
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 02 March 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.641618
 Oliver Hartmann1,2
Oliver Hartmann1,2 Michaela Reissland1,2
Michaela Reissland1,2 Carina R. Maier3Thomas Fischer1,4
Carina R. Maier3Thomas Fischer1,4 Cristian Prieto-Garcia1,2,5
Cristian Prieto-Garcia1,2,5 Apoorva Baluapuri6
Apoorva Baluapuri6 Jessica Schwarz6
Jessica Schwarz6 Werner Schmitz7
Werner Schmitz7 Martin Garrido-Rodriguez8,9,10Nikolett Pahor1,2
Martin Garrido-Rodriguez8,9,10Nikolett Pahor1,2 Clare C. Davies11Florian Bassermann12,13Amir Orian5
Clare C. Davies11Florian Bassermann12,13Amir Orian5 Elmar Wolf6
Elmar Wolf6 Almut Schulze3Marco A. Calzado8,9,10Mathias T. Rosenfeldt2,14
Almut Schulze3Marco A. Calzado8,9,10Mathias T. Rosenfeldt2,14 Markus E. Diefenbacher1,2*
Markus E. Diefenbacher1,2*Lung cancer is the most common cancer worldwide and the leading cause of cancer-related deaths in both men and women. Despite the development of novel therapeutic interventions, the 5-year survival rate for non-small cell lung cancer (NSCLC) patients remains low, demonstrating the necessity for novel treatments. One strategy to improve translational research is the development of surrogate models reflecting somatic mutations identified in lung cancer patients as these impact treatment responses. With the advent of CRISPR-mediated genome editing, gene deletion as well as site-directed integration of point mutations enabled us to model human malignancies in more detail than ever before. Here, we report that by using CRISPR/Cas9-mediated targeting of Trp53 and KRas, we recapitulated the classic murine NSCLC model Trp53fl/fl:lsl-KRasG12D/wt. Developing tumors were indistinguishable from Trp53fl/fl:lsl-KRasG12D/wt-derived tumors with regard to morphology, marker expression, and transcriptional profiles. We demonstrate the applicability of CRISPR for tumor modeling in vivo and ameliorating the need to use conventional genetically engineered mouse models. Furthermore, tumor onset was not only achieved in constitutive Cas9 expression but also in wild-type animals via infection of lung epithelial cells with two discrete AAVs encoding different parts of the CRISPR machinery. While conventional mouse models require extensive husbandry to integrate new genetic features allowing for gene targeting, basic molecular methods suffice to inflict the desired genetic alterations in vivo. Utilizing the CRISPR toolbox, in vivo cancer research and modeling is rapidly evolving and enables researchers to swiftly develop new, clinically relevant surrogate models for translational research.
Lung cancer is the leading cause of cancer-related deaths in the Western world, claiming around 1.8 million lives every year (Bray et al., 2018). While novel treatment options such as immune checkpoint inhibitors or receptor tyrosine kinase inhibitors (RTKi) have revolutionized therapy and improved the survival of patient subgroups, the overall survival remains rather low, with an average 5-year survival rate of approx. 6% (Doroshow et al., 2019; Ruiz et al., 2019; Pottier et al., 2020).
Genetic profiling, conducted by cancer panel “hot spot” sequencing, whole exome, or next-generation sequencing (NGS), has granted deep insights into the genetic diversity of this disease (Cancer Genome Atlas Research Network, 2012, 2014). Despite this knowledge, the development of therapy resistance is prevailing and limits patient survival (Asao et al., 2019; Lim and Ma, 2019). This is frequently observed in patients undergoing either immune checkpoint or RTKi therapy. Here, either tumors acquire additional mutations and thereby render the applied drug ineffective, or a subclone takes center stage and thrives upon the loss of the previously predominant clone. Furthermore, mutations in additional genes, such as tumor suppressors, can lead to tumor recurrence and treatment resistance.
Lung cancer is subdivided in two major cancer types: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) (Travis et al., 2015). NSCLC in itself is not a genetically simple and homogeneous disease. It represents a rather heterogeneous and highly mutated tumor entity, which is divided by histological features and marker expression into adenocarcinoma (ADC) and squamous cell carcinoma (SCC) (Travis et al., 2015; Mengoli et al., 2018). Several alterations have recently been identified to directly interfere with personalized therapy, such as mutations of KEAP1 and STK11/LKB1 (Bonanno et al., 2019; Papillon-Cavanagh et al., 2020). These mutations, either as single or occurring in tandem, negate immunotherapy and negatively affect patient survival. As this is just one example, it highlights the necessity to develop novel surrogate models that allow to swiftly model various somatic mutations found in NSCLC patients, which can be used as therapeutic testbed(s).
Murine models of NSCLC were at the forefront of target validation and drug development for the last decades (Talmadge et al., 2007; Day et al., 2015; Ireson et al., 2019). A steady “workhorse” of these efforts was the NSCLC model established by Tuveson, Jacks, and Berns: the combination of conditional deletion of the whole coding sequence of the tumor suppressor Trp53 (Trp53fl/fl) in combination with the conditional expression of the oncogenic variant of KRas (KRasG12D KRaslsl–G12D/wt) (Jackson et al., 2005; DuPage et al., 2009). Loss of TP53, concomitant with activating mutations in the small GTPase KRAS, is the prime event in NSCLC onset. TP53 is commonly mutated in 51% of ADC and 82% of SCC samples, respectively, while KRAS is predominantly mutant in adenocarcinoma (ADC) but amplified or overexpressed in squamous cell carcinoma (SCC) (Mogi and Kuwano, 2011; Westcott and To, 2013; Wang et al., 2014). In general, KRAS and members of the MAPK pathway are found to be either transcriptionally upregulated or amplified in SCC. Hence, to recapitulate NSCLC in a murine model, mice carrying conditional alleles for Trp53 (Trp53fl/fl) in combination with mutant KRasG12D (Trp53fl/fl:KRaslsl–G12D/wt) represent suitable model systems.
Recognizing the genetic complexity observed in NSCLC and the direct impact mutations have on treatment response highlights the limits of classic mouse models. The introduction of novel genetic alterations requires extensive technical and methodological assets, not available to most research laboratories or facilities. Genetic manipulation by classic murine embryonic stem cell targeting is time and cost intensive; the creation of novel mouse lines (e.g., via blastocyst injection) including testing for germline transmission and breeding into the desired genetic background is challenging. Ultimately, combining the newly generated murine mouse line with mice carrying targeted tumor suppressors or oncogenic drivers, such as KRaslsl–G12D/wt or Trp53fl/fl:KRaslsl–G12D/wt, generates a tremendous amount of excess animals with undesired genotypes. Furthermore, one is limited to the introduced genetic alterations, such as Cre recombinase-mediated exon deletion or activation of a specific point mutant, such as lsl-Trp53R172H/wt.
With the advent of CRISPR/Cas9, research laboratories are now able to recapitulate various genetic mutations identified in patients without the need of an extensive mouse cohort or advanced facilities (Platt et al., 2014; Sanchez-Rivera et al., 2014). Several laboratories have successfully combined classic NSCLC models with CRISPR-mediated targeting of additional tumor suppressors, demonstrating the potential of CRISPR. However, a direct side-by-side comparison between classic and CRISPR-mediated modeling of the “standard” lung cancer model, Trp53fl/fl:KRasG12D/wt, has not been done. In this study, we report that tumors induced by either Cre-mediated recombination of conditional alleles (from here onward referred to as KPGEMM) or CRISPR-mediated gene editing (referred to as KPCRISPR) resulted in highly comparable tumors. Similarities were not restricted to histology but extended also to a transcriptional level. Expanding the repertoire of genetic alterations achieved in vivo only required standard molecular techniques. Furthermore, tumor induction can even be induced in wild-type mouse strains by the use of a dual viral system. Hence, CRISPR is revolutionizing the way we can conduct tumor research and enables laboratories to address and investigate a plethora of genetic alterations at ease while reducing overall animals required in this particular research field.
Loss of TP53, in combination with activating mutations within members of the MAPK pathway, exemplified by alterations in KRAS, is the commonly observed driver of human NSCLC (Figure 1A and Supplementary Figure 1A). To assess if CRISPR/Cas9 gene editing can be utilized to recapitulate NSCLC in vivo, we compared the classic mouse model Trp53fl/fl:KRaslsl–G12D/wt (C57BL6/J background, KPGEMM) infected with an adeno-associated virus (AAV) encoding Cre recombinase with Rosa26Sor–CAG–Cas9–IRES–eGFP mice (C57BL6/J background) infected with an AAV encoding single guide RNA (sgRNA) to target Trp53 and mutate KRas to KRasG12 (KPCRISPR) (Figure 1B and Supplementary Movie 1). Previous studies demonstrated the high degree of fidelity versus off-target mutagenesis by using CRISPR-mediated genome editing in vivo (Sanchez-Rivera et al., 2014; Akcakaya et al., 2018). To ensure optimal viral delivery and infection, several AAV capsids were tested (Supplementary Figure 1B). Packaging with the shuffle capsid AAV-DJ yielded the highest infection efficacy. Twelve weeks post intratracheal instillation of virus-containing solutions, KPCRISPR and KPGEMM developed tumors and analysis of the oral cavity and trachea of infected mice revealed no off-target tumor induction (Figure 1C and Supplementary Figure 2A). Neither tumor burden, as assessed by HE staining, nor tumor cell proliferation, as assessed by % proliferating cell nuclear antigen (PCNA)-positive cells within tumors, revealed significant differences between KPGEMM and KPCRISPR (Figures 1D,E). Next, we wondered if tumor grade differs between KPGEMM and KPCRISPR. Adhering to the established classification system for murine NSCLC revealed no significant differences, as both models presented comparable distributions of all stages, ranging from atypical alveolar hyperplasia (AAH, stage I) to stage IV (Figures 1F,G; Jackson et al., 2001, 2005). Overall animal survival was not affected during the time of analysis by the method used for genetic targeting (12 weeks post i.t., Supplementary Figure 1C). Genetic alterations induced by CRISPR gene editing were confirmed by Sanger sequencing of the respective targeted genomic regions. Successful targeting of KRas was confirmed and the integration of the KRasG12D HDR template detected. For Trp53, CRISPR gene editing led to depletions or various lengths, as identified by Sanger sequencing (Supplementary Figure 1D). Oncogenic mutation of KRas to KRasG12D resulted in downstream activation of the MAPK pathway. Phosphorylation of MAPK1/3 (p-Erk1/2) was readily detectable and increased in KPGEMM and KPCRISPR, compared with adjacent non-tumor tissue (Figure 1H). Analysis of lung lineage-specific marker expression, such as the adenocarcinoma marker thyroid transcription factor-1 (TTF1), the tracheal club cell marker Scgb1a1 (CC10), or the alveolar type II specific surfactant protein C (SftpC), an additional marker for adenocarcinomas, showed similar expression patterns between KPGEMM and KPCRISPR (Figure 1I). One detectable difference was the expression of the basal stem and squamous cell carcinoma marker Krt5. Here, positive tumor cells were only detectable in KPGEMM, while KPCRISPR showed no positive staining at all (Figure 1I). While Krt5 was positive, Sox2, a marker and driver of SCC, was negative in all samples, while positive cells were detected in basal cells of the trachea, suggesting a co-expression rather than squamous differentiation of tumor cells (Figure 1I and Supplementary Figure 1E). Furthermore, we analyzed KPGEMM and KPCRISPR with regard to potential metastasis. Distal organs and prime locations for lung metastasis, such as the liver but also the pancreas and intestine, showed no gross abnormalities nor metastasis in both tumor model systems (Supplementary Figures 2A,B).
