 Igor Dolgalev
Igor Dolgalev Anastasia N. Tikhonova
Anastasia N. Tikhonova
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 12 March 2021
Sec. Stem Cell Research
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.622519
This article is part of the Research Topic The Dynamic Interface between Vascular Blood Vessels to Blood Forming Hematopoietic Stem Cells in Health and Disease View all 8 articles
Single-cell sequencing approaches have transformed our understanding of stem cell systems, including hematopoiesis and its niche within the bone marrow. Recent reports examined the bone marrow microenvironment at single-cell resolution at steady state, following chemotherapy treatment, leukemic onset, and aging. These rapid advancements significantly informed our understanding of bone marrow niche heterogeneity. However, inconsistent representation and nomenclature among the studies hinder a comprehensive interpretation of this body of work. Here, we review recent reports interrogating bone marrow niche architecture and present an integrated overview of the published datasets.
Hematopoiesis is a continuous process of generating blood and immune cells and is one of the best-studied stem cell systems in modern biology (Weissman, 2000). Since the discovery of hematopoietic stem cells (HSCs), the field has been driven by technological advances such as flow cytometry, mass cytometry, high-resolution microscopy, and now single-cell sequencing approaches. In vitro approaches, such as colony-forming assays, defined the intermediate stages between a rare population of multipotent hematopoietic stem cells and the terminally differentiated cell populations. In vivo, early hematopoietic progenitors were further classified as long-term HSCs (LT-HSCs) capable of self-renewal and unlimited differentiation potential and multipotent progenitors (MPPs), characterized by limited self-renewal capacity (Reya et al., 2001). The MPPs have been further separated into myeloid-biased MPP2 and MPP3 as well as lymphoid-biased MPP4 subsets that differentiate to lineage-restricted common myeloid progenitors (CMPs), granulocyte/macrophage progenitors (GMPs), and common lymphoid progenitors (CLPs) (Pietras et al., 2015). Extensive literature continued to define the complexity of hematopoietic differentiation, incorporating further subpopulations and subdivision of downstream progenitors (Cabezas-Wallscheid et al., 2014; Liggett and Sankaran, 2020).
The last decade has witnessed the introduction and wide-scale adoption of high-throughput approaches, including single-cell RNA sequencing (scRNA-seq). Unlike more traditional flow or mass cytometry methods that are limited by predefined markers, scRNA-seq technologies simultaneously capture thousands of genes in each cell, providing an unbiased characterization of their transcriptional diversity even within phenotypically homogenous populations. Notably, a major advantage of single-cell transcriptional profiling is the identification of novel populations and states as well as shifts in their relevant frequencies (Kowalczyk et al., 2015).
In one of the earlier large-scale single-cell studies, Paul et al. (2015) characterized 2,730 myeloid progenitor cells. Clustering analysis based on an expectation maximization (EM)-like procedure resulted in 19 subpopulations, including groups corresponding to known erythrocyte, monocyte, and neutrophil progenitors, as well as more transcriptionally heterogeneous clusters. The data showed that the traditional GMP and CMP definitions include several subpopulations with distinct lineage commitment transcriptional profiles.
Nestorowa et al. (2016) comprehensively profiled individually sorted 1,656 murine hematopoietic stem and progenitor cells (HSPCs), with an average of 6,558 protein-coding genes detected per cell. The study utilized the diffusion maps dimensionality reduction technique (Haghverdi et al., 2015; Angerer et al., 2016) to visualize and interpret the continuous processes of early hematopoietic differentiation. The cells were assigned to 12 commonly sorted HSPC phenotypes based on surface markers. When displayed on the diffusion map, all populations, except CMPs, localized within defined regions. The relative ordering of populations within the diffusion map was in agreement with the established hematopoietic hierarchy and further showed intermediate transition zones containing transcriptionally similar cells that traditional gating assigned to differing phenotypes. Interestingly, the single-cell analysis revealed that the three broad trajectories defined by the diffusion map begin to diverge immediately following the LT-HSC stage.
The findings were extended to the human system. Velten et al. (2017) combined transcriptomic and functional single-cell data to investigate the dynamics of lineage commitment of individual cells. Healthy human HSPCs were defined by the absence of lineage markers and CD34 expression (Lin–CD34+). 1,413 HSPCs from the bone marrow of two individuals were profiled by scRNA-seq and 2,038 cells individually cultured ex vivo were used to determine lineage potential. The transcriptomic and functional datasets were integrated based on surface marker expression. Clustering within the Lin–CD34+CD38– compartment that includes HSCs and their immediate progeny, such as MPPs, was unstable and the cells formed a continuously connected unit. In contrast, more differentiated Lin–CD34+CD38+ progenitors formed clusters corresponding to defined progenitor populations. The scRNA-seq data separated the cells into a continuum of low-primed undifferentiated (CLOUD)-HSPCs and discrete populations of restricted progenitors associated with increased CD38 expression. Analysis of the discrete Lin–CD34+CD38+ sub-populations identified the major branches of hematopoiesis, including B-cell progenitors, megakaryocyte/erythrocyte committed progenitors, neutrophil-primed progenitors, monocyte/dendritic cell progenitors, and eosinophil/basophil/mast cell progenitors. The authors developed a dimensionality reduction technique STEMNET to view the transformation from HSCs to the lineage-restricted progenitors by placing each cell along a trajectory from the least-primed HSCs in the center toward one of the six restricted progenitor populations in the corners of a simplex. The authors proposed a model in which distinct lineages emerge from CLOUD-HSPCs rather than a series of discrete progenitors. Zheng et al. (2018) profiled 21,306 CD34+ progenitor cells from human cord blood across five biological replicates without enrichment or depletion for specific lineages. The analysis revealed previously defined progenitor populations in addition to the intermediate continuous states. The authors additionally integrated the data with the previous bone marrow dataset from Velten et al. to show the high concordance between the two systems.
Pellin et al. (2019) transcriptionally mapped the fates of the early human hematopoietic progenitors using 6,011 CD34+ and 15,401 Lin– single cells profiled with the inDrops protocol. The hierarchical continuum of states that branch out toward cells expressing established lineage signatures was visualized using SPRING (Weinreb et al., 2018a). This two-dimensional force-directed graph layout generates a graph of nodes representing cells connected to their nearest neighbors in high-dimensional gene expression space. The organization of the data broadly segregated the cells into known immunophenotypic subpopulations and detected extensive transcriptional heterogeneity among HSPCs. The analysis suggested a structured hierarchy of fate decisions rather than a single step transition from undifferentiated HSPCs to committed states. This organizational structure was confirmed by inferred transcriptional trajectories as well as by population balance analysis (PBA) (Tusi et al., 2018), which in addition to providing a static continuum description of cell states aims to infer dynamic properties such as fate potential. This work confirmed a continuous hierarchical organizational structure from HSPCs to lineage-committed states not fully defined by traditional immunophenotyping. Importantly, the authors compared the human and mouse HSPCs, revealing a comparable branching structure within the two species despite differences in cell surface markers commonly used to isolate the various subpopulations.
