
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 18 March 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.632946
This article is part of the Research TopicGenetic Mutations Associated with Ocular DiseasesView all 19 articles
 Wei Li1,2,3*†
Wei Li1,2,3*† Ning Qu4†
Ning Qu4† Jian-Kang Li5†
Jian-Kang Li5† Yu-Xin Li1,2,6Dong-Ming Han1,2Yi-Xi Chen7Le Tian1,2,6Kang Shao5Wen Yang5Zhuo-Shi Wang3,8Xuan Chen9Xiao-Ying Jin10
Yu-Xin Li1,2,6Dong-Ming Han1,2Yi-Xi Chen7Le Tian1,2,6Kang Shao5Wen Yang5Zhuo-Shi Wang3,8Xuan Chen9Xiao-Ying Jin10 Zi-Wei Wang1,2Chen Liang6Wei-Ping Qian4*Lu-Sheng Wang5*
Zi-Wei Wang1,2Chen Liang6Wei-Ping Qian4*Lu-Sheng Wang5* Wei He3,8*
Wei He3,8*Aims: To characterize the genetic landscape and mutation spectrum of patients with corneal dystrophies (CDs) in a large Han ethnic Chinese Cohort with inherited eye diseases (IEDs).
Methods: Retrospective study. A large IED cohort was recruited in this study, including 69 clinically diagnosed CD patients, as well as other types of eye diseases patients and healthy family members as controls. The 792 genes on the Target_Eye_792_V2 chip were used to screen all common IEDs in our studies, including 22 CD-related genes.
Results: We identified 2334 distinct high-quality variants on 22 CD-related genes in a large IEDs cohort. A total of 21 distinct pathogenic or likely pathogenic mutations were identified, and the remaining 2313 variants in our IED cohort had no evidence of CD-related pathogenicity. Overall, 81.16% (n = 56/69) of CD patients received definite molecular diagnoses, and transforming growth factor-beta-induced protein (TGFBI), CHTS6, and SLC4A11 genes covered 91.07, 7.14, and 1.79% of the diagnosed cases, respectively. Twelve distinct disease-associated mutations in the TGFBI gene were identified, 11 of which were previously reported and one is novel. Four of these TGFBI mutations (p.D123H, p.M502V, p.P501T, and p.P501A) were redefined as likely benign in our Han ethnic Chinese IED cohort after performing clinical variant interpretation. These four TGFBI mutations were detected in asymptomatic individuals but not in CD patients, especially the previously reported disease-causing mutation p.P501T. Among 56 CD patients with positive detected mutations, the recurrent TGFBI mutations were p.R124H, p.R555W, p.R124C, p.R555Q, and p.R124L, and the proportions were 32.14, 19.64, 14.29, 10.71, and 3.57%, respectively. Twelve distinct pathogenic or likely pathogenic mutations of CHTS6 were detected in 28 individuals. The recurrent mutations were p.Y358H, p.R140X, and p.R205W, and the proportions were 25.00, 21.43, and 14.29%, respectively. All individuals associated with TGFBI were missense mutations; 74.19% associated with CHTS6 mutations were missense mutations, and 25.81% were non-sense mutations. Hot regions were located in exons 4 and 12 of TGFBI individuals and located in exon 3 of CHTS6 individuals. No de novo mutations were identified.
Conclusion: For the first time, our large cohort study systematically described the variation spectrum of 22 CD-related genes and evaluated the frequency and pathogenicity of all 2334 distinct high-quality variants in our IED cohort. Our research will provide East Asia and other populations with baseline data from a Han ethnic population-specific level.
Corneal dystrophies (CDs) are genetically heterogeneous disorders characterized by the gradual accumulation of deposits within different corneal layers, resulting in changes in corneal transparency and refractive index (Bron, 1990).
Clinically, these diseases are divided into anatomical categories according to the specific corneal layer involved. According to the current International Committee for Classification of Corneal Dystrophies (IC3D), CDs can be divided into 4 categories and 22 subcategories. CDs can classify into one of the following anatomical categories (Weiss et al., 2008, 2015): (a) epithelial and subepithelial CDs; (b) epithelial–stromal transforming growth factor-beta-induced protein (TGFBI) CDs; (c) stromal CDs; and (d) endothelial CDs. At present, corneal transplants are the most effective method for the treatment of CDs. Due to lack of clinical symptoms, some patients may be misdiagnosed before phototherapeutic keratectomy (PTK) treatment or neglected before refractive surgery, which highlights the urgent need to understand the disease mechanism of CDs (Zeng et al., 2017).
Genetically, CDs are autosomal dominant, autosomal recessive or X-linked modes. Autosomal dominant inheritance accounts for most cases and is accompanied by a high degree of penetrance (Pieramici and Afshari, 2006). To date, studies have identified disease-causing mutations in 18 genes associated with CDs, many of which have established genotype–phenotype associations. For example, mutations in six genes (CHST6, OMIM 605294; UBIAD1, OMIM 611632; SLC4A11, OMIM 610206; PIKFYVE, OMIM 609414; TACSTD2, OMIM 137290; DCN, and OMIM 125255) have found a direct genetic association with macular corneal dystrophies (MCD), Schnyder CD (SCD), congenital hereditary endothelial dystrophy (CHED), fleck CD (FCD), gelatinous drop-like CD (GDLD), and congenital stromal CD (CSCD), respectively, (Zhang et al., 2013). At the same time, there are significant heterogeneities in distinct mutations of the same gene. For instance, according to the second edition of IC3D, five distinct TGFBI mutations (p.R124H, p.R555W, p.R124C, p.R555Q, and p.R124L) cause different types of CDs, including Granular corneal dystrophy, type 2; Granular corneal dystrophy, type 1; Lattice corneal dystrophy, type 1 (LCD1); Thiel-Behnke corneal dystrophy (TBCD); and Reis-Bücklers corneal dystrophy, respectively, (Weiss et al., 2008, 2015).
Panel-based targeted exon sequencing has proven to have excellent performance in the molecular diagnosis of heterogeneous genetic diseases. Studies have confirmed that targeted enrichment based on multi-gene panels are highly sensitive, accurate, and reproducible (Adams and Eng, 2018). A comprehensive overview of the genetic landscape associated with the CD phenotype have been provided based on sequencing hundreds of potentially related disease-causing genes. In this study, we conducted a comprehensive CD-related genetic mutation spectrum evaluation on a large IED group, including 69 patients with clinically diagnosed CDs. Our results can provide an accurate and reliable diagnosis, and comprehensively describe the detection and carrying of disease-causing mutations in the Chinese population. These findings provide a useful reference for the pre-clinical diagnosis and treatment of patients with suspected CDs, as well as carrier screening before PKT treatment or refractive surgery.
The Ethics Committee of the He Eye Specialists Hospital of He University approved the study, and the studies complied with the tenets of the Declaration of Helsinki. A large IED group and their available family members were recruited from the genetic department between July 2016 and December 2019, including 69 CD patients from 50 unrelated families diagnosed in the He Eye Specialists Hospital (Figure 1A). All members participating in the study collected 5 ml of peripheral blood and signed informed consent forms.
