 Shuangyu Lv
Shuangyu Lv Honggang Wang
Honggang Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 16 March 2021
Sec. Cellular Biochemistry
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.634118
This article is part of the Research Topic Interplay between Autophagy and Metabolic Syndrome: Causes, Consequences and Therapeutic Challenges View all 15 articles
Autophagy is an important and conserved cellular pathway in which cells transmit cytoplasmic contents to lysosomes for degradation. It plays an important role in maintaining the balance of cell composition synthesis, decomposition and reuse, and participates in a variety of physiological and pathological processes. The nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome can induce the maturation and secretion of Interleukin-1 beta (IL-1β) and IL-18 by activating caspase-1. It is involved in many diseases. In recent years, the interplay between autophagy and NLRP3 inflammasome has been reported to contribute to many diseases including metabolic disorders related diseases. In this review, we summarized the recent studies on the interplay between autophagy and NLRP3 inflammasome in metabolic disorders to provide ideas for the relevant basic research in the future.
Autophagy is a closely coordinated process that isolates proteins and damaged or aged organelles in double membrane vesicles called autophagosomes which eventually fuse with lysosomes, leading to the degradation of isolated components (Behrends et al., 2010). It plays an important role in maintaining the balance of cell synthesis, decomposition and reuse (Wei et al., 2016). Abnormal autophagy is involved in the development of the pathological processes such as cardiomyopathy (Mclendon et al., 2014), neurodegenerative diseases (Mclendon et al., 2014), type II diabetes (Sarparanta et al., 2017), and cancer (Amaravadi et al., 2011). The nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome can induce the maturation and secretion of interleukin-1 beta (IL-1β) and IL-18 by activating caspase-1 (Schroder and Tschopp, 2010; Strowig et al., 2012). Studies have shown that the abnormal activation of NLRP3 inflammasome contributes to many diseases including type 2 diabetes, atherosclerosis, and steatohepatitis (Mridha et al., 2017; Van Der Heijden et al., 2017; Zhai et al., 2018). Recently, more and more studies have shown that there is an interplay between autophagy and NLRP3 inflammasome in macrophage and the interplay plays an important role in many diseases including metabolic diseases, the mechanism of which remains to be elucidated (Biasizzo and Kopitar-Jerala, 2020). In this review, the interplay between autophagy and the NLRP3 inflammasome and its mechanism are explored in metabolic disorders to provide ideas for the relevant basic research in the future.
Autophagy is a conserved process of self-sustaining internal environment stability, in which abnormal proteins and organelles are encapsulated by the bilayer membranes and transported to lysosomes for subsequent degradation (Sir et al., 2010; Qiu et al., 2014). Autophagy can be categorized as microautophagy, macroautophagy and chaperone-mediated autophagy according to the inducing signal, action time, target type, and transport pathway into lysosomes (Feng et al., 2014; Gomes et al., 2017). Macroautophagy, the most studied autophagy, is mainly responsible for the degradation of organelles and microorganisms. In this process, the degraded substance is encapsulated by a double membrane vesicle to form autophagosome, which then fuses with lysosome for degradation (Cheon et al., 2019). Microautophagy, without forming autophagosome, mainly degrades cell components by directly engulfing the cytoplasm at the lysosomal membrane through invagination and or septation (Oku and Sakai, 2018). Chaperone-mediated autophagy is a selective autophagy process in which intracellular proteins are transported to lysosomal chambers after binding with chaperones, then digested by lysosomal enzymes (Kaushik and Cuervo, 2012; Figure 1). Moreover, mitochondrial autophagy, also called mitophagy, is a special kind of autophagy, which selectively degrades organelle mitochondria and is largely responsible for mitochondrial quality control (Ashrafi and Schwarz, 2013; Ney, 2015). There are many factors affecting autophagy, such as Ca2+ concentration, immune or inflammatory stimulation, endoplasmic reticulum stress, nutritional deficiency, and accumulation of damaged cells or organelles (Tooze and Yoshimori, 2010; Mizushima et al., 2011). Under physiological conditions, autophagy often occurs at the basic maintenance level. When the body is in a pathological state, significantly enhanced autophagy can clear the abnormal proteins in cells, which is conducive to cell survival (Glick et al., 2010). The effect of autophagy on cells is a “double-edged sword,” because if autophagy is maintained at a high level, autophagy will lead to cell death (Liu and Levine, 2015; Garcia-Huerta et al., 2016). Autophagy plays a key role in many basic physiological processes, including development, protein quality control, innate immunity and cell survival. It is well known that abnormal autophagy contributes to the occurrence of many diseases, including cancer, liver diseases, neurodegenerative diseases, type-II diabetes, and inflammation (Debnath, 2011; Murrow and Debnath, 2013). So far, the mechanism of autophagy in diseases is not fully clear.
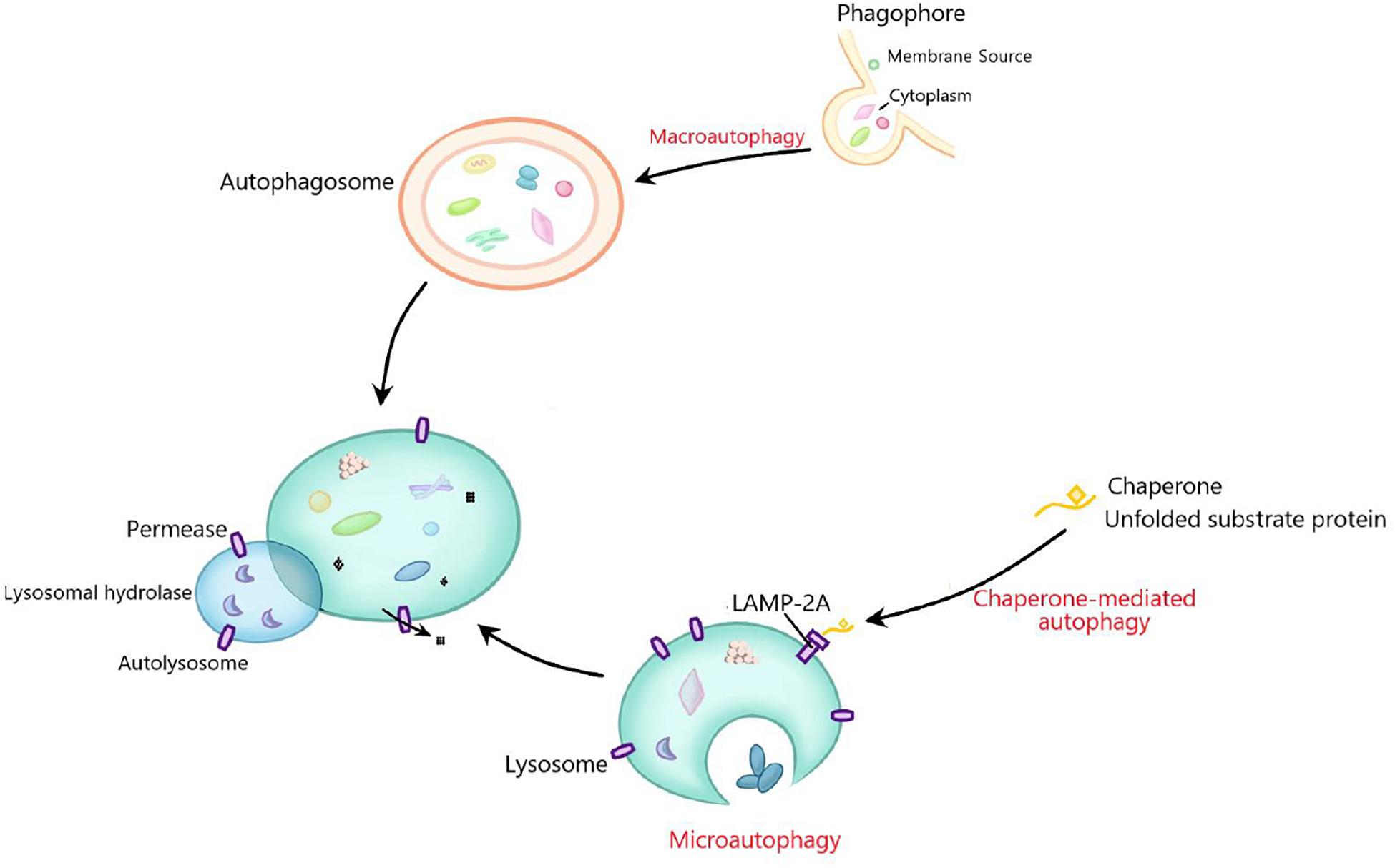
Figure 1. The general process of macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy mainly degrades organelles and microorganisms. The degraded substances are first encapsulated by double membrane vesicles to form autophagosomes, and then fuse with lysosomes for degradation. Microautophagy does not form autophagosomes, but mainly degrades cell components by directly engulfing the cytoplasm at the lysosomal membrane through invagination and or septation. Chaperone-mediated autophagy is a selective autophagy process in which degraded proteins are transported to lysosomal chambers after binding with chaperones, and then digested by lysosomal enzymes.
