
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Cell Dev. Biol. , 30 March 2021
Sec. Epigenomics and Epigenetics
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.651145
This article is part of the Research Topic Epigenomic and Epitranscriptomic Basis of Development and Human Disease View all 14 articles
 Xiaoyu Wei1,2†
Xiaoyu Wei1,2† Hanbo Li2,3†
Hanbo Li2,3† Yang Guo3†Xiaoying Zhao3†Yang Liu1,2Xuanxuan Zou1,2Li Zhou2,3Yue Yuan1,2Yating Qin2,3Chunyan Mao2,3Guodong Huang2
Yang Guo3†Xiaoying Zhao3†Yang Liu1,2Xuanxuan Zou1,2Li Zhou2,3Yue Yuan1,2Yating Qin2,3Chunyan Mao2,3Guodong Huang2 Yeya Yu2,4Qiuting Deng1,2Weimin Feng1,2Jiangshan Xu1,2Mingyue Wang2Shanshan Liu2,3Huanming Yang1,2,5
Yeya Yu2,4Qiuting Deng1,2Weimin Feng1,2Jiangshan Xu1,2Mingyue Wang2Shanshan Liu2,3Huanming Yang1,2,5 Longqi Liu1,2,6
Longqi Liu1,2,6 Chuanyu Liu2,6*
Chuanyu Liu2,6* Ying Gu1,2,7*
Ying Gu1,2,7*Tissue regenerative potential varies significantly across species, tissues, and ages (Yun, 2015; Iismaa et al., 2018). For example, planarian can reconstruct its whole body from small fragments of the original organism (Pellettieri et al., 2010; Zeng et al., 2018); in contrast, many vertebrate organs, such as the heart, can only regenerate primarily through preexisting proliferating cardiomyocytes, like in adult zebrafish and neonatal mice (Vivien et al., 2016). Since Spallanzani first reported the salamander regeneration in 1760s, scientists have been devoted to decipher the codes of such powerful regenerative capability (Dinsmore, 1991). Using different methods to analyze the cellular and molecular phenomena during salamander limb or tail regeneration, researchers revealed complex processes including clotting, immune activation, apoptosis, and reprogramming (Tanaka, 2016). Within such process, a mass of cells called blastema proliferates from the wounded site and fully regenerates the lost body part (McCusker et al., 2015; Haas and Whited, 2017).
Axolotl (Ambystoma mexicanum) is a species of salamander, which has been used as the model animal to investigate key biological processes such as embryo development, limb regeneration, and central nervous system regeneration for nearly 150 years (Pietsch, 1961; Schreckenberg and Jacobson, 1975; Seyedhassantehrani et al., 2017). Although several studies have focused on bulk transcriptome studies (Monaghan et al., 2009; Campbell et al., 2011; Knapp et al., 2013; Stewart et al., 2013; Wu et al., 2013; Bryant et al., 2017), the axolotl genome was not assembled until 2018 with features of large sizes (32 Gb) and abundant repetitive sequences (Nowoshilow et al., 2018). Interestingly, in axolotl, intron size expands 13- to 25-fold in non–developmentally related orthologous genes and 6- to 11-fold in developmentally related orthologous genes as compared to human, mouse, and frog, thus indicating that a more complex regulatory network in non-coding regions may play an important role in both development and regeneration (Nowoshilow et al., 2018). Since the first axolotl genome assembly, multiple studies have been carried out to investigate the transcriptomic patterns of axolotl limb regeneration at single-cell resolution (Gerber et al., 2018; Leigh et al., 2018; Qin et al., 2020). These studies used single-cell gene expression variations to reflect dynamic cell population changes and cell fate transitions, as well as unique immune responses during regeneration (Tsai et al., 2019; Li et al., 2020; Rodgers et al., 2020). Standing on the shoulder of these studies and looking forward, analysis of the epigenetic regulations, which are responsible for the dynamic gene expression changes, will help scientists to better understand the underlying mechanisms of the regenerative process.
To identify crucial regulatory elements and transcription factors (TFs) that drive or support the regenerative response, the assay for transposase-accessible chromatin using sequencing (ATAC-seq) has been used to profile the chromatin accessibility dynamics in multiple species (Buenrostro et al., 2015). For instance, a genome-wide scan for TF binding motifs in thousands of chromatin regions revealed by ATAC-seq highlighted the role of EGR (early growth response) as a pioneer factor to directly activate regeneration-related genes in Hofstenia (Gehrke et al., 2019). In addition to TFs, enhancers also have great significance in regeneration. The conserved teleost regeneration response enhancers in zebrafish and African killifish (Nothobranchius furzeri) were uncovered by histone H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq, a marker for active enhancers), bulk RNA sequencing (RNA-seq), and single-cell RNA sequencing (scRNA-seq) (Wang et al., 2020). These studies suggested that epigenetic regulatory elements play fundamental roles in regeneration. However, how the non-coding axolotl genome responds to wounding to regulate gene expression and consequently drive the process of limb regeneration remains to be elucidated.
