 Takehiko Usui
Takehiko Usui Akira Shinohara*
Akira Shinohara*- Institute for Protein Research, Osaka University, Suita, Japan
Exogenous double-strand breaks (DSBs) induce a DNA damage response during mitosis as well as meiosis. The DNA damage response is mediated by a cascade involving Mec1/Tel1 (ATR/ATM) and Rad53 (Chk2) kinases. Meiotic cells are programmed to form DSBs for the initiation of meiotic recombination. In budding yeast, Spo11-mediated meiotic DSBs activate Mec1/Tel1, but not Rad53; however, the mechanism underlying the insensitivity of Rad53 to meiotic DSBs remains largely unknown. In this study, we found that meiotic cells activate Rad53 in response to exogenous DSBs and that this activation is dependent on an epigenetic marker, Dot1-dependent histone H3K79 methylation, which becomes a scaffold of an Rad53 mediator, Rad9, an ortholog of 53BP1. In contrast, Rad9 is insensitive to meiotic programmed DSBs. This insensitiveness of Rad9 derives from its inability to bind to the DSBs. Indeed, artificial tethering of Rad9 to the meiotic DSBs activated Rad53. The artificial activation of Rad53 kinase in meiosis decreases the repair of meiotic DSBs. These results suggest that the suppression of Rad53 activation is a key event in initiating a meiotic program that repairs programmed DSBs.
Introduction
DNA double-strand breaks (DSBs) are accidentally formed in cells with endogenous and exogenous DNA damage. DSBs induce a DNA damage response enabling the cell to cope with the damage and to coordinate DNA damage tolerance, including the repair of DSBs, throughout progression in the cell cycle. Failure of the DNA damage response to DSBs leads to instability of the genome, which is often associated with the onset of cancer.
DNA damage response is a complex biological pathway that is accompanied by a signaling cascade involving multiple kinases. In mitotic dividing cells, DSBs activate 2 key kinases, Mec1 (ATR in humans), a member of the PI3-kinase family, and Tel1 (ATM in humans) [reviewed in Nyberg et al. (2002))]. Mec1 is recruited to single-stranded DNA (ssDNA) in a Ddc2 (ATRIP)-dependent manner. Ddc2 recognizes replication protein-A (RPA)-coated ssDNA (Paciotti et al., 2000; Zou and Elledge, 2003). Tel1 binds to the ends of DSBs through Xrs2/Nbs1 in the Mre11-Rad50-Xrs2/Nbs1 (MRX/N in humans) complex (Nakada et al., 2003). Activated Mec1/Tel1 then stimulates Rad53 kinase (Chk2 in humans) (Sanchez et al., 1996; Sun et al., 1996) and transduces a signal to downstream effector pathways (Allen et al., 1994). Rad9 (53BP1 in humans) is a key modulator of Rad53 activation (Sun et al., 1998; Schwartz et al., 2002). Rad9 is recruited to the DNA damage area using 2 distinct histone modifications, histone H2AS129 phosphorylation (γ-H2AX in humans) and H3K79 methylation (Giannattasio et al., 2005; Wysocki et al., 2005; Toh et al., 2006; Grenon et al., 2007; Hammet et al., 2007). H2AS129 phosphorylation and H3K79 methylation are recognized by 2 domains of Rad9, that is, BRCT and Tudor, respectively. Histone H2AS129 phosphorylation is DNA damage-dependent and is catalyzed by Mec1 and Tel1 (Shroff et al., 2004). On the other hand, H3K79 methylation is constitutive and is mediated by Dot1 methyltransferase. Rad9 recruited to DSBs is phosphorylated by Mec1/Tel1 (Vialard et al., 1998; Schwartz et al., 2002), leading to phosphorylation-dependent oligomerization of Rad9 (Usui et al., 2009). These upstream events promote a scaffold function of Rad9 for Mec1/Tel1-dependent Rad53 activation (Gilbert et al., 2001; Sweeney et al., 2005). Rad9 facilitates Mec1/Tel1 phosphorylation of Rad53 (Sweeney et al., 2005). This trans-phosphorylation by Mec1/Tel1 appears to function as a priming event for Rad53 activation. Rad9 also mediates the self-phosphorylation of Rad53 (Gilbert et al., 2001). The cis-phosphorylation induces the conversion of Rad53 into a fully activated form (Pellicioli et al., 1999; Gilbert et al., 2001; Sweeney et al., 2005). Moreover, Rad9 undergoes phosphorylation by cyclin-dependent kinase (CDK) to modulate the DNA damage response in a cell-cycle dependent manner (Bonilla et al., 2008; Granata et al., 2010; Pfander and Diffley, 2011). This phosphorylation promotes the binding of Rad9 to the other DDR mediator protein, Dpb11 (Pfander and Diffley, 2011; Puddu et al., 2011; di Cicco et al., 2017), whose recruitment to DSB sites is dependent of the Rad17-Ddc1-Mec3 checkpoint clamp (Rad9-Rad1-Hus1 in other organisms).
Although DSBs are usually an accidental event in cells, several somatic cells also introduce programmed DSBs in their genomes in order to promote cellular differentiation, such as mating type switching in fungi and antigen receptor rearrangement in the immune cells of vertebrates [reviewed in Haber (2012), Alt et al. (2013))]. Moreover, meiotic cells in sexually reproducing eukaryotes develop a program to induce hundreds of DSBs in their genomes (∼160 in the budding yeast, ∼300 in humans/mice, and more than ∼1,000 in the lily), which initiates meiotic recombination (Terasawa et al., 1995; Barlow et al., 1997; Buhler et al., 2007; Kauppi et al., 2011; Pan et al., 2011). Such recombination enables the exchange of paternal and maternal DNA molecules to form a crossover (CO) [reviewed in Bishop and Zickler (2004))]. The CO is converted into an exchange of homologous chromosome axes, which are cytologically visualized as chiasma. The chiasma promotes proper segregation of homologous chromosomes at meiosis I along with cohesion distal to chiasmata. The meiotic recombination is essential for meiosis, and thus gamete formation. Any defects in recombination result in mis-segregation of homologous chromosomes, leading to the formation of aneuploidy gametes.
The formation of meiotic DSBs is catalyzed by Spo11, a meiosis-specific topoisomerase VI-like protein (Bergerat et al., 1997; Keeney et al., 1997). Spo11-induced DSBs are processed to generate a 3′-overhanging ssDNA. This ssDNA is coated with RPA to become a template for homology search by 2 RecA homologs, Rad51 and meiosis-specific Dmc1 (Bishop et al., 1992; Shinohara et al., 1992). After homology search by Rad51 and Dmc1, the ssDNA invades homologous double-stranded DNAs (dsDNAs). The interaction between the ssDNA and dsDNA leads to the formation of a recombination intermediate, single-end invasion (SEI) (Allers and Lichten, 2001b; Hunter and Kleckner, 2001). The SEI is then converted into the next intermediate with double Holliday structures (dHJs) (Schwacha and Kleckner, 1995). The dHJ is predominantly resolved into reciprocal exchange molecules, COs (Allers and Lichten, 2001a; Hunter and Kleckner, 2001; Borner et al., 2004). In meiosis, recombination preferentially occurs between homologous chromosomes rather than between sister chromatids, as seen in mitotic cells (interhomolog bias) (Schwacha and Kleckner, 1997).
Recent studies have shown that canonical DNA damage signaling is non-responsive to ∼160 Spo11-induced DSBs in meiotic cells of the budding yeast (Lydall et al., 1996; Carballo et al., 2008; Cartagena-Lirola et al., 2008). This is quite different from the response of vegetatively growing cells, which are very sensitive even to a single DSB (Lee et al., 1998; Pellicioli et al., 2001). Spo11-induced DSBs are known to activate Mec1/Tel1, which in turn activates a meiosis-specific Rad53 paralog, Mek1/Mre4 kinase in the context of chromatin (Hollingsworth and Gaglione, 2019). Mec1/Tel1-dependent phosphorylation of a Mek1 adaptor protein, Hop1, provides the binding site of Mek1 for oligomerization for the activation (Niu et al., 2005; Carballo et al., 2008). Activated Mek1 promotes interhomolog recombination and CO formation as well as activates the recombination checkpoint during prophase I (Hollingsworth and Gaglione, 2019). Importantly, Spo11-induced DSBs do not activate Rad53 (Carballo et al., 2008; Cartagena-Lirola et al., 2008), suggesting that the upstream activation of the canonical pathway is turned on but that the downstream events are suppressed. Indeed, meiotic cells are not intrinsically inert to Rad53 activation; the treatment of spo11 mutant cells with exogenous DNA damaging agents, such as phleomycin, stimulates Rad53 kinase activity (Cartagena-Lirola et al., 2008). Moreover, the artificial targeting of Rad53, through fusion to Ddc2 (Lee et al., 2004), to ssDNAs of Spo11-induced DSBs induces Rad53 activation in meiosis (Cartagena-Lirola et al., 2008). Previous study also showed that over-expression of Rad53 during meiosis activates Rad53 kinase activity (Usui and Kanehara, 2013). Therefore, it is postulated that meiotic cells develop a mechanism to make Rad53 insensitive to Spo11-dependent DSBs. The mechanism of this insensitiveness of Rad53 to meiotic DSBs remains unknown.
In this study, we showed that, compared to mitotic cells, meiotic cells rely more on Dot1-dependent histone H3K79 methylation to activate Rad53 in response to exogenous DSBs. Moreover, like Rad53 tethering (Cartagena-Lirola et al., 2008), the artificial tethering of Rad9 to meiotic Spo11-mediated DSBs by fusion with Ddc2 stimulated Rad53 kinase activity. Consistent with this, a chromatin-immunoprecipitation (ChIP) assay revealed minimal binding of Rad9 to meiotic DSBs. These results suggest that meiotic DSBs are masked to Rad9 in order to downregulate Rad53 kinase activity in meiotic prophase I. Untimely activation of Rad53 in meiotic prophase I impairs chromosome events such as meiotic recombination. We propose that meiotic cells execute a program that suppresses a part of the DNA damage response to develop meiosis-specific DSB-induced chromosome metabolism.
Materials and Methods
Strains and Plasmids
All the strains described here, with the exception of HO endonuclease-inducible strains, are derivatives of SK1 diploids. The strain list is provided in Supplementary Table 1. The pUS48 plasmid harbored DDC2-3xHA-RAD9 fused with a 0.8-kb DNA fragment containing the DMC1 5′-UTR fragment followed by the kan gene from a pFA6a-KANMX6 plasmid (Longtine et al., 1998). Like the pUS48, the pUS49 plasmid harbored 3xHA-RAD9 fused with the DNA fragment containing the DMC1 5′-UTR fragment followed by the kamMX6 gene. To integrate DMC1p-DDC2-3xHA-RAD9 or -3xHA-RAD9 into the yeast genome, SK1 wild-type cells were transformed with EcoRI-digested pUS48 or pUS49 and were grown on YPAD with 100 μg/mL of G418. The plasmids pUS50, pUS72, pUS73, and pUS70 were used to prepare DDC2-fusion strains carrying rad9-7A (T390A T398A T410A T427A S435A T457A T603A), rad9-Y798A, and rad9-K1088M mutations and a genomic rad9-Y798A mutant, respectively. Yeast strains carrying N-terminal 3xHA-tagged RAD9 (Usui et al., 2009), C-terminal 6xFLAG-tagged RAD53 (Usui and Petrini, 2007), rad53-KD (K227A) (Usui and Petrini, 2007), hht1-K79R, and hht2-K79R were prepared by the 2-step integration method (Kaiser et al., 1994) using the pRS406-based plasmids, pRS406-HA-RAD9, pTAP12, pTAP10, pUS52, and pUS53, respectively. Deletion mutants of the DMC1, DOT1, RAD9, and SPO11 genes and a DDC2-3xHA strain were constructed using a polymerase chain reaction (PCR)-based method (Longtine et al., 1998). The sequences of primers used in the experiments are shown in Supplementary Table 2.
Antibodies
Anti-Flag (M2 Sigma or Wako), anti-HA (12CA5 for western blot, 16B12 for immunostaining, and F-7 for ChIP), anti-α-tubulin (Serotec), rabbit anti-Rad51 (Shinohara et al., 1992), anti-Dmc1 (Hayase et al., 2004), anti-Rad9 (Usui and Petrini, 2007), anti-Zip1 (Shinohara et al., 2008), and anti-H3K79-3me (Abcam) were used for western blot and immunostaining.
