 Xiaohong Meng
Xiaohong Meng Yanling Long
Yanling Long Jiayun Ren
Jiayun Ren Gang Wang
Gang Wang Xin Yin
Xin Yin Shiying Li
Shiying Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 11 March 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.635216
This article is part of the Research TopicGenetic Mutations Associated with Ocular DiseasesView all 19 articles
Bardet–Biedl syndrome (BBS; OMIM 209900) is a rare genetic disease causing damage to multiple organs and affecting patients’ quality of life in late adolescence or early adulthood. In this study, the ocular characteristics including morphology and function, were analyzed in 12 BBS patients from 10 Chinese families by molecular diagnostics. A total of five known and twelve novel variants in four BBS genes (BBS2, 58.33%; BBS4, 8.33%; BBS7, 16.67%; and BBS9, 16.67%) were identified in 10 Chinese families with BBS. All patients had typical phenotypes of retinitis pigmentosa with unrecordable or severely damaged cone and rod responses on full-field flash electroretinography (ffERG). Most of the patients showed unremarkable reactions in pattern visual evoked potential (PVEP) and multifocal electroretinography (mfERG), while their flash visual evoked potentials (FVEP) indicated display residual visual function. Changes in the fundus morphology, including color fundus photography and autofluorescence (AF) imaging, were heterogeneous and not consistent with the patients’ functional tests. Overall, our study expands the variation spectrum of the BBS gene, showing that the ocular characteristics of BBS patients are clinically highly heterogeneous, and demonstrates the usefulness of a combination of the ffERG and FVEP assessments of visual function in the advanced stage of retinopathy in BBS.
Bardet–Biedl syndrome (BBS; OMIM 209900) is a genetic disease causing damage to multiple organs, including early onset progressive retinitis pigmentosa (RP), obesity, hypogonadal hypoplasia, hand and/or foot polydactyly, intellectual disability, abnormal renal development (Bardet, 1995; Biedl, 1995). Therefore, BBS patients’ lives can be seriously threatened. However, while the diagnosis of BBS is often missed, it is usually fully recognized when patients visit an ophthalmologic clinic due to impaired vision or night blindness.
The ocular symptoms of BBS mainly manifest as RP since childhood. Fundus manifestations include optic disk pallor, bone spicule pigment deposits, thinning of the blood vessels, and retinal osteocyte pigmentation (Riise et al., 1996; Moore et al., 2005). Moreover, scotopic rod and maximal responses as well as cone responses are non-detectable in most BBS patients (Berezovsky et al., 2012; Scheidecker et al., 2015). A total of 24 causative genes of BBS have thus far been discovered (Weihbrecht et al., 2017; Bölükbaşı et al., 2018; Mary et al., 2019; Wormser et al., 2019). Most of them encode proteins necessary for the formation of the BBSome multi-subunit complex which localized at the cilia and basal body, and their connecting the cilium in the outer segment of photoreceptor has been thought to play a role in protein trafficking (Wei et al., 2012; Xu et al., 2015). Defects in any one subunit will adversely affect the formation or function of the BBSome, causing the BBS phenotype (Khan et al., 2016; Priya et al., 2016; Weihbrecht et al., 2017). Moreover, rhodopsin in BBS gene mutant mice was incorrectly located in the cells of the inner segment (IS) and outer nuclear layer (ONL), and the OS, IS, and ONL exhibited progressive loss (Nishimura et al., 2004; Datta et al., 2015). Currently, BBS treatment focuses on managing diabetes, hypertension, and metabolic syndrome to minimize the secondary effects of the disease on vulnerable organs.
Most BBS cases reported worldwide are concentrated in Europe and the Middle East (Farag and Teebi, 1988, 1989), whereas the disorder is very rare among the Chinese population. In this study, we conducted clinical and genetic analyses of 10 Chinese families (12 patients) with BBS and comprehensively evaluated visual function and fundus changes in these patients. We also investigated BBS genotype-phenotype differences between Chinese reports and those from abroad.
A total of 12 patients from 10 unrelated families with BBS treated at the Ophthalmology Department, Southwest Hospital, Army Medical University, Chongqing, China from 2012 to 2018 were retrospectively included in this study. The clinical diagnosis of BBS was made based on the potential patients presenting with four primary features or three primary features plus two secondary features. The primary features included retinal dystrophy, obesity, postaxial polydactyly, intellectual disability, gonadal abnormalities, and renal abnormalities. The secondary features included speech disorder/delay, sbtrabismus/cataracts/astigmatism, brachydactyly/syndactyly, developmental delay, polyuria/polydipsia (nephrogenic diabetes insipidus), ataxia/poor coordination/imbalance, mild spasticity (especially in the lower limbs), diabetes mellitus, dental crowding/hypodontia/small roots/high arched palate, left ventricular hypertrophy/congenital heart disease, hepatic fibrosis, short stature and hearing loss (Beales et al., 1999). If a patient was highly suspected to have BBS but one or two cardinal symptoms were missing, genetic analyses were performed for comprehensive analysis. We could clinically exclude the other syndromes such as Usher syndrome, Alström syndrome, Mckusick–Kaufman syndrome, Prader–Willi syndrome, and Joubert syndrome. Available family members of the probands were invited for clinical examination and genetic analysis. The research protocol was approved by the Ethics Review Board of Southwest Hospital (Chongqing, China, KY2020096), and the study was conducted in accordance with the provisions of the Declaration of Helsinki. Written informed consent was obtained from all participants. Assessment of ocular characteristics.
All patients underwent comprehensive ophthalmologic examinations, including best-corrected visual acuity (BCVA) tests with a decimal chart, indirect ophthalmoscopy, and slit lamp biomicroscopy. Color fundus photography, fundus fluorescein angiography (FFA), fundus autofluorescence (FAF; excitation: 488 nm), and Spectralis HRA-optical coherent tomography (OCT, Heidelberg Engineering, Dossenheim, Germany) imaging were used. Full-field flash electroretinography (ffERG; Diagnosys LLC, Lowell, MA, United States) and multifocal electroretinography (mfERG; VERIS Science 6.3.2; Electro-Diagnostic Imaging, Inc., Milpitas, CA, United States) were performed according to the ISCEV standard protocol (Hood et al., 2012; McCulloch et al., 2015). Pattern visual evoked potentials (PVEP) and flash visual evoked potentials (FVEP) were also recorded.
Genomic DNA was extracted from the patients’ peripheral blood using the Tiangen blood kit (Tiangen Biltech, Beijing, China) following the manufacturer’s standard sequencing protocols. Targeted next generation sequencing (NGS) was then performed using a capture panel including 131 known inherited retinal disease (IRD) genes (Supplementary Table 1). All coding exons of these genes were captured using the GenCap Liquid Phase Capture Kit (MyGenostics Inc.) and sequenced on the Illumina NextSeq 500. Then, the sequencing data processing, single nucleotide variants (SNVs) and insertions/deletions (InDels) calling and annotation were performed based on the hg19/GRCh37 human reference genome by using Haplotype Caller in GATK software (V.3.7), VariantFiltration in GATK software(V.3.7) and ANNOVAR software (V.3.4), respectively. Then, the online bioinformatics analysis software used to classify the pathogenicity of the candidate variants according to the American College of Medical Genetics and Genomics (ACMG) guidelines, including Exome Aggregation Consortium (ExAC), 1000Genomes database (1000G), and Genome Aggregation Database (gnomAD) to check the allele frequency; The rs number were found in Single Nucleotide Polymorphism Database (dbSNP); The pathogenicity of missense and synonymous variations were analyzed using ClinVar, HGMD Professional and four software prediction programs: Sorting Intolerant from Tolerance (SIFT), Protein Variation Effect Analyzer (PROVEAN) (Choi and Chan, 2015), Polymorphism Phenotyping v2 (PolyPhen2), and MutationTaster (Schwarz et al., 2014). The predicted effect on the splicing of all missense and synonymous variations was assessed with the Human Splicing finder program version 3.1 (Desmet et al., 2009). Finally, the disease-causing variants were identified while fully considering the clinical phenotypic findings and co-segregation analysis (by Sanger sequencing) of the affected subjects.