Here, we present a comprehensive dataset of chromatin accessibility for eight stages of the axolotl limb regeneration process, including homeostatic [uninjured control, 0 h after amputation (0 hpa)], trauma (3 hpa), wounding healing (1 day after amputation, 1 dpa), early-bud blastema (3 dpa), midbud blastema (7 dpa), late-bud blastema (14 dpa), palette stage (22 dpa), and redifferentiated stages (33 dpa) (Figure 1A). These time points represent the main events during axolotl limb regeneration, making this dataset a valuable platform to understand the complex regulatory network from an overall perspective. We generated 24 samples from the eight stages of limb tissues (three biological replicates per group). Systematic analysis of our dataset identified a total of 342,341 peaks, of which 33,604 showed transient dynamic patterns. We further investigated the occupancy of TFs in clusters with different peaks, which may help to explain the activation and manipulation of these regulatory elements during injury response and regeneration process (Figure 1B).
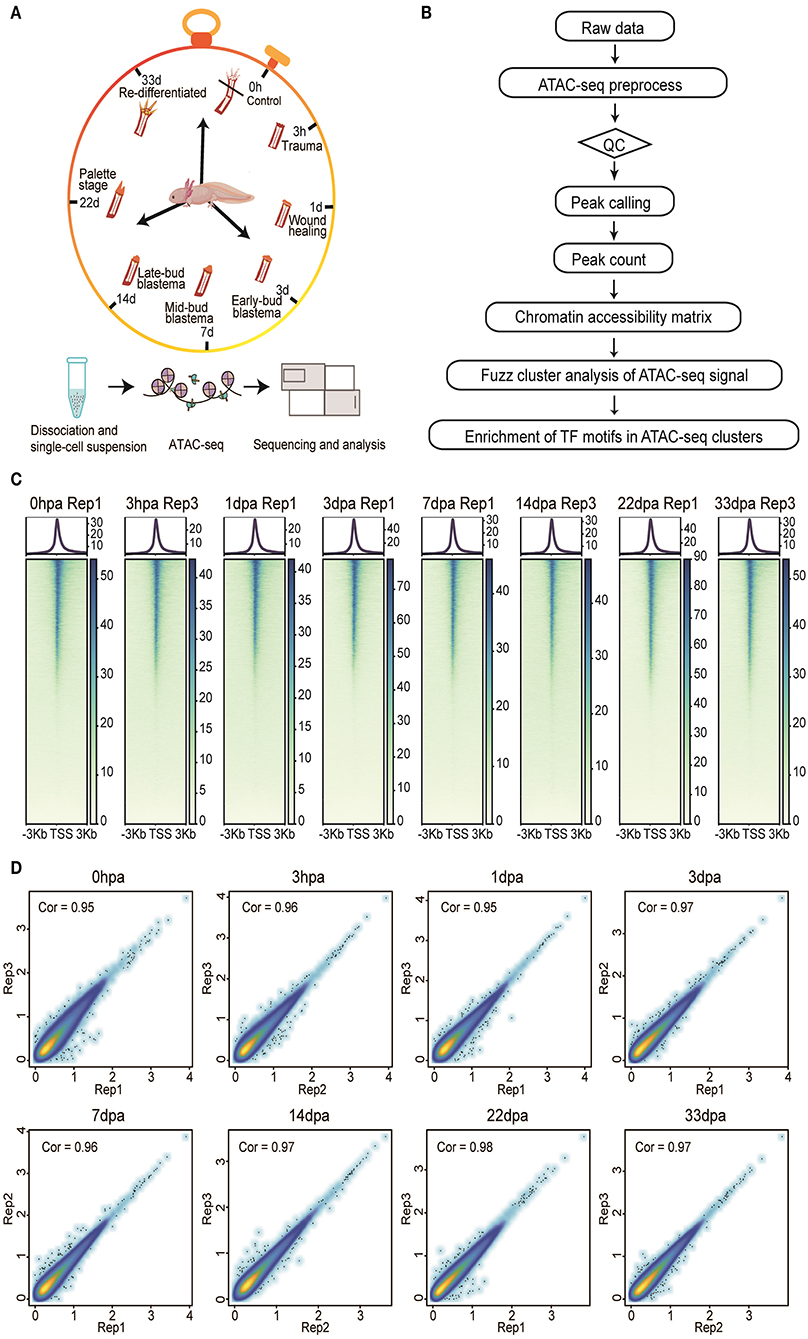
Figure 1. Overview of the experimental, data analysis workflow, and ATAC-seq data quality metrics. (A) Three biological replicates (n > 3) from eight stages of the axolotl limb regeneration process were collected for ATAC-seq profiling. (B) The analysis workflow for ATAC-seq profiles. (C) The ATAC-seq signal enrichment around the transcription start sites (TSSs) for eight representative samples. (D) Scatter plots showing the Pearson correlations between biological replicates.