Meiotic Cell Analyses and DNA Damage Treatment
After SK1 diploid cells entered into meiosis, meiotic cell cycle progression was monitored by 4′,6-Diamidino-2-phenylindole (DAPI) staining as described previously (Hayase et al., 2004). The DNA content of fixed meiotic cells was examined with a FACS Calibur flow cytometer (BD Biosciences) after staining with propidium iodide. Physical analyses for meiotic DSBs and crossover recombinants at the HIS4-LEU2 locus were performed as described previously (Storlazzi et al., 1995; Shinohara et al., 1997). To induce accidental DSBs in meiosis, cells were incubated with 5 μg/ml of phleomycin for 30 min at 3.5 h after incubation with sporulation media (SPM).
Cytological Analysis
Spreads of the meiotic nuclei was prepared as described previously (Bishop, 1994; Shinohara et al., 1997). Immunostained samples were observed as described previously (Shinohara et al., 2000) using an epifluorescent microscope (Olympus BX51 with a 100× objective; NA, 1.4), and images were captured with a CCD camera (Cool Snap, Roper) and processed using iVision (Silicon) and Photoshop (Adobe) applications.
Western Blotting and in situ Autophosphorylation (ISA) Assay
For western blot analysis, trichloroacetic acid (TCA)-precipitated cell extracts were prepared as follows: Meiotic cells (2 × 108) were fixed in 0.5 mL of 20% TCA and centrifuged. The cell pellets were disrupted with glass beads in 0.25 mL of 20% TCA using a bead shocker (5 cycles of 2500 rpm, 60 s ON/OFF, Yasui Kikai). TCA precipitates were collected by centrifugation (4,000 rpm, 3 min) and were suspended in 0.24 mL of the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer supplemented with 0.33 M Tris-HCl (pH 8.0). The ISA assay was performed as described previously (Pellicioli et al., 1999). Briefly, TCA-precipitated cell extracts were fractionated by SDS-PAGE and transferred to a polyvinyldifluoride (PVDF) membrane (Immobilon P, Millipore). The membrane was sequentially treated by denaturation and renaturation solutions, followed by incubation with 32P-γ-ATP. 32P-incorporation to Rad53 was visualized and quantified with BAS2000 (Fujifilm).
HO Induction During Mitosis
To induce HO DSB, cells carrying a single unrepairable HO DSB site (Shroff et al., 2004) were arrested at G2/M by nocodazole (15 μg/mL) in YP-lactate. Galactose was added up to 2% to induce HO endonuclease.
Chromatin Immuno-Precipitation (ChIP) Assay
Meiotic induction was performed as described above. Cells were fixed with 1% formaldehyde for 15 min, and cell extracts were prepared as described previously (Hayase et al., 2004). Cell extracts was prepared using a bead shocker (5 cycles of 2,500 rpm, 60 s ON/OFF, Yasui Kikai). The extract corresponding to 3.6 × 108 cells in pertinent mitotic and meiotic conditions were subjected to immunoprecipitation (IP) using Protein-A coated magnetic beads pre-coated with anti-HA (F-7) or anti-Rad51. DNA precipitated with the immune complex was purified by phenol/chloroform extraction and ethanol precipitation after protease-K treatment and quantified by real-time quantitative PCR (Chromo 4, Bio-Rad) using SyberGreen system (EvaGreen Supermix, Bio-Rad). The ChIP primers used were 5′-GGGTTTATAGTGGTGCCGTTC and 5′-ATGCAACGAAGCTTCCTGGC for HIS4-LEU2, 5′-ATGCTGAAGTACGTGGTGACGGAT and 5′-CCTCCGCC ACGACCACACTCT for 0.05 kb from HO-DSB, and 5′-GGTGTGCGGAGTAATCATTTGAGG and 5′-TTATAGGAGA CAGTTTTTCCATCAA for SMC1.
Results
Histone H3K79 Methylation Is Necessary for Rad9-Dependent Rad53 Activation in Meiotic Cells After Phleomycin Treatment
Although Rad53 is not activated in response to endogenous Spo11-mediated DNA DSBs, Rad53 in meiotic cells is activated by exogenous DSBs (Cartagena-Lirola et al., 2008). Treatment of meiotic spo11 mutant cells, which are defective in meiotic DSB formation (Bergerat et al., 1997; Keeney et al., 1997), with a radiomimetic agent, phleomycin, activates Rad53, and this activation depends on Rad9, as seen in vegetatively growing wild-type cells. This indicates that the Rad9-Rad53 axis is functional during meiosis in response to accidental DSBs (Cartagena-Lirola et al., 2008). We confirmed this by treating wild-type meiotic cells with phleomycin. Treatment of meiotic prophase cells grown in sporulation media (SPM) for 3.5 h with phleomycin for 30 min (by 4 h) induced band shifts of Rad53-Flag on western blots (Figure 1A). The band shifts of Rad53 corresponded with multiple phosphorylation of Rad53 accompanied with the activation of its kinase (Pellicioli et al., 1999). Rad53 kinase activity was tested for autophosphorylation activity of Rad53 using an in situ autophosphorylation (ISA) assay (Pellicioli et al., 1999). This finding suggests that Spo11-mediated endogenous DSBs do not interfere with the Rad53 activation by exogenous DSBs. Moreover, as shown previously (Cartagena-Lirola et al., 2008), untreated wild-type meiotic cells, which have ∼160 DSBs on their genomes, showed little band shifts or little autophosphorylation of Rad53 (Figure 1A and Supplementary Figure 1A).
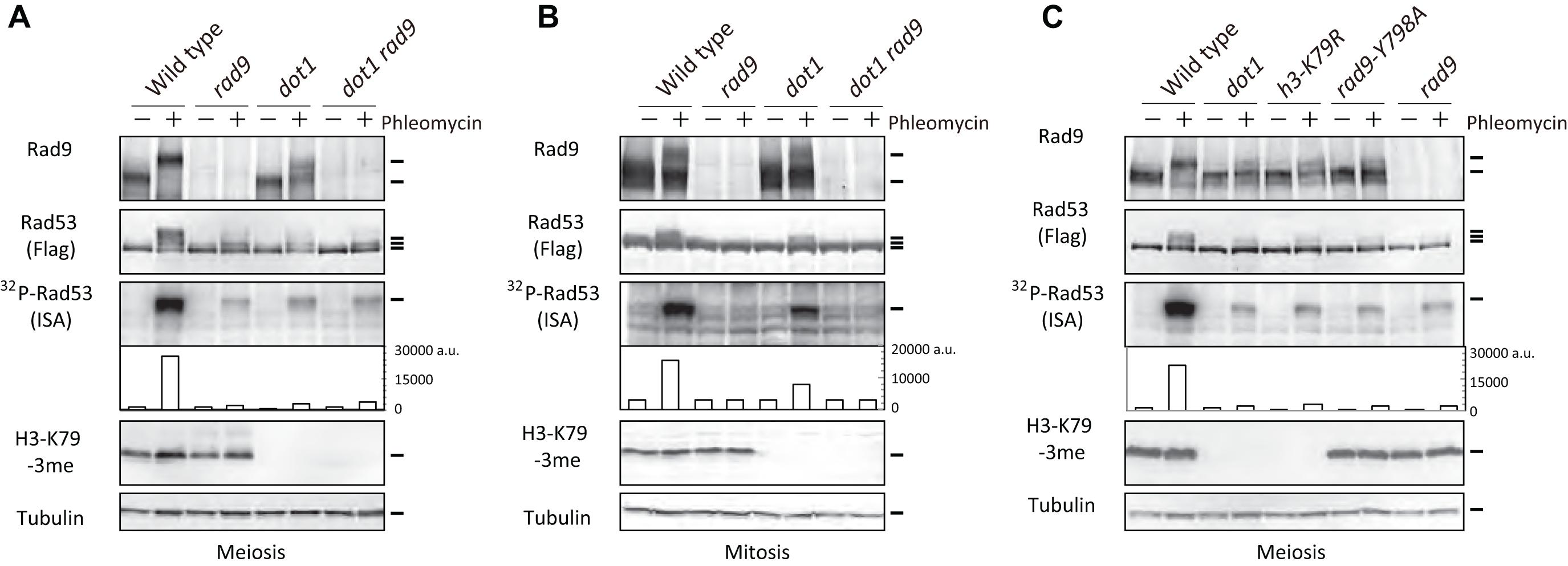
Figure 1. H3K79me is required for Rad9-dependent Rad53 activation in meiosis after phleomycin treatment. (A) TCA-precipitated cell extracts were prepared from wild-type (USY543/544), rad9 (USY524/525), dot1 (USY526/527), and dot1 rad9 (USY522/523) diploid strains, and were analyzed by western blotting using anti-Rad9, anti-Flag (for Rad53), anti-H3K79-3me, and anti-tubulin antibodies. “32P-Rad53” represented 32P-incorporation to Rad53 in the ISA assay. Band intensities were quantified and are shown as arbitrary units without normalization (fourth panels). Cell extracts derived from 4 × 106 cells were loaded for the ISA assay. (A,C) Cell extracts were prepared from 3.5-h meiotic cells before (labeled as “–”) and after 30-min treatment with 5 μg/mL of phleomycin (labeled as “+”). (B) Diploid cells of the indicated strains were arrested at G2/M by nocodazole and treated with 5 μg/mL of phleomycin for 30 min, followed by TCA precipitation. Wild-type (USY543/544), rad9 (USY524/525), dot1 (USY526/527), and dot1 rad9 (USY522/523) were used in the experiment. Results of an independent experiment are shown in Supplementary Figure 1A. (C) Rad53 activation was examined in various strains in meiosis as shown in (A). Wild-type (USY543/544), rad9 (USY524/525), dot1 (USY526/527), h3-K79R (USY693/677), and rad9-Y798A (USY758/759) diploid strains were used. Results of an independent experiment are shown in Supplementary Figure 1B.
As with mitotic cells, the treatment of meiotic wild-type cells with phleomycin leads to Rad53 activation in a Rad9-dependent manner. Meiotic rad9 deletion cells showed reduced levels of Rad53 autophosphorylation with little band-shifts in response to phleomycin treatment (Figure 1A). This finding is consistent with the observed Rad9 dependency of Rad53 activation in meiotic spo11 cells with DNA damage (Cartagena-Lirola et al., 2008). Moreover, the treatment of meiotic wild-type cells with phleomycin induced a band shift of Rad9 (Figure 1A and Supplementary Figure 1A), which is likely to be an activated form with multiple phosphorylation.
Methylation of histone H3K79 (H3K79me) by Dot1 methyltransferase is important, but not essential, for the ability of Rad9 to mediate Rad53 activation in mitosis (Giannattasio et al., 2005; Wysocki et al., 2005; Toh et al., 2006). In order to determine the involvement of Dot1 in Rad9-dependent Rad53 activation in meiosis, we analyzed Rad53 activation in meiotic dot1 cells treated with phleomycin. The dot1 deletion mutation, which abolished H3K79me, substantially decreased Rad53 activation in response to phleomycin; this decrease in activation was detected by reduced band shifts as well as decreased autophosphorylation levels (Figure 1A and Supplementary Figure 1A). This deficiency of Rad53 activation in the dot1 mutant was almost identical to that in the rad9 mutant. Damage-induced Rad9 band shifts were also reduced in the dot1 mutant. The dot1 rad9 double mutant showed similar levels of reduced Rad53 activation to those of either single mutant (Figure 1A), suggesting that Dot1 and Rad9 function in the same pathway for phleomycin-dependent Rad53 activation in meiotic cells.
We confirmed that mitotic dot1 mutant cells are capable of activating Rad53 in response to phleomycin. Previous studies have used only haploid cells, and therefore we used G2/M-arrested diploid cells for treatment with a microtubule polymerization inhibitor, nocodazole. As shown in Figure 1B, wild-type G2/M-arrested diploid cells activated Rad53 in response to phleomycin. In addition, the rad9 deletion completely abolished the activation. The dot1 mutant did show some activation of Rad53, but at half the level of that of the wild type, which was determined by the autophosphorylation activity (Figure 1B and Supplementary Figure 1B). Consistent with this result, like wild-type cells, the dot1 mutant cells exhibited a Rad9 band shift after DNA damage treatment (Figure 1B).