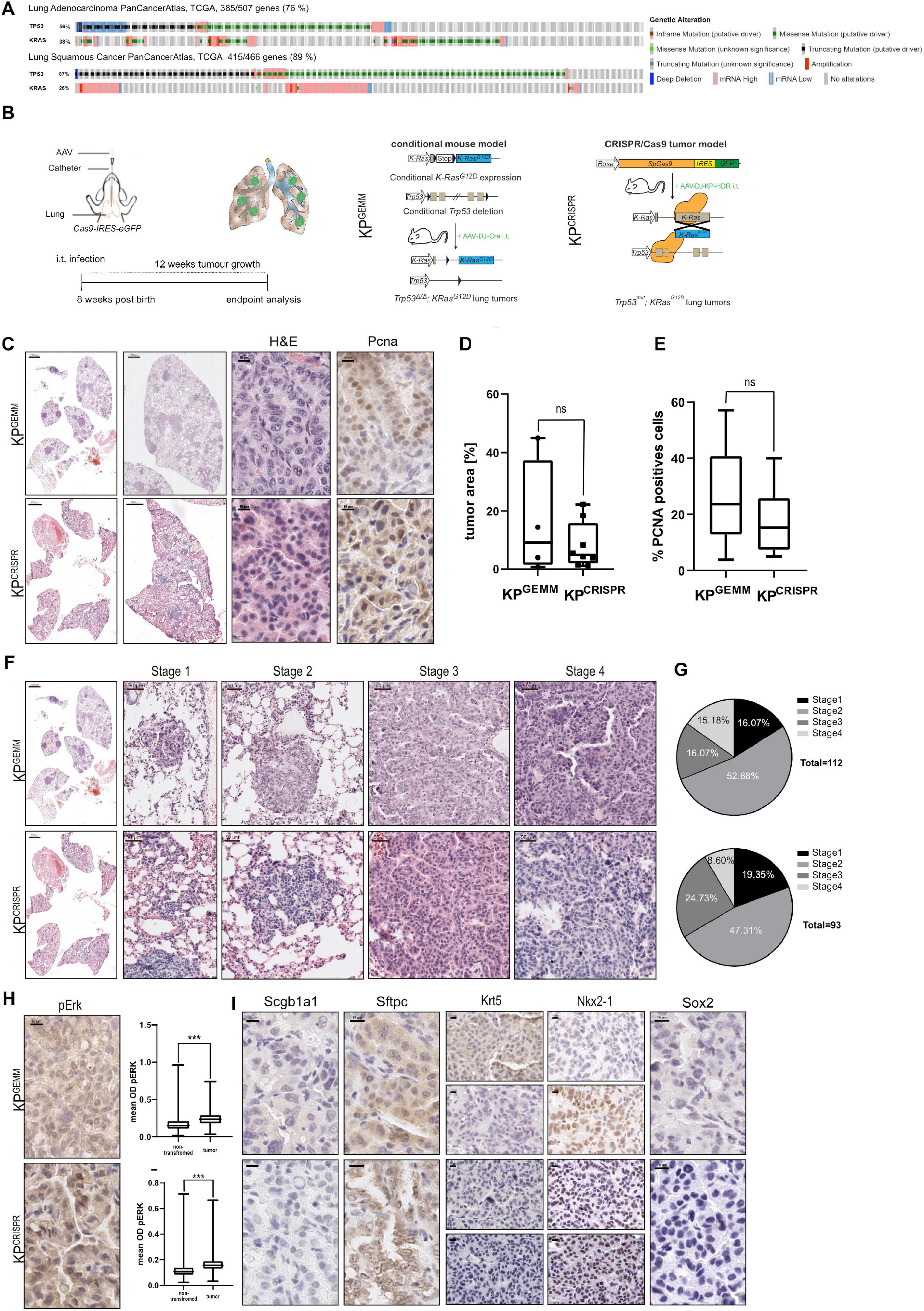
Figure 1. CRISPR/Cas9-mediated editingP of Trp53 and KRasG12D (KPCrispr) induces the formation of NSCLC that is indistinguishable from the classic GEMM model Trp53fl/fl:KRaslsl–G12D/wt (KPGEMM). (A) Analysis of occurring alterations in TP53 and KRAS in lung adenocarcinoma (upper panel) and lung squamous cancer (lower panel) (www.cbioportal.org). (B) Schematic diagram of the intratracheal tumor induction in C57BL6/J Trp53fl/fl:KRaslsl–G12D/wt (KPGEMM) via AAV-DJ-Cre and CRISPR/Cas9-mediated tumor modeling and targeting of p53Δ; KRasG12D in Rosa26Sor–CAGG–Cas9–IRES–GFP mice (KPCRISPR). (C) Representative H&E images of tumor-bearing animals 12 weeks post intratracheal infection (scale bars 2,000, 1,000, and 10 μm) and representative IHC analysis of PCNA. KPGEMM n = 4; KPCRISPR n = 8. (D) Quantification of % tumor area (normalized to total lung area) in KPGEMM n = 4 and KPCRISPR n = 8. The p value (p = 0.3423) was calculated using an unpaired two-tailed t test. (E) Quantification of % PCNA-positive cells in KPGEMM and KPCRISPR tumors with n > 20,000 events, respectively. The p value (p = 0.2027) was calculated using an unpaired two-tailed t test. (F) Representative H&E images of tumor stages in KPGEMM and KPCRISPR according to the WHO guidelines from 2015 (scale bars 2,000 and 50 μm). (G) Quantification of the tumor stages in KPGEMM n = 112 and KPCRISPR n = 93. (H) Representative IHC staining for pERK in KPGEMM and KPCRISPR and the quantification of the mean optical density (OD) of pERK in adjacent non-transformed tissue and tumors. The p value (p < 0.0001) was calculated using an unpaired two-tailed t test. KPGEMM n > 17,000 and KPCRISPR n > 18,000. (I) Representative IHC staining for Scgb1a1/CC10 and the ADC (SFTPC and NKX2-1) and SCC (Krt5 and Sox2) marker expression of KPGEMM and KPCRISPR animals. Please also see Supplementary Figure 1.
These data demonstrate that CRISPR-mediated genome editing of Trp53 and KRas results in tumors which are comparable to the classic GEMM model Trp53fl/fl:KRaslsl–G12D/wt.
Non-small cell lung cancer progression depends on the expression of several oncoproteins, such as the transcription factors cJUN and cMYC (Hartl, 2016; Sanchez-Vega et al., 2018). Furthermore, mutant KRas-driven tumors depend on the expression of NOTCH1/3, and the expression of NOTCH pathway members is associated with overall poor survival (Licciulli et al., 2013; Xu et al., 2014). Additionally, WNT signaling is involved in the maintenance of alveolar stem cells and the WNT effector β-catenin is reported to promote oncogenesis of KRas mutant NSCLC and is associated with poor survival (Pacheco-Pinedo and Morrisey, 2011; Nakayama et al., 2014). Analysis of publicly available expression data of NSCLC identified the common increase in total abundance for these transcription (co-)factors (Figure 2A). We therefore analyzed the protein abundance of these essential oncoproteins in KPGEMM and KPCRISPR and compared the overall abundance relative to non-transformed lung tissue in close proximity to primary tumors (n = 10 tumors and n > 10,000 cells per analysis, Student’s t test, Figures 2B–E). While the expression of the AP-1 transcription factor family member cJun was reduced in tumors compared with adjacent non-tumor tissue, the closely related family members JunB and JunD were significantly upregulated (Figure 2B). This observation was comparable between KPGEMM and KPCRISPR. Next, we analyzed the total protein abundance of the two NSCLC relevant NOTCH receptors–1 and 3. Here, we performed immunohistochemistry of the cleaved and transcriptionally active form of NOTCH1 (NICD1), as well as total and cleaved NOTCH3 (Figure 2C). In both models, KPGEMM and KPCRISPR, NICD1 and NOTCH3 were significantly increased in tumors (Figure 2C). β-Catenin was located predominantly in the cytosol in benign lung epithelial cells, with the exception of alveolar type 2 (AT2) stem cells. In tumors, β-catenin/CTNNB1 was predominantly expressed in the nucleus and significantly enriched (Figure 2D). All the aforementioned factors directly or indirectly regulate the expression of the oncoprotein cMYC and thereby converge on pathways regulating proliferation and transformation. We therefore stained for the oncoprotein cMYC (Figures 2E,F). cMYC was significantly upregulated in tumors compared with benign lung tissue, and overall increases were comparable between KPGEMM and KPCRISPR. We also assessed the protein abundance of the transcription factor ΔNp63, a crucial marker for squamous differentiation (Prieto-Garcia et al., 2020), to re-evaluate the development of SCC in the KPGEMM and KPCRISPR models (Figure 1I). This was required since Krt5+ tumor cells were detected in KPGEMM (Figure 1I). In both tumor entities, ΔNp63 was detectable in its respective stem cell niche, with basal stem cells residing in the trachea and bronchus (Wang et al., 2002; Supplementary Figure 2A). However, tumors were negative, indicating that both model systems developed ADC rather than SCC (Supplementary Figures 2A,B).