The institutional review board approved all experiments in this study on the ethics committee of BGI (permit BGI-IRB 19059). Axolotl breeding, housing, and tissue isolation were performed as previously described (Li et al., 2020). Briefly, we anesthetized the axolotls with 0.2% Tricaine (ethyl 3-aminobenzoate methane sulfonate) before the amputation surgery. The lower forearm tissues were isolated at eight time points including the homeostatic stage (uninjured control, 0 hpa), trauma (3 hpa), wound healing stage (1 dpa), early-bud blastema (3 dpa), midbud blastema (7 dpa), late-bud blastema (14 dpa), palette (22 dpa), and redifferentiation stage (33 dpa), with three replicates for each stage. These eight time points represent the main phases of axolotl limb regeneration. All tissues were washed with amphibian phosphate-buffered saline for three times before further operation. Tissues were enzymatically digested to cell suspension using 0.2% collagenase type I (BBI, cat. #A004194-0001) and 0.2% collagenase type II (BBI, cat. #A004174-0001) at room temperature for 1 h.
Tissues were transferred to the bottom of the Dounce Homogenizer and dounced within 1 mL 1 × Homogenization Buffer Stable Master Mix until resistance goes away (~30 strokes). The cells were then passed through a 100-μm strainer into a clean Dounce Homogenizer and dounced again for 20 strokes. Nuclei were collected through a 40-μm strainer and counted. Around 50,000 nuclei were transferred into a tube containing 1 mL wash buffer (ATAC-RSB+0.1% Tween-20), and then the samples were centrifuged at 500 rcf at 4°C for 5 min. Transposition reaction was performed as the Omni-ATAC-seq method (Corces et al., 2017). Nuclei were then transferred into 50 μL transposition reaction mixture containing 10 μL of 5 × TAG buffer (BGI, cat. #BGE005B01), 2.5 μL of transposase (100 nM final, BGI, cat. #BGE005), 31.5 μL of PBS, 0.5 μL of 1% digitonin, 0.5 μL of 10% Tween-20, and 5 μL of H2O for 30 min at 37°C in a thermomixer by 1,000 rpm.
The transposed DNA was purified with a DNA MinElute kit (Qiagen, Germany) and eluted with 20 μL nuclease-free H2O. The purified DNA was amplified for eight cycles using a reaction mixture containing 2.5 μL of Tn5 Ad153 N5 primer (20 μM), 2.5 μL of Tn5 Ad153 N7 primer (20 μM), 25 μL of NEB Next High-Fidelity 2 × polymerase chain reaction (PCR) Master Mix, with a PCR protocol of 72°C for 5 min, 98°C for 30 s, and then eight cycles of 98°C for 10 s, 63°C for 30 s,72°C for 1 min, finally by 72°C for 10 min and hold at 4°C. The 300- to 500-bp size PCR product was selected using AMPure XP beads (Agencourt, cat. #A63882) according to the manual. All libraries were further prepared based on BGISEQ-500 sequencing platform with pair-end 50-bp read length (Huang et al., 2017).
The data of ATAC-seq were trimmed with SOAPnuke (Chen et al., 2018), and reads were aligned to axolotl genome (Nowoshilow et al., 2018) (https://www.axolotl-omics.org/assemblies) by using Sentieon bwa mem (parameter: -K 100,000,000 -M -t 40) (Li, 2013). Subsequently, we filtered out reads with mapping quality of <30. PCR duplicate reads were discarded by applying Picard's MarkDuplicates (http://broadinstitute.github.io/picard/) (Picard Toolkit, 2019). We next performed model-based analysis of ChIP-seq (MACS2) to identify the peak regions with options -B, -q 0.01 –nomodel, -f BAM (Zhang et al., 2008). The irreproducible discovery rate (IDR) method was employed to identify reproducible high-quality peaks between each two biological replicates (Li et al., 2011). Peak signal can be visualized in IGV by the Broad Institute (http://software.broadinstitute.org/software/igv/). A standard peak list was established by merging reproducible peaks of each two replicates for each time point. The standard peak count matrix was calculated using the intersect function of BedTools (Quinlan and Hall, 2010).
Reads per million mapped reads (RPM) algorithm was used to normalize the raw count matrix (Wei, 2020). Pearson correlations based on the Log10 RPM matrix were used to calculate the coefficients between different biological replicates across every stage. The RPM matrix for biological replicates was aggregated, and peaks with an average of RPM <1 at all time points were removed. Peaks across time with <50 coefficient of variation were filtered out to form a pseudocluster prior to clustering, which reflects the regions with stable accessibility throughout the regeneration. The remaining peaks were then transformed into normalized data using Z score method, followed by performing time course c-means fuzzy clustering with a cluster membership cutoff of 0.8 (Kumar and Futschik, 2007).
Relative genomic region was determined by overlapping each peak with features defined in the custom's annotated genes. The distance to transcription start sites (TSSs) was calculated according to the distance between the peak center and the nearest TSSs using the distanceToNearest function in GenomicRanges packages (Lawrence et al., 2013).