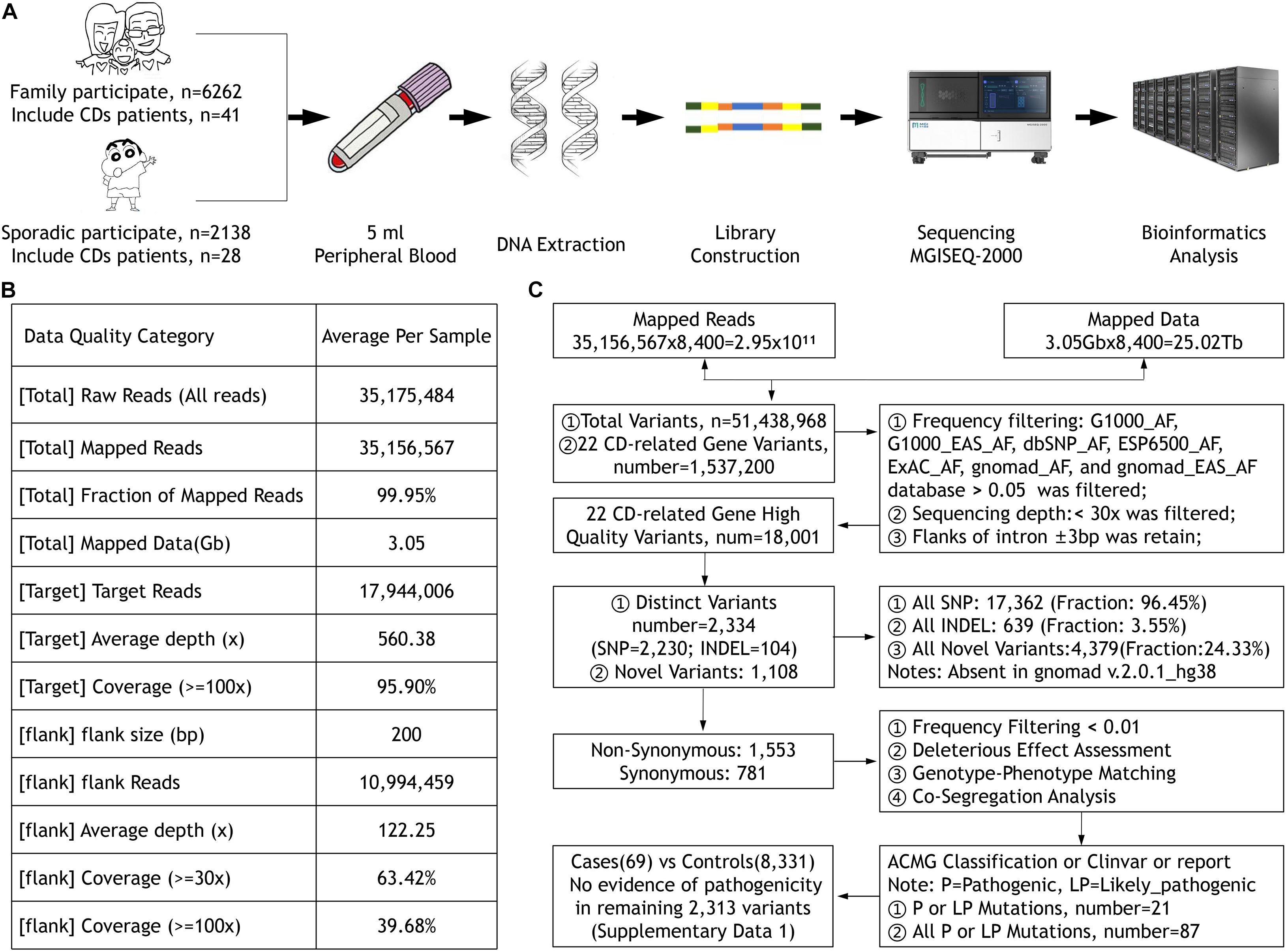
Figure 1. (A) The sample collection, DNA extraction and detection, library construction, and sequencing process. (B) The quality of the data. (C) The workflow of data processing and analysis.
Corneal dystrophies are initially classified according to their corneal phenotypic characteristics and anatomical invasion and then combined with genetic testing results to obtain a more accurate diagnosis. According to the second edition of IC3D classification, individuals who meet the diagnostic criteria of CDs were included in this study (Weiss et al., 2015). All participants received comprehensive, detailed clinical examinations and obtained a family history, previous surgical history, and a description of the patient’s symptoms. Clinical examination includes the following categories: best-corrected visual acuity, slit-lamp biomicroscopic, intraocular pressure (IOP, Goldmann tonometry), fundus autofluorescence, full-field electroretinography, typical in vivo confocal microscopy, and spectral-domain optical coherence tomography images recorded.
The classification of CD subtypes was based on phenotypic appearance and anatomical location, and the precise diagnostic criteria referred to the second edition of IC3D (Weiss et al., 2015). Patients with extraocular somatic cell defects or other ocular or developmental abnormalities were excluded from this study, and 69 patients with suspected congenital CDs were included.
All participants’ blood were collected and DNA were extracted using the FlexiGene DNA Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s protocol. Our high-throughput targeted enrichment method analyzes 792 genes. Supplementary Table 1A lists 792 genes used for common inherited eye diseases (IEDs). A custom-made capture panel (Target_Eye_792_V2 chip) was designed and produced by Beijing Genomics Institute (Shenzhen, China). The panel covers the exon-capture region of 792 genes associated with common IEDs and its flank ± 200 bp. Overall, the average depth of target region was more than 560.38×, that of the flank region was 122.25×, and the coverage of the target region greater than 100× was close to 96% using the MGISEQ-2000 (DNBSEQ-G400) platform (MGI, Inc., BGI-Shenzhen, China; Figure 1B). We used the Burrows–Wheeler aligner version 0.7.10 (BWA-MEM) to align sequence reads to the reference human genome (UCSC hg 38). Previously reported variants were determined using the Human Gene Mutation Database (HGMD; professional version 2019.3), ClinVar, and locus-specific databases. As previously reported (Richards et al., 2015), according to the American Medical Genetics and Genomics guidelines (ACMG), mutations were classified as pathogenic, likely pathogenic, and uncertain of significance. We used Sanger sequencing to verify whether clinically uncertain novel variants that passed the initial filtration were co-segregated among members of the same family. Figure 1C presents the workflow of data processing and analysis. Supplementary Table 1B lists 22 CD-related genes on the Target_Eye_792_V2 chip and their clinical significance.
A total of 8400 individuals were included in this cohort, including 69 clinically diagnosed CD patients, as well as 8331 other types of eye disease patients and healthy family members as controls. All patients were classified into three subgroups according to different clinical characteristics: anterior segment disorders (ASDs, n = 1001), posterior segment disorders (PSDs, n = 3390), and other phenotypes (n = 462). The ASD subgroup includes three types of phenotypic abnormalities of the lens (n = 892), the cornea (n = 69), and iris (n = 40). The PSD subgroup includes choroid dystrophy (n = 40), retina dystrophy (n = 2,570), glaucoma (n = 173), optic neurophenotypes (n = 382), retinoblastoma (n = 40), and vitreoretinopathy (n = 185). All IEDs that do not meet the above two groups are divided into other phenotypes, including myopia, nystagmus, and microphthalmia. Based on these IEDs studies, we collected 69 patients with clinically definite diagnosed CDs, including two patients from two unrelated families with atypical corneal signs but positive family history, and TGFBI mutation was detected. These CD patients come from 50 unrelated families (total patients: 69, total participants: 91). The average age of diagnosis was 44.72 ± 19.97 (range: 3–83, median: 44), and the ratio of male patients was 49.3%.
Collectively, 81.16% (56/69) of patients with a clinical diagnosis of CD received a definite molecular diagnosis, and the detection rate of genetically confirmed diagnosis in families (77.27%, n = 17/22) did not differ greatly from sporadic cases (75%, n = 21/28). TGFBI mutations were most frequently detected. Eight distinct pathogenic mutations account for 91.07% (51/56) of the cases. Five distinct CHTS6 pathogenic or likely pathogenic mutations were detected in 7.14% (4/56) of the cases, including three compound heterozygous mutations (p.D203Y/p.R211W, p.D203Y/p.W232X, and p.R140X/p.R205W) and one homozygous mutation (p.R205W). A homozygous mutation (p.E170K) in the SLC4A11 gene was detected in 1.79% (1/56) of the cases (Figure 2A). No mutations of interest were detected in the remaining 13 cases.
