 Mark Hintze
Mark Hintze Dimitris Katsanos
Dimitris Katsanos Vahid Shahrezaei2
Vahid Shahrezaei2 Michalis Barkoulas
Michalis Barkoulas- 1Department of Life Sciences, Imperial College, London, United Kingdom
- 2Department of Mathematics, Imperial College, London, United Kingdom
Individual cells and organisms experience perturbations from internal and external sources, yet manage to buffer these to produce consistent phenotypes, a property known as robustness. While phenotypic robustness has often been examined in unicellular organisms, it has not been sufficiently studied in multicellular animals. Here, we investigate phenotypic robustness in Caenorhabditis elegans seam cells. Seam cells are stem cell-like epithelial cells along the lateral edges of the animal, which go through asymmetric and symmetric divisions contributing cells to the hypodermis and neurons, while replenishing the stem cell reservoir. The terminal number of seam cells is almost invariant in the wild-type population, allowing the investigation of how developmental precision is achieved. We report here that a loss-of-function mutation in the highly conserved N-acetyltransferase nath-10/NAT10 increases seam cell number variance in the isogenic population. RNA-seq analysis revealed increased levels of mRNA transcript variability in nath-10 mutant populations, which may have an impact on the phenotypic variability observed. Furthermore, we found disruption of Wnt signaling upon perturbing nath-10 function, as evidenced by changes in POP-1/TCF nuclear distribution and ectopic activation of its GATA transcription factor target egl-18. These results highlight that NATH-10/NAT-10 can influence phenotypic variability partly through modulation of the Wnt signaling pathway.
Introduction
Developing organisms face a constant challenge from external (environmental) and internal (genetic) perturbations or stochastic noise in molecular processes. It is therefore remarkable that organisms can produce consistent phenotypes across populations in spite of these perturbations (Bauer et al., 2015; Felix and Barkoulas, 2015). Robustness, first described by Waddington (1959), is the term used to define the invariance of a phenotype when faced with incoming variation (Felix and Barkoulas, 2015). Robustness to various perturbations is particularly important in developing tissues, where cell division and differentiation patterns need to be tightly controlled to generate the required structures and cell types.
Phenotypic robustness has been mostly investigated in single cell organisms such as Saccharomyces cerevisiae, where for example more than 300 genes have been found to modulate morphological variation and suggest a genetic basis for phenotypic robustness (Levy and Siegal, 2008). However, robustness in multicellular animals is much less studied, so it remains a challenge to identify what are the buffering mechanisms in biological systems, as well as their sensitivities across cells and tissues for different perturbations (Felix and Barkoulas, 2015). For example, a recent study in Drosophila melanogaster has shown that neuronal wiring in the thorax is highly variable to genetic and environmental pressure and this variability is under the control of the Hox gene Ultrabithorax (Mellert et al., 2016), suggesting that individual genes can buffer or enhance variability in higher organisms depending on the developmental context.
C. elegans is a highly tractable model for investigating phenotypic robustness due to its well-established cell lineage, fully characterized genome, and isogenic nature, which leads to little genetic variation within a population (Sulston and Horvitz, 1977). Lack of genetic variation removes a key confounder when it comes to studying robustness in phenotypic variation at the population level. Here, we focus on a population of cells in the developing larva that have stem cell properties, the seam cells. Seam cells are found on the lateral sides of the animal in two lines running from anterior to posterior and are labeled as H0-H2 (Head), V1-V6 (Ventral), and T (Tail) (Chisholm and Hsiao, 2012). Seam cells go through rounds of stem cell-like asymmetric and symmetric divisions during larval development. Animals hatch at L1 with 10 seam cells and these expand through a round of symmetric division at the L2 larval stage to a final number of 16. Furthermore, reiterative asymmetric cell divisions throughout larval development contribute the majority of nuclei to the developing hypodermal syncytium and a number of neurons (Figure 1A). In this context, we have recently conducted a mutagenesis screen to find mutations that introduce stochastic variability in terminal seam cell number (Katsanos et al., 2017). A proof-of-concept was established showing that mutations in the Hes-related basic helix-loop-helix transcription factor lin-22 increase seam cell number variability. Loss of seam cells early in post-embryonic development due to ectopic neurogenesis were found to be compensated later in larval development via stochastic symmetrization of cell divisions, thereby causing the seam cell number to be distributed on either side of the mean.
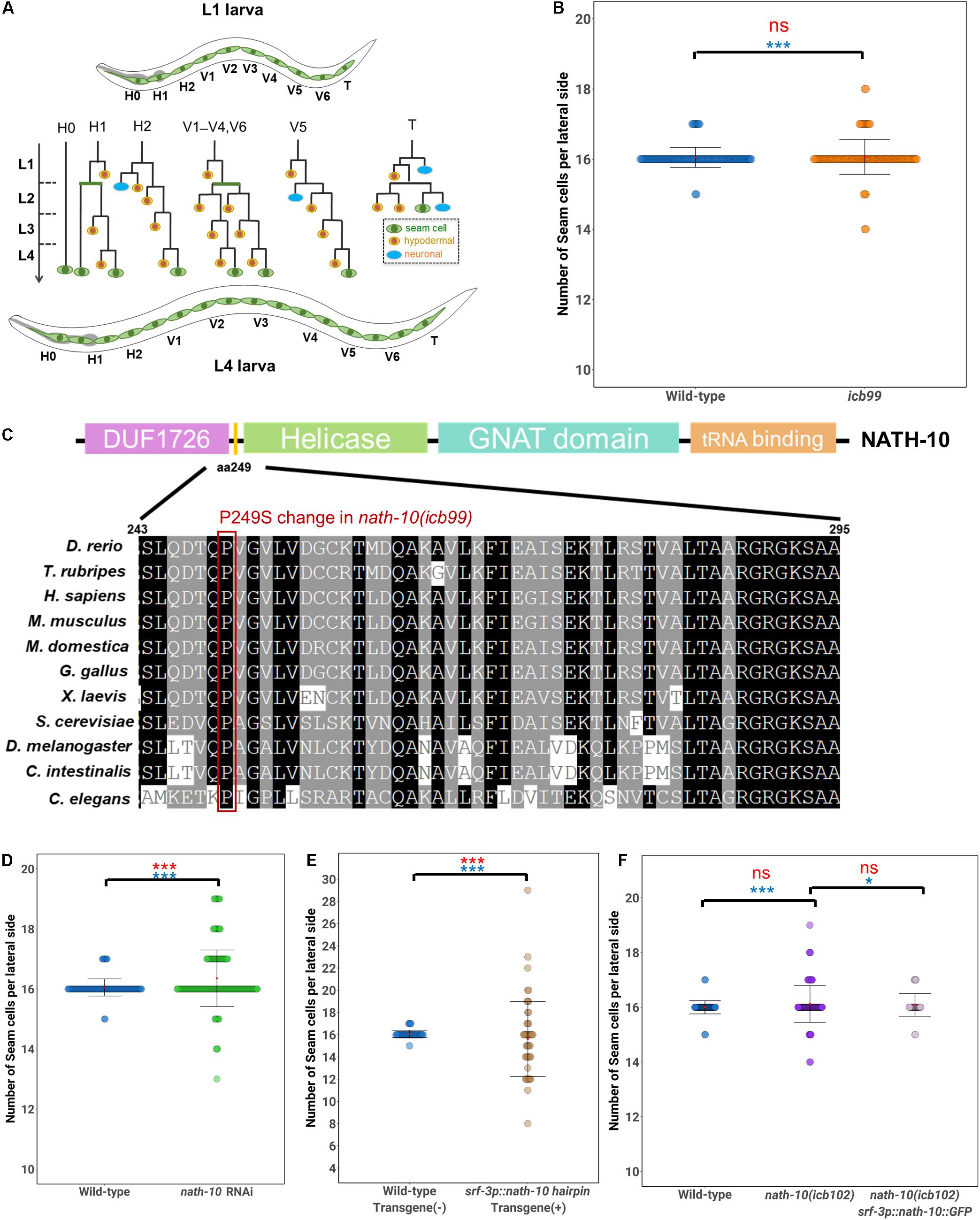
Figure 1. Seam cell number robustness is disrupted by nath-10 loss-of-function. (A) schematic displaying seam cell lineage from L1 to L4, seam cells are labeled green, hypodermal cells are labeled yellow and neurons are labeled blue. (B) Mutants carrying the icb99 mutation display variable seam cell number (SD = 0.28, n = 120 for wild type JR667 animals, SD = 0.50, n = 120 for animals carrying the icb99 mutations. Red ns indicates non-significant difference to the mean by t-test (p > 0.05). Blue stars indicate statistically significant difference to the variance by Levene’s median test (***p < 0.001). (C) Schematic showing protein domains in NATH-10. The P249S change involves a proline to serine shift at amino acid 249, indicated by the yellow bar between the DUF1726 and Helicase domains. Alignment of this region across multiple species shows high conservation of this proline, suggesting it may be of importance to protein function. Red frame shows the conserved proline. (D) nath-10 RNAi phenocopies the icb99 mutation leading to a significant increase in variance and average seam cell number (Levene’s median test ***p < 0.001 and t-test, ***p < 0.001, n = 120 wild type, and n = 183 nath-10 RNAi). (E) Seam cell specific knockdown of nath-10 by a nath-10 hairpin RNAi also causes significant increase in seam cell number and seam cell variance in comparison to wild-type animals not carrying the hairpin transgene (t-test, ***p < 0.001, Levene’s test median ***p < 0.001). (F) A CRISPR replacement mimicking the icb99 mutation phenocopies the original EMS-derived mutant (SD = 0.77, Levene’s test ***p < 0.05). Seam cell specific overexpression of nath-10 (srf-3p:nath-10:GFP) partially rescues this phenotype (SD = 0.41, Levene’s test *p < 0.05 In (B,D–F) mean values are indicated by red dots and error bars correspond to ± Standard deviation. Statistical differences to the mean and variance are indicated above brackets for relevant comparisons in red and blue, respectively, assessed by a t-test or Levene’s median test. Significance levels (ns p > 0.05, *p < 0.05, ***p < 0.001).
