 Sabeeha Malek
Sabeeha Malek Darius V. Köster
Darius V. Köster
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 21 April 2021
Sec. Cell Growth and Division
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.649082
This article is part of the Research Topic Cytoskeletal Dynamics and Mechanics in Cell Growth, Division, Differentiation and Aging View all 12 articles
The Ehlers-Danlos syndromes (EDS) are a group of 13 disorders, clinically defined through features of joint hypermobility, skin hyperextensibility, and tissue fragility. Most subtypes are caused by mutations in genes affecting the structure or processing of the extracellular matrix (ECM) protein collagen. The Hypermobility Spectrum Disorders (HSDs) are clinically indistinguishable disorders, but are considered to lack a genetic basis. The pathogenesis of all these disorders, however, remains poorly understood. Genotype-phenotype correlations are limited, and findings of aberrant collagen fibrils are inconsistent and associate poorly with the subtype and severity of the disorder. The defective ECM, however, also has consequences for cellular processes. EDS/HSD fibroblasts exhibit a dysfunctional phenotype including impairments in cell adhesion and cytoskeleton organization, though the pathological significance of this has remained unclear. Recent advances in our understanding of fibroblast mechanobiology suggest these changes may actually reflect features of a pathomechanism we herein define. This review departs from the traditional view of EDS/HSD, where pathogenesis is mediated by the structurally defective ECM. Instead, we propose EDS/HSD may be a disorder of membrane-bound collagen, and consider how aberrations in cell adhesion and cytoskeleton dynamics could drive the abnormal properties of the connective tissue, and be responsible for the pathogenesis of EDS/HSD.
The Ehlers-Danlos syndromes (EDS) are a group of heritable connective tissue disorders defined by the presence of three clinical features: joint hypermobility, skin hyperextensibility, and tissue fragility (Malfait et al., 2017). The condition is named after two dermatologists, Edvard Lauritz Ehlers and Henri-Alexandre Danlos both of whom independently characterized some of the first clinical descriptions of EDS in the early 20th Century (Parapia and Jackson, 2008). The formal categorization of the EDS subtypes began in the 1960s, and there have since been several reclassifications following advances in our understanding of the molecular and genetic basis of these disorders (Table 1). Most subtypes have now been shown to be caused by mutations in genes affecting the structure or processing of collagen (Malfait et al., 2017). Most recently, the International Consortium on the ehlers-danlos syndromes proposed updated diagnostic criteria in 2017, which recognizes 13 different EDS subtypes (Malfait et al., 2017). Since its publication, a 14th subtype of EDS has been identified (Blackburn et al., 2018). While provisional diagnoses can be made based on clinical major and minor criteria, a definitive diagnosis now relies on the molecular identification of the causative variant(s) in the respective gene. The exception lies with the hypermobile form of EDS (hEDS; formerly EDS type III) which, despite being the most common form, remains as the only subtype without an identified distinct genetic variation, and whose diagnosis remains clinical alone. In addition, it is important to recognize the related Hypermobility Spectrum Disorders (HSD) which are clinically indistinguishable from hEDS, and both diagnoses are often termed together as hEDS/HSD. HSD is a diagnosis the EDS consortium had intended to reflect patients who demonstrate the characteristic feature of hEDS, i.e., joint hypermobility, but lack sufficient clinical evidence to demonstrate a genetic aetiology to their presentation. The segregation of these diagnoses was intended to produce a more homogenous cohort to further aid research efforts to identify the genetic markers of hEDS, but does not differentiate patients in terms of severity of symptoms or disability (Tinkle et al., 2009; Copetti et al., 2019). Collectively, EDS/HSD has a diagnosed prevalence of 1 in 500 (Demmler et al., 2019).
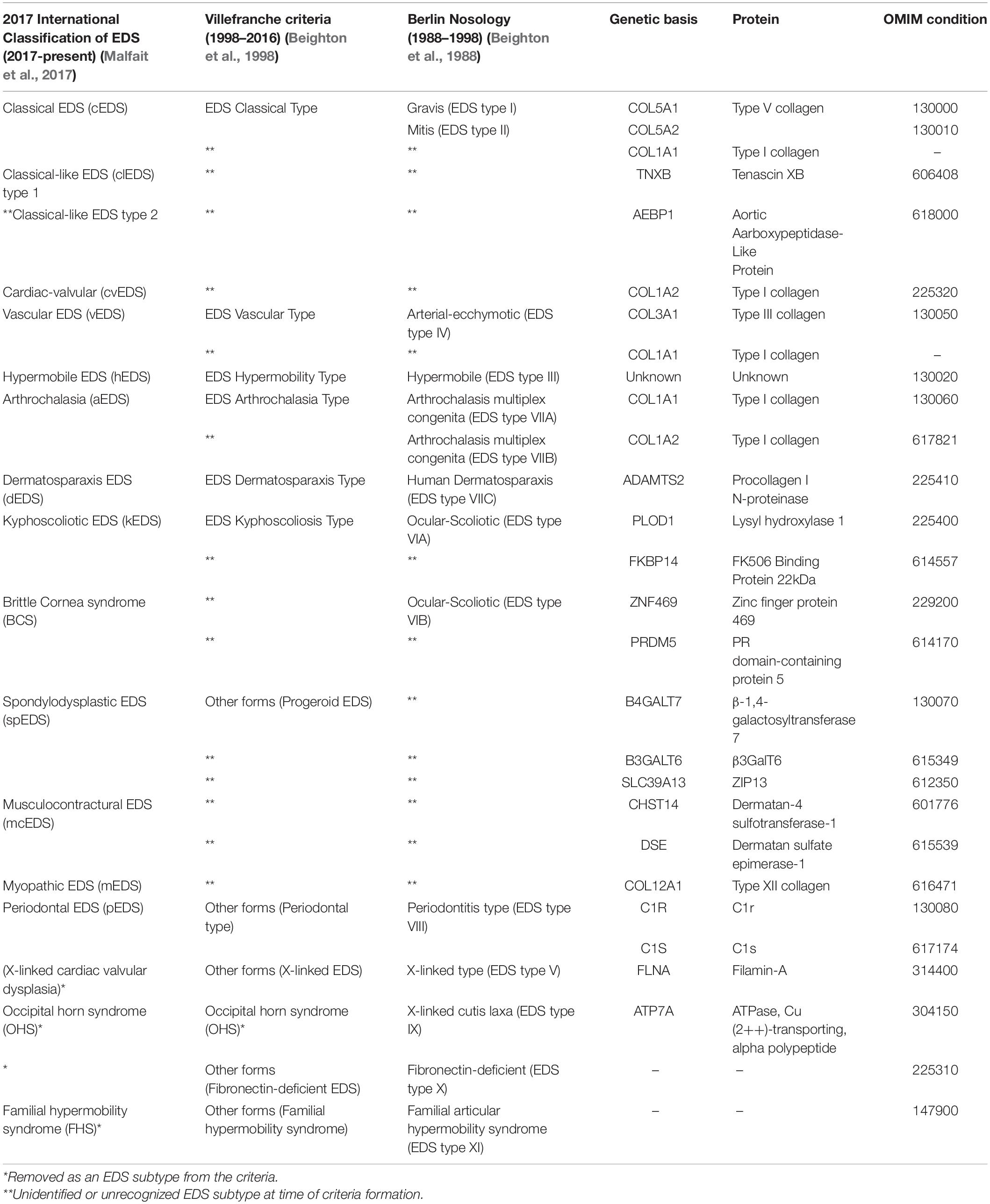
Table 1. Past and present classifications of EDS subtypes and their molecular basis.
Our current understanding of EDS/HSD pathogenesis is informed by the two best characterized subtypes, the classical (cEDS) and vascular (vEDS) forms, which are consequent to mutations in genes encoding the minor collagen proteins, type V (Symoens et al., 2012), and type III (Pepin et al., 2000), respectively. Mutations either cause haploinsufficiency, which is a 50% reduction in protein expression caused by a nonsense-mediated decay of the non-viable RNA transcript (Leistritz et al., 2011; Symoens et al., 2012), or the production of a structurally defective procollagen molecule which is retained within the endoplasmic reticulum (Smith et al., 1997; Symoens et al., 2012). Consequently, in both scenarios, the absence of sufficient functional minor collagen proteins in the extracellular matrix (ECM) then impedes with their role in regulating the formation and organization of the major type I collagen into fibrils (Figure 1; Liu et al., 1997; Wenstrup et al., 2004a,b; D’hondt et al., 2018; Wang et al., 2020). This disrupts the integrity of the ECM and is assumed to be the underlying cause of connective tissue weakness.