To confirm the role of Dot1 through H3K79 methylation in Rad53 activation during meiosis, we created a histone H3K79R mutant strain (hht1-K79R, hht2-K79R; h3-K79R for simplicity) that was defective in Dot1-dependent methylation (Ontoso et al., 2013, Bani Ismail et al., 2014) (Figure 1C). In this strain, the histone H3 mutant proteins were expressed from the endogenous promoters of 2 copies of the histone H3 genes (HHT1 and HHT2) on the genome. In meiosis, the h3-K79R mutant cells showed reduced Rad53 band shifts as well as reduced autophosphorylation when treated with phleomycin (Figure 1C). Decreased levels of activation in the h3-K79R mutant were similar to those observed in the absence of DOT1. In addition, the band shift of Rad9 was compromised in the h3-K79R mutant in a manner similar to that observed in the dot1 mutant. These results suggest that Dot1-dependent H3K79 methylation is more critical for meiotic cells to activate Rad53 in response to exogenous DNA damage than it is for mitotic cells.
Histone H3K79 methylation is recognized by the Tudor domain of Rad9 (Grenon et al., 2007; Hammet et al., 2007). The rad9-Y798A mutation in the Rad9 Tudor domain is known to decrease the interaction between Rad9 Tudor and H3K79me. Therefore, we also investigated the effect of the rad9-Y798A mutation on Rad53 activation in meiotic cells. As shown in Figure 1C, rad9-Y798A mutant cells showed reduced Rad53 activation, as seen in the rad9 null mutant. In addition, by itself, the Rad9-Y798A mutant protein showed minimal band shifting. This finding suggests that, in meiotic cells, Rad9 activates Rad53 in response to DNA damage through the Tudor domain of Rad9, which recognizes Dot1-dependent H3K79 methylation. It is important to stress that exogenous DNA damage slightly activated Rad53 even in the absence of Dot1 or Rad9; however, the residual activation was higher than that in the absence of exogenous DNA damage.
Although exogenous DSBs induce Rad53 activation in meiotic cells, Spo11-induced DSBs do not. Importantly, the rad9, dot1, h3-K79R, and rad9-Y798A mutants are all proficient in the activation of Mec1/Tel1 checkpoint kinases, which are upstream of Rad53-Rad9, in response to Spo11-DSBs (our unpublished results). These results suggest that meiotic cells show distinct responses to programmed and accidental DSBs with respect to activating Rad53 kinase. Moreover, it is likely that Spo11-dependent DSBs are masked for the activation of Rad53, but not for the activation of Mec1/Tel1.
The Ddc2-Rad9 Fusion Protein Activates Rad53 in Response to Meiotic DSBs
Rad53 activation in mitosis is under the control of a multilayered mechanism (Pellicioli and Foiani, 2005). The targeting of Rad9, as well as Rad53, at a DSB site is likely to be crucial for the activation. A previous study showed that tethering Rad53 to meiotic DSBs as a fusion protein to Ddc2 induced Rad53 activation in a Spo11-dependent manner (Cartagena-Lirola et al., 2008). Since Rad9 works upstream of Rad53 in the checkpoint pathway, we questioned whether the tethering of Rad9 to meiotic DSBs might also activate Rad53. To target Rad9 to meiotic DSBs, we placed the Ddc2-3HA-Rad9 fusion protein under the control of a meiosis-specific DMC1 promoter (referred to as Ddc2-Rad9). The diploid strain (referred to as DDC2-RAD9) expressed the Ddc2-Rad9 fusion protein only in meiosis, together with the endogenous (non-tagged) Rad9 protein. Western blots showed that Ddc2-Rad9 was induced in meiotic prophase in a manner similar to the Dmc1 protein (Figure 2A). Ddc2-Rad9 started to appear at 1.5 h and reached a plateau at 3 h. At later periods, Ddc2-Rad9 showed smear bands, suggesting posttranslational modification of Ddc2-Rad9 during meiosis. The expression level of Ddc2-Rad9 from the DMC1 promoter at 4 h was roughly comparable to that of HA-Rad9 from a native promoter (Figure 2B and Supplementary Figure 4). Concomitant with the expression of Ddc2-Rad9 during meiosis, endogenous Rad53 protein started to show reduced mobility, suggesting the activation of Rad53 (Figure 2A and Supplementary Figure 2). Indeed, the ISA assay confirmed robust autophosphorylation activity of Rad53 after 3-h incubation in meiosis, which was not observed at 0 and 1.5 h. The expression of the Ddc2-Rad9 fusion protein with kinase-dead Rad53, Rad53-KD, abolished the autophosphorylation activity, demonstrating that Rad53 activation by DDC2-RAD9 was completely dependent on the Rad53 kinase activity. Interestingly, Ddc2-Rad9 induced a smaller, but significant band-shift of Rad53-KD at 4.5 h relative to wild-type Rad53. These results suggest that Ddc2-Rad9 induces Rad53-kinase independent modification of Rad53.
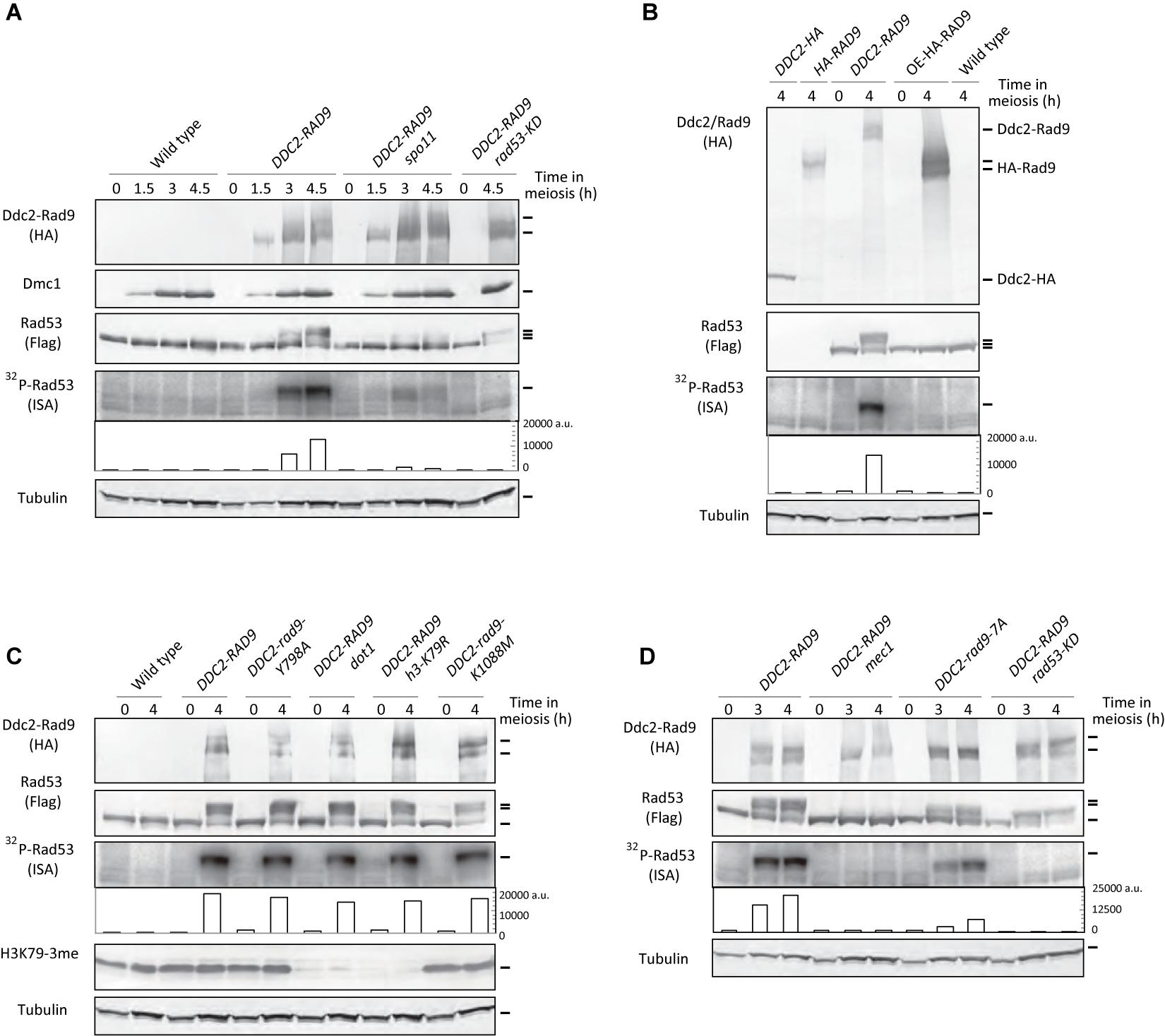
Figure 2. Ddc2-Rad9 fusions activate Rad53 in meiosis. (A) TCA-precipitated meiotic cell extracts of the indicated strains were analyzed by western blotting using anti-HA, anti-Flag, anti-H3K79-3me (tri-methylation), anti-Dmc1, and anti–α-tubulin antibodies.32P-Rad53 represented 32P-incorporation to Rad53 in the ISA assay, and was quantified as shown Figure 1A (fifth panels). Cell extracts derived from 4 × 106 cells were loaded for the ISA assay. Wild-type (USY543/544), DDC2-RAD9 (USY544/661), DDC2-RAD9 spo11 (USY783/414), and DDC2-RAD9 rad53-KD (USY545/720) were examined. (B) Cell extracts were prepared from the strains expressing C-terminal 3x HA-tagged Ddc2 (USY83/84; DDC2-HA) and N-terminal 3x HA-tagged Rad9 (USY20/35; HA-RAD9) from their native promoters; Ddc2-Rad9 (USY544/671; DDC2-RAD9) and 3x HA-tagged Rad9 (USY544/661; OE-HA-RAD9) from the DMC1 promoter; no HA-tagged wild-type strain (USY543/544); wild type. Rad53 was not tagged with Flag in DDC2-HA and HA-RAD9 strains. Flag-Rad53 was expressed from the native promoter. (C) Rad53 activation was examined in various strains as in (A). Wild-type (USY543/544), DDC2-RAD9 (USY544/671), DDC2-rad9-Y798A (USY544/776), DDC2-RAD9 dot1 (USY768/526), DDC2-RAD9 h3-K79R (USY770/693), and DDC2-rad9-K1088M (USY544/797) were examined. Results of an independent experiment are shown in Supplementary Figure 2C. (D) Rad53 activation was examined in various strains as in (A). Wild-type (USY543/544), DDC2-RAD9 (USY544/671), DDC2-RAD9 mec1 (USY495/785), DDC2-rad9-7A (USY544/667), and DDC2-RAD9 rad53-KD (USY4520) were examined. Results of an independent experiment are shown in Supplementary Figure 2D.
Activation of Rad53 by Ddc2-Rad9 fusion still requires meiotic DSBs. The spo11 deletion greatly decreased the band shifts as well as the autophosphorylation activity of Rad53 (Figure 2A). Some band shifts of Rad53 were nonetheless apparent, and a low, but increase in Rad53 autophosphorylation was observed, even in the absence of Spo11. This finding indicates that there might be another mechanism for Rad53 activation by Ddc2-Rad9 in a meiotic-DSB independent manner, e.g., in the S phase, since a slight increase in Rad53 autophosphorylation was observed at 1.5 h, the point at which the meiotic S phase occurs.
An increased amount of Rad9 in the DDC2-RAD9 strain could not explain the activation of Rad53. Expression of non-fusion HA-Rad9 from the DMC1 promoter was approximately 10-fold higher than that from the endogenous promoter, which did not activate Rad53 (Figure 2B and Supplementary Figure 2B). This result reinforces the idea that the tethering of Rad9 to meiotic DSBs is a key event for inducing Rad53 activation in response to Spo11-induced meiotic DSBs.
We also confirmed the activation of Rad53 kinase activity by Ddc2-Rad9 fusion in the background of the dmc1, which is unable to repair meiotic DSBs (Bishop, 1994; Shinohara et al., 2000) and its genetic requirements (see below, with the spo11, mec1, and rad53-KD mutations; Supplementary Figure 3).
Genetic Requirements for Meiotic Rad53 Activation by Ddc2-Rad9
As shown above, in meiotic cells, exogenous DNA damage-dependent Rad53 activation largely relies on the interaction of Rad9’s Tudor domain with Dot1-dependent H3K79me. Therefore, we questioned whether Rad53 activation by Ddc2-Rad9 in meiosis would also depend on the interaction. Even in the background of dot1 deletion and h3-K79R mutants, Ddc2-Rad9 could still fully activate Rad53 (Figure 2C and Supplementary Figure 2C). The band shifts and autophosphorylation levels of Rad53 in the dot1 deletion and h3-K79R mutants were comparable to those in the wild type. Moreover, consistent with the above results, the introduction of a Tudor mutation, rad9-Y798A, in the fusion did not abolish its ability to facilitate the band shifts or autophosphorylation levels of Rad53 (Figure 2C).
