 Chiara Parodi1
Chiara Parodi1 Elisabetta Di Fede1
Elisabetta Di Fede1 Angela Peron2,3,4
Angela Peron2,3,4 Ilaria Viganò1
Ilaria Viganò1 Paolo Grazioli1Silvia Castiglioni1
Paolo Grazioli1Silvia Castiglioni1 Richard H. Finnell5
Richard H. Finnell5 Cristina Gervasini1,6†
Cristina Gervasini1,6† Aglaia Vignoli1†
Aglaia Vignoli1† Valentina Massa1,6*†
Valentina Massa1,6*†- 1Department of Health Sciences, Università degli Studi di Milano, Milan, Italy
- 2Human Pathology and Medical Genetics, ASST Santi Paolo e Carlo, San Paolo Hospital, Milan, Italy
- 3Child Neuropsychiatry Unit–Epilepsy Center, Department of Health Sciences, San Paolo Hospital, ASST Santi Paolo e Carlo, Università degli Studi di Milano, Milan, Italy
- 4Division of Medical Genetics, Department of Pediatrics, University of Utah School of Medicine, Salt Lake City, UT, United States
- 5Departments of Molecular and Cellular Biology, Molecular and Human Genetics and Medicine, Center for Precision Environmental Health, Baylor College of Medicine, Houston, TX, United States
- 6“Aldo Ravelli” Center for Neurotechnology and Experimental Brain Therapeutics, Università degli Studi di Milano, Milan, Italy
Prenatal exposure to valproate (VPA), an antiepileptic drug, has been associated with fetal valproate spectrum disorders (FVSD), a clinical condition including congenital malformations, developmental delay, intellectual disability as well as autism spectrum disorder, together with a distinctive facial appearance. VPA is a known inhibitor of histone deacetylase which regulates the chromatin state. Interestingly, perturbations of this epigenetic balance are associated with chromatinopathies, a heterogeneous group of Mendelian disorders arising from mutations in components of the epigenetic machinery. Patients affected from these disorders display a plethora of clinical signs, mainly neurological deficits and intellectual disability, together with distinctive craniofacial dysmorphisms. Remarkably, critically examining the phenotype of FVSD and chromatinopathies, they shared several overlapping features that can be observed despite the different etiologies of these disorders, suggesting the possible existence of a common perturbed mechanism(s) during embryonic development.
Introduction
Prenatal exposure to antiepileptic drugs (AEDs) are subject to the teratogenic effects associated with all of the frontline AED medications. Most women with epilepsy receiving adequate prenatal care will have uneventful pregnancies, but they are at a well-documented increased risk for having infants with congenital malformations compared to the general population (Viale et al., 2015). In utero AED exposure places their offspring at increased risk not only for major congenital malformations, but also for adverse neurological developmental outcomes. However, many of these risks can be mitigated through comprehensive prenatal maternal care by carefully selecting the type and dose of AEDs prior to conception and continuing to follow a proper therapeutic regimen throughout pregnancy (Tomson et al., 2011). Among all AEDs, valproate (2-propylpentanoic acid, VPA) exposure has been associated with the greatest risks of inducing severe teratogenicity (Lammer et al., 1987; Tomson et al., 2015). Several studies demonstrated a correlation between chronic exposure to VPA treatment and higher risk of displaying fetal anomalies–such as neural tube defects (NTDs), distinctive facial dysmorphia, craniofacial, and skeletal defects–in both in humans and in animal models (Massa et al., 2005, 2006). Among the teratogen-induced congenital malformations, the most commonly observed include spina bifida, atrial septal defects, cleft palate, hypospadias, polydactyly, and craniosynostosis (Macfarlane and Greenhalgh, 2018).
Animal experiments demonstrated morphogenic anomalies throughout the entire axial skeleton and vertebral transformations in rat embryos due to VPA exposure, suggesting a possible compromise of the expression of genes involved in vertebral segments development (Menegola et al., 1998, 1999). In addition, an altered serotonergic differentiation, which correlates with autism-like behavioral abnormalities, was observed both in rodent and zebrafish models in response to prenatal valproate exposure (Dufour-Rainfray et al., 2010; Jacob et al., 2014). The amount of fetal harm appears to be linked to the maternal concentration of the drug (Nau et al., 1981; Nau, 1985), especially when it occurs in the first trimester during fetal organogenesis (Macfarlane and Greenhalgh, 2018). In animal models, such as Xenopus and Hyperolius, the beginning of gastrulation was delayed up to neurulation upon embryonic exposure to VPA, and eventually they displayed NTDs of different types and degree (Oberemm and Kirschbaum, 1992). To date, the correlation between typical dysmorphic facial features and developmental outcomes is unclear (Kini et al., 2006; Nicolini and Fahnestock, 2018).
Fetal valproate syndrome (FVS, OMIM #609442) is a condition resulting from the therapeutic management of epileptic mothers with VPA during their pregnancy, and it is observed in up to 20–30% of children exposed to high VPA dosage in utero (Nau et al., 1991; Ornoy, 2009; Tomson et al., 2011). FVS is characterized by a constellation of congenital malformations and developmental delay, with patients displaying intellectual disability (ID) as well as autism spectrum disorder (ASD), and a distinctive facial appearance strikingly similar to the one described in genetic disorders known as chromatinopathies (DiLiberti et al., 1984). A new term “fetal valproate spectrum disorder” (FVSD) has recently been proposed to describe the range of clinical and developmental effects that are attributed to in utero VPA exposure (Clayton-Smith et al., 2019).
Valproate has been commonly used as an anti-seizure medication for over half a century (Meunier et al., 1963). Given its broad antiepileptic effect, it has also been clinically utilized as a mood stabilizer in the treatment of bipolar disorders and in other neurological conditions–i.e., migraine and neuropathic pain, exposing many more women of reproductive age to this medication (Johannessen and Johannessen, 2003). In addition, this antiepileptic drug has shown anticancer properties for several tumors (Shah and Stonier, 2019), and its use in combination regimens with cytotoxic chemotherapy seems to be promising (Brodie and Brandes, 2014). VPA is also known to be a potent histone deacetylase inhibitor (HDACi) and therefore acts on chromatin. It is known to have dose-related teratogenic properties resulting in altered gene expression and potent inhibition of the histone deacetylases (HDAC) enzymes family (Schölz et al., 2015). Among the various hypotheses that have been proposed for the teratogenicity of VPA, its HDACi effects that is believed to be represent the principle underlying teratogenic mechanism. VPA’s anti-seizure activity can also be explained by its ability to modulate gene expression through the inhibition of HDAC enzymes (Göttlicher et al., 2001; Jacob et al., 2013; Brunton et al., 2018).
Valproate perturbs the cell’s epigenetic machinery controlling its chromatin state. In this context, a group of heterogeneous genetic disorders known as the chromatinopathies, are believed to be caused by mutations in genes that regulate the conformation and function of chromatin, thus acting in concert with epigenetic mechanisms. Defects in the functional network between the complexes associated with chromatin could lead to alterations in gene expression and protein function. As estimated, there are over 80 Mendelian diseases associated with incorrect functioning of the “epigenetic machinery”, the majority of which presents with neurological defects and ID (Fahrner and Bjornsson, 2019). Kabuki syndrome (OMIM #147920 and #300867) (Niikawa et al., 1981) and CHARGE syndrome (OMIM #214800) (Pagon et al., 1981) are among the most well-known and studied chromatinopathies, for the cascading effect of the causative genes on different cell pathways. These syndromes are associated with ID and distinctive craniofacial dysmorphisms that are pathognomonic.
In this review, we explore shared features between FVSD and selected chromatinopathies, leading us to the hypothesis that these disorders, despite divergent etiologies (i.e., environmental or genetic), could operate through a common perturbed mechanism during embryonic development. As such, VPA-induced FVSD is a phenocopy of select chromatinopathies.
Valproate Mechanism of Action
Valproate has multiple cellular mechanisms of action consistent with its broad clinical efficacy. This compound appears to suppress repetitive high-frequency neuronal focus by blocking voltage-dependent sodium channels, but at sites that are different from other AEDs. VPA also appears to increase GABA concentrations in the brain at clinically relevant doses, without having direct effects on the GABA (A) receptors, potentiated by a presynaptic effect of valproate on GABA (B) receptors. In addition, VPA can increase GABA synthesis by activating the enzyme glutamic acid decarboxylase (GAD).
The molecular mechanisms underlying FVSD have not been fully established, although the consequences of in utero VPA exposure have been investigated for several decades. Such effects include apoptotic neurodegeneration observed in the developing rat brain (Bittigau et al., 2002), enhanced synaptic plasticity exhibited in the rat medial prefrontal cortex (Sui and Chen, 2012), and a decrease in folic acid (Wegner and Nau, 1992), suggesting that inadequate embryonic and fetal antioxidant defense mechanisms and consequent oxidative stress could be responsible for brain damage secondary to VPA teratogenicity (Ornoy, 2009).
Despite the fact that VPA has been shown to be neuroprotective in neurons through Bcl-2 upregulation (Chen et al., 1999), its administration in critical developmental stages causes morphological defects and impaired social behavior in rats (Kim et al., 2011). In utero VPA exposure in mouse pups on gestational day 11 leads to dysfunctional pre-weaning social behavior, together with delayed development, impaired olfactory discrimination and reduced cortical Bdnf expression, suggesting that VPA-driven perturbations in neuronal plasticity may underlie the behavioral phenotype (Roullet et al., 2010). Similar to the results of VPA exposure of pregnant rats, neural progenitor cells (NPCs) of murine embryos exposed on gestational day E12 showed a reduced apoptotic cell death, which is fundamental to the proper regulation of NPCs during a developmentally critical period, suggesting another possible mechanism underlying FVSD defects (Go et al., 2011).
Alterations in embryonic gene expression following VPA exposure appears to be one of the primary mechanisms underlying VPA’s teratogenicity. Previous studies showed that VPA alters Wnt signaling by inducing Wnt-dependent gene expression at doses that cause developmental effects (Phiel et al., 2001; Wiltse, 2005). This is due to its role as an HDAC inhibitor, which consists of deregulating class I HDACs, thus counteracting their normal activity of histone acetylation marks removal. This action induces chromatin changes converting segments of heterochromatin into euchromatin. VPA exposure can lead to hyperacetylation of histones and following activation of genes related to cell cycle and apoptosis, possibly explaining its teratogenic action (Göttlicher et al., 2001). For instance, hyperacetylation of all Hoxb developmental genes has been observed in mouse embryonic stem cells exposed to VPA, with increased levels of H3K9ac at upstream, promoter and coding regions across the entire Hoxb cluster (Boudadi et al., 2013).
Previous studies showed how HDACi is involved in early neuronal processes: exposure of neurulation-stage mouse embryos to VPA can cause NTDs and skeletal malformations (Finnell et al., 2002), supported by in vivo studies on chick embryos in which a complete failure of neural tube closure occurred (Murko et al., 2013). Since VPA exposure alters gene expression in the somitic tissues of mouse embryos (Massa et al., 2005) and an increased histone H4 acetylation in the caudal neural tube was observed, modulation of acetylation was hypothesized as mediating the effect of VPA on neurulation (Massa et al., 2005, 2009; Menegola et al., 2005).