Collectively these studies were able to isolate and define highly granular HSPC populations. They highlighted the transitional phases of hematopoietic differentiation, building on the traditional tree-like model of hematopoiesis toward a more fluid path of stem cell differentiation (Laurenti and Göttgens, 2018; Watcham et al., 2019).
Postnatally, HSCs reside in the bone marrow. While substantial progress has been made in understanding the hematopoietic hierarchy, the extrinsic factors guiding HSC self-renewal and differentiation are less defined. The bone marrow is a complex organ composed of numerous cell types that regulate HSC function through physical and biochemical interactions (Pinho and Frenette, 2019). The discovery that SLAM family surface receptors CD150, CD244, and CD48 were differentially expressed among HSPC populations in a way that can track developmental potential made it possible to image and localize those populations within the bone marrow (Kiel et al., 2005). Many HSCs were found associated with sinusoidal endothelial cells (ECs) and the endosteum (Kiel et al., 2005) as well as in contact with Cxcl12-abundant reticular (CAR) cells surrounding ECs (Sugiyama et al., 2006). An extensive network of sinusoids in the central marrow allows the hematopoietic cells to transport in and out of the bone marrow. In addition to serving as a barrier, EC-specific deletion of Scf (Kitl), Cxcl12, Jag1, and Dll4 impacted HSC quiescence, differentiation, and localization (Ding et al., 2012; Ding and Morrison, 2013; Poulos et al., 2013). Stromal cells make up another major constituent and multiple sub-populations have been previously described, including Lepr+ MSPCs, NG2+ (CSPG4, a pericyte marker) cells, and CAR cells. NG2+ cells unsheathe arterioles have been previously proposed to maintain HSC quiescence (Kunisaki et al., 2013). Osteoblasts derived from the bone marrow MSPCs were previously shown to support early lymphoid progenitors (Ding and Morrison, 2013).
Recent studies extended single-cell transcriptomic profiling to the non-hematopoietic compartment of the murine bone marrow to further understand the combination of signals and their cellular source responsible for HSC maintenance and differentiation. As the frequencies of bone marrow cell types vary by orders of magnitude (Gomariz et al., 2018), some groups utilized an approach of depleting abundant hematopoietic cell types to isolate the niche populations in an unbiased manner while others devised a positive selection approach to focus on previously identified subsets (Table 1).
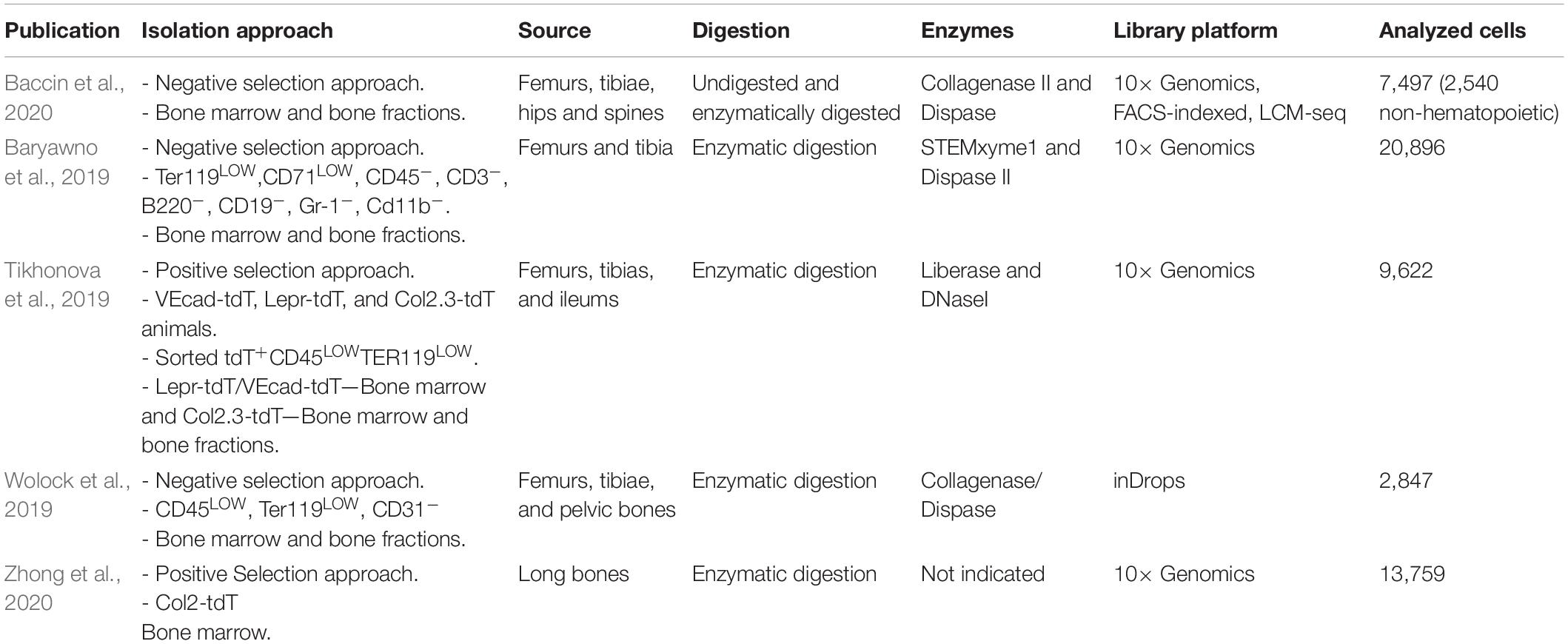
Table 1. Recent scRNA-seq studies profiling the non-hematopoietic compartment of the murine bone marrow.
