
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 25 March 2021
Sec. Stem Cell Research
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.653669
This article is part of the Research Topic Chromatin Regulation in Cell Fate Decisions View all 15 articles
 Puja Agrawal1,2
Puja Agrawal1,2 Sridhar Rao1,2,3*
Sridhar Rao1,2,3*Cell fate decisions are the backbone of many developmental and disease processes. In early mammalian development, precise gene expression changes underly the rapid division of a single cell that leads to the embryo and are critically dependent on autonomous cell changes in gene expression. To understand how these lineage specifications events are mediated, scientists have had to look past protein coding genes to the cis regulatory elements (CREs), including enhancers and insulators, that modulate gene expression. One class of enhancers, termed super-enhancers, is highly active and cell-type specific, implying their critical role in modulating cell-type specific gene expression. Deletion or mutations within these CREs adversely affect gene expression and development and can cause disease. In this mini-review we discuss recent studies describing the potential roles of two CREs, enhancers and binding sites for CTCF, in early mammalian development.
Understanding the molecular underpinnings of cell fate decisions is a central question within developmental biology. For decades it has been assumed that these decisions are mediated almost exclusively by the 2% of DNA that contains protein-coding genes, while the remaining 98% was considered “junk.” Over time, scientists have realized that this 2% of DNA could not exclusively explain all the developmental and disease-related decisions made during early mammalian development. This has given rise to a renewed focus on the remaining 98% of the genome, including non-coding DNA sequences and nuclear architecture and their role(s) in driving cell fate decisions by regulating gene expression. Nuclear architecture is the dynamic, three-dimensional organization of chromatin and its interaction with itself and regulatory proteins within the nucleus. The development of high-resolution imaging and chromosome conformation capture technologies, which permit a direct interrogation of long-range DNA:DNA interactions, has facilitated these important aspects, which regulate gene expression. Two types of cis-regulatory elements (CREs), which play a critical role in modulating gene expression, are enhancers and DNA-binding sites for CTCFs. In this mini-review we will focus on both classes of CREs specifically, as well as their mechanisms and roles in driving early cell fate decisions during early mammalian development. The examples discussed below do not encompass all the literature available by any measure but reflect selected key studies in the field focusing on embryonic stem cells (ESCs). For further information on enhancer and CTCF functions, we suggest these reviews (Ong and Corces, 2014; Shlyueva et al., 2014; Li et al., 2016; Chen et al., 2018; Rowley and Corces, 2018; Agrawal et al., 2019; Braccioli and De Wit, 2019).
Early placental mammalian development is characterized by numerous rapid, distinct cell fate decisions (Arnold and Robertson, 2009). Upon fertilization, an embryo is totipotent: it can form any cell type, both embryonic and extraembryonic tissues. After a few cell divisions the cells become pluripotent: they can form the three germ layers (mesoderm, endoderm, and ectoderm) but not extraembryonic tissue. At the blastocyst stage two distinct populations are formed: trophectoderm and the pluripotent inner cell mass (ICM). The trophectoderm, which eventually gives rise to the placenta, expresses CDX2, which turns off the pluripotency gene Pou5f1, which encodes the pluripotency critical transcription factor Oct3/4 (Strumpf et al., 2005; Rayon et al., 2016). The ICM maintains pluripotency by continuing expression of Oct3/4, which subsequently differentiates into the epiblast (EPI) and the primitive endoderm (PrE). The EPI eventually gives rise to all fetal tissues and maintains pluripotency by expressing Nanog, while the PrE, which will form the yolk sac, differentiates by down-regulating Nanog and expressing Gata6 (Messerschmidt and Kemler, 2010; Schrode et al., 2014). Development of the embryo is thus dependent on the proper temporal and spatial gene expression changes to drive lineage commitment. Experimentally, embryonic stem cells (ESCs) are derived from the early embryo and exist in multiple states, naïve (ICM) and primed (epiblast; Ghimire et al., 2018). These states are distinct with respect to transcription, where primed cells show a relative down-regulation of pluripotency genes and an up-regulation of lineage-specific genes, compared with the naïve embryos, with additional differences, indicating that pluripotency is not a single-cell state.
Enhancers are CREs that contain DNA-binding motifs for transcription factors (TFs) distal to the gene promoter, which are located immediately adjacent to the transcriptional start site (TSS). Binding of these TFs drives enhancers to promote or repress gene expression through multiple mechanisms that are beyond the scope of this review (Li et al., 2016; Chen et al., 2018; Agrawal et al., 2019). Next-generation sequencing-based (NGS) methods have revolutionized the genome-wide identification of enhancers. The combination of chromatin immunoprecipitation (ChIP) with NGS (ChIP-Seq) initially permitted the identification of potential enhancer regions based on p300 and H3K4me1 enrichment (Visel et al., 2009; Rada-Iglesias et al., 2011). These regions can be further subdivided based upon the enrichment of the mutually exclusive marks H3K27me3 and H3K27Ac, indicating whether an enhancer is “poised” or “active,” respectively. Genome-wide study of histone and TF enrichment has led to the identification of a distinct class of enhancers termed super-enhancers. Super-enhancers (SEs, Whyte et al., 2013) are a highly active minority of enhancers defined by their high density of binding lineage-specific TFs and associated cofactors such as the Mediator complex and p300. SEs also exhibit relatively higher levels of H3K27Ac and are larger in size overall compared with enhancers (Parker et al., 2013; Pulakanti et al., 2013). Several studies show that SE differ from classical enhancers due to their stronger ability to drive gene expression than classical enhancers (Hnisz et al., 2015; Huang et al., 2016; Shin et al., 2016; Thomas et al., 2021). SEs are also highly cell-type specific and are often found near key lineage determining genes, implying they are critical to the establishment and/or maintenance of cell identity. For example, in ESCs there are 231 super-enhancers (Whyte et al., 2013), which are defined by their enrichment for the pluripotency TFs Nanog, Oct4, and Sox2, as well as Mediator and high levels of H3K27Ac, and many are in close proximity to pluripotency-promoting genes, including Nanog.