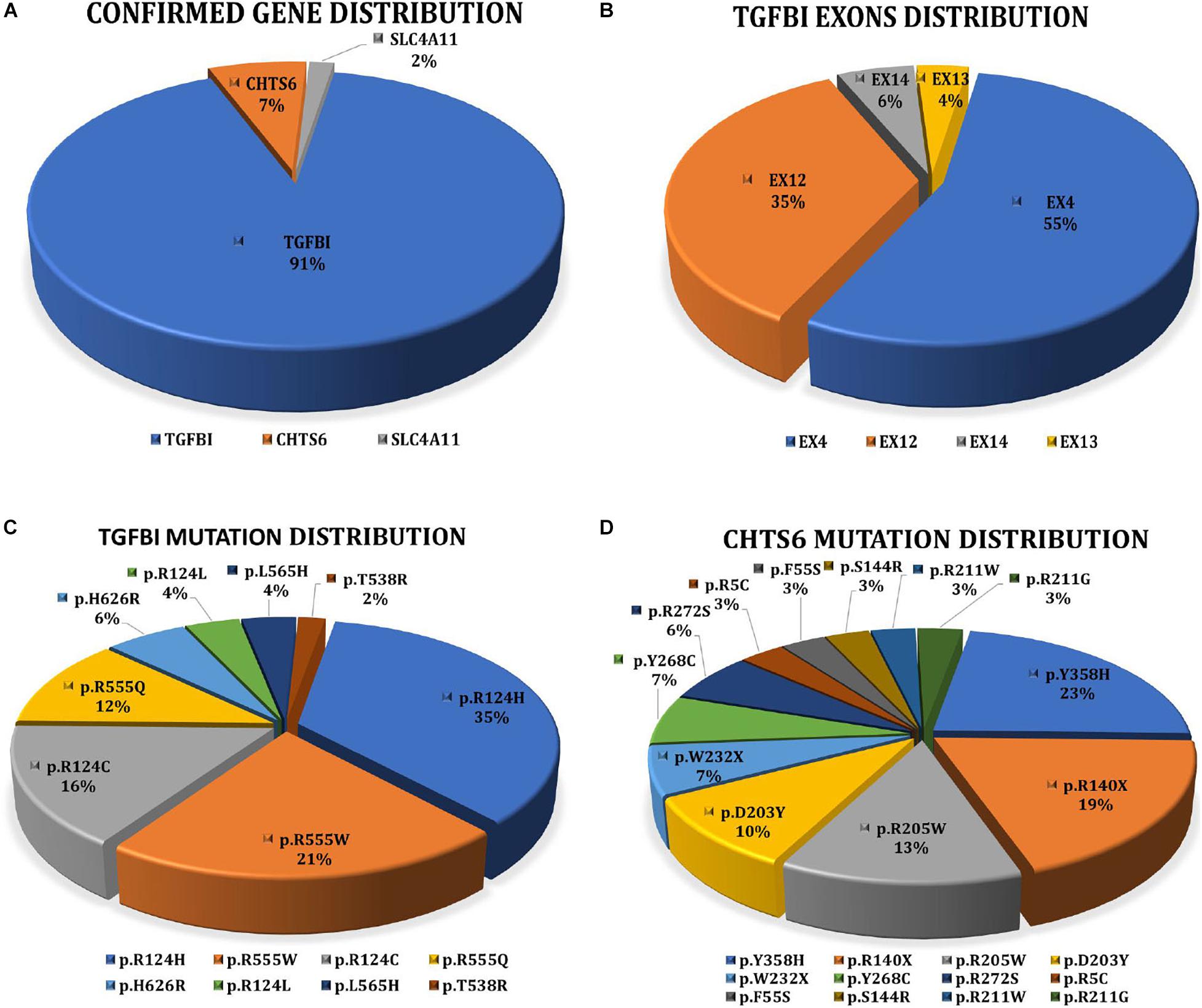
Figure 2. (A) Interpretation rate of three genes detected in 56 clinically diagnosed CD patients with clear molecular diagnosis. 51 patients with TGFBI, four patients with CHST6, and one patient with SLC4A11. (B) Distribution of TGFBI pathogenic mutations in exons. (C) Proportion of eight TGFBI distinct pathogenic mutations detected in 51 CD patients. (D) Proportion of 12 CHST6 distinct pathogenic or likely pathogenic mutations identified in four patients and 24 heterozygous carriers.
Overall, 2334 distinct high-quality variants on 22 CD-related genes in 8400 individuals were identified. 21 distinct pathogenic or likely pathogenic mutations were detected, and the remaining 2313 variants in our IED cohort had no evidence of CD-related pathogenicity (Supplementary Data 1). Of them, 8 had TGFBI mutations, 12 had CHTS6 mutations, and 1 had SLC4A11 mutation. The collective explanation rate in our study was 81.16% (n = 56/69; Table 1). Pathogenic variants in TGFBI individuals were distributed across exons 4, 12, 13, and 14; 90.20% of mutations were distributed across exons 4 (55%, 28/51) and 12 (35%, 18/51; Figure 2B). All CHTS6 mutations were located in exon 3. Eight TGFBI pathogenic mutations were detected in 51 CD patients, and 12 CHTS6 pathogenic or likely pathogenic mutations were detected in 4 CD patients and 24 heterozygous carriers. The frequency of each pathogenic or likely pathogenic mutation is shown in Figures 2C,D.
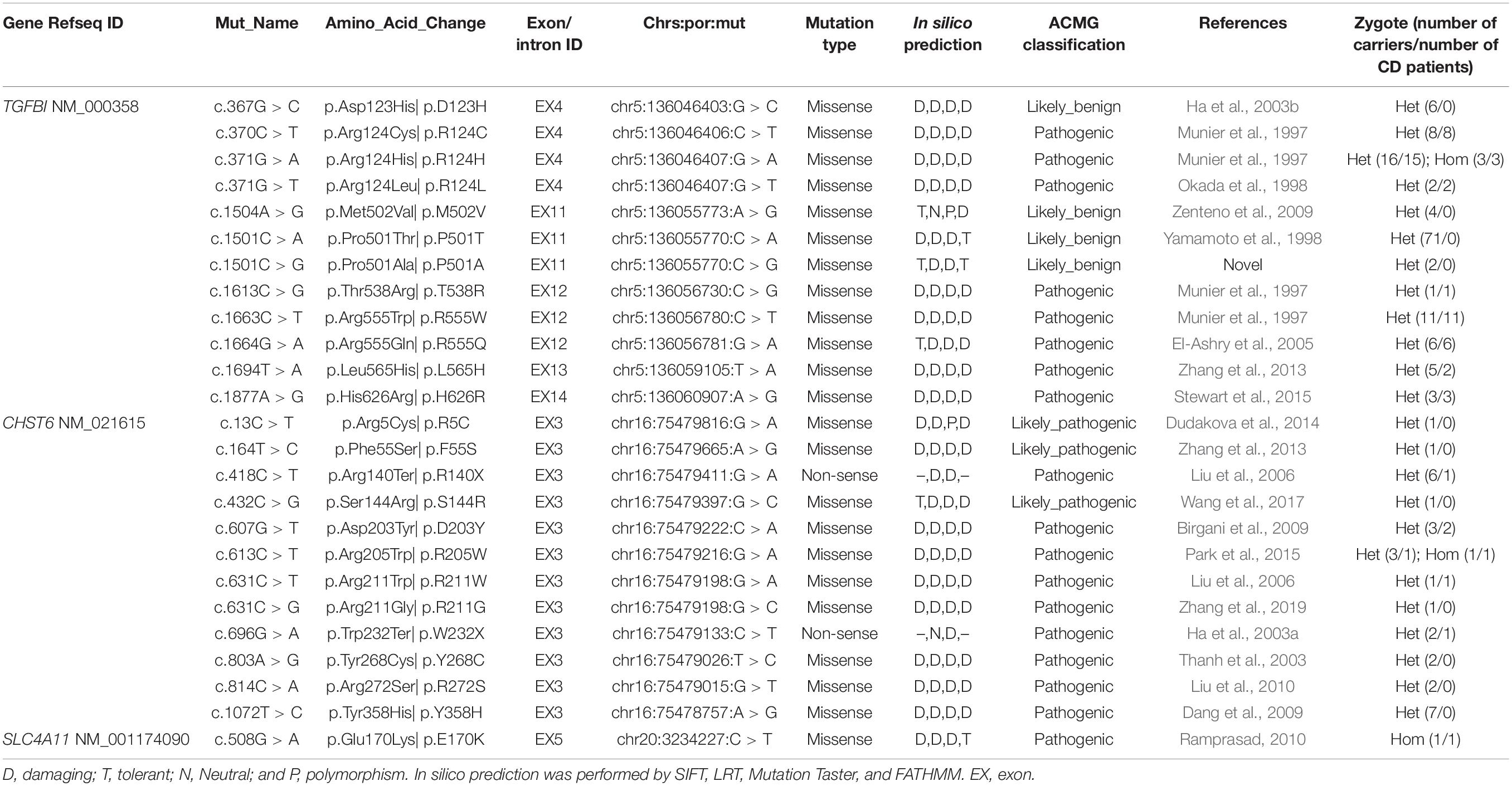
Table 1. Variations included in HGMD or ClinVar or previously reported were identified in this study.
