 Jessica Maiuolo1*
Jessica Maiuolo1* Micaela Gliozzi1
Micaela Gliozzi1 Vincenzo Musolino1Cristina Carresi1Federica Scarano1Saverio Nucera1Miriam Scicchitano1Francesca Bosco1Stefano Ruga1Maria Caterina Zito1
Vincenzo Musolino1Cristina Carresi1Federica Scarano1Saverio Nucera1Miriam Scicchitano1Francesca Bosco1Stefano Ruga1Maria Caterina Zito1 Roberta Macri1Rosamaria Bulotta1
Roberta Macri1Rosamaria Bulotta1 Carolina Muscoli1,2
Carolina Muscoli1,2 Vincenzo Mollace1,2
Vincenzo Mollace1,2- 1IRC-FSH Department of Health Sciences, University “Magna Graecia” of Catanzaro, Catanzaro, Italy
- 2IRCCS San Raffaele, Rome, Italy
Metabolic syndrome is not a single pathology, but a constellation of cardiovascular disease risk factors including: central and abdominal obesity, systemic hypertension, insulin resistance (or type 2 diabetes mellitus), and atherogenic dyslipidemia. The global incidence of Metabolic syndrome is estimated to be about one quarter of the world population; for this reason, it would be desirable to better understand the underlying mechanisms involved in order to develop treatments that can reduce or eliminate the damage caused. The effects of Metabolic syndrome are multiple and wide ranging; some of which have an impact on the central nervous system and cause neurological and neurodegenerative diseases. Autophagy is a catabolic intracellular process, essential for the recycling of cytoplasmic materials and for the degradation of damaged cellular organelle. Therefore, autophagy is primarily a cytoprotective mechanism; even if excessive cellular degradation can be detrimental. To date, it is known that systemic autophagic insufficiency is able to cause metabolic balance deterioration and facilitate the onset of metabolic syndrome. This review aims to highlight the current state of knowledge regarding the connection between metabolic syndrome and the onset of several neurological diseases related to it. Furthermore, since autophagy has been found to be of particular importance in metabolic disorders, the probable involvement of this degradative process is assumed to be responsible for the attenuation of neurological disorders resulting from metabolic syndrome.
Introduction
Metabolic syndrome (MetS), also known as syndrome X, insulin resistance syndrome, or Reaven syndrome; is not a single pathology, but a constellation of cardiovascular disease risk factors. These clinical conditions comprise: (a) central and abdominal obesity, (b) systemic hypertension, (c) insulin resistance (or type 2 diabetes mellitus), and (d) atherogenic dyslipidemia, the so-defined “deadly quartet” (McCracken et al., 2018). There are several definitions to indicate the characteristics of MetS; among these, we point out the one provided by the International Federation of Diabetes (IDF) of 2006 (Saklayen, 2018), which states that metabolic syndrome is represented by glucose in the blood above 5.6 mmol/L (100 mg/dl) (or diabetes already diagnosed) along with the presence of two or more of the following conditions:
• HDL cholesterol (HDL-C) < 1.0 mmol/L (40 mg/dl) in men; < 1.3 mmol/L (50 mg/dl) in women or drug treatment for low HDL-C;
• blood triglycerides > 1.7 mmol/L (150 mg/dl) or drug treatment for elevated triglycerides;
• blood pressure > 130/85 mmHg or drug treatment for hypertension;
• waist > 94 cm (men) or > 80 cm (women). Obesity is diagnosed using waist circumference, which correlates better with visceral adiposity than Body Mass Index (BMI) (Alberti et al., 2005).
The presence of three of the five main risk factors is sufficient to confirm a MetS diagnosis (Altieri et al., 2015). All other MetS definitions are very similar; however, parameters indicated may vary by a few units. The global incidence of MetS is very high, afflicting one third of adults 18 years or older in the United States alone (Aguilar et al., 2015). To date, we can estimate the global prevalence to be about one quarter of the world population (Li et al., 2008). In general, the prevalence of MetS has been found to increase with age, involving about 20% of males and 16% of females under 40 years of age; 41% of males and 37% of females between the ages of 40–59; and 52% of males and 54% of females over 60 years of age (Ervin, 2009; Aoki et al., 2014). During the last 15 years, the prevalence of MetS has increased (Pucci et al., 2017) and the main motivation is related to significant changes in lifestyle; the western lifestyle consists of numerous risk factors such as: high-fat diet, cigarette smoking, alcohol consumption, obesity, and physical inactivity (O’Doherty et al., 2016; Sarmiento Quintero et al., 2016; Finicelli et al., 2019). In particular, the western diet is based on high consumption of salt, refined sugars, and saturated fats that determine significant effects on body composition and metabolism such as: increased BMI, generalized and abdominal obesity, dyslipidemia, and type 2 diabetes (Misra and Khurana, 2008). In addition to lifestyle changes, we should include other concomitant factors such as: chronic inflammation, endothelial dysfunction, genetic susceptibility, hypercoagulability, and chronic stress (Srikanthan et al., 2016; Motamedi et al., 2017; Woo et al., 2019). The main features of MetS are summarized in Figure 1.
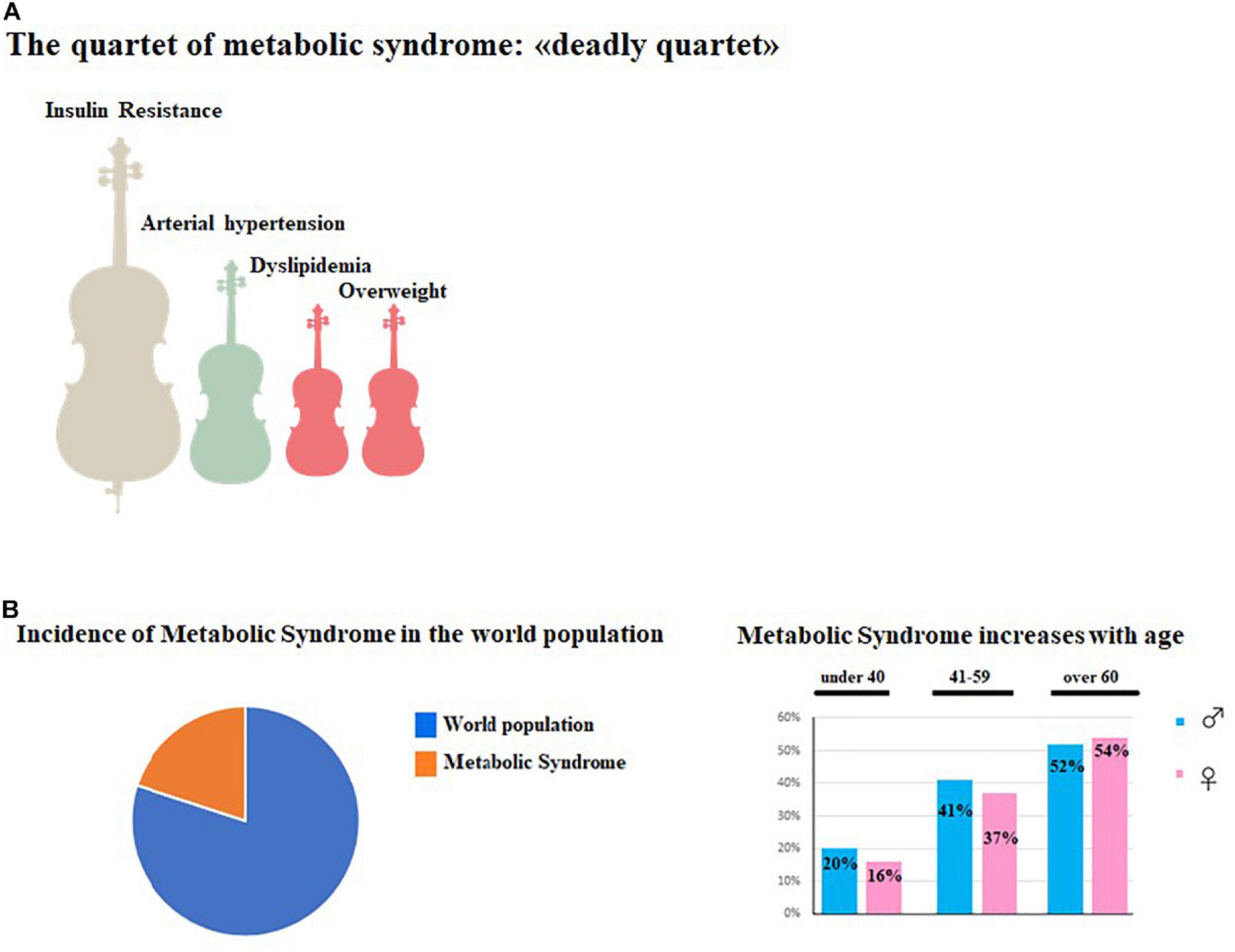
Figure 1. Main features of MetS. (A) represented the symptomatological quartet most widespread in MetS consisting of Insulin Resistance, Hypertension, Dyslipimedia and Obesity. (B) (Left) shows the global prevalence of MetS in the world population, while in (B) (right) the increase of MetS in aging men and women is shown.
The first part of this review, after describing the main features of MetS, deepens the involvement of the vascular endothelium in this metabolic disorder based on current scientific literature. Subsequently, the development of neurological disorders resulting from metabolic syndrome is considered. Lastly, the importance of autophagic function has discussed with the aim of proposing this element as molecular target to reduce and/or eliminate the dangerous symptoms of MetS and the consequent neurological symptoms.
The Role of the Endothelium in Metabolic Syndrome
MetS can, as already stated, increase the risk of developing type 2 diabetes mellitus, obesity, and cardiovascular diseases; contributing, therefore, to high rates of mortality and morbidity (Alberti et al., 2009). The common denominators between MetS and its metabolic disorders are chronic low-grade inflammation and activation of the immune system (Chawla et al., 2011; Ouchi et al., 2011; El-Benna et al., 2016). MetS demonstrates a central role in promoting tissue inflammation in the adipose tissue of the liver, muscles, and pancreas; with concomitant infiltration of macrophages, and production of pro-inflammatory cytokines including Tumor Necrosis Factor alpha (TNFα), Interleukin 6 (IL-6), IL1β, activation of the c-JUN N-terminal kinase (JNK), and nuclear factor-kappa B (NF-κB) pathways (Chawla et al., 2011; Elmarakby and Sullivan, 2012; Grandl and Wolfrum, 2018). Macrophages are classically classified into two distinct subtypes: the activated phenotype secreting pro-inflammatory cytokines (M1), and the alternatively activated phenotype which produces anti-inflammatory cytokines such as IL-10 (M2) (Chawla et al., 2011). Under normal conditions, macrophages have been described as being a mix between M1 and M2 phenotypes, while in MetS, a phenotypic shift from M2 to M1 has been noted, both in mice and in humans (Wentworth et al., 2010) which shows a marked proinflammatory response. The inflammatory process includes increased vascular permeability to immune cells with the endothelium playing a fundamental role in this process (Bañuls et al., 2017; Bruno et al., 2017; Friesen and Cowan, 2019). The endothelium is composed of a layer of cells and constitutes the internal lining of the blood vessels; playing the role of a selectively permeable barrier. Endothelial cells do not play a passive role; instead, they regulate very important physiological functions such as: maintaining homeostatic balance, controlling vasomotor tone, ensuring proper permeability, and managing innate immunity reactions (Godo and Shimokawa, 2017). Precisely for this reason, the endothelium can be considered as an organ. The permeability of the endothelium allows the transport of only a few necessary molecules; this selectivity is ensured by a fine regulation carried out by junction proteins that have the function of keeping endothelial cells closely adjacent; thereby preventing the passage of unwanted molecules or cells (Babushkina et al., 2015). Junction proteins can be classified into two categories: tight junctions, also known as zonula occludens, and adherens junctions. Ensuring the cell adhesions and junctions, cytoskeleton proteins play a particularly key role (Maiuolo et al., 2018). When the expression of these proteins is nullified or reduced, endothelial disassembly and dysfunction occurs (Kiseleva et al., 2018).