We enrolled four women and eight men from 10 unrelated families. Their average age was 20.75 years (range: 8–37 years). The physical examination findings included obesity (12/12), hand and foot polydactyly (7/12), intellectual disability (4/12), abnormal tooth development (8/12), short stature (6/12), hearing loss (3/12), gonadal dysplasia (4/12), epilepsy (2/12), dorsal hemangioma (2/12), renal dysplasia (1/12), spinal dysplasia (1/12), gallstones (1/12), hyperlipidemia (1/12), hypertension (2/12), osteoarthritis-like changes (1/12), and heart abnormalities (1/12) (Table 1 and Figure 1). Five patients had undergone surgical extra toe removal at an early age, and one side ovary was removed from F6-II:1 due to acute torsion of a unilateral polycystic ovary cyst. Three patients dropped out of school at an early age due to intellectual disability.

Table 1. Systemic manifestations of patients with BBS.
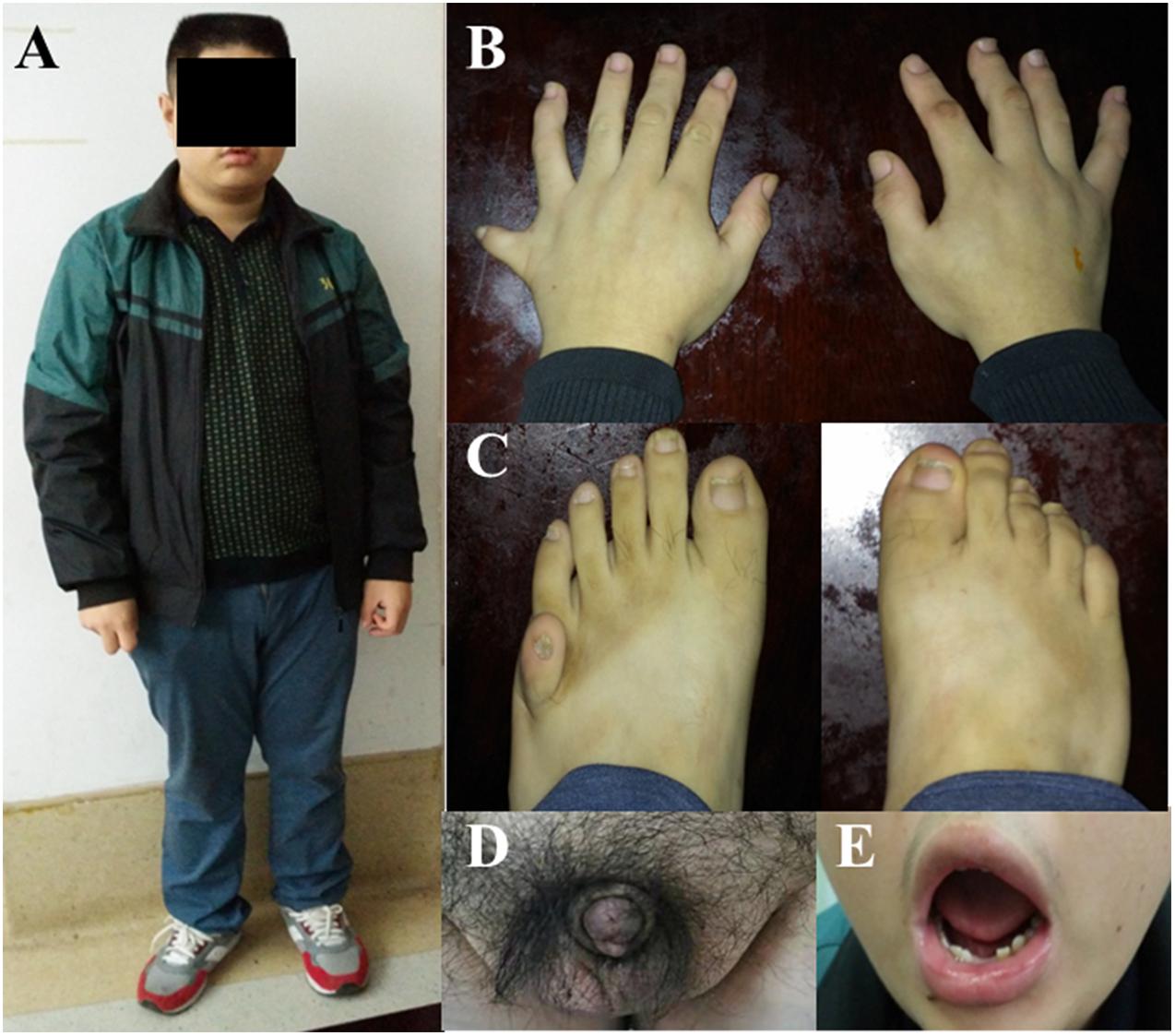
Figure 1. Systemic manifestations of BBS patients. The Wechsler Intelligence Score showed that this patient (F1-II:1) only had an 6-year-old intellectual level, his physical signs are: (A) Obesity (BMI 33.2 Kg/m2). (B) Polydactyly of the right hand and left foot. (C) Sexual organ dysplasia. (D) Dental dysplasia. (E) Abnormal tooth development.
All BBS patients were misdiagnosed with amblyopia upon their first visit to our hospital, and their common initial eye symptom was night blindness, and most of they have vision decline and vision field defect. Three patients (F-II:1, F2-II:2, and F8-II:1) had strabismus, and four (F1-II:1, F4-II:1, F5-II:1, and F8-II:1) had concomitant cataracts. F1-II:1 had nystagmus and concomitant cataract, and F2-II:2 had nystagmus with strabismus. The BCVA ranged from light perception (LP) to 0.3, including low vision (10/24 eyes) and legal blindness (16/24 eyes). All patients presented with typical RP features, including pigmentary changes in their peripheral and midperipheral retina, attenuated arteriolar vessels, pallor of the optic disk, and macular lesions (Figure 2; Fundus). Solitary macular atrophy was found in F3-II:1 and F8-II:1, which was characterized by pigment disorder, retinal thinning, and local retinal pigment epithelial atrophy, with a normal pigment change in the posterior pole retina. The AF of all patients showed plaque hypoautofluorescence in the posterior pole. Plaque hypoautofluorescence in the macular area was enlarged in F1-II:1, F2-II:1, F2-II:2, F3-II:2, F4-II:1, F5-II:1, and F9-II:1 (Figure 2; FAF). Patients’ fundus (F3-II:1 and F8-II:1) showed solitary enlarged macular lesions, while hypoautofluorescence of the macular area seemed to be normal in F10-II:1. OCT showed that the outer structure of the subfovea, including the myoid zone, ellipsoid zone, and external limiting membranes, was completely absent in F1-II:1; while most of the outer structure of the subfovea remained in F6-II:1; the rest of the patients showed different degrees of deletion of the outer structure of the subfovea (Figure 2; OCT). In the objective visual functional test, the ffERG displayed unrecordable waveforms in most of the patients (Figure 3A), and only F3-II:1 exhibited partial residual rod response. The mfERG waves were unrecordable in all patients assessed. The PVEP results showed a severely decreased amplitude and moderate delayed peak time in the P100 wave of F6-II:1, while the other patients had unrecordable PVEP due to nystagmus and fixation loss (Supplementary Figure 2). The FVEP showed mild delayed but visible P2 waves in patients assessed. Interestingly, all of the six patients who showed moderately reduced P2 wave amplitude, also had BCVA better than 0.01 (F2-II:1, F2-II:2, F3-II:1, F6-II:1, F7-II:1, and F8-II:1) (Figure 3B and Table 2).
