
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 18 June 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.645501
This article is part of the Research TopicGenetic Mutations Associated with Ocular DiseasesView all 19 articles
 Yang Liu1†
Yang Liu1† Jin-Jin Zhang2†
Jin-Jin Zhang2† Shun-Yu Piao3
Shun-Yu Piao3 Ren-Juan Shen4
Ren-Juan Shen4 Ya Ma4Zhong-Qi Xue5Wen Zhang3
Ya Ma4Zhong-Qi Xue5Wen Zhang3 Juan Liu1*
Juan Liu1* Zi-Bing Jin4*
Zi-Bing Jin4* Wen-Juan Zhuang3*
Wen-Juan Zhuang3*High myopia (HM) is one of the leading causes of visual impairment worldwide. In order to expand the myopia gene spectrum in the Chinese population, we investigated genetic mutations in a cohort of 27 families with HM from Northwest China by using whole-exome sequencing (WES). Genetic variations were filtered using bioinformatics tools and cosegregation analysis. A total of 201 candidate mutations were detected, and 139 were cosegregated with the disease in the families. Multistep analysis revealed four missense variants in four unrelated families, including c.904C>T (p.R302C) in CSMD1, c.860G>A (p.R287H) in PARP8, c.G848A (p.G283D) in ADAMTSL1, and c.686A>G (p.H229R) in FNDC3B. These mutations were rare or absent in the Exome Aggregation Consortium (ExAC), 1000 Genomes Project, and Genome Aggregation Database (gnomAD), indicating that they are new candidate disease-causing genes. Our findings not only expand the myopia gene spectrum but also provide reference information for further genetic study of heritable HM.
Myopia, also known as shortsightedness, is globally increasing in prevalence. Approximately 108 million people are currently affected, and myopia is expected to become the leading cause of blindness across the globe (Bourne et al., 2013). In East Asian countries, the prevalence of myopia has been reported to be twice as high as in western countries (Pan et al., 2015).
High myopia (HM), which is the extreme form of myopia, is diagnosed in cases with refractive error worse than or equal to −6 diopters (D) and/or ocular axial length (AL) >26.00 mm (Percival, 1987; Young et al., 2007). Recently meta-analyses have estimated that the prevalence of myopia will rise to 49.8% in 2050 and that the prevalence of HM will increase to 9.8% (Holden et al., 2016). The complications associated with HM, such as cataract, glaucoma, retinal detachment, chorioretinal degeneration, and choroidal neovascularization, may lead to severe visual impairment and blindness (Morgan et al., 2012; Tham et al., 2018).
Both environmental and genetic factors are known to contribute to HM (Morgan et al., 2012).
Familial aggregation and twin studies have demonstrated that genetic factors play critical roles in the development of HM (Katz et al., 1997; Hammond et al., 2001). To date, 25 loci (MYP1–MYP3, MYP5–MYP26) associated with HM have been discovered via genome-wide association studies (GWAS), family-based linkage analyses, and whole-exome sequencing (WES) and documented in the Online Mendelian Inheritance in Man (OMIM) (Zhang, 2015; Fan et al., 2016; Cai et al., 2019a). However, only a limited number of causative genes have been identified (Cai et al., 2019a). WES has identified several genes responsible for HM, including two X-linked genes, OPN1LW (Li et al., 2015) and ARR3 (Xiao et al., 2016); six autosomal dominant genes, ZNF644 (Shi et al., 2011a), SCO2 (Tran-Viet et al., 2013), SLC39A5 (Guo et al., 2014), P4HA2 (Guo et al., 2015), BSG (Jin et al., 2017), and CCDC111 (Zhao et al., 2013); and three autosomal recessive genes, LEPREL1 (Mordechai et al., 2011), LRPAP1 (Aldahmesh et al., 2013), and CTSH (Jiang et al., 2015). Nevertheless, these genes explain the pathogenesis of HM in a limited number of patients (Kloss et al., 2017). Therefore, numerous HM-related genes surely remain to be discovered. In this study, we identified candidate mutations and genes in 27 non-syndromic families with HM from Northwest China.
Twenty-seven families with HM were recruited in this study. The study was performed in accordance with the tenets of the Declaration of Helsinki. Written informed consent was obtained from the participants or their statutory guardians, with approval from the institutional review board of the People’s Hospital of Ningxia Hui Autonomous Region, Third Clinical Medical College of Ningxia Medical University. Refractive error and AL of the patients in each family were concisely recorded in Table 1, a collection of the upper and lower limits of clinical data of all patients in each of the 27 families. The pedigree and clinical information of the 27 families were integrated in Supplementary Figure 1. Genomic DNAs were extracted from peripheral blood samples.
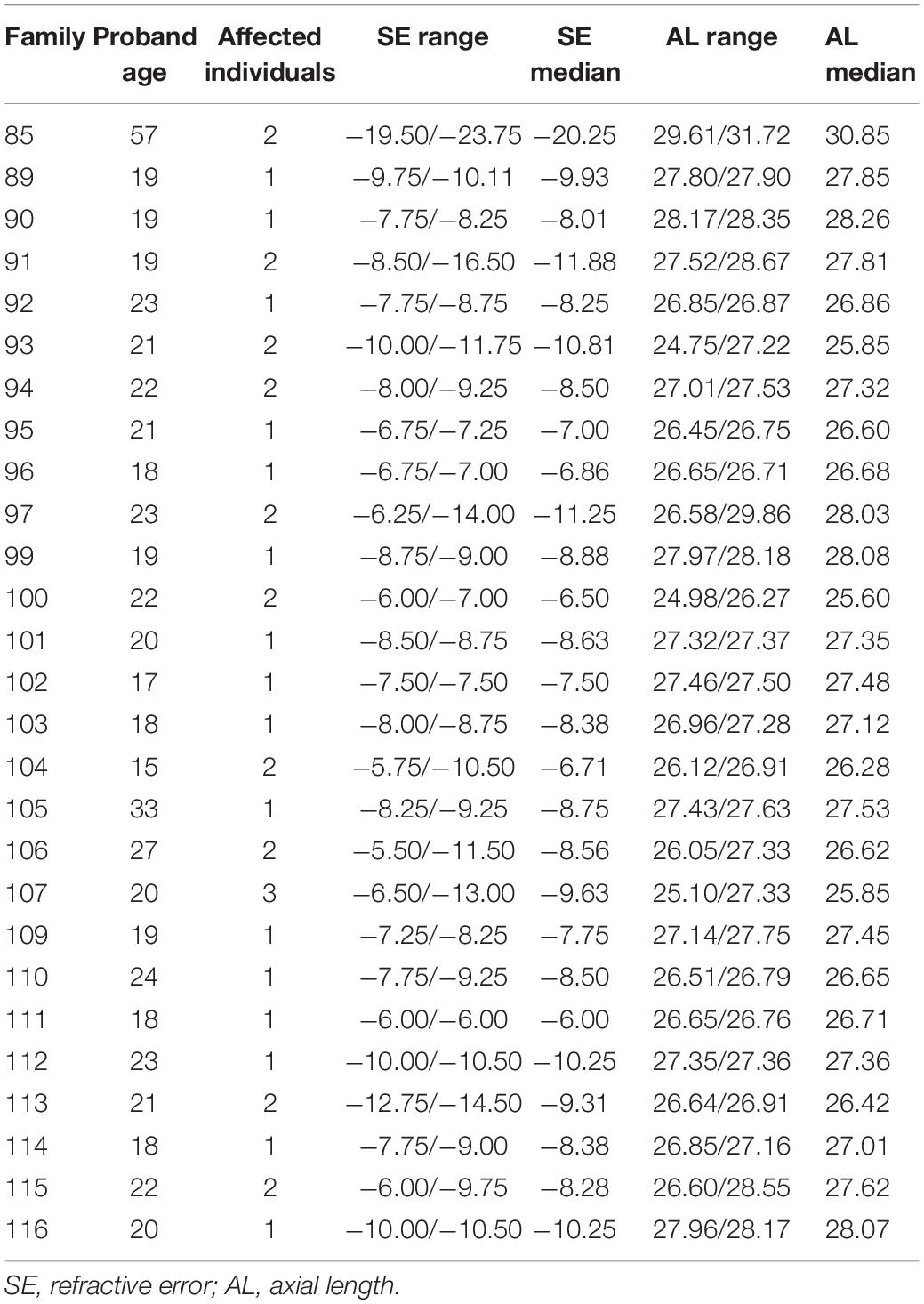
Table 1. Clinical data from 27 families with high myopia.
All 27 families included in this study were from Ningxia Hui Autonomous Region.
Patients were recruited according to the following inclusion criteria: (1) refractive error worse than or equal to −6 D and/or AL >26.00 mm; and (2) no other known ocular diseases or systemic disorders.
Whole-exome sequencing was performed as previously described with the Exome Enrichment V5 Kit (Agilent Technologies, United States) (Jin et al., 2018; Cai et al., 2019b; Zhou et al., 2020). DNA fragments were sequenced on Illumina HiSeq 2000 Analyzers (90 cycles per read). The Illumina libraries were prepared and generated using a sequencing platform (Hiseq2000; Illumina, Inc.), according to the manufacturer’s instructions. Local realignments, quality control text, and variant calling were assembled using the Genome Analysis Toolkit (GATK). All sequencing reads were mapped against a human reference genome (hg19/GRCH37) with BWA-MEM software as described (Kumar et al., 2009). WES yielded a mean read depth of ∼30×. Median coverage of the targeted regions was >95%.