A number of other transcription factors and signaling pathways have been found to regulate seam cell development. GATA transcription factors, such as elt-1 and egl-18, the engrailed homolog ceh-16 and the Runx/CBFβ homologs rnt-1/bro-1 are thought to be key players in the seam cell gene network (Smith et al., 2005; Huang et al., 2009; Gorrepati et al., 2013; Gorrepati and Eisenmann, 2015). During seam cell patterning, the Wnt pathway activates targets necessary for seam cell maintenance, such as egl-18, through the downstream effector POP-1/TCF, while ectopic egl-18 activation can drive ectopic seam cell fate retention (Gorrepati et al., 2013). Activation of Wnt signaling in one of the two daughter cells relies on asymmetric inheritance of Wnt pathway components from polarized mother cells (Mizumoto and Sawa, 2007), while this is overridden during symmetric cell divisions through the repression of pop-1/TCF by the RNT-1/BRO-1 module (Yamamoto et al., 2011; Hughes et al., 2013; van der Horst et al., 2019). Other pathways, such as the heterochronic pathway, control developmental timing of the division patterns (Ambros and Horvitz, 1984; Harandi and Ambros, 2015).
Here, we continue the characterization of mutations from our genetic screen and demonstrate that a mutation in the highly conserved N-acetyltransferase nath-10 disrupts seam cell number robustness. We report that the defect in phenotypic variance is restricted to seam cells and may be associated with an increase in mRNA transcript variance among animal populations. We also demonstrate changes in the distribution of POP-1 and subsequent activation of the Wnt target egl-18 upon impairment of nath-10 function. Therefore, we propose that NATH-10 modulates robustness of seam cell patterning through regulation of the Wnt signaling pathway.
Materials and Methods
C. elegans Culture and Strains
C. elegans strains used in this study were maintained and raised according to standard protocols on NGM plates with the E. coli strain OP50 as a food source (Brenner, 1974). Wild-type animals referred to in this work are the strain JR667 (wIs51) that contains the transgene scm:GFP. A full list of strains used in this study can be found in Supplementary Table 1.
Genetic Screening and Mapping
Mutagenesis of animals using EMS was carried out according to standard protocols (Brenner, 1974). The screen for terminal seam cell number mutants was carried out as described in Katsanos et al. (2017). Whole-genome sequencing was performed using an Illumina platform to reach 20–30-fold genome coverage and mapping was performed using the Cloudmap pipeline on a local version of Galaxy (Minevich et al., 2012). The mutation in the icb99 allele is a C > T change in the third exon of nath-10 (GAGACAAAACCAATCGGACCATTGC) and it was backcrossed 4 times before phenotypic characterization.
Microscopy and Phenotypic Characterization
Animals were mounted under a coverslip on 3% agarose in a droplet of M9 containing 100μM sodium azide for light and fluorescent microscopy. Seam cells on one lateral side were visualized using a Zeiss Compound microscope (AxioScope A1) at 40x and 100x at 44–48 h of development. Lineage analysis was carried out on bleach-synchronized animals carrying the scm:GFP transgene at 10, 17, 24, and 27 h post bleaching. Brood size assays were carried out by bleach synchronizing animals and then isolating single L4s. Single L4s were transferred to new plates every day for 5 days until no evidence of egg laying could be seen and the resulting progeny on each plate was counted. Fluorescence intensity for the egl-18:cherry and POP-1:GFP reporters were calculated in the following manner using ImageJ. A region of interest was drawn around seam cell nuclei and measurements of area and integrated density were taken. Three background readings were taken from areas surrounding the seam cells. Corrected total fluorescence was calculated with the following formula: Integrated Density – (Area (ROI) X Mean fluorescence of background readings.
CRISPR-Cas9 Mediated Genome Editing
The single point mutation from the nath-10(icb99) allele was engineered into an N2 background via injection of Cas9 ribonucleoprotein complexes (Paix et al., 2015) in 10 μl as follows: tracRNA (0.75 nmol – IDT), custom crRNA (nath-10 target sequence CGA GAA AGC AAT GGT CCG AT, IDT) and CAS9 (1 μg/μl -IDT). The co-injection markers myo-2:dsRED at 5 ng/μl and rol-6(su1006) at 40 ng/μl were used to select transgenic animals, and a repair template (6 μM) that introduced a ClaI restriction site. Therefore, nath-10(icb102) contains the point mutation found in nath-10(icb99) and a synonymous mutation that introduces a ClaI site. F1 animals were screened for the co-injection markers, allowed to lay progeny, and screened using a restriction digest for the introduced ClaI site. Positive lines were Sanger-sequenced to identify animals homozygous for SNP insertions.
Molecular Cloning
To produce srf-3p:nath-10:GFP, nath-10 was amplified from N2 cDNA using primers hN53 and hN54, which carried compatible sequences allowing insertion to the XmaJI/PacI digested pDK52 via Gibson assembly creating pHIN9 (srf-3p:nath-10:GFP:unc-54- 3′UTR). The srf-3 promoter used in this study refers to the first intron of the srf-3 locus fused to a minimal pes-10 promoter which drives expression specifically in seam cells.
To produce a platform for quick and efficient assembly of hairpin-RNAi constructs from a single PCR product by golden gate assembly, the following constructs were made. The plasmid pDK102 carrying a dpy-7 promoter and the p10 3′UTR (Pfeiffer et al., 2012) in a pCFJ151 backbone was digested with XmaJI/PacI to linearize and allow for cloning between the promoter and the 3′UTR. A gene fragment (Golden Gate Hairpin) carrying a compatibility arm to the dpy-7 promoter, an outron, 2 inverted repeats of the BpiI enzyme the 5th intron from the srf-3 gene, two inverted repeats of the Esp3I enzyme and compatibility arm to the p10 3′UTR was synthesized (GENEWIZ) and was inserted in the digested pDK102 to form the intermediate plasmid pDK109 (dpy-7p::Golden Gate Hairpin:p10 3′UTR + cb-unc-119). Using oligos DK186 and DK179 the sequence from the promoter to the 3′UTR was amplified and inserted by Gibson assembly in a KpnI/NotI digested pBluescript vector to form pDK110 (dpy-7p::outron::GGBpiI::srf-3a intron5::GGEsp3I::p10 3UTR). To modify the golden gate (GG) enzyme sites such that they leave non-palindromic scars to allow for specific directional cloning the srf-3 intron 5 was amplified from pDK110 using oligos DK212 and DK214. The resulting amplicon was amplified with oligos DK213 and DK215 and was inserted in a BpiI/Esp3I digested pDK110 backbone by Gibson assembly to produce pDK127 (dpy-7p::outron::non-palGGBpiI::srf-3a intron5::non-palGGEsp3I::p10 3′UTR).
To design a GFP hairpin, a fragment from GFP not containing sites for BpiI and Esp3I was amplified from L3135 using oligos DK203 and DK204 and was inserted by Golden gate assembly in pDK127 to create pDK130(dpy-7p::outron:: > GFP-frag > ::srf-3a intron5:: < GFP-frag < ::p10 3UTR). To create a seam cell expressing version the srf-3p promoter was amplified from pDK126 using oligos DK234 and DK244 and was inserted in a Gibson assembly reaction with SalI digested pDK130 to remove the dpy-7 promoter and create pDK134(srf-3p::outron:: > GFP-frag > ::srf-3a intron5:: < GFP-frag < ::p10 3UTR). For pHIN33, 366 bp from the first exon of nath-10 was amplified using the primers hN79 and hN80 with complementary tags which allowed golden gate assembly into a BpiI and Esp3I digested pDK157. Golden gate assembly reactions consisted of adding 2.5 units of T4 DNA Ligase (Thermo Fisher Scientific), 0.5 μl of the BpiI and 0.5 μl of the Esp3I FastDigest enzymes (Thermo Fisher Scientific), 50 ng of the vector plasmid and a 2::1 insert:: vector molar ratio in 1x T4 Ligase buffer (Thermo Fisher Scientific) in a total volume of 10 μl. Reactions were incubated at 37°C for 30 min, 5 min at 50°C and 5 min at 80°C in a thermocycler. All plasmids were verified by Sanger sequencing. A complete list of plasmids and oligos is presented in Supplementary Table 2.
Single Molecule Fluorescence in situ Hybridization
Bleach-synchronized populations of animals were fixed at the appropriate stage as directly monitored by microscopy, smFISH was performed as previously described (Barkoulas et al., 2013) using a pool of 24–48 oligos fluorescently labeled with Cy5 (Biomers, Germany). Imaging was performed using a motorized epifluorescence Ti-eclipse microscope (Nikon) and a DU-934 CCD-17291 camera (Andor Technology, Belfast, United Kingdom) acquiring 0.8 um step z-stacks. Image analysis and spot quantification were performed on raw data using a MATLAB (MathWorks, Natick, MA) routine as previously described (Barkoulas et al., 2013), with the addition of a step to optimize ROI size on the Z-axis in adjacent sections within the area of interest. For images presented in the results section of this study, the probe signal channel was inverted for clarity (black spots correspond to mRNAs) and merged to the seam cell fluorescent marker in ImageJ (NIH, Rockville, MD). A complete list of smFISH oligo probes is presented in Supplementary Table 2.