Vascular endothelium cells (VEC) play a fundamental role in the human body; ensuring the regulation of vasomotor functions, the maintenance of vessel walls, anti-platelet aggregation, and endocrine functions. Numerous harmful stimuli can lead to endothelial cell dysfunction (ECD) with consequential increases in the risk of many diseases including cardiovascular diseases (Vykoukal and Davies, 2011; Khaddaj Mallat et al., 2017; Kaplan et al., 2018; Maiuolo et al., 2020a). In MetS patients, the ECD is a specific early pathophysiological indicator of cardiovascular disorders; its recognition and early intervention are of extreme importance in the prevention, treatment, and prognosis of cardiovascular diseases (Wei et al., 2014; Dalal et al., 2020). In particular, many substances secreted by VEC are considered important indicators of the function of endothelial cells, these include: Plasminogen Activator Inhibitor-1 (PAI-1), von Willebrand factor (vWF), vascular endothelial cadherin (VE-cad), Thrombomodulin (TM), and Vascular endothelial growth factor (VEGF). PAI-1 is mainly produced by the endothelium but is also secreted by other types of tissue, such as adipose tissue. PAI-1 is a fibrinolysis inhibitor; preventing the physiological process that degrades blood clots and contributing to the formation of atherosclerotic plaques (Deng et al., 2012; Yarmolinsky et al., 2016). vWF is a macromolecular plasma glycoprotein involved in hemostasis with important implications in blood viscosity; an increase can predict the risk of thrombosis (Shahidi, 2017). VE-cad is a classic cadherin, belonging to the cadherin superfamily; it is expressed specifically on the endothelial surface and concentrated in existing cell-cell junctions. VE-cad is known to be necessary to maintain a restrictive endothelial barrier; early studies, using blocking antibodies against VE-caderin, resulted in increased endothelial permeability in vitro (Hakanpaa et al., 2018) and in hemorrhage in vivo (Corada et al., 2002). TM is an integral membrane protein expressed on the surface of endothelial cells and serves as a cofactor for thrombin. TM reduces blood clotting by converting thrombin from a pro-clotting factor to an anti-coagulant factor (Loghmani and Conway, 2018). In normal conditions, plasma TM levels are low; but when vascular endothelial cells are damaged, TM increases significantly; highlighting that its level can be used as a marker of endothelial injury (Stadtmann et al., 2011; Kishi et al., 2020). VEGF is a selective signaling protein that promotes the growth of new blood vessels and restores the supply of oxygenated blood to cells and tissues that have been deprived due to impaired blood circulation. VEGF also improves vascular permeability by increasing exudation of blood components and inflammatory cytokines (Sack et al., 2016). Scientific studies have shown that serum levels of PAI-1, vWF, VE-cad, TM, and VEGF were increased in patients with MetS compared to healthy individuals (Georgieva et al., 2004; Hajiluian et al., 2017; Mazidi et al., 2017; Wang et al., 2018). Moreover, an increase has also been found in children and adolescents with MetS, evidencing that these parameters can be considered predictors of early vascular changes (Wei et al., 2014). MetS negatively affects the function of the vascular endothelium; increasing vasoconstriction and prothrombotic state through different mechanisms (Aggoun, 2007; O’Shea et al., 2015; Pomero et al., 2015; Domingueti et al., 2016):
• Hyperglycemia, hyperlipemia, and hypertension increase the release of numerous cytokines, such as: IL-1β, IL-6, and growth factor PDGF; triggering the process of endothelial dysfunction (Varghese et al., 2018).
• Carbohydrate metabolism (hyperinsulinemia/insulin resistance or hyperglycemia/diabetes), dyslipidemia, obesity, and hypertension can all determine vascular contractility (Cuspidi et al., 2018).
• Altered carbohydrate metabolism, dyslipidemia, hypertension, and obesity lead to increased PAI-1, vWF, VE-cad, TM, and VEGF production; a condition responsible for impaired anticoagulant and fibrinolytic activity which can cause hypercoagulability: a low fibrinolytic and high viscosity state, and promote the formation of microthrombi (Wei et al., 2014).
The main stages of endothelial inflammation, involved in Mets are represented in Figure 2. In the light of what has been stated, a clinical evaluation of endothelial involvement should be carried out both in patients with MetS and in people not affected who show clear risk factors related to body weight, metabolism of carbohydrates, and blood pressure. In this way, it could be possible to prevent and control secondary disorders of MetS.
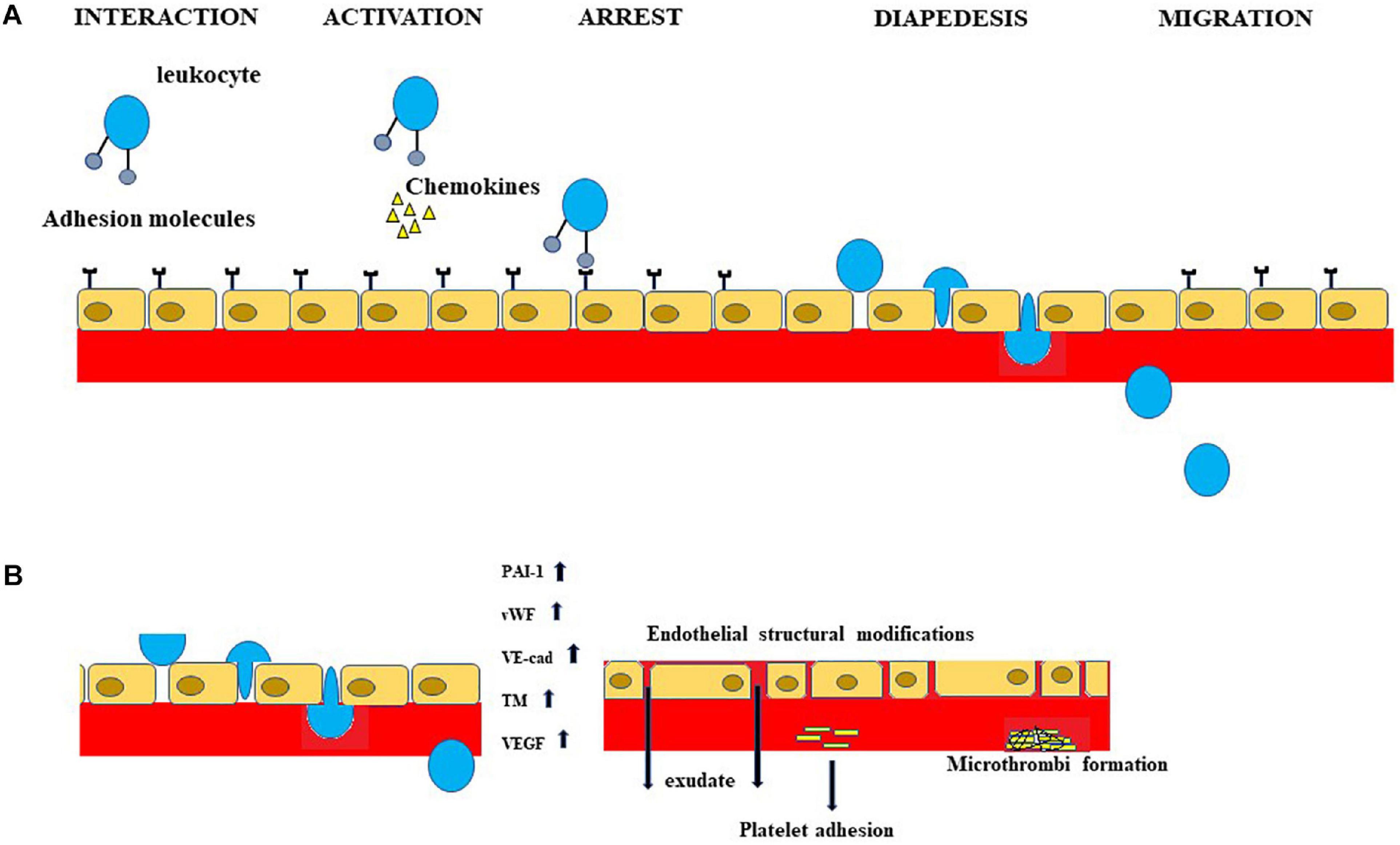
Figure 2. Inflammation of the endothelium is involved in MetS and subsequent neurological manifestations. A portion of the inflammatory process, occurring both in MetS and in neurological disorders related to MetS, is schematized in Figure. In particular, (A) shows the sequence of leukocyte extravasation across the endothelium. (B) Important indicators of the dysfunction of endothelial cells are shown. The increases in PAI-1, vWF, VE-cad, TM and VEGF determine endothelial structural modifications, exudate production, platelet adhesion, and microthrombi formation.
From Metabolic Syndrome to Neurological Diseases
The effects of MetS are multiple and wide ranging; some of which have an impact on the central nervous system (CNS); causing neurodegenerative and neurological diseases (Kathy et al., 2012; Błaszczyk and Gawlik, 2016; Van Dyken and Lacoste, 2018). The CNS is the most complex and organized system in the human body and proper neuron functioning requires a correct biochemical balance (which includes suitable chemical and electrical signaling) in order to support and maintain adequate intracellular and intercellular communication. In addition, the correct concentration of ionic and signaling molecules must be guaranteed to remove catabolites and maintain a low concentration of neurotoxic mediators (Corasaniti et al., 2007; Maiuolo et al., 2018). A key role is played by the Blood Brain Barrier (BBB), which provides all these functions and represents a physical, selective, and highly lipophilic barrier; protecting brain tissue and separating it from systemic circulation (Daneman and Prat, 2015). The basic structure of the BBB consists of endothelial cells, tightly joined by junction proteins; while other constituents, including the basal membrane (BM), pericytes, and astrocytes, perform support and regulation functions (Andreone et al., 2015; O’Brown et al., 2018). The BBB endothelium is rich in specific proteins that act as carriers and receptors; responsible for the passage of metabolites, macronutrients, micronutrients, and junction proteins; which significantly limit their intercellular exchange. Endothelial cells consist of five or six times more mitochondria than other tissues in the human body as these organelles provide the energy required for endothelial cells to maintain cerebral homeostasis (Gentil et al., 2005). Endothelial dysfunction leads to “frailty” of the BBB; characterized by increased vascular permeability, impaired ability to preserve brain tissue homeostasis, infiltration of toxic blood-derived molecules, cells, microbial agents; which together trigger inflammatory and immune neurodegeneration (Maiuolo et al., 2018, 2019). Basement membrane (BM) is composed of collagen, laminin, heparin, and other glycoproteins; it determines an additional barrier in the BBB, although it can be interrupted by metalloproteinases of the matrix (Thomsen et al., 2017). Pericytes are in contact with the BM, juxtaposed to the endothelial cells, with which they are connected and are involved in the regulation of angiogenesis, vascular stability, and BBB control (ElAli et al., 2014). Astrocytes regulate vasomotor responses and the brain blood flow as a result of changes in neural activity; they also release regulating factors for the maturation and maintenance of the BBB (Lacoste and Gu, 2015). Integrity of the BBB is essential to ensure the proper functioning of the CNS and many neurological disorders are caused by its rupture (Zhao et al., 2015; Chakraborty et al., 2017; Maiuolo et al., 2020b). The fundamental cause of the loss of barrier integrity is the inflammatory process that occurs in the CNS as a result of neuronal damage (Varatharaj and Galea, 2017). Under normal conditions, the integrity of the BBB prevents the passage of immune cells into the CNS; however, inflammation induces the opening of the BBB thereby altering the various components. The main consequences are:
• release of cytokines and inflammation mediators (Incalza et al., 2018; Oppedisano et al., 2020);
• leukocyte extravasation (diapedesis) across the endothelium (Rudziak et al., 2019);
• destruction of BBB cells (Chen et al., 2019).
The first important step for the diapedesis is the interaction between the leukocytes and adhesion molecules on endothelial cells, including P-selectin (a cell adhesion molecule on the surfaces of activated endothelial cells); E-selectin (endothelial-leukocyte adhesion molecule 1 expressed only on endothelial cells and activated by cytokines) (Laird et al., 2018); ICAM-1 (intercellular adhesion molecule-1 typically expressed on endothelial cells and cells of the immune system) (Schaefer et al., 2017); and VCAM-1 (vascular cell adhesion molecule-1 that mediates the adhesion of lymphocytes, monocytes, eosinophils, and basophils to vascular endothelium) (Fan et al., 2019). The next step involves the rolling of leukocytes along the wall of the vessel; the release of chemokines that strengthen contact with the endothelium; and the extension of pseudopods, from leukocytes, that allow the attack of endothelial cells (Lutz et al., 2017). The leukocyte crossing of the endothelium is defined as “transendothelial cell migration” and the correct direction to follow is provided by chemotactic and haptotactic stimuli outside of the vascular lumen and beyond. This movement occurs at the same time as the morphological variations of the leukocytes, which guide the cell nucleus through tight endothelial junctions and pores. Transendothelial leukocyte migration can take place through junctions between adjacent endothelial cells (paracellular migration) or through the body of the endothelium (transcellular migration) (Nourshargh and Alon, 2014). Although paracellular migration occurs in 70–80% of cases, the cerebral vascular endothelial cells appear to be an exception to this rule and prefer transcellular migration. This phenomenon is motivated by the particularly narrow specialized junction structures expressed by the brain endothelial cells (Engelhardt and Ransohoff, 2012). It has been widely demonstrated that increased inflammation facilitates BBB breakage (Varatharaj and Galea, 2017); with several mechanisms that include: downregulation of many junction proteins and important amino acid transporters (Yoo et al., 2016), alteration of transcytosis (Ransohoff et al., 2015), up-regulation of transporters for TNF-α, lysosomal degradation enzymes, VCAM-1 P/E-selectin (Varatharaj and Galea, 2017), and the accumulation of insoluble fibrin; responsible for the alteration of the immune response and blood clotting (Davalos and Akassoglou, 2012). Ultimately, these activated inflammatory processes are responsible for the destruction of cellular components of the BBB. In particular, the integrity of astrocytes is compromised, the leukocyte infiltration is increased, and the input of pathogens and toxins in the central nervous system is allowed (Van Dyken and Lacoste, 2018). Astrocytes are very susceptible to oxidative stress and inflammation; becoming unable to perform their role of maintaining ions and neurotransmitters in physiological conditions for BBB integrity (Bhat et al., 2012).
A positive feedback cycle has recently been described where activated microglia produce ROS; leading to cell death and increased levels of local glutamate. This condition results in increased secretion of proinfiammatory cytokines, a further activation of microglia, and destruction of endothelial BBB cells (Mittal et al., 2014; Cai et al., 2017). Increased systemic inflammation impacts many of the systems in the body including the brain, in fact, inflammation is closely associated with neuropathology (Fan et al., 2014). To date, a series of epidemiological studies have shown that MetS increases the risk of developing neurodegenerative diseases, CNS dysfunction (Islam, 2017; Motamedi et al., 2017; Ricci et al., 2017; Arshad et al., 2018; Karaca and Karaca, 2018; Palta et al., 2021), and reduced cognitive performance including deficits in memory, visuospatial abilities, executive functioning, processing speed, and overall intellectual functioning. MetS was found to be a factor of risk for: ischemic stroke, intracranial arteriosclerosis, periventricular white matter hyperintensities, and subcortical white matter lesions (Yates et al., 2012). In this regard, changes in brain metabolism have been shown to be responsible for the onset of neuroinflammation; these brain changes may represent an early associated brain impairment with peripheral metabolic disorders.