Inflammasome, a complex composed of many proteins, is an important part of the innate immune system and used to detect the presence of the infection, the pathogens and the metabolic alarms in cells (Elliott and Sutterwala, 2015; Jo et al., 2016). Five major inflammasomes have been identified: NLRP1, NLRC4, RIG-I, AIM2 and NLRP3. These inflammasomes consist of inflammasome adaptor protein, an active NLRP receptor, caspase-1, and apoptosis-associated speck-like protein containing CARD (ASC) (Barbe et al., 2014). NLRP3 inflammasome is the most thoroughly studied inflammasome which is mainly expressed in myeloid cells such as macrophages (O’connor et al., 2003). When the host is stimulated by the exogenous or endogenous stimuli, NLRP3 is activated to lead to the recruitment of pro-caspase-1 and ASC in macrophages. Stimulated NLRP3 interacts with pro-caspase-1 and ASC to form a large cytoplasmic complex, thus activating caspase-1. Actived caspase-1 cleaves pro-IL-1β and pro-IL-18 into IL-1β and IL-18, which induces inflammation by promoting the production of chemokines, proinflammatory cytokines and growth factors (Prochnicki et al., 2016; Figure 2). NLRP3 inflammasome was activated in two steps. The first signals (signal 1) indicating infection or tissue damage, including toll like receptor 4, a pattern recognition receptor capable of recognizing lipopolysaccharide (LPS) and a series of endogenous risk signals, activate the inflammatory transcription factor NF-κβ and increase the expression of NLRP3, pro-IL-1β, and pro-IL-18. Then, the second activation signal (signal 2) indicating cell damage, including extracellular adenosine triphosphate (ATP), urate and cholesterol crystal, induces the assembly of inflammasome and the autolysis of procaspase-1, activates caspase-1, and cleaves pro-IL-1β and pro-IL-18 into their active forms (Sokolova et al., 2019). Abnormal activation of NLRP3 inflammasome can lead to a variety of diseases including metabolic diseases (Meyers and Zhu, 2020).
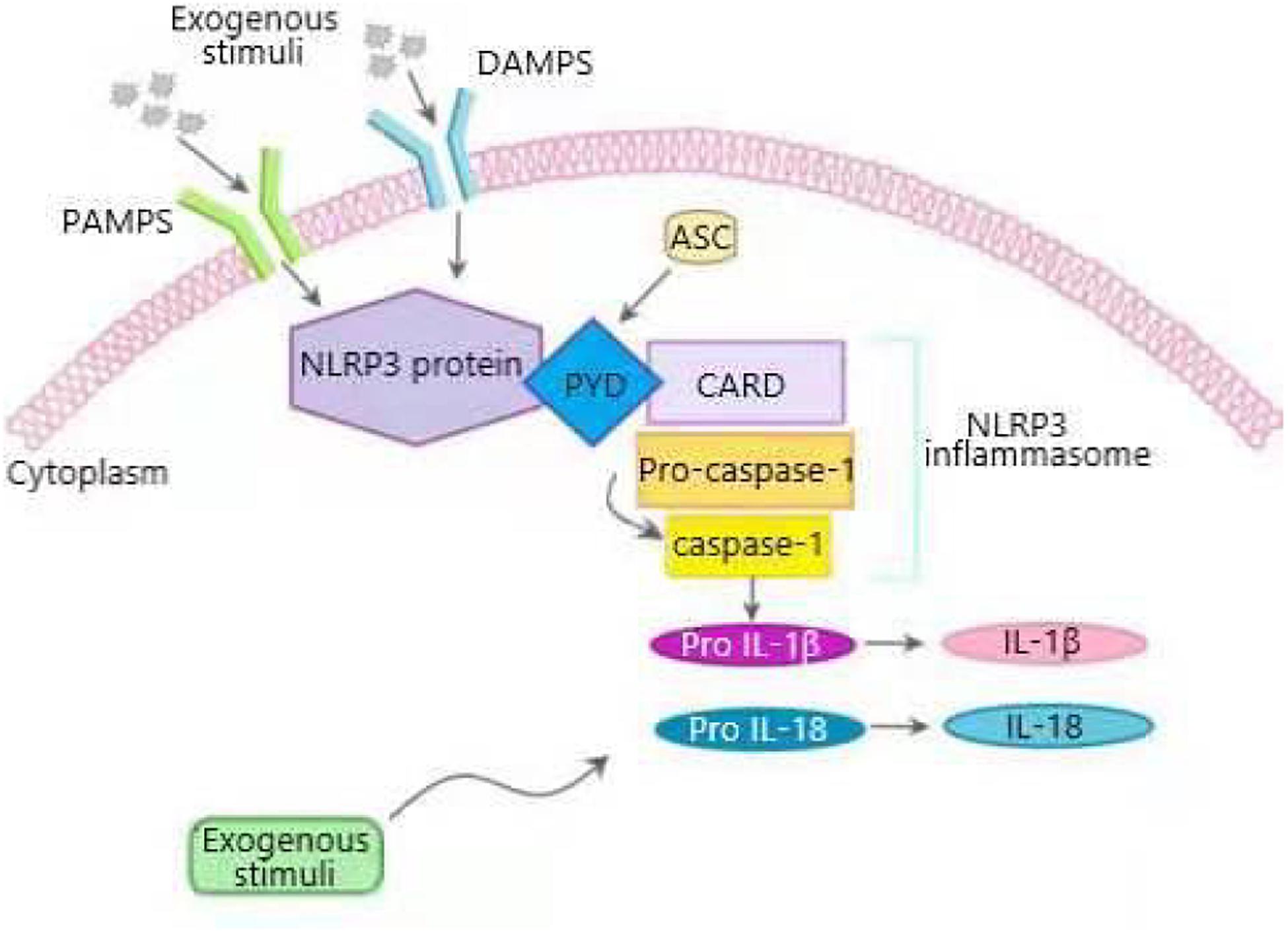
Figure 2. Overview of NLRP3 inflammasome activation process. NLRP3 inflammasome is composed of sensor NLRP3 protein, adapter apoptosis related spot like protein (ASC) containing N-terminal PYRIN-PAAD-DAPIN domain (PYD), C-terminal caspase recruitment domain (CARD), and effector pro-caspase-1. When cells are stimulated by pathogen related molecular patterns (PAMPS) and damage related molecular patterns (DAMPS), NLRP3 inflammasome is activated. Activated NLRP3 interacts with ASC through PYD domain, and pro-caspase-1 binds to ASC through CARD to form a large cytoplasmic complex, thus activating caspase-1. Active caspase-1 cleaves proinflammatory cytokines interleukin-1 β (IL-1β), and IL-18 from their precursors into bioactive forms that induce inflammation.