RNA Sequencing Analysis
Larvae were synchronized by bleaching from 6 plates per replicate and grown to L2 stage (27 h post hatching). The same timepoint was used for RNA collection for wild type and nath-10 mutants since we verified by monitoring seam cell divisions that the animals were growing at the same speed. We used 3 replicates for each condition (we estimate ∼5,000 animals per replicate) and total RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA). Illumina sequencing was performed by BGI (China). The sequencing data were processed and aligned to the C. elegans reference genome using kallisto (Bray et al., 2016). The counts were normalized using the DESeq2 package in R (Anders and Huber, 2010). Differences in gene expression were calculated using the negative binomial test in the DESeq package (FDR = 0.1). Coefficient of variation was calculated by dividing the standard deviation of mean transcript number by the mean number of transcripts. Seam cell-enriched genes based on sci-RNA-seq (single-cell combinatorial indexing RNA sequencing) data were defined by removing genes with fewer than 10 transcripts per million across all tissue clusters, genes enriched only in the germline, as well as ubiquitously expressed genes across all tissue clusters. Seam cell-enriched genes were defined as having a count > 10 transcripts per million in the seam cell tissue cluster and on average less than 100 counts in other tissue clusters. These filtering steps gave rise to 6,014 genes in total. Functional subsets of genes were obtained by downloading gene expression clusters for these terms from Wormbase and WormMine. RNA-seq data are deposited in NCBI GEO under accession number GSE162226.
RNAi by Feeding
Animals were fed with dsRNA expressing bacteria as a food source. Bacteria were grown over-night and then seeded directly onto NGM plates containing 1 μM IPTG, 25 μg/ml ampicillin and 6.25 μg/ml tetracycline. Young adults were bleached to synchronize the population and eggs were plated on RNAi or empty vector plates and counted seam cells at the late L4 stage the nath-10 clone used in this study was obtained from the Ahringer library (Source BioScience).
Statistical Analysis
All statistical analysis was carried out in the R statistical package (R Core Team, 2020) and all plots were carried out using ggplots2 (Wickham, 2016).
Results
A Forward Genetic Screen Identifies a Mutation in nath-10 Affecting Seam Cell Number Robustness
To identify modulators of terminal seam cell number robustness, we carried out a forward genetic screen. We mutagenized L4 larvae carrying the transgene scm::GFP (wIs51) by treating them with EMS, allowed F1 animals to produce progeny and isolated F2 animals that had an extreme departure from the average terminal seam cell number of 16 cells per lateral side in wild type. Phenotypic extremes refer to individuals having either greater than 17 or less than 15 seam cells, which occur very infrequently (less than 1%) in the wild-type population. The selected F2 mutant animals could therefore display at the F3 population either an increase, decrease or variable terminal seam cell number. As seam cell number variance changes as a function of the average number of seam cells (Katsanos et al., 2017), we focused on animals that had an average of 16 seam cells but had a significant increase in variability (identified using a Levene’s test), usually with errors on both sides of the wild-type mean. One mutation that fitted these criteria was icb99, which led to a significant increase in the standard deviation of seam cell number compared to the wild type (Figure 1B).
To identify the molecular change in icb99 we used mapping-by-sequencing (Minevich et al., 2012; Doitsidou et al., 2016). To this end, we crossed the mutant harboring icb99 to the polymorphic C. elegans isolate CB4856. F2 progeny with a similar increase in phenotypic variance were isolated, pooled and whole-genome sequenced. This experiment identified a region on the left arm of chromosome I that contained preferentially N2 sequences. Within this region, we found a C to T change in the third exon of the N-acetyltransferase nath-10, which is the homolog of the human NAT10. This change results in a non-synonymous substitution of a proline to a serine (P249S). Although, this change is not within the known domains of NATH-10, the affected proline is conserved from yeast to humans, suggesting it may affect some structural feature of the protein (Figure 1C).
To validate that the nath-10(icb99) mutation was causative for the seam cell number variance phenotype, we first carried out RNAi against nath-10 in the wild-type population. We found that nath-10 RNAi affected seam cell patterning and significantly increased seam cell number variance (Figure 1D). However, nath-10 RNAi also caused a high rate of sterility, as well as arrest in early larval development, phenotypes which were not observed in the recovered allele from our mutagenesis screen. This is consistent with previously reported nath-10 loss-of-function phenotypes in C. elegans, such as lethality, sterility and defects in vulval development (Duveau and Félix, 2012).
To circumvent these pleiotropic effects and address whether the seam cell phenotype might be a response to global or seam cell-specific impairment of nath-10 function, we knocked down nath-10 in the seam cells using a nath-10 hairpin. To this end, we devised an approach that facilitates cloning of any gene fragment in sense and antisense orientation under an epidermal promoter using a single-step golden gate assembly (Supplementary Figure 1A). Such transgenes result in the production of hairpin dsRNA structures that have been shown to cause silencing of target genes in multiple tissues (Tavernarakis et al., 2000; Timmons et al., 2003). To confirm that the system is effective, we performed control experiments targeting GFP expression with a GFP hairpin. Seam cell expression of a hairpin against GFP in animals expressing scm::GFP led to complete abolishment of GFP signal in 50% of the transgenic animals and strong observable reduction of signal in another 36.2% of animals (Supplementary Figure 1B). A similar effect was observed in the case of hypodermal (hyp7) expression of a GFP hairpin and a hypodermal GFP marker (dpy-7p::GFP), while hypodermal expression of the hairpin resulted in no reduction in the scm::GFP signal (Supplementary Figure 1B). Having established the functionality and specificity of the system, we produced transgenic animals harboring a seam cell driven nath-10 hairpin. We found that seam cell specific knockdown of nath-10 caused a significant increase in seam cell variance (Figure 1E), when compared to animals that did not carry the transgene and did not cause other phenotypes, such as larval lethality. These results therefore suggest that nath-10 is likely to be the causative mutation and that nath-10 may act in a cell autonomous manner to influence seam cell patterning.
To further validate the causative mutation underlying the increase in seam cell number variance, we generated by CRISPR editing a new allele (icb102) mimicking the nucleotide change found in nath-10(icb99). We found that the CRISPR allele phenocopied the original icb99 allele with regard to seam cell number variance (Figure 1F). The increase in variance was again moderate, but it was very reproducible across different days of scoring (Supplementary Figure 2). Furthermore, by introducing nath-10 under a seam cell specific promoter (srf-3p::nath-10::GFP) we could largely rescue the phenotype of icb102 (Figure 1F). Taken together, these results indicate that a mutation in nath-10 breaks down seam cell number robustness. It is of note that the newly recovered nath-10 allele did not show other obvious developmental phenotypes and vulval cell fate errors, but did show a decrease in mean brood size, while the variance in progeny number was not statistically different compared to the wild type (Supplementary Figure 3A). These results suggest that the increase in phenotypic variance upon loss of nath-10 function is likely to be specific to seam cells.
The Mutation in nath-10 Causes Gains and Losses in Seam Cells During Post-embryonic Development
Seam cells belong to well-described cell lineages and take stereotypic positions along the anterior-posterior axis (Figure 1A), allowing us to infer the origin of developmental defects based on changes in the terminal seam cell number observed in a mutant background. In nath-10 loss-of-function mutants, we found that gains and losses of seam cells occur mostly in V2 and mid-body cell lineages (Figures 2A,B). We then sought to understand when such defects occur in development by carrying out seam cell counts at defined intervals. First, we ruled out embryonic defects and developmental delay phenotypes as both wild type and nath-10(icb102) mutants hatched with 10 seam cells (Figure 2C) and the proportion of animals reaching the L4 stage at defined time intervals was not significantly different compared to the wild type (Supplementary Figure 3B). However, we found that 10 h post hatching nath-10(icb102) animals began to show significant increase in seam cell number variability that became even more pronounced at the L2 stage (Figure 2C). These results suggest that nath-10 mutation effects can be seen early in post-embryonic seam cell divisions.