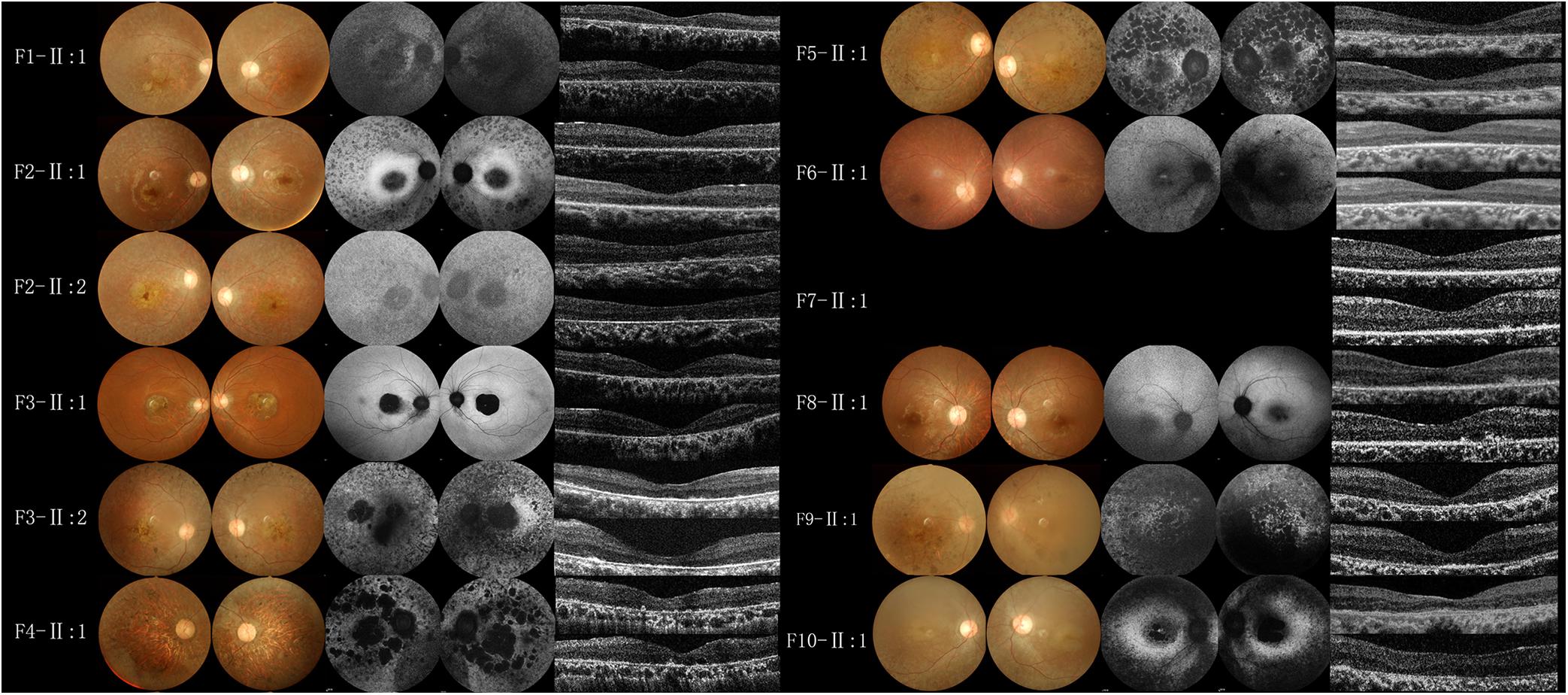
Figure 2. Fundus photography, fundus autofluorescence imaging, and foveal optical coherence tomography scans of probands from the BBS families. All patients had macular lesions, with pigmentary changes in the peripheral and midperipheral retina, attenuated arteriolar vessels, and pallor of the optic disk (Fundus). Plaque hypoautofluorescence in the macular area was enlarged in F1-II:1, F2-II:1, F2-II:2, F3-II:2, F4-II:1, F5-II:1, F9-II:1 (FAF). OCT (OD and OS in the vertical order) showed the outer structure of the subfovea was completely absent in F1-II:1, most of the outer structure remained in F6-II:1, and different degrees of deletion of the outer structure of the subfovea were observed in the rest of the patients (OCT).
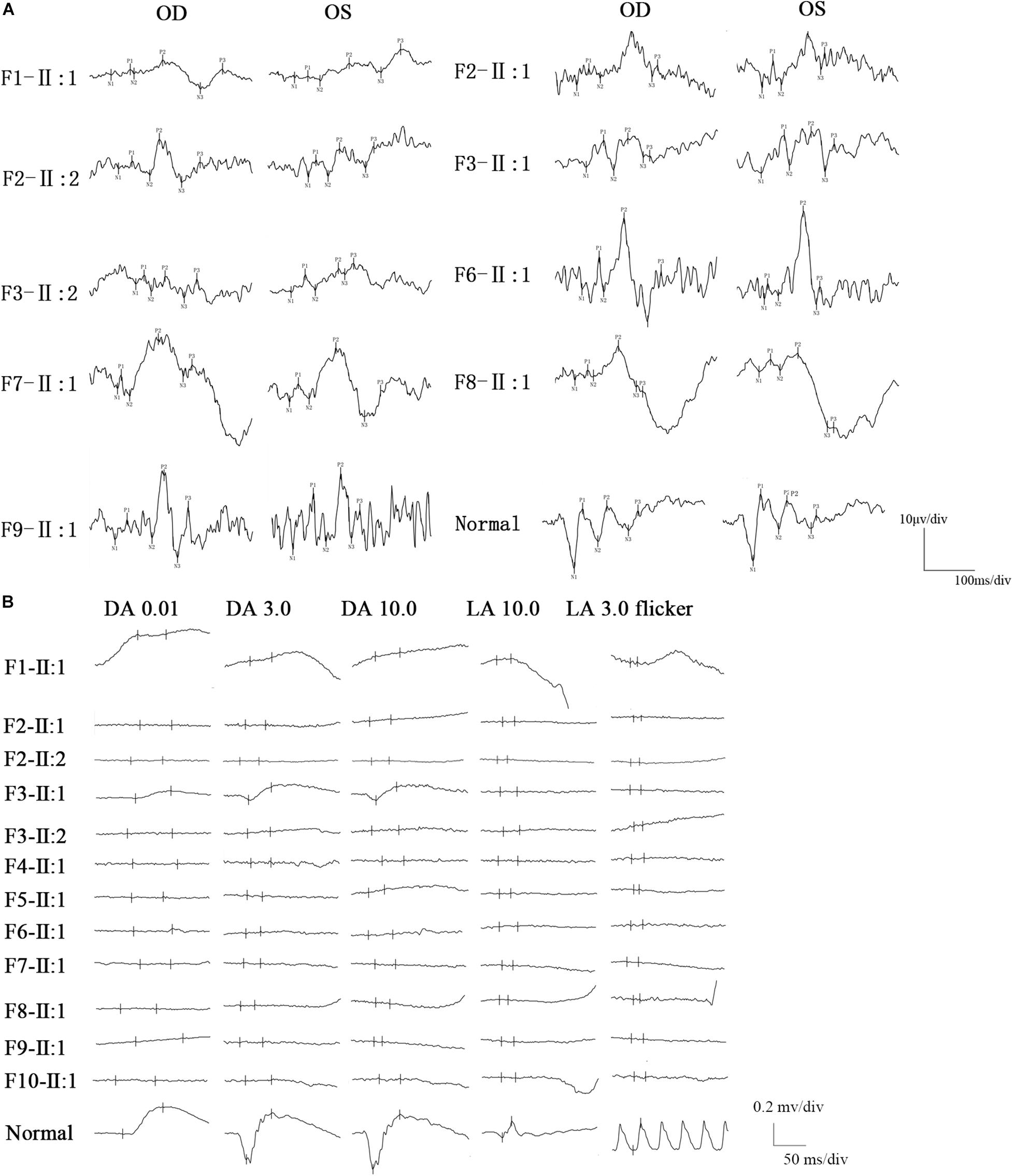
Figure 3. FVEP and ffERG recordings in patients with BBS. (A) The FVEP showed mild delayed but visible P2 waves in patients who performed it. Interestingly, all of six patients who showed moderate reduced P2 wave amplitude, also had BCVA better than 0.01 (F2-II:1, F2-II:2, F3-II:1, F6-II:1, F7-II:1, F8-II:1). (B) The ffERG was unrecordable in most of the patients, only patient (F3-II:1) had partial residual rod response.