The master pluripotency TF Nanog is near three SEs (−5SE, −45SE, +60SE, named based on distance from Nanog promoter in kb; Figure 1A; Pulakanti et al., 2013; Whyte et al., 2013; Blinka et al., 2016; Agrawal et al., 2021). These SEs and the numerous CTCF sites at the locus make it an ideal model system to understand how CREs regulate cell fate decisions by modulating Nanog expression (Blinka and Rao, 2017). Mouse-derived embryonic stem cells (mESCs) are an ideal model for studying enhancer-promoter interactions, which promote Nanog expression. Due to their pluripotent nature, they are sensitive to changes to gene expression with a simple observation of cell fate, differentiation. Tracking eGFP-tagged Nanog and isolating GFP– cells revealed that cells not expressing Nanog could give rise to Nanog expressing cells and continue to self-renew (Chambers et al., 2007). These cells do, however, give rise to fewer undifferentiated colonies, indicating that down-regulating Nanog predisposes but does not proscribe cells to differentiation. At the single-cell level, Nanog-low cells begin to down-regulate pluripotency-associated genes and up-regulate lineage-specific gene markers, while maintaining pluripotency (Abranches et al., 2014). Blastocyst division into the EPI and PrE is dependent on lineage specific gene expression and the location of the cells (Xenopoulos et al., 2015). One of the first markers of this division of cell fates is Nanog, wherein the presence of Nanog denotes EPI cell fate, while the loss of Nanog marks the development of PrE. As the ICM differentiates, there are a few PrE to EPI conversions, but no EPI to PrE conversions, indicating that the EPI state is a distinct form of pluripotency from the ICM. In culture, Nanog is expressed highly in naïve pluripotent ESCs but fluctuates in primed ESCs (Barral et al., 2019). Thus, the modulation of Nanog is important to understand, as the delicate regulation of the gene’s expression is critical to defining pluripotent states. Circular Chromosome Capture (3C) of the three SEs at the Nanog locus demonstrates that they physically interact with the Nanog promoter, but CRISPR-based deletion of each SE differentially alters expression (Blinka et al., 2016). Deletion of the −45SE causes an approximately 50% decrease in Nanog expression, deletion of the −5SE causes a nearly 90% decrease, but deletion of the +60SE causes no change (Figure 1B; Blinka et al., 2016; Agrawal et al., 2021). This leads to the important question: through what mechanism(s) do the −5SE and −45SE regulate Nanog, expression? Interestingly, single-cell RT-qPCR showed that deleting the −5SE altered Nanog expression in all cells demonstrates that it is functional in all cells. Additionally, analysis of RNA Polymerase II (RNAPII) dynamics upon deletion of the −5SE shows a complete loss of RNAPII at the Nanog promoter, indicating that the −5SE modulates Nanog expression by recruiting RNAPII or by promoting its conversion to its initiating form through phosphorylation of Ser 5 of the C-terminal domain (CTD, Figure 1C). Further analysis of the −45SE and its role in regulating Nanog gene expression is necessary to determine whether the two enhancers utilize the same or distinct mechanism(s) from the −5SE (Figure 1D; Henriques et al., 2018; Chen et al., 2018; Bartman et al., 2019; Sheridan et al., 2019; Zatreanu et al., 2019; Noe Gonzalez et al., 2020).
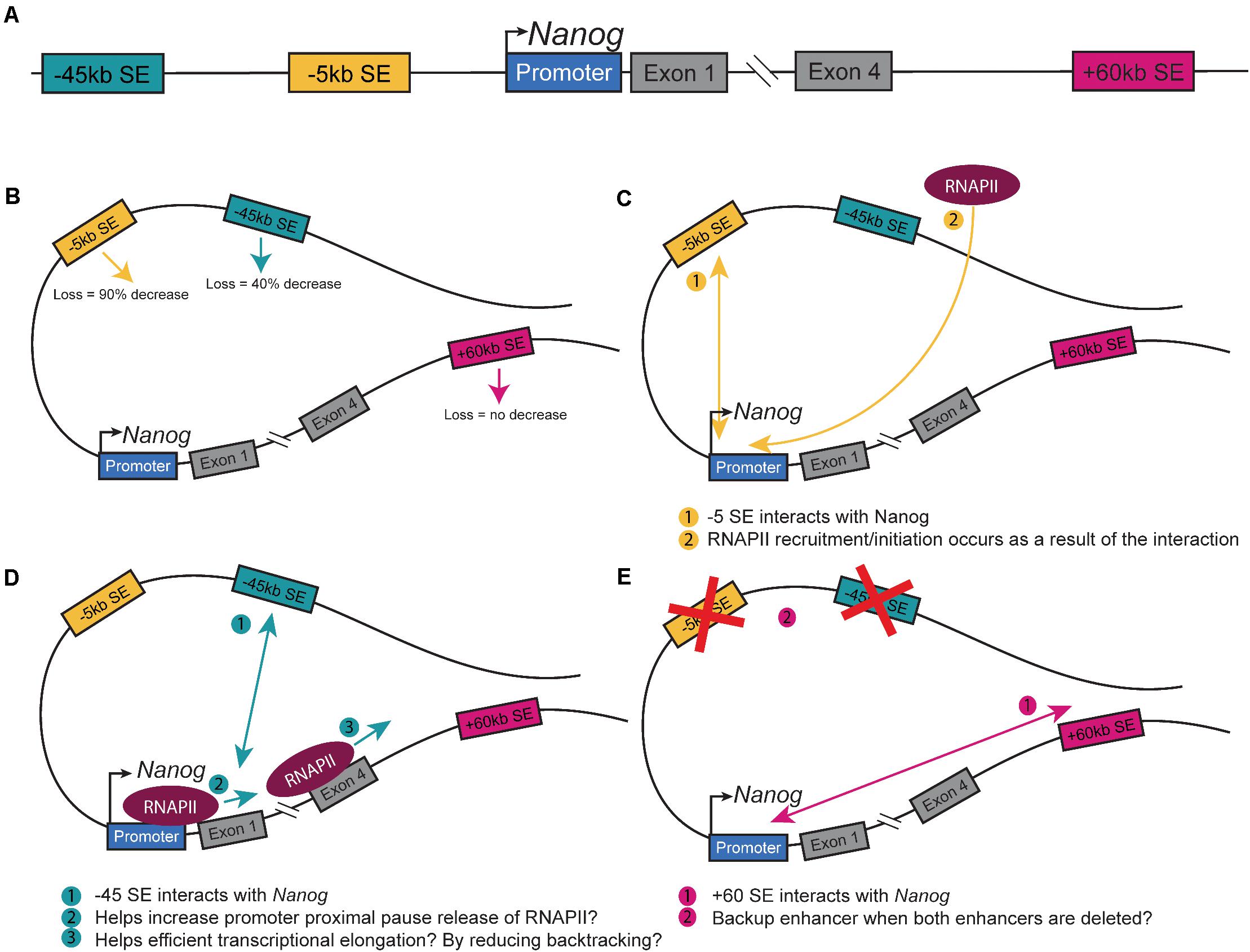
Figure 1. Nanog Super-enhancers. (A) Schematic of the Nanog extended locus with the three SEs. Schematic of a theoretical loop containing all three SEs and Nanog depicting. (B) The changes in Nanog expression upon deletion (Blinka et al., 2016; Agrawal et al., 2021). (C) The interaction between the −5SE and Nanog and how it alters RNAPII dynamics (Agrawal et al., 2021). (D) The interaction between the −45SE and Nanog and its potential mechanisms. (E) The interaction between the +60SE and Nanog and the possible backup role it has in regulating the gene.