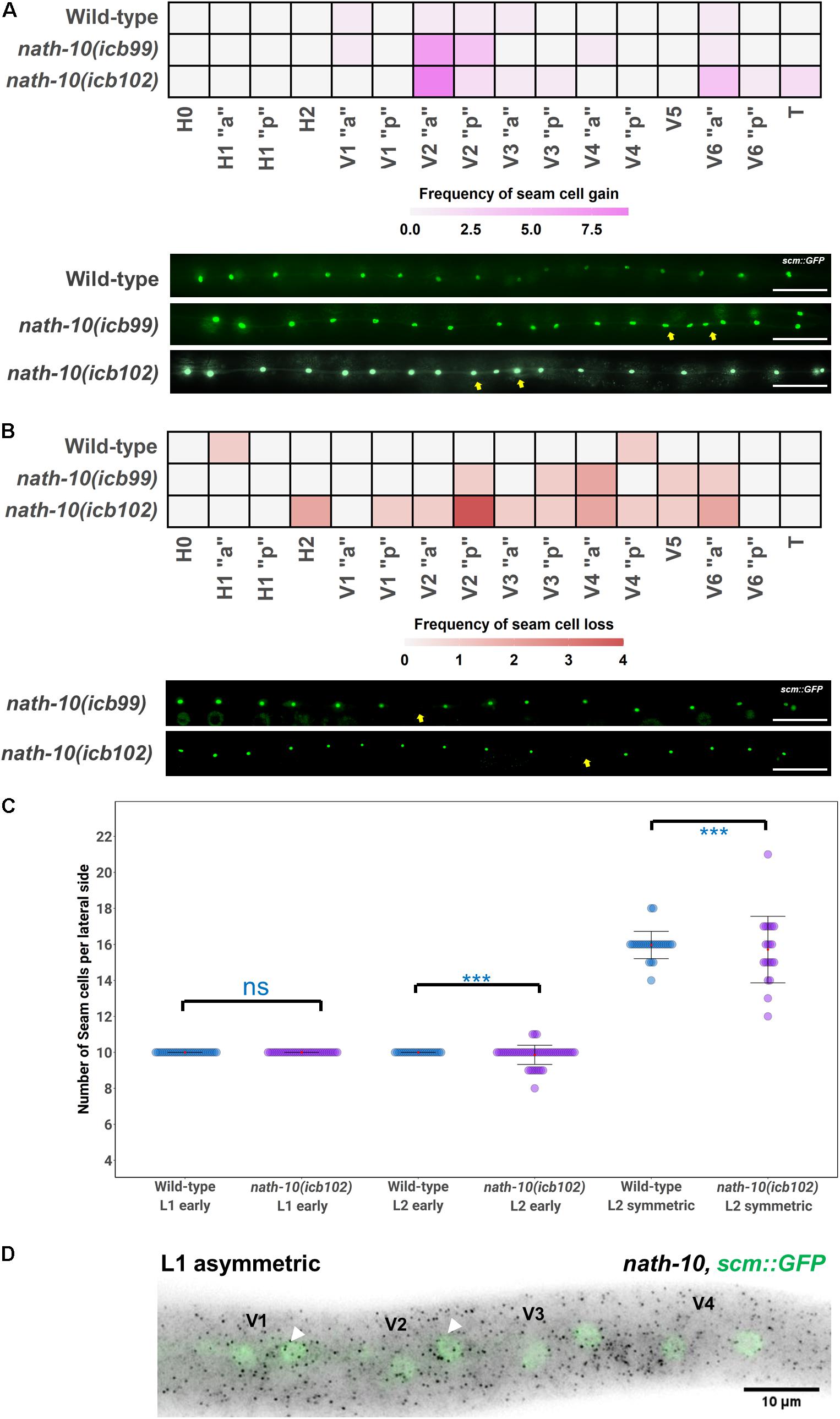
Figure 2. nath-10 loss-of-function causes both gains and losses of seam cells from the early L2 stage. (A) Heat map displaying frequency of seam cell duplications for wild type, nath-10(icb99) and nath-10(icb102). Increased seam cell duplication frequency occurs mostly in the V2 seam cell. Representative images below from wild type, nath-10(icb99) and nath-10(icb102) animals carrying the scm::GFP transgene. Yellow arrows indicate seam cell duplications. (B) Heat maps displaying seam cell losses in nath-10 mutants. Note that losses are found more frequently in the V lineage seam cells. Yellow arrows indicate missing seam cells. In (A,B), “a” and “p” represent the anterior and posterior branch of a seam cell lineage. (C) Seam cell counts at early L1, early L2 and mid L2 stage for wild type and nath-10(icb102) mutants. No difference was found at the early L1 stage as, all animals’ hatch with 10 seam cells, whereas a significant difference in variance of seam cell number was found at the L2 stage (Levene’s test ***p < 0.001). (D) Representative image of nath-10 smFISH shows expression in seam cells at the time errors begin to occur (L1 division). White arrowheads point to posterior daughter cells following the L1 asymmetric division. Nuclei are labeled green because of scm::GFP expression. Scale bars in (A,B) indicate 100 um.
To consolidate the lineaging and genetic results, we studied whether nath-10 is expressed in the seam cells during development. To this end, we carried out nath-10 single molecule fluorescence in situ hybridization (smFISH) and found expression in seam cells, for example in V cell lineages at the L1 asymmetric division stage (Figure 2D). The expression of nath-10 was not specific to the seam, for example it was expressed in the germ line at the L4 stage (Supplementary Figure 3C), which is consistent with the observed brood size defects in nath-10 alleles. Taken together, our results support the idea that nath-10 disruption leads to seam cell number variability through both gains and losses of seam cells during early post-embryonic development and predominantly in the mid-body V lineages.
RNA Sequencing Analysis Identifies an Increase in mRNA Transcript Number Variance in nath-10 Mutants
The human homolog of nath-10, NAT10, has recently been shown to catalyze mRNA acetylation of the N4-acetlycytidine, and loss of this acetylation was shown to decrease mRNA stability and translation efficiency (Arango et al., 2018). To study the molecular basis of seam cell number variability, we carried out RNA sequencing on nath-10 mutants in comparison to wild-type animals. We synchronized animals and harvested them for RNA extraction after 27 h (L2 stage) with three biological replicates for each strain. RNA transcripts were then mapped to the C. elegans mRNA transcriptome using Kallisto (Bray et al., 2016) and analyzed using DESeq2 (Anders and Huber, 2010). While both mutants contain an identical nath-10 mutation, we reasoned that they may still exhibit differences due to the independent origin of the two alleles (one from EMS mutagenesis and the other from CRISPR-mediated genome editing), so we decided to treat them separately for subsequent analysis. Both strains containing nath-10 alleles displayed differentially regulated genes with significant overlap and could be separated from wild-type animals using principal component analysis (Figures 3A–D and Supplementary Figure 4A). We found that phenotype enrichment analysis of the overlapping genes showed enrichment for epidermal development terms such as “epithelial development variant” and “blistered,” or other terms related to previously known developmental and cellular functions of nath-10 in fertility and nuclear morphology (Shen et al., 2009; Supplementary Figures 4B,C). To identify specific candidates that may influence the seam cell phenotype, we compared these overlapping transcripts to seam cell specific transcripts identified in a recent single cell RNA-seq analysis of C. elegans at L2 (Cao et al., 2017). We found that 67 out of 161 genes in the overlap appeared to be expressed in seam cells and these did not show significant enrichment for any GO terms. Furthermore, we prioritized candidates from the differentially expressed genes based on their seam cell expression and putative connection to other core seam cell genes to test their interaction with nath-10 using an RNAi screen. However, we found no enhancement or suppression of the seam cell number variance phenotype in these treatments (Supplementary Figure 5).
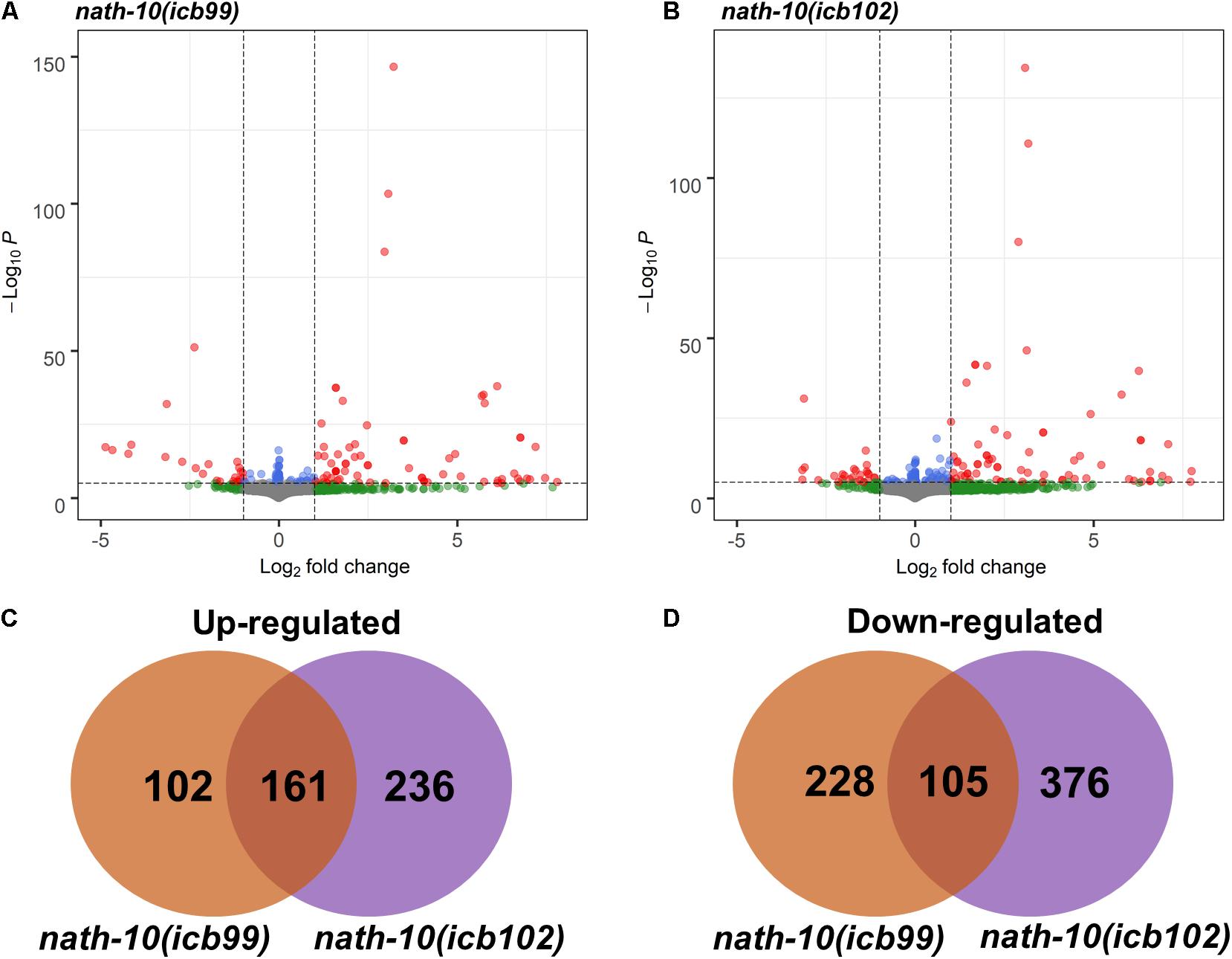
Figure 3. RNA-seq analysis of nath-10 mutants reveals differentially expressed genes. (A,B) Volcano plots displaying summary of nath-10(icb99) and nath-10(icb102) changes in comparison to N2 animals. Green dots indicate > 1.2-fold change, blue dots < 0.01 adjusted p-value and red dots > 1.2-fold change and < 0.01 adjusted p-value. (C) Overlap of significantly differentially upregulated genes in nath-10(icb99) and nath-10(icb102) alleles. (D) Overlap of significantly differentially downregulated genes in nath-10(icb99) and nath-10(icb102) alleles.