Valproate in Clinical Practice
Valproate is a wide-spectrum anti-seizure medication that can be used to treat almost all types of seizure disorders (tonic clonic seizures, absence seizures, myoclonic seizures, less frequently in clonic seizures, tonic seizures and atonic seizures) (Perucca, 2002). It is used as first-line antiepileptic drug in generalized seizures; VPA may also be used in focal seizures, although it is no longer the first choice of neurologists (Tomson et al., 2015).
Valproate is also a mood stabilizer that is used in the treatment of bipolar disorders and other psychiatric conditions, including: anxiety disorders, post-traumatic stress disorder, substance abuse, and schizophrenia. VPA also appears to be an effective treatment for tardive dyskinesia thanks to its GABA-potentiating properties (Swann et al., 2002), for migraine prophylaxis, and for the treatment of neuropathic pain, in particular for trigeminal neuralgia (Johannessen and Johannessen, 2003).
Typically, the initial dose of oral valproate is 10–15 mg/kg per day. If necessary, the dose can be increased with weekly increments of 5–10 mg/kg up to a maximum dose of 60 mg/kg/day. It is recommended to monitor VPA blood level during treatment, as well as blood count, liver enzymes, and coagulation tests, in order to avoid any potential side effects of the drug.
Valproate can alter vitamin D metabolism and affect bone mineral density, therefore 25-hydroxyvitamin D levels should be monitored. It may be useful to obtain serum amylase and lipase levels in cases where symptoms suggestive of pancreatitis, such as abdominal pain, nausea, vomiting and anorexia have occurred. Furthermore, ammonium levels should be monitored in patients receiving VPA who exhibit signs of vomiting or lethargy as the treatment inhibits N-acetyl glutamate, leading to systemic disruption and hyperammonemia (Bruni et al., 1979; Batshaw and Brusilow, 1982; Asconapé et al., 1993; Patsalos et al., 2008).
It is strongly recommended that VPA administration should be avoided during pregnancy; however, if necessary, a slow-release formulation that limits peak concentrations of the drug using the lowest efficacious dose possible should be given along with the administration of a high dose of folic acid. While folic acid has not be shown to be effective in reducing the prevalence of NTDs, it has been shown to be protective in limiting adverse cognitive consequences of VPA treatment, especially with respect to language skills (Meador et al., 2020).
Clinical Features Associated With FVSD
Studies conducted by Robert and Guibaud (1982) first drew attention to the increased risk of spina bifida after exposure to VPA in pregnancy. Subsequently, the initial reports of children suffering from FVSD were published (DiLiberti et al., 1984). FVSD is characterized by major and minor malformations, facial dysmorphia and impaired development with particular risks related to NTDs (Lindhout and Schmidt, 1986), congenital heart disease, ophthalmological, (Glover et al., 2002) and genitourinary abnormalities (DiLiberti et al., 1984; Ozkan et al., 2011), cleft palate (Jackson et al., 2016), overlapping fingers, and scalp defects (DiLiberti et al., 1984; Clayton-Smith and Donnai, 1995; Clayton-Smith et al., 2019).
Neurological development is impaired in many affected individuals. An increased risk of attention deficit hyperactivity disorder (ADHD) and ASD is often observed in these patients (Bromley et al., 2013, 2014, 2019; Christensen et al., 2013).
The typical facial features of FVSD include: swelling of the metopic suture, highly-arched eyebrows, hypertelorism, wide nasal bridge, short nose with anteverted nostrils, small mouth with thin upper lip and flat filament of the inverted lower lip (Ardinger et al., 1988; Clayton-Smith and Donnai, 1995; Kozma, 2001; Schorry et al., 2005; Kini et al., 2006; Chandane and Shah, 2014; Mohd Yunos and Green, 2018) (Figure 1).
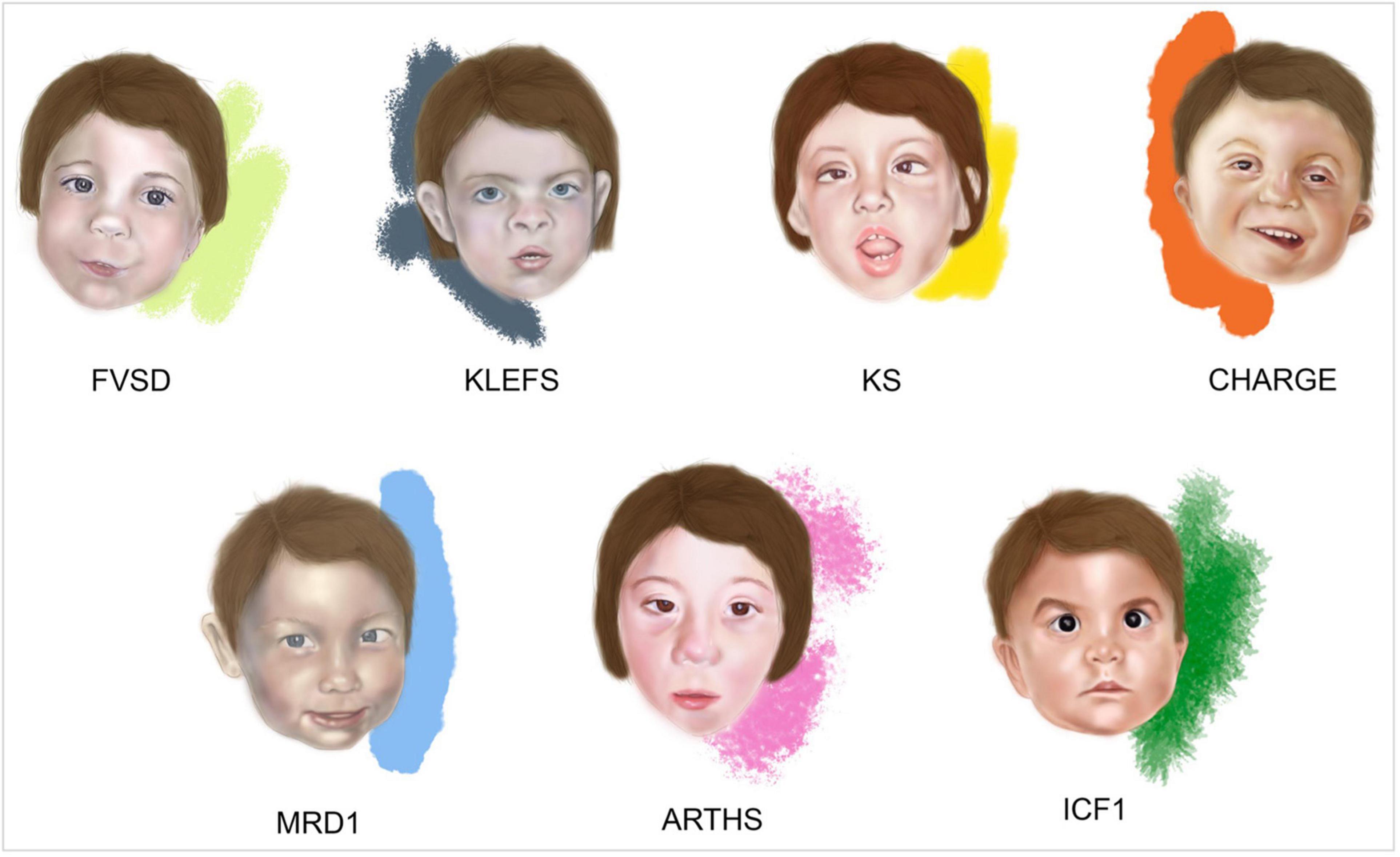
Figure 1. Facies of fetal valproate spectrum disorders (FVSD) and related overlapping chromatinopathies. Distinctive facial phenotypes of patients affected by FVSD (Schorry et al., 2005), KLEFS (Willemsen et al., 2012), KS (Makrythanasis et al., 2013), CHARGE (Hefner and Fassi, 2017), MRD1 (Talkowski et al., 2011), ARTHS (Kennedy et al., 2019), and ICF1 (Gössling et al., 2017).
Differential Diagnoses of FVSD
Fetal valproate spectrum disorder diagnosis has been challenging in many ways, from gathering correct information about prenatal VPA exposure, to obtaining a comprehensive grasp of the clinically varied diagnostic phenotypic signs. Indeed, clinical presentations of affected patients show variability and the prevalence of neurocognitive dysfunction is higher than the prevalence of structural malformations, complicating the path toward reliable diagnosis (Clayton-Smith et al., 2019). In fact, not all the FVSD individuals even display dysmorphisms, which can be age dependent and rather subtle, thus recognizable only by experienced dysmorphologists. When FVSD signs are ascertained, physicians are often challenged by overlapping phenotypes associated with the following syndromes (Figure 1 and Table 1).
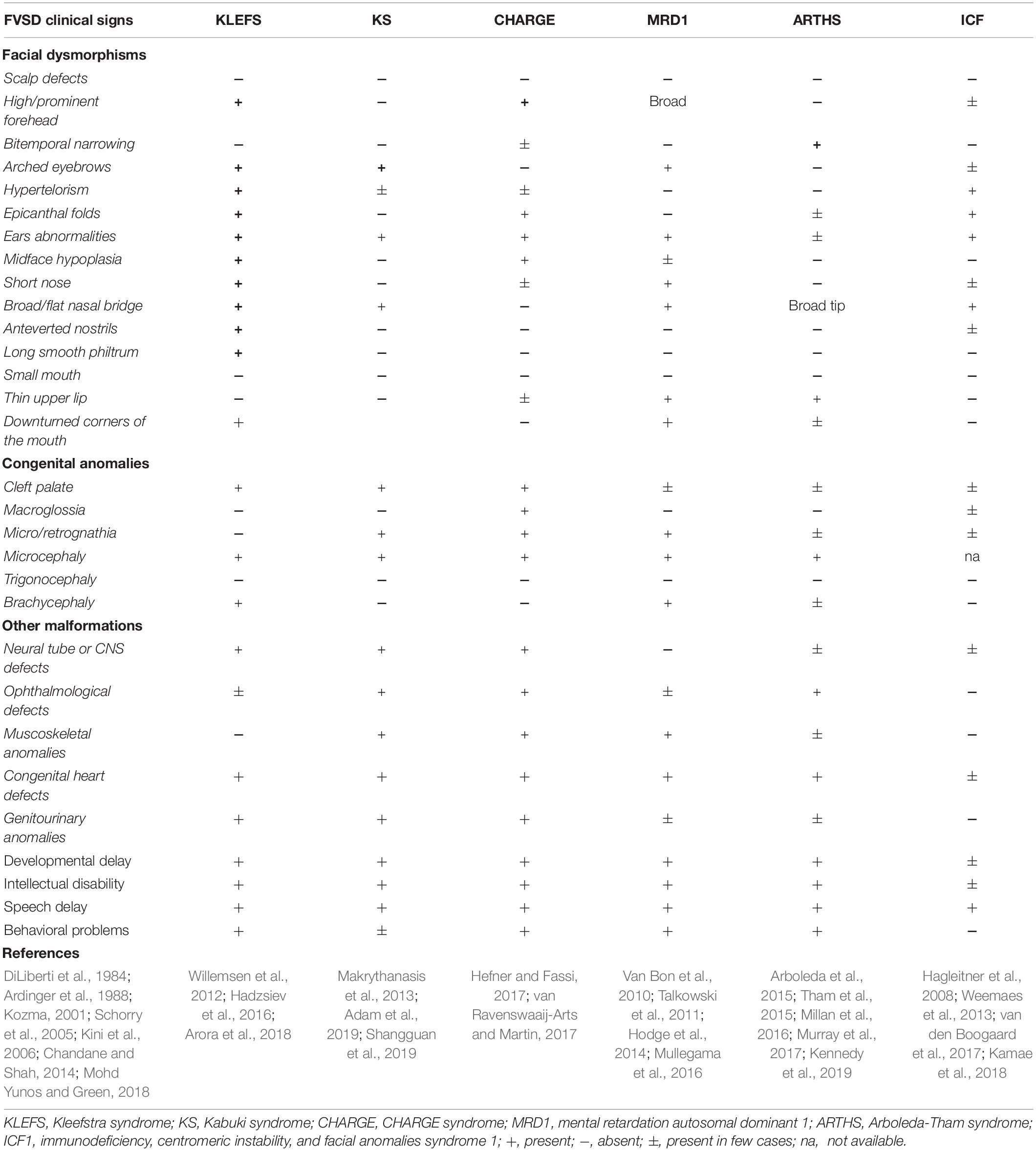
Table 1. Fetal valproate spectrum disorders (FVSD) clinical signs in chromatinopathies.