Other lineage-critical genes are also regulated by enhancers. The expression of Shh, a gene critical at multiple stages of development in many tissues, is critically regulated by enhancers (Anderson and Hill, 2014). In the zone of polarizing activity (ZPA) in the developing limb, mutations in the ZPA regulatory sequence (ZRS) cause polydactyly due to misregulation of Shh in early limb development (Lettice et al., 2003). The ZRS is interesting as it is one megabase away from Shh, displaying just how distant the regulation of some genes can be. The interaction of the ZRS and Shh is specific to E10.5–11.5 mouse embryos, limiting the ZRS activity to limb development stages (Williamson et al., 2016). Interestingly, the ZRS, a highly conserved regulatory region in vertebrates, consists of multiple discrete enhancer elements that regulate gene expression and long-range interactions, emphasizing the multiple and varied roles of enhancers in regulating development (Lettice et al., 2017). In the brain, numerous Shh-Brain-Enhancers (SBEs) modulate the spatiotemporal expression of Shh in the developing midbrain (Jeong et al., 2006; Yao et al., 2016). Curiously, deletion of the SBEs can alter Shh expression in neural progenitor cells and brain development (Jeong et al., 2008; Benabdallah et al., 2016), but 3D-FISH analysis of Shh and SBEs shows an increase in distance between the gene promoter and regulatory element, implying a mechanism independent of looping (Benabdallah et al., 2019).
Sox2, another pluripotency gene, has multiple enhancer regions, some of which have no effect on pluripotency when deleted but have a potential role in neural cells (Ferri et al., 2004). Other regions, specifically the Sox2 Control Regions (SCR) disrupt maintenance of pluripotency in ESCs when deleted (Zhou et al., 2014). Similarly, to Shh, although the SCR is critical to Sox2 transcription, recent live cell imaging shows that in ESCs, the SCR and Sox2 are not in close physical proximity within the nucleus, indicating they operate at a distance (Alexander et al., 2019).
As an example of the interface between development and disease, enhancers play a critical role in the neural-crest-derived craniofacial disorder, the Pierre Robin sequence (PRS), which is classically characterized by mandibular underdevelopment along with other characteristics (Long et al., 2020). PRS is linked to mutations in a Topologically Associated Domain (TAD, described below), containing a single protein coding gene, Sox9 (Gordon et al., 2014). Sox9 plays a critical role during neural crest differentiation and craniofacial development (Lee and Saint-Jeannet, 2011; Dash and Trainor, 2020; Nagakura et al., 2020) and enhancers near Sox9 have been implicated in craniofacial and chondrocyte development (Liu and Lefebvre, 2015; Yao et al., 2015). PRS associated mutations are >1Mb from Sox9 and near a cluster of three enhancers the deletion of which leads to a 50% allele-specific decrease in Sox9 expression (Long et al., 2020). These three clusters are enriched for activating chromatin marks exclusively in the developing human neural crest cells (hNCC), indicating a cell-type specific role for these enhancers. In a mouse model, deletion of one of these enhancer clusters causes a modest 13% decrease in Sox9 expression that is sufficient to cause mandibular developmental changes and adversely effect survival, indicating that a dose-dependent change in Sox9 expression due to enhancer cluster deletion alters mandibular development. Enhancer control of the Shh, Sox2 and Sox9 loci demonstrate how CRE modulation of gene expression allows one gene to play multiple roles at various points in development.
Although all the nuances of how enhancers determine cell fate are still being investigated, we do understand that their role is critical to a range of developmental processes. Two mechanistic insights not discussed here but also under investigation are whether and how enhancers modulate gene expression by regulating transcriptional bursting (Bartman et al., 2016, 2019; Fukaya et al., 2016) and/or phase separation (Cho et al., 2018; Sabari et al., 2018; Zamudio et al., 2019). Analysis of Shh and Sox2 enhancers has shown that physical proximity may not be necessary, further expanding the question of the mechanisms that enhancers use to modulate gene expression.
DNA is organized within the nucleus on multiple levels (Ong and Corces, 2014; Rowley and Corces, 2018). Double-helix DNA is packaged into chromatin, which is further folded into nucleosomes that can be regulated by histone modifications. The development of 3C-based techniques has allowed the study of chromatin-chromatin interactions across large genomic distances (Dekker et al., 2002; de Laat and Dekker, 2012). Hi-C, a technique that studies long range chromatin-chromatin interactions, has led to the definition of Topologically Associated Domains (Figure 2A, TADs). TADs are large regions of chromatin, bounded by CCCCTC binding factor (CTCF) binding sites, in which the majority of interactions remain within the TAD. CTCF is a DNA-binding insulator protein and has binding sites present throughout the genome, within and at the boundaries of TADs. TADs can be further broken down to sub-TADs which are cell-type specific insulated regions within a TAD bounded by CTCF and cohesin (Figure 2A, Phillips-Cremins et al., 2013; Dowen et al., 2014; Hnisz et al., 2016). These sub-TADs are found to contain tissue or development-specific genes. Insulated neighborhoods (INs) are among the types of sub-TAD specifically identified in ESCs described as a CTCF-CTCF loop containing an SE and its target gene (Dowen et al., 2014). Deletion of one boundary CTCF site at key pluripotency genes (i.e., Nanog, Pouf51) is sufficient to alter gene expression, indicating these INs facilitate proper gene expression regulation, perhaps by creating and/or preventing enhancer-promoter interactions. A key point to consider is that INs and TADs were identified using different techniques and therefore may be identifying the same phenomenon but at different scales due to methodological differences.