We then decided to explore the possibility that a change in mRNA transcript variance may associate with the variable seam cell number phenotype in the nath-10 mutant population. Single animal RNA-seq or cell sorting in the epidermis remain experimentally challenging, so we used our RNA-seq data to assess whether a change in transcript variance could be revealed at the population level among replicates. To this end, we calculated the coefficient of variation (CV) for all mapped transcripts that had greater than 5 read counts in two or more of all replicates. While this method would not be able to uncover animal-to-animal differences in variability, we reasoned that it may be possible to uncover population-to-population differences in transcript variance that are beyond what is seen in wild-type populations. Interestingly, we found that both nath-10 alleles had a significant increase in the average CV compared to N2 (Figure 4A), suggesting that transcript variance is increased.
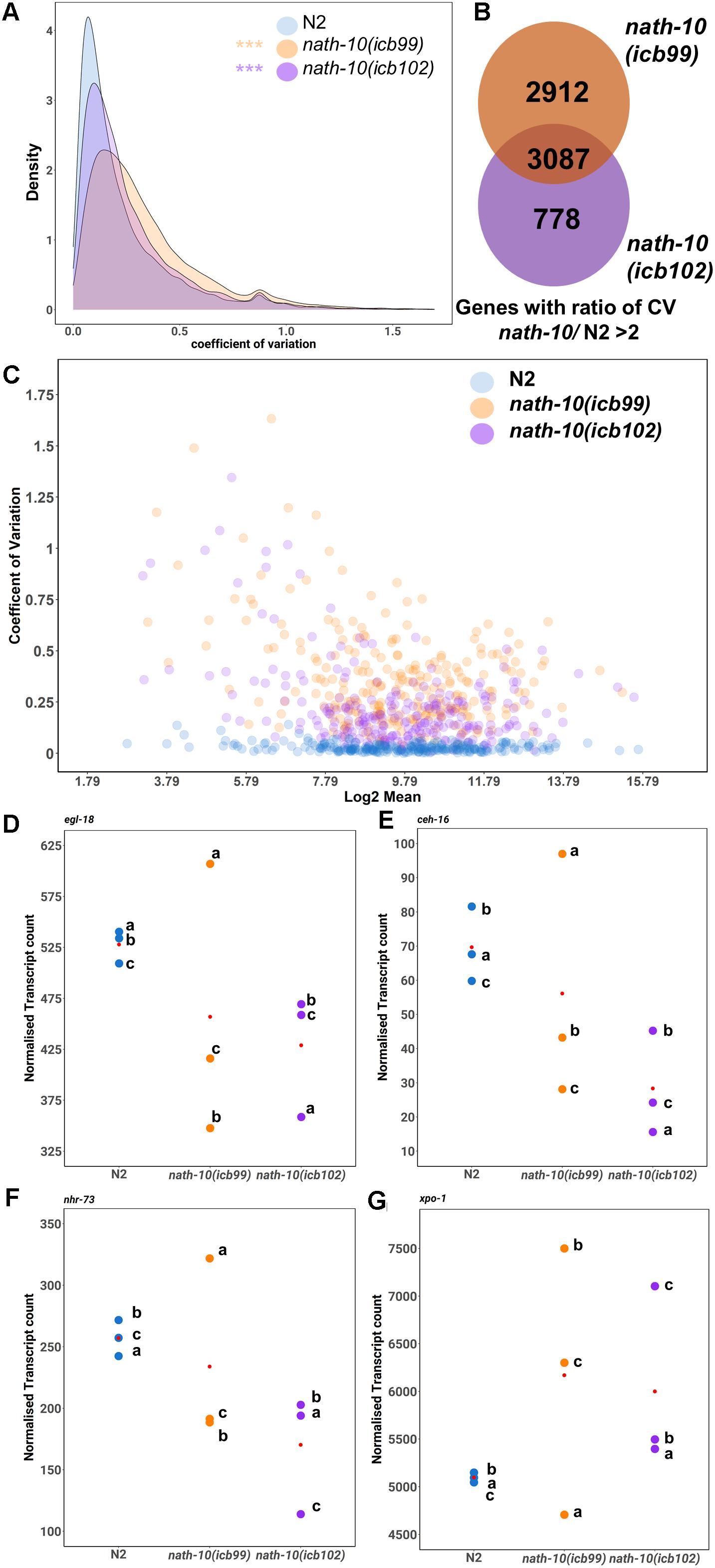
Figure 4. RNA-seq analysis reveals changes in transcript variance at the population level. (A) Density plots of coefficient of variation (CV) in nath-10 mutants and N2. Both nath-10 alleles show significant change in the distribution of the coefficient of variation compared to N2, with a trend toward an increase in transcript variability (Kolmogorov-Smirnov test, ***p < 0.001). (B) Venn diagram displaying the overlap of genes that display a > 2 ratio of CV when compared to N2 for nath-10(icb99) and nath-10(icb102) mutants. Both alleles show a 50% overlap of the most variable genes and this overlap includes core seam cell transcription factors ceh-16, egl-18, and nhr-73. (C) Coefficient of variation plotted against the log2 normalized transcript counts from RNA-seq analysis for seam cell-enriched genes in nath-10 alleles and wild type. (D–G) Normalized transcript counts plotted per replicate (a, b, c) for N2, nath-10(icb99) and nath-10(icb102) showing egl-18, ceh-16, nhr-73 and xpo-1 expression. Note that both nath-10 alleles display a great spread of normalized transcript count compared to the N2 replicates and the variance is not driven consistently by the same replicate for each gene.
To identify gene candidates that may relate to the variable seam cell number phenotype, we found common genes between the two nath-10 alleles that showed higher than twofold increase in transcript variance compared to N2. The overlap contained more than 50% of variable transcripts for each strain (Figure 4B). We compared transcripts that had the highest shift in variance level in our analysis to a seam cell-enriched transcript set (Cao et al., 2017). Interestingly, more than 40% (1,257 transcripts out of 3,087) of the variable genes in the overlap were found to be enriched in the seam cells. Furthermore, this overlap included some transcription factors of the core seam cell gene network, such as egl-18, ceh-16, and nhr-73. For example, egl-18 was in the top 250 of most variable genes together with other genes including xpo-1, for which the human homolog (XPO1/CRM1) has been shown to be linked to NAT10 levels in human cells (Zhang et al., 2014; Figures 4C–E). Plotting of the mean transcript number for each replicate indicates that the increase in variance in nath-10 mutants was not driven by the same underlying pattern for each selected gene, such as a specific replicate being consistently the outlier (Figures 4D–G).
To further investigate whether the transcript variance shown by nath-10 loss-of function was due to animals being sensitized to environmental conditions, we isolated functional subsets of genes that display changes during aging, temperature shifts or exposure to stress and compared the CV pattern between N2 and nath-10 mutants. Interestingly, we found that the nath-10 mutants did not appear different in transcript variance compared to N2 animals for these subsets of genes (Supplementary Figures 6A–C). Furthermore, analysis of highly variable genes found in N2 did not display increased levels of variance beyond their wild-type level of variation in nath-10 alleles (Supplementary Figure 6D), indicating that there is no evidence to suggest that nath-10 alleles were more sensitive to environmental variation. Taken together, these results suggest that nath-10 loss-of-function increases transcript variability across populations for a number of genes expressed in seam cells.
Wnt Pathway Activation Is Disrupted When nath-10 Function Is Impaired
We then sought to uncover a potential seam cell target that may underlie some of the seam cell defects observed in nath-10 mutants. We focused on the GATA transcription factor egl-18 (Koh and Rothman, 2001; Gorrepati et al., 2013) identified in the RNA-seq analysis above, which has been implicated in seam cell development as a direct target of Wnt signaling (Gorrepati et al., 2013).
To test whether egl-18 expression is altered in the nath-10 mutant background, we carried out comparative egl-18 smFISH at the L2 symmetric and asymmetric cell divisions, focusing on V seam cell lineages where most of the developmental errors occur. We found that egl-18 levels were mildly increased in V lineage seam cells during the symmetric and asymmetric cell division (Figures 5A–D). We also found that egl-18 mRNA levels were significantly increased and more variable in anterior seam cell daughters, which normally differentiate in wild type following the asymmetric L2 division (Figures 5B,D). To further confirm these changes in egl-18 expression, we analyzed animals carrying an egl-18 transcriptional reporter, egl-18p::mCherry. In concurrence with the mRNA changes observed by smFISH, we found an increase in the egl-18 reporter expression and variability at the asymmetric division during the L2 stage when animals are exposed to nath-10 RNAi compared to empty vector control (Figures 5E,F). Since egl-18 is known to play a role in seam cell fate acquisition (Gorrepati and Eisenmann, 2015), the increase in egl-18 expression in nath-10 mutants may be indicative of ectopic Wnt target activation and adoption of seam cell fate. This effect is likely to explain seam cell gains, which are somewhat more frequent than seam cell losses in nath-10(icb102) mutant populations (91 errors in 335 animals out of which 67 are seam cell gains and 24 are seam cell losses).