In recent years, the prevalence of diabetes has increased significantly and become an important public health problem in the world (Balakumar et al., 2016). An important characteristic of diabetes is impaired wound healing (Falanga, 2005). It has been reported that NLRP3 inflammasome-mediated inflammatory response in macrophage is one of the key factors of impaired wound healing in diabetic mice (Bitto et al., 2014). Dai et al. (2017) found that high glucose (HG) activated NLRP3 inflammasome, increased the level of IL-1β and ROS production, and suppressed autophagy by decreasing the protein expression of LC3-II and increasing the protein expression of p62 in macrophages from human diabetic wound. Pre-treatment with resveratrol (an autophagy agonist) counteracted the effects of HG, suggesting that autophagy mediated the HG-induced NLRP3 inflammasome and the ROS production was involved in autophagy-associated NLRP3 inflammasome activation. Moreover, the application of ROS scavenger NAC notably inhibited HG-induced NLRP3 inflammasome, indicating that enhancing autophagy supressed HG-induced NLRP3 inflammasome through reducing ROS production. Autophagy can selectively remove the damaged mitochondria from which ROS mainly comes (Ding et al., 2014; Yang et al., 2014), therefore, it can be inferred that autophagy can inhibit ROS-mediated NLRP3 inflammasome activation through clearing damaged mitochondria, which suggests a new treatment strategy for diabetic wound healing. The relationship among autophagy, ROS and NLRP3 inflammasome needs further study.
Consistent with the above, ROS-mediated NLRP3 inflammasome activation is also involved in diabetic nephropathy (DN). DN is one of the major microvascular complications of diabetes, and has become a common cause of end-stage renal disease (Gao et al., 2016). Many studies have shown that autophagy and inflammation play a key role in the pathological process of DN (Kitada et al., 2011; Feng et al., 2016). Gao et al. (2019) found that in glomerular mesangial cells stimulated by HG, the levels of NLRP3 inflammasome, IL-1β and ROS were increased, indicating that ROS-mediated NLRP3 inflammasome was activated to promote the development of DN. HG promoted autophagy in a short period (<12 h), but inhibited autophagy in a long period (72 h). Meanwhile, HG increased the level of ROS, NLRP3 inflammasome and IL-1β in a time-dependent manner, moreover, treatment cells with rapamycin (a specific activator of autophagy) for 72 h decreased the levels of NLRP3 inflammasome, IL-1β and ROS, suggesting that HG-regulated autophagy negatively regulated ROS-mediated NLRP3 inflammasome. In summary, in a short period of treatment, HG-induced autophagy inhibits ROS-mediated NLRP3 inflammasome and improve DN, while a long period of treatment, HG inhibits autophagy to activates ROS-mediated NLRP3 inflammasom to aggravate DN. The reason may be that HG causes mitochondrial damage, which induces autophagy to clear the damaged mitochondria-derived ROS. In a short period of time, the inhibition of HG on autophagy was less than the induction of ROS on autophagy, so the level of autophagy increased. With the extension of time, the induction of autophagy weakens, while high glucose inhibits autophagy to activate ROS-mediated NLRP3 inflammasome.
In addition to the above glomerular mesangial cell inflammation, NLRP3 inflammasome-mediated inflammation of tubulointerstitial also contributes to the development of DN (Chen et al., 2013; Hwang et al., 2017). Optineurin (OPTN), initially considered to be a regulator of NF-κβ signaling, is an important regulator of mitophagy in patients with DN, and reducing OPTN expression leads to premature senescence of renal tubular cells to promote DN (Slowicka et al., 2016; Chen et al., 2018). The results of Chen et al. (2019) showed that the expression of optineurin (OPTN) was reduced and correlated negatively with NLRP3 inflammasome activation in patients with DN. In primary mouse renal tubular epithelial cells (RTECs) treated with HG, the expression level of OPTN significantly decreased, the level of NLRP3 expression, cleaved caspase-1, and IL-1β, and the release of IL-1β increased. Moreover, the expression of mitochondrial fission protein Drp1 increased, and the expression of mitochondrial fusion protein Mitofusin 2 and mitochondrial membrane potential decreased, indicating that HG inhibited OPTN, activated NLRP3 inflammasome-mediated inflammation and promoted mitochondrial dysfunction. While overexpression of OPTN in RTECs counteracted the HG effects on OPTN and NLRP3 inflammasome-mediated inflammation, and silencing of OPTN had the opposite result, suggesting that OPTN inhibited HG-induced activation of NLRP3 inflammasome. HG impaired mitophagy by decreasing the level of LC3II in RTECs. The mitophagy-specific inhibitor (Mdivi-1) could activate NLRP3 inflammasome in the presence of HG, while the autophagy agonist (Torin) had the opposite effect, indicating that mitophagy supressed NLRP3 inflammasome activation induced by HG. In addition, overexpression of OPTN notably increased mitophagy and silencing of OPTN induces mtROS production and activation of NLRP3 inflammasome. Collectively, it can be inferred that OPTN inhibited HG-induced NLRP3 inflammasome by enhancing mitophagy-mediated clearance of ROS. The mechanism of OPTN regulating mitophagy, especially the signal pathway, needs to be further elucidated.
On the contrary to autophagy-mediated regulation of the NLRP3 inflammasome, NLRP3 inflammasome also can regulate autophagy. In podocytes of DN mice induced by high-fat diet/streptozotocin (HFD/STZ), the number of autophagosomes decreased and the level of NLRP3 inflammasome expression and renal proinflammatory cytokines such as IL-1β increased, meanwhile, the similar results were obtained in human DN biopsies, suggesting that podocyte autophagy was inhibited and NLRP3 inflammasome was activated. In podocyte, with the activation of NLRP3 inflammasome by LPS plus ATP, the ratio of LC3II/I, the expression level of beclin 1 and nephrin (which meant podocyte injury), and the formation of autophagosomes decreased, indicating that podocyte autophagy is inhibited by the activation of NLRP3 inflammasome to aggravate the damage of podocyte. While silencing NLRP3 by siRNA had the opposite results. Therefore, appropriate changes of autophagy and inflammasome may potentially improve DN (Hou et al., 2020). The specific mechanism of NLRP3 negatively regulating autophagy needs to be further explored.