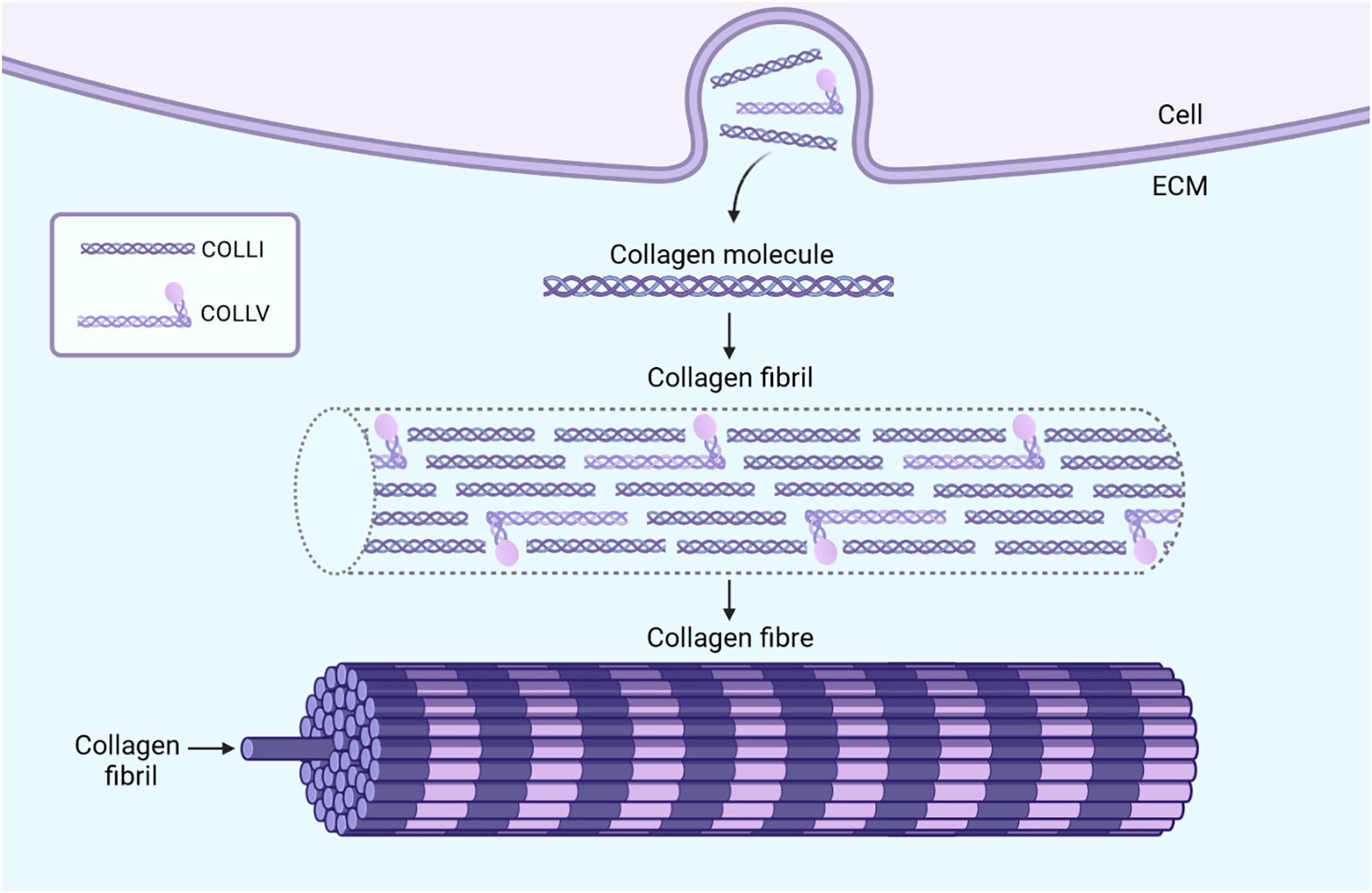
Figure 1. The formation and function of collagen fibrils in connective tissue. Fibroblasts secrete collagen molecules into the ECM which associate in a staggered pattern to form collagen fibrils. These collagen fibrils consist mostly of the major collagen protein, type I, while the minor collagen proteins, such as type III and type V constitute only a fraction of fibril mass (Theocharidis and Connelly, 2019). The importance of these minor collagens, however, lies in their role in regulating the diameter and organization of collagen fibrils. In the example of type V collagen molecules, the presence of a non-collagenous domain which projects outwards introduces steric hindrances when incorporated within collagen fibrils (Wenstrup et al., 2004a). This limits the lateral growth of the fibrils and may also play a role in regulating their diameter (Wenstrup et al., 2004a). Multiple collagen fibrils together form collagen fibers which provide tensile strength to the connective tissue. The absence of sufficient amounts of minor collagen proteins, like in EDS, can result in poorly formed collagen fibrils, and in turn, collagen fibers (Theocharidis and Connelly, 2019). The tissue specific expression and roles of the minor collagen proteins presumably accounts for the characteristic presentation of each specific EDS subtype, all of which differ in the varying presence, manifestation, and degrees of joint hypermobility, skin hyperextensibility, and tissue fragility. Image created with BioRender.com.
However, this notion is not truly reflected in transmission electron microscopy (TEM) analyses of collagen fibril structure and organization in EDS/HSD skin biopsies, though general abnormalities can be found. These abnormalities include reduced and disorganized collagen bundles with abnormal orientations, or non-circular cross-sections of collagen fibrils that are described as “flower-like,” or with variable diameters both larger and smaller than would be typically expected (Holbrook and Byers, 1982; Hausser and Anton-Lamprecht, 1994; Hermanns-Lê and Piérard, 2006). vEDS patients have been characterized as having collagen fibrils that are smaller and more variable, and with an overall reduced density (Smith et al., 1997), while cEDS is associated with the presence of flower-like collagen fibrils (Vogel et al., 1979; de Moraes et al., 2000; Bowen et al., 2017; Angwin et al., 2020).
Yet, these findings are not consistent and this should be considered significant. It has long been noted that the degree of ultrastructural changes observed do not correlate with clinical severity (Proske et al., 2006), or presentation (Kobayasi, 2004; Hermanns-Lê et al., 2012). Further studies have shown that patients can present with no significant collagen abnormalities, despite a clinical presentation and/or confirmation of genetic disorder. In 2019, one study reported two individuals with a likely pathogenic variant of the COL5A1 gene that did not present with the typical collagen flowers expected (Angwin et al., 2019), contradicting a consensus previously reached by the EDS committee in 2017 (Bowen et al., 2017). Another study in vEDS patients found that the size of collagen fibers and bundle characteristics did not discriminate between vEDS and control participants, nor did all vEDS participants present with abnormal fibril diameters (Ong et al., 2012). More recently, a larger study of 177 EDS patients found that no specific TEM finding could be associated with any specific EDS subtype (Angwin et al., 2020). Remarkably, from the 177 patients with a clinical diagnosis, 147 (83%) had a normal TEM report, and from the 24 patients with a genetic confirmation of their subtype, 7 (29%) also had a normal biopsy report (Angwin et al., 2020). Conversely, it has been demonstrated that clinically unaffected family members and control participants can also present with the same EDS-associated ultrastructural abnormalities without displaying features of a connective tissue disorder (Kobayasi, 2004; Hermanns-Lê et al., 2012). As such, the only conclusion that can be derived from these collective TEM studies is that an abnormal biopsy finding is more frequently found in EDS/HSD patients (Angwin et al., 2020).
These findings demonstrate that despite the categorization of EDS/HSD as a disorder of collagen, the reported ultrastructural collagen abnormalities do not appear to be the principal mediators of abnormal connective tissue properties in these disorders. It is therefore plausible to assume that other related factors heavily influence connective tissue properties and mediate the EDS/HSD phenotype. One such factor may be the cellular mechanics of fibroblasts, the predominant cell type that populates the connective tissue.
The ability of fibroblasts to directly exert force and influence the tension in the surrounding environment is significant, as it directly affects the viscoelastic properties of the connective tissue. This has been demonstrated through gel contraction assays (Figure 2) (Dallon and Ehrlich, 2008). Fibroblasts seeded into isolated and recombinant collagen gels generate tractional forces by cytoskeleton contractility which propagate throughout the gel matrix. This compacts the collagen fibrils, eliminating water from between the fibrils which decreases the gel volume. The contraction of this gel is proposed to reflect the ability of fibroblasts to contract an open wound, hence this is used as the typical model assay for the study of cell-ECM interactions in the context of wound healing.
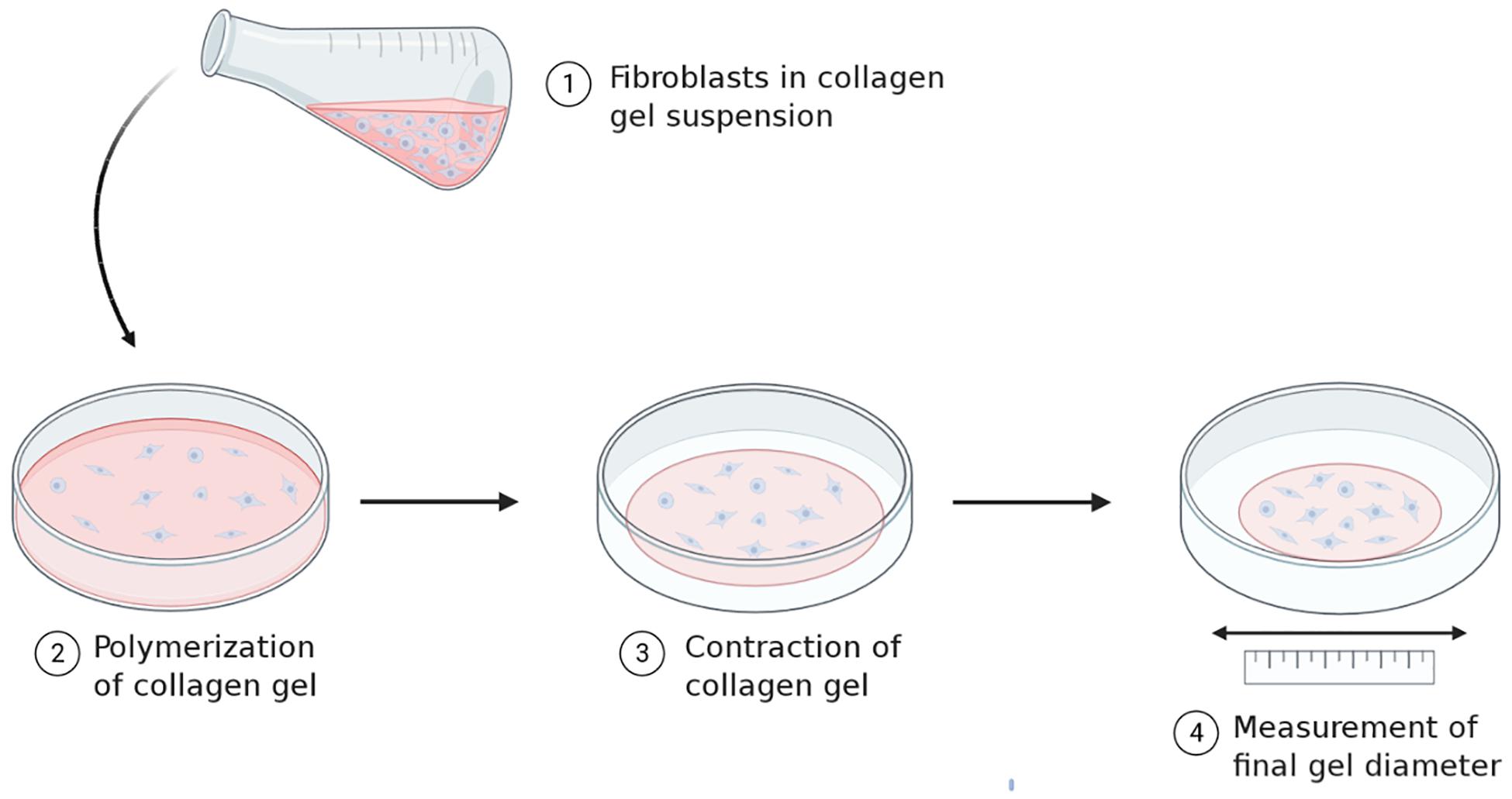
Figure 2. Principles underlying collagen gel contraction assays. Standard gel contraction assays involve seeding fibroblasts into a collagen gel solution and allowing the suspension to polymerize. The freshly polymerized gel is placed in media for a set time period and allowed to contract. Changes in the diameter of the gel are measured before and after and can be used as a parameter to quantify cell contractility. Image created with BioRender.com.