In mitosis, in addition to H3K79me, Rad9 localizes to chromatin adjacent to DSBs via the interaction between the BRCT domain of Rad9 and phosphorylation of S129 of histone H2A by Mec1/Tel1 kinases (Hammet et al., 2007). Histone H2AS129 phosphorylation is induced during meiosis in response to Spo11-mediated DSBs (Eichinger and Jentsch, 2010). During meiosis, expression of the Ddc2-fusion of Rad9 with a BRCT domain mutation, rad9-K1088M, which impairs the binding of Rad9 to phosphorylated H2AS129 (Hammet et al., 2007), activated Rad53, as in the wild-type fusion protein (Figure 2C). Therefore, these data suggest that Rad53 activation by Ddc2-Rad9 does not require the ability of Rad9 to recognize 2 epigenetic markers that play a critical role in the Rad53 activation to exogenous DSBs in mitotic cells.
In mitosis, the kinase activity of Rad53 largely depends on a major DNA damage kinase, Mec1/ATR (Sanchez et al., 1996; Sun et al., 1996). DDC2-RAD9-dependent Rad53 activation in meiosis still requires Mec1. The mec1 sml1 mutant (the sml1 mutation suppresses a lethality of the mec1 deletion) (Zhao et al., 1998) showed little stimulation of Rad53 kinase by DDC2-RAD9 (Figure 2D). Interestingly, only a few band shifts of Rad53 were observed in the mec1 mutant, which is in marked contrast to the results for the DDC2-RAD9 RAD53-KD strain, which showed a clear band shift with little Rad53 activation (Figure 2A). This finding suggests that Mec1-dependent Rad53 phosphorylation might be an initial key event for Rad53 activation by DDC2-RAD9.
Moreover, the Ddc2-Rad9 band shift was compromised in the mec1 mutant. In the wild-type strain, Ddc2-Rad9 exhibited smear bands, often showing 2 major bands (Figures 2C,D), whereas the mec1 (sml1) mutant lacked the major upper band of the Rad9 fusion. Mec1 is known to phosphorylate Rad9 at multiple sites (Schwartz et al., 2002). The rad9-7A mutant, with mutations in the major Mec1-phosphorylation sites, T603, and an additional 6 S/T sites of the SQ/TQ cluster domain (SCD), showed decreased Rad53 band shifts and autophosphorylation activity in mitotic cells with exogenous DNA damage (Schwartz et al., 2002). When the Ddc2-Rad9 fusion protein with the rad9-7A mutation was expressed in meiosis, the band shift greatly decreased, and the autophosphorylation activity of Rad53 was diminished by ∼1/3 relative to that of the wild-type Ddc2-Rad9 protein at 4 h (Figure 2D and Supplementary Figure 2D). The residual activation of Rad53 might have originated from other, as yet unidentified, phosphorylation sites of Rad9 by Mec1/Tel1. The data suggest that Mec1-mediated phosphorylation of Rad9 remains important for Rad53 activation in DDC2-RAD9 meiosis.
The Ddc2-Rad9 Fusion Protein Localizes to Meiotic Chromosomes
The Ddc2 protein shows punctate staining on meiotic chromosomes when ssDNAs are formed (Refolio et al., 2011). We tried to detect the localization of Ddc2-Rad9 on meiotic chromosomes by immunostaining. However, we did not detect any significant signals for the Ddc2-Rad9 fusion protein on meiotic chromosome spreads in the DDC2-RAD9 cells by staining with the anti-HA antibody (Figure 3A). The dmc1 mutant, which cannot repair and accumulate the ssDNA (Bishop et al., 1992), was used for efficient detection of the Ddc2 signal (Refolio et al., 2011). In parallel, staining for Rad51, a marker protein of processed meiotic DSBs (Bishop, 1994), was carried out together with HA-staining. At 6 h in dmc1 meiosis, when Rad51 foci were formed in more than 94.7 ± 6.8% of nuclei, weak, but significant Ddc2-Rad9 signals as a focus were detectable on chromosomes (Figure 3A and Supplementary Figure 5).
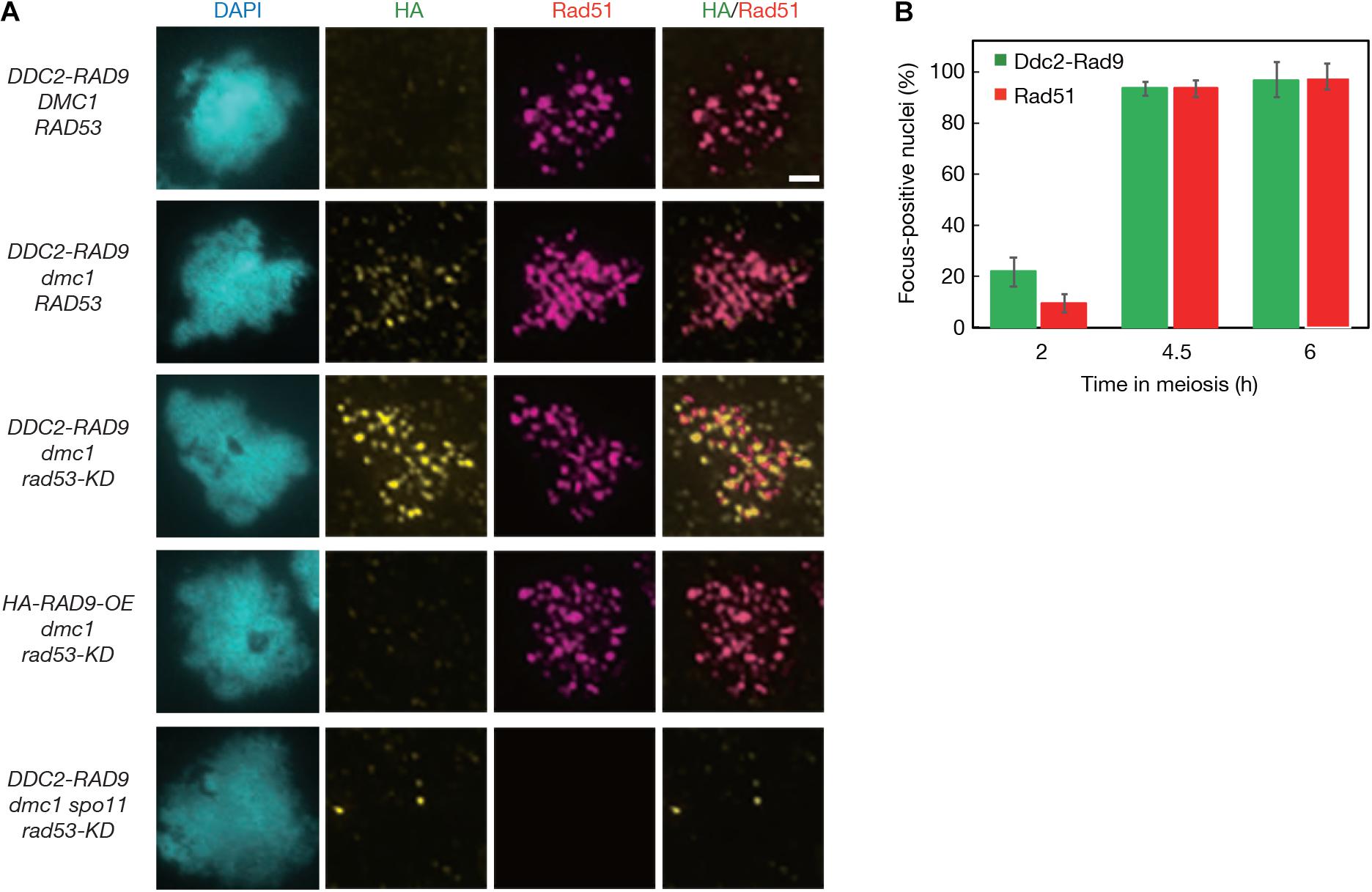
Figure 3. Ddc2-Rad9 localizes to meiotic DSBs. (A) The representative images of anti-HA and anti-Rad51 double immunostaining of spread nuclei prepared from DDC2-RAD9 (USY544/671), DDC2-RAD9 dmc1 RAD53 (USY580/591), DDC2-RAD9 dmc1 rad53-KD (USY582/623), HA-RAD9-OE dmc1 rad53-KD (USY582/674), and DDC2-RAD9 dmc1 spo11 rad53-KD (USY559/666) strains are shown at 4.5 h in meiosis, except for DDC2-RAD9 dmc1 RAD53, which is shown at 6 h in meiosis. Typical DNA images stained with DAPI and merged images by HA- and Rad51-staining are presented. Bar equals 2 μm. (B) The percentage of dmc1Δ rad53-KD DDC2-RAD9 nuclei that had more than 5 HA or Rad51 staining foci is presented at the indicated time points. At least 50 nuclei were counted at each time point. Error bars represent standard deviations obtained from 3 independent time courses.
Given that activated Rad53 kinase negatively regulates Rad9 assembly around DSBs (Usui et al., 2009), we further introduced a kinase-dead rad53-KD mutation, which may stabilize the Ddc2-Rad9 localization to chromatin. We detected brighter Ddc2-Rad9 foci on chromosomes of the dmc1 mutant with the rad53-KD compared to those in the dmc1 mutant with wild-type Rad53 (Figure 3A and Supplementary Figure 5). Ddc2-Rad9 localization to chromosomes was specific to meiotic prophase cells. At 0 and 2 h, there was little focus formation of the fusion in the rad53-KD dmc1 (Figure 3B). Ddc2-Rad9 focus-positive cells appeared concomitantly with Rad51 focus-positive cells (Figure 3B). At 4.5 h, 80% of the cells were positive for Ddc2-Rad9 foci. The average numbers of Ddc2-Rad9 and Rad51 foci in the rad53-KD dmc1 mutant were 59 ± 5.9 and 71 ± 6.8, respectively (n = 18), and 82 ± 4.7% (n = 18) of Ddc2-Rad9 was co-localized with Rad51. This supports the idea that Ddc2-Rad9 does indeed bind to ssDNAs for Rad53 activation.
The focus formation of Ddc2-Rad9 in the dmc1 rad53-KD mutant, like that of Rad51, once again depended on Spo11, and therefore on DSB formation (Figure 3A). Moreover, simple overexpression of the non-fusion version of HA-Rad9 in the dmc1 rad53-KD mutant did not support the localization of the HA-Rad9 protein on meiotic chromosomes (Figure 3A).
Binding of Rad9 to Meiotic DSBs Is Suppressed
The results described above suggested that meiotic cells lack the ability to recruit Rad9 or Rad53 to Spo11-induced DSBs. To confirm this hypothesis, we analyzed the binding of Rad9 to DSBs using a ChIP assay. First, as a control, we carried out ChIP to examine the localization of HA-Rad9 to an HO-endonuclease-induced DSB at the MAT locus in vegetative cells (Shroff et al., 2004; Usui et al., 2009). We used a haploid with GAL1/10-HO. After 1 h induction of the nuclease in G2/M arrested cells (by nocodazole) with the addition of galactose, increased binding of Rad9 to the DSB was observed at the MAT locus, but not at the control locus of SMC1 (Figure 4A). At the same time, robust binding of Rad51 to the break was detected. Then, we analyzed the binding of HA-Rad9 to a strong meiotic recombination hotspot, the HIS4-LEU2 locus. While robust binding of Rad51 to the hotspot was detected at 4 h in meiosis, we could detect little binding of Rad9 to the hotspot at 4 h at the same time (Figure 4B). This suggests that Rad9 is insensitive to Spo11-mediated meiotic DSBs, different from the mitotic DSB.