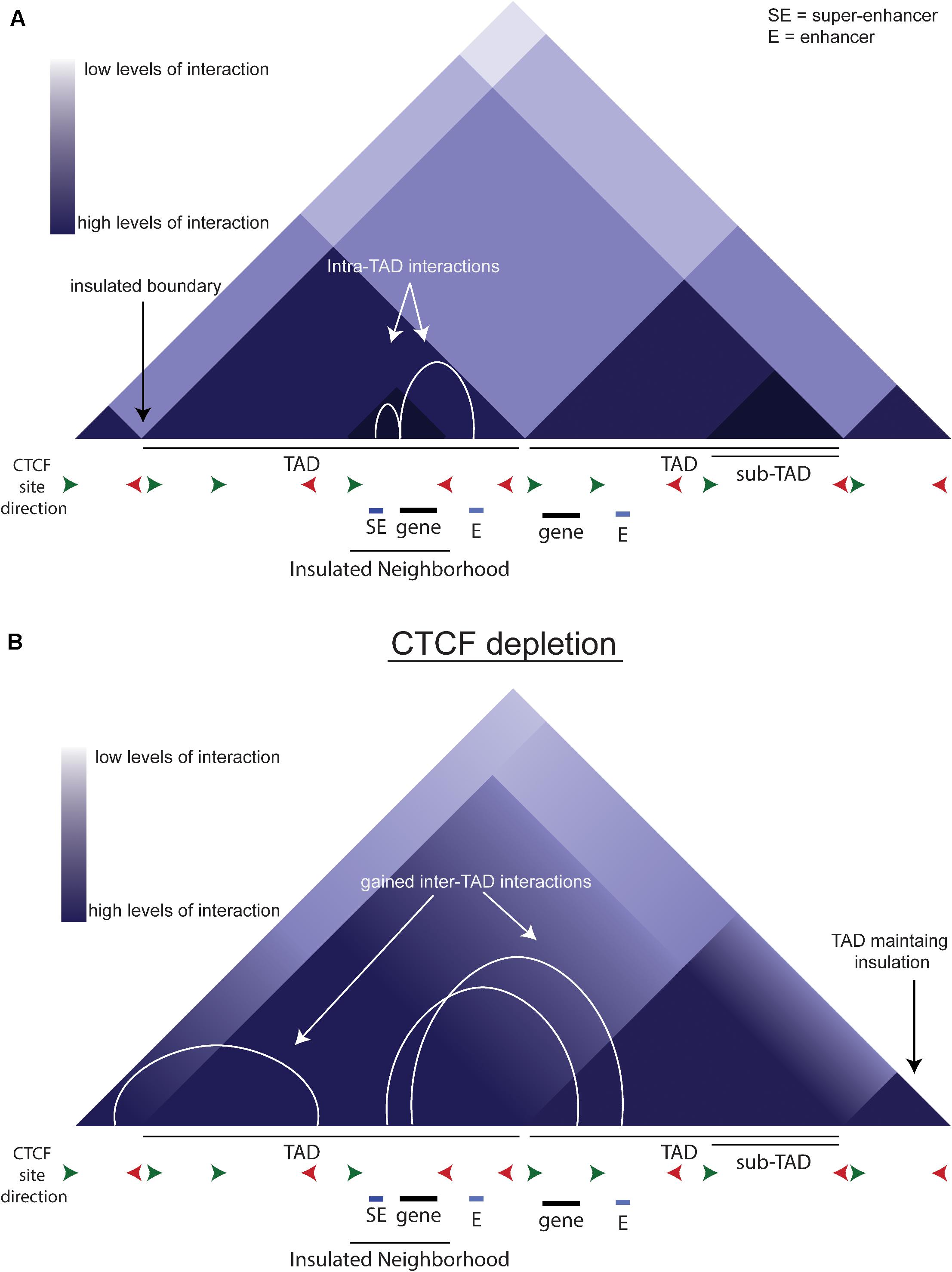
Figure 2. TADs and CTCF. (A) Schematic of chromatin organization. The triangles depict a schematic of a Hi-C contact map. White arcs show the intra-TAD interactions that are insulated by CTCF. (B) Schematic of what a Hi-C contact map may look like upon CTCF depletion. White arcs show the gained interactions across previous TAD boundaries.
The insulator protein CTCF contains 11 highly-conserved zinc-finger DNA binding domains (Phillips and Corces, 2009; Ong and Corces, 2014; Rowley and Corces, 2018). Insulator proteins have numerous functions, including regulating intrachromosomal interactions and the spread of heterochromatin. Knockout of CTCF is embryonic lethal at E5.5, and depletion of maternal CTCF in oocytes adversely impacts blastocyst stage embryos (Moore et al., 2012). CTCF has pleiotropic effects on gene expression, including altering DNA methylation and affecting splicing, as well as the regulation of enhancer-promoter interactions through chromatin looping (Ong and Corces, 2014). Current dogma supports an extrusion model of chromatin looping between two CTCF-bound sites that are in convergent orientation in collaboration with the cohesin complex (Rao et al., 2014; de Wit et al., 2015). The cohesin complex is a ring structure made up of four protein components, Rad21, Smc1, Smc3, and Stag1/2 (Fisher et al., 2017). While its primary function is to stabilize sister chromatids during cell division, cohesin also plays a critical role in maintaining chromatin loops, often in conjunction with CTCF. But 50–80% of CTCF binding sites are co-localized with cohesin. Although depletion of cohesin does not alter CTCF occupancy, there is a loss of looped domains, indicating that CTCF-dependent looping relies on cohesin (Rao et al., 2017). Further analysis of the CTCF protein revealed that mutating the N-terminus reduces TAD insulation (Nora et al., 2019). The orientation of the CTCF binding site positions the N-terminus to interact with cohesin, providing a molecular explanation of the necessity of convergent CTCF sites to create a chromatin loop. The necessity of cohesin and CTCF binding for chromatin looping, however, is currently being questioned, as two recent students have shown that depletion of CTCF and cohesin does not affect a majority of enhancer:promoter interactions (Thiecke et al., 2020; Kubo et al., 2021).