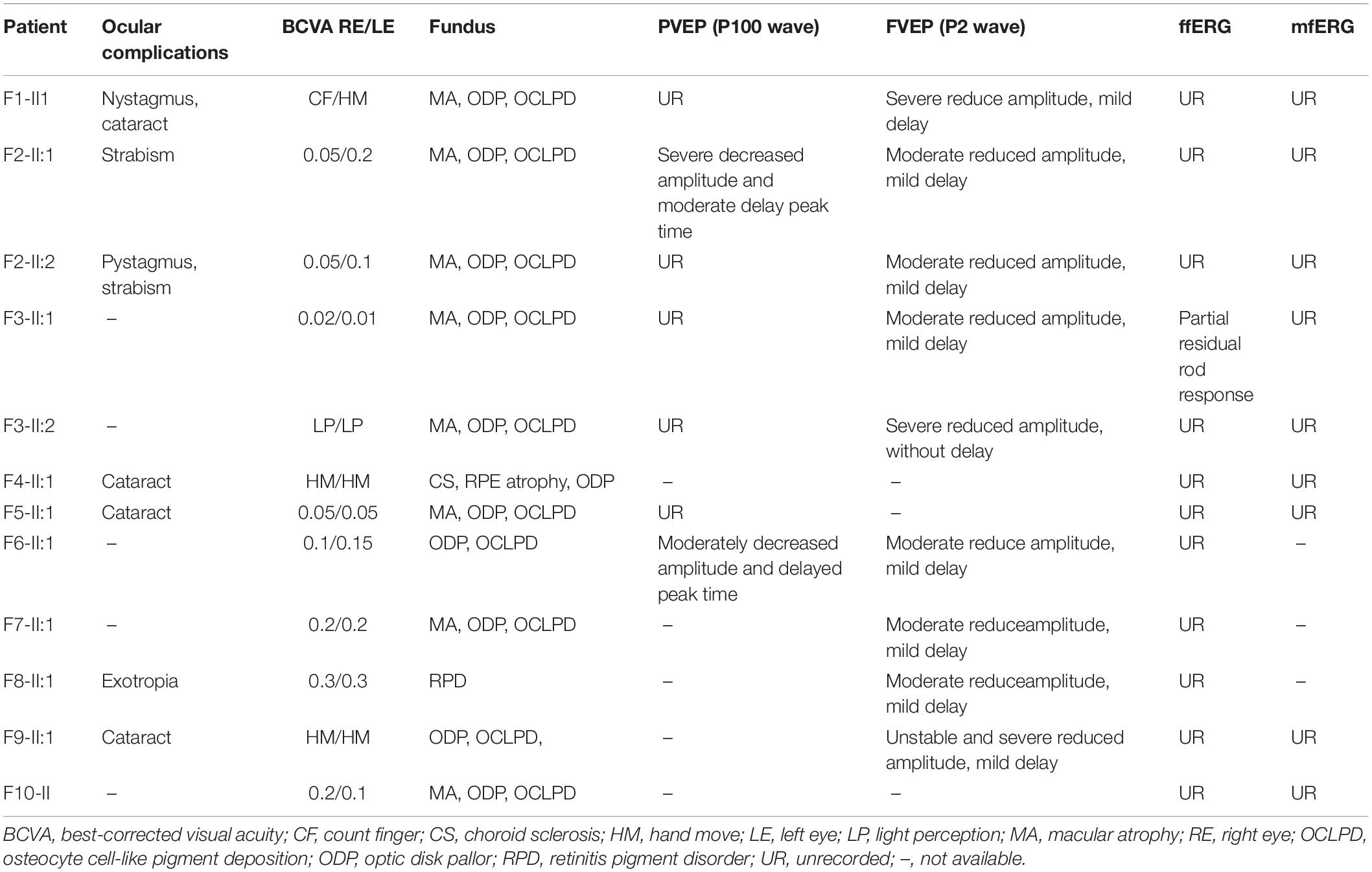
Table 2. Fundus manifestation and evaluation of visual function in patients with BBS.
Consanguineous marriage (F2) was noted in one of the 10 families in our cohort (Figure 4). After variant calling and data filtering, we identified 15 compound variants and two homozygous variants in the 12 patients with BBS2 (F1–F5), BBS4 (F6), BBS7 (F7–F8), and BBS9 (F9–F10) gene (Table 3), including five splice variations (c.263 + 2delT, c.534 + 1G > T, c.1198 + 1G > A, c.2059 + 1G > C, and c.2059 + 1G > T), three frameshift variations c.72delT(p.L25Cfs∗16), c.563delT(p.I188Tfs∗13), and c.1002delT(p.N335Ifs∗47), four nonsense variations c.31C > T(p.Q11∗), c.445C > T(p.R149∗), c.1015C > T(p.R339∗), and c.1395T > A(p.Y465∗), four missense variations c.79A > C(p.T27P), c.728G > A(p.C243Y), c.932G > A(p.G311D), and c.944G > A(p.R315Q), one synonymous variation c.1278A > G(p.E426E). These variants were confirmed as perfectly co-segregated with the disease within the families in which they occurred (Figure 4) based on a recessive pattern of inheritance as ascertained by Sanger sequencing. Among the identified variants, c.563delT(p.I188Tfs∗13), c.944G > A(p.R315Q), and c.1015C > T(p.R339∗) in the BBS2 gene (Katsanis et al., 2001; Xing et al., 2014; Shaheen et al., 2016), c.728G > A(p.C243Y) and c.1002delT(p.N335Ifs∗47) in the BBS7 gene (Chen et al., 2013; Wang et al., 2013) have been reported to be related to RP or BBS, while the rest of the 12 variants in the four genes were never been reported in ClinVar or HGMD Professional, so they were considered novel (BBS2, 41.67%; BBS4, 16.67%; BBS7, 8.33%; and BBS9, 33.33%). Moreover, according to the ACMG guidelines, except for c.79A > C(p.T27P) and c.1278A > G(p.E426E) which were classified as uncertain significance, we classify the other 15 variants as pathogenic. Furthermore, in this study, the novel heterozygous variation c.534 + 1G > T in BBS2 was detected in two families (F3 and F5), and, among the pathogenic BBS genes in the 10 probands, BBS2 was the most common.