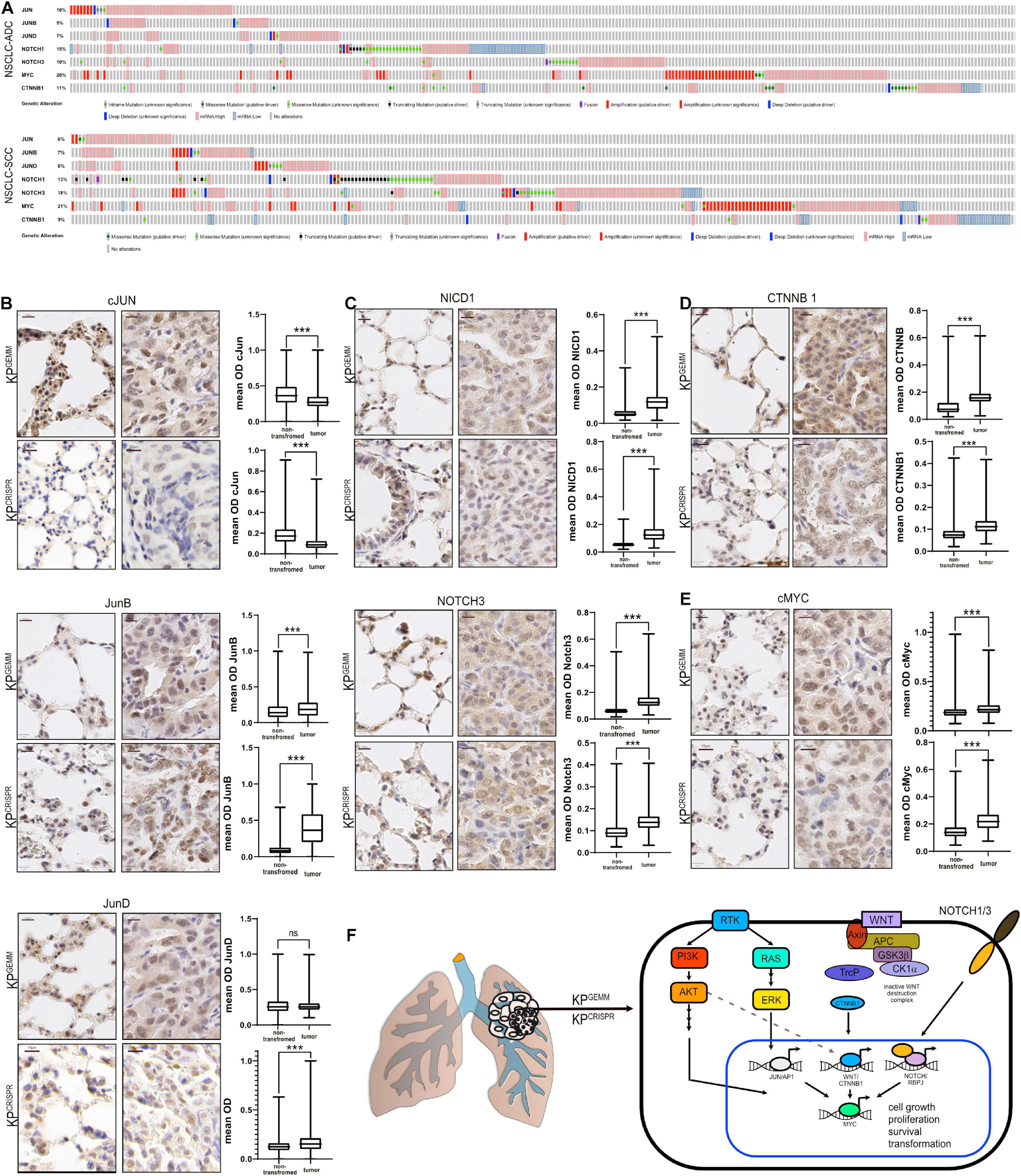
Figure 2. Abundance of NSCLC-associated oncogenic transcription factors in KPGEMM and KPCRISPR shows similarity by immunohistochemistry. (A) Analysis of occurring genetic alterations in lung adenocarcinoma (ADC) and lung squamous cancer (SCC) of associated oncogenes cJun, JUNB, JUND, NOTCH1, NOTCH3, cMYC, and CTNNB1 (www.cbioportal.org). (B) Immunohistochemical staining of cJun, JunB, and JunD in KPGEMM and KPCRISPR. Representative non-transformed trachea and tumor areas. Quantification of relative immunohistochemical staining intensity of cJun, JunB, and JunD in non-transformed lung tissue and tumors. Statistical analysis was performed using unpaired t test. P < 0.0001 (KPGEMM JunD p < 0.8598). Images were quantified using QuPath (version 0.2.8). Boxplots were generated using GraphPad Prism8. KPGEMM n > 5,000; KPCRISPR n > 1,500. P values were calculated using two–tailed t test statistical analysis. (C) Immunohistochemical staining of NICD1 and Notch3 in KPGEMM and KPCRISPR. Representative non-transformed trachea and tumor areas. Quantification of relative immunohistochemical staining intensity of NICD1 and Notch3 in non-transformed lung tissue and tumors. Statistical analysis was performed using unpaired t test. P < 0.0001. Images were quantified using QuPath (version 0.2.8). Boxplots were generated using GraphPad Prism8. KPGEMM n > 8,000; KPCRISPR n > 7,900. P values were calculated using two–tailed t test statistical analysis. (D) Immunohistochemical staining of CTNNB1 in KPGEMM and KPCRISPR. Representative non-transformed trachea and tumor areas. Quantification of relative immunohistochemical staining intensity of CTNNB1 in non-transformed lung tissue and tumors. Statistical analysis was performed using unpaired t test. P < 0.0001. Images were quantified using QuPath (version 0.2.8). Boxplots were generated using GraphPad Prism8. KPGEMM n > 7,000; KPCRISPR n > 5,500. P values were calculated using two–tailed t test statistical analysis. (E) Immunohistochemical staining of cMyc in KPGEMM and KPCRISPR. Representative non-transformed trachea and tumor areas. Quantification of relative immunohistochemical staining intensity of cMyc in non-transformed lung tissue and tumors. Statistical analysis was performed using unpaired t test. P < 0.0001. Images were quantified using QuPath (version 0.2.8). Boxplots were generated using GraphPad Prism8. KPGEMM n > 1,900; KPCRISPR n > 7,000. P values were calculated using two–tailed t test statistical analysis. (F) Schematic of the deregulated oncogenic transcription factors cJun, JunB, JunD, Notch1, Notch3, cMyc, and Ctnnb1 in NSCLC. Please also see Supplementary Figure 2.
Tumors developed via CRISPR-mediated genome editing of Trp53 and KRas activate the pathologically relevant AP-1, NOTCH, WNT, and MYC pathways, to the same extent as KPGEMM.
Since KPGEMM and KPCRISPR-derived primary tumors were indistinguishable on a microscopic scale and are driven by the same oncogenic pathways, we wondered if the similarities extend to the molecular level. To address this question, we isolated primary tumors 12 weeks post induction from KPGEMM and KPCRISPR and generated isogenic cell lines and subjected these to whole transcriptome analysis by RNA-sequencing (Figure 3A). As a control, we used wild-type lung tissue samples. Comparison of the transcriptional profile of two individual KPGEMM and KPCRISPR cells by RNA-sequencing revealed an overall similar transcriptomic profile (Spearman correlation R = 0.83, p < 2.2e–16, Figure 3B and Supplementary Figure 3A). In response to oncogenic transformation, KPGEMM and KPCRISPR tumor cells commonly downregulated ∼3,600 and upregulated ∼700 genes (logFC -2.5/+2.5 cutoff, Figure 3C and Supplementary Figure 3B). Among the commonly upregulated genes were the Hmga1 and 2, which encode for chromatin-associated proteins regulating transcription and being involved in metastasis; the oncogene cMYC; topoisomerase 2a; VGF; Smad3; and Cdc25c, a regulator of cell division. On the other hand, Erbb4 and Dkk3 were commonly downregulated factors (Figure 3D).
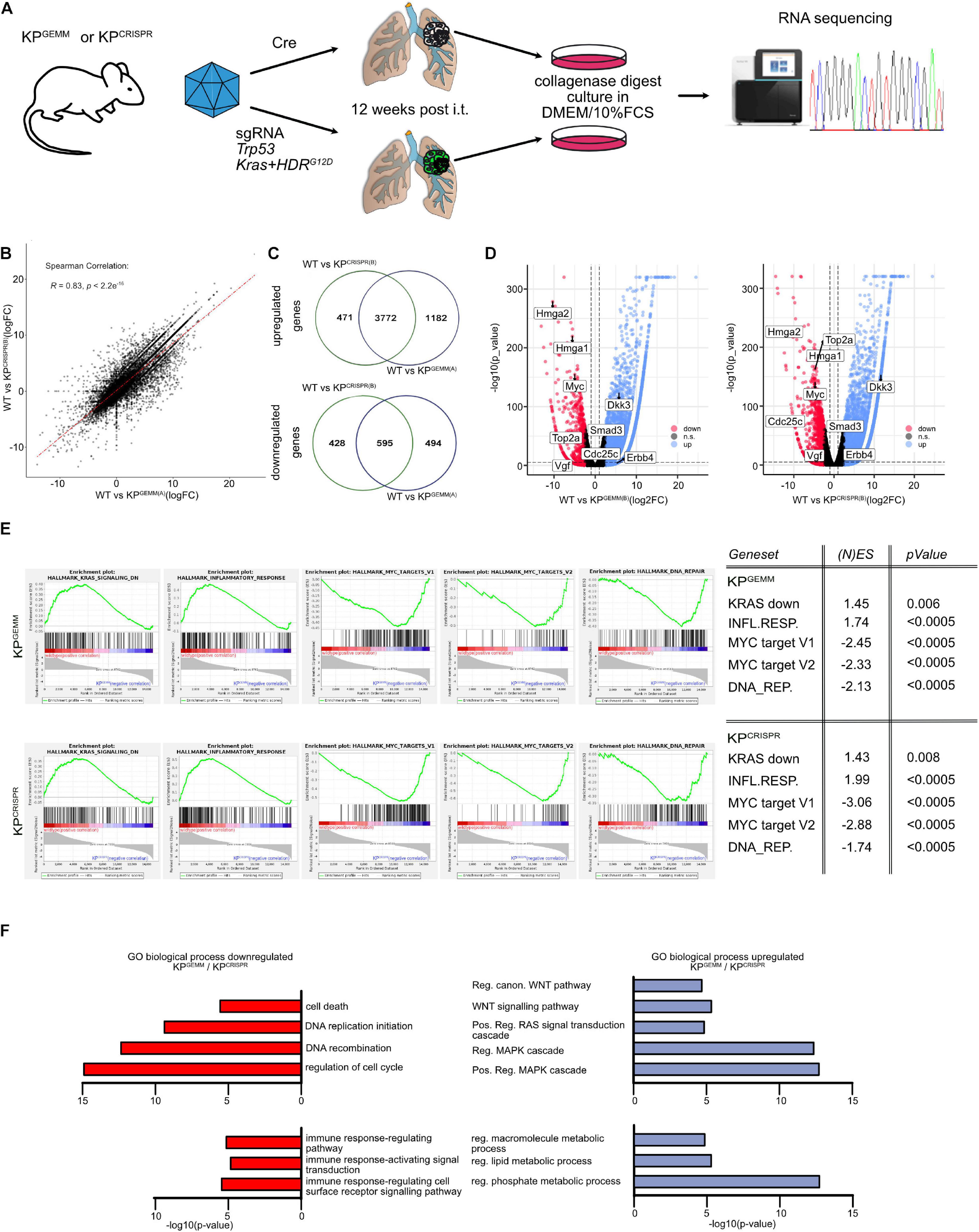
Figure 3. CRISPR-mediated targeting of Trp53 and KRas results in comparable adaptations on the molecular level of tumor cells when compared with KPGEMM. (A) Schematic diagram of the isolation of primary lung tumors from KPGEMM and KPCRISPR, culture, and subsequent RNA-sequencing. (B) Correlation plot of differentially regulated genes in WT versus KPGEMM(A) and WT versus KPCRISPR(A). Spearman correlation of R = 0.83, p ≤ 2.2e– 16. N = 3 each. (C) Venn diagram of individually and commonly up- and downregulated genes between WT versus KPGEMM(A) and WT versus KPCRISPR(A). N = 3 each (http://bioinformatics.psb. ugent.be/webtools/Venn/). (D) Volcano plot of genes up- and downregulated in WT versus KPGEMM(B) and KPCRISPR(B), respectively. Genes were defined as significant up- and downregulated with log2FC > 2.5. N = 3 each. (E) Gene set enrichment analyses of KRAS, inflammatory response, MYC target genes, and DNA REPAIR target gene expression in KPGEMM and KPCRISPR. (N)ES, normalized enrichment score and p values are depicted in the table. N = 3 each. (F) GO term analysis over biological processes of commonly down- and upregulated genes in KPGEMM(A) and KPCRISPR(A), relative to wild-type tissue. N = 3 each. Please also see Supplementary Figure 3.