The transforming growth factor-beta-induced protein (TGFBI; OMIM 601692, also known as βig-H3, and keratoepithelin) is a 68-kD extracellular matrix protein. It has an N-terminal signal peptide (SP), N-terminal cysteine-rich (EMI) domain, four homologous FAS1 domains, and C-terminal arg-gly-asp (RGD) motif. It is autosomal dominant (Ivanov et al., 2008). According to previous reports (Ha et al., 2003b; Zenteno et al., 2009; Groppe, 2013), four TGFBI disease-associated mutations (p.D123H, p.M502V, p.P501T, and p.P501A) were classified as likely benign in this cohort after performing clinical variant interpretation. All these four TGFBI mutations occurred in individuals without clinical symptoms of CDs, especially the previously reported disease-causing mutation p.P501T that was detected in 71 individuals, which include both IEDs and healthy family members without CD symptoms. Most TGFBI mutations (88.25%, 45/51) identified in 51 CD patients were in one of the two recurrent mutations of the first FAS1 domain codon p.R124 or the fourth FAS1 domain p.R555 (Figure 3A). Fifty-one patients with TGFBI corneal dystrophy were detected, including 8 patients with p.R124C, 18 patients with p.R124H (15 patients with heterozygous mutations and 3 patients with homozygous mutations), 2 patients with p.R124L, 1 patient with p.T538R, 11 patients with p.R555W, 6 patients with p.R555Q, 2 patients with p.L565H, and 3 patients with p.H626R. Among them, the heterozygous and homozygous mutations of recurrent p.R124H were detected in 16 individuals and 3 individuals, respectively, and only 1 heterozygous individual showed a normal phenotype. The results indicate that the penetrance of heterozygous carriers of p.R124H mutation was 93.75% (15/16). The other four recurrent mutations (p.R555W, p.R124C, p.R555Q, and p.R124L) were all heterozygous, and the penetrance was 100%. Of the five individuals who carried the heterozygous p.L565H mutation, 3 showed a normal phenotype, indicating that the penetrance of the mutation is 40% in this IED cohort. p.T538R and p.H626R were not detected in individuals with normal phenotype. The p.R124H homozygous mutation was detected in three CD patients.
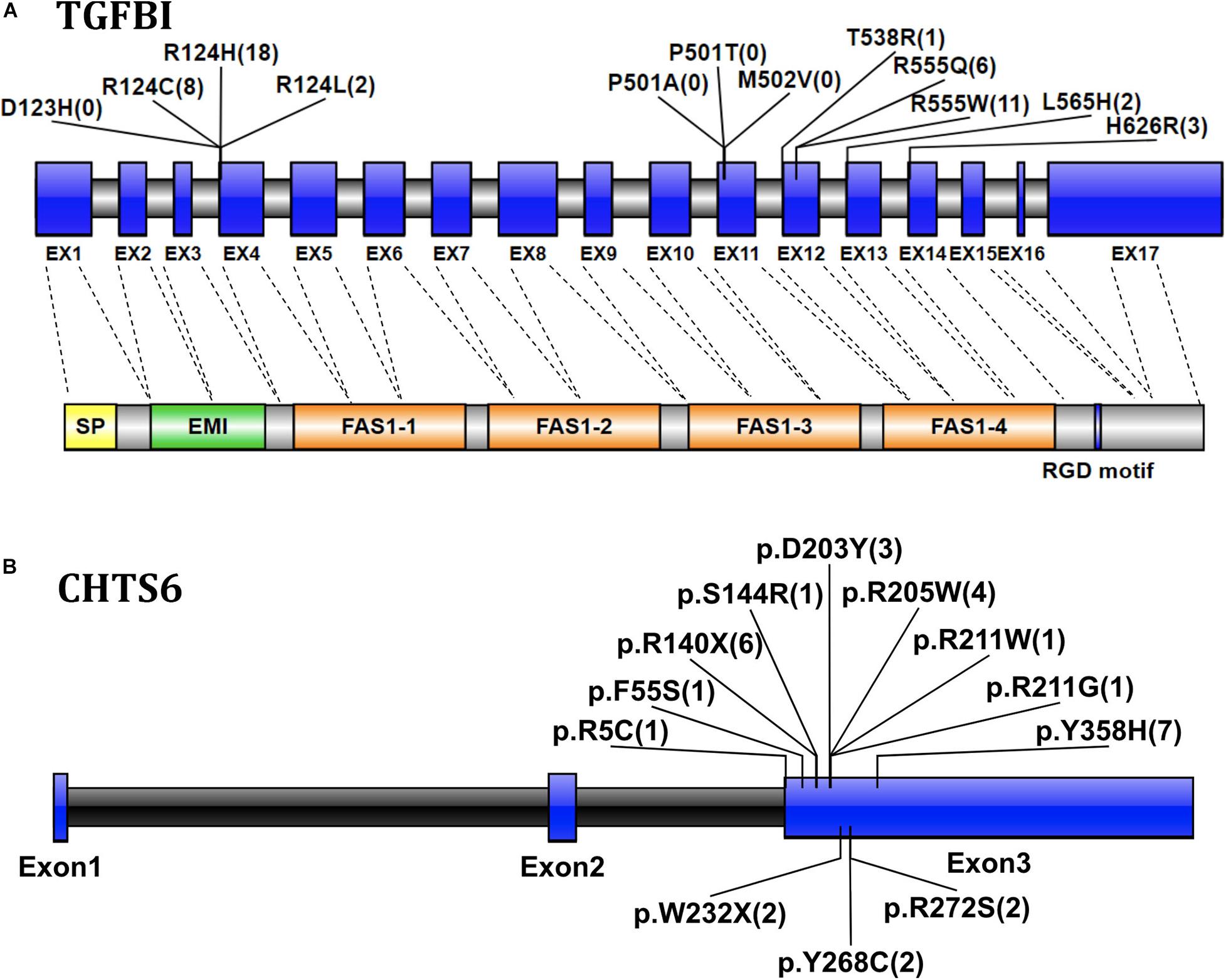
Figure 3. (A) Graphical representation of the TGFBI gene and protein structure, showing the position of the detected mutation exon and the corresponding domain. The numbers in parentheses represent the number of CD patients with mutations detected. Exons are drawn to scale; introns are not drawn to scale, indicating exon–intron boundaries. Transcript NM_000358. (B) The location and number of CHTS6 mutations detected in the IED population, the numbers in parentheses represent the number of individuals with mutations detected, including 4 CD patients and 24 individuals with non-CD phenotype. Transcript NM_021615.