Kleefstra Syndrome
Kleefstra syndrome (KLEFS, OMIM #610253, #617768) is a rare condition characterized by heterozygous genomic deletions at chromosome 9q34.3 removing the EHMT1 gene or EHMT1 point mutations (KLEFS1), or pathogenic variants in KMT2C on chromosome 7q36.1 (KLEFS2), mostly de novo. EHMT1 and KMT2C genes encode two histone methyltransferases. The prevalence is estimated to be 1:120,000 individuals affected by neurodevelopmental disorders. Patients with Kleefstra syndrome exhibit a distinctive phenotype including hypotonia; major anomalies such as congenital heart defects and genitourinary abnormalities; behavioral and developmental manifestations with ID of variable severity, and in some cases severe speech delay. Typical facial dysmorphisms include: microcephaly, arched or straight with synophrys eyebrows, mildly up-slanted palpebral fissures, hypertelorism, short nose with anteverted nares and bulbous nasal tip, thick mouth, and everted lower lip (Kleefstra and De Leeuw, 1993; Kleefstra et al., 2006, 2012; Koemans et al., 2017) (Figure 1).
Fetal valproate spectrum disorders has been recently defined as a “phenocopy” of Kleefstra syndrome by Arora et al. (2018). Despite a preliminary diagnosis of FVSD–due to maternal intake of VPA during pregnancy and with clear facial characteristics that are typically attributable to FVSD–a more thorough examination of the facial features revealed subtle differences. Specific features of the proband included the presence of a broad forehead and brachycephaly in a child with FVSD, who had cephalic deformation due to the premature fusion of the metopic suture, scattered eyebrows, and pointed chin. Genetic testing revealed a de novo deletion on 9q34.3 that is known to cause Kleefstra syndrome. The convergent mechanism present in both conditions is their role in epigenetic modulation that mediates the modification (acetylation, methylation, etc.) of histone proteins and DNA demethylation, which might be responsible for the overlapping phenotype of FVSD and Kleefstra Syndrome (Willemsen et al., 2012; Hadzsiev et al., 2016; Arora et al., 2018) (Table 1).
Kabuki Syndrome
Specific dysmorphisms, postnatal growth delay, skeletal anomalies, and ID are typical features of another chromatinopathy such as Kabuki syndrome (KS, OMIM #147920 and #300867) (Kuroki et al., 1981; Niikawa et al., 1981; Adam et al., 2019). KS is caused by heterozygous pathogenic variants in KMT2D or KDM6A genes (Ng et al., 2010; Banka et al., 2012; Lederer et al., 2012; Miyake et al., 2013), on chromosome 12q13.12 and Xp11.3, causing KS1 and KS2, respectively. These genes altered in KS encode for a histone methyltransferase and a histone demethylase exerting their effect on different histone residues that favor the opening of chromatin and leading to the same downstream effects on gene expression, ultimately resulting in the same condition. Aside from ID, developmental impairment and congenital heart defects, KS shares with FVSD specific craniofacial features such as arched eyebrows, wide nasal bridge, and cleft palate (Makrythanasis et al., 2013; Adam et al., 2019; Shangguan et al., 2019) (Figure 1 and Table 1).
CHARGE Syndrome
CHARGE syndrome (OMIM #214800) is an acronym that summarizes the main clinical manifestations, namely Coloboma of the eye, Heart defect, choanal Atresia, Retardation of psychomotor development and growth, Genital hypoplasia, and Ear abnormalities. This syndrome is caused by heterozygous pathogenic variants in CHD7 (OMIM # 608892), encoding an epigenetic regulator that is involved in the ATP-dependent remodeling of chromatin (Vissers et al., 2004; van Ravenswaaij-Arts and Martin, 2017). Interestingly, Shah et al. (2014) and Jackson et al. (2014) reported on five children affected by FVSD exhibiting unilateral or bilateral ocular coloboma, one of the main manifestations of CHARGE syndrome. Indeed, VPA acting as a HDAC inhibitor reduces the expression of PAX2 and PAX6, which are implicated in ocular development (Pennati et al., 2001; Balmer et al., 2012). Of note, CHARGE syndrome shares autism-like disturbances, congenital anomalies and malformations together with specific facial features with FVSD (Hefner and Fassi, 2017; van Ravenswaaij-Arts and Martin, 2017) (Figure 1 and Table 1).
Mental Retardation Autosomal Dominant 1
Mental retardation autosomal dominant 1 (MRD1, OMIM #156200) or MBD5 haploinsufficiency is a neurodevelopmental disorder caused by heterozygous variants in MBD5 or a deletion encompassing all or part of this gene sequence on chromosome 2q23.1 (Vissers et al., 2003; Talkowski et al., 2011). MBD5 encodes for a methyl-CpG-binding domain protein. MBD5 is part a class of proteins that bind to DNA with a transcriptional repressor activity. In Camarena et al. (2014) MBD5 was shown to act as transcriptional activator in vitro. Hence, MBD5 is considered a “reader” of the epigenetic machinery (Camarena et al., 2014). Patients display ID and developmental delay, sleep disturbances, seizures, severe speech impairment, behavioral problems, feeding difficulties, congenital anomalies mainly affecting the skeletal and cardiovascular systems, and dysmorphic signs. Among them, MRD1 is characterized by broad forehead, highly arched eyebrows, outer ear abnormalities, short nose with broad nasal bridge, thin upper lip, and downturned mouth angles, which are remarkably overlapping with FVSD (Van Bon et al., 2010; Talkowski et al., 2011; Hodge et al., 2014; Mullegama et al., 2016) (Figure 1 and Table 1).
Arboleda-Tham Syndrome
Pathogenetic variants in the KAT6A gene, located on chromosome 8p11.21 cause Arboleda-Tham syndrome (ARTHS, OMIM #616268) or Mental retardation autosomal dominant 32 (MRD32), a recently described disorder affecting neurodevelopment and associated with ID (Arboleda et al., 2015; Tham et al., 2015). KAT6A is a lysine-acetyltransferase involved in chromatin opening, transcriptional regulation, cellular replication and therefore, in multiple developmental programs (Voss et al., 2009). Kennedy et al. (2019) extensively described phenotypes of novel and previously reported ARTHS patients, who display distinctive clinical signs such as ID, developmental and speech delay, cardiac and ophthalmological defects, gastrointestinal problems, sleep disturbance, autism-like behavior and typical dysmorphisms (Arboleda et al., 2015; Tham et al., 2015; Millan et al., 2016; Murray et al., 2017), many of them overlapping with the FVSD phenotype (Figure 1 and Table 1).
Immunodeficiency, Centromeric Instability and Facial Anomalies Syndrome
Immunodeficiency, centromeric instability and facial anomalies syndrome 1 (ICF1, #OMIM 602900) is a rare autosomal recessive disorder characterized by hypogammaglobulinemia leading to severe recurrent infections, instability of pericentromeric regions of chromosomes 1, 9, and 16 in mitogen-stimulated lymphocytes, and facial dysmorphisms (Maraschio et al., 1988; Ehrlich et al., 2006). When the mapping of a locus associated to ICF syndrome on chromosome 20 was performed in 1998 (Wijmenga et al., 1998), pathogenic variants in de novo DNA methyltransferase gene DNMT3B were identified, occurring in about half of ICF patients (Hansen et al., 1999; Xu et al., 1999). DNMT3B is involved in the establishment of DNA methylation patterns in early life and during cell differentiation. Hypomethylation of pericentromeric satellite 2 and 3 repeats represents the molecular hallmark of ICF syndrome (Jeanpierre et al., 1993), making it the first human disorder linked to a constitutive defect in DNA methylation. In addition to distinctive signs such as immunoglobulin deficiency and consequent recurrent infections (mainly respiratory and gastrointestinal), ICF1 patients also display some features that are common to FVSD: hypertelorism, epicanthus, flat nasal bridge, macroglossia, micrognathia, low-set ears, speech, and developmental delay, and–in a minority of affected individuals–CNS anomalies, congenital heart defects and ID (Hagleitner et al., 2008; Weemaes et al., 2013; van den Boogaard et al., 2017; Kamae et al., 2018) (Figure 1 and Table 1).
Other Genetic Disorders
Qiao et al. (2019) recently described a 19 years-old man with ID and distinctive facial features who had a clinical diagnosis of FVSD and was later found to carry a de novo pathogenic variant in the PURA gene on chromosome 5q31. PURA-related neurodevelopmental disorders include Mental Retardation autosomal Dominant 31 (MRD31, #OMIM 616158) or PURA syndrome, caused by heterozygous mutations in the PURA gene or a 5q31.3 deletion affecting completely or partially eliminating the PURA sequence (Brown et al., 2013; Hunt et al., 2014; Lalani et al., 2014; Tanaka et al., 2015). This causative gene encodes for a DNA- and RNA-binding protein critical for survival and development of mammalian hematopoietic and central nervous systems (Daniel and Johnson, 2018). Shared phenotypic features between PURA disorders and FVSD are ID and developmental delay, heart defects, urinary and ophthalmological abnormalities, and distinctive facial dysmorphism such as high/broad forehead, hypertelorism, wide nasal bridge, and thin upper lip (Reijnders et al., 2018). Furthermore, fetal valproate exposure has been reported to cause other malformation complexes such as Baller-Gerold syndrome (BGS, OMIM #218600), an ultra-rare disorder caused by pathogenic variants in the RECQL4 gene on chromosome 8p24, and inherited in an autosomal recessive manner (Baller, 1950; Gerold, 1959). BGS patients display a plethora of phenotypic features (Van Maldergem et al., 1992), some of which are overlapping with FVSD. These include: ID, developmental delay, limb and congenital heart defects, genitourinary anomalies and facial dysmorphisms (Iype et al., 2008). Mutations in the RECQL4 gene that codes for an ATP-dependent DNA helicase essential for genome integrity and involved in DNA replication, recombination and repair (Bachrati and Hickson, 2008) have also been reported in Rothmund-Thomson (RTS, OMIM #268400) families (Kitao et al., 1999). In particular, children affected with type II RTS share a variety of clinical features with BGS patients (Rothmund, 1868; Thomson, 1936; Megarbane et al., 2000; Van Maldergem et al., 2006; Larizza et al., 2010). Considering the similarities among BGS and RTS patients, there are multiple overlapping features with FVSD that can be observed–e.g., head and nose dysmorphisms, developmental delay, cardiac defects and skeletal anomalies. Although these neurodevelopmental disorders are not considered chromatinopathies, it is worthy of note that PURA and RECQL4 are transcriptional regulators, and helicases are considered a guardian of the genome, such that they are involved in proper chromatin maintenance.