Such gel contraction assays have provided several relevant findings of note. Firstly, the ability of fibroblasts to contract the gel significantly, demonstrates how the cellular mechanics of fibroblasts can directly influence the material properties of their environment, e.g. the connective tissue (Delvoye et al., 1991; Eastwood et al., 1994, 1996). These assays have also demonstrated the key cellular features enabling gel contraction. One is cell adhesion to collagen via integrins, as blocking integrin-mediated adhesion with antibodies prevented gel contraction (Klein et al., 1991; Schiro et al., 1991; Kelynack et al., 2000). Equally important is an active cytoskeleton, as using molecular inhibitors that interfere with actin filament and microtubule polymerization abrogates tractional force generation and reduces tension within the gel (Kolodney and Wysolmerski, 1992; Swierczewski et al., 2016; Giannopoulos et al., 2018). The involvement of intermediate filaments has also been indicated in this process, as vimentin-null fibroblasts show a reduced ability to contract the gel (Ridge et al., 2016).
However, the significance of these findings is not just/limited to the context of wound healing, but reflect important processes involved in the ongoing maintenance of connective tissue function. It has been shown that fibroblasts do not simply contract the gel when seeded, but they alter their cytoskeletal contractility to adapt to any external loads applied in order to maintain an overall resting tension in the environment (Delvoye et al., 1991; Brown et al., 1998; Mizutani et al., 2004; Giannopoulos et al., 2018; Zollinger et al., 2018). Indeed, the specific level of resting tension established by fibroblasts has been described as a “tensile setpoint”, and their tendency to actively maintain this equilibrium as “tensional homeostasis” (Brown et al., 1998). Furthermore, it has been shown that the ability to maintain tensional homeostasis is not dependent on the stiffness of the environment, and is only limited by the load force not the displacement (Freyman et al., 2002). This suggests that healthy fibroblasts are also capable of compensating for a materially weak ECM by adjusting the level of contractile force needed to maintain overall normal levels of resting tension (Tomasek et al., 2002). Experiments on mouse subcutaneous connective tissue have also reproduce these findings in a more physiological setting. Stretching connective tissue samples caused a disproportionate change in fibroblast morphology, which could not be explained by simple passive spreading of cells, but indicated an active response (Langevin et al., 2005, 2011). The importance of active processes in mediating tensional homeostasis was shown when inducing cell death or inhibiting cytoskeleton dynamics increased the tension within the connective tissue by 60-80% upon stretch (Langevin et al., 2011).
These findings indicate that fibroblast mechanics play a significant role in mediating the properties of the connective tissue beyond the context of wound healing. Fibroblasts actively alter their contractile forces in response to everyday mechanical strains and loads for the purposes of maintaining appropriate tension, hence, also determining the viscoelastic properties of the connective tissue. Furthermore, these processes are dependent on the key processes of cell adhesion and cytoskeleton dynamics. Impairments in any of these processes, therefore, may affect connective tissue integrity and viscoelasticity, and contribute to the pathogenesis of EDS/HSD.
Indeed, fibroblast dysfunction has been implicated in the pathogenesis of several other connective tissue disorders. Fibroblasts from patients with floppy eyelid syndrome, a hyperelasticity disorder affecting the upper eyelid, demonstrate a significantly higher tensile setpoint compared to control (Ezra et al., 2010). In Dupuytren’s contracture, a condition affecting the facia of the hand, fibroblasts are unable to respond appropriately to mechanical loading, and exert an opposite response and of a greater magnitude compared to control fibroblasts, something which also disturbed the attainment of tensional homeostasis (Bisson et al., 2004, 2009). Of particular significance are findings from Pelvic Organ Prolapse (POP) patients, a condition of weakened connective tissue highly associated with EDS/HSD (Carley and Schaffer, 2000; Lammers et al., 2012). Under static conditions, POP fibroblasts in cell culture demonstrate a significantly higher expression of the cytoskeleton proteins actin, α-tubulin, and vimentin compared to control cells, indicating an increased mechano-response to the stiff substrate of a culture dish (Wang et al., 2015). In response to mechanical strain, however, POP fibroblasts exhibit a significant decrease in the expression of actin, in contrast to control cells that increased actin expression under the same conditions (Wang et al., 2015). Another study also found that POP fibroblasts delayed the alignment of their actin cytoskeleton in the direction of the force in response to mechanical strain compared to control cells (Ruiz-Zapata et al., 2013). These findings indicate that POP fibroblasts are unable to efficiently respond to mechanical forces, which may even overload and impair the integrity of cytoskeletal system. In the context of a whole tissue environment, such a failure would contribute to the weakening of the connective tissue, and similar mechanisms may exist and contribute to the pathogenesis of EDS/HSD.
The culturing of fibroblasts derived from the dermal biopsies of EDS/HSD patients have revealed several relevant molecular changes (Zoppi et al., 2018b; Chiarelli et al., 2019), including an altered integrin expression profile that is shared amongst the main EDS/HSD subtypes (Zoppi et al., 2018b). Integrins are cell surface receptors composed of one α and one β subunit, giving rise to 24 unique integrins which play a central role in cell adhesion, cell signaling, and cell survival (Barczyk et al., 2010). Integrins mediate the attachment between the cell and specific ECM proteins like collagen or fibronectin (Figure 3). Binding of the ECM ligand exposes the cytoplasmic tail of the integrin, which provides a scaffold to allow recruitment of paxilin, vinculin, talin, and other proteins during the formation of multi-protein complexes called focal adhesions constituting signaling complexes (Kanchanawong et al., 2010; Parsons et al., 2010). These in turn, regulate the activity of the Rho family of GTPases, which act as molecular switches to regulate downstream signaling processes and cytoskeleton organization (Kanchanawong et al., 2010; Parsons et al., 2010). These adhesion sites act as a strong anchoring point for the actin cytoskeleton, important for the mechanical feedback between cell and ECM.
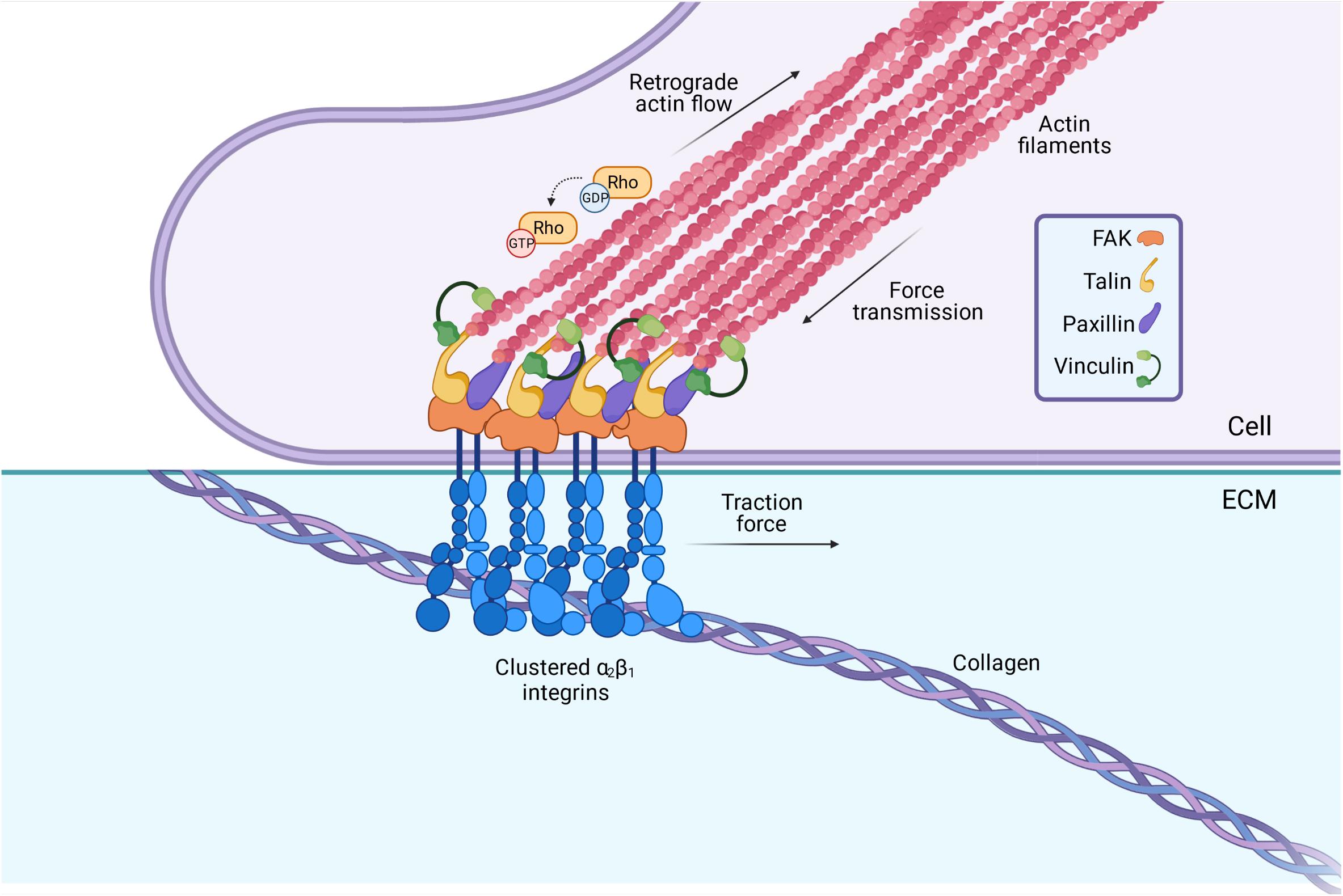
Figure 3. Integrins serve as a vital connection between the cells’ internal cytoskeleton and the ECM. Integrins span the membrane of fibroblasts, with vital intracellular and extracellular domains. Binding of the ECM ligand (collagen, fibronectin etc.) to the extracellular domain leads to the activation of the integrin, and a conformational change in its cytoplasmic tail. This triggers a rapid recruitment of adaptor proteins such as talin and paxillin to the cytoplasmic tail, which in turn, triggers a signaling cascade involving the Rho GTPases. These act as molecular switches to regulate the polymerization of actin, and hence, play an important role in determining the cytoskeletons contractility. The lateral assembly or “clustering” of integrins also occurs upon ligand binding, to form multi-protein complexes termed “focal adhesions” at the cell surface. These act as strong anchoring points and mediate the specific attachment between the cell cytoskeleton and the ECM. Image created with BioRender.com.