The Carbohydrate Sulfotransferase 6 protein (CHST6; OMIM 605294) encodes an enzyme that mediates keratin sulfate in the cornea and plays a role in maintaining corneal transparency. The defects in CHST6 can cause MCD (OMIM 217800), an autosomal recessive genetic disease characterized by bilateral progressive stromal opacity and loss of vision, which ultimately requires corneal transplantation. A total of 12 distinct pathogenic mutations were detected in 28 individuals, including 10 missense, and 2 non-sense mutations. The recurrent mutations were p.Y358H (n = 7), p.R140X (n = 6), p.R205W (n = 4), which represent 25.00, 21.43, and 14.29%, respectively, (Figure 3B). Among 28 individuals with detected CHTS6 mutation, there were 4 CDs patients and 24 heterozygous carriers with non-CD phenotype or normal phenotype. Among these four clinically diagnosed CD patients, three had compound heterozygous mutations p.D203Y and p.R211W, p.R140X and p.R205W, p.D203Y and p.W232X, and one had compound mutation p.R205W. In this IED cohort, the proportion of carriers of CHTS6 pathogenic mutations is 0.39%, so we speculate that the prevalence of CHTS6 pathogenic mutations in the Han ethnic Chinese general population is less than 1.5/100,000.
The Solute Carrier Family 4 Member 11 protein (SLC4A11; OMIM 610206) encodes the membrane transport protein of the basolateral corneal endothelium, causing CHED and Fuchs endothelial corneal dystrophy (FECD). In this study, we detected an SLC4A11 homozygous mutation in one case, and no carrier was detected.
All mutations associated with TGFBI are missense mutations; 74.19% (23/31) with CHTS6 mutations are missense mutations, and 25.81% (8/31) are non-sense mutations. Moreover, of the four patients with CHTS6 mutations, 2 (50%) had missense + missense mutation (M + M), and 2 (50%) had missense + non-sense mutation (M + N). Furthermore, 5.35% (n = 2) of the CD patients who received molecular diagnoses had refinement in the initial clinical diagnoses. These cases showed atypical clinical symptoms, but had positive family history of TGFBI mutations. The remaining 95.65% received definite genetic subtypes of CDs.
Supplementary Data 1 shows the result of 2334 distinct high-quality variants on 22 CD-related genes, including 2230 SNPs and 104 indels, with the total number of variants being 18,001. A total of 781 synonymous mutations and 1553 non-synonymous variants were discovered, of which 1108 novel variants that do not exist in the gnomad database (v.2.0.1.hg38) were discovered in this study. After frequency filtering, deleterious effect assessment, genotype–phenotype matching, and co-segregation analysis, 21 pathogenic mutations were found through ACMG classification, ClinVar, and or were previously reported. The remaining 2313 variants in our IED cohort had no evidence of CD-related pathogenicity. Part of the variants included in HGMD with the symbol “DM or DM?,” or in ClinVar with “likely_pathogenic or pathogenic,” was reported to cause other non-CD phenotype eye disease, but there is no evidence of CD-related pathogenicity in this cohort. Moreover, 111 distinct variants of TGFBI were identified in 919 individuals, and only 8 distinct pathogenic mutations were determined to cause CD disease in 51 individuals. 83 distinct variants of CHST6 were identified in 235 individuals, and only 12 distinct pathogenic or likely pathogenic mutations were detected in 4 CD patients and 24 heterozygous carriers. The allele homogeneity of nine recurrent mutations was evaluated, the frequency of alleles obtained from the gnomAD database (v.2.1.0), and their ethnic group distribution was determined through published literature. Six hotspot mutations and three founder mutations were identified, including two novel suspected founder mutations and one novel suspected hotspot mutation (Table 2).
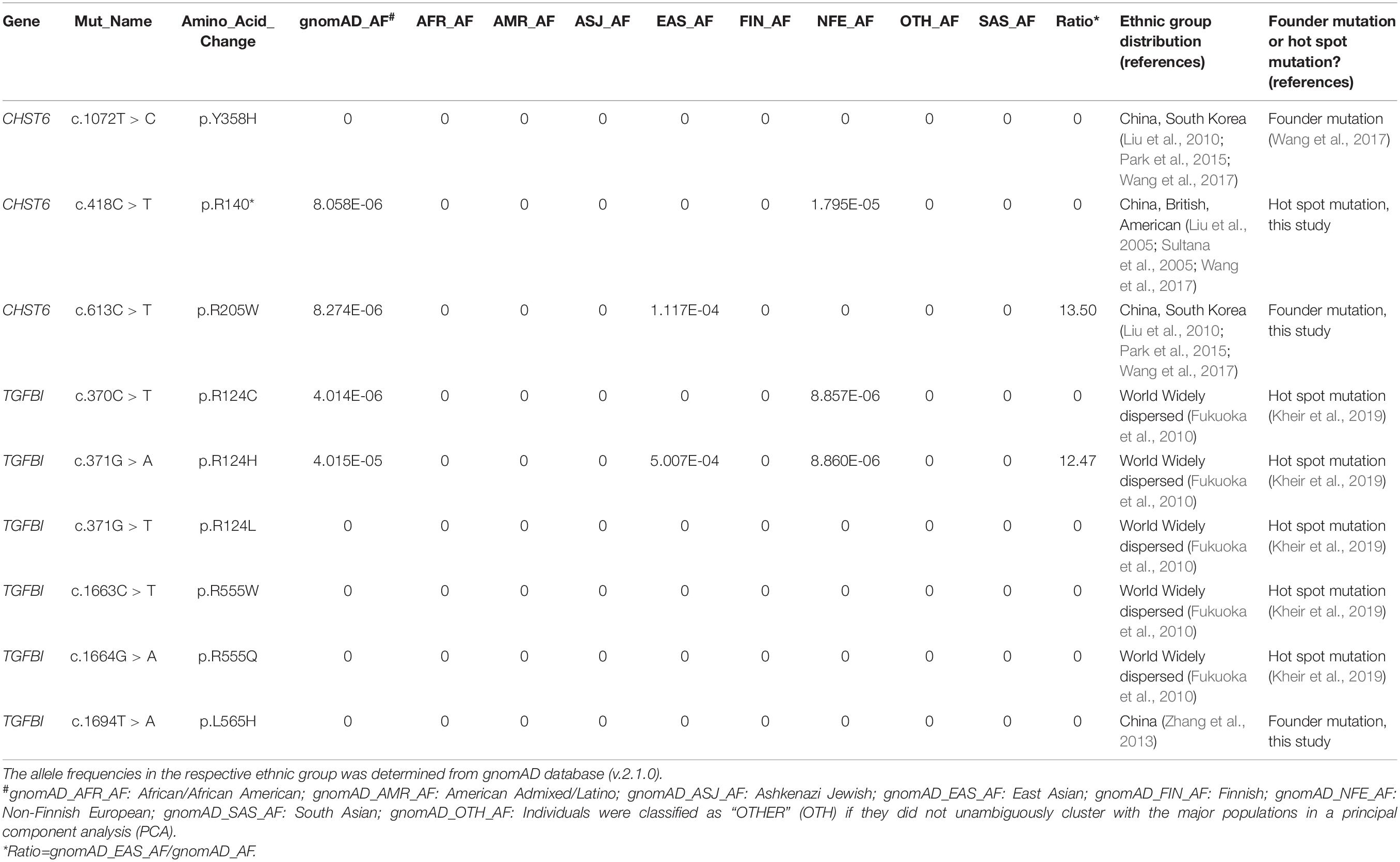
Table 2. The allele homogeneity of nine recurrent mutations was evaluated in this study.
To our knowledge, this is the first mutation spectrum study that focuses on genes associated with CDs in a large IED cohort. The customized panel design contains 792 genes associated with IEDs, which provides us with an accurate molecular diagnosis for identifying cases with overlapping phenotypes. Quick and accurate genetic testing can be used for the early detection and diagnosis of different types of CDs. Considering the correlation between the type of CDs and treatment options, it is essential to perform genetic testing before refractive surgery or PTK. Comprehensive molecular screening might contribute to higher overall mutation detection rate (81.16% n = 56/69) and will contribute to better general understanding of the population carriers of autosomal recessive genetic disease-causing mutations (less than 1.5/100,000). According to the IC3D classification, there are up to 22 distinct clinical classifications of CDs with overlapping clinical manifestations, and a small number of patients displaying no apparent clinical symptoms. These ambiguities pose a huge challenge for clinicians to accurately diagnose patients (Weiss et al., 2008, 2015).