Shared Epigenetic and Gene Expression Alterations
Gene expression deregulation by VPA has been widely investigated over the last few decades (Marchion et al., 2005; Jergil et al., 2009, 2011; Chiu et al., 2013; Shinde et al., 2016; Balasubramanian et al., 2019; Kotajima-Murakami et al., 2019; Lin et al., 2019; Sanaei and Kavoosi, 2019). Interestingly, some of the causative genes of the aforementioned syndromes are dysregulated in different experimental models. Ehmt1 was found to be downregulated in brains of mice exposed to VPA in utero, Kdm6a, and Dnmt3b appeared to be upregulated in the same model (Kotajima-Murakami et al., 2019), while Chd7 was downregulated in embryonal carcinoma cells upon VPA exposure (Jergil et al., 2009). In addition, Ehmt1 and its human orthologs were downregulated in neural stem/progenitor cells of a mouse model for KS1 (Carosso et al., 2019) and in lymphoblastoid cell lines derived from 2q23.1 deletion syndrome patients (Mullegama et al., 2016), respectively. Expression of DNMT3B was decreased in iPSCs derived from KS1 patient (Carosso et al., 2019). Furthermore, Kdm6a and Chd7 have been reported to be interlinked in terms of gene expression regulation (Mansour et al., 2012; Hsu et al., 2020), and in a mouse model expressing catalytically inactive Dnmt3b, they share opposite behavior (Lopusna et al., 2021).
In Table 2, shared pathways with genes deregulated by VPA and downregulated in models of causative genes for KLEFS, KS, CHARGE, MRD1, and ARTHS or ICF1 are summarized (Katsumoto et al., 2006; Issaeva et al., 2007; Min et al., 2007; Fan, 2008; Gupta-Agarwal et al., 2012; Mansour et al., 2012; Balemans et al., 2014; Chen et al., 2014; Kim et al., 2014; Schulz et al., 2014; Turner-Ivey et al., 2014; Gigek et al., 2015; Dhar et al., 2016, 2018; Fang et al., 2016; Mullegama et al., 2016; Sheikh et al., 2016, 2017; Feng et al., 2017a, b; Shpargel et al., 2017; Whittaker et al., 2017; Baell et al., 2018; Marie et al., 2018; Yao et al., 2018, 2020; Carosso et al., 2019; Machado et al., 2019; Nowialis et al., 2019; Cieslar-Pobuda et al., 2020; Frega et al., 2020; Hsu et al., 2020; Kong et al., 2020; Liu et al., 2020; Xu et al., 2020; Ying et al., 2020; Fei et al., 2021; Lopusna et al., 2021). Of note, the most commonly shared pathways involve either morphogenesis signals (for example, beta1 integrin cell surface interactions and extracellular matrix organization), or possible defects of the central nervous system (such as axon guidance and neuronal system). As such, given the recent description of ARTHS, it would be interesting to reassess this matter in the future utilizing state of the art molecular studies.

Table 2. Shared pathways between FVSD and chromatinopathies.
Conclusion
It is well established that the mammalian epigenome can change during embryonic development and be influenced by genetic and/or environmental factors, even though some molecular mechanisms underlying these modifications are yet not clear (Finnell et al., 2002; Xu and Xie, 2018). Chromatinopathies represent a heterogeneous group of Mendelian disorders with defects in the epigenetic apparatus, leading to an imbalance in the chromatin state and consequent aberrant gene expression. As described above, these disorders share several overlapping clinical signs, though with some specific features allowing dysmorphologists to recognize each individual syndrome. We highlighted similarities between the discussed chromatinopathies and FVSD, pointing out shared features in these genetic- and teratogen-induced disorders. As reported in Table 1, overlapping clinical signs are primarily ID and developmental delay, present in 5 out of 5 chromatinopathies described herein (6/6), speech delay (6/6), ASD-like behavior (5/6), microcephaly (5/6), cardiac (6/6) and ophthalmological defects with different degree of severity (5/6), cleft palate (6/6), musculoskeletal anomalies (4/6), and dysmorphic features such as highly arched or thick eyebrows (4/6) and ears abnormalities (6/6).
Intriguingly, despite the different etiology of a FVSD and the chromatinopathies, the action of VPA–i.e., an HDACi acting on chromatin–can suggest a similar pathogenetic mechanism common to the other rare genetic disorders, giving rise to the observed shared phenotypic signs. Furthermore, recent work showed that recognition of FVSD facies can identify individuals with high risk of cognitive deficits, independently of VPA exposure and even in the absence of major malformations (Bromley et al., 2019). Taken together, these pieces of evidence support the hypothesis that FVSD may be considered as a phenocopy of chromatinopathy, caused in this case by environmental factors, and that a further investigation of this aspect could help elucidate the correlation between typical congenital anomalies and neurodevelopment.
Author Contributions
CG, AV, and VM conceived the manuscript. CP and EDF wrote the manuscript. AP reviewed clinical information. IV wrote sections of the manuscript. PG and SC read and edited the manuscript. RHF reviewed and revised the manuscript. All authors contributed to manuscript revision, they have all read and approved the manuscript.
Funding
The authors are grateful to the following funding sources: Fondazione Cariplo (2015-0783 to VM); Intramural funding-Dipartimento DISS, Linea 2, Università degli Studi di Milano (to CG, AV, and VM); Translational Medicine Ph.D. scholarship-Università degli Studi di Milano (to CP and ED); Molecular and Translational Medicine Ph.D. scholarship-Università degli Studi di Milano (to PG); Nickel & Co S.p.A (to VM); and “Aldo Ravelli” Center for Neurotechnology and Experimental Brain Therapeutics-Università degli Studi di Milano (to CG and VM). The authors acknowledge support from the University of Milan through the APC initiative.
Conflict of Interest
RF formerly held a leadership position in TeratOmic Consulting LLC. This now dissolved organization provided expert consulting support in birth defects litigation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Susanna Brusa for graphical support.
References
Adam, M. P., Banka, S., Bjornsson, H. T., Bodamer, O., Chudley, A. E., Harris, J., et al. (2019). Kabuki syndrome: international consensus diagnostic criteria. J. Med. Genet. 56, 89–95. doi: 10.1136/jmedgenet-2018-105625
Arboleda, V. A., Lee, H., Dorrani, N., Zadeh, N., Willis, M., Macmurdo, C. F., et al. (2015). De novo nonsense mutations in KAT6A, a lysine acetyl-transferase gene, cause a syndrome including microcephaly and global developmental delay. Am. J. Hum. Genet. 96, 498–506. doi: 10.1016/j.ajhg.2015.01.017
Ardinger, H. H., Atkin, J. F., Blackston, R. D., Elsas, L. J., Clarren, S. K., Livingstone, S., et al. (1988). Verification of the fetal valproate syndrome phenotype. Am J Med Genet. 29, 171–185. doi: 10.1002/ajmg.1320290123
Arora, V., Joshi, A., Lall, M., Agarwal, S., Bijarnia Mahay, S., Dua puri, R., et al. (2018). Fetal valproate syndrome as a phenocopy of Kleefstra syndrome. Birth Defects Res. 110, 1205–1209. doi: 10.1002/bdr2.1379
Asconapé, J. J., Penry, J. K., Dreifuss, F. E., Riela, A., and Mirza, W. (1993). Valproate-associated pancreatitis. Epilepsia 34, 177–183. doi: 10.1111/j.1528-1157.1993.tb02395.x
Bachrati, C. Z., and Hickson, I. D. (2008). RecQ helicases: guardian angels of the DNA replication fork. Chromosoma 117, 219–233. doi: 10.1007/s00412-007-0142-4
Baell, J. B., Leaver, D. J., Hermans, S. J., Kelly, G. L., Brennan, M. S., Downer, N. L., et al. (2018). Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 560, 253–257. doi: 10.1038/s41586-018-0387-5
Balasubramanian, D., Pearson, J. F., and Kennedy, M. A. (2019). Gene expression effects of lithium and valproic acid in a serotonergic cell line. Physiol. Genomics 51, 43–50. doi: 10.1152/physiolgenomics.00069.2018
Balemans, M. C. M., Ansar, M., Oudakker, A. R., van Caam, A. P. M., Bakker, B., Vitters, E. L., et al. (2014). Reduced Euchromatin histone methyltransferase 1 causes developmental delay, hypotonia, and cranial abnormalities associated with increased bone gene expression in Kleefstra syndrome mice. Dev. Biol. 386, 395–407. doi: 10.1016/j.ydbio.2013.12.016
Balmer, N. V., Weng, M. K., Zimmer, B., Ivanova, V. N., Chambers, S. M., Nikolaeva, E., et al. (2012). Epigenetic changes and disturbed neural development in a human embryonic stem cell-based model relating to the fetal valproate syndrome. Hum. Mol. Genet. 21, 4104–4114. doi: 10.1093/hmg/dds239
Banka, S., Veeramachaneni, R., Reardon, W., Howard, E., Bunstone, S., Ragge, N., et al. (2012). How genetically heterogeneous is Kabuki syndrome: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur. J. Hum. Genet. 20, 381–388. doi: 10.1038/ejhg.2011.220
Batshaw, M. L., and Brusilow, S. W. (1982). Valproate-induced hyperammonemia. Ann. Neurol. 11, 319–321. doi: 10.1002/ana.410110315
Bittigau, P., Sifringer, M., Genz, K., Reith, E., Pospischil, D., Govindarajalu, S., et al. (2002). Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc. Natl. Acad. Sci. U.S.A. 99, 15089–15094. doi: 10.1073/PNAS.222550499
Boudadi, E., Stower, H., Halsall, J. A., Rutledge, C. E., Leeb, M., Wutz, A., et al. (2013). The histone deacetylase inhibitor sodium valproate causes limited transcriptional change in mouse embryonic stem cells but selectively overrides Polycomb-mediated Hoxb silencing. Epigenet. Chromatin 6:11. doi: 10.1186/1756-8935-6-11
Brodie, S. A., and Brandes, J. C. (2014). Could valproic acid be an effective anticancer agent? The evidence so far. Expert Rev. Anticancer Ther. 14, 1097–1100. doi: 10.1586/14737140.2014.940329
Bromley, R., Weston, J., Adab, N., Greenhalgh, J., Sanniti, A., McKay, A. J., et al. (2014). Treatment for epilepsy in pregnancy: neurodevelopmental outcomes in the child. Cochrane Database Syst. Rev. 2014:CD010236. doi: 10.1002/14651858.CD010236.pub2
Bromley, R. L., Baker, G. A., Clayton-Smith, J., and Wood, A. G. (2019). Intellectual functioning in clinically confirmed fetal valproate syndrome. Neurotoxicol. Teratol. 71, 16–21. doi: 10.1016/j.ntt.2018.11.003
Bromley, R. L., Mawer, G. E., Briggs, M., Cheyne, C., Clayton-Smith, J., García-Fiñana, M., et al. (2013). The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. J. Neurol. Neurosurg. Psychiatry 84, 637–643. doi: 10.1136/jnnp-2012-304270
Brown, N., Burgess, T., Forbes, R., Mcgillivray, G., Kornberg, A., Mandelstam, S., et al. (2013). 5q31.3 microdeletion syndrome: clinical and molecular characterization of two further cases. Am. J. Med. Genet. Part A 161, 2604–2608. doi: 10.1002/ajmg.a.36108
Bruni, J., Wilder, B. J., Willmore, L. J., and Barbour, B. (1979). Valproic acid and plasma levels of phenytoin. Neurology 29, 904–905. doi: 10.1212/wnl.29.6.904
Brunton, L. L., Hilal-Dandan, R., and Knollmann, B. C. (2018). Goodman & Gilman’s The Pharmacological Basis of Therapeutics. New York, NY: McGraw-Hill.