The integrin profile demonstrated in EDS/HSD involves the downregulation of the collagen receptor, α2β1 integrin, as well as the fibronectin receptor, α5β1 integrin, while the vitronectin receptor, αvβ3 integrin, is upregulated instead. In EDS/HSD, this αvβ3 integrin functions as an alternative receptor for fibronectin (Zoppi et al., 2018b). The expression of this specific integrin profile is termed the “integrin switch” (Zoppi et al., 2004, 2018b), which can be recreated in control cells with a functional blocking of the α2β1 receptor (Zoppi et al., 2004). This demonstrates that the absence of a structurally organized collagen in the ECM failing to engage with its receptor α2β1 leads to the altered integrin expression profile. This, in turn, will influence other cellular processes that may be critical in the pathogenesis EDS/HSD.
The first of these is mechano-sensitivity, which is an ability of fibroblasts to sense the compliance of their environment and the strain present within it (Schwartz, 2010). This process would be essential to produce the appropriate cytoskeletal response to mechanical strain and requires the cooperative actions of various integrins. A study demonstrating such cooperation of integrins with different mechanosensitive roles showed the α5β1 integrins to be implicated in force generation, whereas αv-class integrins mediated the structural adaptations to forces (Schiller et al., 2013). Fibroblasts in which the interaction between the αvβ3 integrin and fibronectin is blocked, also failed to sense the rigidity of a fibronectin matrix (Jiang et al., 2006). This would suggest that the integrin switch would alter the mechano-sensing ability of EDS/HSD fibroblasts. Indeed, there is already some indication that this process is perturbed in hEDS/HSD. When hEDS/HSD fibroblasts were cultured on a stiff culture substrate, excessive actin stress fibers could be observed compared to control (Zoppi et al., 2018a), however, these were absent in hEDS/HSD fibroblasts that were directly observed within tissue samples (Celli et al., 2020). This demonstrates that excessive stress fiber formation is not a persistent feature of hEDS/HSD fibroblasts, but instead, could be reflective of mechano-sensitivity aberrations. It is possible that this abnormal cytoskeletal response only occurred when fibroblasts adhered to stiff substrates in vitro, but not when they remained within their native soft tissue environment. Such mechano-sensitivity aberrations are physiologically relevant, as conditions of mechanical strain can temporarily stiffen the environment of fibroblasts in vivo, and, similarly, produce an abnormal cytoskeletal response which contributes to abnormal connective tissue properties.
The integrin switch may also impair cell-ECM adhesion, which may be crucial for tissue integrity. Indeed, aberrant adhesion has already been demonstrated in EDS/HSD. In cEDS and vEDS fibroblasts, cell-ECM adhesion has been shown to be mediated specifically between the αvβ3 integrin and fibronectin, whereas control cells show no such critical dependency (Zoppi et al., 2008). The fibronectin organization itself is impaired in EDS/HSD, present only as rare fibrils in the ECM (Zoppi et al., 2018b), further indicating a compromised cell-ECM interaction. The strength of the cell-ECM adhesions themselves, may also be limited. A single bond between the α2β1 integrin and collagen can withstand a mechanical force of 160 pN (Niland et al., 2011), whereas bonds between α5β1 and fibronectin begin to break at 30 pN (Kong et al., 2009). Bonds between αvβ3 and fibronectin break at even lower forces (Roca-Cusachs et al., 2009). This was demonstrated in a study where adhesion between cells and fibronectin-coated beads was mediated either by the αvβ3 integrin or α5β1. Following application of a 1 nN force for 100 s, cells adhering via αvβ3 completely detached from the beads, whereas cells adhering via the α5β1 integrin maintained adhesion (Roca-Cusachs et al., 2009). The same differences in adhesion strength were also observed when clustering of the integrins was promoted (Roca-Cusachs et al., 2009). It is suggested that these weaker αvβ3 bonds facilitate force detection by breaking more easily, and hence, their role is to enable mechano-transduction rather than adhesion. Consequently, these collective findings suggest that the integrin switch, alongside the defective fibronectin organization observed in EDS/HSD, may produce far weaker connections between the cell and ECM. These may break more easily under conditions of mechanical strain, and contribute to the weakening of the integrity of the connective tissue (Roca-Cusachs et al., 2009).
Some impairments in the cytoskeleton of EDS/HSD fibroblasts have also been observed in vitro. cEDS and vEDS fibroblasts demonstrate a disorganized actin cytoskeleton (Zoppi et al., 2008), whereas in hEDS and HSD, the actin cytoskeleton is organized into stress fibers (Zoppi et al., 2018a). Furthermore, cEDS and vEDS fibroblasts show a decreased migratory capacity (Viglio et al., 2008; Zoppi et al., 2018a), whereas cell migration is significantly increased in hEDS and HSD fibroblasts (Zoppi et al., 2018a). This migratory capacity is relevant, as it involves the same mechanical machinery that generates the tractional forces implicated in mediating tensional homeostasis (Pollard and Borisy, 2003; Webster et al., 2014). These findings indicate that this aspect of EDS/HSD fibroblast function may also be aberrant and contribute to abnormal connective tissue properties.
The integrin switch itself may also have further implications for cytoskeleton dynamics. Integrins are also known to influence the activities of the Rho-GTPases, RhoA, Rac1, and Cdc42, which are crucial to cell protrusion formation (Lawson and Burridge, 2014), which in turn, is involved in the tensional homeostasis mechanism (Webster et al., 2014). During this process, the initial extension of the plasma membrane is driven predominantly through Rac-mediated actin polymerization, while RhoA and Cdc42 activity contributes to extension of cell protrusions at later stages of cell spreading (Ridley, 2015). It has been demonstrated that β1 integrin subunit in particular, is required to support RhoA activation at later stages of cell spreading (Danen et al., 2002). Overexpression of the β3 subunit has also been shown to enhance the activity of RhoA and promote stress fiber formation, whereas overexpression of the β1 subunit enhanced Rac activity and cell protrusion formation (Miao et al., 2002). In another study examining cell migration in epithelial cells, adhesion via αvβ3 was shown to support extensive actin cytoskeletal reorganization and the formation of a single broad protrusion at the leading edge, whereas cell adhesion via α5β1 caused the extension of thinner protrusions (Danen et al., 2005). These findings indicate that the integrin profile demonstrated in EDS/HSD cells, may also have direct consequences on the ability of fibroblasts to form and/or maintain cell protrusions under conditions of mechanical strain and maintain connective tissue tension.
It is clear from the molecular changes described that fibroblast dysfunction is to be expected in EDS/HSD, involving aberrations in both cell adhesion and cytoskeleton dynamics. This has been directly examined in gel contraction studies performed with fibroblasts from animals with dermatosparaxis, which is equivalent to the human form of dermatosparaxis EDS (dEDS; formerly EDS type VIIC) (Colige et al., 1999). This disorder is caused by mutations in the ADAMTS2 gene, which encodes the procollagen N-proteinase enzyme, and consequently results in poorly structured and loosely arranged collagen fibrils in the ECM (Bavinton et al., 1985). However, similarly, to the ongoing discrepancies in human EDS/HSD studies, the TEM findings of aberrant collagen fibrils in dermatosparaxis animals do not appear to reflect severity of presentation, and therefore, fail to fully account for and explain the abnormal properties of the connective tissue (Bavinton et al., 1985). The gel contraction abilities, however, do more accurately reflect clinical presentation. Dermal fibroblasts from mildly affected sheep demonstrate a gel contraction profile approaching that of control dermal fibroblasts (Ramshaw et al., 1991), while those from severely affected calf and sheep failed to contract the gel (Delvoye et al., 1983, 1986, 1991; Ramshaw et al., 1991). Furthermore, fibroblasts obtained from the less affected tissues in the severely presenting calf, like the tendons, vena cava, and aorta, also reflect this tissue specific presentation, and contracted the gel, similarly, to control fibroblasts (Delvoye et al., 1983). These finding are highly significant, as the correlation of fibroblast dysfunction with severity of manifestation, both between differently presenting animals, and between tissues within the same animal, likely demonstrates one of the main pathomechanisms mediating the observed connective tissue abnormalities. This provides confirmation that dysfunction of the fibroblast does indeed, constitute a relevant pathomechanism for EDS/HSD, and one that has now been demonstrated in a relevant animal model.