Our group took advantage of lineage-specific Cre-labeling to profile 9,622 cells representing major niche subsets previously shown to play a role in hematopoiesis using the 10× Genomics platform (Tikhonova et al., 2019). Clustering analysis detected two endothelial, four perivascular, and three osteo-lineage subpopulations, validated using orthogonal approaches such as immunofluorescence or flow cytometry-based on the identified biomarkers. The vascular endothelial subset marked by expression of Cdh5 (VE-Cadherin) contained cells representing Stab2+ and Flt4+ sinusoidal capillaries and a smaller (12% of ECs) fraction of Ly6a+ and Cd34+ arterial cells. The Lepr+ perivascular compartment consisted primarily of adipo-primed mesenchymal cells characterized by high expression of Cxcl12 and adipogenic markers such as Adipoq and Lpl as well as osteo-primed cells defined by gradually increasing expression of osteogenic genes such as Bglap and Spp1. This was a surprising finding, given that the bone of young animals contains few adipocytes. The osteo-lineage subset was comprised of three transcriptionally distinct clusters representing osteoblasts, chondrocytes, and fibroblasts. To infer the relationship between the mesenchymal lineage cells, the developmental trajectory was reconstructed using Monocle (Trapnell et al., 2014). Pseudotime ordering of the perivascular and terminally differentiated osteo-lineage cells revealed a continuum of cellular states with progressively increasing levels of expression of adipogenic markers toward one endpoint and osteogenic markers toward the opposite end. The adipo-primed sub-populations were enriched for previously identified human bone marrow mesenchymal stem cell gene signature (Ghazanfari et al., 2016), and functional studies showed higher activity of fibroblastic colony-forming units. In contrast to osteo-rimed progenitors that were tdTomato+ but expressed lower levels of Lepr, adipo-primed progenitors were both high in Lepr expression and tdTomato+. Taken together these findings indicate that early mesenchymal progenitors reside within adipo-gene expressing Lepr+ MSPC fraction.
Wolock et al. (2019) examined the mesenchymal lineage cells to determine the relations and hierarchies of stromal progenitors as they differentiate toward the mature bone, fat, and cartilage cells. 2,847 sorted non-hematopoietic (CD45–Ter119–) and non-endothelial (CD31–) cells were profiled by 3′ droplet-based inDrop scRNA-seq with a median of 736 detected genes per a cell. Spectral clustering identified 7 clusters labeled as MSC, adipocyte progenitors, pre-adipocytes, osteoblast/chondrocyte progenitors, pre-osteoblast/chondrocytes, pro-osteoblasts, and pro-chondrocytes. SPRING visualization exposed a transcriptional continuum across the adipogenic and osteogenic branches. Differentiation trajectories were inferred with Velocyto (La Manno et al., 2018), a method to quantify the relationship between the abundance of precursor and mature mRNA, predicting individual cells’ future state. RNA velocity analysis suggested that the MSC subpopulation was atop the differentiation hierarchy with branches toward the pre-adipocyte, pro-osteoblast, and pro-chondrocyte endpoints. PBA was utilized as an independent methodology to resolve each of the three lineages’ average transcriptional trajectory and order genes based on their expression pattern along those trajectories.
Utilizing a negative selection approach, Baryawno et al. (2019) profiled 20,896 bone marrow Lin- cells and identified 17 clusters originating from bone and bone marrow, including endothelial cells, MSCs, osteolineage cells, chondrocytes, fibroblasts, and pericytes. Multiple methods were utilized to resolve differentiation relationships between the populations, including the correlation of expression profiles between clusters and diffusion map analyses. The clusters’ connectivity was evaluated with partition-based graph abstraction (PAGA) that generates a graph-like map of data maintaining both its continuous and disconnected structure. Three endothelial clusters were identified, spanning a transcriptional continuum from Flt4 + sinusoidal to Ly6a + arteriolar sub-populations. The latter additionally had a sub-population of cells from endosteal and bone marrow arteries, marked by expression of Vwf and Kitl. Mesenchymal cells, classified based on the expression of Lepr and Cxcl12, were further divided into four subsets with varying levels of expression of those markers. One was characterized by osteo-lineage markers Sp7 and Alpl, indicating differentiation toward that lineage. This relationship was further profiled by analyzing the two osteolineage clusters, one covering a range of differentiation states and a transcriptionally distinct subset of mostly cells from the bone fraction. An additional distinct subpopulation of perivascular mesenchymal stromal cells and pericytes was identified with low levels of Lepr but high levels of Nes and NG2 (Cspg4) as well as pericytes markers Acta2, Myh11, and Mcam.
Baccin et al. (2020) combined single-cell with spatially resolved transcriptomics. To compensate for the highly variable abundance of cell types within the bone marrow, undigested bone marrow or enzymatically digested bones were depleted or enriched for populations of interest. The final dataset included 7,497 cells grouped into 32 clusters. The total bone marrow yielded hematopoietic populations. Those populations’ depletion resulted in primarily erythroid progenitors with low expression of Cd45 and 2% non-hematopoietic cells. These rare populations were captured by depleting cells expressing CD45 or an erythroid marker CD71. The identified non-hematopoietic populations included Schwann cells, smooth muscle cells, myofibroblasts, Pdgfra+ mesenchymal cells, and endothelial cells. The endothelial subset was split into arterial and sinusoidal cells. The mesenchymal subset comprised nine sub-populations, including chondrocytes, osteoblasts, three fibroblast clusters, Ng2+Nestin+ MSCs, and two populations similar to Cxcl12-abundant reticular (CAR) cells. The two CAR sub-populations differed primarily in their expression of adipocyte (Adipoq) and osteo-lineage (Sp7, Bglap) genes. RNA velocity analysis of the mesenchymal lineage cells assigned the Ng2+ MSCs atop the differentiation hierarchy.
Baccin et al. (2020) additionally developed a laser-capture microdissection protocol coupled with sequencing (LCM-seq) to overcome spatial transcriptomic approaches that have not been successful in the bone marrow due to dependence on high RNA quality or unfixed tissue material. The method yielded full-length transcriptomic data from bone marrow sections containing 200-300 cells in a layer. LCM-seq was applied to 76 micro-dissected regions from the diaphyseal bone marrow to localize the bone marrow populations to endosteal, sinusoidal, arteriolar, and non-vascular niches. The frequencies of scRNA-seq populations within the spatially resolved transcriptomic data were estimated with CIBERSORT to determine the bone marrow cell types’ localization to candidate niches. Osteoblasts and chondrocytes localized to the endosteal niches. Arterial endothelial cells and smooth muscle cells were found in the arteriolar niches. Sinusoidal cells localized to the sinusoidal niches but were also detected in the (sub)-endosteal niches. Adipo-CAR cells were associated with high sinusoidal areas, but the Osteo-CAR cells were found in arteriolar or non-vascular niches. Additionally, to predict these cell types’ spatial relationships exclusively based on scRNA-seq, the RNA-Magnet method was developed, taking advantage of known cellular adhesion receptors and cognate ligands. The inferred adhesiveness of the identified populations to distinct niches correlated with the localization as measured by spatial transcriptomics. RNA-Magnet was also applied to signaling mediators, such as cytokines and growth factors, expressed by all cells in the dataset. The analysis suggested that the HSPCs were more likely to receive signals from non-hematopoietic populations than the mature immune cells, indicating a shift from mesenchymal to immune signaling. Both Adipo-CAR and Osteo-CAR cells were the primary sources of signals recognized by progenitor populations.