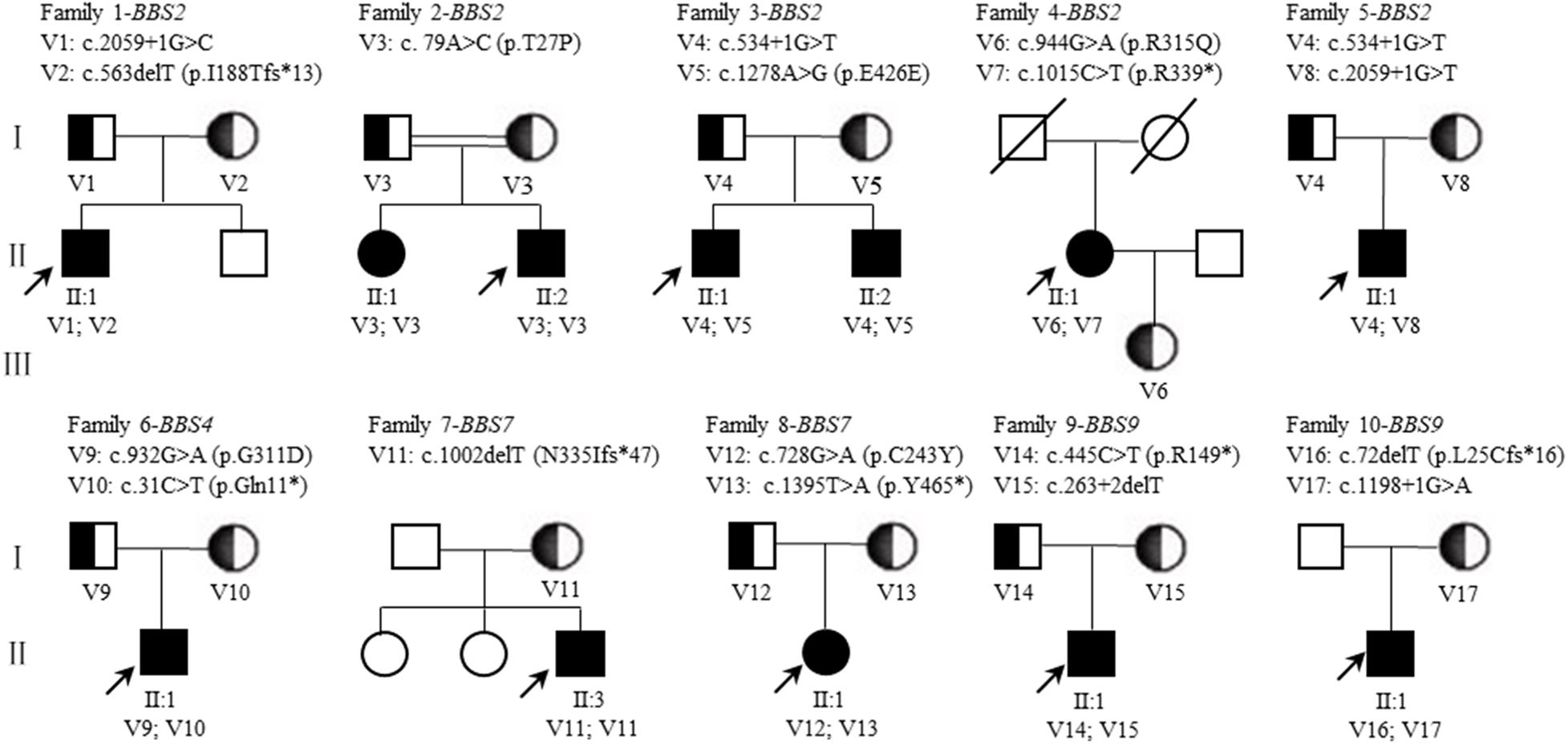
Figure 4. Pedigrees of BBS families. The squares and circles indicate men and women, respectively. The black solid square or circle represent the affected individual. The half black solid circle or square represent variation carrier. The probands are indicated by arrows. V, variant. A double line indicates a consanguineous marriage (The parents of F2-II:1 and F2-II:2 are consanguineous). A diagonal indicates that the individual has died.
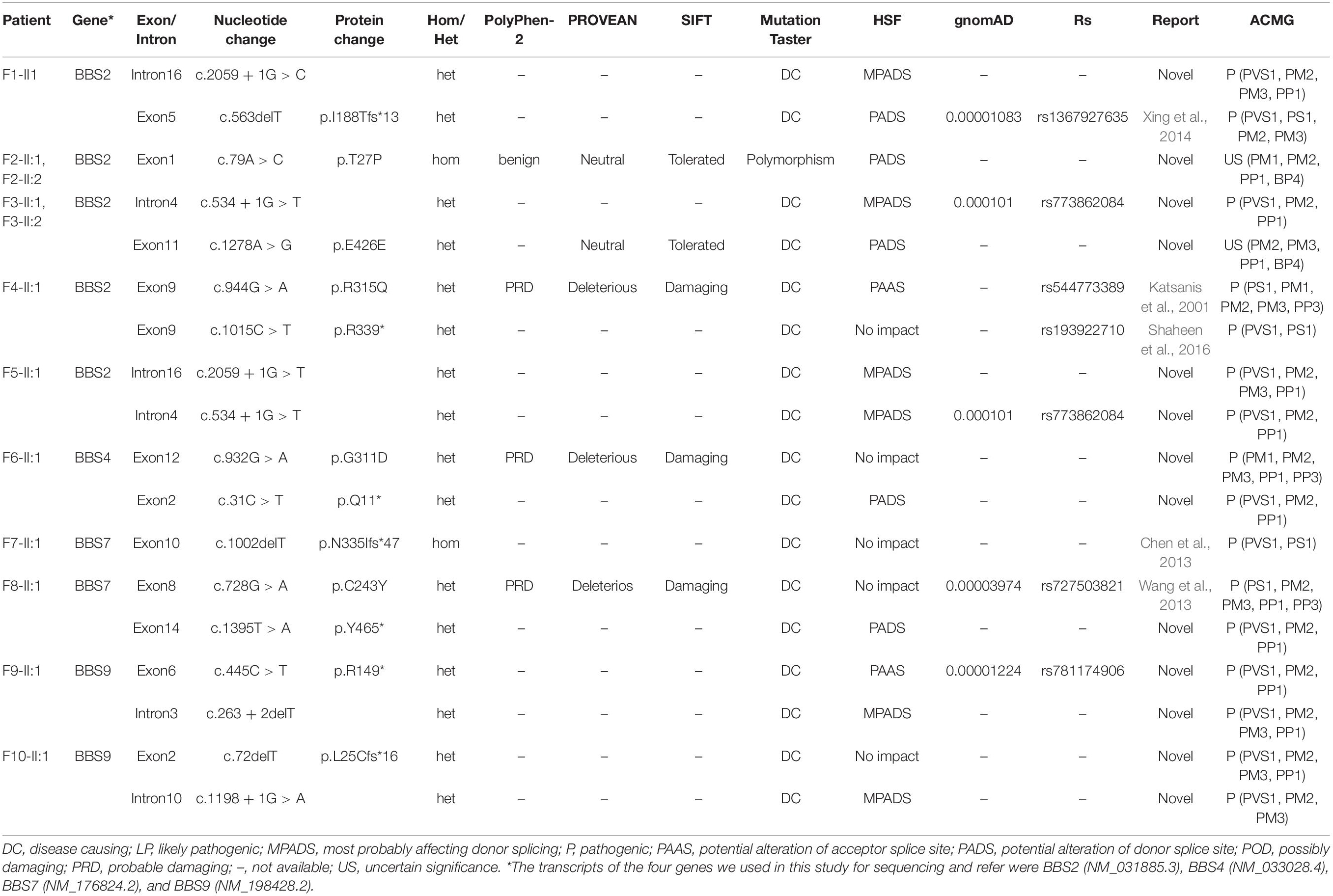
Table 3. Identified variants in patients with BBS.
We performed a literature analysis of the BBS related literatures on both Chinese and foreigner patients (number of cases ≥ 7), focusing on the ocular characteristics shown in Table 4. The first report of a BBS case in the Chinese literature was in 1954. So far, more than 100 BBS patients distributed among 17 provinces in China have been reported (Wei et al., 1998), Among these cases, the initial eye symptoms were most of night blindness, vision decline, so as the foreign reports (Deveault et al., 2011; Berezovsky et al., 2012; Castro-Sánchez et al., 2015; Scheidecker et al., 2015; Esposito et al., 2017). The age at diagnostic exam ranged from 1 to 65 years, and most were later the actual age of onset. The consanguineous mating rate (10% in our study) ranged between 35 and 39% in these previous studies. Among these reports (Table 4), not all BBS cases met the criteria of four main characteristics or three main characteristics plus two secondary characteristics (Hulleman et al., 2016). However, the high penetrance of RP seemed to be universal among all the BBS patients. Moreover, compared with the other five main clinical manifestations, the proportion of renal abnormalities was found to be relatively low (Wei et al., 1998). Variations in BBS1, BBS2, and BBS10 are the most common disease-causing genes among BBS patients in abroad (responsible for 50% of all cases). Thus far, our report, together with other local Chinese patient reports, suggests that the BBS2 (20%) and BBS7 (14.29%) genes are hot spot genes among Chinese BBS patients.