To gain insights into the biological processes that are commonly up- or downregulated, we performed Gene Set Enrichment Analysis and GO term pathway analysis (Figures 3E,F). Here, we identified that gene sets associated with negative regulation upon oncogenic KRAS signaling as well inflammatory responses were downregulated in KPGEMM and KPCRISPR tumor, while tumor cells upregulated MYC target gene signatures and DNA repair pathways (Figure 3E). Not only were these factors deregulated in the murine cell lines but also showed a comparable expression in human NSCLC tumors, as seen by the analysis of publicly available datasets (Supplementary Figure 3D).
GO term pathway analysis identified pathways associated with cell death and cell cycle control that were commonly downregulated in KPGEMM and KPCRISPR tumor cells (Figure 3E). Furthermore, we found that pathways associated with immune responses were also toned down (Figure 3E). This is in line with recent publications demonstrating that mutations in the KRas oncogene contribute to immune-evasive phenotypes (van Maldegem and Downward, 2020). Analyzing pathways upregulated in tumor cells, we could identify that KPGEMM and KPCRISPR are driven by aberrant MAPK signaling and oncogenic Ras, as these pathways were significantly upregulated (Figure 3E). Not only did we identify increased nuclear Ctnnb1 abundance by histopathology, but we also observed an increase in WNT pathway activation by GO term analysis (Figure 3E). To accommodate increased proliferation and oncogenic transformation, tumor cells also upregulated metabolic pathways, such macromolecule synthesis and lipid and phosphate metabolism processes (Figure 3E).
It is noteworthy, despite all the similarities between KPGEMM versus KPCRISP when directly compared (KPGEMM vs. KPCRISP, Spearman correlation R = 0.93, Supplementary Figure 3E), that several genes are differentially regulated between KPGEMM and KPCRISPR (KPCRISPR up n = 1,612, KPGEMM up n = 918, Supplementary Figure 3F). As seen by Sanger sequencing of primary tumors, KPCRISPR predominantly harbored homozygous KRasG12D; in contrast, KPGEMM only carried one mutant allele (Supplementary Figure 1E; Chiosea et al., 2011). While gene expression of major key players in NSCLC, such as Keap1, Stk11/Lkb1, Kras, cJun, cMyc, Notch1, Notch3, or Nkx2-1, was not altered, KPCRISPR showed an increase in the expression of genes involved in Wnt signaling, while KPGEMM upregulated the Egfr signaling pathway (Supplementary Figures 3F,G).
Not only do KPGEMM and KPCRISPR show a high degree of similarity on a macroscopic scale, these data also demonstrate that classic and CRISPR-mediated targeting of Trp53 and KRas results in similar alterations on a molecular level. Furthermore, our data clearly highlighted that CRISPR-mediated genome editing is a suitable replacement of classic mouse models of NSCLC relying on the targeting of Trp53 and KRas.
Non-small cell lung cancer is one of the most mutated solid tumors known (Cancer Genome Atlas Research Network, 2012, 2014). Analysis of publicly available datasets demonstrates the frequent loss of various tumor suppressors. Among the most commonly mutated are E3 ligases, such as APC or KEAP1, key enzymes of the ubiquitin system, as well negative regulators of (stem) cell growth and transformation, such as STK11/LKB1 and PTEN (altered in 403/503 ADC (80%) and 437/477 SCC (94 (Figure 4A and Supplementary Figure 4A). Furthermore, mutations/alterations of these tumor suppressors negatively correlated with patient survival (Figure 4B). While loss of STK11 and PTEN significantly reduced overall survival (32.71 or 46.72 vs. 58.45 months, respectively), mutations in any of the four tumor suppressors negatively affected disease-free and progression-free survival (Figure 4C and Supplementary Figures 4B,C). To assess if CRISPR can be used to swiftly model these particular genetic alterations, we introduced an additional sgRNA cassette, comprising the murine U6 promoter, the guide RNA and trcRNA hybrid, into the KPCRISPR AAV backbone (Supplementary Figure 4D). Twelve weeks post viral application, mice were analyzed by immunohistopathology. Loss of the aforementioned tumor suppressors, APC, KEAP1, STK11/LKB1, and PTEN, significantly enhanced tumor burden and increased tumor cell proliferation when compared with KPCRISPR (Figure 4D and Supplementary Figure 4E). While mutations of Apc and Keap1 led to the development of ADC, targeting of Stk11/Lkb1 and Pten also led to the development of SCC tumors (Krt5+, P63+, TTF1–) (Figure 4D; Prieto-Garcia et al., 2020). Loss of these factors also resulted in an increase in protein abundance of the oncogenes cMYC and cJun (Figure 4D). CRISPR-mediated genome editing and tumor suppressor deletion were confirmed by immunohistochemistry. Apc, Keap1, Lkb1, and Pten were not detectable in the majority of developing tumors at endpoint, confirming successful genomic targeting of the tumor suppressors on the protein level (targeting efficacy as assessed by IHC: Apc: 80%; Keap1: 89%; Lkb1: 86%; Pten: 95%, Figure 4E). Targeting was further confirmed by IHC staining against known downstream target proteins. Loss of Apc resulted in the nuclear accumulation of β-catenin, while deletion of Keap1 led to the accumulation of its bona fide substrate, Nrf2/Nfe2l2 (Figure 4F). Targeting of Stk11/Lkb1 significantly upregulated tumor burden and tumor cell proliferation. These tumors also showed elevated protein abundance of Nrf2/Nfe2l2 (Figure 4E). Loss of Stk11/Lkbn1 impacts tumor cell metabolism and increases ROS production, while elevated abundance of Nrf2 establishes ROS homeostasis, enabling tumor cells to grow under stress conditions (Ma, 2013; Faubert et al., 2014; Kaufman et al., 2014; Galan-Cobo et al., 2019). Lastly, loss of Pten strongly synergized with mutations in Trp53 and KRas. Tumors devoid of Pten accumulated Nrf2/Nfe2l2 as well, which is in line with published data (Figure 4E; Rojo et al., 2014; Best et al., 2018).
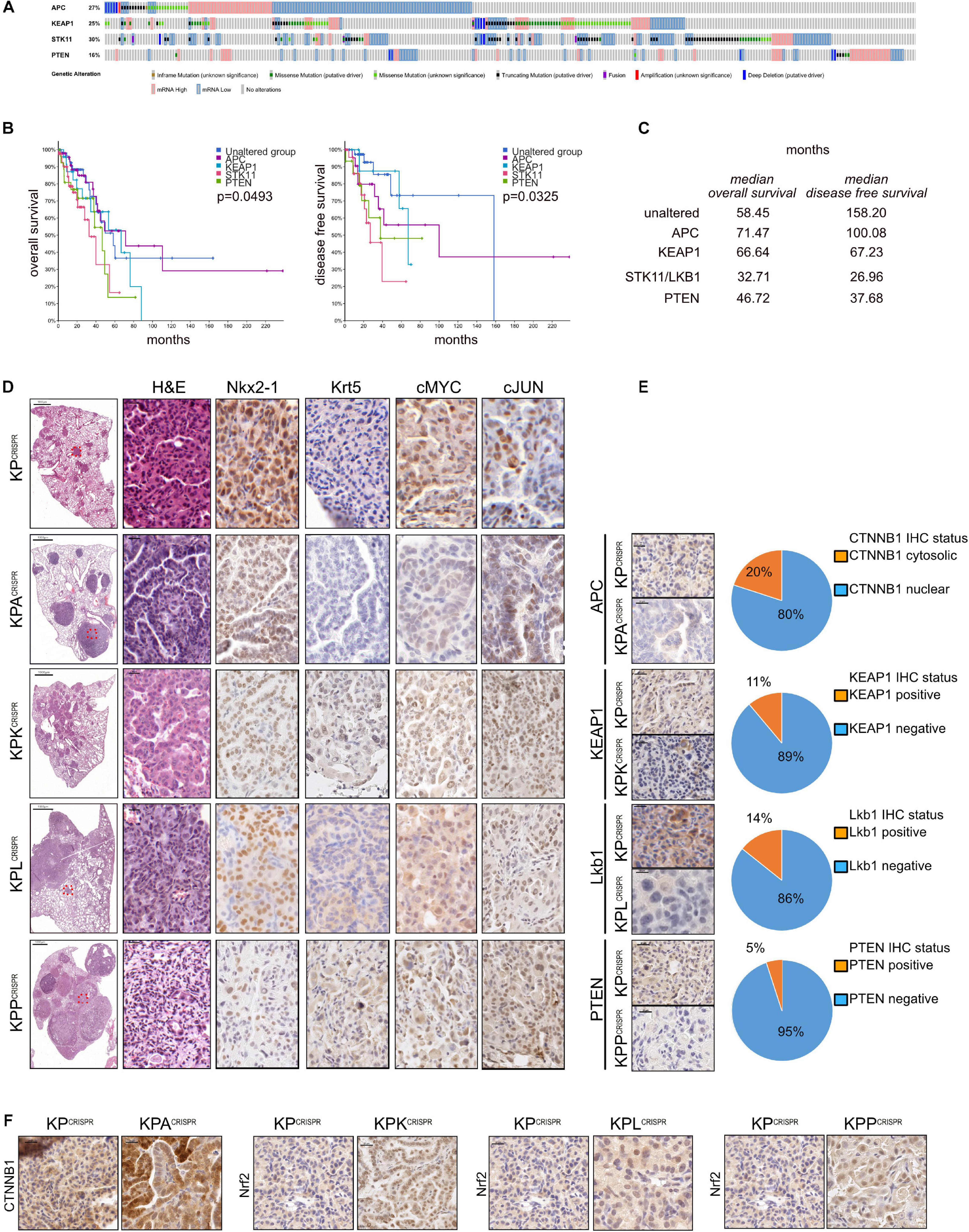
Figure 4. Rapid modeling of common loss-of-function mutations identified in NSCLC patients by CRISPR genome editing. (A) Analysis of occurring alterations in APC, KEAP1, STK11/LKB1, and PTEN in human NSCLC-ADC (www.cbioportal.org). (B) Kaplan–Meier plot of overall and disease-free survival in NSCLC-ADC patients. Shown are the months’ survival. Blue = unaltered group; purple = APC altered; blue = KEAP1 altered; orange = STK11 altered; green = PTEN altered. P < 0.005. (C) Median overall and disease-free survival of patients with the indicated alterations, relative to patients with no mutations in APC, KEAP1, STK11, or PTEN. (D) Representative H&E and immunohistochemical staining against ADC (NKX2-1) and SCC (Krt5) marker expression and the oncogenes cMyc and cJun of mice infected with AAV encoding sgRNA targeting KP, KP-Apc, KP-Keap1, KP-Lkb1, or KP-Pten. Scale bar 20 μm. (E) Representative immunohistochemical staining against encoding sgRNA targets Apc, Keap1, Lkb1, and Pten in KP, KP-Apc, KP-Keap1, KP-Lkb1, or KP-Pten. Scale bar 20 μm. Quantification of CTNNB1 nuclear to cytosolic ratio in KP versus KPA tumors. Quantification of Keap1-positive to negative tumors in mice infected with AAV encoding sgRNA targeting Keap1. Quantification of Lkb1/Stk11-positive to negative tumors in mice infected with AAV encoding sgRNA targeting Lkb1/Stk11. Quantification of Pten-positive to negative tumors in mice infected with AAV encoding sgRNA targeting Pten. (F) Representative immunohistochemical staining against Ctnnb1 in KPCRISPR and KPACRISPR, Nrf2 in KPCRISPR and KPKCRISPR, Nrf2 in KPCRISPR and KPLCRISPR, and Nrf2 in KPCRISPR and KPPCRISPR. Please also see Supplementary Figure 4.