MetS: Obesity, Diabetes, and Cognitive Functions
Obesity is the excess accumulation of body fat caused by an imbalance between energy intake and consumption. The effects of obesity are largely mediated through inflammation and, in these mouse models, neural inflammation can be detected even earlier than weight gain (Van Dyken and Lacoste, 2018). Obesity is directly related to impairment of cognitive function and an increased risk of different forms of dementia. Clinical and experimental evidence indicates that obesity and/or a high-fat diet is associated with impairment of learning, memory, and executive functioning (Anstey et al., 2011; Bremer and Jialal, 2013; Miller and Spencer, 2014; Saltiel and Olefsky, 2017; Costello and Petzold, 2020). Many studies have been carried out on the correlation of BMI-cognitive function and waist circumference-cognitive function. In general, it has been found that BMI is inversely related to cognitive function, including memory and executive functioning. In addition to cognitive performance, obesity can affect brain structure; leading to atrophy (Climie et al., 2015; Gogniat et al., 2018). Moreover, a relationship has also been described regarding particular areas of the brain; the temporal and frontal lobes appear to be particularly vulnerable to the effects of obesity and gray matter volumes of these brain regions are reduced in obese patients; resulting in the reduction of neuronal viability (Gómez-Apo et al., 2018; Lee et al., 2020). The first area of the brain to be affected is the hypothalamus; the subsequent damage reduces the number of synapses on hypothalamic neurons and increases neural apoptosis (Sohn, 2015). The obese mice showed a lower yield than the control group, thus confirming involvement of the hippocampus and cognitive impairment (Pendlebury and Rothwell, 2009; Hargrave et al., 2016; Van Dyken and Lacoste, 2018).
The activation of the inflammatory process, present in obesity, seems to be the fundamental cause of the alteration of the health of the brain (function and structure). In particular, a chain reaction occurs in which the activation of transcription factor NF-kB can be appreciated (Jais and Brüning, 2017), followed by upregulation of pro-inflammatory cytokines, such as: IL-1β, TNF-α, and IL-6 (Lawrence, 2009). It is also important to note that the observed cognitive decline was preceded by the reduction of protein TJ expression and loss of BBB integrity (Gustafson et al., 2007; Davidson et al., 2013; Dorfman and Thaler, 2015).
Scientific evidence has shown, both in animal models and in humans, a close correlation between diabetes mellitus [type 1 (T1DM) and type 2 (T2DM)] and cognitive decline leading to dementia; although T2DM has shown a stronger association with brain disorders (Monette et al., 2014; Zilliox et al., 2016; Black et al., 2018). Among the components of MetS, hyperglycemia has the strongest association with the risk of developing cognitive deterioration (Šmahelová, 2017). Numerous studies have shown reduced performance in cognitive activities in diabetic compared to non-diabetic controls which include memory, mental speed, mental flexibility, and executive function. These dysfunctions have been correlated with a reduced density of gray matter of the prefrontal and temporal cortex (van den Berg et al., 2010). It is not perfectly clear when cognitive impairment occurs in the course of diabetes; in some cases these events can be very early, while in other cases they are later events (Hassing et al., 2004). It is known that insulin signaling improves synaptic plasticity in the hippocampus; playing an important role in memory and learning. In fact, insulin facilitates long-term enhancement of the hippocampus (LTP) and, in the healthy mammalian brain, is associated with learning and memory; increasing the expression of N-methyl-D-aspartate receptors. In addition, insulin regulates the concentration of several important neurotransmitters in memory maintenance such as: acetylcholine, norepinephrine, and epinephrine (Boyd et al., 1985; Figlewicz et al., 1993; Patterson et al., 2016). Insulin dysregulation in patients with diabetes could facilitate cognitive disorders (Kullmann et al., 2016; Denver et al., 2018; Tumminia et al., 2018). Insulin in the brain also has the function of regulating mitochondria (Heras-Sandoval et al., 2012). It has been shown that, in a condition of overt diabetes, highly altered insulin acts on the pre-synaptic terminals causing the mitochondrial DNA mutations responsible for functional and structural changes of the organelles (Lu et al., 2004). Dysfunction of the mitochondria causes the depletion of energy reserves; the enzymatic complexes of the electronic transport chain (complexes I and III) are altered, leading to neuronal synaptic loss and cognitive deficits (Kim et al., 2009; Choi et al., 2014).
MetS: Cognitive Dysfunction and the Possible Role of Brain-Derived Neurotrophic Factor
In the early 1950s, the neurotrophic theory was developed. This theory is based on the functional mechanisms adopted by effecter cells to control growth, survival, differentiation, and neural function through the production of biomolecules, identified as neurotrophins (Lewin and Barde, 1996; Dechant and Neumann, 2002). To date, it is known that the family of these molecules includes: Nerve Growth Factor (NGF), Brain-Derived Neurotrophic Factor (BDNF), Neurotrophin3 (NT3), NT4, NT5, NT6, and NT7 (Emanueli et al., 2014; West et al., 2014; Skup, 2018). BDNF is the most abundant neurotrophin in the mammalian CNS and is synthesized and expressed in different cerebral regions. BDNF is a protein encoded by the BDNF gene found in humans on chromosome 11; it is initially synthesized as a precursor, pro-BDNF, consisting of 129 amino acids in the endoplasmic reticulum. Subsequently, pro-BDNF is split through the action of proconvertase; in a mature form of 118 amino acids in the trans-Golgi. The mature shape dimerizes; forming the BDNF active factor (Hempstead, 2015). BDNF is synthesized in the cytoplasm of neurons and glia; although it is also found in the skeletal and smooth muscles, liver, lymphocytes, endocrine system, pancreas, endothelial cells, and adipose tissue (Ansari et al., 2016). BDNF is involved in many neurological processes such as: neural growth, differentiation, synaptic conductivity, plasticity, neurogenesis, neuroregeneration, cognition, memory, learning, and dendrite growth (Smith et al., 2015; Morales-Marín et al., 2016). For this reason, structural and functional alteration of BDNF, determined by several factors, has been implicated in a number of neurodegenerative diseases and psychiatric disorders such as Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, schizophrenia, intellectual disability, autism, depression, and the development of mood disorders (Björkholm and Monteggia, 2016). It has been shown that neurotrophins exert a metabotropic effect with respect to glucose, lipid, and energy; therefore, they have an important role in MetS (Chaldakov et al., 2014; Kim et al., 2020). Recent studies have shown a correlation between BDNF and MetS. In fact, there is a reduction of BDNF levels, especially in the stages of advancement of MetS (Morales-Marín et al., 2016). BDNF, which is reduced in MetS, performs critical functions within the CNS and alterations determine neurological diseases. Therefore, we can indirectly conclude that cognitive dysfunctions in MetS are correlated with BDNF. However, in order to better understand this topic, further studies should be carried out.
Can Autophagy Interfere with Mets and Associated Neuronal Disorders?
Autophagy is a catabolic intracellular process evolutionarily preserved and finely regulated; essential for the recycling of cytoplasmic materials (proteins, lipids, carbohydrates) and for the degradation of damaged organelles (mitochondria, endoplasmic reticulum and peroxisomes), whose accumulation can be toxic to cells (Lapaquette et al., 2015). In this way, the renewal and proper functioning of intracellular organelles is also guaranteed. In lysosomes, autophagy determines: the recycling of cell portions that can still be used, the reduction of cell waste, the protection and maintenance of cellular energy and cellular adaptation to environmental challenges; all processes that contribute to cell survival (Madrigal-Matute and Cuervo, 2016). Therefore, autophagy is primarily a cytoprotective mechanism; even if excessive self-degradation can be detrimental. To date, it is known that autophagic activity is also connected to other important roles such as: the maintenance of cellular metabolism, control of cell cycle, immune response, development, and differentiation or cell death (Hu et al., 2019). In addition, when cells are subjected to a wide range of stressful conditions (physical, chemical, or metabolic), the autophagic process is activated in order to maintain cellular homeostasis. Since autophagy is critical for the maintenance of cellular metabolic homeostasis of whole body, its dysregulation may be the cause of the onset of pathologies which can affect liver, heart, brain, myopathies, diabetes, obesity, and cancer (Lim et al., 2018). Four main forms of autophagy have been described in mammalians:
• Macroautophagy (herein autophagy) is a catabolic process in which old cytoplasmic proteins, lipids, or damaged organelles are incorporated in double-membrane vesicular structures called autophagosomes. Autophagosomal membranes may derive from a number of sources such as: endoplasmic reticulum, Golgi apparatus, mitochondria, endosomes, and the plasma membrane. The movement of the autophagosomes takes place along the microtubules until reaching the lysosomes where the degradation process occurs (Feng et al., 2015).
• Microautophagy, a non-selective lysosomal process, refers to the direct engulfment of small amounts of cytosolic material (Li and Hochstrasser, 2020).
• Chaperone-mediated autophagy refers to the process in which some protein complexes are recognized by the cytosolic chaperones that deliver them to the surface of the lysosomes (Tekirdag and Cuervo, 2018).
• Selective autophagy that requires the respect of three criteria to ensure an efficient process (Zaffagnini and Martens, 2016): (i) the specific recognition of the cargo to be phagocytized; (ii) an efficient bonding of the cargo to a nascent autophagosome; (iii) the exclusion of components not part of the cargo.
The autophagy mechanism requires the expression of a set of evolutionarily conserved autophagy related genes (ATGs) whose protein products combine to form several useful complexes in the various stages of this process. All ATG genes, originally discovered in yeast, are needed for the efficient formation of autophagosomes that blend with the lysosomes, orchestrating and mediating the intra-cytoplasmic cargo degradation (Mizushima, 2018). The whole autophagic process is divided into six main steps which need to be tightly regulated both spatially and temporally: initiation, nucleation of vesicles, membrane elongation, closure, maturation, and degradation (Grumati and Dikic, 2018). In particular, the induction of autophagy occurs with the recruitment of Atg proteins and other proteic complexes in a specific subcellular position, called Phagophore Assembly Site, and with the nucleation of an insulating membrane forming a structure called phagophore (Yu et al., 2018). The very first autophagy-specific complex is termined Unc-51-Like Kinase 1 (ULK1) and is composed by ULK1 itself, Atg13, Focal adhesion kinase family Interacting Protein (FIP200) and Atg101 (Zachari and Ganley, 2017). When activated, ULK1 phosphorylates other autophagy pathway components, including Beclin1 and Atg9 and the onset of phagophore formation occurs when ULK1 complex is moved to a specific place in the endoplasmic reticulum membrane marked by the activated Atg9 protein (Levine and Kroemer, 2019). Elongation of the autophagosome membrane determines the expansion of the autophagosome in a sphere, around to the portion of the cytosol that must be degraded (Yoshii and Mizushima, 2017). This step involves Atg7 and Atg10 complexes that combine Atg12 and Atg5 proteins. The Atg12-Atg5 conjugate, along with Atg16L1, adds phosphatidylethanolamine monomers, determining the elongation of the autophagosome (Kauffman et al., 2018). The protein Atg9 is partly regulated by the Atg1 complex and other Atg proteins such as Atg17, Atg2, Atg18, Atg14L, Atg8, Atg21, Atg16, Atg12, and Atg5 (Nishimura and Tooze, 2020). The next step is the clearance of most Atgs proteins and the fusion of the autophagosome with the lysosomal membrane to form an autolysosome. Finally, the authophagic load is degraded by the hydrolytic environment into the autolysosome (Yu et al., 2018).
To date it is known that a dysfunction of autophagy is related to the onset of some diseases including cancer (White et al., 2015; Li Y. J. et al., 2017; Li et al., 2020), aging (Wong et al., 2020), neurodegenerative diseases (Menzies et al., 2015; Boland et al., 2018; Kumar et al., 2018; Suresh et al., 2020) and metabolic diseases (Ren and Anversa, 2015; van Niekerk et al., 2018; Wu N. N. et al., 2019; Almeida et al., 2020).
From the structural point of view, the neuron can be divided into three compartments: the soma, the axon and the dendrites. Axons can grow from a few micrometers up to many feet to reach their targets while dendrites are much shorter but can form highly branched nets. Since neurons are post-mitotic and long-lived cells, they must handle external stress removing aggregated and/or damaged proteins and organelles. For this reason, autophagy can perform this function in each neuronal compartment (Stavoe and Holzbaur, 2019). The interruption of homeostasis and physiological functions in the CNS, due to numerous pathophysiological events, can generate a wide range of diseases or pathological consequences. In particular, alteration of the redox state, inflammation, metabolic disorders, and failure in quality control of cellular proteins and organelles have been implicated in neurological and neurodegenerative disorders. In fact, a similar concept is also valid for psychiatric disorders (Menzies et al., 2015; Guo et al., 2018; Tomoda et al., 2020).