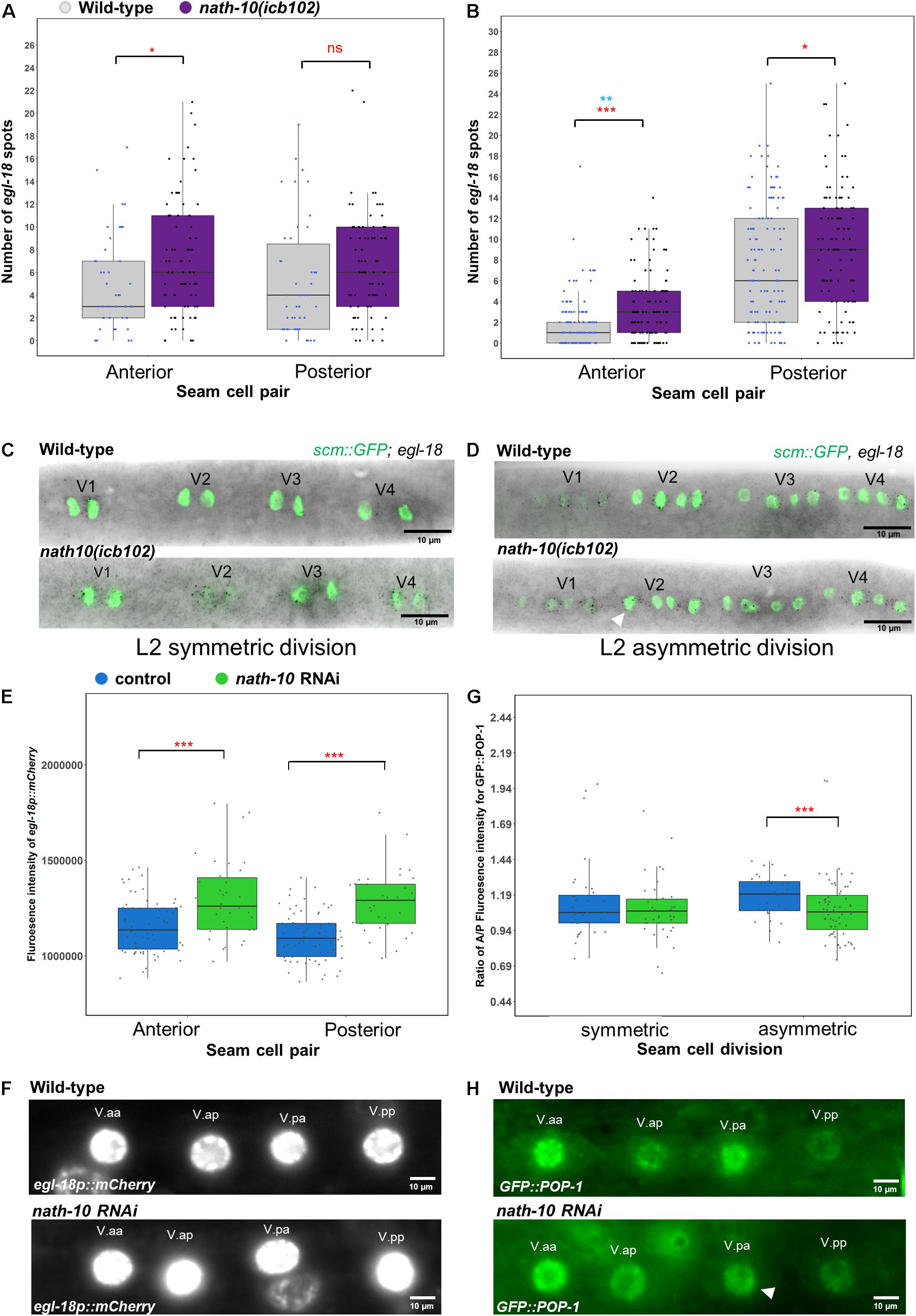
Figure 5. nath-10 loss-of-function leads to changes in Wnt signaling and activation. (A,B) smFISH counts of egl-18 in L2 symmetric (A) and asymmetric (B) cell division of V1-V4 lineage cells in wild type (gray) and nath-10(icb102) (purple). Note that nath-10(icb102) animals show a significant increase in mRNA transcript counts in anterior V lineage daughters [t-test, *p < 0.05, wild type n = 46, nath-10(icb102) n = 78 cells]. (B) smFISH counts of egl-18 in L2 asymmetric V cell lineage division. Anterior and posterior V lineage cells in nath-10(icb102) show a significant increase in average mRNA count and variability [t-test, ***p < 0.001 in red, wild type n = 129, nath-10(icb102) n = 106 cells and Levene’s test **p < 0.01, blue]. (C,D) Representative images of egl-18 smFISH in wild type and nath-10(icb102) animals at L2 symmetric and asymmetric division stage. (E) Quantification of fluorescence intensity from the egl-18::mCherry reporter in the nuclei of V lineage seam cell pairs (V.aa/V.ap and V.pa/V.pp) at the asymmetric L2 division. egl-18::mCherry is significantly increased in the anterior and posterior V lineage seam cell daughters in animals exposed to nath-10 RNAi compared to empty vector control (***p < 0.001, t-test, wild type n = 69, nath-10 RNAi n = 34). (F) Representative images for egl-18p::mCherry in seam cell nuclei pairs (V.aa/V.ap and V.pa/V.pp) at L2 asymmetric division stages for nath-10 RNAi exposed animals and empty vector control. (G) Quantification of anterior/posterior cell ratio of GFP::POP-1 at the symmetric and asymmetric L2 division in V lineage seam cell pairs (V.aa/V.ap and V.pa/V.pp). A significant decrease in this ratio was found when animals were exposed to nath-10 RNAi (***p < 0.001, t-test). (H) Representative GFP::POP-1 images in control and nath-10 RNAi animals, arrowhead indicates representative anterior V lineage cell with lower expression of GFP::POP-1 (wild type n = 31, nath-10 RNAi n = 65).
As egl-18 expression is a consequence of Wnt pathway activation, we sought to understand if the distribution of its upstream activator POP-1 is also changed upon perturbing nath-10 levels. Following wild-type asymmetric seam cell division, POP-1 levels are high in the anterior daughter nucleus and low in the posterior daughter nucleus. In the anterior daughter, high levels of POP-1 are thought to lead to a suppression of Wnt targets, while low levels of POP-1 in the posterior daughter result in Wnt target activation (Lin et al., 1998; Rocheleau et al., 1999; Phillips et al., 2007; Gleason and Eisenmann, 2010). We measured GFP::POP-1 fluorescent intensity in the nucleus of anterior and posterior V lineage cells following the L2 symmetric and asymmetric seam cell divisions in nath-10 RNAi exposed animals and control treatments. We found a lower ratio of anterior to posterior nuclear POP-1 levels in nath-10 RNAi animals compared to control treated animals at the asymmetric division stage, and no significant difference at the symmetric division stage (Figures 5G,H). This finding indicates that a number of V cell lineage daughters in nath-10 RNAi treated animals appear to have more equal POP-1 levels in the nucleus. These results are in line with the increase in egl-18 expression seen in anterior V lineage daughters and suggest that nath-10 loss-of-function may interfere with the pattern of Wnt pathway activation through changes in the cellular distribution of POP-1.
Discussion
Through an unbiased genetic screen followed up by a multifaceted validation approach, we identify here a new role for nath-10 in modulating epidermal stem cell number in C. elegans. The N-acetyltransferase nath-10 is the homolog of the human NAT10, which is known to localize in the nucleolus and is shown to be involved in the regulation of telomerase activity, tRNA transcription and cytokinesis via acetylation of microtubules (Chi et al., 2007; Shen et al., 2009; Cai et al., 2017; Sleiman and Dragon, 2019). In C. elegans, the molecular function of nath-10 has not been previously studied. However, it is known that cryptic genetic variation in nath-10 has been selected in the reference strain N2 for optimal growth under laboratory conditions (Duveau and Félix, 2012). It is conceivable that nath-10, like its human homolog, may have multiple interaction partners and be involved in controlling a number of cellular processes. Therefore, impairment of nath-10 function may have broad phenotypic consequences at the individual and population level.
We report that a partial loss-of-function of nath-10 increases transcript variance across populations. While it is difficult to completely rule out technical variability as a factor contributing to the higher variance observed in nath-10 replicates, we think that this is unlikely to be the case for multiple reasons. For example, we did not observe variable growth in the nath-10 mutant background and we did not find a specific biological replicate being consistently an outlier in the gene expression analysis possibly reflecting variability or error while conducting these experiments. Evidence in yeast suggests that regulators of robustness to stochastic variation can also influence the robustness to environmental variation (Lehner, 2010). It is therefore possible that nath-10 mutations may also sensitize the animals to environmental variation. However, we found that increase in transcript variance does not apply to several temperature or stress-related genes. Furthermore, noisy genes in wild type, which are predicted to be more responsive to environmental variation (Lehner and Kaneko, 2011), do not appear to be hypervariable in nath-10 mutants. One way to explain mRNA transcript variability is through transcript stability, which can readily explain changes in gene expression (Ross, 1995). A recent study showed that the human NAT10 may be responsible for mRNA stability and translation efficiency through N4-actelylation of cytidine present on mRNA transcripts (Ito et al., 2014; Arango et al., 2018). Therefore, it is possible that a change in the ability of NATH-10 to carry out this acetylation may change the stability of mRNA transcripts. Interestingly, the mutation recovered from our screen falls outside the known domains of the protein and therefore may cause a mild structural change, which could compromise the ability of NATH-10 to perform its function. Proline residue mutations have been shown to affect protein structure and subsequent binding efficiency (Davis et al., 2018) so this amino acid change or a decrease in NATH-10 levels may lower acetylation and therefore destabilize mRNA transcripts. It remains unclear at this point what is the biological significance of transcript variance across populations for the variable seam cell number phenotype, but a key prediction from this work is that molecular variation may also occur within single animals in the nath-10 mutant population and this may contribute to the destabilization of seam cell development.