CRISPR-mediated genome editing in vivo allows swift modeling of complex genetic alterations that are clinically relevant and occur in human NSCLC patients. It can therefore serve as a relevant surrogate model and potentially be used for therapy development.
To test if CRISPR-mediated gene editing can be applied in vivo without the need of a constitutive Cas9-expressing mouse strain, we used a dual AAV virus infection protocol (Figure 5A). Here, one AAV virion encoded the SpCas9 enzyme under the control of a short EFS promoter, while the second virion encoded the components necessary to target Trp53 and Stk11/Lkb1 and to oncogenic mutate KRas to KRasG12D, KPCRISPR, or KPLCRISPR, respectively. Balb/C mice were infected via the tracheal route with a mix of 2 ∗ 108 GC (genomic copies) of each virus in a total volume of 60 μl and tumor burden 12 weeks post infection was analyzed. Histopathological analysis of the dual viral-infected animals to single virus KPCRISPR revealed that, while total tumor numbers were significantly reduced (KPCRISPR–double n = x to KPCRISPR n = y), no gross morphological differences with regard to tumor grade were observed as both models displayed the development of stage I to stage IV tumors (Supplementary Figure 5A). The expression of lineage-specific markers, such as Nkx2-1/TTF-1 and Krt5, was comparable and tumors displayed activation of the Erk pathway, as seen by IHC staining against phosphor-Erk1/2 (Supplementary Figures 5B,C). Overall expression and abundance of oncoproteins was comparable with the KPCRISPR model system (Figure 2 and Supplementary Figure 5D). Loss of Lkb1, in combination with deletion of Trp53 and mutation of KRas, resulted in increased tumor incidence and larger tumors, when compared with KP targeting alone (Figure 5B). Proliferation was increased in KPL compared with KP and was comparable with the aforementioned KPL model system (Figures 5C and 4E). Successful targeting of Stk11 was confirmed by immunohistochemistry (Figure 5D). While constitutive SpCas9 enzyme expressing C57Bl6/J mice showed positive immunoreactivity with a SpCas9-specific antibody, the dual AAV-infected animals were devoid of staining (Figure 5E). Targeting of Stk11 also led to the establishment of KRT5+/TTF1– tumors, indicating that loss of this tumor suppressor leads to the simultaneous development of NSCLC-SCC and NSCLC-ADC (Figure 5F). This is in line with a previous observation we made in a constitutive CRISPR-driven mouse model (Prieto-Garcia et al., 2020). cMYC protein levels were elevated in tumors and cMyc and cJun total protein abundance was comparable to KPGEMM and KPCRISPR animals (Figures 4E, 5G).
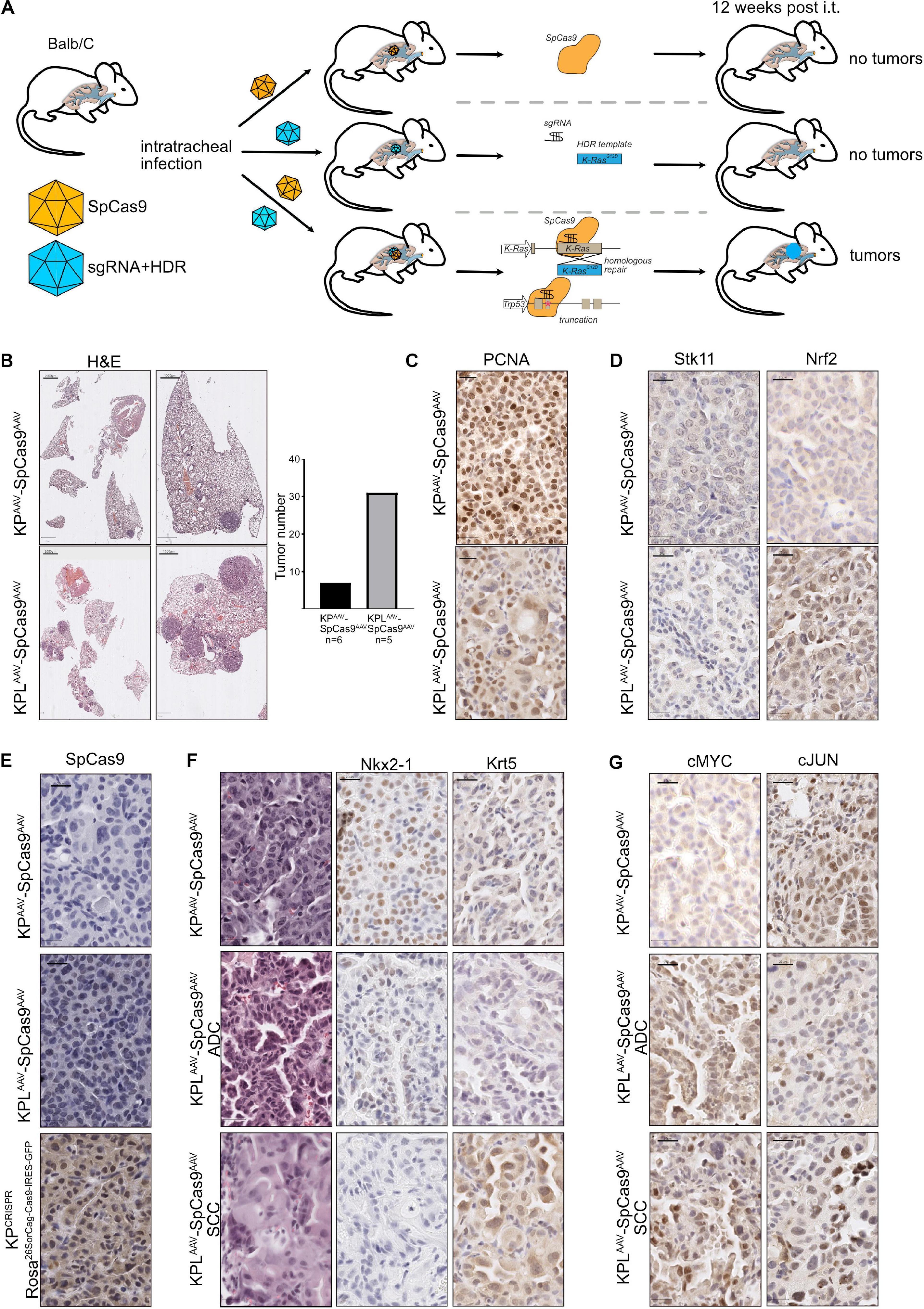
Figure 5. Dual viral approaches enhance the applicability of CRISPR for translational research, irrespective of the mouse strain. (A) Schematic diagram of the dual AAV approach of intratracheal infection of mice to induce spCAS9-mediated lung tumor formation. (B) Representative H&E images of tumor-bearing animals 12 weeks post intratracheal infection (scale bars 2,000 and 1,000 μm). Quantification of absolute tumor numbers in all KP or KPL animals co-infected with an AAV-spCas9. N = 6. (C) Representative IHC images of PCNA in KPAAVSpCas9AAV and KPLAAVSpCas9AAV (scale bar 20 μm). (D) Representative IHC images of Lkb1/Stk11 and Nrf2 in KPAAVSpCas9AAV and KPLAAVSpCas9AAV (scale bar 20 μm). (E) Representative IHC images of spCas9 in KPAAVSpCas9AAV and KPLAAVSpCas9AAV and KPCRISPR (Rosa26SorCag–Cas9–IRES–GFP) (scale bar 20 μm). (F) Representative IHC staining for Nkx2-1 and Krt5 in KPAAVSpCas9AAV and KPLAAVSpCas9AAV (scale bar 20 μm). (G) Representative IHC staining for cMyc and cJun in KPAAVSpCas9AAV and KPLAAVSpCas9AAV (scale bar 20 μm).
Thus, our data shows that CRISPR gene editing enables tumor onset irrespective of the mouse strain and does not require the constitutive expression of the enzyme Cas9.
Genetic modifications or deletions of tumor suppressors, such as Trp53, in combination with activating mutations in proto-oncogenes, such as KRas, are a prerequisite to facilitate a tumor-initiating event (Best et al., 2019). While for the last two decades in mouse models, loss of tumor suppressors was initiated by either germ line deletion or conditional loss (mediated by the activity of a recombinase such as Cre), we wondered if CRISPR-mediated genome editing is capable of facilitating similar effects.
Comparing primary tumors, established by either Cre recombinase-mediated genetic loss of Trp53 and activation of a dominant mutant KRasG12D allele (KPGEMM) versus CRISPR-mediated genomic targeting and mutation (KPCRISPR), we could not detect gross differences on a microscopic level. Mice had a comparable tumor burden and established tumors had a comparable level of proliferative cancer cells. Overall expression of lung and tumor lineage markers was indistinguishable, demonstrating that the genetic alterations inflicted, and not the method used, define the histopathologic features or primary tumors. On the other hand, for primary lung tumors of various grades, ranging from I to IV, we did not detect any metastasis within the analyzed time frame of 12 weeks post tumor induction. This observation is in line with previous reports, as the onset of metastasis, which occurs in Trp53R173H/wt:KRaslsl–G12D/wt conditional mice at a late time point exceeding 12 weeks (Zheng et al., 2007). It will be interesting for future work to extend the time frame to allow and investigate if KPCRISPR animals do form metastasis.