Camarena, V., Cao, L., Abad, C., Abrams, A., Toledo, Y., Araki, K., et al. (2014). Disruption of Mbd5 in mice causes neuronal functional deficits and neurobehavioral abnormalities consistent with 2q23.1 microdeletion syndrome. EMBO Mol. Med. 6, 1003–1015. doi: 10.15252/emmm.201404044
Carosso, G. A., Boukas, L., Augustin, J. J., Nguyen, H. N., Winer, B. L., Cannon, G. H., et al. (2019). Precocious neuronal differentiation and disrupted oxygen responses in Kabuki syndrome. JCI Insight 4:e129375. doi: 10.1172/jci.insight.129375
Chandane, P. G., and Shah, I. (2014). Fetal valproate syndrome. Indian J. Hum. Genet. 20, 187–188. doi: 10.4103/0971-6866.142898
Chen, E. S., Gigek, C. O., Rosenfeld, J. A., Diallo, A. B., Maussion, G., Chen, G. G., et al. (2014). Molecular convergence of neurodevelopmental disorders. Am. J. Hum. Genet. 95, 490–508. doi: 10.1016/j.ajhg.2014.09.013
Chen, G., Zeng, W. Z., Yuan, P. X., Huang, L. D., Jiang, Y. M., Zhao, Z. H., et al. (1999). The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J. Neurochem. 72, 879–882. doi: 10.1046/j.1471-4159.1999.720879.x
Chiu, C. T., Wang, Z., Hunsberger, J. G., and Chuang, D. M. (2013). Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol. Rev. 65, 105–142. doi: 10.1124/pr.111.005512
Christensen, J., Grønborg, T. K., Sørensen, M. J., Schendel, D., Parner, E. T., Pedersen, L. H., et al. (2013). Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 309, 1696–1703. doi: 10.1001/jama.2013.2270
Cieslar-Pobuda, A., Ahrens, T. D., Caglayan, S., Behringer, S., Hannibal, L., and Staerk, J. (2020). DNMT3B deficiency alters mitochondrial biogenesis and α-ketoglutarate levels in human embryonic stem cells. Stem Cells 38, 1409–1422. doi: 10.1002/stem.3256
Clayton-Smith, J., Bromley, R., Dean, J., Journel, H., Odent, S., Wood, A., et al. (2019). Diagnosis and management of individuals with fetal valproate spectrum disorder; a consensus statement from the european reference network for congenital malformations and intellectual disability. Orphanet J. Rare Dis. 14:180. doi: 10.1186/s13023-019-1064-y
Clayton-Smith, J., and Donnai, D. (1995). Fetal valproate syndrome. J. Med. Genet. 32, 724–727. doi: 10.1136/jmg.32.9.724
Daniel, D. C., and Johnson, E. M. (2018). PURA, the gene encoding Pur-alpha, member of an ancient nucleic acid-binding protein family with mammalian neurological functions. Gene 643, 133–143. doi: 10.1016/j.gene.2017.12.004
Dhar, S. S., Lee, S. H., Chen, K., Zhu, G., Oh, W. K., Allton, K., et al. (2016). An essential role for UTX in resolution and activation of bivalent promoters. Nucleic Acids Res. 44, 3659–3674. doi: 10.1093/nar/gkv1516
Dhar, S. S., Zhao, D., Lin, T., Gu, B., Pal, K., Wu, S. J., et al. (2018). MLL4 is required to maintain broad h3k4me3 peaks and super-enhancers at tumor suppressor genes. Mol. Cell 70, 825.e6–841.e6. doi: 10.1016/j.molcel.2018.04.028
DiLiberti, J. H., Farndon, P. A., Dennis, N. R., and Curry, C. J. R. R. (1984). The fetal valproate syndrome. Am. J. Med. Genet. 19, 473–481. doi: 10.1002/ajmg.1320190308
Dufour-Rainfray, D., Vourc’h, P., Le Guisquet, A. M., Garreau, L., Ternant, D., Bodard, S., et al. (2010). Behavior and serotonergic disorders in rats exposed prenatally to valproate: a model for autism. Neurosci. Lett. 470, 55–59. doi: 10.1016/j.neulet.2009.12.054
Ehrlich, M., Jackson, K., and Weemaes, C. (2006). Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF). Orphanet J. Rare Dis. 1:2. doi: 10.1186/1750-1172-1-2
Fahrner, J. A., and Bjornsson, H. T. (2019). Mendelian disorders of the epigenetic machinery: postnatal malleability and therapeutic prospects. Hum. Mol. Genet. 28, R254–R264. doi: 10.1093/hmg/ddz174
Fan, H. (2008). Inhibition of de novo methyltransferase 3B is a potential therapy for hepatocellular carcinoma. Gastroenterol. Res. 1, 33–39. doi: 10.4021/gr2008.10.1240
Fang, X., Poulsen, R. R., Wang-Hu, J., Shi, O., Calvo, N. S., Simmons, C. S., et al. (2016). Knockdown of DNA methyltransferase 3a alters gene expression and inhibits function of embryonic cardiomyocytes. FASEB J. 30, 3238–3255. doi: 10.1096/fj.201600346R
Fei, D., Wang, Y., Zhai, Q., Zhang, X., Zhang, Y., Wang, Y., et al. (2021). KAT6A regulates stemness of aging bone marrow-derived mesenchymal stem cells through Nrf2/ARE signaling pathway. Stem Cell Res. Ther. 12:104. doi: 10.1186/s13287-021-02164-5
Feng, W., Kawauchi, D., Körkel-Qu, H., Deng, H., Serger, E., Sieber, L., et al. (2017a). Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat. Commun. 8:14758. doi: 10.1038/ncomms14758
Feng, W., Shao, C., and Liu, H. K. (2017b). Versatile roles of the chromatin remodeler CHD7 during brain development and disease. Front. Mol. Neurosci. 10:309. doi: 10.3389/fnmol.2017.00309
Finnell, R. H., Gelineau-van Waes, J., Eudy, J. D., and Rosenquist, T. H. (2002). Molecular basis of environmentally induced birth defects. Annu. Rev. Pharmacol. Toxicol. 42, 181–208. doi: 10.1146/annurev.pharmtox.42.083001.110955
Frega, M., Selten, M., Mossink, B., Keller, J. M., Linda, K., Moerschen, R., et al. (2020). Distinct pathogenic genes causing intellectual disability and autism exhibit a common neuronal network hyperactivity phenotype. Cell Rep. 30, 173.e6–186.e6. doi: 10.1016/j.celrep.2019.12.002
Gerold, M. (1959). Frakturheillung bei kongenitaler Anomalie der oberen Gliedmassen. Zentralbl. Chir. 84, 831–834.
Gigek, C. O., Chen, E. S., Ota, V. K., Maussion, G., Peng, H., Vaillancourt, K., et al. (2015). A molecular model for neurodevelopmental disorders. Transl. Psychiatry 5:e565. doi: 10.1038/tp.2015.56
Glover, S. J., Quinn, A. G., Barter, P., Hart, J., Moore, S. J., Dean, J. C. S., et al. (2002). Ophthalmic findings in fetal anticonvulsant syndrome(s). Ophthalmology 109, 942–947. doi: 10.1016/s0161-6420(02)00959-4
Go, H. S., Seo, J. E., Kim, K. C., Han, S. M., Kim, P. K., Kang, Y. S., et al. (2011). Valproic acid inhibits neural progenitor cell death by activation of NF-B signaling pathway and up-regulation of Bcl-XL. J. Biomed. Sci. 18:48. doi: 10.1186/1423-0127-18-48
Gössling, K. L., Schipp, C., Fischer, U., Babor, F., Koch, G., Schuster, F. R., et al. (2017). Hematopoietic stem cell transplantation in an infant with immunodeficiency, centromeric instability, and facial anomaly syndrome. Front. Immunol. 8:773. doi: 10.3389/fimmu.2017.00773
Göttlicher, M., Minucci, S., Zhu, P., Krämer, O. H., Schimpf, A., Giavara, S., et al. (2001). Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 20, 6969–6978. doi: 10.1093/emboj/20.24.6969
Gupta-Agarwal, S., Franklin, A. V., DeRamus, T., Wheelock, M., Davis, R. L., McMahon, L. L., et al. (2012). G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J. Neurosci. 32, 5440–5453. doi: 10.1523/jneurosci.0147-12.2012
Hadzsiev, K., Komlosi, K., Czako, M., Duga, B., Szalai, R., Szabo, A., et al. (2016). Kleefstra syndrome in Hungarian patients: additional symptoms besides the classic phenotype. Mol. Cytogenet. 9:22. doi: 10.1186/s13039-016-0231-2
Hagleitner, M. M., Lankester, A., Maraschio, P., Hultén, M., Fryns, J. P., Schuetz, C., et al. (2008). Clinical spectrum of immunodeficiency, centromeric instability and facial dysmorphism (ICF syndrome). J. Med. Genet. 45, 93–99. doi: 10.1136/jmg.2007.053397
Hansen, R. S., Wijmenga, C., Luo, P., Stanek, A. M., Canfield, T. K., Weemaes, C. M. R., et al. (1999). The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. U.S.A. 96, 14412–14417. doi: 10.1073/pnas.96.25.14412
Hefner, M. A., and Fassi, E. (2017). Genetic Counseling in CHARGE Syndrome: Diagnostic Evaluation Through Follow Up. Malden, MA: Blackwell Publishing Inc.