The Han ethnic group accounts for 91.51% of China’s population and are also the dominant ethnic group in Taiwan, Hong Kong, and Singapore. Accounting for a total population of about 1.5 billion in the world, they are about 19% of the global population (Groppe, 2013). The World Health Organization (WHO) released the first version of the World Report on Vision in October 2019, indicating that moderate to severe distance vision impairment or blindness due to corneal opacities is estimated to be at least 4.2 million worldwide (Bourne et al., 2017; Flaxman et al., 2017; Fricke et al., 2018). Due to clinical manifestations varying widely between the different categories, whenever corneal transparency is lost or corneal opacity occurs spontaneously, especially in two corneas, or in offspring with a positive family history or consanguineous marriage, CD should be suspected. The most common causes of corneal opacity were injuries, vitamin A deficiency, and measles infection (Song P. et al., 2017). Existing data cannot provide accurate statistics on the global prevalence of CD. A retrospective histopathological analysis was performed on the corneal specimens of 3112 patients in China. Among the 637 specimens of non-infectious corneal diseases, 7.85% (50/637) of the cases were caused by CD (Li et al., 2014). Another study analyzed 2068 prospective cases of candidates for refractive surgery. Slit-lamp examinations of four cases with corneal opacity in both eyes and one case without corneal opacity were detected with heterozygous p.R124H mutation of TGFBI. The prevalence rate was 0.24% (5/2068 = 24.2/10,000; Song Y. et al., 2017). Analysis of commercial test data used for pre-refractive surgery screening from 600,000 samples from around the world, most of which are from Korea and Japan, demonstrated that the detection rate of TGFBI CDs in Korea is approximately 15/10,000, which is similar to the previously reported detection rate in the Korean population, which is about 11.5/10,000, and higher than the detection rate in the Japanese population 3/10,000 (Lee et al., 2010; Chao-Shern et al., 2019). In another United States study, almost 8 million eye care visit records in the national managed-care network were analyzed. A total of 27,372 unique individuals have received two or more diagnoses records of some type of corneal dystrophy, and the overall prevalence of corneal dystrophy is 8.97/10,000 (Møller and Sunde, 2013). Considering that our research group focuses on IED, the detection rate is relatively higher, and the overall prevalence is 66.7/10,000 (56/8400). The essential reason for the difference observed in prevalence across different groups is due to different countries’ adoption of different genetic screening strategies. In Korea and Japan, genetic tests are part of the practice guidelines for refractive surgery in clinics/hospitals and are used for screening purposes. In the United States, when performing refractive surgery, some clinics/hospitals use genetic testing for screening, while other clinics/hospitals confirm the clinical diagnosis or exclude TGFBI mutations in individuals with the abnormal cornea. European clinics mainly perform clinical diagnosis (Chao-Shern et al., 2019).
The overall detection rate of molecular diagnoses was 81.16% (56/69), which was obviously higher than the previous study, with a mutation detection rate of 59.2% (42/71; Zhang et al., 2013). There was no significant difference between the two subgroups of family (77.27%, n = 17/22) and sporadic (75%, n = 21/28) cases. A total of 21 distinct pathogenic or likely pathogenic mutations were detected in our IED cohort, including 8 TGFBI mutations, 12 CHTS6 mutations, and 1 SLC4A11 mutation. Moreover, three previously reported disease-associated mutations (p.D123H, p.M502V, and p.P501T) and one novel multiple allele mutation (p.P501A) was detected but was defined as likely_benign mutations in this IED cohort (Ha et al., 2003b; Zenteno et al., 2009; Groppe, 2013). Among them, the p.P501T mutation was detected in 71 individuals with non-CD phenotype or normal phenotype; however, this mutation was found in all 18 patients with Lattice Corneal Dystrophies Type IIIA (LCDIIIA) examined in Japan. LCDIIIA accounts for 11.0% of corneal dystrophy in Japan, while only a few cases of LCDIIIA have been reported in Western countries. Interestingly, the p.P501T mutation was detected in individuals with non-CD phenotype or normal phenotype in China, but not in CD patients, indicating that the pathogenicity may vary between different races (Yamamoto et al., 1998; Tsujikawa et al., 2010; Kheir et al., 2019). Twelve distinct CHTS6 mutations were detected in 28 individuals, including 3 CD patients with M + M mutation, 1 CD patient with M + N mutation, and the remaining 24 heterozygous carriers with non-CD phenotype or normal phenotypes.
We further verified and clarified the distribution of gene mutations for the Han ethnic Chinese population in three genes: TGFBI, CHTS6, and SLC4A11 genes, which account for 91.07% (51/56), 7.14% (4/56), and 1.79% (1/56) of diagnosed cases, respectively. No pathogenic mutations in other 19 CD-related genes were found in this IED cohort. The frequency and pathogenicity of all 2334 distinct high-quality variants in our IED cohort of 8400 individuals were evaluated on 22 CD-related genes, of which 2313 variants had no evidence of CD-related pathogenicity. However, in recent studies of the Chinese Han ethnic, the distribution of gene mutations was mainly observed in five genes: TGFBI, CHTS6, SLC4A11, AGBL1, and COL17A1, and the proportions were 52.38% (22/42), 30.95% (13/42), 9.52% (4/42), 4.76% (2/42), and 2.38% (1/42), respectively, (Zhang et al., 2013). In our study, TGFBI mutations were found in 51 CDs patients, and most of these patients (88.23%, 45/51) harbored mutations in one of two recurrent codons: p.Arg124 of the first FAS1 domain or p.Arg555 of the fourth FAS1 domain. These results are similar to previous studies in the Han ethnic Chinese population (81.82%, 18/22; Zhang et al., 2013). CHST6 mutation is considered to be the most critical genetic factor in MCD (Zhang et al., 2019). In our group of IEDs, we found that the incidence of CHTS6 pathogenic mutations in the general population is less than 1.5/100,000, so this is consistent with our conclusion that CHTS6 mutations account for less than one-tenth of confirmed cases. The CHTS6 mutation has a low incidence in the Han ethnic, despite having a high prevalence in India, Saudi Arabia, and Iceland (Aggarwal et al., 2018). In our study, the proportion of TGFBI (91.07%, 51/56) in confirmed cases is significantly higher than the CHTS6 gene (7.14%, 4/56). It may be due to differences in the types of CDs of recruited patients, and the number of patients with different types of CDs.
Allelic homogeneity is the main factor leading to TGFBI-related corneal dystrophy. It occurs when a few different alleles within the same gene cause similar phenotypic expression (Tsujikawa et al., 2007, 2010). The homogeneity of alleles is explained by two different mechanisms: mutation hotspot and founder mutation. Allelic heterogeneity is a typical feature of CHST6-related corneal dystrophy, which occurs when tens or even hundreds of different alleles within the same gene cause similar phenotypes. A total of 189 distinct disease-causing mutations were found in 375 MCD patients worldwide, including 134 missense mutations, 18 non-sense mutations, and 37 indels (Safari et al., 2020). We evaluated the allele homogeneity of nine recurrent mutations, two novel suspected founder mutations, and one novel hot spot mutation. The p.R124H mutation of the TGFBI gene is significantly higher in East Asian populations than other populations, but it is widely reported by various ethnic groups in the world (Fukuoka et al., 2010). We speculate that it had originated in East Asia and spread to all parts of the world over many years.
In conclusion, our results reveal the mutational spectrum of 22 CD-related genes in a large cohort of IEDs. Our large reference study systematically described the variation spectrum of 22 genes relevant to CD and evaluated the frequency and pathogenicity of all 2334 distinct high-quality variants in our IED cohort of 8,400 individuals. Our research will provide East Asia and other populations with baseline data from a Han ethnic population-specific level. We believe that our work not only provides theoretical guidance and frequent genotype profiles for Han ethnic Chinese population but also provides an effective reference for genetic counseling and accurate and early diagnosis of CD patients.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation. The data that support the findings of this study have been deposited in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with ccession code CNP CNP0000503.