It is noteworthy, however, that, in contrast to KPCRISPR, KPGEMM also developed with low-frequency primary tumors expressing the SCC markers Krt5 while being negative for the pathological relevant markers ΔNp63 and Sox2. It could be possible that these tumor cells are on the trans-differentiation to SCC. We previously reported the expression of SCC markers in this mouse model, and others have reported that trans-differentiation can occur in KRasG12D-driven ADC to SCC (Han et al., 2014; Prieto-Garcia et al., 2020). Despite the extensive use of the KPGEMM model in the past, it was reported to exclusively only give rise to ADC (Jackson et al., 2001). In contrast to other publications, where adenoviruses or lentiviruses were utilized to transmit genomic information, we used AAV virions for Cre recombinase delivery. As loss-of-function mutations in Trp53, concomitant with aberrant MAPK pathway signaling, are common features found in ADC and SCC, we assume that the viral tropism of the AAV shuffle capsid DJ is responsible for this observation.
Similarities between KPCRISPR and KPGEMM are not limited to the expression of histopathologic markers, but extend to the transcriptional programs identified in tumor cells. Irrespective of the targeting strategy, tumor cell programs adapted to transformation by downregulating cell death programs as well the immune response signatures. As the analyzed tumors were driven by oncogenic mutations in KRas, several key pathways were found to be upregulated, including MAPK, RAS, and WNT. To accommodate enhanced proliferation, tumors also readjusted various metabolic pathways. One significant difference we identified between the two NSCLC models is that targeting of KRas by CRISPR led to a homozygous recombination event, resulting in KRasG12D/G12D. This is in contrast to the genetically predefined heterozygous expression of the oncogenic KRasG12D/wt in the KPGEMM models. As homozygous breeding of the conditional mouse model is not feasible due to embryonic lethality observed in biallelic KRaslsl–G12D/lsl–G12D embryos, CRISPR enables us to model and analyze the consequences of homozygous mutations in KRas, as NSCLC patients can present both genetic modifications (Chiosea et al., 2011; Ferrer et al., 2018).
To expand the repertoire of targeted alleles by CRISPR, one only requires to introduce additional sgRNA cassettes with a high on-target, low off-target rates. As we reported in our study, the implementation of CRISPR helps to speed up the time required from target identification to in vivo validation tremendously.
Cloning into available viral vectors encoding for lentivirus or AAV, followed by subsequent virus production, can be completed within one and a half weeks and requires minimal financial investment. This is an improvement in flexibility not seen before which will significantly change the way we will conduct our research in the future. With the development of novel Cas9 systems, the off-target rate can be significantly reduced (Naeem et al., 2020).
One has to consider, however, that the implementation of CRISPR for target gene modification will result in individual and heterogeneous genetic events. Here, the classic conditional targeting of genetic loci will produce genetically identical recombination events. Depending on the scientific question one will address, the classic mouse model can be advantageous.
Our data demonstrated that for gene deletions induced by CRISPR targeting of the tumor suppressor Trp53, the overall molecular changes were comparable among individually tested cell lines, but not identical. Hence, primary tumor cell lines generated by CRISPR should be treated as individual tumor lines and not as clones.
A previous work, pioneered by the Tyler Jacks Laboratory, successfully combined classic conditional Trp53fl/fl:KRaslsl–G12D/wt animals with lentiviral delivery of the CRISPR enzyme Cas9, guide RNA targeting similar tumor suppressor as presented in our study (Apc and Pten), and the Cre recombinase to induce loss of Trp53 and activate the expression of the oncogenic KRasG12D allele (Sanchez-Rivera et al., 2014). This strategy enabled the rapid expansion of already existing in vivo models and also enabled the long-term expression of target genes or encoded additional genetic information, such as shRNAs (Sanchez-Rivera et al., 2014; Xue et al., 2014; Ng et al., 2020). Lentiviruses, in comparison to AAV, have a significantly higher total packaging capacity (10 vs. 4.8 kb); however, they are stably integrated and advertently can result in the disruption of additional genetic loci, including tumor suppressors, while AAV reside as episomal circles in infected cells and integrate with very low frequency (<0.05%) (Inagaki et al., 2008; Yang et al., 2008). A second strategy is the use of adenoviruses, encoding sgRNA and Cre recombinase. Here, the AdEasy I and II recombinant viral production system has enabled researchers for several years to produce high-titer viruses (Luo et al., 2007). Similar to AAV, AdV also reside as episomes in infected cells, but are rapidly cleared. While packaging capacity is higher than in AAV (AdEasy I 7.5 kb, AdEasy II 12 kb), recombination of the pShuttle vector with the AdEasy I backbone can prove difficult and research labs require biosafety level II tissue cultures to produce and propagate adenoviruses and lentivirus (Collins et al., 2017). AAV, on the other hand, are nonpathogenic and high-titer viruses can be produced with ease in biosafety level I tissue cultures.
To investigate the putative tumor-suppressive or oncogenic role of a protein in NSCLC, laboratories would have to generate and establish a novel mouse line, which is a time-consuming endeavor and limited by the availability of the required facilities. Alternatively, one would purchase an available genetically engineered mouse model from commercial suppliers harboring the (available) conditional alleles. Both methods require the combination of the pre-existing mouse strains with the newly acquired lines, necessitating at least two to three generations of breeding until the desired genotype becomes available. Under optimal conditions, this process alone can take up to half a year prior to starting the experiment.
Introducing CRISPR gene targeting, at least for NSCLC, helps to implement the 3R guidelines for in vivo research: it significantly reduces the overall amount of generated animals (Reduce) and, with applied biostatistics, reduces animals used for experiments even further (Refine). Our strategies reported in this study can also be applied to organoid model systems and could therefore also help to partially Replace in vivo tumor models (Figure 6).
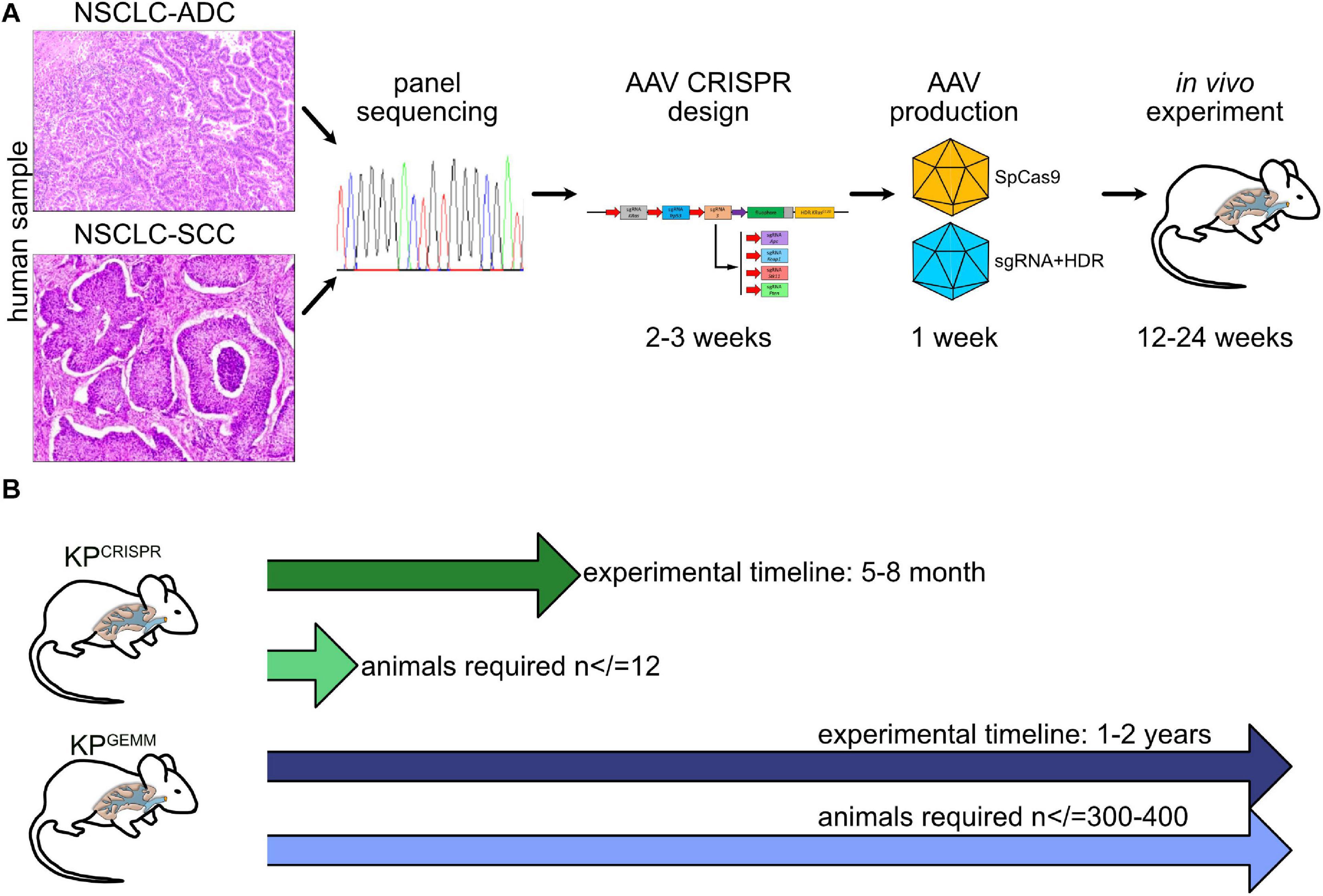
Figure 6. Schematic model of CRISPR NSCLC models. (A) Workflow from “bed” to in vivo tumor model by CRISPR. (B) Comparison of time frame and animals used between KPGEMM and KPCRISPR.
CRISPR has revolutionized the way we conduct our research. It is here to stay and will reshape cancer research for the foreseeable future.
The HEK-293T cell lines were obtained from Agilent. HEK-293T cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (FCS)/1% Pen-Strep. Cell lines were authenticated by STR profiling. Cells were routinely tested for mycoplasma via PCR.
DNA transfection was performed by adding a mix of 2.5 μg plasmid DNA, 200 μl serum-free medium, and 5 μl PEI to the cells seeded in a six-well plate (60% confluence). After 6 h incubation at 37°C, the medium was changed to full supplemented medium, and finally, cells were collected after 48 h for experimental purposes. For viral infection, AAVs or lentiviruses (MOI = 10) were added to the medium in the presence of polybrene (5 μg/ml) and incubated at 37°C for 4 days. The selection of infected cells was performed with 2.5 μg/ml puromycin for 72 h, 250 μg/ml neomycin for 2 weeks, or FACS-sorting RFP/GFP-positive cells (FACS Canto II BD).