We focused on rare variants in ocular diseases and HM; the ocular disease genes were assembled from the databases: genes associated with the myopia phenotype term (HP:0000545) in Human Phenotype Ontology (HPO1) or ocular disease genes from RetNet database2 and OMIM3. The complete list of genes associated with myopia and ocular disease was shown in Supplementary Table 1.
The WES results from patients with HM were filtered as follows: (1) variants outside of the exonic splicing site predicted by the Berkeley Drosophila Genome Project4 were excluded; (2) synonymous variants were excluded without altering splice-site regions; (3) variants with minor allele frequencies (MAFs) greater than or equal to 0.01 in any of the three public population databases [Exome Aggregation Consortium (ExAC), 1000 Genomes Project (1000G), and Genome Aggregation Database (gnomAD)] were excluded; (4) variants that were not heterozygous in all AD families were excluded, as the variants that were not homozygous in all AR families. Meanwhile, de novo variants were also excluded, and these de novo variants were displayed in Supplementary Table 2. After these filtering steps, cosegregation analysis was performed for all family members. Finally, variant scores corresponding to the quality of biological and statistical evidence were assigned. Strong candidates were validated by Sanger sequencing (Figure 1).
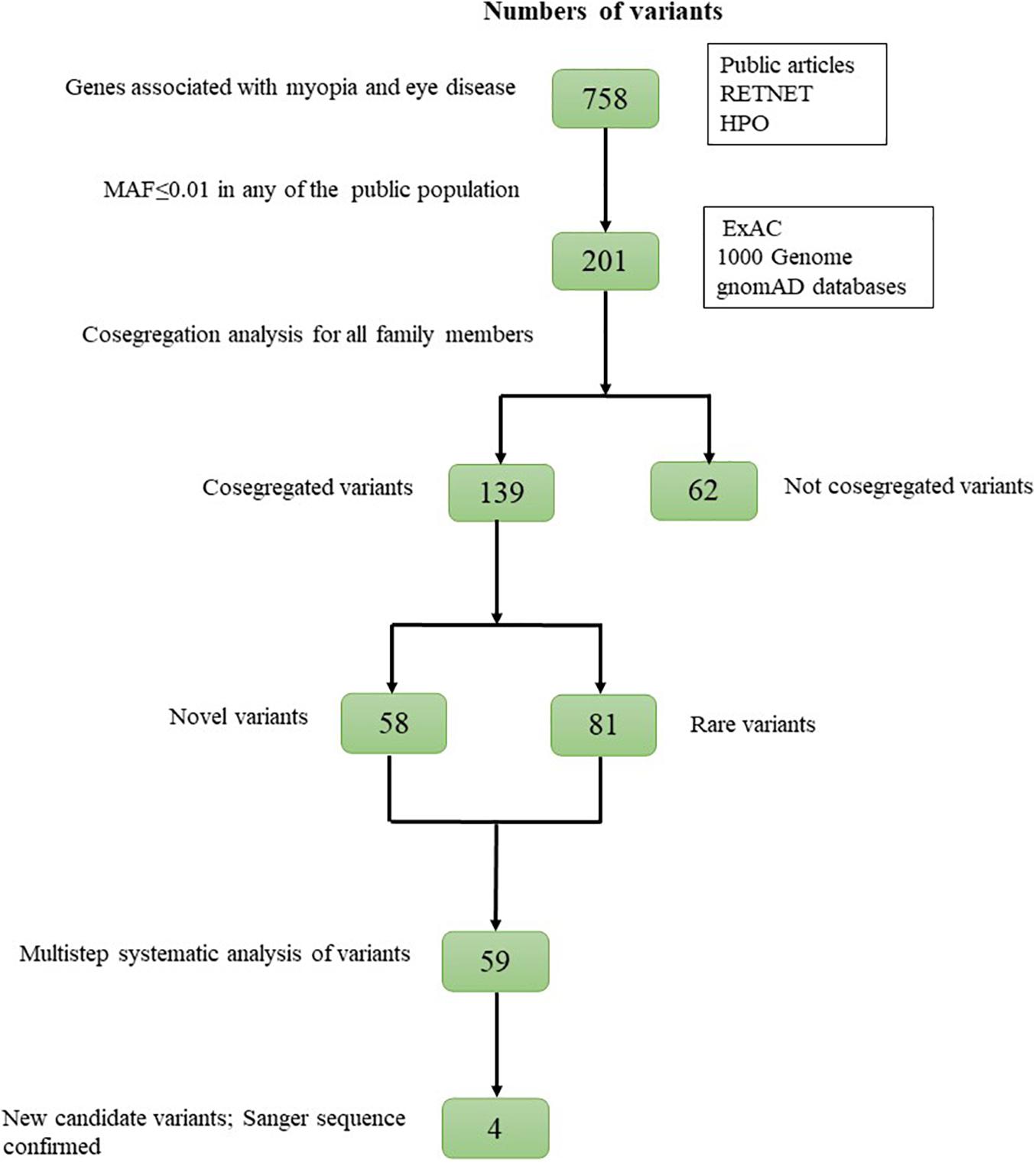
Figure 1. Overview of the variant filtering strategy.
All identified variants were assessed with the following tools and databases. The pathogenicity of gene mutations was predicted with 10 bioinformatics tools:
SIFT (Adzhubei et al., 2010)5, PolyPhen-2-HDIV, PolyPhen-2-HVAR (Schwarz et al., 2010)6, Mutation Taster (Desmet et al., 2009)7, LRT, Mutation Assessor, FATHMM, Radial SVM, LR, and DANN. Splicing mutations were analyzed with Human Splicing Finder software (Huang et al., 2015)8. Mutations with a MAF were evaluated with the ExAC9, 1000G (Bahcall, 2015) (1000G10), and gnomAD11. The RaptorX structure prediction web server12 was used to simulate the wild-type and mutant protein three-dimensional (3D) models. Changes in protein folding and crystal structure were visualized with PyMol software (version 1.5).
A total of 201 potential variants were identified in 27 families: 139 were well cosegregated with the disease in 15 families. Sixty-two variants not cosegregated were excluded (Figure 2). Each variant was reported at a frequency of less than 1 in 100 alleles in the ExAC, 1000G, and gnomAD. For the 139 genes that cosegregated, none of the variants were identified in unaffected family members (refractive error less than 2.00 D). Fifty-eight of the 139 variants were novel changes that were unreported in public databases (Supplementary Table 3). Eighty-one of the 139 variants were rare changes (less than 1 in 1,000 alleles in any population) (Supplementary Table 4). Among 139 variants, 59 had been presented in 10 pathogenicity prediction tools with biological relevance (Supplementary Figure 2).
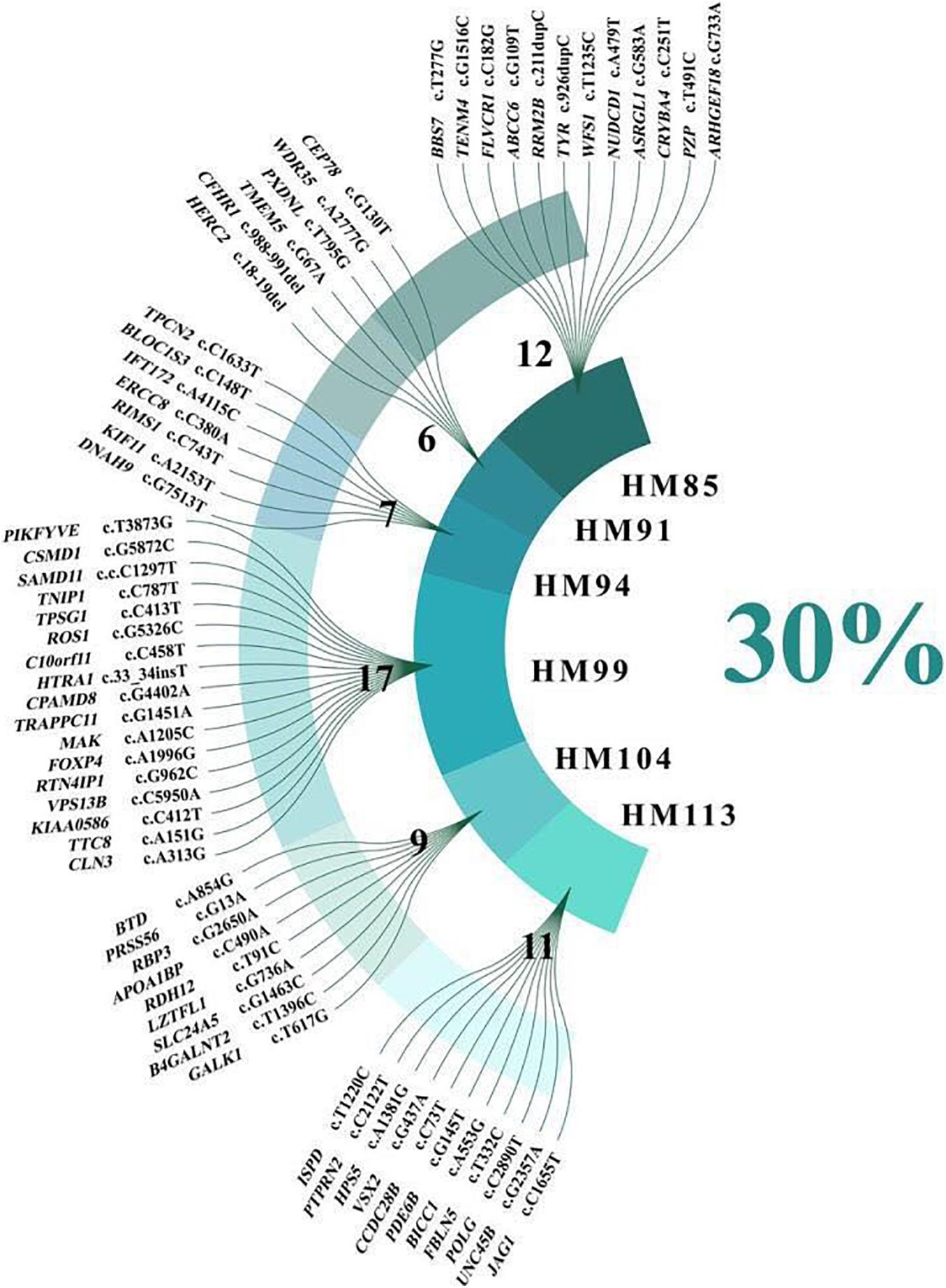
Figure 2. Sixty-two variants related to high myopia and ocular disease that were not cosegregated with high myopia.