The nature of the fibroblasts inability to contract the gel was later shown to be due to the cell surface absence of a 34 kDa collagen binding protein related to anchorin CII (Mauch et al., 1988), which prevented effective cell adhesion of dermatosparatic fibroblasts to collagens type I and IV (Mauch et al., 1986). Consequently, it appears these fibroblasts were unable to attach to the type I collagen in the gel and effectively mediate the cytoskeletal forces needed to contract the collagen gel, as well as maintain homeostatic levels of tension within a restrained gel (Delvoye et al., 1991). These findings, therefore, confirm that impaired cell adhesion is a significant feature of fibroblast dysfunction, reducing integrity of the connective tissue integrity.
These findings highlight the complexity that underlies monogenic disorders. Pathogenesis of dermatosparaxis in this case, may not have been principally mediated by the aberrant collagen molecule itself, but mediated by a reduced cell surface expression of a cell adhesion molecule, that initially appears unrelated to the underlying ADAMTS2 mutation. This demonstrates that far more complex cellular and molecular processes underlie the pathway from initial mutated gene to final pathogenic phenotype and is likely to involve the interplay of various independent environmental and related factors. These independent factors could influence critical pathogenic features, and either promote a less or more severe phenotype in the affected individuals, which may help to explain the vast heterogeneity seen in the presentation of these disorders.
The same gel contraction studies were also performed with human EDS samples at the time, however, they demonstrated unexpected results. Fibroblasts from patients of several EDS subtypes showed no gel contraction abnormalities and behaved similar to control fibroblasts (Delvoye et al., 1986, 1991). Remarkably, this also included dEDS, which has an identical genetic basis to dermatosparaxis. It was concluded from these studies that fibroblast dysfunction did not contribute to the pathogenesis of human EDS/HSD, and research into this pathomechanism discontinued as focus turned toward identifying further EDS-related genes.
However, this conclusion now appears contradictory to our molecular understanding of EDS/HSD. The α2β1 integrin is the main collagen receptor in humans and plays the equivalent role to the anchorin CII-related protein in mediating the attachment between the cell and collagen. The α2β1 integrin is also downregulated in EDS/HSD fibroblasts as part of the integrin switch (Zoppi et al., 2018b). Furthermore, several gel contraction studies performed in control cells while blocking the collagen-α2β1 interaction, prevented contraction of the gel (Klein et al., 1991; Schiro et al., 1991; Kelynack et al., 2000). These findings all highly indicate that the integrin switch seen in EDS/HSD fibroblasts should result in similar impairments in cell-ECM adhesion and prevent gel contraction, while also reflecting their pathological behavior in vivo.
Our understanding of the mechanisms regulating these molecular processes has also progressed. It is now apparent that these gel contraction assays do not truly reflect the EDS/HSD scenario in humans specifically, and hence, the drawn conclusions may not be valid. It has since been demonstrated that adding relevant collagen to the media of cultured human EDS fibroblasts promotes phenotypic correction, which reverses the integrin switch and restores the expression of α2β1 to the cell surface (Zoppi et al., 2004). As such, the collagen supplied in these gel contraction assays may have corrected the phenotype of human EDS fibroblasts, and restored the cell surface expression of α2β1. This would have promoted normal gel contraction, yet not reflect the true in vivo behavior of EDS/HSD fibroblasts. Hence, the human equivalent of this pathomechanism cannot be demonstrated through standard gel contraction assays. The existence of this pathomechanism therefore, has not been excluded in human patients, nor has it yet been truly examined.
This review had the aim to highlight a potential pathomechanism in EDS/HSD involving fibroblast dysfunction. Three principal stages of this pathomechanism can be described (Figure 4).
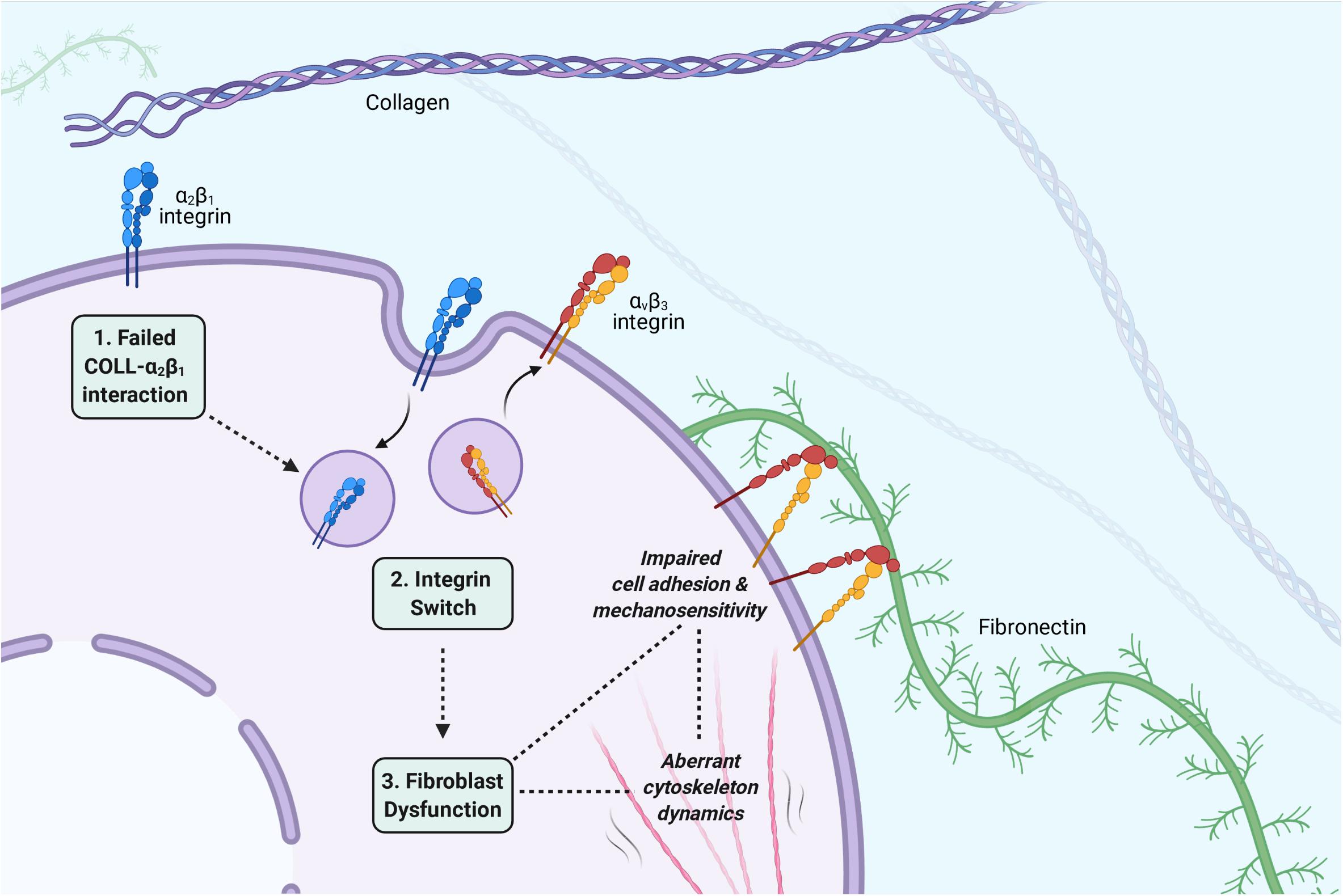
Figure 4. The three principal stages of the proposed pathomechanism for EDS/HSD. (1) A failed interaction between collagen and α2β1: A disorganized or defective collagen-ECM prevents engagement of the collagen ligand with its receptor, the α2β1 integrin, and triggers the integrin switch. (2) The integrin switch: Fibroblasts respond to the defective collagen-ECM by switching their cell adhesion profile to promote adhesion to other ECM ligands. The α2β1 and α5β1 integrins are downregulated, and αvβ3 is upregulated, which then mediates the attachment between cell and ECM via fibronectin. (3) Fibroblast dysfunction: The altered integrin profile also has various consequences for the fibroblasts phenotype, which affects its ability to implement the tensional homeostasis mechanism and maintain integrity of the connective tissue. Impaired cell-ECM adhesion may promote fragility of the connective tissue. Impairments in mechanosensitivity may cause an incorrect interpretation of tension within the connective tissue and facilitate an abnormal cytoskeleton response. Aberrations in the dynamic cytoskeleton response itself, may also facilitate abnormalities in the viscoelastic properties of the connective tissue. All such aspects may contribute to the EDS/HSD phenotype. Image created with BioRender.com.
The first is a failed interaction between collagen and its receptor, the α2β1 integrin (Zoppi et al., 2018b). The underlying cause of this failed interaction is specific for each EDS subtype, and can easily be accounted for in the genetically characterized subtypes of EDS. In cEDS and vEDS for example, mutations in the minor collagen genes prevent the organization of type I collagen into fibrils, which prevents its engagement with α2β1 (Zoppi et al., 2018b). In hEDS and HSD, though the principle underlying molecular defect is unknown, an enhanced expression of the matrix metalloproteinase (MMP), MMP-9, has been observed (Zoppi et al., 2018a). The proteolytic activity of MMP-9 could promote a general disassembly of the ECM, and in turn, prevent the engagement of collagen with α2β1. This stage of the pathomechanism, however, also allows the possibility of an acquired EDS phenotype, consequent to any other disease processes that interferes with collagen-α2β1 binding. Speculatively, any autoimmune or inflammatory condition that promotes a general disassembly of collagen in the ECM could initiate the same pathomechanism and promote connective tissue abnormalities, which then manifest through features of joint hypermobility, skin hyperextensibility, and tissue fragility.