Functional enrichment of peaks in each cluster with distance to TSSs <10,000 bp was performed by using the clusterProfiler R package (Yu et al., 2012), with a q-value threshold of 0.1 for statistical significance.
The findMotifsGenome.pl script of the HOMER software was employed to perform transcript factor enrichment analysis in regeneration dynamic peaks with default settings (Heinz et al., 2010).
To investigate the correlation between chromatin dynamics and gene expression changes, we took advantage of single-cell RNA-seq data of these eight stages we published previously and calculated the average expression of each gene to construct a pseudobulk gene expression matrix (Li et al., 2020). Correlation analysis was done between the chromatin accessibility of promoters (TSS ± 2 kb) and closest genes' expression.
We inspected our ATAC-seq dataset by regularly used statistics, such as the number of total reads, number of mapped reads, percentage of mapped reads, the number of usable reads, the percentage of final usable reads, and the number of peaks (Supplementary Table 1). We generated more than 1,000 million ATAC-seq reads for each replicate on average. Among these reads, we detected strong enrichment around TSSs (Wei, 2020) (Figure 1C). Moreover, size periodicity of the chromatin accessibility fragments corresponding to integer multiples of nucleosomes (Wei, 2020) demonstrated the reliability of our dataset, this being consistent with previously published ATAC-seq profiles (Buenrostro et al., 2013) (Supplementary Figure 1).
To assess the reproducibility of chromatin accessibility regions between biological replicates, we used the IDR method to filter peaks that overlapped between replicates in each regeneration stage. Pearson correlations based on the Log10 RPM matrix were used to calculate the coefficients, showing that a correlation coefficient is higher than 0.9 between each two replicates in each stage, with the exception of replicate 1 from 3 hpa, which was removed for downstream analysis (Figure 1D).
To explore the chromatin accessibility with temporal dynamic features during regeneration, we used normalized ATAC-seq read counts in peaks to perform time-course fuzzy clustering. This approach yielded six separated clusters, which indicate six distinct categories defined by regions: (1) those that become accessible transiently at 22- and 33-dpa stages, cluster 1 (C1, n = 5,930); (2) regions that are close in the intermediate period of regeneration but accessible after 14 dpa, cluster 2 (C2, n = 3,838); (3) regions in which accessibility is established only at 22-dpa stage, cluster 3 (C3, n = 6,797); (4) regions accessible in the control but that exhibit loss of accessibility shortly at 3 hpa and later stages, cluster 4 (C4, n = 2,454); (5) regions that are accessible in specifically at 3 hpa and 14 dpa, cluster 5 (C5, n = 7,727); (6) regions that are stably accessible at the intermediate stages of regeneration, cluster 6 (C6, n = 6,858). This clustering highlighted several characteristics of chromatin reconfiguration during regeneration (Figure 2A). These data collectively demonstrated that the chromatin state is remodeling rapidly in the first few hours following amputation, to prepare for the subsequent regeneration process. The dynamics of chromatin accessibility provides a new perspective to understand the cell fate decision in axolotl limb regeneration process.
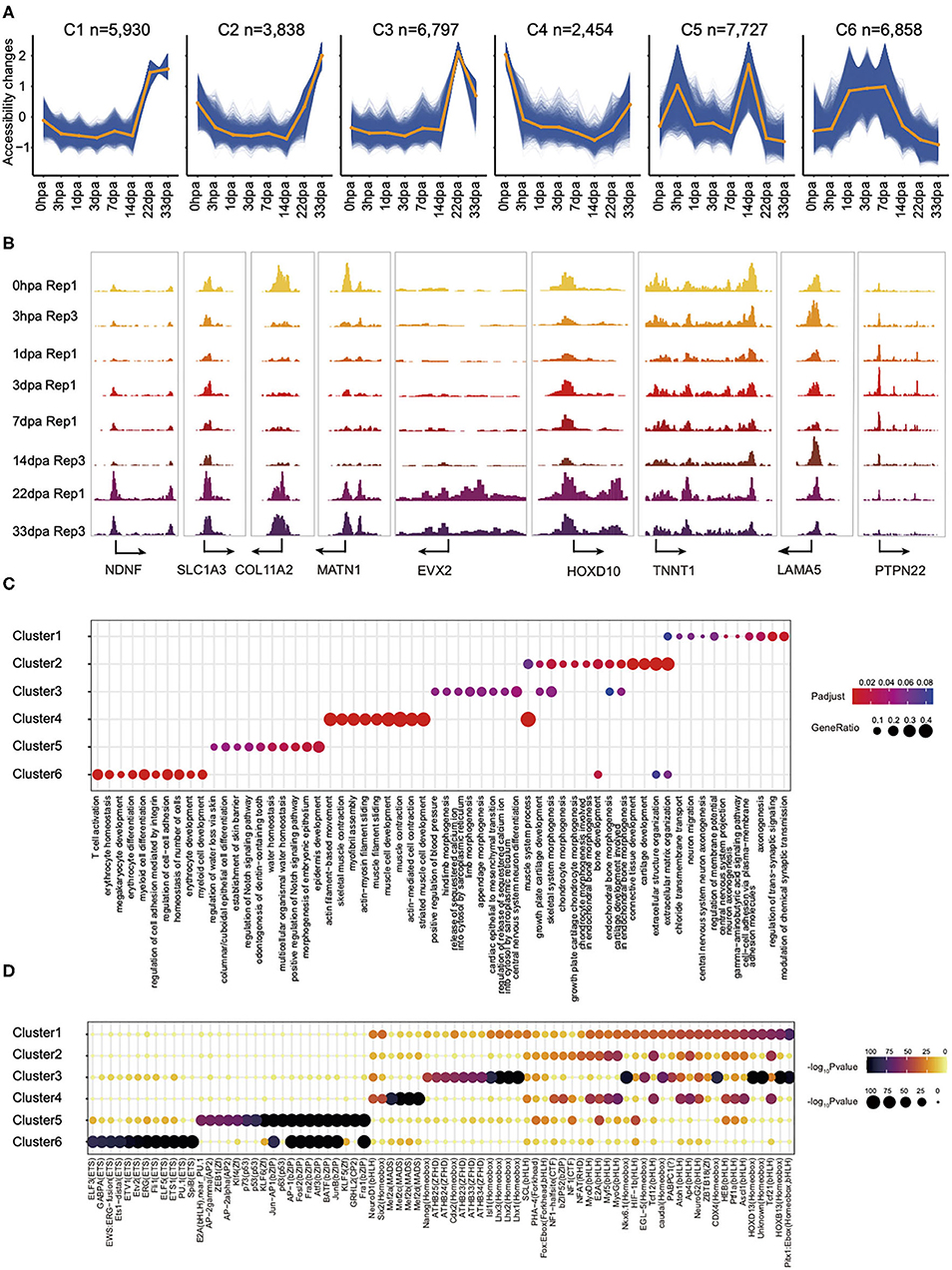