The 20 variants with the highest scores are shown (Figure 3). Four missense variants in the CSMD1, PARP8, ADAMTSL1, and FNDC3B genes in four unrelated families achieved the highest scores, including c.904C>T (p.R302C) in CSMD1, c.860G>A (p.R287H) in PARP8, c.G848A (p.G283D) in ADAMTSL1, and c.686A>G (p.H229R) in FNDC3B. These variants were well cosegregated with the disease. Further analysis was performed according to the guidelines of the American College of Medical Genetics and Genomics (Bahcall, 2015). After these multistep systematic analyses, these genetic variants were estimated as potentially pathogenic and were rare or absent from the ExAC, 1000G, and gnomAD. The scores of DANN in CSMD1, PARP8, ADAMTSL1, and FNDC3B were 0.999, 0.915, 0.998, and 0.98, respectively.
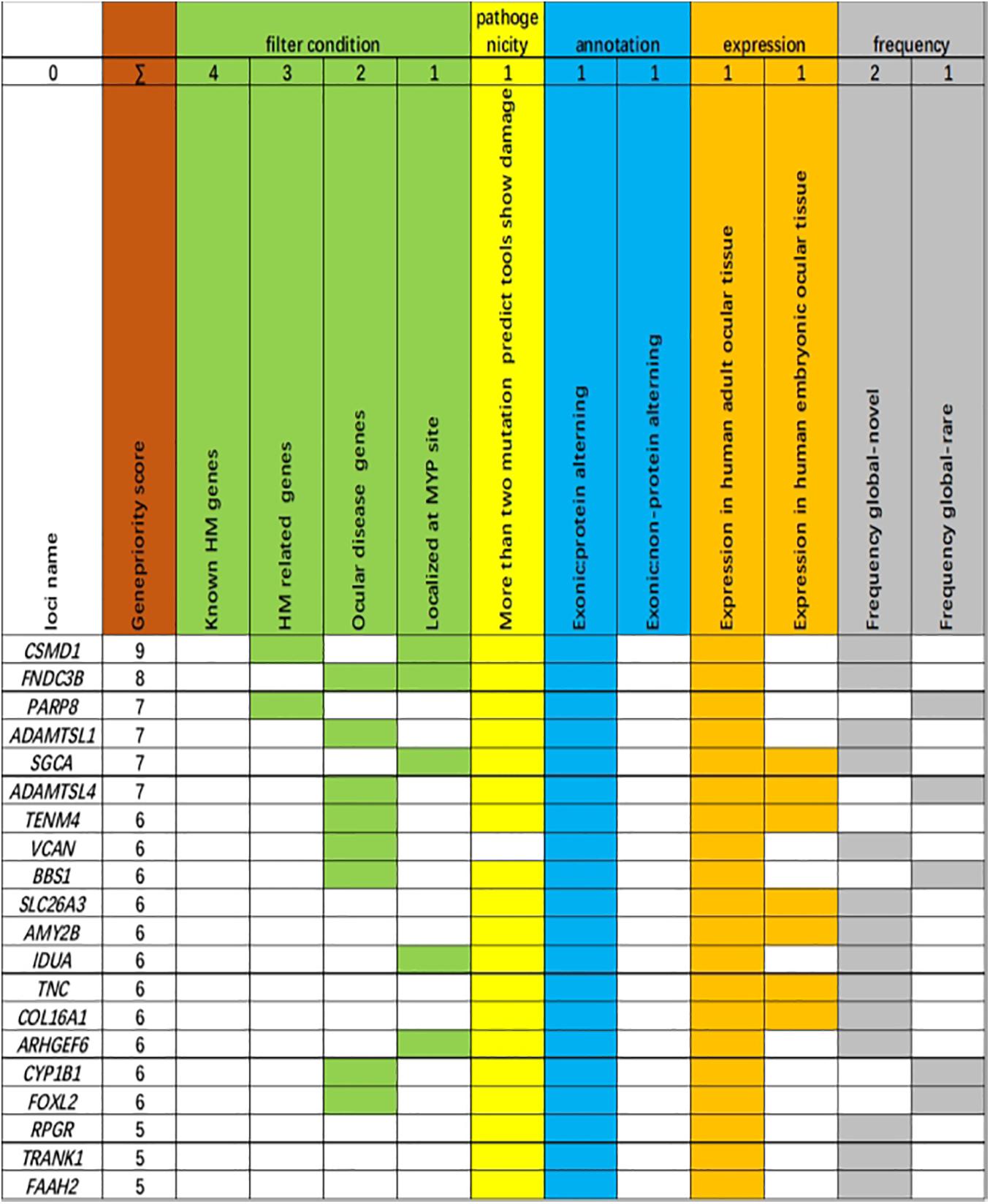
Figure 3. Genes ranked according to biological and statistical evidence. Genes were ranked based on 11 categories that can be divided into five categories: filter condition [green: genes are related to known high myopia (HM) genes, HM-related genes, ocular disease genes, or localized at the MYP site], pathogenicity (light yellow: more than two mutation predict tools show damage), annotation (light blue: genetic variant harboring an exonic protein altering variant or non-protein-altering variant), expression (dark yellow: expression in adult human ocular tissue, expression in human embryonic ocular tissue), and frequency [light dark: frequency global in Exome Aggregation Consortium (ExAC) database, novel; frequency global in ExAC database, rare].
The p.R302C substitution (c.904C>T) in CSMD1 was detected in a 22-year-old man and his father (Figure 4A). The individual and his father had refractive error less than −8.50 D in both eyes, with AL >27 mm, without oculopathy or systemic diseases. Interestingly, proband and their affected family member both had HM before school age according to their self-report. Electroretinogram (ERG) and optical coherence tomography (OCT) examinations revealed the normal retinal appearance of proband and his father (Figure 5 and Supplementary Figure 3), as did his mother in family 94 (Supplementary Figure 4). This mutation was predicted to be pathogenic by eight pathogenicity prediction tools (SIFT, Polyphen-2, Mutation Taster, Mutation Assessor, FATHMM, RadialSVM, LR, and DANN) and listed as novel in the ExAC, 1000G, and gnomAD. The substitution p.R302C occurred at a highly conserved region in multiple orthologous sequence alignments, which demonstrates that the associated protein plays an important role (Figure 6A), and this site may have an important effect on protein function. Three-dimensional modeling demonstrated the absence of a bond between the mutated cysteine at residue 302 and the arginine at residue 301 (Figure 7A).