Diabetic cardiomyopathy (DCM) is an important complication of diabetes and associated with inflammation, oxidative stress, impaired calcium homeostasis and mitochondrial damage (Huynh et al., 2014; Kobayashi and Liang, 2015). Autophagy and NLRP3 inflammasome have been reported to be involved in the pathological process of DCM (Xie et al., 2011; Luo et al., 2014). Visceral adipose tissue derived serine protease inhibitor (vaspin), firstly discovered in 2005, is an adipokine, which has significant anti-inflammatory effect (Li et al., 2008). In STZ-induced diabetes rat model, Vaspin improved cardiac function, cardiomyocyte apoptosis, myocardial tissue morphology and mitochondrial morphology to alleviate DCM. Mechanistic studies of the above effects revealed that LC3II/I ratio decreased and p62 expression increased in diabetic hearts, while Vaspin treatment abrogated the change to promote autophagy. Moreover, Vaspin also suppressed NLRP3 inflammasome through reducing the expression level of NLRP3 inflammasome and IL-1β. In HG-induced H9C2 cells, treatment with the autophagy inhibitor (3-MA) abolished the inhibitory effects of Vaspin on NLRP3 inflammasome, indicating that Vaspin alleviated STZ-induced myocardial injury by suppressing NLRP3 inflammasome activation via promoting autophagy. In addition, Vaspin inhibits ROS production and mitochondrial membrane depolarization in HG-induced H9C2 cells (Li et al., 2019). It can be inferred that there may be two mechanisms of myocardial protection in the above process: one is that Vaspin-induced autophagy can clear the damaged mitochondria to reduce the production of ROS to inhibit NLRP3 inflammasome; another is Vaspin improve damaged mitochondria to reduce the production of ROS to inhibit NLRP3 inflammasome.
Obesity causes chronic low-grade inflammation, leading to insulin resistance, which is characteristic of type 2 diabetes mellitus (Xu et al., 2003; Arkan et al., 2005; Shoelson et al., 2007). It has been reported that NLRP3 inflammasome perceives obesity related risk signals and participate in inflammation and insulin resistance caused by obesity (Vandanmagsar et al., 2011). Berberine (BBR) is a natural plant product and beneficial to diabetes and dyslipidemia (Kong et al., 2004; Turner et al., 2008). Zhou et al. (2017) found that BBR could significantly inhibit the activation of NLRP3 inflammasome and the release of IL-1β induced by saturated fatty acid palmitate (PA) in bone marrow-derived macrophages (BMDMs). Moreover, it also significantly increased the autophagy level and decreased mitochondrial ROS production in LPS plus PA-treated macrophages, while Beclin1 (an autophagy marker gene) knockout and 3-MA reversed the inhibitory effect of BBR on NLRP3 inflammasome and mitochondrial ROS production, indicating that BBR suppressed NLRP3 inflammasome-mediated inflammation and mitochondrial ROS production through enhancing autophagy. In addition, BBR could increase phosphorylation of AMPK (adenosine activated protein kinase) and caused a decrease of mammalian target of rapamycin (mTOR) in LPS plus PA-treated BMDMs, AMPK inhibitor, Ara-A, blocked most of the effects of BBR, suggesting that AMPK signal may be involved in BBR-induced autophagy. The similar effects of BBR were obtained in a HFD induced insulin resistance model. In conclusion, BBR inhibit obesity-induced inflammation and insulin resistance by activating AMPK dependent autophagy through suppressing mitochondrial ROS production in adipose tissue macrophages, which needs to be further studied. Targeting the effect of autophagy on NLRP3 with oral small molecule compound BBR might improve insulin resistance. Similar to the above results, our previous studies have shown that enhancing autophagy by hydrogen sulfide (H2S) suppresses NLRP3 inflammasome through AMPK-mTOR pathway in L02 cells induced by oleic acid (Wang et al., 2019). AMPK-mTOR pathway is an important signaling pathway for the interaction between autophagy and NLRP3 inflammasome.
High energy diets rich in fat and high sugar leads to low-grade systemic inflammatory response and increases the risk of cardiovascular disease (CVD) (Minihane et al., 2015). NLRP3 inflammasome-mediated inflammation plays an important role in the cardiomyopathy of rats treated with HFD and STZ (Luo et al., 2014). Genetic ablation of NLRP3 or inhibition of NLRP3 by MCC950 could relieve high-sugar diet (HSD), HFD or high sugar/fat diet induced obesity, cardiomyocyte apoptosis and inflammation, and improve the antioxidant capacity to ameliorate cardiac injury. Mechanistic studies revealed that genetic ablation of NLRP3 or inhibition of NLRP3 by MCC950 could induce autophagy by increasing LC3-II level and decreasing p62 level (Pavillard et al., 2017). HFD-induced obesity can impair autophagy in cardiomyocytes (He et al., 2012), while the inhibition of NLRP3-inflammasome and promotion of autophagy can improve HFD-induced cardiac injury (Abderrazak et al., 2015). Collectively, it can be inferred that inhibition of NLRP3-inflammasome improve HSD, HFD, or high sugar/fat diet induced heart damage through promoting autophagy, which needs to be further explored. It is predicted that therapeutic targetting of autophagy/NLRP3 inflammasomes can improve obesogenic diet-induced heart injury.
Uric acid nephropathy (UAN) is one of the most common metabolic diseases, which leads to kidney damage (Wu et al., 2012; Hou et al., 2014). Urate can activate NLRP3 inflammasome and NLRP3 inflammasome-mediated inflammation is involved in kidney injury (Wang et al., 2012). Hu et al. (2018) found that uric acid activated NLRP3 inflammasome, promoted caspase-1 activation, and induced renal inflammation, leading to the generation and development of UAN. Weicao, a traditional Chinese medicine preparation, could reduce uric acid, improve proteinuria and renal insufficiency and reduce renal tissue crystals to avoid renal interstitial fibrosis. It also reduced the levels of NLRP3 inflammasome, IL-18 and IIL-1β, and significantly increased the levels of beclin-1, LC3-II, and LC3-II/LC3-I ratio, suggesting that Weicao inhibited NLRP3 inflammasome-mediated inflammation and promoted autophagy to improve UAN. In addition, autophagy inhibitor 3-MA abolished the effects of Weicao on NLRP3 inflammasome, suggesting autophagy mediated the inhibitory effect of Weicao on NLRP3 inflammasome activation. NLRP3 inflammasome activator, ATP, blocked the effects of Weicao capsule on autophagy, indicating that NLRP3 inflammasome participated in Weicao induction of autophagy. The similar results were obtained in vitro. Collectively, Weicao could ameliorate renal injury of UAV rats through inhibiting NLRP3 inflammasome and inducing autophagy. It is predicted that therapeutic targetting of autophagy/NLRP3 inflammasomes can ameliorate UAN. The relationship between autophagy and NLRP3 inflammasome is very complex, which needs to be further studied.
Endothelial cell senescence is an important factor in the pathogenesis of senile vascular diseases, which has been observed in diabetic CVDs (Yokoi et al., 2006). Studies have shown that autophagy is closely related to the accelerated aging of vascular endothelial cells, and enhanced autophagy may have a strong anti-aging effect (Wohlgemuth et al., 2014; Lee et al., 2015). Purple sweet potato pigment (PSPC), a flavonoid compound isolated from purple sweet potato, can inhibit the aging of endothelial cells of diabetic patients (Sun et al., 2015). Sun et al. (2019) found that PSPC promoted autophagy by increasing LC3 II and decreasing p62 level and suppressed the premature senescence of endothelial cells induced by HG. The similar results were obtained in diabetic mice in vivo. Rapamycin, an autophagy promotor, inhibited HG-induced endothelial cell senescence, while 3-MA accelerated endothelial cell senescence, indicating that autophagy mediated PSPC suppression of endothelial cell senescence. Moreover, PSPC and Rapamycin suppressed HG-induced NLRP3 inflammasome, and 3-MA abolished the inhibitory effect of PSPC on NLRP3 inflammasome, suggesting that PSPC suppressed NLRP3 inflammasome by promoting autophagy. P62, an autophagic receptor, acts as a molecular bridge to deliver substrates to autophagosomes (Seibenhener et al., 2004). P62 binded to NLRP3 inflammasome and transported it to lysosome for degradation. In the senescent endothelium of diabetic mice, HG impaired autophagy to inhibit P62-NLRP3 inflammasome degradation in lysosome, while PSPC had the opposite effects, which was the mechanism that PSPC inhibits NLRP3 inflammasome by promoting autophagy (Sun et al., 2019).
Nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 inflammasome-mediated inflammation disorder is closely related to aging-related metabolic disorders (Finkel, 2015; Cordero et al., 2018). Marin-Aguilar et al. (2020) found that MCC950, a specific NLRP3 inhibitor, could reduce body weight, increase insulin sensitivity, improve hepatic metabolic dysfunction by reducing hepatic transaminases, lactate dehydrogenase, and creatine phosphokinase and alleviate steatosis and fibrosis in aged mice. Mechanistic studies revealed that MCC950 inhibited PI3K/Akt/mTOR pathway and enhanced autophagy by increasing LC3-II level and decreasing p62 level to improve the metabolism and liver dysfunction of old mice in vivo and in vitro. It can be inferred that NLRP3 inflammasome inhibition with MCC950 improves metabolism disorder via promoting autophagy through inhibiting PI3K/Akt/mTOR pathway in aged mice, which needs to be further studied by using autophagy inhibitors and the activator of PI3K/Akt/mTOR pathway.
Evidence indicates that mild chronic inflammation, including adipose tissue inflammation, contributes to metabolic disorders and is associated with many metabolic diseases, such as type 2 diabetes mellitus, insulin resistance and obesity (Weisberg et al., 2003; Lumeng and Saltiel, 2011; Harte et al., 2013). IL-1β activated by NLRP3 inflammasome can worse insulin resistance in human adipocytes (Gao et al., 2014), moreover, inhibition of IL-1β can reduce hyperglycemia in obese/diabetic rats (Ehses et al., 2009). So, inhibition of NLRP3 inflammasome-mediated production of IL-1β can improve the metabolic disorder associated with adipose tissue inflammation. AEDC is a cycloartane triterpenoid isolated from the whole plant of Amanita vaginata (Fang et al., 2019). Zhang et al. (2020) found that AEDC suppressed LPS plus ATP-induced NLRP3 inflammasome activation and IL-1β secretion in THP-1 macrophages. Moreover, LPS plus ATP notably decreased the protein levels of Atg5, Atg7, beclin 1 and the ratio of LC3-II/LC3-I and increased the level of p62, while AEDC had opposite effects, indicating that AEDC restored autophagy impaired by LPS plus ATP. 3-MA abolished the AEDC suppression of NLRP3 inflammasome activation induced by LPS plus ATP, suggesting that AEDC inhibited LPS plus ATP-induced NLRP3 inflammasome activation by promoting autophagy. Mechanistic studies showed that AEDC increased the expression level of NAD+-dependent deacetylase sirtuin-3 (SIRT3) deacetylase and enhanced its deacetylation activity to restore mitochondrial dysfunction induced by LPS plus ATP. 3-TYP, a selective SIRT3 inhibitor, reversed AEDC effect on autophagy, indicating that AEDC inhibited NLRP3 inflammasome activation to ameliorate inflammation-related metabolic disorders by promoting autophagy via SIRT3. Here, autophagy still suppresses NLRP3 inflammasome by clearing damaging mitochondria derived ROS.
The interplay between autophagy and NLRP3 inflammasome plays an important role in metabolic disorders. In this review, there are two mechanisms of the effects of autophagy on NLRP3 inflammasome in metabolic disorders: One is that autophagy suppresses NLRP3 inflammasome by inhibiting ROS production through scavenging damaged mitochondria. ROS released from mitochondria can activate the NF-κβ pathway to promote the transcription of NLRP3 and pro-IL-1β, thus activating NLRP3 inflammasome. The other is that autophagy can isolate and promote the degradation of inflammasome components such as pro-IL-1β, NLRP3, caspase-1, and ASC (Figure 3). Generally speaking, in addition to the above two ways, autophagy also inhibits NLRP3 inflammasome by phosphorylating NLRP3 (Cao et al., 2019). Whether autophagy can influence NLRP3 inflammasome by phosphorylating NLRP3 in metabolic disorders remains to be clarified.
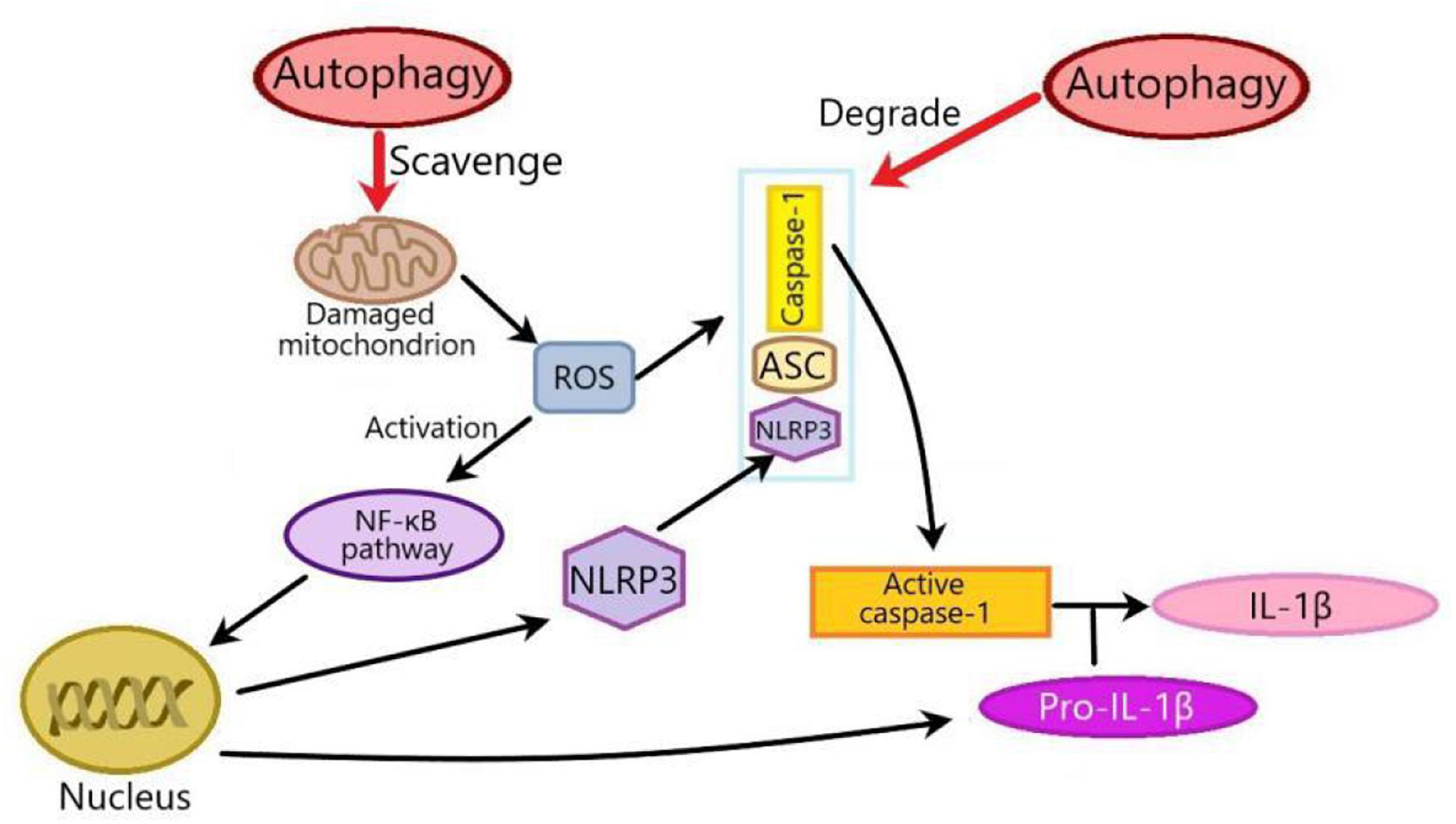
Figure 3. Two mechanisms of the effects of autophagy on NLRP3 inflammasome in metabolic disorders. There are two mechanisms of the effects of autophagy on NLRP3 inflammasome: one is that autophagy inhibits NLRP3 inflammasome by scavenging ROS from damaged mitochondria. Mitochondria-derived ROS can activate NF-κβ pathway to promote the transcription of NLRP3 and pro-IL-1β, thus activating NLRP3 inflammasome. The other is that autophagy inhibits NLRP3 inflammasome through degrading inflammasome components such as pro-IL-1β, NLRP3, caspase-1, or ASC. ROS, Reactive oxygen species; NF-κβ, nuclear factor kappa-B; ASC, apoptosis-associated speck-like protein.