Numerous scientific studies have shown that the basal activity of autophagy is fundamental for the maintenance of homeostasis and neuronal vitality: in fact, it is known that neurons are particularly vulnerable in case of altered and/or switched off autophagy (Yamamoto and Yue, 2014; Kulkarni et al., 2018). It is important to stress that autophagy in CNS is important not only to maintain neuronal homeostasis, but also to ensure neurodevelopment (Stavoe and Holzbaur, 2019). Studies conducted in cultured embryonic peripheral neurons showed a retrogradely transport of large acidified vesicles from axons toward the soma along microtubules, suggesting that autophagosomes formed in the distal axon and were subsequently transported to the neuronal soma. The neuronal soma is rich in lysosomes and late endosomes with which autophagosomes fuse to lead autolysosomes (Maday and Holzbaur, 2014). Maintaining the integrity of proteins, neurotransmitters, receptors (localized at the synaptic level), and organelles (synaptic vesicles, mitochondria) is essential to support neuronal functionality (Lu et al., 2017; Conway et al., 2020).
Recent studies have revealed that autophagosomes are not only present in axons but also in the site of synaptic activation and these data suggest the possibility that neuronal autophagy regulates synaptic functions and neuroplasticity of the nervous system (Wang et al., 2015; Vanhauwaert et al., 2017; Tomoda et al., 2020). Synaptic plasticity requires intense biochemical activity that includes the synthesis of presynaptic neurotransmitters, postsynaptic receptor, signal transduction activity, gene expression, proper regulation of synapse, synaptic vesicle formation, and proper control of mitochondria (Binotti et al., 2015; Alvarez-Castelao and Schuman, 2015; Cohen and Ziv, 2017; Wang et al., 2017). All of these synaptic components are susceptible to wear and damage due to the frequencies of neuronal activation. Consequently, synapses are a site of great demand for cellular catabolic activities, for which an efficient degradation to support synaptic functions is indispensable (Vijayan and Verstreken, 2017). BDNF is also an inductor of long-term potentiation (LTP), considered one of the major cellular mechanisms that underlies learning and memory. Scientific evidence has shown that a receptor for BDNF is localized on the autophagosomic membrane, supporting the role of autophagy in transduction of BDNF signals (Nikoletopoulou et al., 2017; Nikoletopoulou and Tavernarakis, 2018). To date, it is known that failure of autophagy functions causes neuronal death, while impaired autophagy functions are associated with neurodegenerative disorders such as: Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease (Menzies et al., 2017; Conway et al., 2020). Studies in mice with ablation of autophagy genes ATG5 and ATG7 showed degeneration of axonic terminals, progressive axonal swelling, and no signs of the autophagosome formation (Komatsu et al., 2006; Komatsu et al., 2007).
In addition, a recent study conducted on an in vivo model of knockout mice conditionally lacking the essential autophagy protein Atg5, showed that the loss of this neuronal mechanism led to a selective accumulation of the endoplasmic reticulum in axons. Increased endoplasmic reticulum leads to increased excitatory neurotransmission due to high release of calcium from the organelle and compromises neuronal viability. These results have suggested, therefore, that neuronal autophagy could control the axonal calcium reserves of endoplasmic reticulum to regulate neurotransmission in healthy neurons and in the brain (Kuijpers et al., 2021).
Autophagy and Neurodegenerative Diseases
To date, it is known that failure of autophagy causes neuronal death, while impaired autophagy is associated with neurodegenerative disorders such as: Alzheimer’s Disease (AD), Huntington’s Disease (HD), Parkinson’s Disease (PD), and Amyotrophic Lateral Sclerosis (ALS) (Menzies et al., 2017; Conway et al., 2020). These pathologies are characterized by the formation of intracellular aggregates that result from misfolding and oligomerization of proteins. AD, the most common neurodegenerative disease, is characterized by extracellular β-amyloid plaques and intracellular neurofibrillary tangles composed of aggregated hyperphosphorylated tau protein (Hanzel et al., 2014; Cheignon et al., 2018). A dysfunctional autophagy is present in AD suffering patients, characterized by the loss of lysosome acidification, lysosomal alteration, failure to fusion between autophagosome and lysosomes and accumulation of autophagosomes (Yoon and Kim, 2016). Beclin1 is a key factor in the formation of autophagosomes and has been shown to be transcriptionally suppressed in the brain of AD (Salminen et al., 2013); in addition, caspase-3, an executive enzyme in the apoptosis pathway, can split the beclin1 protein and lead to the destruction of autophagy (Guo et al., 2018). Based on these observations, it is reasonable to assume that restoring lysosome function can increase the removal of protein aggregations. Finally, in vitro treatment with rapamycin, an enhancer of autophagy, has been shown to significantly increase the fusion of the autophagosomes with the lysosomes, improving the autophagic result (Li Q. et al., 2017).
PD, the second most common neurodegenerative disease, is characterized by selective loss of dopamine neurons in substantia nigra and accumulation of Lewy bodies, composed in misfolded and aggregated α-synuclein protein (Dauer and Przedborski, 2003; Lashuel et al., 2013). A close connection between this neurodegenerative disease and autophagy was indicated by the finding of dysfunctional lysosomes and accumulation of autophagosomes in the post-mortem brain samples of PD patients (Dehay et al., 2010; Xilouri et al., 2016). Additionally, if lysosomes are inhibited the levels of α-synuclein are increased, suggesting a link between α-synuclein degradation and autophagy (Guo et al., 2018). Lysosomal hydrolases, the enzymes responsible for degradation in lysosome, can be altered by autosomal recessive mutations: the consequence is the induction of defects in autophagosome-lysosome pathway and the aggregation of α-synuclein. Finally depletion of ATP6AP2, a transmembrane protein essential for lysosomal acidification, has been associated with PD (Abeliovich and Gitler, 2016).
HD is an autosomal dominant neurodegenerative disease, characterized by the repetition of trinucleotide CAG in the gene related to the protein huntingtin which leads to the expansion and pathogenic aggregation of the protein (Saudou and Humbert, 2016; Jimenez-sanchez et al., 2017). Huntingtin protein plays a key role in the autophagic process: in fact, its reduction results in an abnormal accumulation of autophagosomes (Wong and Holzbaur, 2014) and non-mutant huntingtin binds P62 to interact with ULK1 and activate it (Ravikumar et al., 2005).
ALS is a fatal disease characterized by the selective loss of motor neurons in the brain and spinal cord that causes weakness and muscle atrophy (Guo et al., 2018). The main cause of ALS is a mutation in superoxide dismutase 1 and other genes that produce dysfunctional proteins, toxic cellular effects and oxidative stress (Medinas et al., 2018). To date, it is known that autophagy is closely associated with ALS (Morimoto et al., 2007); in fact, autophagic processes are also activated in degenerated motor neurons, although a reduced digestion of lysosomal load has been suggested (Sasaki, 2011). Finally, it has been highlighted that mutated and autophagy-related proteins are involved in the onset of ALS (Filimonenko et al., 2007). Scientific evidences indicate that dysregulated autophagy plays a key role in the neurodegenerative diseases; for this reason, the regulation of autophagy is a potential therapeutic strategy to improve the course of neurodegenerative diseases.
Autophagy is also involved in higher brain functions, such as learning, memory, mood, social interaction, and cognition. In fact, it has been pointed out that deregulated autophagy is responsible for the predisposition to neurological disorders such as schizophrenia, bipolar disorder, psychosis, attention deficit disorder, hyperactivity, autism, cognitive decline, and depression (Tomoda et al., 2020). In addition, autophagic activity is known to gradually decrease during aging, in conjunction with the reduction of these brain functions (Vilchez et al., 2014). This apparent correlation has been demonstrated by studies that have shown that the intake of substances that enhance autophagy, are responsible for an increase in life span. The same substances may reduce memory deterioration associated with aging (Eisenberg et al., 2009). Since autophagic regulation has been shown to alleviate the deficit in synaptic plasticity and improve cognition, its regulation can be therapeutic and diagnostic in neurological and neuropsychiatric disorders.
In summary, we can say that the strategies used by neuronal autophagy to ensure the proper functioning of the CNS are basically five (Tomoda et al., 2020):
• degradation of dysfunctional cytosolic proteins;
• degradation of damaged organelles;
• selective surface presentation of neurotransmitter receptors;
• degradation of neurotransmitter activity and their receptors;
• morphological and functional regulation of synapses.
Autophagy and Metabolic Syndrome
Inflammatory diseases can trigger autophagy dysfunction (Deretic and Klionsky, 2018). It has been highlighted that autophagy is fundamental for the maintenance of cellular metabolic homeostasis and that it plays a crucial role in the control of body metabolism, whose dysregulation could participate in the onset of Mets (Lim et al., 2018). The role of autophagy in MetS was widely studied using animal models in which genetic alterations were induced (Lim et al., 2018). An example of this can be observed in a scientific study that highlighted how knockout mice of ATG7, an essential autophagy gene in pancreatic β-cells, showed structural and functional defects of these cells; with subsequent glucose intolerance and increased predisposition to develop diabetes (Ebato et al., 2008; Jung et al., 2008; Quan et al., 2012). Additionally, in another study, overexpression of ATG5 another essential autophagic gene, improved the metabolic profile of older mice (Pyo et al., 2013). A further example of the effects of autophagy is confirmed by important results showing how systemic autophagic insufficiency is able to cause deterioration of adaptation to metabolic stress and facilitate progression from obesity to diabetes (Lim et al., 2014). Recently it has been shown that Mets is characterized by a dysfunctional mitophagy (Wu H. et al., 2019) and present alterations which manifest as inadequate acidification in lysosomes (Yamamoto et al., 2017; Park et al., 2020; Yamamoto et al., 2020).
Conclusion
MetS is a clustering of several disorders including dyslipidemia, obesity and hyperglycemia/insulin resistance and every component is close connected with a high risk of developing atherosclerotic cardiovascular disease and type 2 diabetes. In addition, Mets has been shown to develop secondary disorders that affect the nervous system; the altered physiological state, due to Mets, can generate neurological deficits and a cognitive metabolic syndrome (Gasparova et al., 2018).
A physiopathological mechanism that links Mets and the neurological disorders is the endothelial dysfunction. In fact, in both cases, we can find an alteration of the endothelium of the cardiocirculatory system and of the BBB, respectively. In this review we wanted to better characterize the important correlation MetS-Neuronal disorders associated with another key process of cellular homeostasis: the autophagy. It has been highlighted that autophagy is fundamental for the maintenance of cellular metabolic homeostasis and that its dysregulation could participate in the onset of Mets (Lim et al., 2018). Moreover, neuronal autophagy is important also for the maintenance of homeostasis and vitality of neurons and these cells are particularly vulnerable in case of altered and/or switched off autophagy (Yamamoto and Yue, 2014; Kulkarni et al., 2018). It is important to stress that neuronal autophagy regulates also presynaptic excitatory neurotransmission by controlling the axonal cellular organelle ER. In particular, scientific experiments have shown that the deprivation of the Atg5, essential autophagic protein, leads to a selective accumulation of the tubular endoplasmic reticulum in axons, an increased release of the calcium ion, increased excitatory neurotransmission and impairment of postnatal viability in vivo. Therefore, the main consequence is an altered control of neuronal neurotransmission that, to occur properly, needs physiological autophagy (Kuijpers et al., 2021). Dysregulated autophagy can occur for multiple reasons but current scientific literature highlighted a common link in both Mets and neurological or neurodegenerative disorders. In fact, dysfunctional lysososomal acidification may be involved, leading to consequent failure of the fusion between autophagosomes and lysosomes (Yamamoto et al., 2017; Park et al., 2020; Yamamoto et al., 2020). Although this hypothesis is very fascinating, further studies are needed to confirm this link. In this direction, autophagy may be considered a target to reduce MetS risk factors and related disorders.
In summary, the model we propose is as follows:
• MetS involves, in addition to the risks already mentioned, the predisposition to the onset of neurological disorders;
• The inflammatory process is constantly present in MetS and participates in the onset of the neurological component. In fact, the subsequent dysfunction of the vascular endothelium involves the suffering of the nerve cells and the loss of integrity of the BBB;
• The autophagic process, notoriously involved in MetS, is a fundamental actor also in neurological disorders;
• Dysfunctional lysososomal acidification occurs in both MetS and neurological pathologies;
• Maintaining proper autophagic regulation, could reduce the constellation of primary and secondary MetS risk factors.
A summary cartoon of this described model is shown in Figure 3.
