates PLCγ2 variants mediating ibrutinib resistance in human chronic lymphocytic leukemia cells. J. Biol. Chem. 295, 5717–5736. doi: 10.1074/jbc.RA119.011946
Woyach, J. A., Furman, R. R., Liu, T.-M., Ozer, H. G., Zapatka, M., Ruppert, A. S., et al. (2014). Resistance Mechanisms for the bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 370, 2286–2294. doi: 10.1056/nejmoa1400029
Wu, J., Zhang, M., and Liu, D. (2016). Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor. J. Hematol. Oncol. 9:21. doi: 10.1186/s13045-016-0250-9
Wu, W. I., Voegtli, W. C., Sturgis, H. L., Dizon, F. P., Vigers, G. P. A., and Brandhuber, B. J. (2010). Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One 5:e0012913. doi: 10.1371/journal.pone.0012913
Xie, Q., Joseph, R. E., Fulton, D. B., and Andreotti, A. H. (2013). Substrate recognition of PLCγ1 via a specific docking surface on Itk. J. Mol. Biol. 425, 683–696. doi: 10.1016/j.jmb.2012.10.023
Xie, T., Saleh, T., Rossi, P., and Kalodimos, C. G. (2020). Conformational states dynamically populated by a kinase determine its function. Science 370:eabc2754. doi: 10.1126/science.abc2754
Xu, L., Tsakmaklis, N., Yang, G., Chen, J. G., Liu, X., Demos, M., et al. (2017). Acquired mutations associated with ibrutinib resistance in Waldenström macroglobulinemia. Blood 129, 2519–2525. doi: 10.1182/blood-2017-01-761726
Yang, S., Zhang, Y., Shen, F., Ma, X., Zhang, M., Hou, Y., et al. (2019). The flavonoid baicalin improves glucose metabolism by targeting the PH domain of AKT and activating AKT/GSK3β phosphorylation. FEBS Lett. 593, 175–186. doi: 10.1002/1873-3468.13305
Yoon, Y. (2014). Small chemicals with inhibitory effects on PtdIns(3,4,5)P3 binding of Btk PH domain. Bioorg. Med. Chem. Lett. 24, 2334–2339. doi: 10.1016/j.bmcl.2014.03.068
Zhang, M., Ma, X., Xu, H., Wu, W., He, X., Wang, X., et al. (2020). A natural AKT inhibitor swertiamarin targets AKT-PH domain, inhibits downstream signaling, and alleviates inflammation. FEBS J. 287:e15112. doi: 10.1111/febs.15112
Zhou, Q., Lee, G.-S., Brady, J., Datta, S., Katan, M., Sheikh, A., et al. (2012). A hypermorphic missense mutation in PLCG2, encoding phospholipase Cγ2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am. J. Hum. Genet. 91, 713–720. doi: 10.1016/j.ajhg.2012.08.006
Keywords: Bruton’s tyrosine kinase, B cell lymphoma, tyrosine kinase regulation, drug resistance, drug targeting approaches, BTK autoinhibition, Ibrutinib, BTK inhibitors
Citation: Kueffer LE, Joseph RE and Andreotti AH (2021) Reining in BTK: Interdomain Interactions and Their Importance in the Regulatory Control of BTK. Front. Cell Dev. Biol. 9:655489. doi: 10.3389/fcell.2021.655489
Received: 18 January 2021; Accepted: 02 June 2021;
Published: 23 June 2021.
Edited by:
Serge Roche, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Cynthia L. Bristow, Alpha-1 Biologics, United StatesCopyright © 2021 Kueffer, Joseph and Andreotti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amy H. Andreotti, YW15YW5kQGlhc3RhdGUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.