The studies involving human participants were reviewed and approved by The Ethics Committee of the He Eye Specialists Hospital of He University. The patients/participants provided their written informed consent to participate in this study.
WL, NQ, J-KL, W-PQ, L-SW, and WH: conception and design. WL, NQ, W-PQ, WH, and Z-SW: clinical examinations and interpretation. WL, NQ, J-KL, Y-XL, D-MH, Z-WW, Y-XC, LT, XC, X-YJ, and CL: acquisition of data. WL, KS, and WY: analysis and interpretation of data. WL, NQ, and J-KL: writing, review, and revision of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant NSFC 61972329), the Project of Shenyang Science and Technology Bureau (Grant Number: 20-301-4-00), the Project for Research Team of Female Reproductive Health and Fertility Preservation (SZSM201612065), the Project for Medical Discipline Advancement of Health and Family Planning commission of Shenzhen Municipality (SZXJ2017003), the Project for Exploration of New method of Non-invasive Fertility Evaluation and Establishment of National Standard (2018YFC1002104), and grants from the Research Grants Council of the Hong Kong Special Administrative Region, China (CityU 11206120 and CityU 11210119).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We sincerely thank all of the patients and families who agreed to participate in this study. In addition, we would like to thank BGI Shenzhen for their technical support and the staff at He Eye Specialists Hospital of He University for their assistance. We also acknowledge the English language revision assistance provided by David Cao (Biomedical Engineering, Harvard College). Finally, we are grateful to WH, L-SW, and W-PQ for their invaluable contributions to this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.632946/full#supplementary-material
Adams, D. R., and Eng, C. M. (2018). Next-Generation sequencing to diagnose suspected genetic disorders. N. Eng. J. Med. 379, 1353–1362. doi: 10.1056/NEJMra1711801
Aggarwal, S., Peck, T., Golen, J., and Karcioglu, Z. A. (2018). Macular corneal dystrophy: a review. Survey Ophthalmol. 63, 609–617. doi: 10.1016/j.survophthal.2018.03.004
Birgani, S. A., Salehi, Z., Houshmand, M., Mohamadi, M. J., Promehr, L. A., Mozafarzadeh, Z., et al. (2009). Novel mutations of CHST6 in Iranian patients with macular corneal dystrophy. Mol. Vis. 15, 373–377.
Bourne, R. R. A., Flaxman, S. R., Braithwaite, T., Cicinelli, M. V., Das, A., Jonas, J. B., et al. (2017). Magnitude, temporal trends, and projections of the global prevalence of blindness and distance and near vision impairment: a systematic review and meta-analysis. Lancet Global Health. 5, e888–e897. doi: 10.1016/S2214-109X(17)30293-0
Bron, A. J. (1990). The corneal dystrophies. Curr. Opin. Ophthalmol. 1, 333–346. doi: 10.1097/00055735-199008000-00003
Chao-Shern, C., Dedionisio, L. A., Jang, J. H., Chan, C., Thompson, V., Christie, K., et al. (2019). Evaluation of TGFBI corneal dystrophy and molecular diagnostic testing. Eye 33, 874–881. doi: 10.1038/s41433-019-0346-x
Dang, X., Zhu, Q., Wang, L., Su, H., Lin, H., Zhou, N., et al. (2009). Macular corneal dystrophy in a Chinese family related with novel mutations of CHST6. Mol. Vis. 15, 700–705.
Dudakova, L., Palos, M., Svobodova, M., Bydzovsky, J., Huna, L., Jirsova, K., et al. (2014). Macular corneal dystrophy and associated corneal thinning. Eye 28:1201. doi: 10.1038/eye.2014.164
El-Ashry, M. F., Abd El-Aziz, M. M., Hardcastle, A. J., Bhattacharya, S. S., and Ebenezer, N. D. (2005). A clinical and molecular genetic study of autosomal-dominant stromal corneal dystrophy in British population. Ophthalmic Res. 37, 310–317. doi: 10.1159/000087791
Flaxman, S. R., Bourne, R. R. A., Resnikoff, S., Ackland, P., Braithwaite, T., Cicinelli, M. V., et al. (2017). Global causes of blindness and distance vision impairment 1990–2020: a systematic review and meta-analysis. Lancet Global Health 5, e1221–e1234. doi: 10.1016/S2214-109X(17)30393-5
Fricke, T. R., Tahhan, N., Resnikoff, S., Papas, E., Burnett, A., Ho, S. M., et al. (2018). Global prevalence of presbyopia and vision impairment from uncorrected presbyopia: systematic review, meta-analysis, and modelling. Ophthalmology 125, 1492–1499. doi: 10.1016/j.ophtha.2018.04.013
Fukuoka, H., Kawasaki, S., Yamasaki, K., Matsuda, A., Fukumoto, A., Murakami, A., et al. (2010). Lattice corneal dystrophy type IV (p.Leu527Arg) is caused by a founder mutation of the TGFBI gene in a single Japanese ancestor. Invest. Ophthalmol. Visual Sci. 51:4523. doi: 10.1167/iovs.10-5343
Groppe, A. (2013). Critical han studies: the history, representation, and identity of China’s majority. Asian Ethnicity 14, 483–491. doi: 10.1080/14631369.2013.759770
Ha, N. T., Chau, H. M., Cung, L. X., Thanh, T. K., Fujiki, K., Murakami, A., et al. (2003a). Identification of novel mutations of the CHST6 gene in vietnamese families affected with macular corneal dystrophy in two generations. Cornea 22:508. doi: 10.1097/00003226-200308000-00004
Ha, N. T., Cung le, X., Chau, H. M., Thanh, T. K., Fujiki, K., Murakami, A., et al. (2003b). A novel mutation of the TGFBI gene found in a vietnamese family with atypical granular corneal dystrophy. Jpn. J. Ophthalmol. 47, 246–248. doi: 10.1016/S0021-5155(03)00019-4
Ivanov, S. V., Ivanova, A. V., Salnikow, K., Timofeeva, O., Subramaniam, M., and Lerman, M. I. (2008). Two novel VHL targets, TGFBI (BIGH3) and its transactivator KLF10, are up-regulated in renal clear cell carcinoma and other tumors. Biochem. Biophys. Res. Commun. 370, 536–540. doi: 10.1016/j.bbrc.2008.03.066
Kheir, V., Cortés-González, V., Zenteno, J. C., and Schorderet, D. F. (2019). Mutation update: TGFBI pathogenic and likely pathogenic variants in corneal dystrophies. Hum. Mutat. 40, 675–693. doi: 10.1002/humu.23737
Lee, J. H., Cristol, S. M., Kim, W. C., Chung, E. S., Tchah, H., Kim, M. S., et al. (2010). Prevalence of Granular Corneal Dystrophy Type 2 (Avellino Corneal Dystrophy) in the Korean Population. Ophthalmic Epidemiol. 17, 160–165. doi: 10.3109/09286581003624939
Li, X., Wang, L., Dustin, L., and Wei, Q. (2014). Age distribution of various corneal diseases in china by histopathological examination of 3112 surgical specimens. Invest. Ophthalmol. Vis. 55, 3022–3028. doi: 10.1167/iovs.13-13805
Liu, N. P., Bao, W., Smith, C. F., Vance, J. M., and Klintworth, G. K. (2005). Different mutations in carbohydrate sulfotransferase 6 (CHST6) gene cause macular corneal dystrophy types I and II in a single sibship. Am. J. Ophthalmol. 139, 1118–1120. doi: 10.1016/j.ajo.2004.11.054
Liu, N. P., Smith, C. F., Bowling, B. L., Jonasson, F., and Klintworth, G. K. (2006). Macular corneal dystrophy types I and II are caused by distinct mutations in the CHST6 gene in Iceland. Mol. Vis. 12, 1148–1152.