The second stage of the pathomechanism involves the integrin switch itself, which is a shared feature of the main EDS/HSD subtypes (Zoppi et al., 2018b). Following the failed collagen-α2β1 interaction, a downregulation of the α2β1 integrin itself occurs, which is accompanied by a downregulation of the fibronectin receptor α5β1, and an upregulation of the alternative fibronectin receptor αvβ3. It is possible that fibroblasts are able to sense the compromised collagen-ECM via the α2β1 integrin, and recognize a potential risk of anoikis, a specific form of cell death that occurs upon cell detachment from the ECM. To avoid such a fate, it is possible that fibroblasts respond by switching their integrin profile to promote cell adhesion to other ECM ligands, such as fibronectin, in order to maintain some form of attachment to the ECM and survive. Indeed, this notion is supported by apoptosis assays performed in cEDS and vEDS fibroblasts, where the αvβ3 integrin was specifically shown to rescue EDS cells from anoikis (Zoppi et al., 2008), indicating that this integrin switch is indeed, a critical cell survival response. The importance of cell-ECM adhesion, however, extends beyond the transmission of cell survival signals, and we have described in this review its importance in maintaining connective tissue integrity. The integrin switch, therefore, may form a key stage of this pathomechanism, due to the consistent nature of this finding across all of the main EDS/HSD subtypes, as well as the critical consequences in weakening cell-ECM adhesion and its contribution to overall fibroblast dysfunction.
The final stage of this pathomechanism is fibroblast dysfunction itself, which ultimately mediates the abnormal properties of the connective tissue. The critical features of this dysfunction are impairments in cell adhesion and cytoskeleton dynamics. As already described, impaired cell-ECM adhesion weakens the integrity of the connective tissue, however, it also prevents fibroblasts from mediating their cytoskeletal forces to the surrounding environment and implement the tensional homeostasis mechanism. The integrin switch itself, has also been shown to directly affect cytoskeleton dynamics, impairing the cytoskeletal forces that are transferred through the weakened cell-ECM adhesions. The integrin switch may also cause abnormalities in the mechanosensitivity of fibroblasts, which may prevent accurate interpretations of the surrounding ECM properties, and hence, may produce an inappropriate response by fibroblasts. As such, key connective tissue properties such as viscoelasticity and integrity are all affected consequent the integrin switch and may contribute to the pathogenesis of EDS/HSD.
The contribution of this specific pathomechanism to the overall EDS/HSD phenotype could potentially be significant. It has to be highlighted that the current TEM analyses of collagen fibril structure and organization in EDS/HSD are highly inconsistent, and fail to associate with any EDS subtype, specific connective tissue abnormality, or severity of the manifestation (Angwin et al., 2020). Indeed, even the most significant abnormality associated with EDS/HSD, in the form of collagen flowers in cEDS, is one that cannot explain the disproportionate changes in skin viscoelasticity seen in this subtype, and is also one that is found in unaffected individuals (Hermanns-Lê et al., 2012). To our knowledge, the studies into fibroblast mechanics in dermatosparaxis animals, are the only non-genetic findings to have correlated with severity of connective tissue manifestation (Delvoye et al., 1983; Ramshaw et al., 1991), yet the possible significance of this appears to have been overlooked.
These findings suggest that what unites and defines the EDS/HSD subtypes may not be defects in the structure and/or organization of collagen itself, but in cell adhesion to collagen. If such a notion is correct, this may provide an ideal basis for the development of a universal or non-specific diagnostic test that truly captures all EDS/HSD subtypes, irrespective of the principal underlying defect. An assessment for membrane-bound collagen for example, would highly indicate that cell-ECM adhesion has switched in vivo from collagen to other ECM ligands, reflecting the occurrence of the initial stages of this pathomechanism in patients presenting with connective tissue ailments. This is of importance, since the most common forms of hEDS and HSD, are currently without any diagnostic biomarker, and it is likely that further unidentified subtypes remain. This may also have implications for other related disorders such as fibromyalgia, and Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS), which demonstrate significant overlaps with hypermobility and connective tissue abnormalities, though the association is poorly understood (Bragée et al., 2020; Eccles et al., 2020). It is plausible therefore, that similar pathomechanisms involving fibroblast dysfunction may be contributing to the pathogenesis of these related disorders, which could be explored through their associations with membrane-bound collagen.
Of additional importance is to determine if this pathomechanism contributes to arterial fragility in vEDS, a lethal subtype of EDS with a median life expectancy of 40 years (Eagleton, 2016). Death commonly occurs due to complications associated with spontaneous vascular or hollow organ ruptures, however, the development of these are also poorly understood (Eagleton, 2016). If such a pathomechanism demonstrates itself to be significant, the development of a therapeutic agent that promotes arterial integrity via these processes could provide hope for a number of patients.
SM and DK conceived this manuscript. SM wrote the initial draft of the manuscript. DK reviewed, added, and edited the content of the manuscript. Both authors contributed to and approved the final manuscript for submission.
SM was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) and University of Warwick funded Midlands Integrative Biosciences Training Partnership (MIBTP) (grant number BB/M01116X/1). DK was supported by the Wellcome-Warwick Quantitative Biomedical Program (RMRCB0058).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank the Midlands Integrative Biosciences Training Partnership (MIBTP), Biotechnology and Biological Sciences Research Council (BBSRC), Dr. Emma Reinhold, and Dr. Gemma Pearce for supporting our EDS/HSD research.
Angwin, C., Brady, A. F., Colombi, M., Ferguson, D. J. P., Pollitt, R., Pope, F. M., et al. (2019). Absence of Collagen Flowers on Electron Microscopy and Identification of (Likely) Pathogenic COL5A1 Variants in Two Patients. Genes 10, 762. doi: 10.3390/genes10100762
Angwin, C., Ghali, N., Baker, D., Brady, A. F., Pope, F. M., Vandersteen, A., et al. (2020). Electron microscopy in the diagnosis of Ehlers-Danlos syndromes: correlation with clinical and genetic investigations. The British journal of dermatology 182, 698–707. doi: 10.1111/bjd.18165
Barczyk, M., Carracedo, S., and Gullberg, D. (2010). Integrins. Cell and tissue research 339, 269–280.
Bavinton, J. H., Peters, D. E., and Ramshaw, J. A. (1985). A morphologic study of a mild form of ovine dermatosparaxis. The Journal of investigative dermatology 84, 391–395. doi: 10.1111/1523-1747.ep12265475
Beighton, P., de Paepe, A., Danks, D., Finidori, G., Gedde-Dahl, T., Goodman, R., et al. (1988). ‘International Nosology of Heritable Disorders of Connective Tissue, Berlin, 1986. American Journal of Medical Genetics 29, 581–594.
Beighton, P., De Paepe, A., Steinmann, B., Tsipouras, P., and Wenstrup, R. J. (1998). Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). American Journal of Medical Genetics 77, 31–37. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o
Bisson, M. A., Beckett, K. S., McGrouther, D. A., Grobbelaar, A. O., and Mudera, V. (2009). Transforming growth factor-beta1 stimulation enhances Dupuytren’s fibroblast contraction in response to uniaxial mechanical load within a 3-dimensional collagen gel. The Journal of hand surgery 34, 1102–1110. doi: 10.1016/j.jhsa.2009.02.008
Bisson, M. A., Mudera, V., McGrouther, D. A., and Grobbelaar, A. O. (2004). The contractile properties and responses to tensional loading of Dupuytren’s disease–derived fibroblasts are altered: a cause of the contracture? Plastic and reconstructive surgery 113, 611–621. doi: 10.1097/01.prs.0000101527.76293.f1
Blackburn, P., Xu, Z., Tumelty, K., Zhao, R., Monis, W., Harris, K., et al. (2018). Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome. American journal of human genetics 102, 696–705. doi: 10.1016/j.ajhg.2018.02.018
Bowen, J. M., Glenda, J. S., Nigel, P. B., Marina, C., Mark, E. L., Fransiska, M., et al. (2017). ‘Ehlers–Danlos syndrome, classical type’. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 175, 27–39.
Bragée, B., Michos, A., Drum, B., Fahlgren, M., Szulkin, R., and Bertilson, B. C. (2020). Signs of Intracranial Hypertension, Hypermobility, and Craniocervical Obstructions in Patients With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Frontiers in neurology 11:828. doi: 10.3389/fneur.2020.00828
Brown, R. A., Prajapati, R., McGrouther, D. A., Yannas, I. V., and Eastwood, M. (1998). Tensional homeostasis in dermal fibroblasts: mechanical responses to mechanical loading in three-dimensional substrates. Journal of cellular physiology 175, 323–332. doi: 10.1002/(sici)1097-4652(199806)175:3<323::aid-jcp10>3.0.co;2-6
Carley, M. E., and Schaffer, J. (2000). Urinary incontinence and pelvic organ prolapse in women with Marfan or Ehlers Danlos syndrome. American journal of obstetrics and gynecology 182, 1021–1023. doi: 10.1067/mob.2000.105410
Celli, M., Iacovino, C., Febbo, A., Lotti, L. V., Miraglia, E., Celli, L., et al. (2020). Ultrastructure study of skin fibroblasts in patients with Ehlers-Danlos Syndrome (EDS): preliminary results. La Clinica terapeutica 171, e431–e436.
Chiarelli, N., Ritelli, M., Zoppi, N., and Colombi, M. (2019). Cellular and Molecular Mechanisms in the Pathogenesis of Classical, Vascular, and Hypermobile Ehlers-Danlos Syndromes. Genes 10, 609. doi: 10.3390/genes10080609
Colige, A., Al Sieron, S. W., Li, U., Schwarze, E., Petty, W., Wertelecki, W., et al. (1999). Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. American Journal of Human Genetics 65, 308–317. doi: 10.1086/302504
Copetti, M., Morlino, S., Colombi, M., Grammatico, P., Fontana, A., and Castori, M. (2019). Severity classes in adults with hypermobile Ehlers-Danlos syndrome/hypermobility spectrum disorders: a pilot study of 105 Italian patients. Rheumatology 58, 1722–1730.