Although the interplay between autophagy and NLRP3 inflammasome has potential therapeutic value in metabolic disorders, its mechanism has not been fully explained. For example, in metabolic disorders, does autophagy promote the activation of the NLRP3 inflammasome? What is the mechanism of NLRP3 inflammasome acting on autophagy? And are there any side effects of targeting autophagy/NLRP3 inflammasome to improve metabolic disorder? With the deepening of the research, targeting autophagy/NLRP3 inflammasome will provide a new method for the treatment of metabolic disorders related diseases.
HW devised, wrote, and funded the review. SL wrote and funded the review. XL drew the pictures of the review.
This work was supported by grants from Key Scientific and Technological Projects in Henan Province, China (Grant No. 202102310153) and the National Natural Science Foundation of China (Grant No. 81971280).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abderrazak, A., Couchie, D., Mahmood, D. F. D., Elhage, R., Vindis, C., Laffargue, M., et al. (2015). Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE(2).Ki mice fed a high-fat diet. Circulation 131, 1061–1070. doi: 10.1161/circulationaha.114.013730
Amaravadi, R. K., Lippincott-Schwartz, J., Yin, X. M., Weiss, W. A., Takebe, N., Timmer, W., et al. (2011). Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 17, 654–666. doi: 10.1158/1078-0432.ccr-10-2634
Arkan, M. C., Hevener, A. L., Greten, F. R., Maeda, S., Li, Z. W., Long, J. M., et al. (2005). IKK-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 11, 191–198. doi: 10.1038/nm1185
Ashrafi, G., and Schwarz, T. L. (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20, 31–42. doi: 10.1038/cdd.2012.81
Balakumar, P., Maung-U, K., and Jagadeesh, G. (2016). Prevalence and prevention of cardiovascular disease and diabetes mellitus. Pharmacol. Res. 113, 600–609. doi: 10.1016/j.phrs.2016.09.040
Barbe, F., Douglas, T., and Saleh, M. (2014). Advances in Nod-like receptors (NLR) biology. Cytokine Growth Factor Rev. 25, 681–697. doi: 10.1016/j.cytogfr.2014.07.001
Behrends, C., Sowa, M. E., Gygi, S. P., and Harper, J. W. (2010). Network organization of the human autophagy system. Nature 466, 68–76. doi: 10.1038/nature09204
Biasizzo, M., and Kopitar-Jerala, N. (2020). Interplay between NLRP3 inflammasome and autophagy. Front. Immunol. 11:591803. doi: 10.3389/fimmu.2020.591803
Bitto, A., Altavilla, D., Pizzino, G., Irrera, N., Pallio, G., Colonna, M. R., et al. (2014). Inhibition of inflammasome activation improves the impaired pattern of healing in genetically diabetic mice. Br. J. Pharmacol. 171, 2300–2307. doi: 10.1111/bph.12557
Cao, Z., Wang, Y., Long, Z., and He, G. (2019). Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim. Biophys. Sin. (Shanghai) 51, 1087–1095. doi: 10.1093/abbs/gmz098
Chen, K., Dai, H., Yuan, J., Chen, J., Lin, L., Zhang, W., et al. (2018). Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 9:105.
Chen, K., Feng, L., Hu, W., Chen, J., Wang, X., Wang, L., et al. (2019). Optineurin inhibits NLRP3 inflammasome activation by enhancing mitophagy of renal tubular cells in diabetic nephropathy. FASEB J. 33, 4571–4585. doi: 10.1096/fj.201801749rrr
Chen, K., Zhang, J., Zhang, W., Zhang, J., Yang, J., Li, K., et al. (2013). ATP-P2X4 signaling mediates NLRP3 inflammasome activation: a novel pathway of diabetic nephropathy. Int. J. Biochem. Cell Biol. 45, 932–943. doi: 10.1016/j.biocel.2013.02.009
Cheon, S. Y., Kim, H., Rubinsztein, D. C., and Lee, J. E. (2019). Autophagy, cellular aging and age-related human diseases. Exp. Neurobiol. 28, 643–657. doi: 10.5607/en.2019.28.6.643
Cordero, M. D., Williams, M. R., and Ryffel, B. (2018). AMP-activated protein kinase regulation of the NLRP3 inflammasome during aging. Trends Endocrinol. Metab. 29, 8–17. doi: 10.1016/j.tem.2017.10.009
Dai, J., Zhang, X., Li, L., Chen, H., and Chai, Y. (2017). Autophagy inhibition contributes to ROS-producing NLRP3-dependent inflammasome activation and cytokine secretion in high glucose-induced macrophages. Cell. Physiol. Biochem. 43, 247–256. doi: 10.1159/000480367
Debnath, J. (2011). The multifaceted roles of autophagy in tumors-implications for breast cancer. J. Mammary Gland Biol. Neoplasia 16, 173–187. doi: 10.1007/s10911-011-9223-3
Ding, Z., Liu, S., Wang, X., Dai, Y., Khaidakov, M., Deng, X., et al. (2014). LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: implications in atherogenesis. Cardiovasc. Res. 103, 619–628. doi: 10.1093/cvr/cvu114
Ehses, J. A., Lacraz, G., Giroix, M. H., Schmidlin, F., Coulaud, J., Kassis, N., et al. (2009). IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proc. Natl. Acad. Sci. U.S.A. 106, 13998–14003. doi: 10.1073/pnas.0810087106
Elliott, E. I., and Sutterwala, F. S. (2015). Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 265, 35–52. doi: 10.1111/imr.12286
Falanga, V. (2005). Wound healing and its impairment in the diabetic foot. Lancet 366, 1736–1743. doi: 10.1016/s0140-6736(05)67700-8
Fang, Z. J., Zhang, T., Chen, S. X., Wang, Y. L., Zhou, C. X., Mo, J. X., et al. (2019). Cycloartane triterpenoids from Actaea vaginata with anti-inflammatory effects in LPS-stimulated RAW264.7 macrophages. Phytochemistry 160, 1–10. doi: 10.1016/j.phytochem.2019.01.003
Feng, H., Gu, J., Gou, F., Huang, W., Gao, C., Chen, G., et al. (2016). High glucose and lipopolysaccharide prime NLRP3 inflammasome via ROS/TXNIP pathway in mesangial cells. J. Diabetes Res. 2016:6973175.
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. doi: 10.1038/cr.2013.168
Gao, C., Chen, J., Fan, F., Long, Y., Tang, S., Jiang, C., et al. (2019). RIPK2-mediated autophagy and negatively regulated ROS-NLRP3 inflammasome signaling in GMCs stimulated with high glucose. Mediators Inflamm. 2019:6207563.