NAT10 has also been implicated in the rapid cellular aging disease Hutchinson-Gilford progeria (HGP). A recent study showed that cells affected by HGP, chemical inhibition of NAT10 through application of a small molecule caused the rapid aging of cells to be reversed (Larrieu et al., 2018). It was further shown that an interaction with the microtubule network and the disruption of a nuclear import pathway and nuclear pore complexes was responsible for this phenotype. During seam cell division, the import and export of factors in and out of the nucleus is crucial for correct cell fate patterning. This is exemplified by the nuclear accumulation of the TCF/LEF factor POP-1, which changes in Wnt responding cells and was found to be perturbed upon nath-10 knockdown. It is possible that impairment of nath-10 may interfere with nuclear import and export pathways, as seen for example in the variability of expression of the exportin 1 homolog xpo-1, which may further disrupt protein localization during stem cell patterning.
Seam cell division patterns and polarity are predominantly under the control of Wnt signaling (Takeshita and Sawa, 2005; Wildwater et al., 2011; Yamamoto et al., 2011; Gorrepati et al., 2013; van der Horst et al., 2019). We describe a new link between NATH-10 and Wnt signaling, based on the impaired distribution of a Wnt effector and subsequent activation of a Wnt pathway target in nath-10 RNAi treated animals. Strikingly, anterior V cell daughters following the second L2 division displayed an increase in average number and variance of egl-18 mRNA transcripts. These observations were reproduced with a transcriptional egl-18::mCherry marker, suggesting that changes in the nath-10 background reflect transcriptional activity of egl-18, likely through changes in upstream regulators. This is consistent with the changes found in POP-1 distribution in nath-10 RNAi treated animals. POP-1 is the TCF/LEF factor that is responsible for activating Wnt target genes including egl-18, and seam cell fate through asymmetric nuclear accumulation (Lin et al., 1995; Rocheleau et al., 1999; Mizumoto and Sawa, 2007; Phillips et al., 2007; Gorrepati et al., 2013). It is of note that POP-1 requires acetylation at three specific sites to increase its nuclear export efficiency (Gay et al., 2003). NAT10 has been shown to have a role in regulating nuclear export machinery and is able to acetylate multiple substrates, so it is therefore possible that nath-10 may play a direct role in the control of POP-1 or other core Wnt components (Larrieu et al., 2018). Future work is required to be able to understand how NATH-10 mechanistically interacts with the Wnt signaling pathway to facilitate robust seam cell fate patterning.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
MH carried out the majority of experiments and data analysis. DK developed the hairpin construction platform and contributed to mutation mapping. VS advised on gene expression variability analysis. MB supervised the work. MH, DK and MB wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the European Research Council (ROBUSTNET-639485). DK received a President’s Ph. D. scholarship from Imperial College London.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Barkoulas lab for discussions and Diane Cheng for technical help. We also thank Wormbase. Some C. elegans strains were provided by the CGC, which was funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.640856/full#supplementary-material
Supplementary Figure 1 | Design for hairpin construction and validation of the system in the epidermis. (A) A schematic of the hairpin RNAi construct. The final construct carries restriction sites flanking the promoter site, to allow for insertion of any promoter of interest, such as a seam cell and hypodermis-specific promoter. It also carries two entry sites (forward and reverse) for the insertion of the inverted fragments, flanking intron 5 of srf-3 isoform a, followed by a 3′ UTR. The insertion sites contain inverted repeats of recognition sites for the Type IIS restriction enzymes BpiI in the forward entry site and Esp3I in the reverse. These enzymes cut asymmetrically 2–6 bp away from the recognition sequence creating 5′ 4 bp long overhangs. The sequences cleaved for both enzymes create incompatible non-palindromic overhangs, so that a fragment with compatible overhangs can be inserted in the two sites in opposite orientations. Transgenic animals in all panels carry myo-2p:dsRED marker. (B) Seam cell specific knockdown of targets was validated by creating a hairpin against GFP and expressing this under the srf-3 promoter. Note that scm:GFP was strongly reduced in seam cells in transgenic animals (left panel). A hypodermal specific GFP hairpin showed strong knockdown of dpy-7p:GFP in hypodermal cells (middle panel) but did not affect scm:GFP expression in seam cells.
Supplementary Figure 2 | Increase of seam cell number variance in nath-10(icb102) mutants is reproducible. Seam cell counts for wild type and nath-10(icb102) animals carried out on separate days show similar average seam cell counts and reproducibly significant increase in variability (Levene’s test ∗∗∗p < 0.001).
Supplementary Figure 3 | Characterization of nath-10 mutant phenotypes and expression outside the seam cells. (A) Brood size assay for N2, nath-10(icb102) and nath-10(icb99). There was a significant decrease in the number of progeny for both strains carrying nath-10 alleles (t-test, ∗∗∗p < 0.001, red), but there was no significant difference in the variability of progeny number (P > 0.05, Levene’s test, blue). (B) Proportion of L4 animals in N2 and nath-10 alleles at defined time intervals. No significant delay in developmental timing was observed for the nath-10 alleles compared to wild-type animals. (C) Wild-type animals show nath-10 expression by smFISH within the gonad, outlined in yellow, at the late L4 stage.
Supplementary Figure 4 | PCA of all replicates used in the RNA-seq analysis and GO term analysis of differentially expressed genes. (A) PCA analysis of samples used for RNA-seq analysis indicates nath-10 and wild-type replicates can be separated on the PC1 axis. (B,C) GO analysis of upregulated genes (A) or downregulated genes (B). Significance was calculated using a hypergeometric probability test and corrected by the Benjamini-Hochberg step-up algorithm with a q-value threshold of 0.1.
Supplementary Figure 5 | RNAi screen on candidate genes to test interaction with the nath-10 seam cell phenotype. (A–E) Seam cell counts for selected candidates showed no significant interaction with the nath-10(icb102) seam cell number variability. Candidate genes for the RNAi screen were selected as top on the list of differentially expressed genes using the following filtering criteria: fold change, seam cell expression, seam cell phenotype and predicted interaction with either core seam cell network genes (ceh-16, egl-18 and elt-1) or nath-10.
Supplementary Figure 6 | Analysis of variance within functional subsets of genes for N2 and nath-10 alleles. (A–C) Plots of the coefficient of variation against the log2 average normalized transcript count for N2 and nath-10 alleles. (D) Genes with the highest coefficient of variation in N2 were extracted from the RNA-seq data. Note that nath-10 alleles do not show variation beyond what is found in N2 in all of these cases. Functional subsets were generated from the following gene expression clusters: WBPaper00038118:stress_upregulated_N2 (A), WBPaper0032062:age_regulated_genes (B), and WBPaper00039792:TempShift (C).
Supplementary Table 1 | List of strains used in this study.
Supplementary Table 2 | List of plasmids, oligos and smFISH probes used in the study.
Supplementary Table 3 | RNA-seq analysis and gene set comparisons.