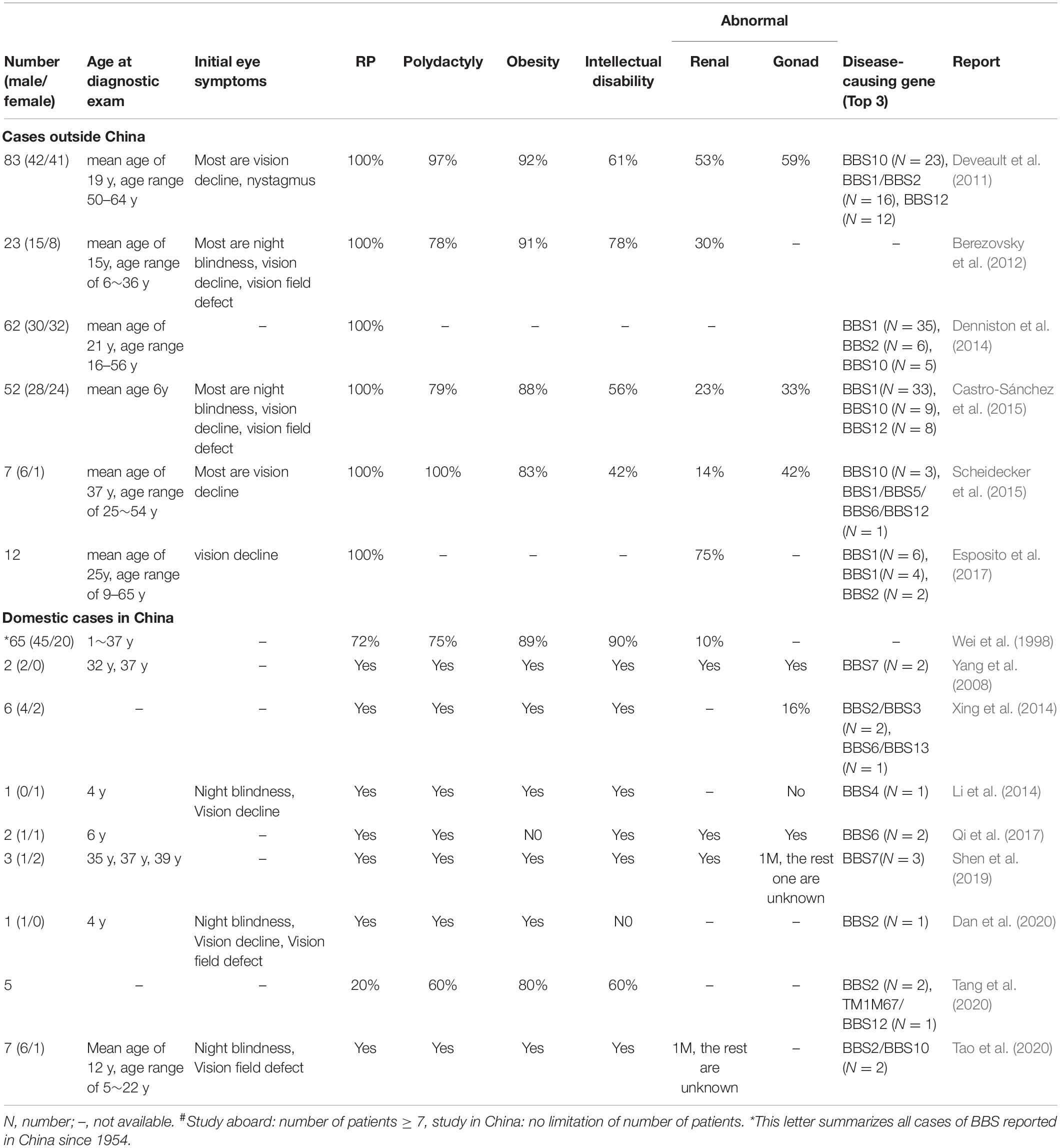
Table 4. Clinical manifestations and disease-causing genes of patients with BBS from previous reports#.
The detailed clinical and genetic characteristics of a cohort of 12 affected subjects from 10 Chinese families with BBS are illustrated in this study. To our knowledge, this is largest cohort of BBS patients by diagnosis using comprehensive clinical and genetic analysis to date in China. Although BBS has similar features and molecular overlap with Usher syndrome, Alström syndrome (OMIM 203800), McKusick-Kauffman (OMIM 604896) syndrome, Joubert syndrome (OMIM 213300, 608091, 608629, 609583, 610688, 611560, 61229, 612285, and 300804) or Meckel Gruber syndrome (OMIM 249000, 603294, 607361, and 611134), they are considered distinct clinical diseases, such as Alström syndrome (Pereiro et al., 2010) and Usher syndrome (Tsang et al., 2018) no polydactyly, or hypogonadism. Advanced gene sequencing technology has played an important role in helping distinguish between these similar genetic diseases. Although, ALMS1 gene which belong to Alström syndrome was not tested genetically specially by the patients (F1-II:1, F3-II:1, and F3-II:2) which have hearing disturbances, the disease-causing gene of BBS we identified was co-segregated with the family members and phenotype. The BBS patients in our study did not all fully meet the clinical diagnosis criteria for BBS, especially with the low proportion of renal abnormalities. Wei et al. (1998) reviewed 65 Chinese cases published from 1956 to 1998, finding that renal abnormalities of patients accounted for only 10.6%, and only 44.6% of patients had five main features. Furthermore, the ratio of renal abnormalities in cases reported abroad has been very different (Table 4) (Imhoff et al., 2011), as the phenotypes of BBS cases in different regions have differed, including the unexpected high frequency of tooth anomalies in our cohort, which further confirmed the high heterogeneity phenotype of BBS patients.
In this study, we focus on the ocular characteristics of BBS patients and explore the similarities and differences between the cohort we reported and previous cases reported in China and abroad. It is always challenging for doctors to access the complete information regarding the development of the retina of BBS patients, since patients present with visual symptoms at a young age and their visit to an ocular department usually occurs later than their actual onset. Impaired vision and night blindness have been found to be the most common initial eye symptom in patients worldwide. In our study, 12 patients exhibited low vision and night blindness which was independent age and gender. Weihbrecht et al. (2017) reported more than 90% of individuals with BBS displayed rod-cone dystrophy with general early macular involvement. They also observed the retina morphology by fundus, FAF, and OCT, and accessed the retinal function via ffERG and mfERG. We found that the visual field could not be measured accurately in these BBS patients because the vision of these patients was considered too poor to perform the kinetic visual field testing. And in the objective vision function assessment via visual electrophysiology, the ffERG wave was mostly unrecordable. Scheidecker et al. (2015) reported macular atrophy in all BBS patients, and severely reduced and delayed ERG-light-adapted responses were found in most BBS patients. In general, FVEP testing can appraise the residual visual function of BBS patients, and FVEP changes seem to be consistent with BCVA impairment, suggesting the feasibility of using FVEP to assess residual visual function in patients with rod-cone dystrophy without detection of FERG response.
Researcher made effort to find the genotype-phenotype correlations in BBS families (Table 4) (Deveault et al., 2011; Daniels et al., 2012; Niederlova et al., 2019), but actually, it is high diversity among affected patients that existed the same genotype from one family (F2 and F3 in our study). In our study, we found that the clinical manifestations of the different genes did not differ significantly, and five patients with the same disease-causing gene in BBS2 had completely different fundus morphological and functional changes. This indicated that the BBS genes and phenotypes are highly heterogeneous, which is consistent with previous findings (Table 4). In our study, we identified four pathogenic BBS genes (BBS2, BBS4, BBS7, and BBS9). Of those, BBS2 was the most common in our cohort. We also identified a novel BBS2 heterozygous variant c.534 + 1G > T in two families. Due to the small number of cases included in this study, it was not clear whether this is a hot spot variant of BBS in the Chinese population. However, the gene and its regional distributions, and even the distributions of hot spot variants have differed among studies (Mykytyn et al., 2002; Katsanis, 2004; Stoetzel et al., 2006; Ajmal et al., 2013). Yet variation in BBS1, BBS2, and BBS10 are the most common disease-causing genes among BBS patients abroad (responsible for 50% of all cases).
Overall, our study had some limitations. First, the number of enrolled patients was small. Second, we did not perform a longitudinal study regarding visual impairment. The changes in morphology and function of the retina should be followed up and correlated with different genes. Therefore, future studies with a larger sample size of BBS patients with BBS variants should be conducted using longitudinal clinical assessments and genotype-phenotype correlation analyses, not only in retina but also nephrology and endocrinology investigations and others to find some late syndromatic symptoms and signs (follow up by ophthalmologist, nephrologist and internist).