Zhong et al. (2020) performed additional profiling of mesenchymal lineage cells in young, adult, and aging mice. The study used a Col2-Cre mouse model as a strategy to capture the most primitive mesenchymal lineage cells. 13,759 cells with a median of 2,686 detected genes were captured from young (1-1.5-month-old) animals. Clustering analysis identified 22 populations, of which 9 were mesenchymal, 11 hematopoietic, 1 endothelial, and 1 mural. The 7,585 mesenchymal linage cells contained osteoblast, osteocyte, adipocyte, and chondrocyte populations as well as four progenitor subsets. Early mesenchymal progenitors (EMPs) were determined as the most primitive cells in the dataset based on the expression of stemness markers Sca1, Cd34, and Thy1. Intermediate mesenchymal progenitors and late mesenchymal progenitors expressed progressively higher levels osteo-lineage genes such as Sp7 and Col1a1. Differentiation trajectory inference with Monocle placed EMPs at the terminal end of one of the three branches, with terminally differentiated osteocytes and adipocytes at the other ends. Lineage committed progenitors were placed at the point of bifurcation. Pseudotemporal reconstruction was additionally performed with Slingshot, which builds minimum spanning trees from clusters as opposed to single cells with Monocle. UMAP cell embeddings were used for trajectory inference, resulting in an analogous branching structure.
The single-cell studies have been able to reveal extensive heterogeneity in the populations thought to be homogeneous. One of the challenges associated with the rapid flow of this new information is an understanding of how the populations described in these studies are related. Furthermore, inconsistent nomenclature further complicates one’s ability to compare and contrast the data. To address this gap, we reanalyzed the five discussed scRNA-seq datasets combined (Table 1). We calculated the cluster markers for the populations identified within each dataset and performed hierarchical clustering based on the scaled expression correlation distances of those genes (Figure 1A). To better assess the heterogeneity of the described populations and their transcriptional relationships, we additionally performed a joint analysis of 32,743 cells from the three datasets encompassing the most diverse subsets. Using the Seurat anchor-based data integration pipeline (Stuart et al., 2019), we were able to overlay them and visualize the populations using a consistent representation (Figure 1B). Despite distinct sample preparation strategies (enzyme cocktails, digestion time, sorting and sorting strategies), assayed fraction (bone marrow or a combination of bone marrow and bone), and computational analysis approaches (summarized in Table 2), the findings are largely in agreement among the experiments carried out by independent groups. Expectantly, the relative abundances of various subpopulations fluctuated across the datasets (Figure 1C). For example, Baryawno dataset contained a large fraction of chondrocytes and fibroblasts, whereas Baccin subset uniquely captured myofibroblasts and Schwann cells.
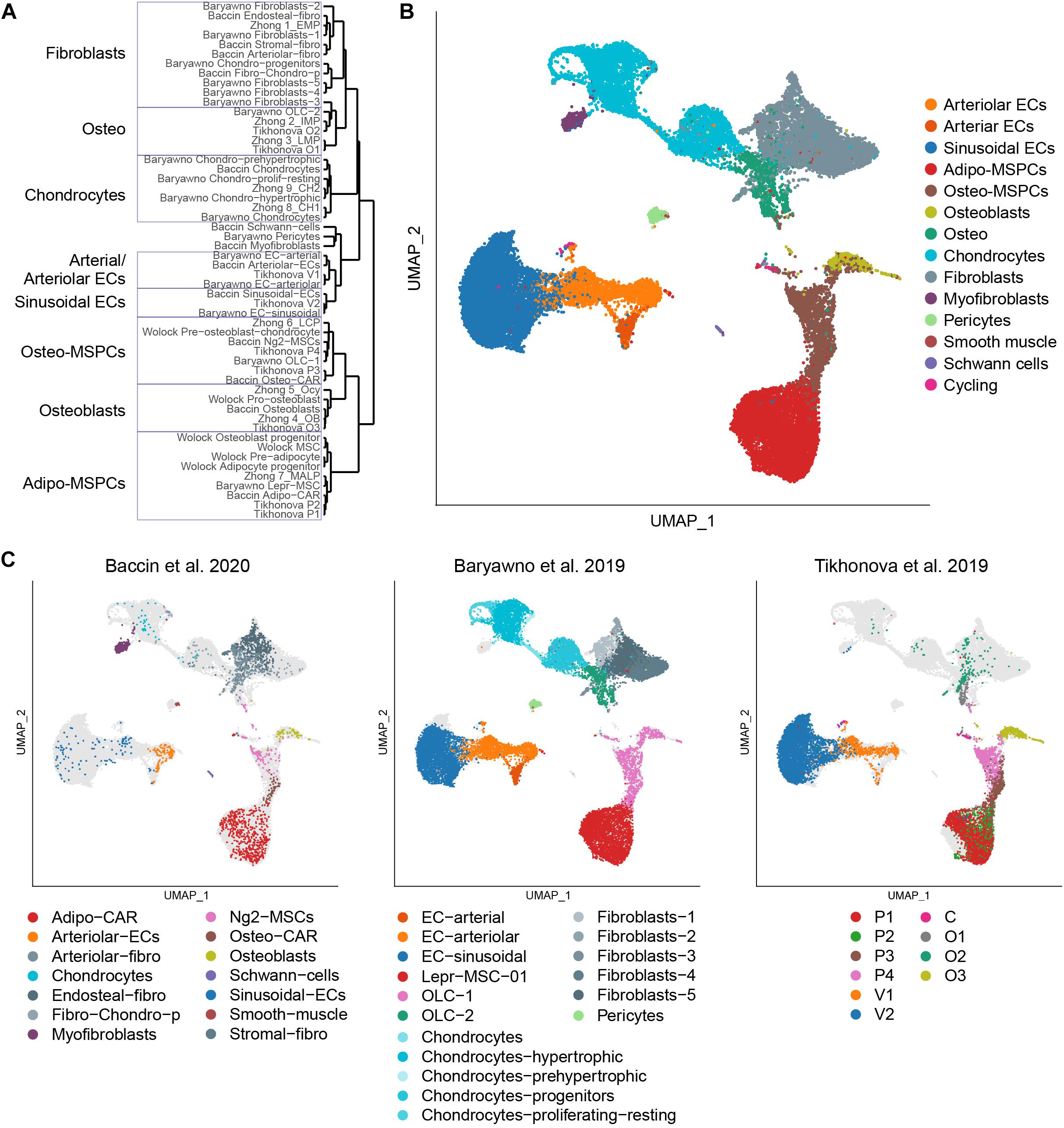
Figure 1. Integrated analysis of the bone marrow niche datasets. (A) Hierarchical clustering of the populations profiled in the discussed studies based on the scaled expression correlation distances of cluster marker genes. (B) UMAP visualization of all 32,743 cells from the integrated analysis, color-coded based on the harmonized population labels. (C) UMAP visualization of each of the datasets, color-coded based on the original population labels. Cells from the non-represented datasets are shown in light gray.