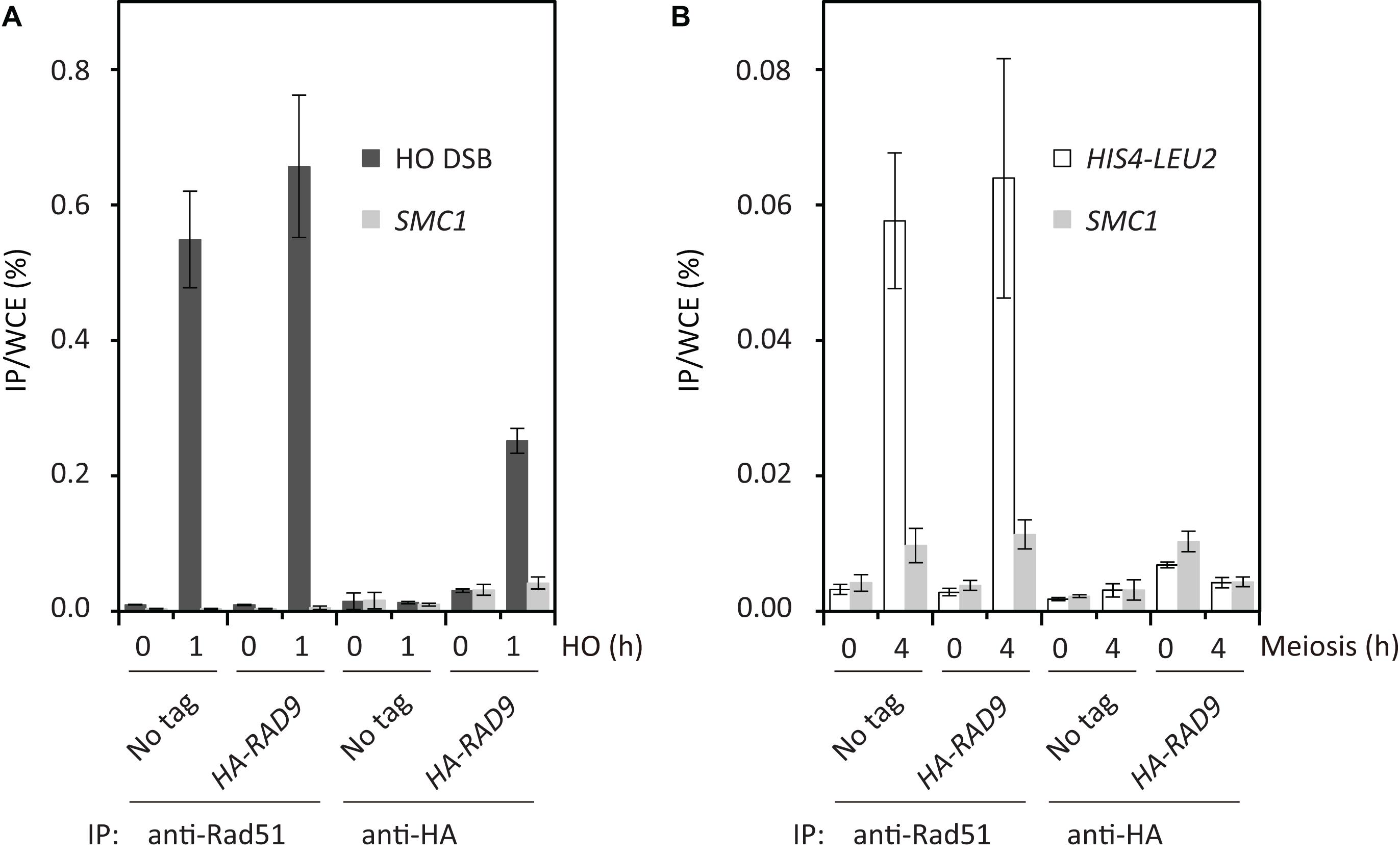
Figure 4. Rad9 is insensitive to meiotic DSBs. Rad9 binding to chromatin adjacent to mitotic HO-induced DSBs (A) and to meiotic programmed DSBs at the HIS4-LEU2 locus (B) was tested by ChIP. Formaldehyde-fixed chromatin extracts were prepared at 0 and 1 h after HO DSB induction in G2/M-arrested cells, and at 0 and 4 h after meiotic induction, followed by anti-HA and anti-Rad51 ChIP. Anti-HA- or anti-Rad51-bound DNAs were quantified by real-time PCR. Error bars represent standard deviations obtained from 3 independent cultures. The non-tagged wild-type and HA-RAD9 strains examined were USY99 and USY100 in (A) and NKY1551 and USY2035 in (B), respectively.
Ddc2-Rad9 Fusion Delays DSB Repair and Cell Cycle Progression During Meiosis
We next examined the effect of Ddc2-Rad9 expression, and thus Rad53 activation, on meiosis. The DDC2-RAD9 diploid showed wild-type spore viability levels (97.9% vs. 98.4% in the wild type; 48 asci). DDC2-RAD9 cells delayed MI entry by approximately 2 h compared to the wild type (Figure 5A), without any delay in the pre-meiotic S phase (Supplementary Figure 6A).
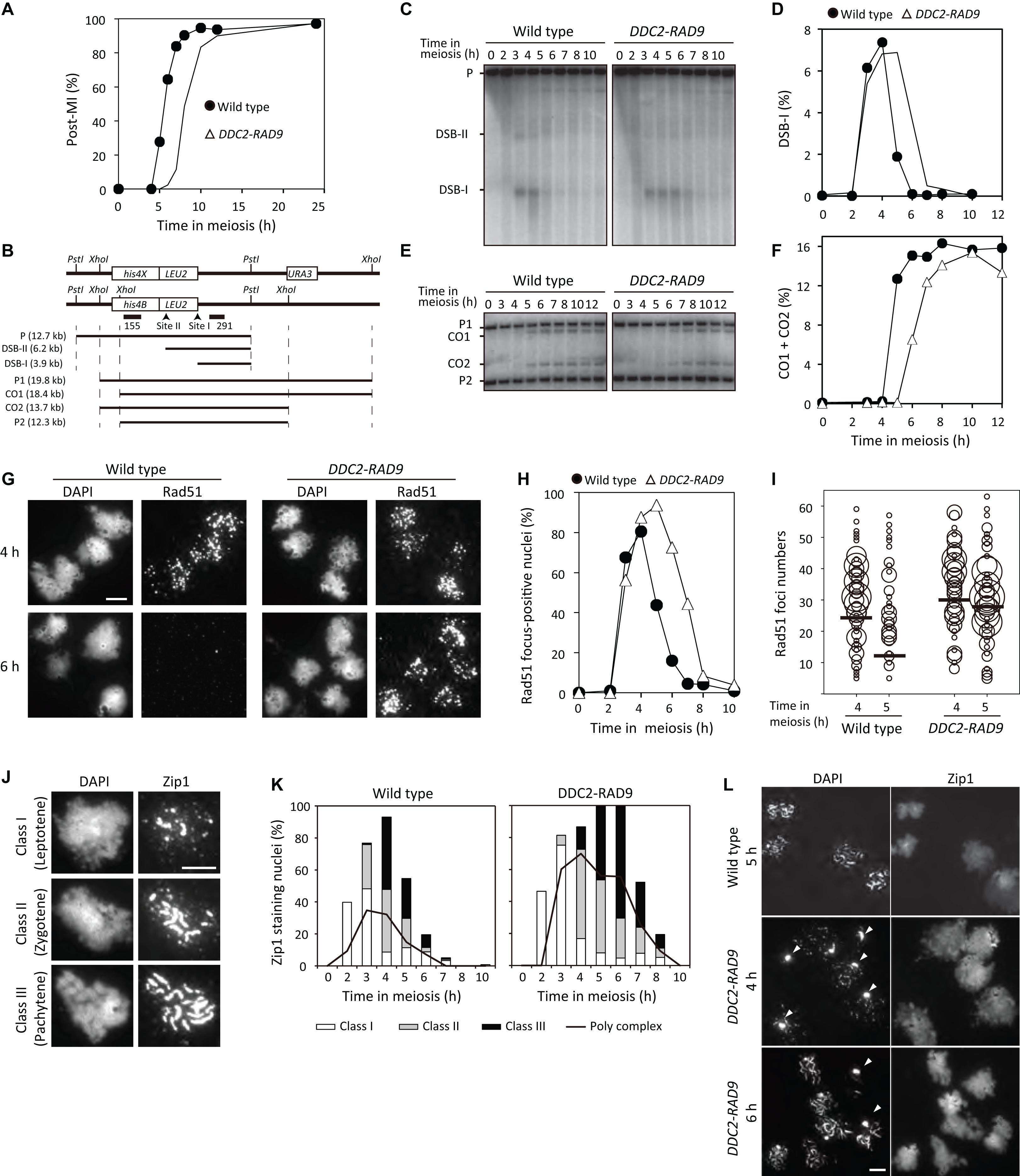
Figure 5. DDC2-RAD9 impedes cell cycle progression and meiotic recombination. (A) Meiotic nuclear divisions were monitored in wild-type (USY543/544) and DDC2-RAD9 (USY544/671) strains by DAPI staining. The y-axis represents the percentage of cells that completed meiosis I and II (Post-MI). At least 200 cells were counted at each time point. (B) Schematic presentation of the HIS4-LEU2 DSB site. (C,E) Genomic DNAs were prepared from the indicated strains during the meiotic time course. Meiotic DSBs (DSB-I and DSB-II) (C) or crossover recombinants (CO1 and CO2) (E) were detected by Southern blot using the probes pNK291 or pNKY155 after genomic DSBs were digested with PstI or XhoI, respectively. (D,F) Quantitative data of DSB-I (C) and CO1 + CO2 (E) obtained from Southern blot are plotted. (G–L) DDC2-RAD9 influences localization of Rad51 and Zip1 on meiotic chromosome. (G) The representative images of Rad51 immunostaining nuclear foci in spread nuclei prepared from the indicated strains at 4 and 6 h in meiosis are shown. (H) The percentage of the indicated cells’ nuclei that had more than 5 Rad51 foci was plotted at the indicated time points. (I) Distribution of nuclei according to Rad51 foci number per nucleus at the indicated time is presented using the same data samples as in (H). Only nuclei that had more than 5 Rad51 foci were plotted. The size of the bubbles reflects the percentage of nuclei that had a certain foci number of Rad51 among all nuclei examined. Bars represent the average numbers of Rad51 foci. (J) Representative images of Zip1 staining classification are shown. (K) Classifications of Zip1 staining at the indicated time in the indicated strains are shown. The line represents the Zip1 polycomplex (PC). At least 100 nuclei were examined at each time point. (L) Representative images of Zip1 staining in DDC2-RAD9 at 4 h and 6 h are shown. Zip1 PCs are indicated by arrowheads. Experiments were performed at least twice, and representative data are shown. Bar equals 5 μm.
We investigated meiotic DSB repair and CO formation at the HIS4-LEU2, by Southern blotting (Figure 5B) (Cao et al., 1990; Storlazzi et al., 1995). In the wild type, meiotic DSBs formed at 3 h, peaked at 4 h, and then gradually decreased after 5 h (Figures 5C,D). Concomitant with the disappearance of DSBs, COs started to appear at 5 h, and reached a plateau by 6 h in meiosis (Figures 5E,F). DDC2-RAD9 cells formed DSBs in a manner similar to wild-type cells, but the disappearance of the DSBs was delayed by 1.5 h compared to the wild type (Figures 5C,D). Consistent with the kinetics of DSBs, COs in DDC2-RAD9 started to appear at 6 h, and reached a plateau by 8 h (Figures 5E,F). Although delayed, the final level of COs in DDC2-RAD9 cells was comparable to that in the wild type. These data suggest that meiotic DSB repair is delayed in DDC2-RAD9 meiosis.
To confirm delayed DSB repair in DDC2-RAD9 meiosis, we observed focus formation of Rad51 on meiotic chromosomes by immunostaining (Bishop, 1994; Shinohara et al., 2000). In the wild type, Rad51 focus-positive nuclei started to appear at 3 h in meiosis, reached a peak at 4 h, and disappeared gradually during further incubation with kinetics similar to those of meiotic DSBs (Figures 5G,H). Rad51 focus-positive nuclei in DDC2-RAD9 appeared at 3 h and peaked at 5 h. However, the fusion induced a 2-h delay in the disappearance of Rad51 foci (Figures 5G,H). These data support the delay in DSB repair, particularly after Rad51 loading, in the DDC2-RAD9 strain. Consistent with delayed repair, the average number of Rad51 foci in DDC2-RAD9 was 30 ± 16 at 4 h (per total nucleus; n = 112; Figure 5I), which was higher than that observed in the wild type (24 ± 16, n = 113; P = 0.009, Mann–Whitney U test). Dmc1-focus kinetics were similar to those of the Rad51 focus in DDC2-RAD9 meiosis (Supplementary Figure 6B). These data demonstrate that Ddc2-Rad9 expression impedes a recombination step after the loading of Rad51/Dmc1. The delayed DSB repair during meiosis of DDC2-RAD9 cells is consistent with the result that the over-expression of Rad53 delays meiotic DSB repair (Usui and Kanehara, 2013).