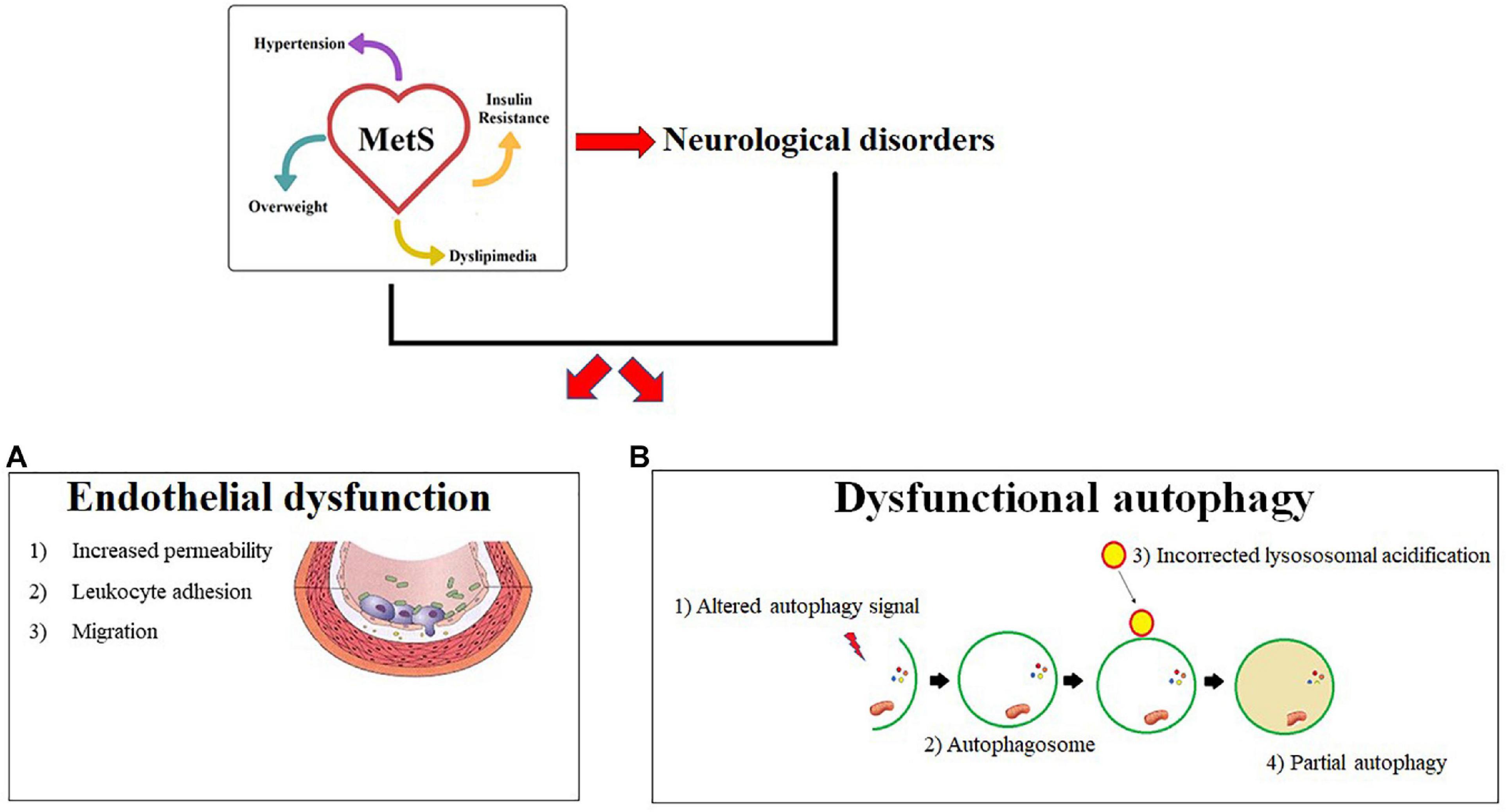
Figure 3. Common mechanisms in Mets and neurological diseases. MetS and its neurological complications are characterized by two common pathophysiological mechanisms described in panels a and b. In particular, (A) endothelial dysfunction is shown with the main characteristics: (1) Increased permeability (2) Leukocyte adhesion (3) Cellular Migration. (B) Shows a dysfunctional autophagy due to an altered signal. An incorrect lysosomal acidification is responsible for the changed formation of the autofagosoma-lisosome complex and a consequent partial autophagy.
Author Contributions
JM and VMo conceptualized, designed, and wrote the manuscript. MG, CC, VMu, SN, FB, FS, RM, SR, MZ, and CM revised the manuscript critically. All authors contributed to the article and approved the submitted version.
Funding
The work has been supported by the public resources from the Italian Ministry of Research. This work has been supported by PON-MIUR 03PE000_78_1 and PON-MIUR 03PE000_78_2.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.651021/full#supplementary-material
References
Abeliovich, A., and Gitler, A. D. (2016). Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539, 207–216. doi: 10.1038/nature20414
Aggoun, Y. (2007). Obesity, metabolic syndrome, and cardiovascular disease. Pediatr. Res. 61, 653–656.
Aguilar, M., Bhuket, T., Torres, S., Liu, B., and Wong, R. J. (2015). Prevalence of the metabolic syndrome in the United States, 2003-2012. JAMA 313, 1973–1974. doi: 10.1001/jama.2015.4260
Alberti, K. G., Zimmet, P., and Shaw, J. (2005). The metabolic syndrome–a new worldwide definition. Lancet. 366, 1059–1062.
Alberti, K. G., Eckel, R. H., Grundy, S. M., Zimmet, P. Z., Cleeman, J., Donato, K. A., et al. (2009). Harmonizing the metabolic syndrome: a joint interim statement of the international diabetes federation task force on epidemiology and prevention; national heart, lung, and blood institute; american heart association; world heart federation; international atherosclerosis society; and international association for the study of obesity. Circulation 120, 1640–1645. doi: 10.1161/circulationaha.109.192644
Almeida, M. F., Bahr, B. A., and Kinsey, S. T. (2020). Endosomal-lysosomal dysfunction in metabolic diseases and Alzheimer’s disease. Int. Rev. Neurobiol. 154, 303–324. doi: 10.1016/bs.irn.2020.02.012
Altieri, P. I., Marcial, J., Banchs, H. L., Escobales, N., and Crespo, Ml (2015). The metabolic syndrome in Hispanics–the role of inflammation. Glob. J. Obes. Diabetes Metab. Syndr. 2, 12–17.
Alvarez-Castelao, B., and Schuman, E. M. (2015). The regulation of synaptic protein turnover. J. Biol. Chem. 290, 28623–28630. doi: 10.1074/jbc.r115.657130
Andreone, B. J., Lacoste, B., and Gu, C. (2015). Neuronal and vascular interactions. Annu. Rev. Neurosci. 38, 25–46. doi: 10.1146/annurev-neuro-071714-033835
Ansari, S., Djalali, M., Mohammadzadeh, N., Mohammadzadeh Honarvar, N., Mazaherioun, M., Zarei, M., et al. (2016). Assessing the effect of omega-3 fatty acids supplementation on serum BDNF (brain derived neurotrophic factor) in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled study. Int. Res. J. Appl. Basic Sci. 10, 380–383.
Anstey, K. J., Cherbuin, N., Budge, M., and Young, J. (2011). Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obes. Rev. 12:e426-e437.
Aoki, Y., Yoon, S. S., Chong, Y., and Carroll, M. D. (2014). Hypertension, abnormal cholesterol, and high body mass index among non-Hispanic Asian adults: United States, 2011–2012. NCHS Data Brief. 140, 1–8.
Arshad, N., Lin, T. S., and Yahaya, M. F. (2018). Metabolic syndrome and its effect on the brain: possible mechanism. CNS Neurol. Disord. Drug Targets 17, 595–603. doi: 10.2174/1871527317666180724143258
Babushkina, I. V., Sergeeva, A. S., Pivovarov, I., Kuril’skaia, T. E., and Koriakina, L. B. (2015). Structural and functional properties of vascular endothelium. Kardiologiia. 55, 82–86.
Bañuls, C., Rovira-Llopis, S., Martinez de Marañon, A., Veses, S., Jover, A., Gomez, M., et al. (2017). Metabolic syndrome enhances endoplasmic reticulum, oxidative stress and leukocyte-endothelium interactions in PCOS. Metabolism 71, 153–162. doi: 10.1016/j.metabol.2017.02.012
Bhat, R., Crowe, E. P., Bitto, A., Moh, M., Katsetos, C. D., Garcia, F. U., et al. (2012). Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7:e45069. doi: 10.1371/journal.pone.0045069
Binotti, B., Pavlos, N. J., Riedel, D., Wenzel, D., Vorbrüggen, G., Schalk, A. M., et al. (2015). The GTPase Rab26 links synaptic vesicles to the autophagy pathway. Elife 4:e05597.
Björkholm, C., and Monteggia, L. M. (2016). BDNF - a key transducer of antidepressant effects. Neuropharmacology. 102, 72–79. doi: 10.1016/j.neuropharm.2015.10.034
Black, S., Kraemer, K., Shah, A., Simpson, G., Scogin, F., and Smith, A. (2018). Diabetes, depression, and cognition: a recursive cycle of cognitive dysfunction and glycemic dysregulation. Curr. Diab. Rep. 18:118.
Błaszczyk, E., and Gawlik, A. (2016). Neurotrophins, VEGF and matrix metalloproteinases: new markers or causative factors of metabolic syndrome components? Pediatr. Endocrinol. Diab. Metab. 22. doi: 10.18544/PEDM-22.03.0060
Boland, B., Yu, W. H., Corti, O., Mollereau, B., Henriques, A., Bezard, E., et al. (2018). Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 17, 660–688. doi: 10.1038/nrd.2018.109
Boyd, F. T. Jr., Clarke, D. W., Muther, T. F., and Raizada, M. K. (1985). Insulin receptors and insulin modulation of norepinephrine uptake in neuronal cultures from rat brain. J. Biol. Chem. 260, 15880–15884. doi: 10.1016/s0021-9258(17)36340-8
Bremer, A. A., and Jialal, I. (2013). Adipose tissue dysfunction in nascent metabolic syndrome. J. Obes. 2013:393192.
Bruno, R. M., Reesink, K. D., and Ghiadoni, L. (2017). Advances in the non-invasive assessment of vascular dysfunction in metabolic syndrome and diabetes: focus on endothelium, carotid mechanics and renal vessels. Nutr. Metab. Cardiovasc. Dis. 27, 121–128. doi: 10.1016/j.numecd.2016.09.004
Cai, W., Zhang, K., Li, P., Zhu, L., Xu, J., Yang, B., et al. (2017). Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: an aging effect. Ageing Res. Rev. 34, 77–87. doi: 10.1016/j.arr.2016.09.006
Chakraborty, A., de Wit, N. M., van der Flier, W. M., and de Vries, H. E. (2017). The blood brain barrier in Alzheimer’s disease. Vascul. Pharmacol. 89, 12–18.
Chaldakov, G. N., Fiore, M., Ranćić, G., Beltowski, J., Tunçel, N., and Aloe, L. (2014). An integrated view: neuroadipocrinology of diabesity. Ser. J. Expclin. Res. 15, 61–69. doi: 10.2478/sjecr-2014-0008
Chawla, A., Nguyen, K. D., and Goh, Y. P. (2011). Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 11, 738–749. doi: 10.1038/nri3071
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 14, 450–464.
Chen, A. Q., Fang, Z., Chen, X. L., Yang, S., Zhou, Y. F., Mao, L., et al. (2019). Microglia-derived TNF-alpha mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis. 10:487.
Choi, J., Chandrasekaran, K., Demarest, T. G., Kristian, T., Xu, S., Vijaykumar, K., et al. (2014). Brain diabetic neurodegeneration segregates with low intrinsic aerobic capacity. Ann. Clin. Transl. Neurol. 1, 589–604. doi: 10.1002/acn3.86
Climie, R. E., Moran, C., Callisaya, M., Blizzard, L., Sharman, J. E., Venn, A., et al. (2015). Abdominal obesity and brain atrophy in type 2 diabetes mellitus. PLoS One 10:e0142589. doi: 10.1371/journal.pone.0142589
Cohen, L. D., and Ziv, N. E. (2017). Recent insights on principles of synaptic protein degradation. F1000Res 6:675. doi: 10.12688/f1000research.10599.1
Conway, O., Akpinar, H. A., Rogov, V. V., and Kirkin, V. (2020). Selective autophagy receptors in neuronal health and disease. J. Mol. Biol. 432, 2483–2509. doi: 10.1016/j.jmb.2019.10.013
Corada, M., Zanetta, L., Orsenigo, F., Breviario, F., Lampugnan, M. G., Bernasconi, S., et al. (2002). A monoclonal antibody to vascular endothelial-cadherin inhibits tumor angiogenesis without side effects on endothelial permeability. Blood 100, 905–911. doi: 10.1182/blood.v100.3.905
Corasaniti, M. T., Maiuolo, J., Maida, S., Fratto, V., Navarra, M., Russo, R., et al. (2007). Cell signaling pathways in the mechanisms of neuroprotection afforded by bergamot essential oil against NMDA-induced cell death in vitro. Br. J. Pharmacol. 151, 518–529. doi: 10.1038/sj.bjp.0707237
Costello, F., and Petzold, A. (2020). Weighting evidence in MS: obesity and neurodegeneration. Mult. Scler. 26, 748–750. doi: 10.1177/1352458520912171
Cuspidi, C., Sala, C., Provenzano, F., Tadic, M., Gherbesi, E., Grassi, G., et al. (2018). Metabolic syndrome and subclinical carotid damage: a meta-analysis from population-based studies. J. Hypertens. 36, 23–30. doi: 10.1097/hjh.0000000000001575
Dalal, P. J., Muller, W. A., and Sullivan, D. P. (2020). Endothelial cell calcium signaling during barrier function and inflammation. Am. J. Pathol. 190, 535–542. doi: 10.1016/j.ajpath.2019.11.004
Daneman, R., and Prat, A. (2015). The blood-brain barrier. Cold Spring Harb Perspect Biol. 7:a020412.
Dauer, W., and Przedborski, S. (2003). Parkinson’s disease: mechanisms and models. Neuron. 39, 889–909.