Table 2. Computational approaches utilized by scRNA-seq studies profiling the bone marrow.
Based on the expression patterns of cluster markers and visualization of integrated datasets, we harmonized the various sub-populations identified in the original studies using a consistent nomenclature (Figure 1B). Bone marrow vascular ECs were represented by sinusoidal (blue) and arteriolar (orange). By sampling a higher number of cells, Baryawno et al. identified an additional smaller sub-population of cells from endosteal and bone marrow arteries (dark orange). The mesenchymal stem cells give rise to a diverse range of cells such as adipocytes, osteoblasts, and chondrocytes. Previous studies took advantage of either Cxcl12 or Lepr to isolate perivascular cells, but the extent of the overlap of the two populations was not clear. The scRNA-seq work revealed a broad transcriptional heterogeneity within these subsets. Bone marrow mesenchymal progenitors split into adipo-MSPCs (red) and osteo-MS (brown). Adipogenesis-primed mesenchymal cells were characterized by high expression of both Lepr and Cxcl12. These key markers were gradually decreased as the cells progressed toward osteogenesis-primed subpopulations. Osteoblasts (olive), chondrocytes (cyan) and a variety of fibroblast populations (gray) were represented across different dataset. Single cell studies are limited by the efficiency of digestion protocols and rare populations may not be sufficiently sampled. A more granular deconvolution of smaller bone subsets will require focused functional studies. To resolve transcriptional relationships between distinctly labeled clusters, the readers can refer to Figure 1A. To confirm that the gene expression of the integrated dataset corresponded to the populations identified in the individual studies, we assessed most differentially expressed marker genes (Figures 2A,B) and mapped expression patters of key pro-hematopoietic factors in the bone (Figure 2C). Indeed, we found the expression of Cxcl12 and Kitl to be enriched in adipo-MSCPs progenitors and arteriolar ECs, while Il7 and Il34 were expressed primarily in adipo-MSPCs (Figure 2C). Collectively, our brief survey highlights a concordance between five different bone marrow niche single-cell datasets and resolves inconsistent nomenclature offered by distinct scientific groups.
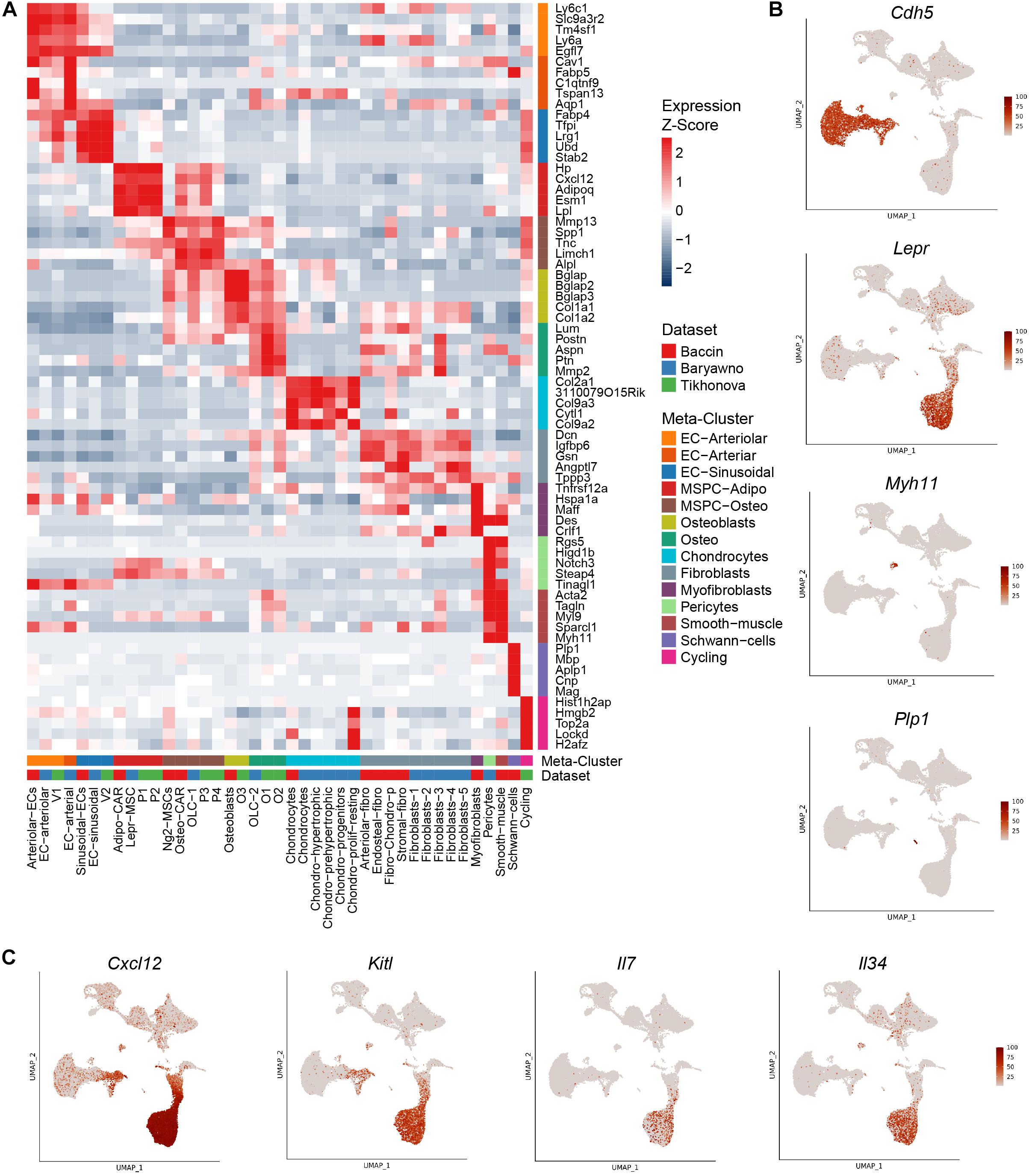
Figure 2. Expression patterns of marker genes and pro-hematopoietic factors. (A) Relative expression levels of the markers of the combined clusters within the populations defined in the original studies. (B) Expression levels of population marker genes Cdh5 (ECs), Lepr (perivascular cells), Myh11 (pericytes/smooth muscle), Plp1 (Schwann cells) overlaid on the UMAP. (C) Expression levels of pro-hematopoietic factors Cxcl12, Kitl, Il7, and Il34 overlaid on the UMAP.