Figure 2. The landscape of chromatin accessibility dynamic changes in axolotl limb regeneration and potential function of dynamic elements identification. (A) Fuzzy cluster analysis of ATAC-seq signal. Line plots show standardized ATAC-seq signal, with individual blue lines representing individual loci and the orange line representing the cluster center's values. (B) Genome browser views of ATAC-seq signal for the dynamic peaks. (C) Enrichment of GO terms in ATAC-seq clusters. Where a point is present, a significant enrichment for the go term of biological process (x axis) was found in the ATAC-seq clusters (y axis). Point size represents the gene ratio in the go term, and color represents the adjusted P-value. (D) Enrichment of the indicated TF motifs in each ATAC-seq cluster. The size and color of each point represent the motif enrichment P-value (–log10 P-value).
Peaks in C1 are highly enriched in Gene Ontology (GO) terms related to axonogenesis. Examples of C1 include a promoter in the Ndnf locus, which is a novel neurotrophic factor derived from neurons that may be useful in the treatment of neuronal degeneration diseases and nerve injuries (Kuang et al., 2010). C2 consists of elements that are highly accessible in the extracellular matrix organization and connective tissue development. For example, Col11a2, a fibril-forming collagen found mainly in the cartilage extracellular matrix, is important for the integrity and development of the skeleton (Lui et al., 1996). We also found some genes associated to limb morphogenesis in C3 such as the Hoxbox gene, Evx2, and Hoxd10 (Herault et al., 1996; Tarchini and Duboule, 2006). GO terms enriched in C4 were related to muscle cell development, whereas those in C5 were related to epidermis development. Interestingly, we observed some immune response–related GO terms in C6, such as T-cell activation and myeloid cell differentiation (Figures 2B,C), which is consistent with the inflammatory process following injury (Supplementary Figure 2).
Our analysis also indicated that the binding site for TF that bound to C1 is enriched for NeuroG2 (Figure 2D). NeuroG2 is a TF that can specify a neuronal fate and expressed in neural progenitor cells within the developing central and peripheral nervous systems (Dennis et al., 2019). Notably, we also observed major binding events for the pioneer factor PU.1 in C6, this being a master transcriptional regulator in activating many target genes during both myeloid and B-lymphoid development (Turkistany and DeKoter, 2011). In addition, transcript factors from the MyoG, MyoD, and Mef families, which are essential for the development of functional skeletal muscle, were found in C4 (Al-Khalili et al., 2004; Ganassi et al., 2020). Taken together, we provide a high-quality comprehensive dataset to study the regenerative epigenomic dynamics of axolotl limb regeneration.
To summarize, by applying the state-of-the-art technique ATAC-seq, we provide the first chromatin accessibility landscape in axolotl regenerative limb tissues from the immediate response stage to the complete recovery stage. These data will be of great importance to the studies of various scientific disciplines such as development, cell reprogramming, and mechanisms underlying regeneration. Further analysis of these datasets by focusing on the differentially regulated regions may help deduce key regulatory elements that are critical for regeneration initiation in the axolotl limb in the future.