Many studies have shown that CTCF plays a role during development (reviewed in Arzate-Mejía et al., 2018). At the HoxA locus, deletion of CTCF sites within the locus alters the enrichment of the repressive H3K27me3 mark, permitting aberrant HoxA gene expression during the differentiation of ESCs to motor neurons (Narendra et al., 2015). In the developing heart, deletion of the CTCF gene in cardiac progenitor cells causes embryonic lethality at E12.5, through loss of chromatin interactions and, potentially, enhancer:promoter interactions at key developmental genes (Gomez-Velazquez et al., 2017). As a distinct example, inhibiting CTCF function prevents Schwann cell differentiation and causes hypomyelination in vivo (Wang et al., 2020). These are a few examples of the variety of roles CTCF plays in gene expression regulation, but little is known on how to identify what role CTCF is playing modulating nearby genes and whether there are biological signs that differentiate one CTCF site’s function from another.
In an auxin-inducible depletion system in mESCs, loss of CTCF reduces proliferation, indicating that CTCF is integral to the maintenance of pluripotency (Nora et al., 2017). The acute depletion of CTCF causes an overall loss of looping anchored by CTCF and cohesin (Figure 2B). There is also an increase in inter-TAD interactions that is reversed upon auxin wash-off, implying that CTCF normally prevents these interactions. Interestingly, while CTCF depletion causes loss of insulation on most boundaries, some boundaries remain intact (Figure 2B), indicating that the role of CTCF is not universal across the genome. CTCF sites are more likely to be near genes up-regulated by CTCF depletion, and these genes are more likely to be near active enhancers, so they are normally separated by a TAD boundary. Thus, the loss of the TAD boundary likely permits aberrant enhancer-promoter interaction(s) causing the up-regulation of these genes. In conclusion, CTCF is critical to the precise regulation of gene expression by modulating enhancer:promoter contacts.
Several studies have focused on the interaction between CTCF and enhancers, specifically SEs. CTCF has been found to be bound near most SEs and within some SEs (Dowen et al., 2014; Ing-Simmons et al., 2015; Huang et al., 2017). One study finds that a mammary gland specific SE modulates not only the mammary gland specific gene Wap, but also the non-mammary-specific Ramp3. Ramp3 lies outside of the chromatin loop formed by two CTCF sites flanking Wap and the associated SE. Deletion of the separating CTCF sites greatly increases the interaction between the SE and Ramp3 and Ramp3 expression, thus showing that the CTCF sites insulate against this interaction to maintain proper expression (Willi et al., 2017). In a variety of cancers, SEs are gained near MYC to up-regulate MYC and drive oncogenic transformation. These SEs lay between two CTCF sites that form an IN neighborhood and modulate expression (Schuijers et al., 2018). These studies, and others (Hay et al., 2016; Hanssen et al., 2017; Shin, 2019), support the theory that CTCF is critical to SE-mediated gene expression regulation.
The full potential of CTCF sites and enhancers in regulating gene expression is still being investigated and a number of questions remain. As an example, in view of the evidence that CTCF sites modulate SE function, at the Nanog locus, what is CTCF’s role? Is the IN truly the two sites just 5′ and 3′ of the gene, as implied in Dowen et al., and can other nearby CTCF sites compensate? And are these CTCF sites encouraging and/or preventing enhancer:promoter interactions? Beyond Nanog, the question still remains whether there are different classes of CTCF binding sites that can be identified through a biological signature. Given that loss of CTCF can cause an up-regulation and down-regulation of gene expression, how can we identify which role it plays for any given gene? Similarly, there are still questions regarding enhancers and their role in cell fate decisions: specifically are all super-enhancers and the same? It is clear from the data regarding the Nanog +60E that this cannot be true, because although it has all the defining features of a super-enhancer, in ESCs, at least, it does not play a significant role. Perhaps it is a redundant enhancer, becoming fully functional if the −5SE and −45SE are out of commission or is utilized in other cell types such as primordial germ cells (Figure 1E). Can we identify different features of SEs and assign them to different categories? The field of nuclear architecture remains open to investigation, and as we further study chromatin-chromatin interactions, the minute control of gene expression control will clarify how cell fate decisions are made and how this process breaks down in disease pathology.
PA wrote the first draft of the manuscript. SR and PA edited the manuscript together. Both authors contributed to the article and approved the submitted version.
This work was supported in part by funding from NIDDK (DK120152) to PA and the MCW MSTP T32 (NIGMS, GM080202) to PA. Additional support came from NIH (CA204231) to SR.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abranches, E., Guedes, A. M. V., Moravec, M., Maamar, H., Svoboda, P., Raj, A., et al. (2014). Stochastic NANOG fluctuations allow mouse embryonic stem cells to explore pluripotency. Development 141, 2770–2779. doi: 10.1242/dev.108910
Agrawal, P., Blinka, S., Pulakanti, K., Reimer, M. H., Stelloh, C., Meyer, A. E., et al. (2021). Genome editing demonstrates that the -5 kb Nanog enhancer regulates Nanog expression by modulating RNAPII initiation and/or recruitment. JBC 296:100189. doi: 10.1074/jbc.RA120.015152