Hodge, J. C., Mitchell, E., Pillalamarri, V., Toler, T. L., Bartel, F., Kearney, H. M., et al. (2014). Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol. Psychiatry 19, 368–379. doi: 10.1038/mp.2013.42
Hsu, J., Huang, H. T., Lee, C. T., Choudhuri, A., Wilson, N. K., Abraham, B. J., et al. (2020). CHD7 and Runx1 interaction provides a braking mechanism for hematopoietic differentiation. Proc. Natl. Acad. Sci. U.S.A. 117, 23626–23635. doi: 10.1073/pnas.2003228117
Hunt, D., Leventer, R. J., Simons, C., Taft, R., Swoboda, K. J., Gawne-Cain, M., et al. (2014). Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J. Med. Genet. 51, 806–813. doi: 10.1136/jmedgenet-2014-102798
Issaeva, I., Zonis, Y., Rozovskaia, T., Orlovsky, K., Croce, C. M., Nakamura, T., et al. (2007). Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol. Cell. Biol. 27, 1889–1903. doi: 10.1128/mcb.01506-06
Iype, M., Henry, P., Aravind, C., and Arun, K. (2008). Baller-Gerold syndrome: further evidence for association with prenatal exposure to valproate. Ann. Indian Acad. Neurol. 11, 52–55. doi: 10.4103/0972-2327.40228
Jackson, A., Bromley, R., Morrow, J., Irwin, B., and Clayton-Smith, J. (2016). In utero exposure to valproate increases the risk of isolated cleft palate. Arch. Dis. Child. Fetal Neonatal Ed. 101, F207–F211. doi: 10.1136/archdischild-2015-308278
Jackson, A., Fryer, A., Clowes, V., and Clayton-Smith, J. (2014). Ocular coloboma and foetal valproate syndrome: four further cases and a hypothesis for aetiology. Clin. Dysmorphol. 23, 74–75. doi: 10.1097/MCD.0000000000000028
Jacob, J., Ribes, V., Moore, S., Constable, S., Wilkinson, D., and Briscoe, J. (2013). A Chemical-Genetics Approach to Study the Molecular Pathology of Central Serotonin Abnormalities in Fetal Valproate Syndrome. Available online at: www.thelancet.com (accessed April 1, 2020).
Jacob, J., Ribes, V., Moore, S., Constable, S. C., Sasai, N., Gerety, S. S., et al. (2014). Valproic acid silencing of ascl1b/Ascl1 results in the failure of serotonergic differentiation in a zebrafish model of fetal valproate syndrome. DMM Dis. Model. Mech. 7, 107–117. doi: 10.1242/dmm.013219
Jeanpierre, M., Turleau, C., Aurias, A., Prieur, M., Ledeist, F., Fischer, A., et al. (1993). An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum. Mol. Genet. 2, 731–735. doi: 10.1093/hmg/2.6.731
Jergil, M., Forsberg, M., Salter, H., Stockling, K., Gustafson, A. L., Dencker, L., et al. (2011). Short-time gene expression response to valproic acid and valproic acid analogs in mouse embryonic stem cells. Toxicol. Sci. 121, 328–342. doi: 10.1093/toxsci/kfr070
Jergil, M., Kultima, K., Gustafson, A. L., Dencker, L., and Stigson, M. (2009). Valproic acid-induced deregulation In vitro of genes associated in vivo with neural tube defects. Toxicol. Sci. 108, 132–148. doi: 10.1093/toxsci/kfp002
Johannessen, C. U., and Johannessen, S. I. (2003). Valproate: past, present, and future. CNS Drug Rev. 9, 199–216. doi: 10.1111/j.1527-3458.2003.tb00249.x
Kamae, C., Imai, K., Kato, T., Okano, T., Honma, K., Nakagawa, N., et al. (2018). Clinical and immunological characterization of ICF syndrome in Japan. J. Clin. Immunol. 38, 927–937. doi: 10.1007/s10875-018-0559-y
Katsumoto, T., Aikawa, Y., Iwama, A., Ueda, S., Ichikawa, H., Ochiya, T., et al. (2006). MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 20, 1321–1330. doi: 10.1101/gad.1393106
Kennedy, J., Goudie, D., Blair, E., Chandler, K., Joss, S., McKay, V., et al. (2019). KAT6A Syndrome: genotype–phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet. Med. 21, 850–860. doi: 10.1038/s41436-018-0259-2
Kim, J. H., Sharma, A., Dhar, S. S., Lee, S. H., Gu, B., Chan, C. H., et al. (2014). UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 74, 1705–1717. doi: 10.1158/0008-5472.CAN-13-1896
Kim, K. C., Kim, P., Go, H. S., Choi, C. S., Yang, S. I. L., Cheong, J. H., et al. (2011). The critical period of valproate exposure to induce autistic symptoms in Sprague-Dawley rats. Toxicol. Lett. 201, 137–142. doi: 10.1016/j.toxlet.2010.12.018
Kini, U., Adab, N., Vinten, J., Fryer, A., and Clayton-Smith, J. (2006). Dysmorphic features: an important clue to the diagnosis and severity of fetal anticonvulsant syndromes. Arch. Dis. Child. Fetal Neonatal Ed. 91, 90–95. doi: 10.1136/adc.2004.067421
Kitao, S., Shimamoto, A., Goto, M., Miller, R. W., Smithson, W. A., Lindor, N. M., et al. (1999). Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 22, 82–84. doi: 10.1038/8788
Kleefstra, T., Brunner, H. G., Amiel, J., Oudakker, A. R., Nillesen, W. M., Magee, A., et al. (2006). Loss-of-function mutations in Euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 79, 370–377. doi: 10.1086/505693
Kleefstra, T., and De Leeuw, N. (1993). Kleefstra Syndrome Synonyms: 9q Subtelomeric Deletion Syndrome, 9q34.3 Microdeletion Syndrome, 9qSTDS. Available online at: https://www.ncbi.nlm.nih.gov/books/ (accessed January 10, 2021).
Kleefstra, T., Kramer, J. M., Neveling, K., Willemsen, M. H., Koemans, T. S., Vissers, L. E. L. M., et al. (2012). Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am. J. Hum. Genet. 91, 73–82. doi: 10.1016/j.ajhg.2012.05.003
Koemans, T. S., Kleefstra, T., Chubak, M. C., Stone, M. H., Reijnders, M. R. F., de Munnik, S., et al. (2017). Functional convergence of histone methyltransferases EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. PLoS Genet. 13:e1006864. doi: 10.1371/journal.pgen.1006864
Kong, Q., Yu, M., Zhang, M., Wei, C., Gu, H., Yu, S., et al. (2020). Conditional Dnmt3b deletion in hippocampal dCA1 impairs recognition memory. Mol. Brain 13:42. doi: 10.1186/s13041-020-00574-9
Kotajima-Murakami, H., Kobayashi, T., Kashii, H., Sato, A., Hagino, Y., Tanaka, M., et al. (2019). Effects of rapamycin on social interaction deficits and gene expression in mice exposed to valproic acid in utero. Mol. Brain 12:3. doi: 10.1186/s13041-018-0423-2
Kozma, C. (2001). Valproic acid embryopathy: report of two siblings with further expansion of the phenotypic abnormalities and a review of the literature. Am. J. Med. Genet. 98, 168–175.
Kuroki, Y., Suzuki, Y., Chyo, H., Hata, A., and Matsui, I. (1981). A new malformation syndrome of long palpebralfissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J. Pediatr. 99, 570–573. doi: 10.1016/S0022-3476(81)80256-9
Lalani, S. R., Zhang, J., Schaaf, C. P., Brown, C. W., Magoulas, P., Tsai, A. C. H., et al. (2014). Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am. J. Hum. Genet. 95, 579–583. doi: 10.1016/j.ajhg.2014.09.014
Lammer, E. J., Sever, L. E., and Oakley, G. P. (1987). Teratogen update: valproic acid. Teratology 35, 465–473. doi: 10.1002/tera.1420350319
Larizza, L., Roversi, G., and Volpi, L. (2010). Rothmund-thomson syndrome. Orphanet J. Rare Dis. 5:2. doi: 10.1186/1750-1172-5-2
Lederer, D., Grisart, B., Digilio, M. C., Benoit, V., Crespin, M., Ghariani, S. C., et al. (2012). Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with kabuki syndrome. Am. J. Hum. Genet. 90, 119–124. doi: 10.1016/j.ajhg.2011.11.021
Lin, Y. L., Bialer, M., Cabrera, R. M., Finnell, R. H., and Wlodarczyk, B. J. (2019). Teratogenicity of valproic acid and its constitutional isomer, amide derivative valnoctamide in mice. Birth Defects Res. 111, 1013–1023. doi: 10.1002/bdr2.1406
Lindhout, D., and Schmidt, D. (1986). In-utero exposure to valproate and neural tube defects. Lancet 1, 1392–1393. doi: 10.1016/s0140-6736(86)91711-3
Liu, C., Li, Q., Xiao, Q., Gong, P., and Kang, N. (2020). CHD7 regulates osteogenic differentiation of human dental follicle cells via PTH1R signaling. Stem Cells Int. 2020:8882857. doi: 10.1155/2020/8882857
Lopusna, K., Nowialis, P., Opavska, J., Abraham, A., Riva, A., and Opavsky, R. (2021). Dnmt3b catalytic activity is critical for its tumour suppressor function in lymphomagenesis and is associated with c-Met oncogenic signalling. EBioMedicine 63:e103191. doi: 10.1016/j.ebiom.2020.103191
Macfarlane, A., and Greenhalgh, T. (2018). Sodium valproate in pregnancy: what are the risks and should we use a shared decision-making approach? BMC Preg. Childbirth 18:200. doi: 10.1186/s12884-018-1842-x
Machado, R. A. C., Schneider, H., DeOcesano-Pereira, C., Lichtenstein, F., Andrade, F., Fujita, A., et al. (2019). CHD7 promotes glioblastoma cell motility and invasiveness through transcriptional modulation of an invasion signature. Sci. Rep. 9:3952. doi: 10.1038/s41598-019-39564-w
Makrythanasis, P., van Bon, B. W., Steehouwer, M., Rodríguez-Santiago, B., Simpson, M., Dias, P., et al. (2013). MLL2 mutation detection in 86 patients with Kabuki syndrome: a genotype-phenotype study. Clin. Genet. 84, 539–545. doi: 10.1111/cge.12081
Mansour, A. A., Gafni, O., Weinberger, L., Zviran, A., Ayyash, M., Rais, Y., et al. (2012). The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature 488, 409–413. doi: 10.1038/nature11272
Maraschio, P., Zuffardi, O., Dalla Fior, T., and Tiepolo, L. (1988). Immunodeficiency, centromeric heterochromatin instability of chromosomes 1, 9, and 16, and facial anomalies: the ICF syndrome. J. Med. Genet. 25, 173–180. doi: 10.1136/jmg.25.3.173
Marchion, D. C., Bicaku, E., Daud, A. I., Sullivan, D. M., and Munster, P. N. (2005). Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res. 65, 3815–3822. doi: 10.1158/0008-5472.CAN-04-2478
Marie, C., Clavairoly, A., Frah, M., Hmidan, H., Yan, J., Zhao, C., et al. (2018). Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc. Natl. Acad. Sci. U.S.A. 115, E8246–E8255. doi: 10.1073/pnas.1802620115
Massa, V., Cabrera, R. M., Menegola, E., Giavini, E., and Finnell, R. H. (2005). Valproic acid-induced skeletal malformations: associated gene expression cascades. Pharmacogenet. Genomics 15, 787–800. doi: 10.1097/01.fpc.0000170914.11898.3a
Massa, V., Greene, N. D. E., and Copp, A. J. (2009). Do cells become homeless during neural tube closure? Cell Cycle 8, 2479–2480. doi: 10.4161/cc.8.16.9272
Massa, V., Wlodarczyk, B., Giavini, E., and Finnell, R. H. (2006). Myo-inositol enhances teratogenicity of valproic acid in the mouse. Birth Defects Res. Part A Clin. Mol. Teratol. 76, 200–204. doi: 10.1002/bdra.20228
Meador, K. J., Pennell, P. B., May, R. C., Brown, C. A., Baker, G., Bromley, R., et al. (2020). Effects of periconceptional folate on cognition in children of women with epilepsy: NEAD study. Neurology 94, e729–e740. doi: 10.1212/WNL.0000000000008757
Megarbane, A., Melki, I., Souraty, N., Gerbaka, J., El Ghouzzi, V., Bonaventure, J., et al. (2000). Overlap between Baller-Gerold and Rothmund-Thomson syndrome. Clin. Dysmorphol. 9, 303–305. doi: 10.1097/00019605-200009040-00018
Menegola, E., Broccia, M., Prati, M., and Giavini, E. (1999). Morphological alterations induced by sodium valproate on somites and spinal nerves in rat embryos. Teratology 59, 110–119.