All raw data have been submitted to the CNGB (Nucleotide Sequence Archive) (https://db.cngb.org/search/project/CNP0001445/), and also the NCBI (SRA) (https://www.ncbi.nlm.nih.gov/search/all/?term=PRJNA682840).
The animal study was reviewed and approved by the Ethics Committee of BGI.
XW, YGu, HL, CL, YGuo, and XZh conceived the idea. YGuo and XZh collected samples. XZh and YGuo generated the data. HL, YYua, YQ, CM, JX, MW, YYu, and QD assisted with the experiments. XW analyzed the data with the assistance from YL, XZo, LZ, GH, and WF. XW wrote the manuscript with the input of HL, YGuo, and XZh. YGu and CL supervised the study and revised the manuscript. LL, HY, and SL provided helpful comments on this study. All authors reviewed and approved the final manuscript.
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA16010000) and Guangdong Provincial Key Laboratory of Genome Read and Write (2017B030301011).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Center for Digitizing Cells, Institute of SuperCells, BGI-Shenzhen for helpful comments. This work is supported by China National GeneBank for providing sequencing service. We also thank Liang Chen from Hubei Key Laboratory of Cell Homeostasis, College of Life Sciences, Wuhan University and Giacoma Volpe from GIBH for valuable advice.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.651145/full#supplementary-material
Supplementary Table 1. ATAC-seq metadata and mapping statistics. *Mapped reads: total number of reads minus number of unaligned reads. *Usable reads: number of mapped reads minus number of low mapping quality and duplicate reads.
Supplementary Figure 1. The insert size distribution of ATAC-seq profiles for the same samples shown in Figure 2A.
Supplementary Figure 2. Decomposition of chromatin accessibility and gene expression dynamics during axolotl limb regeneration. (A) Chromatin accessibility dynamics of promoters (TSS ± 2 kb) in six clusters. (B) Pseudobulk gene expression in (A). (C) Heatmap shows the correlation between bulk ATAC-seq and pseudobulk RNA for each gene in C6. (D) Chromatin accessibility and gene expression for genes in C6.
Al-Khalili, L., Chibalin, A. V., Yu, M., Sjodin, B., Nylén, C., Zierath, J. R., et al. (2004). MEF2 activation in differentiated primary human skeletal muscle cultures requires coordinated involvement of parallel pathways. Am. J. Physiol. Cell Physiol. 286, C1410–C1416. doi: 10.1152/ajpcell.00444.2003
Bryant, D. M., Johnson, K., DiTommaso, T., Tickle, T., Couger, M. B., Payzin-Dogru, D., et al. (2017). A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 18, 762–776. doi: 10.1016/j.celrep.2016.12.063
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., and Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10:1213. doi: 10.1038/nmeth.2688
Buenrostro, J. D., Wu, B., Chang, H. Y., and Greenleaf, W. J. (2015). ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21–29. doi: 10.1002/0471142727.mb2129s109
Campbell, L. J., Suárez-Castillo, E. C., Ortiz-Zuazaga, H., Knapp, D., Tanaka, E. M., and Crews, C. M. J. D.D. (2011). Gene expression profile of the regeneration epithelium during axolotl limb regeneration. Dev. Dyn. 240, 1826–1840. doi: 10.1002/dvdy.22669
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2018). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7:gix120. doi: 10.1093/gigascience/gix120
Corces, M. R., Trevino, A. E., Hamilton, E. G., Greenside, P. G., Sinnott-Armstrong, N. A., Vesuna, S., et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. doi: 10.1038/nmeth.4396
Dennis, D. J., Han, S., and Schuurmans, C. (2019). bHLH transcription factors in neural development, disease, and reprogramming. Brain Res. 1705, 48–65. doi: 10.1016/j.brainres.2018.03.013
Dinsmore, C. E. (1991). A History of Regeneration Research: Milestones in the Evolution of a Science. Cambridge: Cambridge University Press.