Gao, D., Madi, M., Ding, C., Fok, M., Steele, T., Ford, C., et al. (2014). Interleukin-1 beta mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am. J. Physiol. Endocrinol. Metab. 307, E289–E304.
Gao, H. X., Regier, E. E., and Close, K. L. (2016). International diabetes federation world diabetes congress 2015. J. Diabetes 8, 300–304.
Garcia-Huerta, P., Troncoso-Escudero, P., Jerez, C., Hetz, C., and Vidal, R. L. (2016). The intersection between growth factors, autophagy and ER stress: a new target to treat neurodegenerative diseases? Brain Res. 1649, 173–180. doi: 10.1016/j.brainres.2016.02.052
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Gomes, L. R., Menck, C. F. M., and Cuervo, A. M. (2017). Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 13, 928–940. doi: 10.1080/15548627.2017.1293767
Harte, A. L., Tripathi, G., Piya, M. K., Barber, T. M., Clapham, J. C., Al-Daghri, N., et al. (2013). NF kappa B as a potent regulator of inflammation in human adipose tissue, Influenced by depot, adiposity, T2DM status, and TNF alpha. Obesity 21, 2322–2330. doi: 10.1002/oby.20336
He, C., Bassik, M. C., Moresi, V., Sun, K., Wei, Y., Zou, Z., et al. (2012). Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481, 511–515. doi: 10.1038/nature10758
Hou, S. X., Zhu, W. J., Pang, M. Q., Jeffry, J., and Zhou, L. L. (2014). Protective effect of iridoid glycosides from Paederia scandens (LOUR.) MERRILL (Rubiaceae) on uric acid nephropathy rats induced by yeast and potassium oxonate. Food Chem. Toxicol. 64, 57–64. doi: 10.1016/j.fct.2013.11.022
Hou, Y., Lin, S., Qiu, J., Sun, W., Dong, M., Xiang, Y., et al. (2020). NLRP3 inflammasome negatively regulates podocyte autophagy in diabetic nephropathy. Biochem. Biophys. Res. Commun. 521, 791–798. doi: 10.1016/j.bbrc.2019.10.194
Hu, J., Wu, H., Wang, D., Yang, Z., Zhuang, L., Yang, N., et al. (2018). Weicao capsule ameliorates renal injury through increasing autophagy and NLRP3 degradation in UAN rats. Int. J. Biochem. Cell Biol. 96, 1–8. doi: 10.1016/j.biocel.2018.01.001
Huynh, K., Bernardo, B. C., Mcmullen, J. R., and Ritchie, R. H. (2014). Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol. Ther. 142, 375–415. doi: 10.1016/j.pharmthera.2014.01.003
Hwang, S., Park, J., Kim, J., Jang, H. R., Kwon, G. Y., Huh, W., et al. (2017). Tissue expression of tubular injury markers is associated with renal function decline in diabetic nephropathy. J. Diabetes Complications 31, 1704–1709. doi: 10.1016/j.jdiacomp.2017.08.009
Jo, E. K., Kim, J. K., Shin, D. M., and Sasakawa, C. (2016). Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 13, 148–159. doi: 10.1038/cmi.2015.95
Kaushik, S., and Cuervo, A. M. (2012). Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 22, 407–417. doi: 10.1016/j.tcb.2012.05.006
Kitada, M., Takeda, A., Nagai, T., Ito, H., Kanasaki, K., and Koya, D. (2011). Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp. Diabetes Res. 2011:908185.
Kobayashi, S., and Liang, Q. (2015). Autophagy and mitophagy in diabetic cardiomyopathy. Biochim. Biophys. Acta Mol. Basis Dis. 1852, 252–261. doi: 10.1016/j.bbadis.2014.05.020
Kong, W. J., Wei, J., Abidi, P., Lin, M. H., Inaba, S., Li, C., et al. (2004). Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat. Med. 10, 1344–1351. doi: 10.1038/nm1135
Lee, M. J., Kim, E. H., Lee, S. A., Kang, Y. M., Jung, C. H., Yoon, H. K., et al. (2015). Dehydroepiandrosterone prevents linoleic acid-induced endothelial cell senescence by increasing autophagy. Metab. Clin. Exp. 64, 1134–1145. doi: 10.1016/j.metabol.2015.05.006
Li, Q., Chen, R., Moriya, J., Yamakawa, J., Sumino, H., Kanda, T., et al. (2008). A novel adipocytokine, visceral adipose tissue-derived serine protease inhibitor (vaspin), and obesity. J. Int. Med. Res. 36, 625–629. doi: 10.1177/147323000803600402
Li, X., Ke, X., Li, Z., and Li, B. (2019). Vaspin prevents myocardial injury in rats model of diabetic cardiomyopathy by enhancing autophagy and inhibiting inflammation. Biochem. Biophys. Res. Commun. 514, 1–8. doi: 10.1016/j.bbrc.2019.04.110
Liu, Y., and Levine, B. (2015). Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 22, 367–376. doi: 10.1038/cdd.2014.143
Lumeng, C. N., and Saltiel, A. R. (2011). Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 121, 2111–2117. doi: 10.1172/jci57132
Luo, B., Li, B., Wang, W., Liu, X., Xia, Y., Zhang, C., et al. (2014). NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One 9:e104771. doi: 10.1371/journal.pone.0104771
Marin-Aguilar, F., Castejon-Vega, B., Alcocer-Gomez, E., Lendines-Cordero, D., Cooper, M. A., De La Cruz, P., et al. (2020). NLRP3 inflammasome inhibition by MCC950 in aged mice improves health via enhanced autophagy and PPAR alpha activity. J. Gerontol. A Biol. Sci. Med. Sci. 75, 1457–1464. doi: 10.1093/gerona/glz239
Mclendon, P. M., Ferguson, B. S., Osinska, H., Bhuiyan, M. S., James, J., Mckinsey, T. A., et al. (2014). Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy. Proc. Natl. Acad. Sci. U.S.A. 111, E5178–E5186.
Meyers, A. K., and Zhu, X. (2020). The NLRP3 inflammasome: metabolic regulation and contribution to inflammaging. Cells 9:1808. doi: 10.3390/cells9081808
Minihane, A. M., Vinoy, S., Russell, W. R., Baka, A., Roche, H. M., Tuohy, K. M., et al. (2015). Low-grade inflammation, diet composition and health: current research evidence and its translation. Br. J. Nutr. 114, 999–1012. doi: 10.1017/s0007114515002093
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2011). “The role of Atg proteins in autophagosome formation,” in Annual Review Of Cell And Developmental Biology, Vol. 27, eds R. Schekman, L. Goldstein, and R. Lehmann, (Palo Alto, CA: Annual reviews), 107–132. doi: 10.1146/annurev-cellbio-092910-154005
Mridha, A. R., Wree, A., Robertson, A. A. B., Yeh, M. M., Johnson, C. D., Van Rooyen, D. M., et al. (2017). NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 66, 1037–1046. doi: 10.1016/j.jhep.2017.01.022
Murrow, L., and Debnath, J. (2013). Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Ann. Rev. Pathol. 8, 105–137. doi: 10.1146/annurev-pathol-020712-163918
Ney, P. A. (2015). Mitochondrial autophagy: origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta Mol. Cell Res. 1853, 2775–2783. doi: 10.1016/j.bbamcr.2015.02.022
O’connor, W., Harton, J. A., Zhu, X. S., Linhoff, M. W., and Ting, J. P. Y. (2003). Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF-kappa B suppressive properties. J. Immunol. 171, 6329–6333. doi: 10.4049/jimmunol.171.12.6329
Oku, M., and Sakai, Y. (2018). Three distinct types of microautophagy based on membrane dynamics and molecular machineries. Bioessays 40:e1800008.