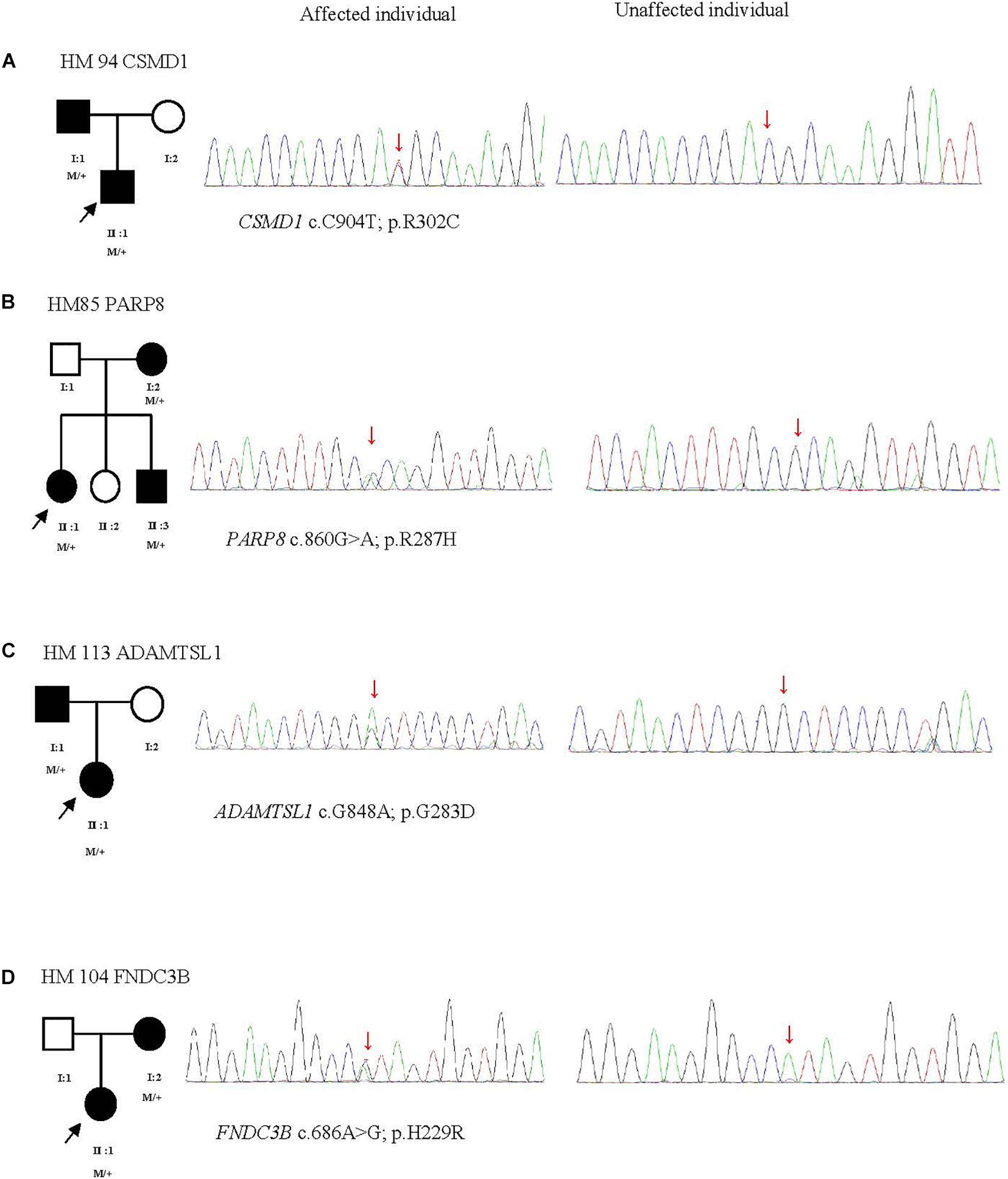
Figure 4. Four potentially pathogenic variants detected in this study. The black arrow represents the patient. +, wild-type; M, mutation. From left to right: pedigree plots of mutations, sequences from affected individual with identified mutation, sequences from unaffected control (A–D).
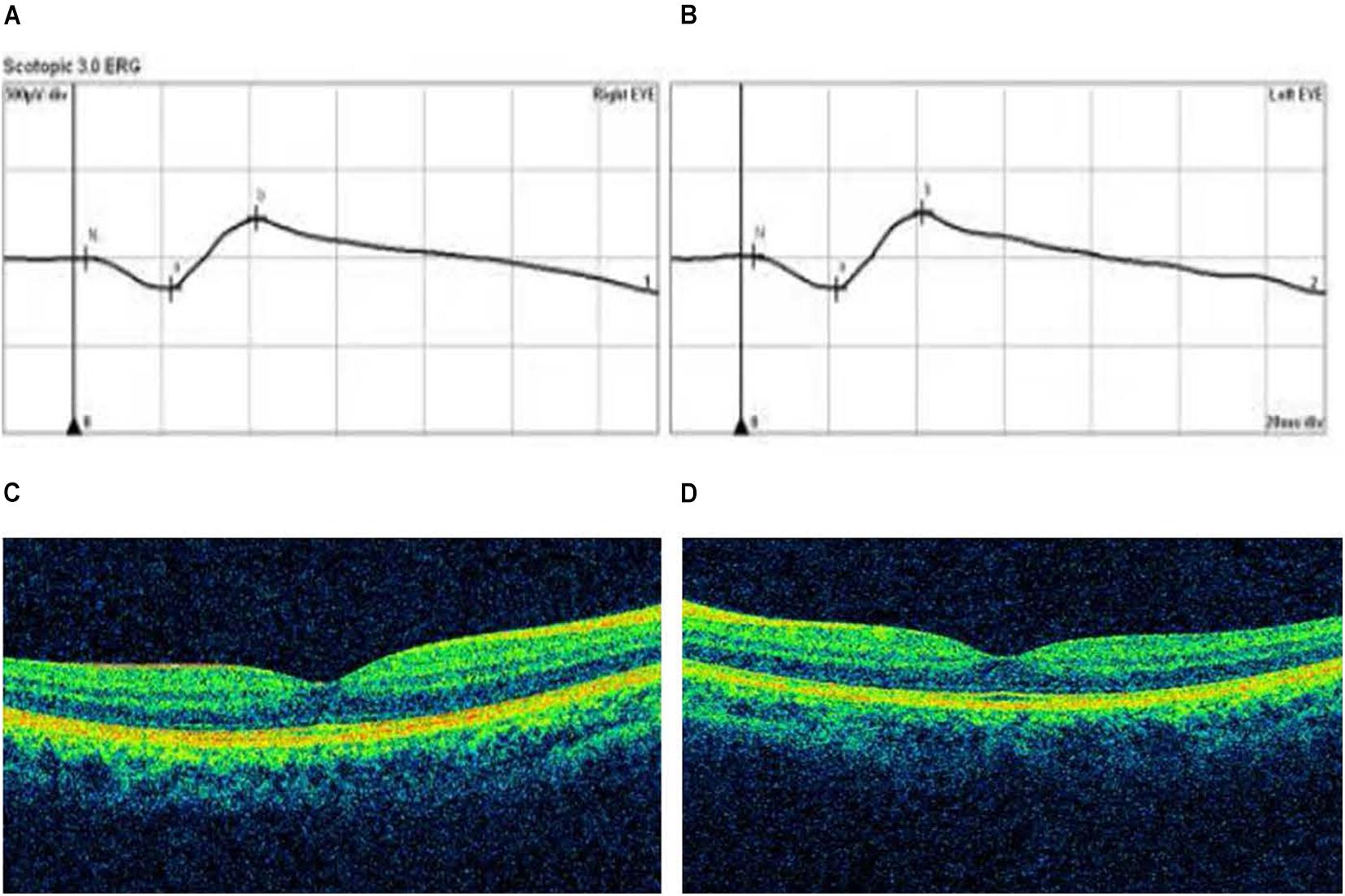
Figure 5. Optical coherence tomography (OCT) and electroretinogram (ERG) results of proband in Family 94. (A,B) Standard scotopic response of full-field ERG of the right and left eye showing normal b-wave amplitude. (C,D) OCT images of the right and left eye reveal the normal retinal appearance.
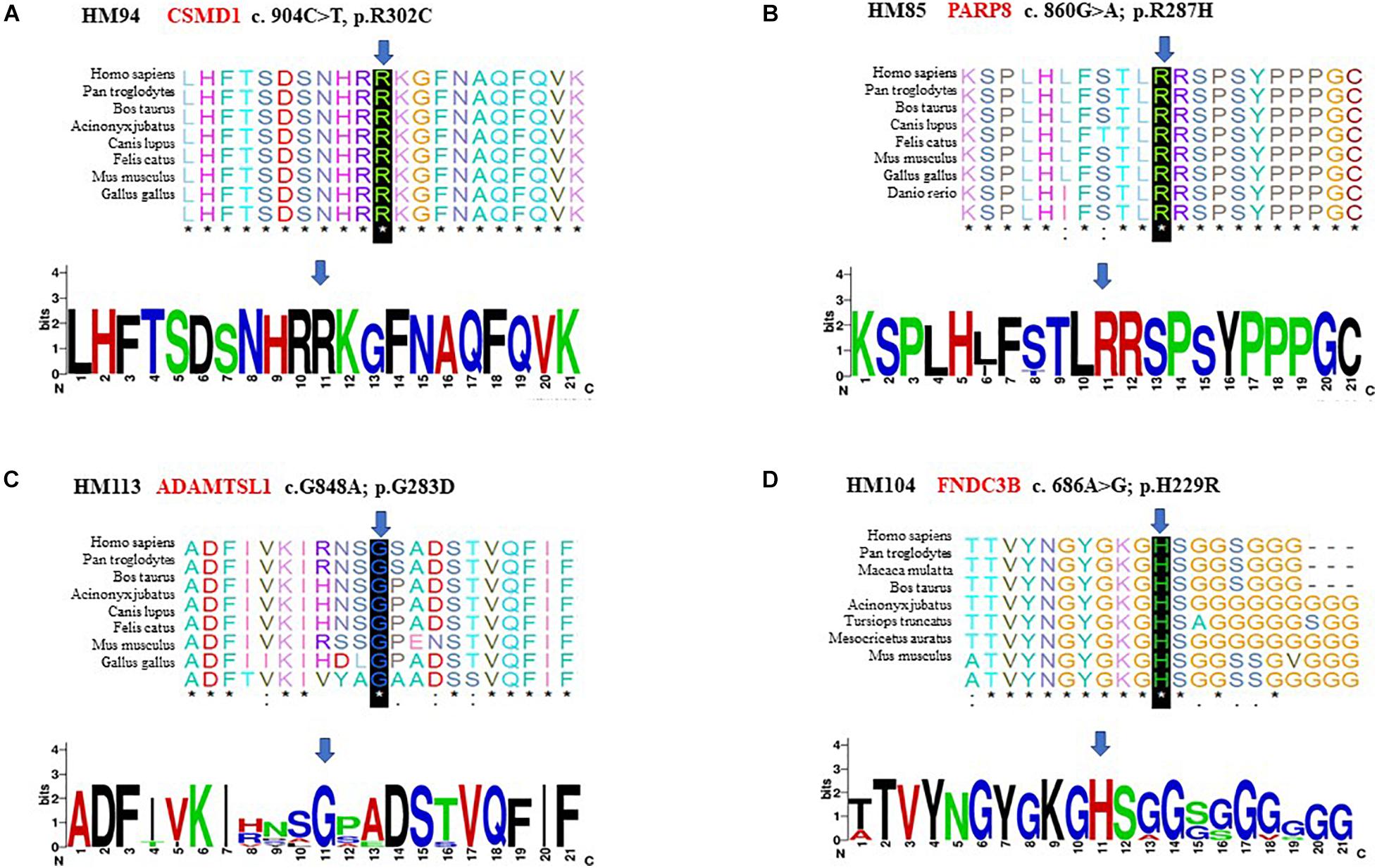
Figure 6. Conservation analysis revealed evolutionary conservation of the mutation. It shows multiple alignments of the amino acids from different species. The arrow indicates the location of the mutation (A–D).