In our study, it demonstrated heterogeneous visual function and retinal morphology changes in these BBS patients. Our study expands the variation spectrum of BBS, and indicates that a combination of ffERG and FVEP may be helpful in evaluating the residual bioelectric activity of retina and visual pathway. To our knowledge, this is the largest cohort study with BBS patients in the Chinese population until now, we may conduct a large-scale screening BBS clinical cohort study among RP patients in China in the future.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Ethics Review Board of Southwest Hospital, Army Medical University (Third Military Medical University) (Chongqing, China, KY2020096). Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
XM collected cases, evaluated clinical characteristics, molecular diagnosis, and wrote the manuscript. YL made the figures and tables, and management data. GW helped in data evaluating. JR and XY collected and organized the data. SL collected cases, and wrote and revised the manuscript.
This work was supported by grants from the National Natural Science Foundation of China (81974138) and National Basic Research Program of China (2018YFA0107301).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge all the investigated family members for their cooperation, and very appreciate Minfang Zhang, Cheng Sun, Ziyang Li, and Min Wang for their help in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.635216/full#supplementary-material
Supplementary Figure 1 | The mfERG recordings in patients with BBS. The mfERG waves were unrecordable in all patients accessed.
Supplementary Figure 2 | PVEP recordings in patients with BBS. The PVEP results showed a severely decreased amplitude and moderate delayed peak time in the P100 wave of BBS patient F6-II:1, while the other patients had unrecordable PVEP due to nystagmus and fixation loss.
Supplementary Figure 3 | Sanger sequencing of disease-causing variants were identified in BBS patients.
Supplementary Table 1 | 131 inherited retinal disease genes analyzed by targeted NGS diagnostic testing.
Ajmal, M., Khan, M. I., Neveling, K., Tayyab, A., Jaffar, S., Sadeque, A., et al. (2013). Exome sequencing identifies a novel and a recurrent BBS1 mutation in Pakistani families with Bardet-Biedl syndrome. Mol. Vis. 19, 644–653.
Bardet, G. (1995). On congenital obesity syndrome with polydactyly and retinitis pigmentosa (a contribution to the study of clinical forms of hypophyseal obesity). Obes. Res. 3, 387–399. doi: 10.1002/j.1550-8528.1995.tb00165.x
Beales, P. L., Elcioglu, N., Woolf, A. S., Parker, D., and Flinter, F. A. (1999). New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J. Med. Genet. 36, 437–446.
Berezovsky, A., Rocha, D. M., Sacai, P. Y., Watanabe, S. S., Cavascan, N. N., and Salomão, S. R. (2012). Visual acuity and retinal function in patients with Bardet-Biedl syndrome. Clinics 67, 145–149. doi: 10.6061/clinics/2012(02)09
Biedl, A. (1995). A pair of siblings with adiposo-genital dystrophy. Obes. Res. 3:404. doi: 10.1136/bjo.81.5.378
Bölükbaşı, E. Y., Mumtaz, S., Afzal, M., Woehlbier, U., Malik, S., and Tolun, A. (2018). Homozygous mutation in CEP19, a gene mutated in morbid obesity, in Bardet-Biedl syndrome with predominant postaxial polydactyly. J. Med. Genet. 55, 189–197. doi: 10.1136/jmedgenet-2017-104758
Castro-Sánchez, S., Álvarez-Satta, M., Cortón, M., Guillén, E., Ayuso, C., and Valverde, D. (2015). Exploring genotype-phenotype relationships in Bardet-Biedl syndrome families. J. Med. Genet. 52:503. doi: 10.1136/jmedgenet-2015-103099
Chen, X., Zhao, K., Sheng, X., Li, Y., Gao, X., Zhang, X., et al. (2013). Targeted sequencing of 179 genes associated with hereditary retinal dystrophies and 10 candidate genes identifies novel and known mutations in patients with various retinal diseases. Invest. Ophthalmol. Vis. Sci. 54, 2186–2197. doi: 10.1167/iovs.12-10967
Choi, Y., and Chan, A. P. (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. doi: 10.1093/bioinformatics/btv195
Dan, H., Huang, X., Xing, Y., and Shen, Y. (2020). Application of targeted panel sequencing and whole exome sequencing for 76 Chinese families with retinitis pigmentosa. Mol. Genet. Genomic. Med. 8:e1131. doi: 10.1002/mgg3.1131
Daniels, A. B., Sandberg, M. A., Chen, J., Weigel-DiFranco, C., Fielding, H. J., and Berson, E. L. (2012). Genotype-phenotype correlations in Bardet-Biedl syndrome. Arch. Ophthalmol. 130, 901–907. doi: 10.1001/archophthalmol.2012.89
Datta, P., Allamargot, C., Hudson, J. S., Andersen, E. K., Bhattarai, S., Drack, A. V., et al. (2015). Accumulation of non-outer segment proteins in the outer segment underlies photoreceptor degeneration in BardetBiedl syndrome. Proc. Natl. Acad. Sci. U.S.A. 112, E4400–E4409.
Denniston, A. K., Beales, P. L., Tomlins, P. J., Good, P., Langford, M., and Foggensteiner, L. (2014). Evaluation of visual function and needs in adult patients with bardet-biedl syndrome. Retina 34, 2282–2289. doi: 10.1097/IAE.0000000000000222
Desmet, F. O., Hamroun, D., Lalande, M., Collod-Béroud, G., Claustres, M., and Béroud, C. (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic. Acids. Res. 37:e67. doi: 10.1093/nar/gkp215
Deveault, C., Billingsley, G., Duncan, J. L., Bin, J., Theal, R., Vincent, A., et al. (2011). BBS genotype–phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum. Mutat. 32, 610–619. doi: 10.1002/humu.21480
Esposito, G., Testa, F., Zacchia, M., Crispo, A. A., Di-Iorio, V., Capolongo, G., et al. (2017). Genetic characterization of Italian patients with Bardet-Biedl syndrome and correlation to ocular, renal and audio-vestibular phenotype: identification of eleven novel pathogenic sequence variants. BMC. Med. Genet. 17:10. doi: 10.1186/s12881-017-0372-0
Farag, T. I., and Teebi, A. S. (1988). Bardet-Biedl and laurence-moon syndromes in a mixed arab population. Clin. Genet. 33, 78–82. doi: 10.1111/j.1399-0004.1988.tb03414.x
Farag, T. I., and Teebi, A. S. (1989). High incidence of Bardet Biedl syndrome among the Bedouin. Clin. Genet. 36, 463–464. doi: 10.1111/j.1399-0004.1989.tb03378.x
Hood, D. C., Bach, M., Brigell, M., Keating, D., Kondo, M., Lyons, J. S., et al. (2012). ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc. Ophthalmol. 124, 1–13. doi: 10.1007/s10633-011-9296-8
Hulleman, J. D., Nguyen, A., Ramprasad, V. L., Murugan, S., Gupta, R., Mahindrakar, A., et al. (2016). A novel H395R mutation in MKKS/BBS6 causes retinitis pigmentosa and polydactyly without other findings of Bardet-Biedl or McKusick-Kaufman syndrome. Mol. Vis. 22, 73–81.
Imhoff, O. V., Marion, C., Stoetzel, M., Durand, M., Holder, S., Sigaudy, P., et al. (2011). Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin. J. Am. Soc. Nephrol. 6, 22–29. doi: 10.2215/CJN.03320410
Katsanis, N. (2004). The oligogenic properties of Bardet–Biedl syndrome. Hum. Mol. Genet. 13, 65–71. doi: 10.1093/hmg/ddh092
Katsanis, N., Ansley, S. J., Badano, J. L., Eichers, E. R., Lewis, R. A., Hoskins, B. E., et al. (2001). Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 293, 2256–2259. doi: 10.1126/science.1063525
Khan, S. A., Muhammad, N., Khan, M. A., Kamal, A., Rehman, Z. U., and Khan, S. (2016). Genetics of human Bardet-Biedl syndrome, an updates. Clin. Genet. 90, 3–15. doi: 10.1111/cge.12737
Li, Q., Zhang, Y., Jia, L., and Peng, X. (2014). A novel nonsense mutation in BBS4 gene identified in a Chinese family with Bardet-Biedl syndrome. Chin. Med. J. 127, 4190–4196.