Synaptonemal complex (SC) formation was also investigated in the DDC2-RAD9 strain. We observed the localization of the Zip1 protein, which is the central component of the SC (Sym et al., 1993). As shown in Figures 5J,K, in the wild type, dotty staining was initially observed for Zip1 (class I, leptonema), and the nuclei containing a mixture of dotty and short linear localizations of Zip1 increased (class II, zygonema). Finally, Zip1 was fully extended along chromosomes, which is indicative of a matured SC and complete synapsis (class III, pachynema), and then disappeared subsequently. In DDC2-RAD9 cells, dotty-staining of Zip1 appeared at 2 h, similar to the wild type (Figure 5K). However, the appearance of short and long linear localization of Zip1 was delayed in DDC2-RAD9 (Figures 5K,L). At 6 h, DDC2-RAD9 showed a maximum fraction of Zip1-long lines, whereas the wild type showed the maximum at 4 h. Disassembly of Zip1 lines in the DDC2-RAD9 strain was delayed by 2 h relative to the wild type. These results suggest that SC elongation is partially defective in DDC2-RAD9. Consistent with this, DDC2-RAD9 cells showed an increased fraction of aggregates (polycomplex) of Zip1 (Figures 5K,L). These results indicate that Ddc2-Rad9 has negative impacts on SC assembly, probably due to delayed DSB repair.
Discussion
Rad53 is the central effector kinase in the DNA damage checkpoint in mitosis but is not activated during meiosis (Cartagena-Lirola et al., 2008), even in the presence of ∼160 DSBs induced by Spo11 (Buhler et al., 2007; Pan et al., 2011), suggesting that Rad53 activation is masked in response to meiotic programmed DSBs. However, the molecular mechanism underlying the masking of Rad53 is largely unknown. Previous study showed that over-expression of Rad53 during meiosis delays DSB repair, thus the onset of meiosis I, suggesting the presence of silencing mechanism of Rad53 in meiotic cells (Usui and Kanehara, 2013). Here, we showed that Rad9, which mediates Mec1-dependent Rad53 activation (Gilbert et al., 2001; Schwartz et al., 2002; Sweeney et al., 2005), is a key regulator for this masking. Rad9 cannot bind to Spo11-mediated DSBs in meiosis, whereas it does bind to HO-induced DSBs in mitosis. Indeed, tethering of Rad9 to meiotic DSBs by Ddc2-fusion induces the activation of Rad53 kinase.
The inability of Rad9 to bind to meiotic DSB may result from meiosis-specific modulation of Rad9 in order to inhibit the activity, either by posttranslational modification or through interaction with a meiosis-specific inhibitor. However, this latter possibility is less likely, given that Ddc2-Rad9 can activate Rad53 in response to meiotic programmed DSBs. If meiotic cells impose inhibition on Rad9 directly, Ddc2-Rad9 would also be inhibited in meiosis. Alternatively, in contrast to exogenous DSBs as well as HO-mediated DSBs, meiotic DSBs are intrinsically inert to Rad9 binding. Rad9 binding to chromosomes is promoted by 2 epigenetic markers in mitosis, histone H2AS129 phosphorylation and H3K79 methylation, which are recognized by 2 domains of Rad9, that is, BRCT and Tudor, respectively (Figure 6). H2AS129 phosphorylation is DNA damage-dependent and is mediated by Mec1/Tel1 checkpoint kinases, whereas H3K79 methylation is constitutive and is catalyzed by Dot1 methyltransferase. H2AS129 phosphorylation is induced by Spo11-mediated DSBs in meiosis in a Mec1-dependent manner (Eichinger and Jentsch, 2010). Interestingly, recent genome-wide mapping of H3K79 methylation showed that promoter regions of the yeast genome, where most of the meiotic DSBs occur (Buhler et al., 2007; Pan et al., 2011), tend to exhibit reduced levels of this modification (Zhang et al., 2011). One likely possibility is that this reduction of H3K79 methylation around DSB sites may partially explain the weak binding of Rad9 to meiotic DSBs (Figure 6). Consistent with this hypothesis, Rad53 activation by artificial targeting of Rad9 to the DSBs is independent of Dot1 or H3K79 methylation. However, previous studies have shown that meiotic cell cycle arrest in the zip1 mutant, which is defective in chromosome synapsis and meiotic recombination, requires Dot1-dependent H3K79 methylation, suggesting that the methylation plays a role in response to meiotic DSBs, at least in the zip1 mutant (Ontoso et al., 2013). We propose a simple model in which meiosis-specific chromosome structures play a role in making meiotic DSBs insensitive to Rad9, and thus to Rad53. This model is supported by a previous study that showed that Rad53 was activated when DSBs escaped from the recombination checkpoint and were present in meiosis-II, where meiosis-specific chromosome structures are dismantled (Cartagena-Lirola et al., 2008).
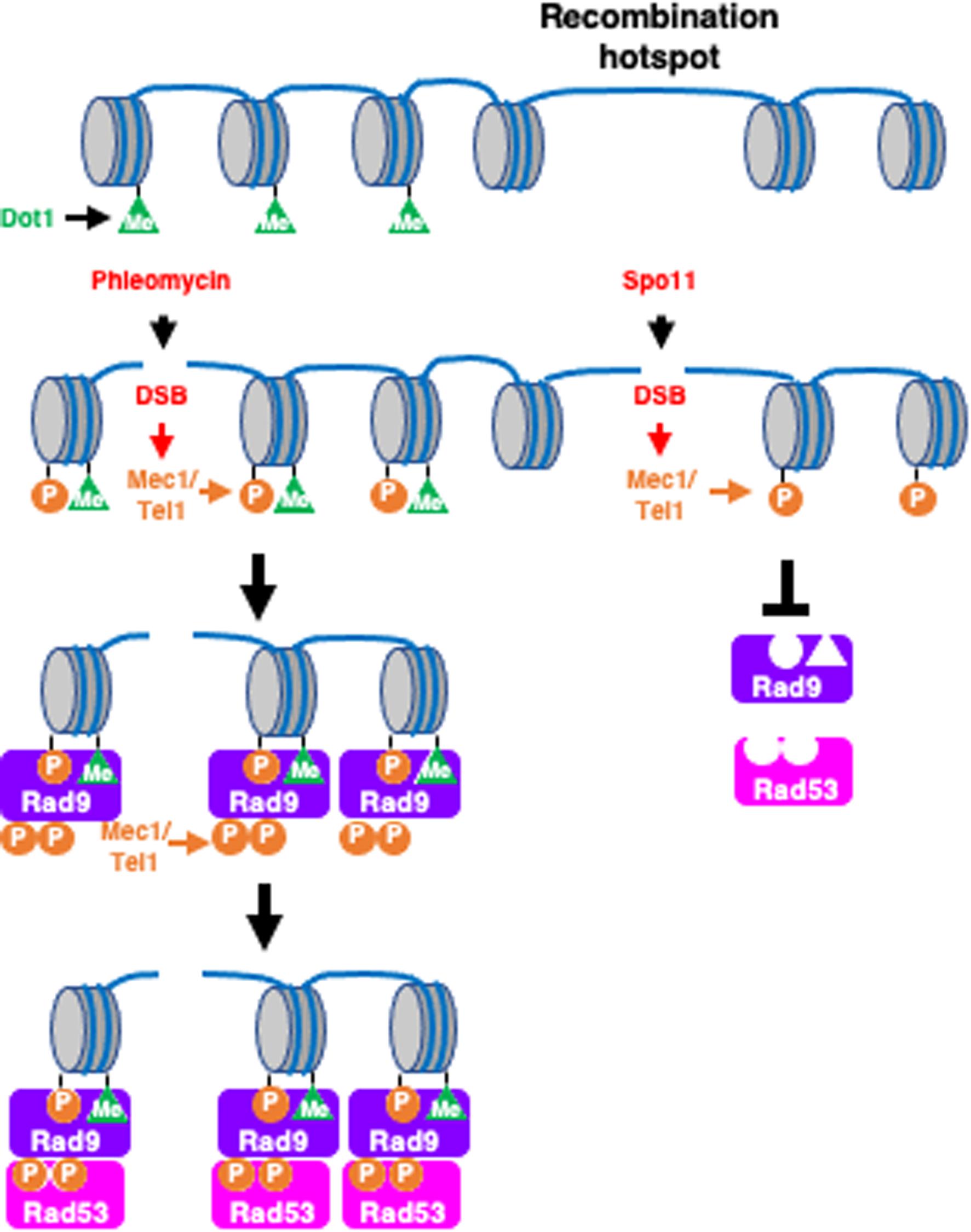
Figure 6. Rad9 is incompetent to meiotic DSBs. Exogenous DSBs often occur in nucleosome-rich gene body where Dot1-mediated histone H3K79 methylation marks are enriched (left pathway). The DSBs activates Mec1/Tel1, which in turn phosphorylates histone H2AS129. The two epigenetics marks, H3K79 methylation and H129S phosphorylation, provides multiple binding sites to Rad9. Rad9 is phosphorylated by Mec1/Tel1 and activates Rad53 kinase. At recombination hotspots is located in nucleosome-free region, which shows less H3K79 methylation compared to gene bodies right pathway. This chromatin features make the region insensitive to Rad9 binding.
Why do meiotic cells mask Rad53 activation in response to Spo11-mediated DSBs? In this study, we found that the untimely activation of Rad53 kinase during meiosis has a negative impact on meiotic chromosome metabolism. The Ddc2-Rad9 fusion delayed meiotic repair of DSBs, suggesting that Rad53 activation is inhibitory to meiotic DSB repair, and thus meiotic recombination. Meiotic recombination preferentially occurs between homologous chromosomes (Schwacha and Kleckner, 1997). On the other hand, intersister recombination is preferred in mitotic recombination (Kadyk and Hartwell, 1992; Bzymek et al., 2010). One simple idea is that Rad53 activation inhibits interhomolog recombination or activates intersister recombination. However, this is less likely since DDC2-RAD9 cells, in which Rad53 is activated, still show normal levels of crossover formation in meiosis (Figures 5E,F).
Although Rad53 activation was inhibited in wild-type meiosis, we did not observe drastic defects in artificial activation of the kinase by Ddc2-Rad9. The effect of Rad53 activation by Ddc2-Rad9 on meiotic events was weak. This finding might be attributable to the fusion protein restricting the activation of Rad53 very close to the DSBs. Rad9-Rad53 is generally more dynamic in nuclei for signaling, at least in mitotic cells with exogenous DSBs (Melo et al., 2001; Usui et al., 2009).
There are several downstream targets of the Rad53 pathway, including Dbf4-dependent Cdc7 kinase (DDK) (Lopez-Mosqueda et al., 2010; Zegerman and Diffley, 2010). In meiosis, DDK plays multiple roles such as in the formation of DSBs, expression of middle pachytene genes, monopolar spindle attachment, and efficient cleavage of meiosis-specific cohesion (Lo et al., 2008; Matos et al., 2008; Sasanuma et al., 2008; Katis et al., 2010). Thus, it is likely that, in meiosis, DDK should be constitutively activated during meiotic prophase-I by avoiding inhibitory Rad53 activation. Moreover, a recent study clearly showed that meiotic prophase develops a program to guarantee a longer prophase (G2) compared to the mitotic phase (Okaz et al., 2012). It is also possible that DSB-dependent Rad53 activation might inhibit the operation of this program in a timely manner. We speculate that meiotic cells ensure a longer prophase (G2) through the inactivation of the Rad53 checkpoint kinase.
In mitotic cells, in addition to Rad9, the other DDR mediator protein, Dpb11, plays a role for efficient activation of Rad53 (Pfander and Diffley, 2011; Puddu et al., 2011; di Cicco et al., 2017). The recruitment of Dpb11 to DSB sites is dependent of the phosphorylation of a C-terminal tail of Ddc1 in the Rad17-Ddc1-Mec3 checkpoint clamp (Rad9-Rad1-Hus1 in other organisms). Then, Dpb11 recruits Rad9 through CDK-dependent phosphorylation of Rad9. We have known little about the regulation of Dpb11 during meiosis. However, the involvement of the checkpoint clamp in meiotic DNA damage response is controversial (Shinohara et al., 2003; Shinohara et al., 2015).
In mammals, 53BP1, a Rad9 homolog, is recruited to DSB sites through multiple interactions, including various histone modifications (Huyen et al., 2004; Botuyan et al., 2006; Mailand et al., 2007; Stewart et al., 2009; Fradet-Turcotte et al., 2013). These interactions function in the positive regulation of 53BP1 recruitment. The present study suggests that negative control of Rad9 to the DSB is also a key process for modulating the DNA damage response under different physiological conditions.