Dallon, J., and Ehrlich, H. (2008). A review of fibroblast-populated collagen lattices. Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 16, 472–479. doi: 10.1111/j.1524-475x.2008.00392.x
Danen, E. H., Sonneveld, P., Brakebusch, C., Fassler, R., and Sonnenberg, A. (2002). The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. The Journal of cell biology 159, 1071–1086. doi: 10.1083/jcb.200205014
Danen, E. H., van Rheenen, J., Franken, W., Huveneers, S., Sonneveld, P., Jalink, K., et al. (2005). Integrins control motile strategy through a Rho-cofilin pathway. The Journal of cell biology 169, 515–526. doi: 10.1083/jcb.200412081
de Moraes, A. M., Cintra, M. L., Sampaio, A., de, S., Sotto, M. N., and Sesso, A. (2000). The ultrastructural and histophotometric study of elastic and collagen fibers in type II Ehlers-Danlos syndrome and subclinical forms. Ultrastructural pathology 24, 129–134. doi: 10.1080/01913120050132859
Delvoye, P., Mauch, C., Krieg, T., and Lapiere, C. M. (1986). Contraction of collagen lattices by fibroblasts from patients and animals with heritable disorders of connective tissue. The British journal of dermatology 115, 139–146. doi: 10.1111/j.1365-2133.1986.tb05709.x
Delvoye, P., Nusgens, B., and Lapière, C. M. (1983). The capacity of retracting a collagen matrix is lost by dermatosparactic skin fibroblasts. The Journal of investigative dermatology 81, 267–270. doi: 10.1111/1523-1747.ep12518288
Delvoye, P., Wiliquet, P., Levêque, J. L., Nusgens, B. V., and Lapière, C. M. (1991). Measurement of mechanical forces generated by skin fibroblasts embedded in a three-dimensional collagen gel. The Journal of investigative dermatology 97, 898–902. doi: 10.1111/1523-1747.ep12491651
Demmler, J. C., Mark, D. A., Reinhold, E. J., Choy, E., Lyons, R. A., and Brophy, S. T. (2019). Diagnosed prevalence of Ehlers-Danlos syndrome and hypermobility spectrum disorder in Wales, UK: a national electronic cohort study and case–control comparison. BMJ 9, 58961.
D’hondt, S., Guillemyn, B., Syx, D., Symoens, S., De Rycke, R., Vanhoutte, L., et al. (2018). Type III collagen affects dermal and vascular collagen fibrillogenesis and tissue integrity in a mutant Col3a1 transgenic mouse model. Matrix biology : journal of the International Society for Matrix Biology 70, 72–83. doi: 10.1016/j.matbio.2018.03.008
Eagleton, M. J. (2016). Arterial complications of vascular Ehlers-Danlos syndrome. Journal of vascular surgery 64, 1869–1880. doi: 10.1016/j.jvs.2016.06.120
Eastwood, M., McGrouther, D. A., and Brown, R. A. (1994). A culture force monitor for measurement of contraction forces generated in human dermal fibroblast cultures: evidence for cell-matrix mechanical signalling. Biochimica et biophysica acta 1201, 186–192. doi: 10.1016/0304-4165(94)90040-x
Eastwood, M., Porter, R., Khan, U., McGrouther, G., and Brown, R. (1996). Quantitative analysis of collagen gel contractile forces generated by dermal fibroblasts and the relationship to cell morphology. Journal of cellular physiology 166, 33–42. doi: 10.1002/(sici)1097-4652(199601)166:1<33::aid-jcp4>3.0.co;2-h
Eccles, J. A., Thompson, B., Themelis, K., Amato, M., Stocks, R., Pound, A., et al. (2020). Beyond Bones - The Relevance of Variants of Connective Tissue (Hypermobility) to Fibromyalgia, ME/CFS and Controversies Surrounding Diagnostic Classification: An Observational Study. Clin Med (Lond) 21, 53–58. doi: 10.7861/clinmed.2020-0743
Ezra, D. G., Ellis, J. S., Beaconsfield, M., Collin, R., and Bailly, M. (2010). Changes in fibroblast mechanostat set point and mechanosensitivity: an adaptive response to mechanical stress in floppy eyelid syndrome. Investigative ophthalmology & visual science 51, 3853–3863. doi: 10.1167/iovs.09-4724
Freyman, T. M., Yannas, I. V., Yokoo, R., and Gibson, L. J. (2002). Fibroblast contractile force is independent of the stiffness which resists the contraction. Experimental cell research 272, 153–162. doi: 10.1006/excr.2001.5408
Giannopoulos, A., Svensson, R. B., Heinemeier, K. M., Schjerling, P., Kadler, K. E., Holmes, D. F., et al. (2018). Cellular homeostatic tension and force transmission measured in human engineered tendon. Journal of Biomechanics 78, 161–165. doi: 10.1016/j.jbiomech.2018.07.032
Hausser, I., and Anton-Lamprecht, I. (1994). Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Human genetics 93, 394–407.
Hermanns-Lê, T., and Piérard, G. E. (2006). Collagen fibril arabesques in connective tissue disorders. American Journal of Clinical Dermatology 7, 323–326. doi: 10.2165/00128071-200607050-00006
Hermanns-Lê, T., Reginster, M.-A., Piérard-Franchimont, C., Delvenne, P., Piérard, G. E., and Manicourt, D. (2012). Dermal ultrastructure in low Beighton score members of 17 families with hypermobile-type Ehlers-Danlos syndrome. Journal of Biomedicine & Biotechnology 2012, 878107.
Holbrook, K. A., and Byers, P. H. (1982). ‘Structural abnormalities in the dermal collagen and elastic matrix from the skin of patients with inherited connective tissue disorders’. The Journal of investigative dermatology 79(Suppl.), 1.
Jiang, G., Huang, A. H., Cai, Y., Tanase, M., and Sheetz, M. P. (2006). Rigidity sensing at the leading edge through alphavbeta3 integrins and RPTPalpha. Biophysical Journal 90, 1804–1809. doi: 10.1529/biophysj.105.072462
Kanchanawong, P., Shtengel, G., Pasapera, A. M., Ramko, E. B., Davidson, M. W., Hess, H. F., et al. (2010). ‘Nanoscale architecture of integrin-based cell adhesions’. Nature 468, 580–584. doi: 10.1038/nature09621
Kelynack, K. J., Hewitson, T. D., Nicholls, K. M., Darby, I. A., and Becker, G. J. (2000). Human renal fibroblast contraction of collagen I lattices is an integrin-mediated process. Nephrology, dialysis, transplantation 15, 1766–1772. doi: 10.1093/ndt/15.11.1766
Klein, C. E., Dressel, D., Steinmayer, T., Mauch, C., Eckes, B., Krieg, T., et al. (1991). Integrin alpha 2 beta 1 is upregulated in fibroblasts and highly aggressive melanoma cells in three-dimensional collagen lattices and mediates the reorganization of collagen I fibrils. The Journal of cell biology 115, 1427–1436. doi: 10.1083/jcb.115.5.1427
Kobayasi, T. (2004). Abnormality of dermal collagen fibrils in Ehlers Danlos syndrome. Anticipation of the abnormality for the inherited hypermobile disorders. European journal of dermatology : EJD 14, 221–229.
Kolodney, M. S., and Wysolmerski, R. B. (1992). Isometric contraction by fibroblasts and endothelial cells in tissue culture: a quantitative study. The Journal of cell biology 117, 73–82. doi: 10.1083/jcb.117.1.73
Kong, F., García, A. J., Mould, A. P., Humphries, M. J., and Zhu, C. (2009). Demonstration of catch bonds between an integrin and its ligand. The Journal of cell biology 185, 1275–1284. doi: 10.1083/jcb.200810002
Lammers, K., Lince, S. L., Spath, M. A., van Kempen, L. C., Hendriks, J. C., Vierhout, M. E., et al. (2012). Pelvic organ prolapse and collagen-associated disorders. International urogynecology journal 23, 313–319.
Langevin, H. M., Bouffard, N. A., Badger, G. J., Iatridis, J. C., and Howe, A. K. (2005). Dynamic fibroblast cytoskeletal response to subcutaneous tissue stretch ex vivo and in vivo. American journal of physiology. Cell physiology 288, C747–C756.
Langevin, H. M., Bouffard, N. A., Fox, J. R., Palmer, B. M., Wu, J., Iatridis, J. C., et al. (2011). ‘Fibroblast Cytoskeletal Remodeling Contributes to Connective Tissue Tension’. J Cell Physiol 226, 1166–1175. doi: 10.1002/jcp.22442
Lawson, C. D., and Burridge, K. (2014). The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 5, e27958. doi: 10.4161/sgtp.27958
Leistritz, D. F., Pepin, M. G., Schwarze, U., and Byers, P. H. (2011). COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genetics in medicine : official journal of the American College of Medical Genetics 13, 717–722. doi: 10.1097/gim.0b013e3182180c89
Liu, X., Wu, H., Byrne, M., Krane, S., and Jaenisch, R. (1997). Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proceedings of the National Academy of Sciences of the United States of America 94, 1852–1856. doi: 10.1073/pnas.94.5.1852
Malfait, F., Francomano, C., Byers, P., Belmont, J., Berglund, B., Black, J., et al. (2017). ‘The 2017 international classification of the Ehlers-Danlos syndromes’. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics 175, 8–26.