Mary, L., Chennen, K., Stoetzel, C., Antin, M., Leuvrey, A., Nourisson, E., et al. (2019). Bardet-Biedl syndrome: antenatal presentation of forty-five fetuses with biallelic pathogenic variants in known Bardet-Biedl syndrome genes. Clin. Genet. 95, 384–397. doi: 10.1111/cge.13500
McCulloch, D. L., Marmor, M. F., Brigell, M. G., Hamilton, R., Holder, G. E., Tzekov, R., et al. (2015). ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 130, 1–12. doi: 10.1007/s10633-014-9473-7
Moore, S. J., Green, J. S., Fan, Y., Bhogal, A. K., Dicks, E., Fernandez, B. A., et al. (2005). Clinical and genetic epidemiology of Bardet–Biedl syndrome in newfoundland: a 22-year prospective, population-based, cohort study. Am. J. Med. Genet. A 132, 352–360. doi: 10.1002/ajmg.a.30406
Mykytyn, K., Nishimura, D. Y., Searby, C. C., Shastri, M., Yen, H. J., Beck, J. S., et al. (2002). Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat. Genet. 31, 435–438. doi: 10.1038/ng935
Niederlova, V., Modrak, M., Tsyklauri, O., Huranova, M., and Stepanek, O. (2019). Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum. Mutat. 40, 2068–2087. doi: 10.1002/humu.23862
Nishimura, D. Y., Fath, M., Mullins, R. F., Searby, C., Andrews, M., Davis, R., et al. (2004). Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 23, 16588–16593. doi: 10.1073/pnas.0405496101
Pereiro, I., Hoskins, B. E., Marshall, J. D., Collin, G. B., Naggert, J. K., Piñeiro-Gallego, T., et al. (2010). Arrayed primer extension technology simplifies mutation detection in Bardet-Biedl and Alström syndrome. Eur. J. Hum. Genet. 19, 485–488. doi: 10.1038/ejhg.2010.207
Priya, S., Nampoothiri, S., Sen, P., and Sripriya, S. (2016). Bardet–Biedl syndrome: Genetics, molecular pathophysiology, and disease management. Indian. J. Ophthalmol. 64, 620–627. doi: 10.4103/0301-4738
Qi, Z., Shen, Y., Fu, Q., Li, W., Yang, W., Xu, W., et al. (2017). Whole-exome sequencing identified compound heterozygous variants in MMKS in a Chinese pedigree with Bardet-Biedl syndrome. Sci. China. Life. Sci. 60, 739–745. doi: 10.1007/s11427-017-9085-7
Riise, R., Andréasson, S., and Tornqvist, K. (1996). Full-field electroretinograms in individuals with the laurence-moon-Bardet-Biedl syndrome. Acta Ophthalmol. Scand. 74, 618–620. doi: 10.1111/j.1600-0420.1996.tb00747.x
Scheidecker, S., Hull, S., Perdomo, Y., Studer, F., Pelletier, V., Muller, J., et al. (2015). Predominantly cone-system dysfunction as rare form of retinal degeneration in patients with molecularly confirmed Bardet-Biedl syndrome. Am. J. Ophthalmol. 160, 364–372. doi: 10.1016/j.ajo.2015.05.007
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. doi: 10.1038/nmeth.2890
Shaheen, R., Szymanska, K., Basu, B., Patel, N., Ewida, N., Faqeih, E., et al. (2016). Characterizing the morbid genome of ciliopathies. Genome. Biol. 17:242. doi: 10.1186/s13059-016-1099-5
Shen, T., Gao, J. M., Shou, T., Li, L., Zhang, J. P., Zhao, Q., et al. (2019). Identification of a homozygous BBS7 frameshift mutation in two (related) Chinese miao families with Bardet-Biedl syndrome. J. Chin. Med. Assoc. 82, 110–114. doi: 10.1097/jcma.0000000000000011
Stoetzel, C., Laurier, V., Davis, E. E., Muller, J., Rix, S., Badano, J. L., et al. (2006). BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 38, 521–524. doi: 10.1038/ng1771
Tang, X., Liu, C., Liu, X., Chen, J., Fan, X., Liu, J., et al. (2020). Phenotype and genotype spectra of a Chinese cohort with nephronophthisis-related ciliopathy. J. Med. Genet. 1–8. doi: 10.1136/jmedgenet-2020-107184
Tao, T., Wang, L., Chong, W., Yang, L., and Li, G. (2020). Characteristics of genotype and phenotype in Chinese patients with Bardet-Biedl syndrome. Int. Ophthalmol. 40, 2325–2343. doi: 10.1007/s10792-020-01415-3
Tsang, S. H., Aycinena, A. R. P., and Sharma, T. (2018). Ciliopathy: usher syndrome. Adv. Exp. Med. Biol. 1085, 167–170. doi: 10.1007/978-3-319-95046-4_32
Wang, X., Wang, H., Sun, V., Tuan, H. F., Keser, V., and Wang, K. (2013). Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J. Med. Genet. 50, 674–688. doi: 10.1136/jmedgenet-2013-101558
Wei, L. J., Pang, X., Duan, C., and Pang, X. (1998). Bardet-Biedl syndrome: a review of Chinese literature and a report of two cases. Ophthalmic. Genet. 19, 107–109. doi: 10.1076/opge.19.2.107.2315
Wei, Q., Zhang, Y., Li, Y., Zhang, Q., Ling, K., and Hu, J. (2012). The BBSome controls IFT assembly and turnaround in cilia. Nat. Cell. Biol. 14, 950–957. doi: 10.1038/ncb2560
Weihbrecht, K., Goar, W. A., Pak, T., Garrison, J. E., DeLuca, A. P., Stone, E. M., et al. (2017). Keeping an eye on Bardet-Biedl syndrome: a comprehensive review of the role of Bardet-Biedl syndrome genes in the eye. Med. Res. Arch. 5:10.18103/mra.v5i9.1526. doi: 10.18103/mra.v5i9.1526
Wormser, O., Gradstein, L., Yogev, Y., Perez, Y., Kadir, R., Goliand, I., et al. (2019). SCAPER localizes to primary cilia and its mutation affects cilia length, causing Bardet-Biedl syndrome. Eur. J. Hum. Genet. 27, 928–940. doi: 10.1038/s41431-019-0347-z
Xing, D. J., Zhang, H. X., Huang, N., Wu, K. C., Huang, X. F., Huang, F., et al. (2014). Comprehensive molecular diagnosis of Bardet-Biedl Syndrome by high-throughput targeted exome sequencing. PLos One 9:e90599. doi: 10.1371/journal.pone.0090599
Xu, Q., Zhang, Y., Wei, Q., Huang, Y., Li, Y., Ling, K., et al. (2015). BBS4 and BBS5 show functional redundancy in the BBSome to regulate the degradative sorting of ciliary sensory receptors. Sci. Rep. 5:11855. doi: 10.1038/srep11855
Keywords: Bardet–Biedl syndrome, ocular characteristics, morphology, visual function, gene variation
Citation: Meng X, Long Y, Ren J, Wang G, Yin X and Li S (2021) Ocular Characteristics of Patients With Bardet–Biedl Syndrome Caused by Pathogenic BBS Gene Variation in a Chinese Cohort. Front. Cell Dev. Biol. 9:635216. doi: 10.3389/fcell.2021.635216
Received: 30 November 2020; Accepted: 08 February 2021;
Published: 11 March 2021.
Edited by:
Minzhong Yu, Case Western Reserve University, United StatesReviewed by:
Jean Muller, INSERM U1112 Laboratoire de Génétique Médicale, FranceCopyright © 2021 Meng, Long, Ren, Wang, Yin and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiying Li, c2hpeWluZ19saUAxMjYuY29t
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.