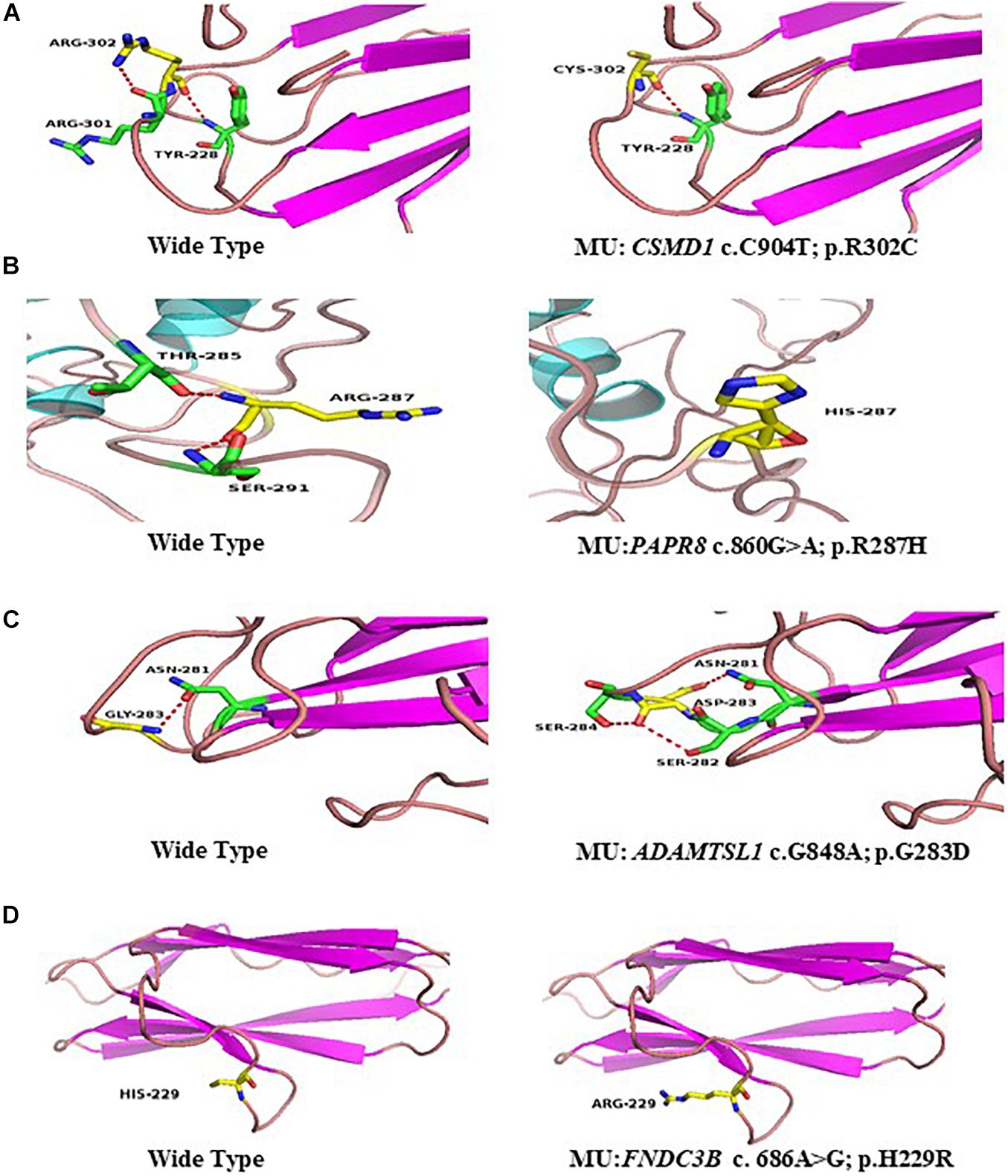
Figure 7. Predicted three-dimensional (3D) structure of proteins. Predicted crystal structures of wild-type (left) and mutant (right) proteins. Yellow shows residue of wild-type and mutant; green represents residues interact with wild-type (left), and mutant residue (right) (A–D).
A heterozygous mutation in PARP8 was detected in a 25-year-old woman and her mother (Figure 4B), both of whom had extreme myopia, with average spherical equivalent refractive error greater than −20 D and AL >29 mm. This mutation was also detected in the proband’s younger brother, who had AL of 25.06 mm in the right eye and 25.14 mm in the left eye. But there were no other symptoms of oculopathy or systemic diseases. The c.860G>A mutation is a rare single-nucleotide polymorphism (SNP) (rs142224685) located in exon 11. The pathogenicity of the p.R287H mutation in PARP8 (c.860G>A) was verified by SIFT, Mutation Taster, and PolyPhen-2. Furthermore, the p.R287H variant was detected as an SNP with a MAF of 0.0014 in ExAC. This amino acid mutation is evolutionarily conserved among species (Figure 6B). Three-dimensional protein modeling demonstrated the absence of a bond between mutated residue 287 and residues 285 and 291 (Figure 7B).
The c.848G>A, p.G283D variant in ADAMTSL1 was identified in a 21-year-old female (Figure 4C), whose spherical equivalent refractive error was −15.25 D in the right eye, with an AL of 26.64 mm and −13.50 D in the left eye with AL 26.91 mm. This mutation in ADAMTSL1 was a novel conservative mutation (Figure 6C) that was predicted to be disease-causing by SIFT, Polphen-2, and Mutation Taster. Three-dimensional structural modeling revealed a newly formed bond between residue 283 and residues 282 and 284 (Figure 7C), and this mutation is absent in the ExAC, 1000G, and gnomAD, indicating its disease causality.
A heterozygous mutation (c.686A>G; p.H229R) in FNDC3B was found in a proband from Family 104 and her mother (Figure 4D). The proband, a 15-year-old female, presented with concomitant severe anisometropia. Her spherical equivalent refractive error was −6.25 D in the right eye and −10.50 D in the left eye. According to the results of the corneal topography, the thickness of the thinnest point is 504 μm, and Km is 56.2 D for her left eye (Supplementary Figure 5). The p.H229R mutation in FNDC3B (c.686A>G) was predicted to be pathogenic by six in silico tools (SIFT, Polphen-2, Mutation Taster, LRT, Mutation Assessor, and DANN). The mutation was in a region extremely conserved across eight homologous species (Figure 6D), in line with the changes of 3D protein folding (Figure 7D).
The screening of 11 known HM genes revealed four variants to be highly pathogenic, including c.518A>T (p.D173V) in SCO2, c.3266A>G (p.Y1089C) in ZNF644, c.758C>A (p. A253E) in SLC39A5 (Family 89), and c.577G>A (p. D193N) in SLC39A5 (Family 95). Notably, Families 89 and 95 exhibited an AR mode of inheritance. We detected the same mutation in the mother of the proband from Family 89 and the father of the proband from Family 95. The c.758C>A mutation in SLC39A5, which was predicted to be deleterious by eight of the 10 computational tools, was absent from the 1000G, ExAC, and gnomAD East Asian databases. The c.577G>A mutation in SLC39A5, which was predicted to be deleterious by Polyphen2_HDIV and MutationTaster, was extremely rare in healthy populations. The 1000G and ExAC databases listed MAF as 0.001 and 0.00007462 and MAF in gnomAD is 0.00008761, well below the Popmax Filtering AF 0.0001781 in the East Asian population.