Pavillard, L. E., Canadas-Lozano, D., Alcocer-Gomez, E., Marin-Aguilar, F., Pereira, S., Robertson, A. A. B., et al. (2017). NLRP3-inflammasome inhibition prevents high fat and high sugar diets-induced heart damage through autophagy induction. Oncotarget 8, 99740–99756. doi: 10.18632/oncotarget.20763
Prochnicki, T., Mangan, M. S., and Latz, E. (2016). Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Research 5, F1000FacultyRev–1469.
Qiu, D. M., Wang, G. L., Chen, L., Xu, Y. Y., He, S., Cao, X. L., et al. (2014). The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer 14:327. doi: 10.1186/1471-2407-14-327
Sarparanta, J., Garcia-Macia, M., and Singh, R. (2017). Autophagy and mitochondria in obesity and type 2 diabetes. Curr. Diabetes Rev. 13, 352–369.
Seibenhener, M. L., Babu, J. R., Geetha, T., Wong, H. C., Krishna, N. R., and Wooten, M. W. (2004). Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 24, 8055–8068. doi: 10.1128/mcb.24.18.8055-8068.2004
Shoelson, S. E., Herrero, L., and Naaz, A. (2007). Obesity, inflammation, and insulin resistance. Gastroenterology 132, 2169–2180.
Sir, D., Tian, Y., Chen, W. L., Ann, D. K., Yen, T. S. B., and Ou, J. H. J. (2010). The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. U.S.A. 107, 4383–4388. doi: 10.1073/pnas.0911373107
Slowicka, K., Vereecke, L., and Van Loo, G. (2016). Cellular functions of optineurin in health and disease. Trends Immunol. 37, 621–633. doi: 10.1016/j.it.2016.07.002
Sokolova, M., Ranheim, T., Louwe, M. C., Halvorsen, B., Yndestad, A., and Aukrust, P. (2019). NLRP3 inflammasome: a novel player in metabolically induced inflammation-potential influence on the myocardium. J. Cardiovasc. Pharmacol. 74, 276–284. doi: 10.1097/fjc.0000000000000704
Strowig, T., Henao-Mejia, J., Elinav, E., and Flavell, R. (2012). Inflammasomes in health and disease. Nature 481, 278–286. doi: 10.1038/nature10759
Sun, C., Diao, Q., Lu, J., Zhang, Z., Wu, D., Wang, X., et al. (2019). Purple sweet potato color attenuated NLRP3 inflammasome by inducing autophagy to delay endothelial senescence. J. Cell. Physiol. 234, 5926–5939. doi: 10.1002/jcp.28003
Sun, C., Fan, S., Wang, X., Lu, J., Zhang, Z., Wu, D., et al. (2015). Purple sweet potato color inhibits endothelial premature senescence by blocking the NLRP3 inflammasome. J. Nutr. Biochem. 26, 1029–1040. doi: 10.1016/j.jnutbio.2015.04.012
Tooze, S. A., and Yoshimori, T. (2010). The origin of the autophagosomal membrane. Nat. Cell Biol. 12, 831–835. doi: 10.1038/ncb0910-831
Turner, N., Li, J. Y., Gosby, A., To, S. W. C., Cheng, Z., Miyoshi, H., et al. (2008). Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 57, 1414–1418. doi: 10.2337/db07-1552
Van Der Heijden, T., Kritikou, E., Venema, W., Van Duijn, J., Van Santbrink, P. J., Slutter, B., et al. (2017). NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler. Thromb. Vasc. Biol. 37, 1457–1461. doi: 10.1161/atvbaha.117.309575
Vandanmagsar, B., Youm, Y. H., Ravussin, A., Galgani, J. E., Stadler, K., Mynatt, R. L., et al. (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188. doi: 10.1038/nm.2279
Wang, C., Pan, Y., Zhang, Q. Y., Wang, F. M., and Kong, L. D. (2012). Quercetin and allopurinol ameliorate kidney injury in STZ-treated rats with regulation of renal NLRP3 inflammasome activation and lipid accumulation. PLoS One 7:e38285. doi: 10.1371/journal.pone.0038285
Wang, H., Zhong, P., and Sun, L. (2019). Exogenous hydrogen sulfide mitigates NLRP3 inflammasome-mediated inflammation through promoting autophagy via the AMPK-mTOR pathway. Biol. Open 8:bio043653. doi: 10.1242/bio.043653
Wei, C., Gao, J., Li, M., Li, H., Wang, Y., Li, H., et al. (2016). Dopamine D2 receptors contribute to cardioprotection of ischemic post-conditioning via activating autophagy in isolated rat hearts. Int. J. Cardiol. 203, 837–839.
Weisberg, S. P., Mccann, D., Desai, M., Rosenbaum, M., Leibel, R. L., and Ferrante, A. W. (2003). Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808.
Wohlgemuth, S. E., Calvani, R., and Marzetti, E. (2014). The interplay between autophagy and mitochondrial dysfunction in oxidative stress-induced cardiac aging and pathology. J. Mol. Cell. Cardiol. 71, 62–70.
Wu, X., Liu, L., Xie, H., Liao, J., Zhou, X., Wan, J., et al. (2012). Tanshinone IIA prevents uric acid nephropathy in rats through NF-kappa B inhibition. Planta Med. 78, 866–873.
Xie, Z., Lau, K., Eby, B., Lozano, P., He, C., Pennington, B., et al. (2011). Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 60, 1770–1778.
Xu, H. Y., Barnes, G. T., Yang, Q., Tan, Q., Yang, D. S., Chou, C. J., et al. (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 112, 1821–1830.
Yang, S., Xia, C., Li, S., Du, L., Zhang, L., and Zhou, R. (2014). Defective mitophagy driven by dysregulation of rheb and KIF5B contributes to mitochondrial reactive oxygen species (ROS)-induced nod-like receptor 3 (NLRP3) dependent proinflammatory response and aggravates lipotoxicity. Redox Biol. 3, 63–71.
Yokoi, T., Fukuo, K., Yasuda, O., Hotta, M., Miyazaki, J., Takemura, Y., et al. (2006). Apoptosis signal-regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes 55, 1660–1665.
Zhai, Y., Meng, X., Ye, T., Xie, W., Sun, G., and Sun, X. (2018). Inhibiting the NLRP3 inflammasome activation with MCC950 ameliorates diabetic encephalopathy in db/db mice. Molecules 23:522.
Zhang, T., Fang, Z., Linghu, K. G., Liu, J., Gan, L., and Lin, L. (2020). Small molecule-driven SIRT3-autophagy-mediated NLRP3 inflammasome inhibition ameliorates inflammatory crosstalk between macrophages and adipocytes. Br. J. Pharmacol. 177, 4645–4665.
Keywords: autophagy, NLRP3 inflammasome, glucose metabolic disorders, uric acid metabolic disorders, inflammation-related metabolic disorders
Citation: Lv S, Wang H and Li X (2021) The Role of the Interplay Between Autophagy and NLRP3 Inflammasome in Metabolic Disorders. Front. Cell Dev. Biol. 9:634118. doi: 10.3389/fcell.2021.634118
Received: 27 November 2020; Accepted: 03 February 2021;
Published: 16 March 2021.
Edited by:
Allen Liu, University of Michigan, United StatesReviewed by:
Neil J. Grimsey, University of Georgia, United StatesCopyright © 2021 Lv, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Honggang Wang, d2hnMTk3MTY3QHZpcC5oZW51LmVkdS5jbg==; d2hnMTk3NTMxNkBzaW5hLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.