Ganassi, M., Badodi, S., Wanders, K., Zammit, P. S., and Hughes, S. M. (2020). Myogenin is an essential regulator of adult myofibre growth and muscle stem cell homeostasis. Elife 9:e60445. doi: 10.7554/eLife.60445.sa2
Gehrke, A. R., Neverett, E., Luo, Y.-J., Brandt, A., Ricci, L., Hulett, R. E., et al. (2019). Acoel genome reveals the regulatory landscape of whole-body regeneration. Science 363:6173. doi: 10.1126/science.aau6173
Gerber, T., Murawala, P., Knapp, D., Masselink, W., Schuez, M., Hermann, S., et al. (2018). Single-cell analysis uncovers convergence of cell identities during axolotl limb regeneration. Science 362:681. doi: 10.1126/science.aaq0681
Haas, B. J., and Whited, J. L. (2017). Advances in decoding axolotl limb regeneration. Trends Genet. 33, 553–565. doi: 10.1016/j.tig.2017.05.006
Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y. C., Laslo, P., et al. (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. doi: 10.1016/j.molcel.2010.05.004
Herault, Y., Hraba-Renevey, S., Van der Hoeven, F., and Duboule, D. (1996). Function of the Evx-2 gene in the morphogenesis of vertebrate limbs. EMBO J. 15, 6727–6738. doi: 10.1002/j.1460-2075.1996.tb01062.x
Huang, J., Liang, X., Xuan, Y., Geng, C., Li, Y., Lu, H., et al. (2017). A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience 6:gix024. doi: 10.1093/gigascience/gix024
Iismaa, S. E., Kaidonis, X., Nicks, A. M., Bogush, N., Kikuchi, K., Naqvi, N., et al. (2018). Comparative regenerative mechanisms across different mammalian tissues. NPJ Regen. Med. 3, 1–20. doi: 10.1038/s41536-018-0044-5
Knapp, D., Schulz, H., Rascon, C. A., Volkmer, M., Scholz, J., Nacu, E., et al. (2013). Comparative transcriptional profiling of the axolotl limb identifies a tripartite regeneration-specific gene program. PLoS ONE 8:e61352. doi: 10.1371/journal.pone.0061352
Kuang, X.-L., Zhao, X.-M., Xu, H.-F., Shi, Y.-Y., Deng, J.-B., and Sun, G.-T. (2010). Spatio-temporal expression of a novel neuron-derived neurotrophic factor (NDNF) in mouse brains during development. BMC Neurosci. 11, 1–11. doi: 10.1186/1471-2202-11-137
Kumar, L., and Futschik, M. E. (2007). Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2:5. doi: 10.6026/97320630002005
Lawrence, M., Huber, W., Pages, H., Aboyoun, P., Carlson, M., Gentleman, R., et al. (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9:e1003118. doi: 10.1371/journal.pcbi.1003118
Leigh, N. D., Dunlap, G. S., Johnson, K., Mariano, R., Oshiro, R., Wong, A. Y., et al. (2018). Transcriptomic landscape of the blastema niche in regenerating adult axolotl limbs at single-cell resolution. Nat. Commun. 9, 1–14. doi: 10.1038/s41467-018-07604-0
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [Preprint]. arXiv:1303.3997. doi: 10.6084/M9.FIGSHARE.963153.V1
Li, H., Wei, X., Zhou, L., Zhang, W., Wang, C., Guo, Y., et al. (2020). Dynamic cell transition and immune response landscapes of axolotl limb regeneration revealed by single-cell analysis. Protein Cell 12, 57–66. doi: 10.1007/s13238-020-00763-1
Li, Q., Brown, J. B., Huang, H., and Bickel, P. J. (2011). Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 5, 1752–1779. doi: 10.1214/11-AOAS466
Lui, V. C., Ng, L. J., Sat, E. W., and Cheah, K. S. (1996). The human α2 (XI) collagen gene (COL11A2): completion of coding information, identification of the promoter sequence, and precise localization within the major histocompatibility complex reveal overlap with the KE5 gene. Genomics 32, 401–412. doi: 10.1006/geno.1996.0135
McCusker, C., Bryant, S. V., and Gardiner, D. M. (2015). The axolotl limb blastema: cellular and molecular mechanisms driving blastema formation and limb regeneration in tetrapods. Regeneration 2, 54–71. doi: 10.1002/reg2.32
Monaghan, J. R., Epp, L. G., Putta, S., Page, R. B., Walker, J. A., Beachy, C. K., et al. (2009). Microarray and cDNA sequence analysis of transcription during nerve-dependent limb regeneration. BMC Biol. 7:1. doi: 10.1186/1741-7007-7-1
Nowoshilow, S., Schloissnig, S., Fei, J.-F., Dahl, A., Pang, A. W., Pippel, M., et al. (2018). The axolotl genome and the evolution of key tissue formation regulators. Nature 554, 50–55. doi: 10.1038/nature25458
Pellettieri, J., Fitzgerald, P., Watanabe, S., Mancuso, J., Green, D. R., and Alvarado, A. S. (2010). Cell death and tissue remodeling in planarian regeneration. Dev. Biol. 338, 76–85. doi: 10.1016/j.ydbio.2009.09.015