Primary lung cancer cell lines were obtained from 12-week-old mice as previously described (Prieto-Garcia et al., 2020). At the endpoint of the experiment, mice were sacrificed and lung tumors isolated. Tissue was digested in collagenase I (100 U/ml in PBS for 30 min at 37°C), and after stopping the reaction with FCS, the mixture was centrifuged and resuspended in DMEM (Gibco) supplemented with 10% fetal bovine serum (FCS) and 1% Pen-Strep. Fibroblasts were counter-selected by selective trypsinization and homogeneous cell clusters were clonally expanded.
Primary lung cancer samples were obtained from 12-week-old mice as previously described (Prieto-Garcia et al., 2020) and embedded in paraffin and sectioned at 4 μm using a microtome (Leica). Before staining, slides were deparaffinized and rehydrated using the following protocol: 3 × 5 min in Histo-Clear®, 2 × 3 min in EtOH (100%), 2 × 3 min in EtOH (95%), 2 × 3 min in EtOH (70%), 3 min in EtOH (50%), and 3 min in H2O. Slides were stained with hematoxylin and eosin. For IHC, primary antibodies were incubated at 4°C or for 3 h at 37°C, followed by subsequent incubation with the secondary antibody for 1 h at room temperature. After antibody exposure, slides were washed twice with TBS and stained with the DAB staining solution. The stained samples were mounted with CytosealTM 60, and IHC images were recorded using either Pannoramic DESK scanner DW II or Roche Ventana DP 200 slide scanner and analyzed with the CaseViewer software (3DHISTECH) and QuPath1 ,2 as indicated at the website3.
sgRNAs were designed using the CRISPRtool4.
Viruses were synthetized in HEK-293T cells. For AAV production, cells were co-transfected with the plasmid of interest (10 μg), pHelper (15 μg), and pAAV-DJ (10 μg) using PEI (70 μg/15 cm dish). AAV virus isolation from transfected cells was performed as previously described (Prieto-Garcia et al., 2020).
In detail, for the production of AAVs, 5 *106 HEK-293T cells were seeded in 15 cm cell culture dishes and cultivated for 24 h or until a confluence of ∼60–70% was achieved. In order to gain a high titer, three to four plates for one virus were used, respectively, and pooled during harvest.
Cells were transfected with the pRepCap (pRC), the cis-plasmid (pAAV), and the pAdDeltaF6 (Table 1) in a 1:1:2 molar ratio. Therefore, the DNA was mixed in 2 ml DMEM (w/o FCS), and polyethylenimine (PEI) (DNA:PEI ratio of 1:2, e.g., 45 μg DNA = 90 μg PEI) was added. The mixture was incubated 15 min at room temperature and added dropwise to the plates.
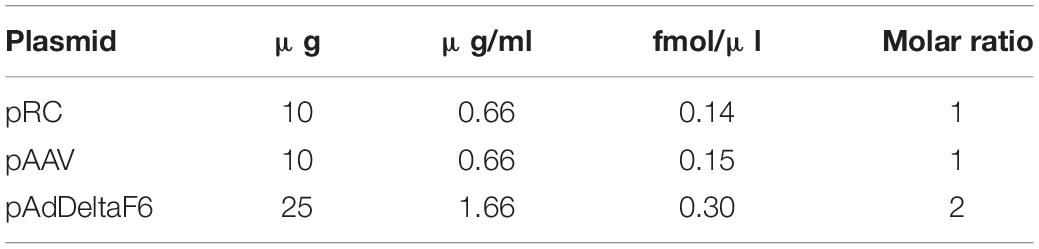
Table 1. DNA amount for AAV plasmids.
To harvest the AAV, cells and the supernatant were collected after 96 h and transferred into a 50-ml conical tube. At first, NaCl was added (f.c. 0.5 M) and slowly mixed for 1 h at 4°C. Next, chloroform was added (f.c. 10%) and slowly mixed for 30 min at 4°C. Eventually, the suspension was centrifuged at 2,000 × g for 30 min at 4°C. The water phase was transferred into a new conical tube and PEG 8,000 was added (f.c. 10%) and mixed well. The AAV can be precipitated for 90 min or overnight at 4°C.
After the centrifugation at 2,000 × g for 20 min at 4°C, the pellet was dissolved in PBS Mg2+/Ca2+ or AAV resuspension buffer (PBS + 0.001% Pluronic F68 + 200 mM NaCl) (∼100 μl/15 cm dish used), and protease inhibitor and DNase/RNase were added (f.c. 1 ×). Then, it was incubated for 2 h at 37°C, added with chloroform (1:1 ratio), and centrifuged at 12,000 × g for 5 min at 4°C. The chloroform step was repeated and the water phase was collected. Finally, the product was titrated or stored at −80°C.
All in vivo experiments were approved by the Regierung Unterfranken and the ethics committee under license numbers 2532-2-362, 2532-2-367, 2532-2-374, and 2532-2-1003. The mouse strains used for this publication are listed. All animals are housed in standard cages in pathogen−free facilities on a 12−h light/dark cycle with ad libitum access to food and water. FELASA2014 guidelines were followed for animal maintenance.
Adult mice were anesthetized with isoflurane and intratracheally intubated with 60 μl AAV virus (3 × 107 PFU) as previously described (Prieto-Garcia et al., 2020). Viruses were quantified using the AAV Titration by qPCR protocol from Addgene (Aurnhammer et al., 2012). Animals were sacrificed by cervical dislocation and lungs were fixed using 5% NBF.
RNA concentration was determined with a NanoDrop spectrophotometer and analyzed on a Fragment Analyzer (Advanced Analytical) for RNA quality; 400 ng of RNA was then used and enriched for poly(A) mRNA using the NEBNext Poly(A) mRNA Magnetic Isolation Module (#E7490L) according to the manufacturer’s instructions. The fragmentation time was 15 min at 94°C for every sample [RNA quality number (RQN) > 8], except for the three wild-type replicates that showed an RQN between 4 and 7. For those samples, the fragmentation time was reduced to 7.5 min. After poly(A) mRNA enrichment and fragmentation, the NEBNext® UltraTM II Directional RNA Library Prep Kit for Illumina® (#E7760L) was used for library preparation according to the manufacturer’s instructions with 9 cycles of PCR amplification.
RNA-sequencing was performed with Illumina NextSeq 500 as described previously (Buchel et al., 2017).
Base calling was performed using Illumina’s BaseSpace platform and overall sequencing quality was analyzed using the multiQC script. Reads were aligned to the murine genome (mm10) using Bowtie2 v2.4.1 (Langdon, 2015), and samples were normalized to the number of mapped reads in the smallest sample. For differential gene expression analysis, reads per gene were counted with the “featureCounts” function from the R (version 4.0.3) package “GenomicFeatures” (version 1.40.1), and none or weakly expressed genes were removed (mean read count over all samples <6). Differentially expressed genes were called using edgeR (Robinson et al., 2010) and the resulting p values were corrected for multiple testing by false discovery rate (FDR) calculations by the Benjamini–Hochberg method. GSEA analyses (Subramanian et al., 2007) were done with Signal2Noise metric and 1,000 permutations against C2, C5, C6, and hallmark MsigDB. Reactome analysis was performed with PANTHER (Mi et al., 2013) using the “Statistical overrepresentation test” tool with default settings. Genes were considered significantly downregulated for reactome analysis when Log2FC > 2.5 and FDR p value < 0.05.
All publicly available data and software used for this publication are listed (Supplementary Material Table). OncoPrints were generated using cBioPortal (Gao et al., 2013). Briefly, OncoPrints generate graphical representations of genomic alterations, somatic mutations, copy number alterations, and mRNA expression changes. TCGA data was used for the different analyses.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE163087.
The animal study was reviewed and approved by the Regierung Unterfranken and the Ethics Committee under the license numbers 2532-2-362, 2532-2-367, 2532-2-374, and 2532-2-1003.
OH and MD: conceptualization. OH (in vitro), AB, JS, MR (RNA-Seq), CP-G (Biochemistry), CM, and WS (Mass Spec): methodology. MR (Bioinformatics), OH, MC, MTR, and MD (Pathology): formal analysis. OH, MR, TF, NP, MTR, and MD: investigation. AO, CD, MC, FB, MTR, and MD: resources. MD: writing—original draft. OH, MR, MTR, CD, AO, AS, and MD: writing—review and editing. MD: supervision and funding acquisition.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
CP-G and OH are supported by the German Cancer Aid via grant 70112491. MR was funded by the DFG-GRK 2243 and IZKF B335. MD and MR are funded by the German Israeli Foundation grant 1431. TF was funded by the IZKF program Z2/CS-1. NP was funded by the MSNZ research center Würzburg.
We are grateful to the animal facility and to Barbara Bauer from the Biocenter, University Würzburg.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.641618/full#supplementary-material
Akcakaya, P., Bobbin, M. L., Guo, J. A., Malagon-Lopez, J., Clement, K., Garcia, S. P., et al. (2018). In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 561, 416–419. doi: 10.1038/s41586-018-0500-9
Asao, T., Takahashi, F., and Takahashi, K. (2019). Resistance to molecularly targeted therapy in non-small-cell lung cancer. Respir. Investig. 57, 20–26. doi: 10.1016/j.resinv.2018.09.001
Aurnhammer, C., Haase, M., Muether, N., Hausl, M., Rauschhuber, C., Huber, I., et al. (2012). Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods 23, 18–28. doi: 10.1089/hgtb.2011.034
Best, S. A., De Souza, D. P., Kersbergen, A., Policheni, A. N., Dayalan, S., Tull, D., et al. (2018). Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. Cell Metab. 27, 935–43e4.
Best, S. A., Ding, S., Kersbergen, A., Dong, X., Song, J. Y., Xie, Y., et al. (2019). Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat. Commun. 10:4190.
Bonanno, L., Zulato, E., Pavan, A., Attili, I., Pasello, G., Conte, P., et al. (2019). LKB1 and tumor metabolism: the interplay of immune and angiogenic microenvironment in lung cancer. Int. J. Mol. Sci. 20, 1874. doi: 10.3390/ijms20081874
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Buchel, G., Carstensen, A., Mak, K. Y., Roeschert, I., Leen, E., Sumara, O., et al. (2017). Association with Aurora-A Controls N-MYC-dependent promoter escape and pause release of RNA polymerase II during the cell cycle. Cell Rep. 21, 3483–3497. doi: 10.1016/j.celrep.2017.11.090
Cancer Genome Atlas Research Network (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525. doi: 10.1038/nature11404
Cancer Genome Atlas Research Network (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. doi: 10.1038/nature13385
Chiosea, S. I., Sherer, C. K., Jelic, T., and Dacic, S. (2011). KRAS mutant allele-specific imbalance in lung adenocarcinoma. Mod. Pathol. 24, 1571–1577.
Collins, D. E., Reuter, J. D., Rush, H. G., and Villano, J. S. (2017). Viral vector biosafety in laboratory animal research. Comp. Med. 67, 215–221.