Vascular ECs comprise a highly organized network throughout the body, facilitating the distribution of oxygen and nutrients. In addition to their delivery function, ECs enable immune cell trafficking allowing for cell migration and immune surveillance. Vascular ECs display tissue-specific morphology and have been shown in some cases to carry organ-specific functions. Blood-brain barrier and kidney glomeruli vasculature have been areas of active research, but the specifics of vascular cells in other tissues are less understood. High-throughput transcriptional profiling studies compared molecular architecture of ECs across various organs, revealing a marked transcriptional heterogeneity (Chi et al., 2003; Nolan et al., 2013). In addition to variability driven by tissues, each organ contains several vessel types, including arteries, veins, and capillaries. Recent single-cell RNA-seq analysis of > 32,000 ECs isolated from 11 different mouse tissues at the steady-state showed that vascular heterogeneity is largely dictated by the anatomical location of ECs rather than the vessel type (Kalucka et al., 2020), suggesting tissues-mediated molding of the vasculature and tissues specific functions.
In the bone marrow, ECs serve multiple roles, including HSC maintenance and leukocyte trafficking. Highly branched sinusoidal capillaries (SECs) carry venous blood and make up the vast majority of vascular cells. Arteriole vessels (AECs) carry arterial blood into the bone, have few branches, and constitute around ∼10% of the total bone marrow vasculature (Xu et al., 2018; Tikhonova et al., 2019). Here, we specifically focused on the vascular subset from the combined single-cell bone marrow niche datasets (Figure 3A). We calculated module scores for ECs based on the sinusoidal and arterial gene expression signatures obtained from the bulk tissue analysis (Xu et al., 2018) and found that 67.3% of cells were enriched for sinusoidal and 23.5% for the arterial signatures (Figure 3B). To further confirm the identity of the cells, we assessed the expression levels of population marker genes and found Flt4, Tfpi, Stab2, and Vcam1 to be highly specific to sinusoids (Figure 3C) and Cxl12, Kitl, Gja4, Vegfc to arterioles (Figure 3D). Previous studies indicated that sinusoids and arterioles possess different permeability properties (Itkin et al., 2016). Notably, hematopoietic stem cell adhesion, rolling and trans-endothelial events were reported exclusively in SECs (Itkin et al., 2016). Indeed, sinusoids are enriched for expression of adhesion molecules such as E-selectin (Sele), Vcam-1 (Vcam1), ICAM-1 (Icam1), while arterioles expressed ICAM-2 (Icam2) (Figure 3E), consistent with the idea of differential trans-endothelial migration. It is not clear if all sinusoids can support trans-endothelial migration and it is tempting to speculate that there might be specialized sinusoid subtypes facilitating leukocyte trafficking.
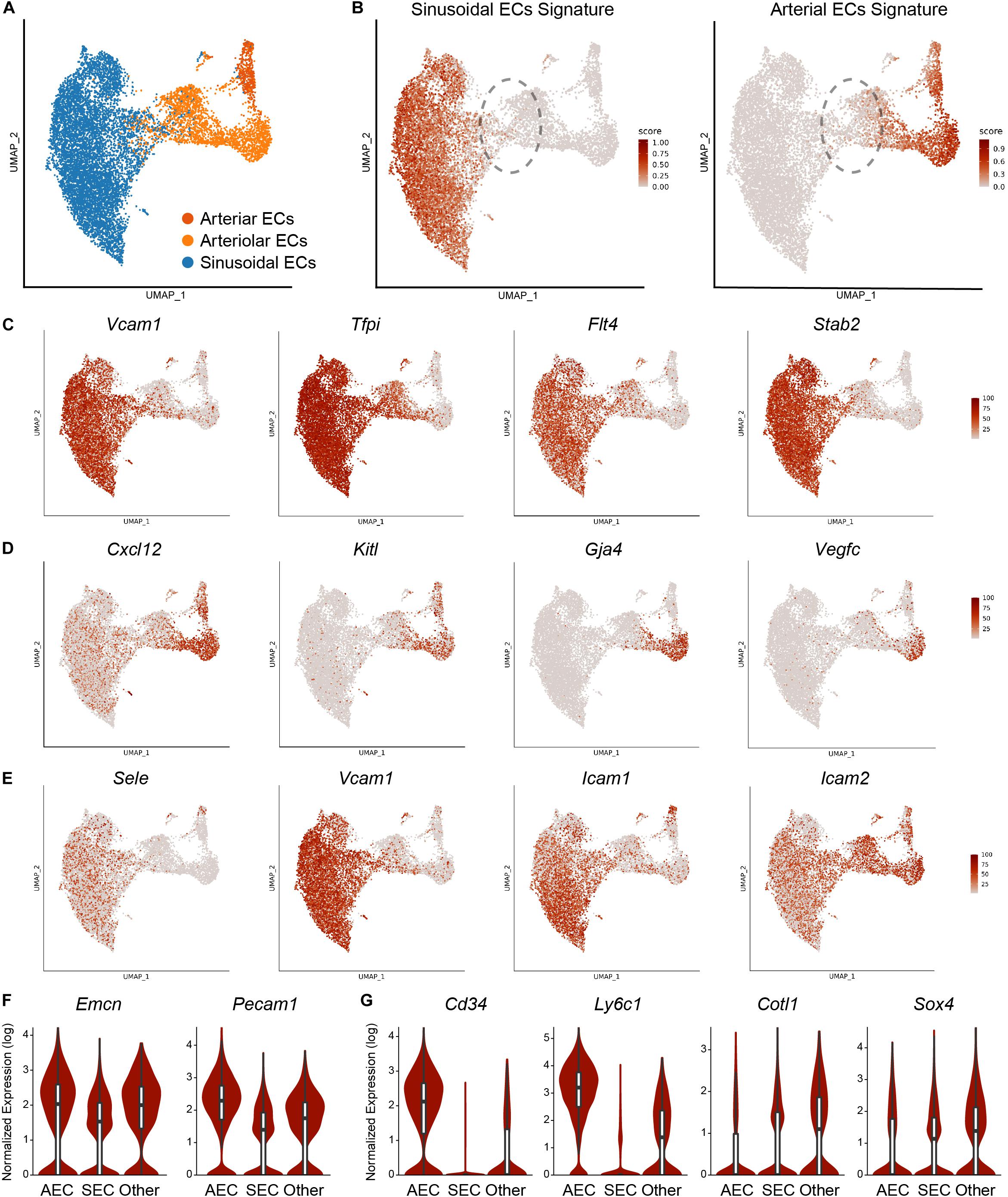
Figure 3. Analysis of the EC subset. (A) UMAP visualization of 10,578 ECs from the integrated analysis, color-coded based on the harmonized population labels. (B) Module scores for sinusoidal and arterial gene signatures overlaid on the UMAP. Dashed outline highlights the population not enriched for sinusoidal or arterial signatures. Expression of sinusoidal (C) and arteriolar (D) marker genes as well as adhesion molecules (E) overlaid on the UMAP representation. (F) Expression of transitional H vessel cell-surface markers within the populations defined by sinusoidal and arterial EC signatures. (G) Expression of marker genes within the populations defined by sinusoidal and arterial EC signatures.