Liu, Z., Tian, X., Iida, N., Fujiki, K., Xie, P., Wang, W., et al. (2010). Mutation analysis of CHST6 gene in chinese patients with macular corneal dystrophy. Cornea 29, 883–888. doi: 10.1097/ICO.0b013e3181ca2e74
Møller, H. U., and Sunde, L. (2013). Prevalence of corneal dystrophies in the united states: estimates from claims data. Invest. Ophthalmol. Vis. Sci. 54:387. doi: 10.1167/iovs.12-11211
Munier, F. L., Korvatska, E., Djema, A., Le Paslier, D., Zografos, L., Pescia, G., et al. (1997). Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat. Genet. 15, 247–251. doi: 10.1038/ng0397-247
Okada, M., Yamamoto, S., Watanabe, H., Inoue, Y., Tsujikawa, M., Maeda, N., et al. (1998). Granular corneal dystrophy with homozygous mutations in the kerato-epithelin gene. Am. J. Ophthalmol. 129, 411–412. doi: 10.1016/S0002-9394(98)00075-0
Park, S. H., Ahn, Y. J., Chae, H., Kim, Y., Kim, M. S., Kim, M., et al. (2015). Molecular analysis of the CHST6 gene in Korean patients with macular corneal dystrophy: identification of three novel mutations. Mol. Vis. 21, 1201– 1209.
Pieramici, S. F., and Afshari, N. A. (2006). Genetics of corneal dystrophies: the evolving landscape. Curr. Opin. Ophthalmol. 17, 361–366. doi: 10.1097/01.icu.0000233955.94347.84
Ramprasad, V. L. (2010). Novel SLC4A11 mutations in patients with recessive congenital hereditary endothelial dystrophy (CHED2). Mutation in brief #958. Online. Hum. Mutat. 28, 522–523. doi: 10.1002/humu.9487
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Safari, I., Baradaran-Rafii, A., Issazadeh-Navikas, S., and Elahi, E. (2020). CHST6 mutations identified in Iranian MCD patients and CHST6 mutations reported worldwide identify targets for gene editing approaches including the CRISPR/Cas system. Int. Ophthalmol. 40, 2223–2235. doi: 10.1007/s10792-020-01401-9
Song, P., Wang, J., Wei, W., Chang, X., Wang, M., and An, L. (2017). The prevalence of vitamin a deficiency in chinese children: a systematic review and bayesian meta-analysis. Nutrients 9:1285. doi: 10.3390/nu9121285
Song, Y., Sun, M., Wang, N., Zhou, X., Zhao, J., Wang, Q., et al. (2017). Prevalence of transforming growth factor β–induced gene corneal dystrophies in Chinese refractive surgery candidates. J. Cataract Refract. Surg. 43, 1489–1494. doi: 10.1016/j.jcrs.2017.07.038
Stewart, H. S., Ridgway, A. E., Dixon, M. J., Bonshek, R., Parveen, R., Black, G., et al. (2015). Heterogeneity in granular corneal dystrophy: identification of three causative mutations in the TGFBI (BIGH3) gene—Lessons for corneal amyloidogenesis. Hum. Mutat. 14, 126–132. doi: 10.1002/(SICI)1098-1004(1999)14:2<126::AID-HUMU4>3.0.CO;2-W
Sultana, A., Sridhar, M. S., Klintworth, G. K., Balasubramanian, D., and Kannabiran, C. (2005). Allelic heterogeneity of the carbohydrate sulfotransferase-6 gene in patients with macular corneal dystrophy. Clin. Genet. 68, 454–460. doi: 10.1111/j.1399-0004.2005.00517.x
Thanh, H. N., Minh, C. H., Xuan, C. L., Thanh, T. K., Fujiki, K., Murakami, A., et al. (2003). Mutation analysis of the carbohydrate sulfotransferase gene in vietnamese with macular corneal dystrophy. Invest. Ophthalmol. Visual Sci. 44:3310. doi: 10.1167/iovs.03-0031
Tsujikawa, K., Tsujikawa, M., Watanabe, H., Maeda, N., Inoue, Y., Fujikado, T., et al. (2007). Allelic homogeneity in Avellino corneal dystrophy due to a founder effect. J. Hum. Genet. 52, 92–97. doi: 10.1007/s10038-006-0083-4
Tsujikawa, K., Tsujikawa, M., Yamamoto, S., Maeda, N., Inoue, Y., Fujikado, T., et al. (2010). Allelic homogeneity due to a founder mutation in Japanese patients with lattice corneal dystrophy type IIIA. Am. J. Med. Genet. 113, 20–22. doi: 10.1002/ajmg.10709
Wang, L., Tang, X., Lv, X., Sun, E., Wu, D., Wang, C., et al. (2017). CHST6 mutation screening and endoplasmatic reticulum stress in macular corneal dystrophy. Oncotarget 8, 96301–96312. doi: 10.18632/oncotarget.22028
Weiss, J. S., Møller, H. U., Aldave, A. J., Seitz, B., Bredrup, C., Kivelä, T., et al. (2015). IC3D classification of corneal dystrophies-edition 2. Cornea 34, 117–159. doi: 10.1097/ICO.0000000000000307
Weiss, J. S., Møller, H. U., Lisch, W., Kinoshita, S., Aldave, A. J., Belin, M. W., et al. (2008). The IC3D classification of the corneal dystrophies. Cornea 27, S1–S42. doi: 10.1097/ICO.0b013e31817780fb
Yamamoto, S., Okada, M., Tsujikawa, M., Shimomura, Y., Nishida, K., Inoue, Y., et al. (1998). A kerato-epithelin (betaig-h3) mutation in lattice corneal dystrophy type IIIA. Am. J. Hum. Genet. 62, 719–722. doi: 10.1086/301765
Zeng, L., Zhao, J., Chen, Y., Zhao, F., Li, M., Chao-Shern, C., et al. (2017). TGFBI gene mutation analysis of clinically diagnosed granular corneal dystrophy patients prior to PTK: a pilot study from Eastern China. Sci. Rep. 7:596. doi: 10.1038/s41598-017-00716-5
Zenteno, J. C., Correa-Gomez, V., Santacruz-Valdez, C., Suarez-Sanchez, R., and Villanueva-Mendoza, C. (2009). Clinical and genetic features of TGFBI-linked corneal dystrophies in Mexican population: description of novel mutations and novel genotype-phenotype correlations. Exp. Eye Res. 89, 172–177. doi: 10.1016/j.exer.2009.03.004
Zhang, J., Wu, D., Li, Y., Fan, Y., Chen, H., Hong, J., et al. (2013). Novel mutations associated with various types of corneal dystrophies in a han chinese population. Front. Genet. 10:881. doi: 10.3389/fgene.2019.00881
Keywords: corneal dystrophy, NGS-panel, mutation spectrum, population-specific level, baseline data
Citation: Li W, Qu N, Li J-K, Li Y-X, Han D-M, Chen Y-X, Tian L, Shao K, Yang W, Wang Z-S, Chen X, Jin X-Y, Wang Z-W, Liang C, Qian W-P, Wang L-S and He W (2021) Evaluation of the Genetic Variation Spectrum Related to Corneal Dystrophy in a Large Cohort. Front. Cell Dev. Biol. 9:632946. doi: 10.3389/fcell.2021.632946
Received: 24 November 2020; Accepted: 02 February 2021;
Published: 18 March 2021.
Edited by:
Gavin Arno, University College London, United KingdomReviewed by:
Anna Marina Pandolfi, Politecnico di Milano, ItalyCopyright © 2021 Li, Qu, Li, Li, Han, Chen, Tian, Shao, Yang, Wang, Chen, Jin, Wang, Liang, Qian, Wang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Ping Qian, cWlhbndlaXBpbmdzekAxMjYuY29t; Lu-Sheng Wang, Y3N3YW5nbEBjaXR5dS5lZHUuaGs=; Wei He, aGV3ZWlAaHVoLmVkdS5jbg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.