SCO2, located at MYP6, was identified as an AD gene. The c.518A>T (p.D173V) mutation in SCO2 was detected in Family 111, with a recessive mode of inheritance.
This variant was listed as novel in 1000G and gnomAD, associated with (MAF) 0.0000269 in the ExAC database. The SIFT and FATHMM predicted that this mutation would be deleterious. The MAF of c.3266A>G (p.Y1089C) in ZNF644, identified in the proband and his father of Family 115, was 0.001in 1000G, 0.0003 in the ExAC, and 0.002456 in gnomAD. This mutation was predicted to be deleterious by five of 10 computational tools.
Up to now, only a few genes responsible for non-syndromic myopia have been discovered. In our previous study of large-scale screening of eight causative genes in 731 patients with HM, we merely identified mutations in 6.16% cases (Cai et al., 2019c). In this study, we identified four new candidate genes associated with HM. All mutations in these genes affect the function of the coding region according to the 3D structure model, and CSMD1, ADAMTSL1, and FNDC3B were absent in three examined databases (ExAC, 1000G, gnomAD). SIFT, PolyPhen-2-HDIV, PolyPhen-2-HVAR, Mutation Taster, LRT, Mutation Assessor, FATHMM, RadialSVM, and LR were employed to assess the mutation changes in protein function and mutation changes in protein spatial conformation, which further cause harmful changes in physiological functions (Mahfuz and Khan, 2020).
CSMD1, located on MYP10 and mapped to chromosome 8p23.2, was highly expressed in the central nervous system and epithelial tissues. Furthermore, Wan et al. (2018) found that the CSMD1 gene is expressed at extremely high levels in peripheral retina and the area surrounding the macula. It seems that CSMD1 plays a critical role in the growth of cones, including signal transduction and matrix adhesion (Kraus et al., 2006). The de novo mutations of CSMD1 have been reported in a Chinese family with early-onset high myopia (EOHM) (Jin et al., 2017). We hypothesize CSMD1 is also a candidate gene for HM in addition to neurological effects. In our study, a heterozygous missense mutation (c.904C>T) in CSMD1 was found in two patients of Family 94, which shows the characteristics of dominant inheritance and is consistent with family cosegregation. However, there were no significant retina changes from the OCT and ERG results of the proband and his father. Further molecular mechanism study about how CSMD1 plays a role in the development of HM is needed. Meanwhile, our results provide more genetic evidence for the hypothesis that CSMD1 is the causative gene for HM.
PARP8 is a critical regulator of eukaryotic physiology, localized to the nuclear envelope, and resulted in cell morphology defects that showed potential functions for PARPs in the assembly or maintenance of membranous organelles (Vyas et al., 2013). As reported, PARP is the major risk factor associated with age-related cataract in central India (Ughade et al., 1998). Fan et al. (2012) first investigated the SNPs rs282544, rs2404958, rs32396, rs12055210, and rs11954386 on PARP8, which is closely related to the AL with p-values < 1 × 10–5.
Dongyan (2019) subsequently identified SNPs in PARP8 gene including rs1195438, rs2404958, rs282544, and rs32396, which are significantly associated with HM in the Han southwest Chinese population. In this study, we also found a rare SNP (rs142224685) as a heterozygous missense mutation c.860G>A in PARP8. The research on the association between SNP on PARP8 and HM needs to be further explored.
A recent study reported a heterozygous c.124T>C (p. Trp42Arg) mutation in the first thrombospondin type 1 (TSR) motif of ADAMTSL1, which is responsible for a complex disorder with features including developmental glaucoma, myopia, and/or retinal defects (Hendee et al., 2017). Moreover, ADAMTSL1 caused the dislocation of microspherophakic lens that causes severe myopia, glaucoma, or cataract. Two modes of inheritance have been reported: autosomal dominant and autosomal recessive (Delhon et al., 2016).
The c.848G>A (p.G283D) variant in ADAMTSL1, identified in the proband and her father in Family 113 accompanied by extremely HM, shows a dominant inheritance model. This result is consistent with the previous study in that ADAMTSL1 accounts for the myopia, especially for severe HM (Hendee et al., 2017).
FNDC3B, located on MYP8, which maps to chromosome 3q26.31, plays an important role in central corneal thickness (CCT), intraocular pressure, and anterior chamber angle depth (Rong et al., 2017). As we know, previous studies mainly focused on the association of FNDC3B with glaucoma and keratoconus (Shi et al., 2011b). CCT as one of the most heritable human traits was related to many eye diseases, especially for myopia (Pedersen et al., 2005). CCT reduction related to myopia was also reported (Hao et al., 2015; Rong et al., 2017). In our study, we identified a heterozygous mutation (c.686A>G; p.H229R) in FNDC3B from Family 104, and the left eye of the proband with higher refractive error showed typical keratoconus symptoms (−6.25 D/−10.50 D). Based on our findings, we speculate that the clinical phenotype of mutations in FNDC3B may be HM for some people, and it may also be keratoconus for other people, so whether it is an HM candidate disease-causing gene remains to be further evaluated.
The association of known HM genes with HM has been previously investigated in several studies (Jiang et al., 2015; Rong et al., 2017). For this study, we examined the associations within our own HM cohort. The screening of 11 known HM genes revealed four variants in SCO2, ZNF644, and SLC39A5 to be highly potentially pathogenic.
SCO2 was found to be related to HM in a large three-generation family in Europe, with nine affected individuals with HM (average spherical refractive error of −22.00 D). Four heterozygous mutations, p.G53∗, p.A114H, p.G140L, and p.A259V, in SCO2 were identified; these mutations may truncate or destabilize the protein structure and lead to retinal neuronal thinning (Tran-Viet et al., 2013). Moreover, R120W, R112W, and A97V were reported in subsequent studies (Jiang et al., 2015; Wakazono et al., 2016).Tran-Viet et al. (2013) observed Sco2 protein localization in the retina, retinal pigment epithelium (RPE), and scleral wall in mouse ocular tissues. We further identified mutations A201P and I221V in SCO2 in 2019 (Cai et al., 2019c). Here, a new variant c.518A>T (p.D173V) in SCO2 is detected.
ZNF644, which is located at 1p22.2, can inhibit histone methyltransferase complex through G9a/GLP, functioning as a regulator of histone methyltransferase complex (Bian et al., 2015). ZNF644 is widely expressed in eye, placenta, and liver and is assumed to play a pivotal role in changes in ocular wall. Mutations in this gene have been reported in previous studies to be associated with HM (Shi et al., 2011a; Jin et al., 2017). In this study, we detected a missense mutation c.3266A>G (p.Y1089C) in ZNF644 from the proband and his father. Interestingly, while the son had HM, the father had normal vision. It seems that this situation does not correspond to the cosegregation of the clinical phenotype. Further functional studies are needed to elucidate this.
SLC39A5, located at 12q13.3, encodes zinc transporter (Xia et al., 2020). A heterozygous truncation mutation in the SLC39A5 gene p.Y47∗ was first reported in 2014 (Guo et al., 2014). Subsequently, the missense mutation p.M304T was detected in the same family; p.G413A (Jiang et al., 2015), p.A84T, p.P87L, p.A319T, and p.A243fs∗140 were found to be responsible for HM (Feng et al., 2017; Liu et al., 2020). Other studies have shown that the bone morphogenetic protein (BMP)/transforming growth factor-beta (TGF-β) pathway is related to myopia, and c.141C>G, p.Y47∗ mutation may cause the loss of function (Jobling et al., 2009; Guo et al., 2014). It has been demonstrated that destruction of BMP/TGF-β pathway may lead to refractive errors (Wu et al., 2018). In the present study, two missense mutations, c.758C>A (p. A253E) and c.577G>A (p.D193N), in SLC39A5 were detected in two unrelated families. The relationship between these mutations and functions needs to be further studied.
In our study, 62 variants that were highly potentially pathogenic and related to ocular disease or HM were incompatible with cosegregation; they may be used as references for subsequent research. Notably, our technology failed to detect variants in 12 families. On one hand, we set stringent screening criteria, which may have reduced the number of genes for selection. On the other hand, most published articles on HM genetics are focused on families with dominant patterns of inheritance, which means that families with dominant genetic patterns of inheritance may be easier to screen for genes associated with HM. In our study, 18 of 27 families had a recessively inherited clinical phenotype. HM is a very clinically heterogeneous disease, even between patients in the same family. Refractive status and AL are different between patients. This point was also verified in our investigation of the four families in the sibships (Families 85, 99, 100, and 107). In Family 85, the 14-year-old brother of the patient showed the eye AL above 25 mm, and his diopter was less than 3.00 D; with the increase in age, the patient’s brother was very likely to develop into HM characterized by excessive AL. The younger brother of the proband in Family 99 is now 13 years old with refractive error −1.75 D in the right eye and −2.25 D in the left eye. On the other hand, the AL of his eyes is about 25 mm. There is also a risk of developing axial HM. Interestingly, the situation of Family 107 is opposite to that of Families 85 and 99. The three sisters are patients, and all showed HM with high diopter and low AL.