References
Ambros, V., and Horvitz, H. R. (1984). Heterochronic mutants of the nematode Caenorhabditis elegans. Science 226, 409–416. doi: 10.1126/science.6494891
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Arango, D., Sturgill, D., Alhusaini, N., Meier, J. L., Coller, J., Correspondence, S. O., et al. (2018). Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175, 1–15. doi: 10.1016/j.cell.2018.10.030
Barkoulas, M., van Zon, J. S., Milloz, J., van Oudenaarden, A., and Felix, M. A. (2013). Robustness and epistasis in the C. elegans vulval signaling network revealed by pathway dosage modulation. Dev. Cell 24, 64–75. doi: 10.1016/j.devcel.2012.12.001
Bauer, C. R., Li, S., and Siegal, M. L. (2015). Essential gene disruptions reveal complex relationships between phenotypic robustness, pleiotropy, and fitness. Mol. Syst. Biol. 11:773. doi: 10.15252/msb.20145264
Bray, N. L., Pimentel, H., Melsted, P., and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi: 10.1038/nbt.3519
Cai, S., Liu, X., Zhang, C., Xing, B., and Du, X. (2017). Autoacetylation of NAT10 is critical for its function in rRNA transcription activation. Biochem. Biophys. Res. Commun. 483, 624–629. doi: 10.1016/j.bbrc.2016.12.092
Cao, J., Packer, J. S., Ramani, V., Cusanovich, D. A., Huynh, C., Daza, R., et al. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667. doi: 10.1126/science.aam8940
Chi, Y.-H., Haller, K., Peloponese, J.-M. Jr., and Jeang, K.-T. (2007). Histone acetyltransferase hALP and nuclear membrane protein hsSUN1 function in de-condensation of mitotic chromosomes. J. Biol. Chem. 282, 27447–27458. doi: 10.1074/jbc.M703098200
Chisholm, A. D., and Hsiao, T. I. (2012). The Caenorhabditis elegans epidermis as a model skin. I: development, patterning, and growth. Wiley Interdiscip. Rev. Dev. Biol. 1, 861–878. doi: 10.1002/wdev.79
Davis, J. E., Alghanmi, A., Gundampati, R. K., Jayanthi, S., Fields, E., Armstrong, M., et al. (2018). Probing the role of proline -135 on the structure, stability, and cell proliferation activity of human acidic fibroblast growth factor. Arch. Biochem. Biophys. 654, 115–125. doi: 10.1016/j.abb.2018.07.017
Doitsidou, M., Jarriault, S., and Poole, R. J. (2016). Next-generation sequencing-based approaches for mutation mapping and identification in Caenorhabditis elegans. Genetics 204, 451–474. doi: 10.1534/genetics.115.186197
Duveau, F., and Félix, M. A. (2012). Role of pleiotropy in the evolution of a cryptic developmental variation in Caenorhabditis elegans. PLoS Biol. 10:e1001230. doi: 10.1371/journal.pbio.1001230
Felix, M. A., and Barkoulas, M. (2015). Pervasive robustness in biological systems. Nat. Rev. Genet. 16, 483–496. doi: 10.1038/nrg3949
Gay, F., Calvo, D., Lo, M. C., Ceron, J., Maduro, M., Lin, R., et al. (2003). Acetylation regulates subcellular localization of the Wnt signaling nuclear effector POP-1. Genes Dev. 17, 717–722. doi: 10.1101/gad.1042403
Gleason, J. E., and Eisenmann, D. M. (2010). Wnt signaling controls the stem cell-like asymmetric division of the epithelial seam cells during C. elegans larval development. Dev. Biol. 348, 58–66. doi: 10.1016/j.ydbio.2010.09.005
Gorrepati, L., and Eisenmann, D. M. (2015). The C. elegans embryonic fate specification factor EGL-18 (GATA) is reutilized downstream of Wnt signaling to maintain a population of larval progenitor cells. Worm 4:e996419. doi: 10.1080/23723556.2014.996419
Gorrepati, L., Thompson, K. W., and Eisenmann, D. M. (2013). C. elegans GATA factors EGL-18 and ELT-6 function downstream of Wnt signaling to maintain the progenitor fate during larval asymmetric divisions of the seam cells. Development 140, 2093–2102. doi: 10.1242/dev.091124
Harandi, O. F., and Ambros, V. R. (2015). Control of stem cell self-renewal and differentiation by the heterochronic genes and the cellular asymmetry machinery in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 112, E287–E296. doi: 10.1073/pnas.1422852112
Huang, X., Tian, E., Xu, Y., and Zhang, H. (2009). The C. elegans engrailed homolog ceh-16 regulates the self-renewal expansion division of stem cell-like seam cells. Dev. Biol. 333, 337–347. doi: 10.1016/j.ydbio.2009.07.005
Hughes, S., Brabin, C., Appleford, P. J., and Woollard, A. (2013). CEH-20/Pbx and UNC-62/Meis function upstream of rnt-1/Runx to regulate asymmetric divisions of the C. elegans stem-like seam cells. Biol. Open 2, 718–727. doi: 10.1242/bio.20134549
Ito, S., Horikawa, S., Suzuki, T., Kawauchi, H., Tanaka, Y., Suzuki, T., et al. (2014). Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J. Biol. Chem. 289, 35724–35730. doi: 10.1074/jbc.C114.602698
Katsanos, D., Koneru, S. L., Mestek Boukhibar, L., Gritti, N., Ghose, R., Appleford, P. J., et al. (2017). Stochastic loss and gain of symmetric divisions in the C. elegans epidermis perturbs robustness of stem cell number. PLoS Biol. 15:e2002429. doi: 10.1371/journal.pbio.2002429
Koh, K., and Rothman, J. H. (2001). ELT-5 and ELT-6 are required continuously to regulate epidermal seam cell differentiation and cell fusion in C. elegans. Development 128, 2867–2880.
Larrieu, D., Vire, E., Robson, S., Breusegem, S. Y., Kouzarides, T., and Jackson, S. P. (2018). Inhibition of the acetyltransferase NAT10 normalizes progeric and aging cells by rebalancing the Transportin-1 nuclear import pathway. Sci. Signal. 11:eaar5401. doi: 10.1126/scisignal.aar5401
Lehner, B. (2010). Genes confer similar robustness to environmental, stochastic, and genetic perturbations in yeast. PLoS One 5:e9035. doi: 10.1371/journal.pone.0009035
Lehner, B., and Kaneko, K. (2011). Fluctuation and response in biology. Cell. Mol. Life Sci. 68, 1005–1010. doi: 10.1007/s00018-010-0589-y
Levy, S. F., and Siegal, M. L. (2008). Network hubs buffer environmental variation in Saccharomyces cerevisiae. PLoS Biol. 6:e264. doi: 10.1371/journal.pbio.0060264
Lin, R., Hill, R. J., and Priess, J. R. (1998). POP-1 and anterior-posterior fate decisions in C. elegans embryos. Cell 92, 229–239.
Lin, R., Thompson, S., and Priess, J. R. (1995). pop-1 encodes an HMG box protein required for the specification of a mesoderm precursor in early C. elegans embryos. Cell 83, 599–609.
Mellert, D. J., Williamson, W. R., Shirangi, T. R., Card, G. M., and Truman, J. W. (2016). Genetic and environmental control of neurodevelopmental robustness in Drosophila. PLoS One 11:e0155957. doi: 10.1371/journal.pone.0155957
Minevich, G., Park, D. S., Blankenberg, D., Poole, R. J., and Hobert, O. (2012). CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–1269. doi: 10.1534/genetics.112.144204
Mizumoto, K., and Sawa, H. (2007). Cortical beta-catenin and APC regulate asymmetric nuclear beta-catenin localization during asymmetric cell division in C. elegans. Dev. Cell 12, 287–299. doi: 10.1016/j.devcel.2007.01.004
Paix, A., Folkmann, A., Rasoloson, D., and Seydoux, G. (2015). High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics 201, 47–54. doi: 10.1534/genetics.115.179382
Pfeiffer, B. D., Truman, J. W., and Rubin, G. M. (2012). Using translational enhancers to increase transgene expression in Drosophila. Proc. Natl. Acad. Sci. 109, 6626–6631. doi: 10.1073/pnas.1204520109
Phillips, B. T., Kidd, A. R. III, King, R., Hardin, J., and Kimble, J. (2007). Reciprocal asymmetry of SYS-1/beta-catenin and POP-1/TCF controls asymmetric divisions in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 104, 3231–3236. doi: 10.1073/pnas.0611507104
R Core Team. (2020). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rocheleau, C. E., Yasuda, J., Shin, T. H., Lin, R., Sawa, H., Okano, H., et al. (1999). WRM-1 activates the LIT-1 protein kinase to transduce anterior/posterior polarity signals in C. elegans. Cell 97, 717–726. doi: 10.1016/s0092-8674(00)80784-9
Shen, Q., Zheng, X., McNutt, M. A., Guang, L., Sun, Y., Wang, J., et al. (2009). NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp. Cell Res. 315, 1653–1667. doi: 10.1016/j.yexcr.2009.03.007
Sleiman, S., and Dragon, F. (2019). Recent advances on the structure and function of RNA acetyltransferase Kre33/NAT10. Cells 8:1035.
Smith, J. A., McGarr, P., and Gilleard, J. S. (2005). The Caenorhabditis elegans GATA factor elt-1 is essential for differentiation and maintenance of hypodermal seam cells and for normal locomotion. J. Cell Sci. 118(Pt 24), 5709–5719. doi: 10.1242/jcs.02678
Sulston, J. E., and Horvitz, H. R. (1977). Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110–156. doi: 10.1016/0012-1606(77)90158-0
Takeshita, H., and Sawa, H. (2005). Asymmetric cortical and nuclear localizations of WRM-1/beta-catenin during asymmetric cell division in C. elegans. Genes Dev. 19, 1743–1748. doi: 10.1101/gad.1322805
Tavernarakis, N., Wang, S. L., Dorovkov, M., Ryazanov, A., and Driscoll, M. (2000). Heritable and inducible genetic interference by double-stranded RNA encoded by transgenes. Nat. Genet. 24, 180–183. doi: 10.1038/72850
Timmons, L., Tabara, H., Mello, C. C., and Fire, A. Z. (2003). Inducible systemic RNA silencing in Caenorhabditis elegans. Mol. Biol. Cell 14, 2972–2983. doi: 10.1091/mbc.e03-01-0858
van der Horst, S. E. M., Cravo, J., Woollard, A., Teapal, J., and van den Heuvel, S. (2019). C. elegans Runx/CBFbeta suppresses POP-1 TCF to convert asymmetric to proliferative division of stem cell-like seam cells. Development 146:dev180034. doi: 10.1242/dev.180034
Waddington, C. H. (1959). Canalization of development and genetic assimilation of acquired characters. Nature 183, 1654–1655.
Wildwater, M., Sander, N., de Vreede, G., and van den Heuvel, S. (2011). Cell shape and Wnt signaling redundantly control the division axis of C. elegans epithelial stem cells. Development 138, 4375–4385. doi: 10.1242/dev.066431
Yamamoto, Y., Takeshita, H., and Sawa, H. (2011). Multiple Wnts redundantly control polarity orientation in Caenorhabditis elegans epithelial stem cells. PLoS Genet. 7:e1002308. doi: 10.1371/journal.pgen.1002308
Keywords: developmental robustness, seam cells, epidermis, stem cells, C. elegans, nath-10, NAT10, Wnt signaling
Citation: Hintze M, Katsanos D, Shahrezaei V and Barkoulas M (2021) Phenotypic Robustness of Epidermal Stem Cell Number in C. elegans Is Modulated by the Activity of the Conserved N-acetyltransferase nath-10/NAT10. Front. Cell Dev. Biol. 9:640856. doi: 10.3389/fcell.2021.640856
Received: 12 December 2020; Accepted: 19 April 2021;
Published: 18 May 2021.
Edited by:
Rodrigo Young, University College London, United KingdomReviewed by:
Rob Jelier, KU Leuven, BelgiumMirko Francesconi, École Normale Supérieure de Lyon, Université de Lyon, France
Copyright © 2021 Hintze, Katsanos, Shahrezaei and Barkoulas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michalis Barkoulas, bS5iYXJrb3VsYXNAaW1wZXJpYWwuYWMudWs=