Davalos, D., and Akassoglou, K. (2012). Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 34, 43–62. doi: 10.1007/s00281-011-0290-8
Davidson, T. L., Hargrave, S. L., Swithers, S. E., Sample, C. H., Fu, X., Kinzig, K. P., et al. (2013). Inter-relationships among diet, obesity and hippocampaldependent cognitive function. Neuroscience 253, 110–122. doi: 10.1016/j.neuroscience.2013.08.044
Dehay, B., Bové, J., Rodríguez-Muela, N., Perier, C., Recasens, A., Boya, P., et al. (2010). Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 30, 12535–12544. doi: 10.1523/jneurosci.1920-10.2010
Deng, J., Liu, S., Zou, L., Xu, C., Geng, B., and Xu, G. (2012). Lipolysis response to endoplasmic reticulum stress in adipose cells. J. Biol. Chem. 2012, 6240–6249. doi: 10.1074/jbc.m111.299115
Denver, P., English, A., and McClean, P. L. (2018). Inflammation, insulin signaling and cognitive function in aged APP/PS1 mice. Brain Behav. Immun. 70, 423–434. doi: 10.1016/j.bbi.2018.03.032
Deretic, V., and Klionsky, D. J. (2018). Autophagy and inflammation: a special review issue. Autophagy 14, 179–180. doi: 10.1080/15548627.2017.1412229
Domingueti, C. P., Dusse, L. M., Carvalho, M., de Sousa, L. P., Gomes, K. B., and Fernandes, A. P. (2016). Diabetes mellitus: the linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complications. 30, 738–745. doi: 10.1016/j.jdiacomp.2015.12.018
Dorfman, M. D., and Thaler, J. P. (2015). Hypothalamic inflammation and gliosis in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 22, 325–330. doi: 10.1097/med.0000000000000182
Ebato, C., Uchida, T., Arakawa, M., Komatsu, M., Ueno, T., Komiya, K., et al. (2008). Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 8:32.
Eisenberg, T., Knauer, H., Schauer, A., Büttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., et al. (2009). Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314.
ElAli, A., Thériault, P., and Rivest, S. (2014). The role of pericytes in neurovascular unit remodeling in brain disorders. Int. J. Mol. Sci. 15, 6453–6474. doi: 10.3390/ijms15046453
El-Benna, J., Hurtado-Nedelec, M., Marzaioli, V., Marie, J. C., Gougerot-Pocidalo, M. A., and Dang, P. M. (2016). Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol. Rev. 273, 180–193. doi: 10.1111/imr.12447
Elmarakby, A. A., and Sullivan, J. C. (2012). Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc. Ther. 30, 49–59. doi: 10.1111/j.1755-5922.2010.00218.x
Emanueli, C., Meloni, M., Hasan, W., and Habecker, B. A. (2014). The biology of neurotrophins: cardiovascular function. Handb. Exp Pharmacol. 220, 309–328. doi: 10.1007/978-3-642-45106-5_12
Engelhardt, B., and Ransohoff, R. M. (2012). Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. 33, 579–589. doi: 10.1016/j.it.2012.07.004
Ervin, R. B. (2009). Prevalence of metabolic syndrome among adults 20 years of age and over, by sex, age, race and ethnicity, and body mass index: United States, 2003-2006. Natl. Health Stat. Report 13, c1–c7.
Fan, L., Wang, T., Chang, L., Song, Y., Wu, Y., and Ma, D. (2014). Systemic inflammation induces a profound long term brain cell injury in rats. Acta Neurobiol. Exp. 74, 298–306.
Fan, X., Chen, X., Feng, Q., Peng, K., Wu, Q., Passerini, A. G., et al. (2019). Downregulation of GATA6 in mTOR-inhibited human aortic endothelial cells: effects on TNF-alpha-induced VCAM-1 expression and monocytic cell adhesion. Am. J. Physiol. Heart Circ. Physiol. 316, H408–H420.
Feng, Y., Yao, Z., and Klionsky, D. J. (2015). How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 25, 354–363. doi: 10.1016/j.tcb.2015.02.002
Figlewicz, D. P., Bentson, K., and Ocrant, I. (1993). The effect of insulin on norepinephrine uptake by PC12 cells. Brain Res. Bull. 32, 425–431. doi: 10.1016/0361-9230(93)90210-3
Filimonenko, M., Stuffers, S., Raiborg, C., Yamamoto, A., Malerød, L., Fisher, E. M. C., et al. (2007). Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. cell Biol. 179, 485–500. doi: 10.1083/jcb.200702115
Finicelli, M., Squillaro, T., Di Cristo, F., Di Salle, A., Melone, M. A. B., Galderisi, U., et al. (2019). Metabolic syndrome, mediterranean diet, and polyphenols: evidence and perspectives. J. Cell Physiol. 234, 5807–5826.
Friesen, M., and Cowan, C. A. (2019). Adipocyte metabolism and insulin signaling perturbations: insights from genetics. Trends Endocrinol. Metab. 30, 396–406. doi: 10.1016/j.tem.2019.03.002
Gasparova, Z., Janega, P., Weismann, P., El Falougy, H., Tyukos Kaprinay, B., Liptak, B., et al. (2018). Effect of metabolic syndrome on neural plasticity and morphology of the hippocampus: correlations of neurological deficits with physiological status of the rat. Gen. Physiol. Biophys. 37, 619–632. doi: 10.4149/gpb_2018016
Gentil, B. J., Benaud, C., Delphin, C., Remy, C., Berezowski, V., Cecchelli, R., et al. (2005). Specific AHNAK expression in brain endothelial cells with barrier properties. J. Cell Physiol. 203, 362–371. doi: 10.1002/jcp.20232
Georgieva, A. M., Cate, H. T., Keulen, E. T., van Oerle, R., Govers-Riemslag, J. W., Hamulyák, K., et al. (2004). Prothrombotic markers in familial combined hyperlipidemia: evidence of endothelial cell activation and relation to metabolic syndrome. Atherosclerosis 175, 345–351. doi: 10.1016/j.atherosclerosis.2004.04.006
Godo, S., and Shimokawa, H. (2017). Endothelial functions. Arterioscler. Thromb. Vasc. Biol. 37, e108–e114.
Gogniat, M. A., Robinson, T. L., Mewborn, C. M., Jean, K. R., and Miller, L. S. (2018). Body mass index and its relation to neuropsychological functioning and brain volume in healthy older adults. Behav. Brain Res. 348, 235–240. doi: 10.1016/j.bbr.2018.04.029
Gómez-Apo, E., García-Sierra, A., Silva-Pereyra, J., Soto-Abraham, V., Mondragón-Maya, A., Velasco-Vales, V., et al. (2018). A postmortem study of frontal and temporal gyri thickness and cell number in human obesity. Obesity 26, 94–102. doi: 10.1002/oby.22036
Grandl, G., and Wolfrum, C. (2018). Hemostasis, endothelial stress, inflammation, and the metabolic syndrome. Semin. Immunopathol. 40, 215–224. doi: 10.1007/s00281-017-0666-5
Grumati, P., and Dikic, I. (2018). Ubiquitin signaling and autophagy. J. Biol. Chem. 293, 5404–5413. doi: 10.1074/jbc.tm117.000117
Guo, F., Liu, X., Cai, H., and Le, W. (2018). Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathol. 28, 3–13. doi: 10.1111/bpa.12545
Gustafson, D. R., Karlsson, C., Skoog, I., Rosengren, L., Lissner, L., and Blennow, K. (2007). Mid-life adiposity factors relate to blood-brain barrier integrity in late life. J. Intern. Med. 262, 643–650. doi: 10.1111/j.1365-2796.2007.01869.x
Hajiluian, G., Abbasalizad Farhangi, M., and Jahangiry, L. (2017). Mediterranean dietary pattern and VEGF +405 G/C gene polymorphisms in patients with metabolic syndrome: an aspect of gene-nutrient interaction. PLoS One. 12:e0171637. doi: 10.1371/journal.pone.0171637
Hakanpaa, L., Kiss, E. A., Jacquemet, G., Miinalainen, I., Lerche, M., Guzmán, C., et al. (2018). Targeting β1-integrin inhibits vascular leakage in endotoxemia. Proc. Natl. Acad. Sci. U.S.A. 115, E6467–E6476.
Hanzel, C. E., Pichet-Binette, A., Pimentel, L. S., Iulita, M. F., Allard, S., Ducatenzeiler, A., et al. (2014). Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 35, 2249–2262. doi: 10.1016/j.neurobiolaging.2014.03.026
Hargrave, S. L., Davidson, T. L., Zheng, W., and Kinzig, K. P. (2016). Western diets induce blood-brain barrier leakage and alter spatial strategies in rats. Behav. Neurosci. 130, 123–135. doi: 10.1037/bne0000110
Hassing, L. B., Grant, M. D., Hofer, S. M., Pedersen, N. L., Nilsson, S. E., Berg, S., et al. (2004). Type 2 diabetes mellitus contributes to cognitive decline in old age: a longitudinal population-based study. J. Int. Neuropsychol. Soc. 10, 599–607. doi: 10.1017/s1355617704104165
Hempstead, B. L. (2015). Brain-Derived neurotrophic factor: three ligands, many actions. Trans. Am. Clin. Climatol. Assoc. 126, 9–19.
Heras-Sandoval, D., Ferrera, P., and Arias, C. (2012). Amyloid-beta protein modulates insulin signaling in presynaptic terminals. Neurochem. Res. 37, 1879–1885. doi: 10.1007/s11064-012-0800-7
Hu, Y. X., Han, X. S., and Jing, Q. (2019). Autophagy in development and differentiation. Adv. Exp. Med. Biol. 1206, 469–487. doi: 10.1007/978-981-15-0602-4_22
Incalza, M. A., D’Oria, R., Natalicchio, A., Perrini, S., Laviola, L., and Giorgino, F. (2018). Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 100, 1–19. doi: 10.1016/j.vph.2017.05.005
Islam, M. T. (2017). Oxidative stress and mitochondrial dysfunction linked neurodegenerative disorders. Neurol. Res. 39, 73–82. doi: 10.1080/01616412.2016.1251711
Jais, A., and Brüning, J. C. (2017). Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Invest. 127, 24–32. doi: 10.1172/jci88878
Jimenez-sanchez, M., Licitra, F., Underwood, B. R., and Rubinsztein, D. C. (2017). Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med. 7:a024240.
Jung, H. S., Chung, K. W., Kim, J. W., Kim, J., Komatsu, M., Tanaka, K., et al. (2008). Loss of autophagy diminishes pancreatic b-cell mass and function with resultant hyperglycemia. Cell. Metab. 8, 318–324. doi: 10.1016/j.cmet.2008.08.013
Kaplan, H. M., Kuyucu, Y., Polat, S., Pazarci, P., Yegani, A. A., Şingirik, E., et al. (2018). Molecular basis of vascular damage caused by cigarette smoke exposure and a new approach to the treatment: alpha-linolenic acid. Biomed Pharmacother. 102, 458–463. doi: 10.1016/j.biopha.2018.03.112
Karaca, C., and Karaca, Z. (2018). Beyond hyperglycemia, evidence for retinal neurodegeneration in metabolic syndrome. Invest. Ophthalmol. Vis. Sci. 59, 1360–1367. doi: 10.1167/iovs.17-23376
Kathy, F. Y., Ma, V. S., Po Lai, Y., Turchianoa, M. M., and Convit, A. (2012). Impact of metabolic syndrome on cognition and brain: a selected review of the literature. Arterioscler. Thromb. Vasc. Biol. 32, 2060–2067. doi: 10.1161/atvbaha.112.252759
Kauffman, K. J., Yu, S., Jin, J., Mugo, B., Nguyen, N., O’Brien, A., et al. (2018). Delipidation of mammalian Atg8-family proteins by each of the four ATG4 proteases. Autophagy 14, 992–1010.
Khaddaj Mallat, R., Mathew John, C., Kendrick, D. J., and Braun, A. P. (2017). The vascular endothelium: a regulator of arterial tone and interface for the immune system. Crit. Rev. Clin. Lab. Sci. 54, 458–470. doi: 10.1080/10408363.2017.1394267
Kim, B., Backus, C., Oh, S., Hayes, J. M., and Feldman, E. L. (2009). Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 150, 5294–5301. doi: 10.1210/en.2009-0695
Kim, H. W., Shi, H., Winkler, M. A., Lee, R., and Weintraub, N. L. (2020). Perivascular adipose tissue and vascular perturbation/atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 40, 2569–2576. doi: 10.1161/atvbaha.120.312470
Kiseleva, R. Y., Glassman, P. M., Greineder, C. F., Shuvaev, V. V., and Muzykantov, V. R. (2018). Targeting therapeutics to endothelium: are we there yet? Drug Deliv. Transl. Res. 8, 883–902. doi: 10.1007/s13346-017-0464-6
Kishi, T., Chipman, J., Evereklian, M., Nghiem, K., Stetler-Stevenson, M., Rick, M. E., et al. (2020). Endothelial activation markers as disease activity and damage measures in juvenile dermatomyositis. J. Rheumatol. 47, 1011–1018. doi: 10.3899/jrheum.181275
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Komatsu, M., Wang, Q. J., Holstein, G. R., Friedrich, V. L. Jr., Iwata, J., Kominami, E., et al. (2007). Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. U.S.A. 104, 14489–14494. doi: 10.1073/pnas.0701311104
Kuijpers, M., Kochlamazashvili, G., Stumpf, A., Puchkov, D., Swaminathan, A., Lucht, M. T., et al. (2021). Neuronal autophagy regulates presynaptic neurotransmission by controlling the axonal endoplasmic reticulum. Neuron 109, 299–313.e9.