Menegola, E., Broccia, M. L., Prati, M., and Giavini, E. (1998). Stage-dependent skeletal malformations induced by valproic acid in rat. Int. J. Dev. Biol. 42, 99–102. doi: 10.1387/IJDB.9496792
Menegola, E., Di Renzo, F., Broccia, M. L., Prudenziati, M., Minucci, S., Massa, V., et al. (2005). Inhibition of histone deacetylase activity on specific embryonic tissues as a new mechanism for teratogenicity. Birth Defects Res. Part B Dev. Reprod. Toxicol. 74, 392–398. doi: 10.1002/bdrb.20053
Meunier, H., Carraz, G., Neunier, Y., Eymard, P., and Aimard, M. (1963). Pharmacodynamic properties of N-dipropylacetic acid. Therapie 18, 435–438.
Millan, F., Cho, M. T., Retterer, K., Monaghan, K. G., Bai, R., Vitazka, P., et al. (2016). Whole exome sequencing reveals de novo pathogenic variants in KAT6A as a cause of a neurodevelopmental disorder. Am. J. Med. Genet. Part A 170, 1791–1798. doi: 10.1002/ajmg.a.37670
Min, G. L., Villa, R., Trojer, P., Norman, J., Yan, K. P., Reinberg, D., et al. (2007). Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318, 447–450. doi: 10.1126/science.1149042
Miyake, N., Mizuno, S., Okamoto, N., Ohashi, H., Shiina, M., Ogata, K., et al. (2013). KDM6A point mutations cause kabuki syndrome. Hum. Mutat. 34, 108–110. doi: 10.1002/humu.22229
Mohd Yunos, H., and Green, A. (2018). Fetal valproate syndrome: the Irish experience. Ir. J. Med. Sci. 187, 965–968. doi: 10.1007/s11845-018-1757-6
Mullegama, S. V., Mendoza-Londono, R., and Elsea, S. H. (2016). MBD5 Haploinsufficiency. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/27786435 (accessed December 2, 2020).
Murko, C., Lagger, S., Steiner, M., Seiser, C., Schoefer, C., and Pusch, O. (2013). Histone deacetylase inhibitor Trichostatin A induces neural tube defects and promotes neural crest specification in the chicken neural tube. Differentiation 85, 55–66. doi: 10.1016/j.diff.2012.12.001
Murray, C., Abel, S., McClure, M., Foster, J., Walke, M., Jayakar, P., et al. (2017). Novel causative variants in DYRK1A, KARS, and KAT6A associated with intellectual disability and additional phenotypic features. J. Pediatr. Genet. 06, 077–083. doi: 10.1055/s-0037-1598639
Nau, H. (1985). Teratogenic valproic acid concentrations: infusion by implanted minipumps vs conventional injection regimen in the mouse. Toxicol. Appl. Pharmacol. 80, 243–250. doi: 10.1016/0041-008X(85)90081-X
Nau, H., Hauck, R. S., and Ehlers, K. (1991). Valproic acid-induced neural tube defects in mouse and human: aspects of chirality, alternative drug development, pharmacokinetics and possible mechanisms. Pharmacol. Toxicol. 69, 310–321. doi: 10.1111/j.1600-0773.1991.tb01303.x
Nau, H., Zierer, R., Spielmann, H., Neubert, D., and Gansau, C. (1981). A new model for embryotoxicity testing: teratogenicity and pharmacokinetics of valproic acid following constant-rate administration in the mouse using human therapeutic drug and metabolite concentrations. Life Sci. 29, 2803–2813. doi: 10.1016/0024-3205(81)90541-5
Ng, S. B., Bigham, A. W., Buckingham, K. J., Hannibal, M. C., McMillin, M. J., Gildersleeve, H. I., et al. (2010). Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42, 790–793. doi: 10.1038/ng.646
Nicolini, C., and Fahnestock, M. (2018). The valproic acid-induced rodent model of autism. Exp. Neurol. 299, 217–227. doi: 10.1016/j.expneurol.2017.04.017
Niikawa, N., Matsuura, N., Fukushima, Y., Ohsawa, T., and Kajii, T. (1981). Kabuki make-up syndrome: a syndrome of mentalretardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr. 99, 565–569. doi: 10.1016/S0022-3476(81)80255-7
Nowialis, P., Lopusna, K., Opavska, J., Haney, S. L., Abraham, A., Sheng, P., et al. (2019). Catalytically inactive Dnmt3b rescues mouse embryonic development by accessory and repressive functions. Nat. Commun. 10:4374. doi: 10.1038/s41467-019-12355-7
Oberemm, A., and Kirschbaum, F. (1992). Valproic acid induced abnormal development of the central nervous system of three species of amphibians: implications for neural tube defects and alternative experimental systems. Teratog. Carcinog. Mutagen. 12, 251–262. doi: 10.1002/tcm.1770120603
Ornoy, A. (2009). Valproic acid in pregnancy: how much are we endangering the embryo and fetus? Reprod. Toxicol. 28, 1–10. doi: 10.1016/J.REPROTOX.2009.02.014
Ozkan, H., Cetinkaya, M., Köksal, N., and Yapici, S. (2011). Severe fetal valproate syndrome: combination of complex cardiac defect, multicystic dysplastic kidney, and trigonocephaly. J. Matern. Fetal. Neonatal Med. 24, 521–524. doi: 10.3109/14767058.2010.501120
Pagon, R. A., Graham, J. M., Zonana, J., and Yong, S. L. (1981). Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J. Pediatr. 99, 223–227. doi: 10.1016/S0022-3476(81)80454-4
Patsalos, P. N., Berry, D. J., Bourgeois, B. F. D., Cloyd, J. C., Glauser, T. A., Johannessen, S. I., et al. (2008). Antiepileptic drugs - best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ilae commission on therapeutic strategies. Epilepsia 49, 1239–1276. doi: 10.1111/j.1528-1167.2008.01561.x
Pennati, R., Groppelli, S., De Bernardi, F., and Sotgia, C. (2001). Action of valproic acid on Xenopus laevis development: teratogenic effects on eyes. Teratog. Carcinog. Mutagen. 21, 121–133.
Perucca, E. (2002). Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience. CNS Drugs 16, 695–714. doi: 10.2165/00023210-200216100-00004
Phiel, C. J., Zhang, F., Huang, E. Y., Guenther, M. G., Lazar, M. A., and Klein, P. S. (2001). Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 276, 36734–36741. doi: 10.1074/jbc.M101287200
Qiao, Y., Bagheri, H., Tang, F., Badduke, C., Martell, S., Lewis, S. M. E., et al. (2019). Exome sequencing identified a de novo mutation of PURA gene in a patient with familial Xp22.31 microduplication. Eur. J. Med. Genet. 62, 103–108. doi: 10.1016/j.ejmg.2018.06.010
Reijnders, M. R. F., Janowski, R., Alvi, M., Self, J. E., Van Essen, T. J., Vreeburg, M., et al. (2018). PURA syndrome: clinical delineation and genotype-phenotype study in 32 individuals with review of published literature. J. Med. Genet. 55, 104–113. doi: 10.1136/jmedgenet-2017-104946
Robert, E., and Guibaud, P. (1982). Maternal valproic acid and congenital neural tube defects. Lancet 2:937. doi: 10.1016/s0140-6736(82)90908-4
Rothmund, A. (1868). Ueber Cataracten in Verbindung mit einer eigenthümlichen Hautdegeneration. Arch. für Opthalmol. 14, 159–182. doi: 10.1007/BF02720945
Roullet, F. I., Wollaston, L., deCatanzaro, D., and Foster, J. A. (2010). Behavioral and molecular changes in the mouse in response to prenatal exposure to the anti-epileptic drug valproic acid. Neuroscience 170, 514–522. doi: 10.1016/j.neuroscience.2010.06.069
Sanaei, M., and Kavoosi, F. (2019). Effect of 5-aza-2’-deoxycytidine in comparison to valproic acid and trichostatin a on histone deacetylase 1, dna methyltransferase 1, and cip/kip family (p21, p27, and p57) genes expression, cell growth inhibition, and apoptosis induction in colon cancer sw480 cell line. Adv. Biomed. Res. 8:52. doi: 10.4103/abr.abr_91_19
Schölz, C., Weinert, B. T. B., Wagner, S. A. S., Beli, P., Miyake, Y., Qi, J., et al. (2015). Acetylation site specificities of lysine deacetylase inhibitors in human cells. Cells 33, 415–423.