Agrawal, P., Heimbruch, K. E., and Rao, S. (2019). Genome-wide maps of transcription regulatory elements and transcription enhancers in development and disease. Compr. Physiol. 9, 439–455. doi: 10.1002/cphy.c180028
Alexander, J. M., Guan, J., Li, B., Maliskova, L., Song, M., Shen, Y., et al. (2019). Live-cell imaging reveals enhancer-dependent Sox2 transcription in the absence of enhancer proximity. Elife 8:e41769. doi: 10.7554/eLife.41769
Anderson, E., and Hill, R. E. (2014). Long range regulation of the sonic hedgehog gene. Curr. Opin. Genet. Dev. 27, 54–59. doi: 10.1016/j.gde.2014.03.011
Arnold, S. J., and Robertson, E. J. (2009). Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nat. Rev. Mol. Cell Biol. 10, 91–103. doi: 10.1038/nrm2618
Arzate-Mejía, R. G., Recillas-Targa, F., and Corces, V. G. (2018). Developing in 3D: the role of CTCF in cell differentiation. Development 145:dev137729. doi: 10.1242/dev.137729
Barral, A., Rollan, I., Sanchez-Iranzo, H., Jawaid, W., Badia-Careaga, C., Menchero, S., et al. (2019). Nanog regulates Pou3f1 expression at the exit from pluripotency during gastrulation. Biol. Open 8:bio046367. doi: 10.1242/bio.046367
Bartman, C. R., Hamagami, N., Keller, C. A., Giardine, B., Hardison, R. C., Blobel, G. A., et al. (2019). Transcriptional burst initiation and polymerase pause release are key control points of transcriptional regulation. Mol. Cell 73, 519–532.e4. doi: 10.1016/j.molcel.2018.11.004
Bartman, C. R., Hsu, S. C., Hsiung, C. C. S., Raj, A., and Blobel, G. A. (2016). Enhancer regulation of transcriptional bursting parameters revealed by forced chromatin looping. Mol. Cell 62, 237–247. doi: 10.1016/j.molcel.2016.03.007
Benabdallah, N. S., Gautier, P., Hekimoglu-Balkan, B., Lettice, L. A., Bhatia, S., and Bickmore, W. A. (2016). SBE6: a novel long-range enhancer involved in driving sonic hedgehog expression in neural progenitor cells. Open Biol. 6:160197. doi: 10.1098/rsob.160197
Benabdallah, N. S., Williamson, I., Illingworth, R. S., Kane, L., Boyle, S., Sengupta, D., et al. (2019). Decreased enhancer-promoter proximity accompanying enhancer activation. Mol. Cell 76, 473–484.e7. doi: 10.1016/j.molcel.2019.07.038
Blinka, S., and Rao, S. (2017). Nanog expression in embryonic stem cells - an ideal model system to dissect enhancer function. BioEssays 39:1700086. doi: 10.1002/bies.201700086
Blinka, S., Reimer, M. H., Pulakanti, K., and Rao, S. (2016). Super-Enhancers at the nanog locus differentially regulate neighboring pluripotency-associated genes. Cell Rep. 17, 19–28. doi: 10.1016/j.celrep.2016.09.002
Braccioli, L., and De Wit, E. (2019). CTCF: a Swiss-army knife for genome organization and transcription regulation. Essays Biochem. 63, 157–165. doi: 10.1042/EBC20180069
Chambers, I., Silva, J., Colby, D., Nichols, J., Nijmeijer, B., Robertson, M., et al. (2007). Nanog safeguards pluripotency and mediates germline development. Nature 450, 1230–1234. doi: 10.1038/nature06403
Chen, F. X., Smith, E. R., and Shilatifard, A. (2018). Born to run: control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 19, 464–478. doi: 10.1038/s41580-018-0010-5
Cho, W.-K. K., Spille, J.-H. H., Hecht, M., Lee, C., Li, C., Grube, V., et al. (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 4199:eaar4199. doi: 10.1126/science.aar4199
Dash, S., and Trainor, P. A. (2020). The development, patterning and evolution of neural crest cell differentiation into cartilage and bone. Bone 137:115409. doi: 10.1016/j.bone.2020.115409
de Laat, W., and Dekker, J. (2012). 3C-based technologies to study the shape of the genome. Methods 58, 189–191. doi: 10.1016/j.ymeth.2012.11.005
de Wit, E., Vos, E. S. M., Holwerda, S. J. B., Valdes-Quezada, C., Verstegen, M. J. A. M., Teunissen, H., et al. (2015). CTCF binding polarity determines chromatin looping. Mol. Cell 60, 676–684. doi: 10.1016/j.molcel.2015.09.023
Dekker, J., Rippe, K., Dekker, M., and Kleckner, N. (2002). Capturing chromosome conformation. Science 295, 1306–1311. doi: 10.1126/science.1067799
Dowen, J. M., Fan, Z. P., Hnisz, D., Ren, G., Abraham, B. J., Zhang, L. N., et al. (2014). Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387. doi: 10.1016/j.cell.2014.09.030
Ferri, A. L. M., Cavallaro, M., Braida, D., Di Cristofano, A., Canta, A., Vezzani, A., et al. (2004). Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 131, 3805–3819. doi: 10.1242/dev.01204
Fisher, J. B., McNulty, M., Burke, M. J., Crispino, J. D., and Rao, S. (2017). Cohesin mutations in myeloid malignancies. Trends Cancer 3, 282–293. doi: 10.1016/j.trecan.2017.02.006
Fukaya, T., Lim, B., and Levine, M. (2016). Enhancer control of transcriptional bursting. Cell 166, 358–368. doi: 10.1016/j.cell.2016.05.025
Ghimire, S., Van Der Jeught, M., Neupane, J., Roost, M. S., Anckaert, J., Popovic, M., et al. (2018). Comparative analysis of naive, primed and ground state pluripotency in mouse embryonic stem cells originating from the same genetic background. Sci. Rep. 8:5884. doi: 10.1038/s41598-018-24051-5
Gomez-Velazquez, M., Badia-Careaga, C., Lechuga-Vieco, A. V., Nieto-Arellano, R., Tena, J. J., Rollan, I., et al. (2017). CTCF counter-regulates cardiomyocyte development and maturation programs in the embryonic heart. PLoS Genet. 13:e1006985. doi: 10.1371/journal.pgen.1006985
Gordon, C. T., Attanasio, C., Bhatia, S., Benko, S., Ansari, M., Tan, T. Y., et al. (2014). Identification of novel craniofacial regulatory domains located far upstream of SOX9 and disrupted in pierre robin sequence. Hum. Mutat. 35, 1011–1020. doi: 10.1002/humu.22606
Hanssen, L. L. P., Kassouf, M. T., Oudelaar, A. M., Biggs, D., Preece, C., Downes, D. J., et al. (2017). Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nat. Cell Biol. 19, 952–961. doi: 10.1038/ncb3573