Kulkarni, A., Chen, J., and Maday, S. (2018). Neuronal autophagy and intercellular regulation of homeostasis in the brain. Curr. Opin. Neurobiol. 51, 29–36. doi: 10.1016/j.conb.2018.02.008
Kullmann, S., Heni, M., Hallschmid, M., Fritsche, A., Preissl, H., and Häring, H. U. (2016). Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol. Rev. 96, 1169–1209. doi: 10.1152/physrev.00032.2015
Kumar, A., Dhawan, A., Kadam, A., and Shinde, A. (2018). Autophagy and mitochondria: targets in neurodegenerative disorders. CNS Neurol. Disord. Drug Targets 17, 696–705. doi: 10.2174/1871527317666180816100203
Lacoste, B., and Gu, C. (2015). Control of cerebrovascular patterning by neural activity during postnatal development. Mech. Dev. 138(Pt. 1), 43–49. doi: 10.1016/j.mod.2015.06.003
Laird, C. T., Hassanein, W., O’Neill, N. A., French, B. M., Cheng, X., Fogler, W. E., et al. (2018). P- and E-selectin receptor antagonism prevents human leukocyte adhesion to activated porcine endothelial monolayers and attenuates porcine endothelial damage. Xenotransplantation 25:e12381. doi: 10.1111/xen.12381
Lapaquette, P., Guzzo, J., Bretillon, L., and Bringer, M. A. (2015). Cellular and molecular connections between autophagy and inflammation. Mediators Inflamm. 2015:398483.
Lashuel, H. A., Overk, C. R., Oueslati, A., and Masliah, E. (2013). The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48. doi: 10.1038/nrn3406
Lawrence, T. (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1:a001651.
Lee, S., Joo, Y. J., Kim, R. Y., Hwang, J., Lim, S. M., Yoon, S., et al. (2020). Obesity may connect insulin resistance to decreased neuronal viability in human diabetic brain. Obesity 28, 1626–1630. doi: 10.1002/oby.22869
Levine, B., and Kroemer, G. (2019). Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42. doi: 10.1016/j.cell.2018.09.048
Lewin, G. R., and Barde, Y. A. (1996). Physiology of the neurotrophins. Annu. Rev. Neurosci. 19, 289–317. doi: 10.1146/annurev.ne.19.030196.001445
Li, J., and Hochstrasser, M. (2020). Microautophagy regulates proteasome homeostasis. Curr. Genet. 66, 683–687. doi: 10.1007/s00294-020-01059-x
Li, Q., Liu, Y., and Sun, M. (2017). Autophagy and Alzheimer’s disease. Cell Mol. Neurobiol. 37, 377–388.
Li, W. J., Xue, H., Sun, K., Song, X. D., Wang, Y. B., Zhen, Y. S., et al. (2008). Cardiovascular risk and prevalence of metabolic syndrome by differing criteria. Chin. Med. J. 121, 1532–1536. doi: 10.1097/00029330-200808020-00006
Li, X., He, S., and Ma, B. (2020). Autophagy and autophagy-related proteins in cancer. Mol. Cancer 19:12.
Li, Y. J., Lei, Y. H., Yao, N., Wang, C. R., Hu, N., Ye, W. C., et al. (2017). Autophagy and multidrug resistance in cancer. Chin. J. Cancer 36:52.
Lim, H., Lim, Y. M., Kim, K. H., Jeon, Y. E., Park, K., Kim, J., et al. (2018). A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat. Commun. 9:1438.
Lim, Y.-M., Lim, H., Hur, K. H., Quan, W., Lee, H. Y., and Cheon, H. (2014). Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat. Commun. 5:4934.
Loghmani, H., and Conway, E. M. (2018). Exploring traditional and nontraditional roles for thrombomodulin. Blood. 132, 148–158. doi: 10.1182/blood-2017-12-768994
Lu, K., den Brave, F., and Jentsch, S. (2017). Receptor oligomerizationguides pathway choice between proteasomal and autophagic degradation. Nat. Cell Biol. 19, 732–739. doi: 10.1038/ncb3531
Lu, T., Pan, Y., Kao, S. Y., Li, C., Kohane, I., Chan, J., et al. (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891. doi: 10.1038/nature02661
Lutz, S. E., Smith, J. R., Kim, D. H., Olson, C. V. L., Ellefsen, K., Bates, J. M., et al. (2017). Caveolin1 is required for Th1 cell infiltration, but not tight junction remodeling, at the blood-brain barrier in autoimmune neuroinflammation. Cell Rep. 21, 2104–2117. doi: 10.1016/j.celrep.2017.10.094
Maday, S., and Holzbaur, E. L. (2014). Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev. Cell 30, 71–85. doi: 10.1016/j.devcel.2014.06.001
Madrigal-Matute, J., and Cuervo, A. M. (2016). Regulation of liver metabolism by autophagy. Gastroenterology 150, 328–339. doi: 10.1053/j.gastro.2015.09.042
Maiuolo, J., Gliozzi, M., Musolino, V., Carresi, C., Nucera, S., Macrì, R., et al. (2019). The role of endothelial dysfunction in peripheral blood nerve barrier: molecular mechanisms and pathophysiological implications. Int. J. Mol. Sci. 20:3022. doi: 10.3390/ijms20123022
Maiuolo, J., Gliozzi, M., Musolino, V., Carresi, C., Nucera, S., Scicchitano, M., et al. (2020b). Environmental and nutritional “Stressors” and oligodendrocyte dysfunction: role of mitochondrial and endoplasmatic reticulum impairment. Biomedicines 8:553. doi: 10.3390/biomedicines8120553
Maiuolo, J., Gliozzi, M., Musolino, V., Scicchiatano, M., Carresi, C., Scarano, F., et al. (2018). The “Frail” brain blood barrier in neurodegenerative diseases: role of early disruption of endothelial Cell-to-Cell connections. Int. J. Mol. Sci. 19:2693. doi: 10.3390/ijms19092693
Maiuolo, J., Mollace, R., Gliozzi, M., Musolino, V., Carresi, C., Paone, S., et al. (2020a). The contribution of endothelial dysfunction in systemic injury subsequent to SARS-Cov-2 infection. Int. J. Mol. Sci. 21:9309. doi: 10.3390/ijms21239309
Mazidi, M., Rezaie, P., Kengne, A. P., Stathopoulou, M. G., Azimi-Nezhad, M., and Siest, S. (2017). VEGF, the underlying factor for metabolic syndrome; fact or fiction? Diab. Metab. Syndr. 11(Suppl. 1), S61–S64.
McCracken, E., Monaghan, M., and Sreenivasan, S. (2018). Pathophysiology of the metabolic syndrome. Clin. Dermatol. 36, 14–20.
Medinas, D. B., Rozas, P., Martinez Traub, F., Woehlbier, U., Brown, R. H., Bosco, D. A., et al. (2018). Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 115, 8209–8214. doi: 10.1073/pnas.1801109115
Menzies, F. M., Fleming, A., Caricasole, A., Bento, C. F., Andrews, S. P., Ashkenazi, A., et al. (2017). Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034. doi: 10.1016/j.neuron.2017.01.022
Menzies, F. M., Fleming, A., and Rubinsztein, D. C. (2015). Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 16, 345–357. doi: 10.1038/nrn3961
Miller, A. A., and Spencer, S. J. (2014). Obesity and neuroinflammation: a pathway to cognitive impairment. Brain Behav. Immun. 42, 10–21. doi: 10.1016/j.bbi.2014.04.001
Misra, A., and Khurana, L. (2008). Obesity and the metabolic syndrome in developing countries. J. Clin. Endocrinol. Metab. 93, S9–S30.
Mittal, M., Siddiqui, M. R., Tran, K., Reddy, S. P., and Malik, A. B. (2014). Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 20, 1126–1167. doi: 10.1089/ars.2012.5149
Mizushima, N. (2018). A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 20, 521–527. doi: 10.1038/s41556-018-0092-5
Monette, M. C., Baird, A., and Jackson, D. L. (2014). A meta-analysis of cognitive functioning in nondemented adults with type 2 diabetes mellitus. Can. J. Diabetes 38, 401–408. doi: 10.1016/j.jcjd.2014.01.014
Morales-Marín, M. E., Genis-Mendoza, A. D., Tovilla-Zarate, C. A., Lanzagorta, N., Escamilla, M., and Nicolini, H. (2016). Association between obesity and the brain-derived neurotrophic factor gene polymorphism Val66Met in individuals with bipolar disorder in Mexican population. Neuropsychiatr. Dis. Treat. 12, 1843–1848. doi: 10.2147/ndt.s104654
Morimoto, N., Nagai, M., Ohta, Y., Miyazaki, K., Kurata, T., Morimoto, M., et al. (2007). Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1167, 112–117. doi: 10.1016/j.brainres.2007.06.045
Motamedi, S., Karimi, I., and Jafari, F. (2017). The interrelationship of metabolic syndrome and neurodegenerative diseases with focus on brain-derived neurotrophic factor (BDNF): kill two birds with one stone. Metab. Brain Dis. 32, 651–665. doi: 10.1007/s11011-017-9997-0
Nikoletopoulou, V., Sidiropoulou, K., Kallergi, E., Dalezios, Y., and Tavernarakis, N. (2017). Modulation of autophagy by BDNF underlies synaptic plasticity. Cell Metab. 26, 230–242.e5.
Nikoletopoulou, V., and Tavernarakis, N. (2018). Regulation and roles of autophagy at synapses. Trends Cell Biol. 28, 646–661. doi: 10.1016/j.tcb.2018.03.006
Nishimura, T., and Tooze, S. A. (2020). Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 6:32.
Nourshargh, S., and Alon, R. (2014). Leukocyte migration into inflamed tissues. Immunity 41, 694–707. doi: 10.1016/j.immuni.2014.10.008
O’Brown, N. M., Pfau, S. J., and Gu, C. (2018). Bridging barriers: a comparative look at the blood–brain barrier across organisms. Genes Dev. 32, 466–478. doi: 10.1101/gad.309823.117
O’Shea, J. J., Schwartz, D. M., Villarino, A. V., Gadina, M., McInnes, I. B., and Laurence, A. (2015). The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu. Rev. Med. 66, 311–328. doi: 10.1146/annurev-med-051113-024537
O’Doherty, M. G., Cairns, K., O’Neill, V., Lamrock, F., Jørgensen, T., Brenner, H., et al. (2016). Effect of major lifestyle risk factors, independent and jointly, on life expectancy with and without cardiovascular disease: results from the Consortium on Health and Ageing Network of Cohorts in Europe and the United States (CHANCES). Eur. J. Epidemiol. 31, 455–468. doi: 10.1007/s10654-015-0112-8
Oppedisano, F., Maiuolo, J., Gliozzi, M., Musolino, V., Carresi, C., Nucera, S., et al. (2020). The potential for natural antioxidant supplementation in the early stages of neurodegenerative disorders. Int. J. Mol. Sci. 21:2618. doi: 10.3390/ijms21072618
Ouchi, N., Parker, J. L., Lugus, J. J., and Walsh, K. (2011). Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 11, 85–97. doi: 10.1038/nri2921
Palta, P., Rippon, B., Tahmi, M., Sherwood, G., Soto, L., Ceballos, F., et al. (2021). Metbolic syndrome and its components in relation to in vivo brain amyloid and neurodegeneration in late middle age. Neurobiol. Aging. 97, 89–96. doi: 10.1016/j.neurobiolaging.2020.09.023
Park, H. S., Song, J. W., Park, J. H., Lim, B. K., Moon, O. S., Son, H. Y., et al. (2020). TXNIP/VDUP1 attenuates steatohepatitis via autophagy and fatty acid oxidation. Autophagy 16, 1–16. doi: 10.1080/15548627.2020.1834711
Patterson, S., Irwin, N., Guo-Parke, H., Moffett, R. C., Scullion, S. M., Flatt, P. R., et al. (2016). Evaluation of the role of N-methyl-D-aspartate (n.d.) receptors in insulin secreting beta-cells. Eur. J. Pharmacol. 771, 107–113. doi: 10.1016/j.ejphar.2015.12.015
Pendlebury, S. T., and Rothwell, P. M. (2009). Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 8, 1006–1018. doi: 10.1016/s1474-4422(09)70236-4
Pomero, F., Di Minno, M. N., Fenoglio, L., Gianni, M., Ageno, W., and Dentali, F. (2015). Is diabetes a hypercoagulable state? A critical appraisal. Acta Diabetol. 52, 1007–1016. doi: 10.1007/s00592-015-0746-8
Pucci, G., Alcidi, R., Tap, L., Battista, F., Mattace-Raso, F., and Schillaci, G. (2017). Sex- and gender-related prevalence, cardiovascular risk and therapeutic approach in metabolic syndrome: a review of the literature. Pharmacol. Res. 120, 34–42. doi: 10.1016/j.phrs.2017.03.008
Pyo, J. O., Yoo, S. M., Ahn, H. H., Nah, J., Hong, S. H., Kam, T. I., et al. (2013). Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 4:2300.
Quan, W., Hur, K. Y., Lim, Y., Oh, S. H., Lee, J.-C., Kim, K. H., et al. (2012). Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 55, 392–403. doi: 10.1007/s00125-011-2350-y
Ransohoff, R. M., Schafer, D., Vincent, A., Blachère, N. E., and Bar-Or, A. (2015). Neuroinflammation: ways in which the immune system affects the brain. Neurotherapeutics 12, 896–909. doi: 10.1007/s13311-015-0385-3
Ravikumar, B., Acevedo-Arozena, A., Imarisio, S., Berger, Z., Vacher, C., O’Kane, C. J., et al. (2005). Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 37, 771–776. doi: 10.1038/ng1591
Ren, J., and Anversa, P. (2015). The insulin-like growth factor I system: physiological and pathophysiological implication in cardiovascular diseases associated with metabolic syndrome. Biochem. Pharmacol. 93, 409–417. doi: 10.1016/j.bcp.2014.12.006
Ricci, G., Pirillo, I., Tomassoni, D., Sirignano, A., and Grappasonni, I. (2017). Metabolic syndrome, hypertension, and nervous system injury: epidemiological correlates. Clin. Exp. Hypertens. 39, 8–16. doi: 10.1080/10641963.2016.1210629
Rudziak, P., Ellis, C. G., and Kowalewska, P. M. (2019). Role and molecular mechanisms of pericytes in regulation of leukocyte diapedesis in inflamed tissues. Mediators Inflamm. 2019:4123605.