Within the bone marrow, transitional zone vessels connect bone marrow sinusoids and arterioles. Based on the cell-surface marker expression and phenotypic characteristics, Kasumbe et al. identified two subtypes of SECs: CD31lowEMCNlow L-vessels form highly branched capillary networks throughout the bone marrow and represent the majority of the SECs. CD31highEMCNhigh H-vessels were found in the metaphysis, organized as vessel columns, and in transitional zones bridging arterioles with the sinusoids (Kusumbe et al., 2014). H-vessels are of a substantial clinical interest, as they were shown to regulate osteogenesis (Kusumbe et al., 2014, 2016; Ramasamy et al., 2014). We were not able to clearly identify transitional vessels with our own transcriptional dataset (Tikhonova et al., 2019). However, exploring the combined analysis based on the three studies allowed us to pinpoint a subset of ECs that were not enriched for either arterial or sinusoidal signatures. Further examination revealed that these transition cells expressed higher levels of Emcn and Cd31 compared to sinusoids, suggesting that they might represent transitional H vessels (Figure 3F). We found this population to be enriched for arterial-associated genes Cd34 and Ly6c1 but also express unique markers such as Cotl1 and Sox4 (Figure 3G). Collectively, this integrated analysis allows for deeper exploration of BM EC heterogeneity. Although transitional EC population was not clearly revealed by each individual analysis, combining the datasets allowed for their further exploration, underscoring the utility of publicly available data to the community.
A recent study reports even further specification among bone marrow SECs. Chen and Liu et al. demonstrated that Apelin+ (Apln) bone marrow ECs control vascular regeneration following irradiation-induced vascular damage. These specialized SECs make up approximately 0.03% of the total bone marrow and are located in the metaphysis, endosteum, and the transition area between the metaphysis and diaphysis. Bulk RNA-seq of Apln+ cells revealed an enrichment of pathways related to angiogenesis and vascular endothelial growth factor (VEGF) signaling. Importantly, irradiation triggers the expansion of Apln+ ECs and vascular regeneration. Spatial organization of hematopoietic differentiation is another open question. Elegant work by Zhang et al. revealed that following acute systemic infection, monocyte–dendritic cell progenitors localize to a subset of blood vessels expressing colony-stimulating factor 1 (Csf1), known to be a key regulator of myelopoiesis. Indeed, EC-specific deletion of Csf1, results in a loss of non-classical monocytes and dendritic cells during steady-state and following infection (Zhang et al., 2021). It remains to be elucidated if specialized ECs also support progenitors of lymphoid and erythroid lineages. Further mining of single-cell bone marrow niche datasets, single-cell sequencing of ECs following stress scenarios, combined with receptor-ligand interaction analysis (Browaeys et al., 2019; Cabello-Aguilar et al., 2020; Efremova et al., 2020) and clever functional studies will reveal further specialization of vascular ECs.
The precise identity of the vascular cells that support HSCs continues to be debated. Combining computational modeling with whole-mount confocal immunofluorescence imaging techniques, Kunisaki et al. found quiescent HSCs to be closely associated with AECs (Kunisaki et al., 2013). Optically clearing studies of the bone marrow, that allowed to perform deep confocal imaging, demonstrate that independently of their cycling status, approximately 80% of HSCs are closely associated with SECs, with another 10% of HSCs being adjacent to AECs, and 10% being adjacent to transition zone vessels (Acar et al., 2015). In contrast, Kunisaki et al., using whole-mount confocal immunofluorescence imaging, showed that quiescent HSCs associate specifically with small arterioles that are preferentially found in endosteal bone marrow. Furthermore, Itkin et al. demonstrated that arterioles maintain hematopoietic stem cells in a low reactive species oxygen state (Itkin et al., 2016), perpetuating the debate regarding the precise identity of vascular cells that support quiescent HSCs. Just as novel technologies have been driving forward our understanding of hematopoietic differentiation, further advances combining multi-modal single-cell approaches, such as pairing gene expression data with spatial tissue context, and in vivo imaging will shape our understanding of hematopoietic cell interactions with their niches and resolve standing questions.
In a span of less than two years, our understanding of the hematopoietic differentiation and bone marrow microenvironment has made a major leap forward. Despite these advances, key questions remain unanswered (Tikhonova et al., 2020). For example, cellular identity of the bone marrow mesenchymal stem cells remains to be elucidated. Despite highly comparable data, pseudo-temporal ordering revealed conflicting differential MSPC hierarchies and led to distinct conclusions. We believe that scRNA-seq analyses are able to guide our understanding of MSPC biology and their exploration can inspire creativity in our scientific inquiry. However, they cannot replace functional experiments required to answer complex biological questions.