Data Availability Statement
The original contributions generated for this study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
TU and AS conceived and designed the experiments and prepared the manuscript. TU performed the experiments. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by a Grant-in-Aid from the JSPS KAKENHI Grant Numbers 22125001, 22125002, 15H05973, 16H04742, and 19H00981 to AS and 21770006 and 26440189 to TU.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. John Petrini for the antibodies and strains. We are also indebted to the members of the Shinohara laboratory for the stimulating discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.635383/full#supplementary-material
Supplementary Figure 1 | H3K79me is required for Rad9-dependent Rad53 activation in meiosis after phleomycin treatment. (A) TCA-precipitated cell extracts from meiotic cells were prepared from wild-type (USY543/544), dot1 (USY526/527), and h3-K79R (USY693/677) diploid strains, and were analyzed by western blotting using anti-Rad9, anti-Flag (for Rad53), anti-Histone H3K79- tri-methyl and anti-tubulin antibodies. “32P-Rad53” represented 32P-incorporation to Rad53 in the ISA assay. Cell extracts derived from 4 × 106 cells were loaded for the ISA assay. Cell extracts were prepared at indicated time points after the treatment of 3.5-h meiotic cells with 5 μg/mL of phleomycin. (B) Diploid cells of the indicated strains were arrested at G2/M by nocodazole and treated with 5 μg/mL of phleomycin for 30 min, followed by TCA precipitation. Wild-type (USY543/544), rad9 (USY524/525), dot1 (USY526/527), and dot1 rad9 (USY522/523) were used in the experiment.
Supplementary Figure 2 | Ddc2-Rad9 fusions activate Rad53 in meiosis. (A) Cells in meiotic time course at indicated times were collected and TCA-precipitated meiotic cell extracts were analyzed by western blotting using anti-Flag, and anti–α-tubulin antibodies. Wild-type (USY543/544), DDC2-RAD9 (USY544/661), DDC2-rad9-7A (USY544/667), and DDC2-RAD9 rad53-KD (USY545/720) were examined. (B) Cell extracts were prepared from the strains and analyzed by western blotting using anti-HA, anti-Flag and anti–α-tubulin antibodies; DDC2-RAD9 (USY544/661), DDC2-RAD9 rad53-KD (USY545/720), 3x HA-tagged Rad9 (USY544/661; OE-HA-RAD9) from the DMC1 promoter, and DDC2-RAD9 spo11 rad53-KD (USY545/720). (C) Cell extracts were prepared from the strains and analyzed by western blotting using anti-HA, anti-Flag, anti-histone H3K79-tri-methyl and anti–α-tubulin antibodies; Wild-type (USY543/544), DDC2-RAD9 (USY544/661), DDC2-rad9-Y798A (USY544/776), DDC2-RAD9 dot1 (USY768/526), and DDC2-RAD9 h3-K79R (USY770/693). (D) Cell extracts at 0, 3, and 4 h time points were prepared from the strains and analyzed by western blotting using anti-HA, anti-Flag, and anti–α-tubulin antibodies and also by ISA (third panel); Wild-type (USY543/544), DDC2-RAD9 (USY544/661) and DDC2-rad9-7A (USY510/512). Cell extracts derived from 4 × 106 cells were loaded for the ISA assay.
Supplementary Figure 3 | Ddc2-Rad9 fusions activate Rad53 in dmc1 meiosis. (A) Cell extracts at 0, 3, and 4.5 h time points of meiosis were prepared from the dmc1strains and analyzed by western blotting using anti-Flag and by ISA assay. Results from two independent time course are shown. Cell extracts derived from 4 × 106 cells were loaded for the ISA assay. HA-Rad9 and Ddc1-HA-Rad9 protein were expressed from the DMC1 promoter. HA-RAD9 dmc1 (USY528/529), DDC2-RAD9 dmc1 (USY528/529), and DDC2-RAD9 dmc1 rad53-KD (USY508/509). (B) Cell extracts at 0, 3, and 4.5 h time points of meiosis of the dmc1 mutant were prepared from the strains and analyzed by western blotting using anti-HA and anti-Flag, and by ISA assay. HA-RAD9 dmc1 (USY528/529), DDC2-RAD9 dmc1 (USY528/529), and DDC2-RAD9 dmc1 mec1 (USY458/459). (C,D) Cell extracts at 0, 1.5, 3, and 4.5 h time points of meiosis of the dmc1 mutant were prepared from the strains and analyzed by western blotting using anti-HA and anti-Flag, and by ISA assay. (C) DDC2-RAD9 dmc1 (USY528/529), and DDC2-RAD9 dmc1 mec1 (USY458/459) and DDC2-RAD9 dmc1 spo11 (USY514/515); (D) HA-RAD9 dmc1 (USY528/529), DDC2-RAD9 dmc1 (USY528/529), and DDC2-RAD9 dmc1 rad53-KD (USY508/509).
Supplementary Figure 4 | TCA-precipitated extracts prepared from cells expressing Ddc2-3xHA (USY8384) or 3xHA-Rad9 (USY2035) from their native promoter and the indicated dilutions of DDC2-RAD9 (USY544/671) and HA-RAD9-OE (USY544/561) extracts were examined. All cell extracts were prepared from 4 h meiotic cells. Cell extracts derived from 1 × 107 or 4 × 106 cells were loaded for anti-HA or anti-Flag/tubulin Western blot, respectively. Dilutions were prepared accordingly.
Supplementary Figure 5 | The representative images of immunostaining of nuclear spreads prepared from dmc1 RAD53 DDC2-RAD9 (USY580/591) at 6 h and dmc1 rad53-KD DDC2-RAD9 (USY582/623) at 4.5 h in meiosis are shown with anti-HA and anti-Rad51. DNA images stained with DAPI are also presented. Bar equals 5 μm.
Supplementary Figure 6 | (A) DNA contents of wild type (USY543/544) and DDC2-RAD9 (USY544/671) strains were examined by FACS analysis. The representative profiles were shown. (B) The percentage of nuclei of the indicated strains that had more than five Rad51 or Dmc1 foci was plotted each time point.
Abbreviations
ChIP, chromatin immuno-precipitation; DAPI, 4 ′, 6-diamidino-2-phenylindole; DSBs, DNA double-strand breaks; ISA, in situ autophosphorylation; SC, synaptonemal complex; WCE, whole cell extract.
References
Allen, J. B., Zhou, Z., Siede, W., Friedberg, E. C., and Elledge, S. J. (1994). The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 8, 2401–2415.
Allers, T., and Lichten, M. (2001a). Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106, 47–57.
Allers, T., and Lichten, M. (2001b). Intermediates of yeast meiotic recombination contain heteroduplex DNA. Mol. Cell 8, 225–231.
Alt, F. W., Zhang, Y., Meng, F. L., Guo, C., and Schwer, B. (2013). Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 152, 417–429. doi: 10.1016/j.cell.2013.01.007
Bani Ismail, M., Shinohara, M., and Shinohara, A. (2014). Dot1-dependent histone H3K79 methylation promotes meiotic double-strand break formation in the absence of histone H3K4 methylation in budding yeast. PLoS One 9:e96648. doi: 10.1371/journal.pone.0096648
Barlow, A. L., Benson, F. E., West, S. C., and Hulten, M. A. (1997). Distribution of the Rad51 recombinase in human and mouse spermatocytes. EMBO J. 16, 5207–5215. doi: 10.1093/emboj/16.17.5207
Bergerat, A., de Massy, B., Gadelle, D., Varoutas, P. C., Nicolas, A., and Forterre, P. (1997). An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature 386, 414–417. doi: 10.1038/386414a0
Bishop, D. K. (1994). RecA homologs Dmc1 and Rad51 interact to form multiple nuclear complexes prior to meiotic chromosome synapsis. Cell 79, 1081–1092.
Bishop, D. K., Park, D., Xu, L., and Kleckner, N. (1992). DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69, 439–456.
Bishop, D. K., and Zickler, D. (2004). Early decision; meiotic crossover interference prior to stable strand exchange and synapsis. Cell 117, 9–15.
Bonilla, C. Y., Melo, J. A., and Toczyski, D. P. (2008). Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol. Cell 30, 267–276. doi: 10.1016/j.molcel.2008.03.023
Borner, G. V., Kleckner, N., and Hunter, N. (2004). Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell 117, 29–45.
Botuyan, M. V., Lee, J., Ward, I. M., Kim, J. E., Thompson, J. R., Chen, J., et al. (2006). Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373. doi: 10.1016/j.cell.2006.10.043
Buhler, C., Borde, V., and Lichten, M. (2007). Mapping meiotic single-strand DNA reveals a new landscape of DNA double-strand breaks in Saccharomyces cerevisiae. PLoS Biol. 5:e324. doi: 10.1371/journal.pbio.0050324
Bzymek, M., Thayer, N. H., Oh, S. D., Kleckner, N., and Hunter, N. (2010). Double Holliday junctions are intermediates of DNA break repair. Nature 464, 937–941. doi: 10.1038/nature08868
Cao, L., Alani, E., and Kleckner, N. (1990). A pathway for generation and processing of double-strand breaks during meiotic recombination in S. cerevisiae. Cell 61, 1089–1101.
Carballo, J. A., Johnson, A. L., Sedgwick, S. G., and Cha, R. S. (2008). Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell 132, 758–770. doi: 10.1016/j.cell.2008.01.035
Cartagena-Lirola, H., Guerini, I., Manfrini, N., Lucchini, G., and Longhese, M. P. (2008). Role of the Saccharomyces cerevisiae Rad53 checkpoint kinase in signaling double-strand breaks during the meiotic cell cycle. Mol. Cell Biol. 28, 4480–4493. doi: 10.1128/MCB.00375-08
di Cicco, G., Bantele, S. C. S., Reusswig, K. U., and Pfander, B. (2017). A cell cycle-independent mode of the Rad9-Dpb11 interaction is induced by DNA damage. Sci. Rep. 7:11650. doi: 10.1038/s41598-017-11937-z
Eichinger, C. S., and Jentsch, S. (2010). Synaptonemal complex formation and meiotic checkpoint signaling are linked to the lateral element protein Red1. Proc. Natl. Acad. Sci. U.S.A. 107, 11370–11375. doi: 10.1073/pnas.1004248107
Fradet-Turcotte, A., Canny, M. D., Escribano-Diaz, C., Orthwein, A., Leung, C. C., Huang, H., et al. (2013). 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54. doi: 10.1038/nature12318
Giannattasio, M., Lazzaro, F., Plevani, P., and Muzi-Falconi, M. (2005). The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J. Biol. Chem. 280, 9879–9886. doi: 10.1074/jbc.M414453200
Gilbert, C. S., Green, C. M., and Lowndes, N. F. (2001). Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol. Cell 8, 129–136.
Granata, M., Lazzaro, F., Novarina, D., Panigada, D., Puddu, F., Abreu, C. M., et al. (2010). Dynamics of Rad9 chromatin binding and checkpoint function are mediated by its dimerization and are cell cycle-regulated by CDK1 activity. PLoS Genet. 6:1001047. doi: 10.1371/journal.pgen.1001047
Grenon, M., Costelloe, T., Jimeno, S., O’Shaughnessy, A., Fitzgerald, J., Zgheib, O., et al. (2007). Docking onto chromatin via the Saccharomyces cerevisiae Rad9 Tudor domain. Yeast 24, 105–119. doi: 10.1002/yea.1441
Haber, J. E. (2012). Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics 191, 33–64. doi: 10.1534/genetics.111.134577
Hammet, A., Magill, C., Heierhorst, J., and Jackson, S. P. (2007). Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 8, 851–857. doi: 10.1038/sj.embor.7401036
Hayase, A., Takagi, M., Miyazaki, T., Oshiumi, H., Shinohara, M., and Shinohara, A. (2004). A protein complex containing Mei5 and Sae3 promotes the assembly of the meiosis-specific RecA homolog Dmc1. Cell 119, 927–940. doi: 10.1016/j.cell.2004.10.031
Hollingsworth, N. M., and Gaglione, R. (2019). The meiotic-specific Mek1 kinase in budding yeast regulates interhomolog recombination and coordinates meiotic progression with double-strand break repair. Curr. Genet. 65, 631–641. doi: 10.1007/s00294-019-00937-3
Hunter, N., and Kleckner, N. (2001). The single-end invasion: an asymmetric intermediate at the double-strand break to double-holliday junction transition of meiotic recombination. Cell 106, 59–70.
Huyen, Y., Zgheib, O., Ditullio, R. A. Jr., Gorgoulis, V. G., Zacharatos, P., Petty, T. J., et al. (2004). Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432, 406–411. doi: 10.1038/nature03114
Kadyk, L. C., and Hartwell, L. H. (1992). Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 132, 387–402.