Sack, K. D., Teran, M., and Nugent, M. A. (2016). Extracellular matrix stiffness controls VEGF signaling and processing in endothelial cells. J. Cell Physiol. 231, 2026–2039. doi: 10.1002/jcp.25312
Saklayen, M. G. (2018). The global epidemic of the metabolic syndrome. Curr. Hypertens. Reports 20:12.
Salminen, A., Kaarniranta, K., Kauppinen, A., Ojala, J., Haapasalo, A., Soininen, H., et al. (2013). Impaired autophagy and APP processing in Alzheimer’s disease: the potential role of Beclin 1 interactome. Prog. Neurobiol. 10, 33–54. doi: 10.1016/j.pneurobio.2013.06.002
Saltiel, A. R., and Olefsky, J. M. (2017). Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest. 127, 1–4. doi: 10.1172/jci92035
Sarmiento Quintero, F., Ariza, A. J., Barboza García, F., Canal de Molano, N., Castro Benavidesm, M., Cruchet Muñoz, S., et al. (2016). Overweight and obesity: review and update. Acta Gastroenterol. Latinoam. 46, 131–159.
Sasaki, S. (2011). Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 70, 349–359. doi: 10.1097/nen.0b013e3182160690
Saudou, F., and Humbert, S. (2016). The biology of huntingtin. Neuron 89, 910–926. doi: 10.1016/j.neuron.2016.02.003
Schaefer, A., van Duijn, T. J., Majolee, J., Burridge, K., and Hordijk, P. L. (2017). Endothelial CD2AP binds the receptor ICAM-1 to control mechanosignaling, leukocyte adhesion, and the route of leukocyte diapedesis in vitro. J. Immunol. 198, 4823–4836. doi: 10.4049/jimmunol.1601987
Skup, M. (2018). Neurotrophins: evolution of concepts on rational therapeutic approaches. Postepy Biochem. 64, 231–241.
Šmahelová, A. (2017). Diabetes mellitus and cognitive disorders from the diabetologists perspective. Vnitr Lek. 63, 717–720.
Smith, A. J., Malan, L., Uys, A. S., Malan, N. T., Harvey, B. H., and Ziemssen, T. (2015). Attenuated brain-derived neurotrophic factor and hypertrophic remodelling: the SABPA study. J. Hum. Hypertens. 29, 33–39. doi: 10.1038/jhh.2014.39
Sohn, J. W. (2015). Network of hypothalamic neurons that control appetite. BMB Rep. 48, 229–233. doi: 10.5483/bmbrep.2015.48.4.272
Srikanthan, K., Feyh, A., Visweshwar, H., Shapiro, J. I., and Sodhi, K. (2016). Systematic review of metabolic syndrome biomarkers: a panel for early detection, management, and risk stratification in the west virginian population. Int. J. Med. Sci. 13, 25–38. doi: 10.7150/ijms.13800
Stadtmann, A., Brinkhaus, L., Mueller, H., Rossaint, J., Bolomini-Vittori, M., Bergmeier, W., et al. (2011). Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur. J. Immunol. 41, 2074–2085. doi: 10.1002/eji.201041196
Stavoe, A. K. H., and Holzbaur, E. L. F. (2019). Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 35, 477–500.
Suresh, S. N., Chakravorty, A., Giridharan, M., Garimella, L., and Manjithaya, R. (2020). Pharmacological tools to modulate autophagy in neurodegenerative diseases. J. Mol. Biol. 432, 2822–2842. doi: 10.1016/j.jmb.2020.02.023
Tekirdag, K., and Cuervo, A. M. (2018). Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J. Biol. Chem. 293, 5414–5424. doi: 10.1074/jbc.r117.818237
Thomsen, M. S., Routhe, L. J., and Moos, T. (2017). The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 37, 3300–3317. doi: 10.1177/0271678x17722436
Tomoda, T., Yang, K., and Sawa, A. (2020). Neuronal autophagy in synaptic functions and psychiatric disorders. Biol. Psychiat. 87, 787–796. doi: 10.1016/j.biopsych.2019.07.018
Tumminia, A., Vinciguerra, F., Parisi, M., and Frittitta, L. (2018). Type 2 diabetes mellitus and Alzheimer’s Disease: role of insulin signalling and therapeutic implications. Int. J. Mol. Sci. 19:3306. doi: 10.3390/ijms19113306
van den Berg, E., Reijmer, Y. D., de Bresser, J., Kessels, R. P., Kappelle, L. J., and Biessels, G. J. (2010). A 4 year followup study of cognitive functioning in patients with type 2 diabetes mellitus. Diabetologia 53, 58–65.
Van Dyken, P., and Lacoste, B. (2018). Impact of metabolic syndrome on neuroinflammation and the blood–brain barrier. Front. Neurosci. 12:930. doi: 10.3389/fnins.2018.00930
van Niekerk, G., du Toit, A., Loos, B., and Engelbrecht, A. M. (2018). Nutrient excess and autophagic deficiency: explaining metabolic diseases in obesity. Metabolism. 82, 14–21. doi: 10.1016/j.metabol.2017.12.007
Vanhauwaert, R., Kuenen, S., Masius, R., Bademosi, A., Manetsberger, J., Schoovaerts, N., et al. (2017). The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 36, 1392–1411. doi: 10.15252/embj.201695773
Varatharaj, A., and Galea, I. (2017). The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 60, 1–12. doi: 10.1016/j.bbi.2016.03.010
Varghese, J. F., Patel, R., and Yadav, U. C. S. (2018). Novel insights in the metabolic syndrome-induced oxidative stress and inflammation-mediated atherosclerosis. Curr. Cardiol. Rev. 14, 4–14. doi: 10.2174/1573403x13666171009112250
Vijayan, V., and Verstreken, P. (2017). Autophagy in the presynaptic compartment in health and disease. J. Cell Biol. 216, 1895–1906. doi: 10.1083/jcb.201611113
Vilchez, D., Saez, I., and Dillin, A. (2014). The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 5:5659.
Vykoukal, D., and Davies, M. G. (2011). Vascular biology of metabolic syndrome. J. Vasc. Surg. 54, 819–831. doi: 10.1016/j.jvs.2011.01.003
Wang, L., Chen, L., Liu, Z., Liu, Y., Luo, M., Chen, N., et al. (2018). PAI-1 exacerbates white adipose tissue dysfunction and metabolic dysregulation in high fat diet-induced obesity. Front. Pharmacol. 9:1087. doi: 10.3389/fphar.2018.01087
Wang, S., Livingston, M. J., Su, Y., and Dong, Z. (2015). Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy 11, 607–616. doi: 10.1080/15548627.2015.1023983
Wang, Y. C., Lauwers, E., and Verstreken, P. (2017). Presynaptic protein homeostasis and neuronal function. Curr. Opin. Genet. Dev. 44, 38–46. doi: 10.1016/j.gde.2017.01.015
Wei, Y., Liu, G. L., Yang, J. Y., Zheng, R. Z., Jiang, L. H., Li, Y. P., et al. (2014). Association between metabolic syndrome and vascular endothelium dysfunction in children and adolescent. Genet. Mol. Res. 13, 8671–8678. doi: 10.4238/2014.october.27.7
Wentworth, J. M., Naselli, G., Brown, W. A., Doyle, L., Phipson, B., Smyth, G. K., et al. (2010). Proinflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes 59, 1648–1656. doi: 10.2337/db09-0287
West, A. E., Pruunsild, P., and Timmusk, T. (2014). Neurotrophins: transcription and translation. Handb. Exp. Pharmacol. 220, 67–100. doi: 10.1007/978-3-642-45106-5_4
White, E., Mehnert, J. M., and Chan, C. S. (2015). Autophagy, metabolism, and cancer. Clin. Cancer Res. 21, 5037–5046. doi: 10.1158/1078-0432.ccr-15-0490
Wong, S. Q., Kumar, A. V., Mills, J., and Lapierre, L. R. (2020). Autophagy in aging and longevity. Hum. Genet. 139, 277–290.
Wong, Y. C., and Holzbaur, E. L. (2014). The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 34, 1293–1305. doi: 10.1523/jneurosci.1870-13.2014
Woo, C. Y., Jang, J. E., Lee, S. E., Koh, E. H., and Lee, K. U. (2019). Mitochondrial dysfunction in adipocytes as a primary cause of adipose tissue inflammation. Diabetes Metab. J. 43, 247–256. doi: 10.4093/dmj.2018.0221
Wu, H., Wang, Y., Li, W., Chen, H., Du, L., and Liu, D. (2019). Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy 15, 1882–1898. doi: 10.1080/15548627.2019.1596482
Wu, N. N., Zhang, Y., and Ren, J. (2019). Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid Med Cell Longev. 2019:9825061.
Xilouri, M., Brekk, O. R., and Stefanis, L. (2016). Autophagy and alpha-synuclein: relevance to Parkinson’s Disease and related synucleopathies. Mov. Disord. 31, 178–192. doi: 10.1002/mds.26477
Yamamoto, A., and Yue, Z. (2014). Autophagy and its normal and pathogenic states in the brain. Annu. Rev. Neurosci. 37, 55–78. doi: 10.1146/annurev-neuro-071013-014149
Yamamoto, T., Takabatake, Y., Takahashi, A., Kimura, T., Namba, T., Matsuda, J., et al. (2017). High-fat diet-induced lysosomal dysfunction and impaired autophagic flux contribute to lipotoxicity in the kidney. J. Am. Soc. Nephrol. 28, 1534–1551. doi: 10.1681/asn.2016070731
Yamamoto, T., Takabatake, Y., Minami, S., Sakai, S., Fujimura, R., Takahashi, A., et al. (2020). Eicosapentaenoic acid attenuates renal lipotoxicity by restoring autophagic flux. Autophagy. 28, 1–14. doi: 10.1080/15548627.2020.1782034
Yarmolinsky, J., Barbieri, N. B., Weinmann, T., Ziegelmann, P. K., Duncan, B. B., and Schmidt, M. I. (2016). Plasminogen activator inhibitor-1 and type 2 diabetes: a systematic review and meta-analysis of observational studies. Sci. Rep. 6:17714.
Yates, K. F., Sweat, V., Po Lai Yau, Turchiano, M. M., and Convit, A. (2012). Impact of metabolic syndrome on cognition and brain: a selected review of the literature. Arterioscler. Thromb. Vasc. Biol. 32, 2060–2067.
Yoo, D. Y., Yim, H. S., Jung, H. Y., Nam, S. M., Kim, J. W., Choi, J. H., et al. (2016). Chronic type 2 diabetes reduces the integrity of the blood-brain barrier by reducing tight junction proteins in the hippocampus. J. Vet. Med. Sci. 78, 957–962. doi: 10.1292/jvms.15-0589
Yoon, S. Y., and Kim, D. H. (2016). Alzheimer’s disease genes and autophagy. Brain Res. 1649(Pt B), 201–209.
Yoshii, S. R., and Mizushima, N. (2017). Monitoring and measuring autophagy. Int. J. Mol. Sci. 18:1865. doi: 10.3390/ijms18091865
Yu, L., Chen, Y., and Tooze, S. A. (2018). Autophagy pathway: cellular and molecular mechanisms. Autophagy. 14, 207–215. doi: 10.1080/15548627.2017.1378838
Zachari, M., and Ganley, I. G. (2017). The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 61, 585–596. doi: 10.1042/ebc20170021
Zaffagnini, G., and Martens, S. (2016). Mechanisms of selective autophagy. J. Mol. Biol. 428 (9 Pt. A), 1714–1724. doi: 10.1016/j.jmb.2016.02.004
Zhao, Z., Nelson, A. R., Betsholtz, C., and Zlokovic, B. V. (2015). Establishment and dysfunction of the blood-brain barrier. Cell 163, 1064–1078. doi: 10.1016/j.cell.2015.10.067
Keywords: metabolic syndrome, vascular endothelium, neurological disorders, autophagy, brain-derived neurotrophic factor
Citation: Maiuolo J, Gliozzi M, Musolino V, Carresi C, Scarano F, Nucera S, Scicchitano M, Bosco F, Ruga S, Zito MC, Macri R, Bulotta R, Muscoli C and Mollace V (2021) From Metabolic Syndrome to Neurological Diseases: Role of Autophagy. Front. Cell Dev. Biol. 9:651021. doi: 10.3389/fcell.2021.651021
Received: 08 January 2021; Accepted: 26 February 2021;
Published: 19 March 2021.
Edited by:
Stephen Fernandes Rodrigues, University of São Paulo, BrazilReviewed by:
Aurore Claude-Taupin, Université Paris Descartes, FranceVijay Karkal Hegde, Texas Tech University, United States
Copyright © 2021 Maiuolo, Gliozzi, Musolino, Carresi, Scarano, Nucera, Scicchitano, Bosco, Ruga, Zito, Macri, Bulotta, Muscoli and Mollace. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica Maiuolo, amVzc2ljYW1haXVvbG9AdmlyZ2lsaW8uaXQ=