Hay, D., Hughes, J. R., Babbs, C., Davies, J. O. J., Graham, B. J., Hanssen, L. L. P., et al. (2016). Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet. 48, 895–903. doi: 10.1038/ng.3605
Henriques, T., Scruggs, B. S., Inouye, M. O., Muse, G. W., Williams, L. H., Burkholder, A. B., et al. (2018). Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 32, 26–41. doi: 10.1101/gad.309351.117.GENES
Hnisz, D., Day, D. S., and Young, R. A. (2016). Insulated neighborhoods: structural and functional units of mammalian gene control. Cell 167, 1188–1200. doi: 10.1016/J.CELL.2016.10.024
Hnisz, D., Schuijers, J., Lin, C. Y., Weintraub, A. S., Abraham, B. J., Lee, T. I., et al. (2015). Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 58, 362–370. doi: 10.1016/j.molcel.2015.02.014
Huang, J., Li, K., Cai, W., Liu, X., Zhang, Y., Orkin, S. H., et al. (2017). Dissecting super-enhancer hierarchy based on chromatin interactions. bioRxiv [Preprint] doi: 10.1101/149583
Huang, J., Liu, X., Li, D., Shao, Z., Cao, H., Zhang, Y., et al. (2016). Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev. Cell 36, 9–23. doi: 10.1016/j.devcel.2015.12.014
Ing-Simmons, E., Seitan, V. C., Faure, A. J., Flicek, P., Carroll, T., Dekker, J., et al. (2015). Spatial enhancer clustering and regulation of enhancer-proximal genes by cohesin. Genome Res. 25, 504–513. doi: 10.1101/gr.184986.114
Jeong, Y., El-Jaick, K., Roessler, E., Muenke, M., and Epstein, D. J. (2006). A functional screen for sonic hedgehog regulatory elements across a 1 Mb interval identifies long-range ventral forebrain enhancers. Development 133, 761–772. doi: 10.1242/dev.02239
Jeong, Y., Leskow, F. C., El-Jaick, K., Roessler, E., Muenke, M., Yocum, A., et al. (2008). Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat. Genet. 40, 1348–1353. doi: 10.1038/ng.230
Kubo, N., Ishii, H., Xiong, X., Bianco, S., Meitinger, F., Hu, R., et al. (2021). Promoter-proximal CTCF binding promotes distal enhancer-dependent gene activation. Nat. Struct. Mol. Biol. 28, 152–161. doi: 10.1038/s41594-020-00539-5
Lee, Y. H., and Saint-Jeannet, J. P. (2011). Sox9 function in craniofacial development and disease. Genesis 49, 200–208. doi: 10.1002/dvg.20717
Lettice, L. A., Devenney, P., De Angelis, C., and Hill, R. E. (2017). The conserved sonic hedgehog limb enhancer consists of discrete functional elements that regulate precise spatial expression. Cell Rep. 20, 1396–1408. doi: 10.1016/j.celrep.2017.07.037
Lettice, L. A., Heaney, S. J. H., Purdie, L. A., Li, L., de Beer, P., Oostra, B. A., et al. (2003). A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 12, 1725–1735. doi: 10.1093/hmg/ddg180
Li, W., Notani, D., and Rosenfeld, M. G. (2016). Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat. Rev. Genet. 17, 207–223. doi: 10.1038/nrg.2016.4
Liu, C. F., and Lefebvre, V. (2015). The transcription factors SOX9 and SOX5/SOX6 cooperate genome-wide through super-enhancers to drive chondrogenesis. Nucleic Acids Res. 43, 8183–8203. doi: 10.1093/nar/gkv688
Long, H. K., Osterwalder, M., Welsh, I. C., Hansen, K., Davies, J. O. J., Liu, Y. E., et al. (2020). Loss of extreme long-range enhancers in human neural crest drives a craniofacial disorder. Cell Stem Cell 27, 765–783.e14. doi: 10.1016/j.stem.2020.09.001
Messerschmidt, D. M., and Kemler, R. (2010). Nanog is required for primitive endoderm formation through a non-cell autonomous mechanism. Dev. Biol. 344, 129–137. doi: 10.1016/j.ydbio.2010.04.020
Moore, J. M., Rabaia, N. A., Smith, L. E., Fagerlie, S., Gurley, K., Loukinov, D., et al. (2012). Loss of maternal CTCF is associated with peri-implantation lethality of Ctcf null embryos. PLoS One 7:e34915. doi: 10.1371/journal.pone.0034915
Nagakura, R., Yamamoto, M., Jeong, J., Hinata, N., Katori, Y., Chang, W. J., et al. (2020). Switching of Sox9 expression during musculoskeletal system development. Sci. Rep. 10:8425. doi: 10.1038/s41598-020-65339-9
Narendra, V., Rocha, P. P., An, D., Raviram, R., Skok, J. A., Mazzoni, E. O., et al. (2015). CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021. doi: 10.1126/science.1262088
Noe Gonzalez, M., Blears, D., and Svejstrup, J. Q. (2020). Causes and consequences of RNA polymerase II stalling during transcript elongation. Nat. Rev. Mol. Cell Biol. 22, 3–21. doi: 10.1038/s41580-020-00308-8
Nora, E., Caccianini, L., Fudenberg, G., Kameswaran, V., Nagle, A., Uebersohn, A., et al. (2019). Molecular basis of CTCF binding polarity in genome folding. bioRxiv [Preprint] doi: 10.1101/2019.12.13.876177
Nora, E. P., Goloborodko, A., Valton, A. L., Gibcus, J. H., Uebersohn, A., Abdennur, N., et al. (2017). Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e22. doi: 10.1016/j.cell.2017.05.004
Ong, C.-T., and Corces, V. G. (2014). CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Genet. 15, 234–246. doi: 10.1038/nrg3663
Parker, S. C. J., Stitzel, M. L., Taylor, D. L., Orozco, J. M., Erdos, M. R., Akiyama, J. A., et al. (2013). Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. U.S.A. 110, 17921–17926. doi: 10.1073/pnas.1317023110
Phillips, J. E., and Corces, V. G. (2009). CTCF: master weaver of the genome. Cell 137, 1194–1211. doi: 10.1016/j.cell.2009.06.001
Phillips-Cremins, J. E., Sauria, M. E. G., Sanyal, A., Gerasimova, T. I., Lajoie, B. R., Bell, J. S. K., et al. (2013). Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295. doi: 10.1016/j.cell.2013.04.053
Pulakanti, K., Pinello, L., Stelloh, C., Blinka, S., Allred, J., Milanovich, S., et al. (2013). Enhancer transcribed RNAs arise from hypomethylated, Tet-occupied genomic regions. Epigenetics 8, 1303–1320. doi: 10.4161/epi.26597
Rada-Iglesias, A., Bajpai, R., Swigut, T., Brugmann, S. A., Flynn, R. A., and Wysocka, J. (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–285. doi: 10.1038/nature09692