Mauch, C., Aumailley, M., Paye, M., Lapière, C. M., Timpl, R., and Krieg, T. (1986). Defective attachment of dermatosparactic fibroblasts to collagens I and IV. Experimental cell research 163, 294–300. doi: 10.1016/0014-4827(86)90060-1
Mauch, C., van der Mark, K., Helle, O., Mollenhauer, J., Pfäffle, M., and Krieg, T. (1988). A defective cell surface collagen-binding protein in dermatosparactic sheep fibroblasts. The Journal of cell biology 106, 205–211. doi: 10.1083/jcb.106.1.205
Miao, H., Li, S., Hu, Y. L., Yuan, S., Zhao, Y., Chen, B. P., et al. (2002). Differential regulation of Rho GTPases by beta1 and beta3 integrins: the role of an extracellular domain of integrin in intracellular signaling. Journal of cell science 115, 2199–2206.
Mizutani, T., Haga, H., and Kawabata, K. (2004). Cellular stiffness response to external deformation: tensional homeostasis in a single fibroblast. Cell motility and the cytoskeleton 59, 242–248. doi: 10.1002/cm.20037
Niland, S., Westerhausen, C., Schneider, S. W., Eckes, B., Schneider, M. F., and Eble, J. A. (2011). Biofunctionalization of a generic collagenous triple helix with the α2β1 integrin binding site allows molecular force measurements. The international journal of biochemistry & cell biology 43, 721–731. doi: 10.1016/j.biocel.2011.01.013
Ong, K. T., Plauchu, H., Peyrol, S., Roux, E., Errazuriz, E., Khau Van Kien, P., et al. (2012). Ultrastructural scoring of skin biopsies for diagnosis of vascular Ehlers-Danlos syndrome. Virchows Archiv : an international journal of pathology 460, 637–649. doi: 10.1007/s00428-012-1233-z
Parapia, L. A., and Jackson, C. (2008). Ehlers-Danlos syndrome–a historical review. British Journal of Haematology 141, 32–35.
Parsons, J. T., Horwitz, A. R., and Schwartz, M. A. (2010). Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nature Reviews. Molecular Cell Biology 11, 633–643. doi: 10.1038/nrm2957
Pepin, M., Schwarze, U., Superti-Furga, A., and Byers, P. H. (2000). Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. The New England Journal of Medicine 342, 673–680.
Pollard, T. D., and Borisy, G. G. (2003). Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465.
Proske, S., Hartschuh, W., Enk, A., and Hausser, I. (2006). Ehlers-Danlos syndrome - 20 years experience with diagnosis and classification at the university skin clinic of Heidelberg. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG 4, 308–318.
Ramshaw, J. A., Mitrangas, K., and Bateman, J. F. (1991). Heterogeneity in dermatosparaxis is shown by contraction of collagen gels. Connective tissue research 25, 295–300.
Ridge, K., Shumaker, D., Robert, A., Hookway, C., Gelfand, V., Janmey, P., et al. (2016). Methods for Determining the Cellular Functions of Vimentin Intermediate Filaments. Methods in enzymology 568, 389–426.
Ridley, A. J. (2015). Rho GTPase signalling in cell migration. Current opinion in cell biology 36, 103–112.
Roca-Cusachs, P., Gauthier, N. C., Del Rio, A., and Sheetz, M. P. (2009). Clustering of alpha(5)beta(1) integrins determines adhesion strength whereas alpha(v)beta(3) and talin enable mechanotransduction. Proceedings of the National Academy of Sciences of the United States of America 106, 16245–16250.
Ruiz-Zapata, A. M., Kerkhof, M. H., Zandieh-Doulabi, B., Brölmann, H. A., Smit, T. H., and Helder, M. N. (2013). Fibroblasts from women with pelvic organ prolapse show differential mechanoresponses depending on surface substrates. International urogynecology journal 24, 1567–1575.
Schiller, H. B., Hermann, M. R., Polleux, J., Vignaud, T., Zanivan, S., Friedel, C. C., et al. (2013). β1- and αv-class integrins cooperate to regulate myosin II during rigidity sensing of fibronectin-based microenvironments. Nature cell biology 15, 625–636.
Schiro, J. A., Chan, B. M., Roswit, W. T., Kassner, P. D., Pentland, A. P., Hemler, M. E., et al. (1991). Integrin alpha 2 beta 1 (VLA-2) mediates reorganization and contraction of collagen matrices by human cells. Cell 67, 403–410.
Schwartz, M. A. (2010). Integrins and extracellular matrix in mechanotransduction. Cold Spring Harbor perspectives in biology 2, 5264.
Smith, L. T., Schwarze, U., Goldstein, J., and Byers, P. H. (1997). Mutations in the COL3A1 gene result in the Ehlers-Danlos syndrome type IV and alterations in the size and distribution of the major collagen fibrils of the dermis. The Journal of investigative dermatology 108, 241–247.
Swierczewski, R., Hedley, J., and Redfern, C. P. (2016). High-resolution micromechanical measurement in real time of forces exerted by living cells. Cell adhesion & migration 10, 322–330.
Symoens, S., Syx, D., Malfait, F., Callewaert, B., De Backer, J., Vanakker, O., et al. (2012). Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Human mutation 33, 1485–1493.
Theocharidis, G., and Connelly, J. (2019). Minor collagens of the skin with not so minor functions. Journal of anatomy 235, 418–429.
Tinkle, B. T., Bird, H. A., Grahame, R., Lavallee, M., Levy, H. P., and Sillence, D. (2009). The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). American Journal of Medical Genetics. Part A 149A, 2368–2370.
Tomasek, J. J., Gabbiani, G., Hinz, B., Chaponnier, C., and Brown, R. A. (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nature Reviews. Molecular Cell Biology 3, 349–363.
Viglio, S., Zoppi, N., Sangalli, A., Gallanti, A., Barlati, S., Mottes, M., et al. (2008). Rescue of Migratory Defects of Ehlers-Danlos Syndrome Fibroblasts in Vitro by Type V Collagen but Not Insulin-Like Binding protein-1. The Journal of investigative dermatology 128, 1915–1919.
Vogel, A., Holbrook, K. A., Steinmann, B., Gitzelmann, R., and Byers, P. H. (1979). Abnormal collagen fibril structure in the gravis form (type I) of Ehlers-Danlos syndrome. Laboratory Investigation; a Journal of Technical Methods and Pathology 40, 201–206.
Wang, C., Brisson, B. K., Terajima, M., Li, Q., Hoxha, K., Han, B., et al. (2020). Type III collagen is a key regulator of the collagen fibrillar structure and biomechanics of articular cartilage and meniscus. Matrix biology : journal of the International Society for Matrix Biology 20, 85–86.
Wang, S., Zhang, Z., Lü, D., and Xu, Q. (2015). Effects of mechanical stretching on the morphology and cytoskeleton of vaginal fibroblasts from women with pelvic organ prolapse. International journal of molecular sciences 16, 9406–9419.
Webster, K. D., Ng, W. P., and Fletcher, D. A. (2014). Tensional homeostasis in single fibroblasts. Biophysical Journal 107, 146–155.
Wenstrup, R. J., Florer, J. B., Brunskill, E. W., Bell, S. M., Chervoneva, I., and Birk, D. E. (2004a). Type V collagen controls the initiation of collagen fibril assembly. The Journal of biological chemistry 279, 53331–53337.
Wenstrup, R. J., Florer, J. B., Cole, W. G., Willing, M. C., and Birk, D. E. (2004b). Reduced type I collagen utilization: a pathogenic mechanism in COL5A1 haplo-insufficient Ehlers-Danlos syndrome. Journal of cellular biochemistry 92, 113–124.
Zollinger, A. J., Xu, H., Figueiredo, J., Paredes, J., Seruca, R., Stamenović, D., et al. (2018). Dependence of Tensional Homeostasis on Cell Type and on Cell-Cell Interactions. Cellular and molecular bioengineering 11, 175–184.
Zoppi, N., Barlati, S., and Colombi, M. (2008). FAK-independent alphavbeta3 integrin-EGFR Complexes Rescue From Anoikis Matrix-Defective Fibroblasts. Biochimica et biophysica acta 1783, 1177–1188.
Zoppi, N., Chiarelli, N., Binetti, S., Ritelli, M., and Colombi, M. (2018a). ‘Dermal fibroblast-to-myofibroblast transition sustained by αvß3 integrin-ILK-Snail1/Slug signaling is a common feature for hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders’. Biochimica Et Biophysica Acta. Molecular Basis of Disease 1864, 1010–1023.
Zoppi, N., Chiarelli, N., Ritelli, M., and Colombi, M. (2018b). Multifaced Roles of the αvβ3 Integrin in Ehlers-Danlos and Arterial Tortuosity Syndromes’ Dermal Fibroblasts. International journal of molecular sciences 19, 982.
Zoppi, N., Gardella, R., De Paepe, A., Barlati, S., and Colombi, M. (2004). Human Fibroblasts With Mutations in COL5A1 and COL3A1 Genes Do Not Organize Collagens and Fibronectin in the Extracellular Matrix, Down-Regulate alpha2beta1 Integrin, and Recruit alphavbeta3 Instead of alpha5beta1 Integrin. The Journal of biological chemistry 279, 18157–18168.
Keywords: Ehlers-Danlos syndrome, hypermobility spectrum disorder, fibroblasts, integrins, cytoskeleton, mechanobiology
Citation: Malek S and Köster DV (2021) The Role of Cell Adhesion and Cytoskeleton Dynamics in the Pathogenesis of the Ehlers-Danlos Syndromes and Hypermobility Spectrum Disorders. Front. Cell Dev. Biol. 9:649082. doi: 10.3389/fcell.2021.649082
Received: 03 January 2021; Accepted: 22 March 2021;
Published: 21 April 2021.
Edited by:
Ting Gang Chew, Zhejiang University-University of Edinburgh Institute, ChinaReviewed by:
Yaming Jiu, Institut Pasteur of Shanghai (CAS), ChinaCopyright © 2021 Malek and Köster. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabeeha Malek, cy5tYWxla0B3YXJ3aWNrLmFjLnVr; Darius V. Köster, ZC5rb2VzdGVyQHdhcndpY2suYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.