Kaiser, C., Michaelis, S., and Mitchell, A. Cold Spring Harbor Laboratory (1994). Methods in Yeast Genetics: a Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Katis, V. L., Lipp, J. J., Imre, R., Bogdanova, A., Okaz, E., Habermann, B., et al. (2010). Rec8 phosphorylation by casein kinase 1 and Cdc7-Dbf4 kinase regulates cohesin cleavage by separase during meiosis. Dev. Cell 18, 397–409. doi: 10.1016/j.devcel.2010.01.014
Kauppi, L., Barchi, M., Baudat, F., Romanienko, P. J., Keeney, S., and Jasin, M. (2011). Distinct properties of the XY pseudoautosomal region crucial for male meiosis. Science 331, 916–920. doi: 10.1126/science.1195774
Keeney, S., Giroux, C. N., and Kleckner, N. (1997). Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384.
Lee, S. E., Moore, J. K., Holmes, A., Umezu, K., Kolodner, R. D., and Haber, J. E. (1998). Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94, 399–409.
Lee, S. J., Duong, J. K., and Stern, D. F. (2004). A Ddc2-Rad53 fusion protein can bypass the requirements for RAD9 and MRC1 in Rad53 activation. Mol. Biol. Cell 15, 5443–5455. doi: 10.1091/mbc.E04-07-0608
Lo, H. C., Wan, L., Rosebrock, A., Futcher, B., and Hollingsworth, N. M. (2008). Cdc7-Dbf4 regulates NDT80 transcription as well as reductional segregation during budding yeast meiosis. Mol. Biol. Cell 19, 4956–4967. doi: 10.1091/mbc.E08-07-0755
Longtine, M. S., McKenzie, A. III, Demarini, D. J., Shah, N. G., Wach, A., Brachat, A., et al. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961.
Lopez-Mosqueda, J., Maas, N. L., Jonsson, Z. O., Defazio-Eli, L. G., Wohlschlegel, J., and Toczyski, D. P. (2010). Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature 467, 479–483. doi: 10.1038/nature09377
Lydall, D., Nikolsky, Y., Bishop, D. K., and Weinert, T. (1996). A meiotic recombination checkpoint controlled by mitotic checkpoint genes. Nature 383, 840–843. doi: 10.1038/383840a0
Mailand, N., Bekker-Jensen, S., Faustrup, H., Melander, F., Bartek, J., Lukas, C., et al. (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900. doi: 10.1016/j.cell.2007.09.040
Matos, J., Lipp, J. J., Bogdanova, A., Guillot, S., Okaz, E., Junqueira, M., et al. (2008). Dbf4-dependent CDC7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell 135, 662–678. doi: 10.1016/j.cell.2008.10.026
Melo, J. A., Cohen, J., and Toczyski, D. P. (2001). Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev. 15, 2809–2821. doi: 10.1101/gad.903501
Nakada, D., Matsumoto, K., and Sugimoto, K. (2003). ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 17, 1957–1962. doi: 10.1101/gad.1099003
Niu, H., Wan, L., Baumgartner, B., Schaefer, D., Loidl, J., and Hollingsworth, N. M. (2005). Partner choice during meiosis is regulated by Hop1-promoted dimerization of Mek1. Mol. Biol. Cell 16, 5804–5818. doi: 10.1091/mbc.e05-05-0465
Nyberg, K. A., Michelson, R. J., Putnam, C. W., and Weinert, T. A. (2002). Toward maintaining the genome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36, 617–656. doi: 10.1146/annurev.genet.36.060402.113540
Okaz, E., Arguello-Miranda, O., Bogdanova, A., Vinod, P. K., Lipp, J. J., Markova, Z., et al. (2012). Meiotic prophase requires proteolysis of M phase regulators mediated by the meiosis-specific APC/CAma1. Cell 151, 603–618. doi: 10.1016/j.cell.2012.08.044
Ontoso, D., Acosta, I., van Leeuwen, F., Freire, R., and San-Segundo, P. A. (2013). Dot1-dependent histone H3K79 methylation promotes activation of the Mek1 meiotic checkpoint effector kinase by regulating the Hop1 adaptor. PLoS Genet. 9:e1003262. doi: 10.1371/journal.pgen.1003262
Paciotti, V., Clerici, M., Lucchini, G., and Longhese, M. P. (2000). The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev. 14, 2046–2059.
Pan, J., Sasaki, M., Kniewel, R., Murakami, H., Blitzblau, H. G., Tischfield, S. E., et al. (2011). A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 144, 719–731. doi: 10.1016/j.cell.2011.02.009
Pellicioli, A., and Foiani, M. (2005). Signal transduction: how rad53 kinase is activated. Curr. Biol. 15, R769–R771. doi: 10.1016/j.cub.2005.08.057
Pellicioli, A., Lee, S. E., Lucca, C., Foiani, M., and Haber, J. E. (2001). Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol. Cell 7, 293–300.
Pellicioli, A., Lucca, C., Liberi, G., Marini, F., Lopes, M., Plevani, P., et al. (1999). Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J. 18, 6561–6572. doi: 10.1093/emboj/18.22.6561
Pfander, B., and Diffley, J. F. (2011). Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J. 30, 4897–4907. doi: 10.1038/emboj.2011.345
Puddu, F., Piergiovanni, G., Plevani, P., and Muzi-Falconi, M. (2011). Sensing of replication stress and Mec1 activation act through two independent pathways involving the 9-1-1 complex and DNA polymerase ε. PLoS Genet. 7:e1002022. doi: 10.1371/journal.pgen.1002022
Refolio, E., Cavero, S., Marcon, E., Freire, R., and San-Segundo, P. A. (2011). The Ddc2/ATRIP checkpoint protein monitors meiotic recombination intermediates. J. Cell Sci. 124(Pt 14), 2488–2500. doi: 10.1242/jcs.081711
Sanchez, Y., Desany, B. A., Jones, W. J., Liu, Q., Wang, B., and Elledge, S. J. (1996). Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271, 357–360.
Sasanuma, H., Hirota, K., Fukuda, T., Kakusho, N., Kugou, K., Kawasaki, Y., et al. (2008). Cdc7-dependent phosphorylation of Mer2 facilitates initiation of yeast meiotic recombination. Genes Dev. 22, 398–410. doi: 10.1101/gad.1626608
Schwacha, A., and Kleckner, N. (1995). Identification of double Holliday junctions as intermediates in meiotic recombination. Cell 83, 783–791.
Schwacha, A., and Kleckner, N. (1997). Interhomolog bias during meiotic recombination: meiotic functions promote a highly differentiated interhomolog-only pathway. Cell 90, 1123–1135.
Schwartz, M. F., Duong, J. K., Sun, Z., Morrow, J. S., Pradhan, D., and Stern, D. F. (2002). Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol. Cell 9, 1055–1065.
Shinohara, A., Gasior, S., Ogawa, T., Kleckner, N., and Bishop, D. K. (1997). Saccharomyces cerevisiae recA homologues RAD51 and DMC1 have both distinct and overlapping roles in meiotic recombination. Genes Cells 2, 615–629.
Shinohara, A., Ogawa, H., and Ogawa, T. (1992). Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 69, 457–470.
Shinohara, M., Gasior, S. L., Bishop, D. K., and Shinohara, A. (2000). Tid1/Rdh54 promotes colocalization of rad51 and dmc1 during meiotic recombination. Proc. Natl. Acad. Sci. U.S.A. 97, 10814–10819.
Shinohara, M., Hayashihara, K., Grubb, J. T., Bishop, D. K., and Shinohara, A. (2015). DNA damage response clamp 9-1-1 promotes assembly of ZMM proteins for formation of crossovers and synaptonemal complex. J. Cell Sci. 128, 1494–1506. doi: 10.1242/jcs.161554
Shinohara, M., Oh, S. D., Hunter, N., and Shinohara, A. (2008). Crossover assurance and crossover interference are distinctly regulated by the ZMM proteins during yeast meiosis. Nat. Genet. 40, 299–309. doi: 10.1038/ng.83
Shinohara, M., Sakai, K., Ogawa, T., and Shinohara, A. (2003). The mitotic DNA damage checkpoint proteins Rad17 and Rad24 are required for repair of double-strand breaks during meiosis in yeast. Genetics 164, 855–865.
Shroff, R., Arbel-Eden, A., Pilch, D., Ira, G., Bonner, W. M., Petrini, J. H., et al. (2004). Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 14, 1703–1711.
Stewart, G. S., Panier, S., Townsend, K., Al-Hakim, A. K., Kolas, N. K., Miller, E. S., et al. (2009). The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434. doi: 10.1016/j.cell.2008.12.042
Storlazzi, A., Xu, L., Cao, L., and Kleckner, N. (1995). Crossover and noncrossover recombination during meiosis: timing and pathway relationships. Proc. Natl. Acad. Sci. U.S.A. 92, 8512–8516.
Sun, Z., Fay, D. S., Marini, F., Foiani, M., and Stern, D. F. (1996). Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev. 10, 395–406.
Sun, Z., Hsiao, J., Fay, D. S., and Stern, D. F. (1998). Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 281, 272–274.
Sweeney, F. D., Yang, F., Chi, A., Shabanowitz, J., Hunt, D. F., and Durocher, D. (2005). Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr. Biol. 15, 1364–1375. doi: 10.1016/j.cub.2005.06.063
Sym, M., Engebrecht, J. A., and Roeder, G. S. (1993). ZIP1 is a synaptonemal complex protein required for meiotic chromosome synapsis. Cell 72, 365–378.
Terasawa, M., Shinohara, A., Hotta, Y., Ogawa, H., and Ogawa, T. (1995). Localization of RecA-like recombination proteins on chromosomes of the lily at various meiotic stages. Genes Dev. 9, 925–934.
Toh, G. W., O’Shaughnessy, A. M., Jimeno, S., Dobbie, I. M., Grenon, M., Maffini, S., et al. (2006). Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair. 5, 693–703. doi: 10.1016/j.dnarep.2006.03.005
Usui, T., Foster, S. S., and Petrini, J. H. (2009). Maintenance of the DNA-damage checkpoint requires DNA-damage-induced mediator protein oligomerization. Mol. Cell 33, 147–159. doi: 10.1016/j.molcel.2008.12.022
Usui, T., and Kanehara, Y. (2013). Elevated Rad53 kinase activity influences formation and interhomolog repair of meiotic DNA double-strand breaks in budding yeast. Biochem. Biophys. Res. Commun. 441, 593–599. doi: 10.1016/j.bbrc.2013.10.111
Usui, T., and Petrini, J. H. (2007). The Saccharomyces cerevisiae 14-3-3 proteins Bmh1 and Bmh2 directly influence the DNA damage-dependent functions of Rad53. Proc. Natl. Acad. Sci. U.S.A. 104, 2797–2802. doi: 10.1073/pnas.0611259104
Vialard, J. E., Gilbert, C. S., Green, C. M., and Lowndes, N. F. (1998). The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J. 17, 5679–5688. doi: 10.1093/emboj/17.19.5679
Wysocki, R., Javaheri, A., Allard, S., Sha, F., Cote, J., and Kron, S. J. (2005). Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol. Cell Biol. 25, 8430–8443. doi: 10.1128/MCB.25.19.8430-8443.2005
Zegerman, P., and Diffley, J. F. (2010). Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467, 474–478. doi: 10.1038/nature09373
Zhang, L., Ma, H., and Pugh, B. F. (2011). Stable and dynamic nucleosome states during a meiotic developmental process. Genome Res. 21, 875–884. doi: 10.1101/gr.117465.110
Zhao, X., Muller, E. G., and Rothstein, R. (1998). A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell 2, 329–340.
Keywords: Rad9/53BP1, Rad53, DDR (DNA damage response), checkpoint, meiosis, recombination
Citation: Usui T and Shinohara A (2021) Rad9, a 53BP1 Ortholog of Budding Yeast, Is Insensitive to Spo11-Induced Double-Strand Breaks During Meiosis. Front. Cell Dev. Biol. 9:635383. doi: 10.3389/fcell.2021.635383
Received: 30 November 2020; Accepted: 25 February 2021;
Published: 25 March 2021.
Edited by:
Eesin Chen, National University of Singapore, SingaporeReviewed by:
Marcus Smolka, Cornell University, United StatesArmin Michael Gamper, University of Alberta, Canada
Copyright © 2021 Usui and Shinohara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akira Shinohara, YXNoaW5vQHByb3RlaW4ub3Nha2EtdS5hYy5qcA== orcid.org/0000-0003-4207-8247