Especially the two sisters of the proband showed the diopter was greater than −6.00 D with the AL less than 26 mm. The two patients in Family 100 are of similar age; the older sister is 22 years old and the younger brother is 21 years old. The refractive error and AL of their eyes are relatively similar, in addition to the common genetic factors. The consistency of their living background may also play an important role in the development of their myopia for HM is a disease in which environmental and genetic factors work together. Different life backgrounds, dietary habits, and near-work habits may all contribute to the development of HM.
This study had some limitations. Because environmental and genetic factors determine the manifestation of HM, environmental factors may be an important influencing factor that causes inconsistency between the genotype and the clinical phenotype. This study did not focus on the impact of environmental factors, which may explain our observations of non-cosegregated genes, and we did not carry out functional verification tests. However, analysis of the pathogenicity of gene mutations based on the interpretation standards and guidelines of gene mutations developed by the American College of Medical Genetics and Genomics (ACMG) showed that four new candidate genes met the evidence criteria for possibly pathogenic mutations. The mechanisms underlying the effects of these genetic variants in the context of HM require further elucidation through additional studies. In addition, we have not investigated the impact of copy number variation (Huang et al., 2017) and somatic mosaicism (Jin et al., 2007) in these patients. Finally, targeted exome sequencing including a panel of disease-causing genes, likewise the strategy of inherited retinal dystrophy (Huang et al., 2013, 2015; Xing et al., 2014; Chen et al., 2020), might be more appropriate for future genetic diagnosis of genetic HM.
In conclusion, we identified 201 genetic variants related to the development of HM by WES and bioinformatics analysis. To our knowledge, this is the first study of WES in patients with HM from Northwest China. The findings presented four new candidate genes associated with HM, providing additional evidence of heritable HM in Chinese patients. These findings expand the mutation spectrum of myopia genes and provide clues for further genetic study of HM.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://figshare.com/articles/dataset/Whole-exome_sequencing_in_a_cohort_of_high_myopia_patients_in_northwest_China/14480316, accession doi: 10.6084/m9.figshare.14480316.
The studies involving human participants were reviewed and approved by the Institutional Review Board of People’s Hospital of Ningxia Hui Autonomous Region, Third Clinical Medical College of Ningxia Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
W-JZ, Z-BJ, and JL designed and supervised the whole study. YL, J-JZ, and S-YP performed the experiments. YL, R-JS, YM, Z-QX, and WZ interpreted the results. YL drafted the manuscript. Z-BJ and W-JZ revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (82060182 to WZ) and Beijing Natural Science Foundation (Z200014 to Z-BJ). This work was supported by the Ningxia Natural Science Foundation (2021AAC03301).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.645501/full#supplementary-material
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Aldahmesh, M. A., Khan, A. O., Alkuraya, H., Adly, N., Anazi, S., Al-Saleh, A. A., et al. (2013). Mutations in LRPAP1 are associated with severe myopia in humans. Am. J. Hum. Genet. 93, 313–320. doi: 10.1016/j.ajhg.2013.06.002
Bahcall, O. G. (2015). Genetic testing. ACMG guides on the interpretation of sequence variants. Nat. Rev. Genet. 16, 256–257.
Bian, C., Chen, Q., and Yu, X. (2015). The zinc finger proteins ZNF644 and WIZ regulate the G9a/GLP complex for gene repression. Elife 4:e05606.
Bourne, R. R., Stevens, G. A., White, R. A., Smith, J. L., Flaxman, S. R., and Price, H. (2013). Causes of vision loss worldwide, 1990–2010: a systematic analysis. Lancet Glob. Health 1, e339–e349.
Cai, X. B., Shen, S. R., Chen, D. F., Zhang, Q., and Jin, Z. B. (2019a). An overview of myopiagenetics. Exp. Eye Res. 188:107778.
Cai, X. B., Wu, K. C., Zhang, X., Lv, J. N., Jin, G. H., Xiang, L., et al. (2019b). Whole-exome sequencing identified ARL2 as a novel candidate genefor MRCS (microcornea, rod-cone dystrophy, cataract, and posterior staphyloma) syndrome. Clin. Genet. 96, 61–71.
Cai, X. B., Zheng, Y. H., Chen, D. F., Zhou, F. Y., Xia, L. Q., Wen, X. R., et al. (2019c). Expanding the phenotypic and genotypic landscape of nonsyndromic high myopia: a cross-sectional study in 731 Chinese patients. Invest. Ophthalmol. Vis. Sci. 60, 4052–4062.
Chen, Z. J., Lin, K. H., Lee, S. H., Shen, R. J., Feng, Z. K., Wang, X. F., et al. (2020). Mutation spectrum and genotype-phenotype correlation of inherited retinal dystrophy in Taiwan. Clin. Exp. Ophthalmol. 48, 486–499. doi: 10.1111/ceo.13708
Delhon, L., Cormier-Daire, V., and Le Goff, C. (2016). Metalloproteinases and their inhibitors in the pathophysiology of heritable connective tissue disorders: current evidence. Metalloproteinases Med. 3, 1–9. doi: 10.2147/mnm.s63634
Desmet, F.-O., Hamroun, D., Lalande, M., Collod-Béroud, G., Claustres, M., and Béroud, C. (2009). Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37:e67. doi: 10.1093/nar/gkp215
Dongyan, L. (2019). “Association of PARP8 gene Polymorphisms as genetic risk factors with High Myopia in a Chinese population,” in Proceedings of the 2nd China Conference on Clinical Molecular Diagnosis, (Chengdu, China).
Fan, Q., Barathi, V. A., Cheng, C. Y., Zhou, X., Meguro, A., Nakata, I., et al. (2012). Genetic variants on chromosome 1q41 influence ocular axial length and high myopia. PLoS Genet. 8:e1002753. doi: 10.1371/journal.pgen.1002753
Fan, Q., Guo, X., Tideman, J. W., Williams, K. M., Yazar, S., Hosseini, S. M., et al. (2016). Childhood gene-environment interactions and age-dependent effects ofgenetic variants associated with refractive error and myopia: the CREAM consortium. Sci. Rep. 6:25853.
Feng, C. Y., Huang, X. Q., Cheng, X. W., Wu, R. H., Lu, F., and Jin, Z. B. (2017). Mutational screening of SLC39A5, LEPREL1 and LRPAP1 in a cohort of 187 high myopia patients. Sci. Rep. 7:1120.
Guo, H., Jin, X., Zhu, T., Wang, T., Tong, P., Tian, L., et al. (2014). SLC39A5 mutations interfering with the BMP/TGF-β pathway in non-syndromic high myopia. J. Med. Genet. 51, 518–525. doi: 10.1136/jmedgenet-2014-102351
Guo, H., Tong, P., Liu, Y., Xia, L., Wang, T., Tian, Q., et al. (2015). Mutations of P4HA2 encoding prolyl 4-hydroxylase 2 are associated with nonsyndromic high myopia. Genet. Med. 17, 300–306. doi: 10.1038/gim.2015.28
Hammond, C. J., Snieder, H., Gilbert, C. E., and Spector, T. D. (2001). Genes and environment in refractive error: the twin eye study. Invest. Ophthalmol. Vis. Sci. 42, 1232–1236.
Hao, X. D., Chen, P., Chen, Z. L., Li, S. X., and Wang, Y. (2015). Evaluating the association between keratoconus and reported genetic loci in a Han Chinese population. Ophthalmic Genet. 36, 132–136. doi: 10.3109/13816810.2015.1005317
Hendee, K., Wang, L. W., Reis, L. M., Rice, G. M., Apte, S. S., and Semina, E. V. (2017). Identification and functional analysis of an ADAMTSL1 variant associated with a complex phenotype including congenital glaucoma, craniofacial, and other systemic features in a three-generation human pedigree. Hum. Mut. 38, 1485–1490. doi: 10.1002/humu.23299
Holden, B. A., Fricke, T. R., Wilson, D. A., Jong, M., Naidoo, K. S., Sankaridurg, P., et al. (2016). Global prevalence of myopia and high myopia and temporal trendsfrom 2000 through 2050. Ophthalmology 123, 1036–1042. doi: 10.1016/j.ophtha.2016.01.006
Huang, X.-F., Huang, F., Wu, K. C., Wu, J., Chen, J., Pang, C. P., et al. (2015). Genotype–phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 17, 271–278. doi: 10.1038/gim.2014.138
Huang, X. F., Mao, J. Y., Huang, Z. Q., Rao, F. Q., Cheng, F. F., Li, F. F., et al. (2017). Genome-wide detection of copy number variations in unsolved inherited retinal disease. Invest. Ophthalmol. Vis. Sci. 58, 424–429. doi: 10.1167/iovs.16-20705
Huang, X. F., Xiang, P., Chen, J., Xing, D. J., Huang, N., Min, Q., et al. (2013). Targeted exome sequencing identified novel USH2A mutations in Usher syndrome families. PLoS One 8:e63832. doi: 10.1371/journal.pone.0063832
Jiang, D., Li, J., Xiao, X., Li, S., Jia, X., Sun, W., et al. (2015). Detection of mutations in LRPAP1, CTSH, LEPREL1, ZNF644, SLC39A5, and SCO2 in 298 families with early-onset high myopia by exome sequencing. Invest. Ophthalmol. Vis. Sci. 56, 339–345. doi: 10.1167/iovs.14-14850
Jin, Z. B., Gu, F., Matsuda, H., Yukawa, N., Ma, X., and Nao-i, N. (2007). Somatic and gonadal mosaicism in X-linked retinitis pigmentosa. Am. J. Med. Genet. A 143A, 2544–2548. doi: 10.1002/ajmg.a.31984
Jin, Z. B., Li, Z., Liu, Z., Jiang, Y., Cai, X. B., and Wu, J. (2018). Identification of de novo germline mutations and causal genes for sporadic diseases using trio-based whole-exome/genome sequencing. Biol. Rev. 93, 1014–1031. doi: 10.1111/brv.12383
Jin, Z.-B., Wu, J., Huang, X. F., Feng, C. Y., Cai, X. B., Mao, J. Y., et al. (2017). Trio-based exome sequencing arrests de novo mutations in early-onset high myopia. Proc. Natl. Acad. Sci. U.S.A. 114, 4219–4224. doi: 10.1073/pnas.1615970114
Jobling, A. I., Wan, R., Gentle, A., Bui, B. V., and McBrien, N. A. (2009). Retinal and choroidal TGF-β in the tree shrew model of myopia: isoform expression, activation and effects on function. Exp. Eye Res. 88, 458–466.