Day, C. P., Merlino, G., and Van Dyke, T. (2015). Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163, 39–53. doi: 10.1016/j.cell.2015.08.068
Doroshow, D. B., Sanmamed, M. F., Hastings, K., Politi, K., Rimm, D. L., Chen, L., et al. (2019). Immunotherapy in non-small cell lung cancer: facts and hopes. Clin. Cancer Res. 25, 4592–4602. doi: 10.1158/1078-0432.ccr-18-1538
DuPage, M., Dooley, A. L., and Jacks, T. (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc. 4, 1064–1072. doi: 10.1038/nprot.2009.95
Faubert, B., Vincent, E. E., Griss, T., Samborska, B., Izreig, S., Svensson, R. U., et al. (2014). Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc. Natl. Acad. Sci. U.S.A. 111, 2554–2559.
Ferrer, I., Zugazagoitia, J., Herbertz, S., John, W., Paz-Ares, L., and Schmid-Bindert, G. (2018). KRAS – Mutant non-small cell lung cancer: from biology to therapy. Lung Cancer 124, 53–64. doi: 10.1016/j.lungcan.2018.07.013
Galan-Cobo, A., Sitthideatphaiboon, P., Qu, X., Poteete, A., Pisegna, M. A., Tong, P., et al. (2019). LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 79, 3251–3267. doi: 10.1158/0008-5472.can-18-3527
Gao, J., Aksoy, B. A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S. O., et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6:l1.
Han, X., Li, F., Fang, Z., Gao, Y., Li, F., Fang, R., et al. (2014). Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat. Commun. 5:3261.
Hartl, M. (2016). The quest for targets executing MYC-dependent cell transformation. Front. Oncol. 6:132. doi: 10.3389/fonc.2016.00132
Inagaki, K., Piao, C., Kotchey, N. M., Wu, X., and Nakai, H. (2008). Frequency and spectrum of genomic integration of recombinant adeno-associated virus serotype 8 vector in neonatal mouse liver. J. Virol. 82, 9513–9524. doi: 10.1128/jvi.01001-08
Ireson, C. R., Alavijeh, M. S., Palmer, A. M., Fowler, E. R., and Jones, H. J. (2019). The role of mouse tumour models in the discovery and development of anticancer drugs. Br. J. Cancer 121, 101–108. doi: 10.1038/s41416-019-0495-5
Jackson, E. L., Olive, K. P., Tuveson, D. A., Bronson, R., Crowley, D., Brown, M., et al. (2005). The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 65, 10280–10288. doi: 10.1158/0008-5472.can-05-2193
Jackson, E. L., Willis, N., Mercer, K., Bronson, R. T., Crowley, D., Montoya, R., et al. (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248. doi: 10.1101/gad.943001
Kaufman, J. M., Amann, J. M., Park, K., Arasada, R. R., Li, H., Shyr, Y., et al. (2014). LKB1 Loss induces characteristic patterns of gene expression in human tumors associated with NRF2 activation and attenuation of PI3K-AKT. J. Thorac. Oncol. 9, 794–804. doi: 10.1097/jto.0000000000000173
Langdon, W. B. (2015). Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData Min. 8:1.
Licciulli, S., Avila, J. L., Hanlon, L., Troutman, S., Cesaroni, M., Kota, S., et al. (2013). Notch1 is required for Kras-induced lung adenocarcinoma and controls tumor cell survival via p53. Cancer Res. 73, 5974–5984. doi: 10.1158/0008-5472.can-13-1384
Lim, Z. F., and Ma, P. C. (2019). Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 12:134.
Luo, J., Deng, Z. L., Luo, X., Tang, N., Song, W. X., Chen, J., et al. (2007). A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protoc. 2, 1236–1247. doi: 10.1038/nprot.2007.135
Ma, Q. (2013). Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426. doi: 10.1146/annurev-pharmtox-011112-140320
Mengoli, M. C., Longo, F. R., Fraggetta, F., Cavazza, A., Dubini, A., Ali, G., et al. (2018). The 2015 World Health Organization classification of lung tumors: new entities since the 2004 Classification. Pathologica. 110, 39–67.
Mi, H., Muruganujan, A., Casagrande, J. T., and Thomas, P. D. (2013). Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–1566. doi: 10.1038/nprot.2013.092
Mogi, A., and Kuwano, H. (2011). TP53 mutations in nonsmall cell lung cancer. J. Biomed. Biotechnol. 2011:583929.
Naeem, M., Majeed, S., Hoque, M. Z., and Ahmad, I. (2020). Latest developed strategies to minimize the off-target effects in CRISPR-Cas-mediated genome editing. Cells 9:1608. doi: 10.3390/cells9071608
Nakayama, S., Sng, N., Carretero, J., Welner, R., Hayashi, Y., Yamamoto, M., et al. (2014). beta-catenin contributes to lung tumor development induced by EGFR mutations. Cancer Res. 74, 5891–5902. doi: 10.1158/0008-5472.can-14-0184
Ng, S. R., Rideout, W. M. III, Akama-Garren, E. H., Bhutkar, A., Mercer, K. L., Schenkel, J. M., et al. (2020). CRISPR-mediated modeling and functional validation of candidate tumor suppressor genes in small cell lung cancer. Proc. Natl. Acad. Sci. U.S.A. 117, 513–521. doi: 10.1073/pnas.1821893117
Pacheco-Pinedo, E. C., and Morrisey, E. E. (2011). Wnt and Kras signaling-dark siblings in lung cancer. Oncotarget 2, 569–574. doi: 10.18632/oncotarget.305
Papillon-Cavanagh, S., Doshi, P., Dobrin, R., Szustakowski, J., and Walsh, A. M. (2020). STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open 5:e000706. doi: 10.1136/esmoopen-2020-000706
Platt, R. J., Chen, S., Zhou, Y., Yim, M. J., Swiech, L., Kempton, H. R., et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. doi: 10.1016/j.cell.2014.09.014
Pottier, C., Fresnais, M., Gilon, M., Jerusalem, G., Longuespee, R., and Sounni, N. E. (2020). Tyrosine kinase inhibitors in cancer: breakthrough and challenges of targeted therapy. Cancers (Basel) 12:731. doi: 10.3390/cancers12030731
Prieto-Garcia, C., Hartmann, O., Reissland, M., Braun, F., Fischer, T., Walz, S., et al. (2020). Maintaining protein stability of Np63 via USP28 is required by squamous cancer cells. EMBO Mol. Med. 12:e11101.
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Rojo, A. I., Rada, P., Mendiola, M., Ortega-Molina, A., Wojdyla, K., Rogowska-Wrzesinska, A., et al. (2014). The PTEN/NRF2 axis promotes human carcinogenesis. Antioxid. Redox Signal. 21, 2498–2514. doi: 10.1089/ars.2014.5843
Ruiz, E. J., Diefenbacher, M. E., Nelson, J. K., Sancho, R., Pucci, F., Chakraborty, A., et al. (2019). LUBAC determines chemotherapy resistance in squamous cell lung cancer. J. Exp. Med. 216, 450–465. doi: 10.1084/jem.20180742
Sanchez-Rivera, F. J., Papagiannakopoulos, T., Romero, R., Tammela, T., Bauer, M. R., Bhutkar, A., et al. (2014). Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431. doi: 10.1038/nature13906
Sanchez-Vega, F., Mina, M., Armenia, J., Chatila, W. K., Luna, A., La, K. C., et al. (2018). Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–37.e10.
Subramanian, A., Kuehn, H., Gould, J., Tamayo, P., and Mesirov, J. P. (2007). GSEA-P: a desktop application for gene set enrichment analysis. Bioinformatics 23, 3251–3253. doi: 10.1093/bioinformatics/btm369
Talmadge, J. E., Singh, R. K., Fidler, I. J., and Raz, A. (2007). Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am. J. Pathol. 170, 793–804. doi: 10.2353/ajpath.2007.060929
Travis, W. D., Brambilla, E., Burke, A. P., Marx, A., and Nicholson, A. G. (2015). Introduction to The 2015 World Health Organization classification of tumors of the Lung, Pleura, Thymus, and Heart. J. Thorac. Oncol. 10, 1240–1242.
van Maldegem, F., and Downward, J. (2020). Mutant KRAS at the heart of tumor immune evasion. Immunity 52, 14–16. doi: 10.1016/j.immuni.2019.12.013
Wang, B. Y., Gil, J., Kaufman, D., Gan, L., Kohtz, D. S., and Burstein, D. E. (2002). P63 in pulmonary epithelium, pulmonary squamous neoplasms, and other pulmonary tumors. Hum. Pathol. 33, 921–926. doi: 10.1053/hupa.2002.126878
Wang, R., Wang, L., Li, Y., Hu, H., Shen, L., Shen, X., et al. (2014). FGFR1/3 tyrosine kinase fusions define a unique molecular subtype of non-small cell lung cancer. Clin. Cancer Res. 20, 4107–4114. doi: 10.1158/1078-0432.ccr-14-0284
Westcott, P. M., and To, M. D. (2013). The genetics and biology of KRAS in lung cancer. Chin. J. Cancer 32, 63–70. doi: 10.5732/cjc.012.10098
Xu, X., Huang, L., Futtner, C., Schwab, B., Rampersad, R. R., Lu, Y., et al. (2014). The cell of origin and subtype of K-Ras-induced lung tumors are modified by Notch and Sox2. Genes Dev. 28, 1929–1939. doi: 10.1101/gad.243717.114
Xue, W., Chen, S., Yin, H., Tammela, T., Papagiannakopoulos, T., Joshi, N. S., et al. (2014). CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 514, 380–384. doi: 10.1038/nature13589
Yang, S. H., Cheng, P. H., Sullivan, R. T., Thomas, J. W., and Chan, A. W. (2008). Lentiviral integration preferences in transgenic mice. Genesis 46, 711–718.
Keywords: non-small cell lung cancer, CRISPR-Cas9, mouse model, lung cancer, MYC, JUN, KRAS, TP53
Citation: Hartmann O, Reissland M, Maier CR, Fischer T, Prieto-Garcia C, Baluapuri A, Schwarz J, Schmitz W, Garrido-Rodriguez M, Pahor N, Davies CC, Bassermann F, Orian A, Wolf E, Schulze A, Calzado MA, Rosenfeldt MT and Diefenbacher ME (2021) Implementation of CRISPR/Cas9 Genome Editing to Generate Murine Lung Cancer Models That Depict the Mutational Landscape of Human Disease. Front. Cell Dev. Biol. 9:641618. doi: 10.3389/fcell.2021.641618
Received: 14 December 2020; Accepted: 21 January 2021;
Published: 02 March 2021.
Edited by:
Paolo Ceppi, University of Southern Denmark, DenmarkReviewed by:
Mohammad Aarif Siddiqui, University of Southern Denmark, DenmarkCopyright © 2021 Hartmann, Reissland, Maier, Fischer, Prieto-Garcia, Baluapuri, Schwarz, Schmitz, Garrido-Rodriguez, Pahor, Davies, Bassermann, Orian, Wolf, Schulze, Calzado, Rosenfeldt and Diefenbacher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus E. Diefenbacher, bWFya3VzLmRpZWZlbmJhY2hlckB1bmktd3VlcnpidXJnLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.