Schorry, E. K., Oppenheimer, S. G., and Saal, H. M. (2005). Valproate embryopathy: clinical and cognitive profile in 5 siblings. Am. J. Med. Genet. 133A, 202–206. doi: 10.1002/ajmg.a.30494
Schulz, Y., Wehner, P., Opitz, L., Salinas-Riester, G., Bongers, E. M. H. F., van Ravenswaaij-Arts, C. M. A., et al. (2014). CHD7, the gene mutated in CHARGE syndrome, regulates genes involved in neural crest cell guidance. Hum. Genet. 133, 997–1009. doi: 10.1007/s00439-014-1444-2
Shah, K. H., Shailaja, S., and Girisha, K. M. (2014). Is coloboma a feature of fetal valproate syndrome? Clin. Dysmorphol. 23, 24–25. doi: 10.1097/MCD.0000000000000018
Shah, R. R., and Stonier, P. D. (2019). Repurposing old drugs in oncology: opportunities with clinical and regulatory challenges ahead. J. Clin. Pharm. Ther. 44, 6–22. doi: 10.1111/jcpt.12759
Shangguan, H., Su, C., Ouyang, Q., Cao, B., Wang, J., Gong, C., et al. (2019). Kabuki syndrome: novel pathogenic variants, new phenotypes and review of literature. Orphanet J. Rare Dis. 14:255. doi: 10.1186/s13023-019-1219-x
Sheikh, B. N., Yang, Y., Schreuder, J., Nilsson, S. K., Bilardi, R., Carotta, S., et al. (2016). MOZ (KAT6A) is essential for the maintenance of classically defined adult hematopoietic stem cells. Blood 128, 2307–2318. doi: 10.1182/blood-2015-10-676072
Sheikh, M. A., Malik, Y. S., and Zhu, X. (2017). RA-induced transcriptional silencing of checkpoint kinase-2 through promoter methylation by Dnmt3b is required for neuronal differentiation of P19 cells. J. Mol. Biol. 429, 2463–2473. doi: 10.1016/j.jmb.2017.07.005
Shinde, V., Perumalinivasan, S., Henry, M., Rotshteyn, T., Hescheler, J., Rahnenführer, J., et al. (2016). Comparison of a teratogenic transcriptome-based predictive test based on human embryonic versus inducible pluripotent stem cells. Stem Cell Res. Ther. 7:190. doi: 10.1186/s13287-016-0449-2
Shpargel, K. B., Starmer, J., Wang, C., Ge, K., and Magnuson, T. (2017). UTX-guided neural crest function underlies craniofacial features of Kabuki syndrome. Proc. Natl. Acad. Sci. U.S.A. 114, E9046–E9055. doi: 10.1073/pnas.1705011114
Sui, L., and Chen, M. (2012). Prenatal exposure to valproic acid enhances synaptic plasticity in the medial prefrontal cortex and fear memories. Brain Res. Bull. 87, 556–563. doi: 10.1016/J.BRAINRESBULL.2012.01.011
Swann, A. C., Bowden, C. L., Calabrese, J. R., Dilsaver, S. C., and Morris, D. D. (2002). Pattern of response to divalproex, lithium, or placebo in four naturalistic subtypes of mania. Neuropsychopharmacology 26, 530–536. doi: 10.1016/S0893-133X(01)00390-6
Talkowski, M. E., Mullegama, S. V., Rosenfeld, J. A., Van Bon, B. W. M., Shen, Y., Repnikova, E. A., et al. (2011). Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 89, 551–563. doi: 10.1016/j.ajhg.2011.09.011
Tanaka, A. J., Bai, R., Cho, M. T., Anyane-Yeboa, K., Ahimaz, P., Wilson, A. L., et al. (2015). De novo mutations in PURA are associated with hypotonia and developmental delay. Mol. Case Stud. 1:a000356. doi: 10.1101/mcs.a000356
Tham, E., Lindstrand, A., Santani, A., Malmgren, H., Nesbitt, A., Dubbs, H. A., et al. (2015). Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am. J. Hum. Genet. 96, 507–513. doi: 10.1016/j.ajhg.2015.01.016
Thomson, M. S. (1936). POIKILODERMA CONGEXITALE. Br. J. Dermatol. 48, 221–234. doi: 10.1111/j.1365-2133.1936.tb10332.x
Tomson, T., Battino, D., Bonizzoni, E., Craig, J., Lindhout, D., Perucca, E., et al. (2015). Dose-dependent teratogenicity of valproate in mono-and polytherapy. Neurology 85, 866–872. doi: 10.1212/WNL.0000000000001772
Tomson, T., Battino, D., Bonizzoni, E., Craig, J., Lindhout, D., Sabers, A., et al. (2011). Dose-dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 10, 609–617. doi: 10.1016/S1474-4422(11)70107-7
Turner-Ivey, B., Guest, S. T., Irish, J. C., Kappler, C. S., Garrett-Mayer, E., Wilson, R. C., et al. (2014). KAT6A, a chromatin modifier from the 8p11-p12 amplicon is a candidate oncogene in luminal breast cancer. Neoplasia 16, 644–655. doi: 10.1016/j.neo.2014.07.007
Van Bon, B. W. M., Koolen, D. A., Brueton, L., McMullan, D., Lichtenbelt, K. D., Adès, L. C., et al. (2010). The 2q23.1 microdeletion syndrome: clinical and behavioural phenotype. Eur. J. Hum. Genet. 18, 163–170. doi: 10.1038/ejhg.2009.152
van den Boogaard, M. L., Thijssen, P. E., Aytekin, C., Licciardi, F., Kıykım, A. A., Spossito, L., et al. (2017). Expanding the mutation spectrum in ICF syndrome: evidence for a gender bias in ICF2. Clin. Genet. 92, 380–387. doi: 10.1111/cge.12979
Van Maldergem, L., Siitonen, H. A., Jalkh, N., Chouery, E., De Roy, M., Delague, V., et al. (2006). Revisiting the craniosynostosis-radial ray hypoplasia association: baller-gerold syndrome caused by mutations in the RECQL4 gene. J. Med. Genet. 43, 148–152. doi: 10.1136/jmg.2005.031781
Van Maldergem, L., Verloes, A., Lejeune, L., and Gillerot, Y. (1992). The Baller-Gerold syndrome. J. Med. Genet. 29, 266–268. doi: 10.1136/jmg.29.4.266
van Ravenswaaij-Arts, C., and Martin, D. M. (2017). New insights and advances in CHARGE syndrome: diagnosis, etiologies, treatments, and research discoveries. Am. J. Med. Genet. Part C Semin. Med. Genet. 175, 397–406. doi: 10.1002/ajmg.c.31592
Viale, L., Allotey, J., Cheong-See, F., Arroyo-Manzano, D., McCorry, D., Bagary, M., et al. (2015). Epilepsy in pregnancy and reproductive outcomes: a systematic review and meta-analysis. Lancet 386, 1845–1852. doi: 10.1016/S0140-6736(15)00045-8
Vissers, L. E. L. M., De Vries, B. B. A., Osoegawa, K., Janssen, I. M., Feuth, T., Choy, C. O., et al. (2003). Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 73, 1261–1270. doi: 10.1086/379977
Vissers, L. E. L. M., Van Ravenswaaij, C. M. A., Admiraal, R., Hurst, J. A., De Vries, B. B. A., Janssen, I. M., et al. (2004). Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957. doi: 10.1038/ng1407
Voss, A. K., Collin, C., Dixon, M. P., and Thomas, T. (2009). Moz and retinoic acid coordinately regulate H3K9 acetylation, hox gene expression, and segment identity. Dev. Cell 17, 674–686. doi: 10.1016/j.devcel.2009.10.006
Weemaes, C. M., Van Tol, M. J., Wang, J., Van Ostaijen-Ten Dam, M. M., Van Eggermond, M. C., Thijssen, P. E., et al. (2013). Heterogeneous clinical presentation in icf syndrome: correlation with underlying gene defects. Eur. J. Hum. Genet. 21, 1219–1225. doi: 10.1038/ejhg.2013.40
Wegner, C., and Nau, H. (1992). Alteration of embryonic folate metabolism by valproic acid during organogenesis: implications for mechanism of teratogenesis - PubMed. Neurology 42, 17–24.
Whittaker, D. E., Riegman, K. L. H., Kasah, S., Mohan, C., Yu, T., Sala, B. P., et al. (2017). The chromatin remodeling factor CHD7 controls cerebellar development by regulating reelin expression. J. Clin. Invest. 127, 874–887. doi: 10.1172/JCI83408
Wijmenga, C., Van Den Heuvel, L. P. W. J., Strengman, E., Luyten, J. A. F. M., Van Der Burgt, I. J. A. M., De Groot, R., et al. (1998). Localization of the ICF syndrome to chromosome 20 by homozygosity mapping. Am. J. Hum. Genet. 63, 803–809. doi: 10.1086/302021
Willemsen, M. H., Vulto-Van Silfhout, A. T., Nillesen, W. M., Wissink-Lindhout, W. M., Van Bokhoven, H., Philip, N., et al. (2012). Update on Kleefstra syndrome. Mol. Syndromol. 2, 202–212. doi: 10.1159/000335648
Wiltse, J. (2005). Mode of action: inhibition of histone deacetylase, altering WNT-dependent gene expression, and regulation of beta-catenin—developmental effects of valproic acid. Crit. Rev. Toxicol. 35, 727–738. doi: 10.1080/10408440591007403
Xu, B., Mulvey, B., Salie, M., Yang, X., Matsui, Y., Nityanandam, A., et al. (2020). UTX/KDM6A suppresses AP-1 and a gliogenesis program during neural differentiation of human pluripotent stem cells. Epigenet. Chromatin 13:38. doi: 10.1186/s13072-020-00359-3
Xu, G. L., Bestor, T. H., BourC’His, D., Hsieh, C. L., Tommerup, N., Bugge, M., et al. (1999). Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 402, 187–191. doi: 10.1038/46052
Xu, Q., and Xie, W. (2018). Epigenome in early mammalian development: inheritance, reprogramming and establishment. Trends Cell Biol. 28, 237–253. doi: 10.1016/j.tcb.2017.10.008
Yao, H., Hannum, D. F., Zhai, Y., Hill, S. F., Albanus, R. D., Lou, W., et al. (2020). CHD7 promotes neural progenitor differentiation in embryonic stem cells via altered chromatin accessibility and nascent gene expression. Sci. Rep. 10:17445. doi: 10.1038/s41598-020-74537-4
Yao, H., Hill, S. F., Skidmore, J. M., Sperry, E. D., Swiderski, D. L., Sanchez, G. J., et al. (2018). CHD7 represses the retinoic acid synthesis enzyme ALDH1A3 during inner ear development. JCI Insight 3:e97440. doi: 10.1172/jci.insight.97440
Keywords: fetal valproate syndrome, chromatinopathies, anti-epileptic drugs, neurodevelopment, HDAC inhibitor
Citation: Parodi C, Di Fede E, Peron A, Viganò I, Grazioli P, Castiglioni S, Finnell RH, Gervasini C, Vignoli A and Massa V (2021) Chromatin Imbalance as the Vertex Between Fetal Valproate Syndrome and Chromatinopathies. Front. Cell Dev. Biol. 9:654467. doi: 10.3389/fcell.2021.654467
Received: 16 January 2021; Accepted: 01 April 2021;
Published: 20 April 2021.
Edited by:
Xiajun Li, ShanghaiTech University, ChinaReviewed by:
Taiping Chen, University of Texas MD Anderson Cancer Center, United StatesAlejandro Vaquero, Josep Carreras Leukaemia Research Institute (IJC), Spain
Copyright © 2021 Parodi, Di Fede, Peron, Viganò, Grazioli, Castiglioni, Finnell, Gervasini, Vignoli and Massa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Massa, dmFsZW50aW5hLm1hc3NhQHVuaW1pLml0
†These authors have contributed equally to this work and share last authorship