Picard Toolkit (2019). Broad Institute, GitHub Repository. Available online at: http://broadinstitute.github.io/picard/
Pietsch, P. (1961). Differentiation in regeneration I. The development of muscle and cartilage following deplantation of regenerating limb blastemata of Amblystoma larvae. Dev. Biol. 3, 255–264. doi: 10.1016/0012-1606(61)90046-X
Qin, T., Fan, C.-m., Wang, T.-z., Sun, H., Zhao, Y.-y., Yan, R.-j., et al. (2020). Single-cell RNA-seq reveals novel mitochondria-related musculoskeletal cell populations during adult axolotl limb regeneration process. Cell Death Diff. 28, 1110–1125. doi: 10.1038/s41418-020-00640-8
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rodgers, A. K., Smith, J. J., and Voss, S. R. (2020). Identification of immune and non-immune cells in regenerating axolotl limbs by single-cell sequencing. Exp. Cell Res. 394:112149. doi: 10.1016/j.yexcr.2020.112149
Schreckenberg, G., and Jacobson, A. (1975). Normal stages of development of the axolotl, Ambystoma mexicanum. Dev. Biol. 42, 391–399. doi: 10.1016/0012-1606(75)90343-7
Seyedhassantehrani, N., Otsuka, T., Singh, S., and Gardiner, D. M. (2017). The axolotl limb regeneration model as a discovery tool for engineering the stem cell niche. Curr. Stem Cell Rep. 3, 156–163. doi: 10.1007/s40778-017-0085-5
Stewart, R., Rascón, C. A., Tian, S., Nie, J., Barry, C., Chu, L. F., et al. (2013). Comparative RNA-seq analysis in the unsequenced axolotl: the oncogene burst highlights early gene expression in the blastema PLoS Comput. Biol. 9:e1002936. doi: 10.1371/journal.pcbi.1002936
Tanaka, E. M. (2016). The molecular and cellular choreography of appendage regeneration. Cell 165, 1598–1608. doi: 10.1016/j.cell.2016.05.038
Tarchini, B., and Duboule, D. (2006). Control of Hoxd genes' collinearity during early limb development. Dev. Cell 10, 93–103. doi: 10.1016/j.devcel.2005.11.014
Tsai, S. L., Baselga-Garriga, C., and Melton, D. A. (2019). Blastemal progenitors modulate immune signaling during early limb regeneration. Development 146:e169128. doi: 10.1242/dev.169128
Turkistany, S. A., and DeKoter, R. P. (2011). The transcription factor PU. 1 is a critical regulator of cellular communication in the immune system. Arch. Immunol. Ther. Exp. 59, 431–440. doi: 10.1007/s00005-011-0147-9
Vivien, C. J., Hudson, J. E., and Porrello, E. R. (2016). Evolution, comparative biology and ontogeny of vertebrate heart regeneration. NPJ Regen. Med. 1, 1–14. doi: 10.1038/npjregenmed.2016.12
Wang, W., Hu, C.-K., Zeng, A., Alegre, D., Hu, D., Gotting, K., et al. (2020). Changes in regeneration-responsive enhancers shape regenerative capacities in vertebrates. Science 369:3090. doi: 10.1126/science.aaz3090
Wei, X. (2020). An ATAC-seq Dataset Uncovers the Regulatory Landscape Axolotl Limb Regeneration. Available online at: https://doi.org/10.6084/m9.figshare.13370468.v1
Wu, C. H., Tsai, M. H., Ho, C. C., Chen, C. Y., and Lee, H. S. J. B.G. (2013). De novo transcriptome sequencing of axolotl blastema for identification of differentially expressed genes during limb regeneration. BMC Genomics 14:434. doi: 10.1186/1471-2164-14-434
Yu, G., Wang, L.-G., Han, Y., and He, Q.-Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. doi: 10.1089/omi.2011.0118
Yun, M. H. (2015). Changes in regenerative capacity through lifespan. Int. J. Mol. Sci. 16, 25392–25432. doi: 10.3390/ijms161025392
Zeng, A., Li, H., Guo, L., Gao, X., McKinney, S., Wang, Y., et al. (2018). Prospectively isolated tetraspanin(+) neoblasts are adult pluripotent stem cells underlying planaria regeneration. Cell 173, 1593–1608. doi: 10.1016/j.cell.2018.05.006
Keywords: limb, regenertion, axolotl, ATAC-seq, regulatory element
Citation: Wei X, Li H, Guo Y, Zhao X, Liu Y, Zou X, Zhou L, Yuan Y, Qin Y, Mao C, Huang G, Yu Y, Deng Q, Feng W, Xu J, Wang M, Liu S, Yang H, Liu L, Liu C and Gu Y (2021) An ATAC-seq Dataset Uncovers the Regulatory Landscape During Axolotl Limb Regeneration. Front. Cell Dev. Biol. 9:651145. doi: 10.3389/fcell.2021.651145
Received: 08 January 2021; Accepted: 26 February 2021;
Published: 30 March 2021.
Edited by:
Zhao-Qian Teng, Chinese Academy of Sciences (CAS), ChinaCopyright © 2021 Wei, Li, Guo, Zhao, Liu, Zou, Zhou, Yuan, Qin, Mao, Huang, Yu, Deng, Feng, Xu, Wang, Liu, Yang, Liu, Liu and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Gu, Z3V5aW5nQGdlbm9taWNzLmNu; Chuanyu Liu, bGl1Y2h1YW55dUBnZW5vbWljcy5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.