Katz, J., Tielsch, J. M., and Sommer, A. (1997). Prevalence and risk factors for refractive errors in an adult inner city population. Invest. Ophthalmol. Vis. Sci. 38, 334–340.
Kloss, B. A., Tompson, S. W., Whisenhunt, K. N., Quow, K. L., Huang, S. J., Pavelec, D. M., et al. (2017). Exome sequence analysis of 14 families with high myopia. Invest. Ophthalmol. Vis. Sci. 58, 1982–1990. doi: 10.1167/iovs.16-20883
Kraus, D. M., Elliott, G. S., Chute, H., Horan, T., Pfenninger, K. H., Sanford, S. D., et al. (2006). CSMD1 is a novel multiple domain complement-regulatory protein highly expressed in the central nervous system and epithelial tissues. J. Immunol. 176, 4419–4430. doi: 10.4049/jimmunol.176.7.4419
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4:1073. doi: 10.1038/nprot.2009.86
Li, J., Gao, B., Guan, L., Xiao, X., Zhang, J., Li, S., et al. (2015). Unique variants in OPN1LW cause both syndromic and nonsyndromic X-linked high myopia mapped to MYP1. Invest. Ophthalmol. Vis. Sci. 56, 4150–4155. doi: 10.1167/iovs.14-16356
Liu, F., Wang, J., Xing, Y., and Li, T. (2020). Mutation screening of 17 candidate genes in a cohort of 67 probands with early−onset high myopia. Ophthalmic Physiol. Opt. 40, 271–280. doi: 10.1111/opo.12683
Mahfuz, A., and Khan, M. A. (2020). Identification of deleterious single nucleotide polymorphism (SNP) s in the human TBX5 gene & prediction of their structural & functional consequences: an in silico approach. bioRxiv [Preprint] doi: 10.1101/2020.05.16.099648
Mordechai, S., Gradstein, L., Pasanen, A., Ofir, R., El Amour, K., Levy, J., et al. (2011). High myopia caused by a mutation in LEPREL1, encoding prolyl 3-hydroxylase 2. Am. J. Hum. Genet. 89, 438–445. doi: 10.1016/j.ajhg.2011.08.003
Pan, C.-W., Dirani, M., Cheng, C.-Y., Wong, T.-Y., and Saw, S.-M. (2015). The age-specific prevalence of myopia in Asia: a meta-analysis. Optom. Vis. Sci. 92, 258–266. doi: 10.1097/opx.0000000000000516
Pedersen, L., Hjortdal, J., and Ehlers, N. (2005). Central corneal thickness in high myopia. Acta Ophthalmol. Scand. 83, 539–5425. doi: 10.1111/j.1600-0420.2005.00498.x
Percival, S. (1987). Redefinition of high myopia: the relationship of axial length measurement to myopic pathology and its relevance to cataract surgery. Dev. Ophthalmol. 14:42. doi: 10.1159/000414364
Rong, S. S., Ma, S. T. U., Yu, X., Ma, L., Chu, W. K., Chan, T. C. Y., et al. (2017). Genetic associations for keratoconus: a systematic review and meta-analysis. Sci. Rep. 7:4620.
Schwarz, J. M., Rödelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi: 10.1038/nmeth0810-575
Shi, Y., Li, Y., Zhang, D., Zhang, H., Li, Y., Lu, F., et al. (2011a). Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet. 7:e1002084. doi: 10.1371/journal.pgen.1002084
Shi, Y., Qu, J., Zhang, D., Zhao, P., Zhang, Q., Tam, P. O. S., et al. (2011b). Genetic variants at 13q12. 12 are associated with high myopia in the Han Chinese population. Am. J. Hum. Genet. 88, 805–813.
Tham, Y.-C., Lim, S. H., Shi, Y., Chee, M. L., Zheng, Y. F., Chua, J., et al. (2018). Trends of visual impairment and blindness in the Singapore Chinese population over a decade. Sci. Rep. 8:12224.
Tran-Viet, K.-N., Powell, C., Barathi, V. A., Klemm, T., Maurer-Stroh, S., Limviphuvadh, V., et al. (2013). Mutations in SCO2 are associated with autosomal-dominant high-grade myopia. Am. J. Hum. Genet. 92, 820–826. doi: 10.1016/j.ajhg.2013.04.005
Ughade, S. N., Zodpey, S. P., and Khanolkar, V. A. (1998). Risk factors for cataract: a case control study. Indian J. Ophthalmol. 46, 221–227.
Vyas, S., Chesarone-Cataldo, M., Todorova, T., Huang, Y. H., and Chang, P. (2013). A systematic analysis of the PARPprotein family identifies new functions critical for cell physiology. Nat. Commun. 4:2240.
Wakazono, T., Miyake, M., Yamashiro, K., Yoshikawa, M., and Yoshimura, N. (2016). Association between SCO2 mutation and extreme myopia in Japanese patients. Jpn. J. Ophthalmol. 60, 319–325. doi: 10.1007/s10384-016-0442-4
Wan, L., Deng, B., Wu, Z., and Chen, X. (2018). Exome sequencing study of 20 patients with high myopia. PeerJ 6:e5552. doi: 10.7717/peerj.5552
Wu, H., Jiang, L., Zheng, R., Luo, D., Liu, X., Hao, F., et al. (2018). Genetic association study between the COL11A1 and COL18A1 genes and high myopia in a Han Chinese population. Genet. Test. Mol. Biomark. 22, 359–365. doi: 10.1089/gtmb.2017.0235
Xia, Z., Bi, X., Lian, J., Dai, W., He, X., Zhao, L., et al. (2020). Slc39a5-mediated zinc homeostasis plays an essential role in venous angiogenesis in zebrafish. Open Biol. 10:200281. doi: 10.1098/rsob.200281
Xiao, X., Li, S., Jia, X., Guo, X., and Zhang, Q. (2016). X-linked heterozygous mutations in ARR3 cause female-limited early onset high myopia. Mol. Vis. 22, 1257–1266.
Xing, D. J., Zhang, H. X., Huang, N., Wu, K. C., Huang, X. F., Huang, F., et al. (2014). Comprehensive molecular diagnosis of Bardet-Biedl syndrome by high-throughput targeted exome sequencing. PLoS One 9:e90599. doi: 10.1371/journal.pone.0090599
Young, T. L., Metlapally, R., and Shay, A. E. (2007). Complex trait genetics of refractive error. Arch. Ophthalmol. 125, 38–48. doi: 10.1001/archopht.125.1.38
Zhang, Q. (2015). Genetics of refraction and myopia. Prog. Mol. Biol. Transl. Sci. 134, 269–279. doi: 10.1016/bs.pmbts.2015.05.007
Zhao, F., Wu, J., Xue, A., Su, Y., Wang, X., Lu, X., et al. (2013). Exome sequencing reveals CCDC111 mutation associated with high myopia. Hum. Genet. 132, 913–921. doi: 10.1007/s00439-013-1303-6
Keywords: high myopia, cohort, mutation, gene, Northwest China
Citation: Liu Y, Zhang J-J, Piao S-Y, Shen R-J, Ma Y, Xue Z-Q, Zhang W, Liu J, Jin Z-B and Zhuang W-J (2021) Whole-Exome Sequencing in a Cohort of High Myopia Patients in Northwest China. Front. Cell Dev. Biol. 9:645501. doi: 10.3389/fcell.2021.645501
Received: 23 December 2020; Accepted: 27 May 2021;
Published: 18 June 2021.
Edited by:
Wei He, He Eye Specialist Hospital, ChinaReviewed by:
Colin Anfimov Johnson, University of Leeds, United KingdomCopyright © 2021 Liu, Zhang, Piao, Shen, Ma, Xue, Zhang, Liu, Jin and Zhuang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Liu, cnl1a2VuMDUxOEAxNjMuY29t; Zi-Bing Jin, amluemliaW5nQGZveG1haWwuY29t; Wen-Juan Zhuang, emhfd2VuakAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.