Rao, S. S. P., Huang, S. C., Glenn St Hilaire, B., Engreitz, J. M., Perez, E. M., Kieffer-Kwon, K. R., et al. (2017). Cohesin loss eliminates all loop domains. Cell 171, 305–320.e24. doi: 10.1016/j.cell.2017.09.026
Rao, S. S. P., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Rayon, T., Menchero, S., Rollán, I., Ors, I., Helness, A., Crespo, M., et al. (2016). Distinct mechanisms regulate Cdx2 expression in the blastocyst and in trophoblast stem cells. Sci. Rep. 6:27139. doi: 10.1038/srep27139
Rowley, M. J., and Corces, V. G. (2018). Organizational principles of 3D genome architecture. Nat. Rev. Genet. 19, 789–800. doi: 10.1038/s41576-018-0060-8
Sabari, B. R., Agnese, A. D., Boija, A., Klein, I. A., Coffey, E. L., Shrinivas, K., et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361:eaar3958. doi: 10.1126/science.aar3958
Schrode, N., Saiz, N., Di Talia, S., and Hadjantonakis, A. K. (2014). GATA6 levels modulate primitive endoderm cell fate choice and timing in the mouse blastocyst. Dev. Cell 29, 454–467. doi: 10.1016/j.devcel.2014.04.011
Schuijers, J., Manteiga, J. C., Weintraub, A. S., Day, D. S., Zamudio, A. V., Hnisz, D., et al. (2018). Transcriptional dysregulation of MYC reveals common enhancer-docking mechanism. Cell Rep. 23, 349–360. doi: 10.1016/j.celrep.2018.03.056
Sheridan, R. M., Fong, N., D’Alessandro, A., and Bentley, D. L. (2019). Widespread backtracking by RNA Pol II Is a major effector of gene activation, 5′ pause release, termination, and transcription elongation rate. Mol. Cell 73, 107–118.e4. doi: 10.1016/j.molcel.2018.10.031
Shin, H. Y. (2019). The structural and functional roles of CTCF in the regulation of cell type-specific and human disease-associated super-enhancers. Genes Genomics 41, 257–265. doi: 10.1007/s13258-018-0768-z
Shin, H. Y., Willi, M., Yoo, K. H., Zeng, X., Wang, C., Metser, G., et al. (2016). Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 48, 904–911. doi: 10.1038/ng.3606
Shlyueva, D., Stampfel, G., and Stark, A. (2014). Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 15, 272–286. doi: 10.1038/nrg3682
Strumpf, D., Mao, C. A., Yamanaka, Y., Ralston, A., Chawengsaksophak, K., Beck, F., et al. (2005). Cdx2 is required for correct cell fate specification and differentiation of trophectoderm in the mouse blastocyst. Development 132, 2093–2102. doi: 10.1242/dev.01801
Thiecke, M. J., Wutz, G., Muhar, M., Tang, W., Bevan, S., Malysheva, V., et al. (2020). Cohesin-dependent and -independent mechanisms mediate chromosomal contacts between promoters and enhancers. Cell Rep. 32:107929. doi: 10.1016/j.celrep.2020.107929
Thomas, H. F., Kotova, E., Jayaram, S., Halbritter, F., Wysocka, J., Correspondence, C. B., et al. (2021). Temporal dissection of an enhancer cluster reveals distinct temporal and functional contributions of individual elements. Mol. Cell 81, 969–982.e13. doi: 10.1016/j.molcel.2020.12.047
Visel, A., Blow, M. J., Li, Z., Zhang, T., Akiyama, J. A., Holt, A., et al. (2009). ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858. doi: 10.1038/nature07730
Wang, J., Wang, J., Yang, L., Zhao, C., Wu, L. N., Xu, L., et al. (2020). CTCF-mediated chromatin looping in EGR2 regulation and SUZ12 recruitment critical for peripheral myelination and repair. Nat. Commun. 11:4133. doi: 10.1038/s41467-020-17955-2
Whyte, W. A., Orlando, D. A., Hnisz, D., Abraham, B. J., Lin, C. Y., Kagey, M. H., et al. (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. doi: 10.1016/j.cell.2013.03.035
Willi, M., Yoo, K. H., Reinisch, F., Kuhns, T. M., Lee, H. K., Wang, C., et al. (2017). Facultative CTCF sites moderate mammary superenhancer activity and regulate juxtaposed gene in non-mammary cells. Nat. Commun. 8:16069. doi: 10.1038/ncomms16069
Williamson, I., Lettic, L. A., Hill, R. E., and Bickmore, W. A. (2016). Shh and ZRS enhancer colocalisation is specific to the zone of polarising activity. Development 143, 2994–3001. doi: 10.1242/dev.139188
Xenopoulos, P., Kang, M., Puliafito, A., DiTalia, S., and Hadjantonakis, A. K. (2015). Heterogeneities in nanog expression drive stable commitment to pluripotency in the mouse blastocyst. Cell Rep. 10, 1508–1520. doi: 10.1016/j.celrep.2015.02.010
Yao, B., Wang, Q., Liu, C. F., Bhattaram, P., Li, W., Mead, T. J., et al. (2015). The SOX9 upstream region prone to chromosomal aberrations causing campomelic dysplasia contains multiple cartilage enhancers. Nucleic Acids Res. 43, 5394–5408. doi: 10.1093/nar/gkv426
Yao, Y., Minor, P. J., Zhao, Y. T., Jeong, Y., Pani, A. M., King, A. N., et al. (2016). Cis-regulatory architecture of a brain signaling center predates the origin of chordates. Nat. Genet. 48, 575–580. doi: 10.1038/ng.3542
Zamudio, A. V., Dall’Agnese, A., Henninger, J. E., Manteiga, J. C., Afeyan, L. K., Hannett, N. M., et al. (2019). Mediator condensates localize signaling factors to key cell identity genes. Mol. Cell 76, 753–766.e6. doi: 10.1016/j.molcel.2019.08.016
Zatreanu, D., Han, Z., Mitter, R., Tumini, E., Williams, H., Gregersen, L., et al. (2019). Elongation factor TFIIS prevents transcription stress and R-Loop accumulation to maintain genome stability. Mol. Cell 76, 57–69.e9. doi: 10.1016/j.molcel.2019.07.037
Keywords: CTCF, enhancer, nanog, pluripotency, embryonic stem cell
Citation: Agrawal P and Rao S (2021) Super-Enhancers and CTCF in Early Embryonic Cell Fate Decisions. Front. Cell Dev. Biol. 9:653669. doi: 10.3389/fcell.2021.653669
Received: 14 January 2021; Accepted: 18 February 2021;
Published: 25 March 2021.
Edited by:
Justin Brumbaugh, University of Colorado Boulder, United StatesReviewed by:
Konstantinos Chronis, University of Illinois at Chicago, United StatesCopyright © 2021 Agrawal and Rao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sridhar Rao, c3JpZGhhci5yYW9AdmVyc2l0aS5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.