Single-cell profiling of hematopoietic hierarchy highlighted a fluidic nature of differentiation. Although we understand some of the intrinsic signals implementing these decisions, we do not yet grasp which extrinsic factors mediate hematopoietic differentiation. We speculate that specific temporospatial combinations of bone marrow niche signals guide hematopoietic progenitors toward gradual commitment to a lineage choice. Novel computational approaches and genetic strategies are required to connect these dots.
An interactive browser of the integrated analysis presented in this study is available at the Broad Institute Single Cell Portal (https://singlecell.broadinstitute.org/single_cell/study/SCP1248). The data from the integrated analysis is available through the Open Science Framework (https://osf.io/ne9vj).
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by American Society of Hematology (A.N.T.).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acar, M., Kocherlakota, K. S., Murphy, M. M., Peyer, J. G., Oguro, H., Inra, C. N., et al. (2015). Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature 526, 126–130. doi: 10.1038/nature15250
Angerer, P., Haghverdi, L., Büttner, M., Theis, F. J., Marr, C., and Buettner, F. (2016). destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243. doi: 10.1093/bioinformatics/btv715
Baccin, C., Al-Sabah, J., Velten, L., Helbling, P. M., Grünschläger, F., Hernández-Malmierca, P., et al. (2020). Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol. 22, 38–48. doi: 10.1038/s41556-019-0439-6
Baryawno, N., Przybylski, D., Kowalczyk, M. S., Kfoury, Y., Severe, N., Gustafsson, K., et al. (2019). A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell 177, 1915–1932.e16. doi: 10.1016/j.cell.2019.04.040
Becht, E., McInnes, L., Healy, J., Dutertre, C.-A. A., Kwok, I. W. H. H., Ng, L. G., et al. (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–47. doi: 10.1038/nbt.4314
Browaeys, R., Saelens, W., and Saeys, Y. (2019). NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162. doi: 10.1038/s41592-019-0667-5
Butler, A., Hoffman, P., Smibert, P., Papalexi, E., and Satija, R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. doi: 10.1038/nbt.4096
Cabello-Aguilar, S., Alame, M., Kon-Sun-Tack, F., Fau, C., Lacroix, M., and Colinge, J. (2020). SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 48:e55. doi: 10.1093/nar/gkaa183
Cabezas-Wallscheid, N., Klimmeck, D., Hansson, J., Lipka, D. B., Reyes, A., Wang, Q., et al. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522. doi: 10.1016/j.stem.2014.07.005
Chi, J. T., Chang, H. Y., Haraldsen, G., Jahnsen, F. L., Troyanskaya, O. G., Chang, D. S., et al. (2003). Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. U.S.A. 100, 10623–10628. doi: 10.1073/pnas.1434429100
Ding, L., and Morrison, S. J. (2013). Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235. doi: 10.1038/nature11885
Ding, L., Saunders, T. L., Enikolopov, G., and Morrison, S. J. (2012). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462. doi: 10.1038/nature10783
Efremova, M., Vento-Tormo, M., Teichmann, S. A., and Vento-Tormo, R. (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15, 1484–1506. doi: 10.1038/s41596-020-0292-x
Ghazanfari, R., Li, H., Zacharaki, D., Lim, H. C., and Scheding, S. (2016). Human non-hematopoietic CD271 pos/CD140a low/neg bone marrow stroma cells fulfill stringent stem cell criteria in serial transplantations. Stem Cells Dev. 25, 1652–1658. doi: 10.1089/scd.2016.0169
Gomariz, A., Helbling, P. M., Isringhausen, S., Suessbier, U., Becker, A., Boss, A., et al. (2018). Quantitative spatial analysis of haematopoiesis-regulating stromal cells in the bone marrow microenvironment by 3D microscopy. Nat. Commun. 9:2532. doi: 10.1038/s41467-018-04770-z
Haghverdi, L., Buettner, F., and Theis, F. J. (2015). Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 31, 2989–2998. doi: 10.1093/bioinformatics/btv325
Itkin, T., Gur-Cohen, S., Spencer, J. A., Schajnovitz, A., Ramasamy, S. K., Kusumbe, A. P., et al. (2016). Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 532, 323–328. doi: 10.1038/nature17624
Kalucka, J., de Rooij, L. P. M. H., Goveia, J., Rohlenova, K., Dumas, S. J., Meta, E., et al. (2020). Single-cell transcriptome atlas of murine endothelial cells. Cell 180, 764–779.e20. doi: 10.1016/j.cell.2020.01.015
Kiel, M. J., Yilmaz, ÖH., Iwashita, T., Yilmaz, O. H., Terhorst, C., Morrison, S. J., et al. (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. doi: 10.1016/j.cell.2005.05.026
Kowalczyk, M. S., Tirosh, I., Heckl, D., Rao, T. N., Dixit, A., Haas, B. J., et al. (2015). Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 25, 1860–1872. doi: 10.1101/gr.192237.115
Kunisaki, Y., Bruns, I., Scheiermann, C., Ahmed, J., Pinho, S., Zhang, D., et al. (2013). Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643. doi: 10.1038/nature12612
Kusumbe, A. P., Ramasamy, S. K., and Adams, R. H. (2014). Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507, 323–328. doi: 10.1038/nature13145
Kusumbe, A. P., Ramasamy, S. K., Itkin, T., Mäe, M. A., Langen, U. H., Betsholtz, C., et al. (2016). Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature 532, 380–384. doi: 10.1038/nature17638
La Manno, G., Soldatov, R., Zeisel, A., Braun, E., Hochgerner, H., Petukhov, V., et al. (2018). RNA velocity of single cells. Nature 560, 494–498. doi: 10.1038/s41586-018-0414-6
Laurenti, E., and Göttgens, B. (2018). From haematopoietic stem cells to complex differentiation landscapes. Nature 553, 418–426. doi: 10.1038/nature25022
Liggett, L. A., and Sankaran, V. G. (2020). Unraveling hematopoiesis through the lens of genomics. Cell 182, 1384–1400. doi: 10.1016/j.cell.2020.08.030
McInnes, L., Healy, J., and Melville, J. (2018). UMAP: uniform manifold approximation and projection for dimension reduction. arXiv [Preprint]. Available online at: http://arxiv.org/abs/1802.03426 (accessed February 10, 2021).
Nestorowa, S., Hamey, F. K., Pijuan Sala, B., Diamanti, E., Shepherd, M., Laurenti, E., et al. (2016). A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–e31. doi: 10.1182/blood-2016-05-716480
Nolan, D. J., Ginsberg, M., Israely, E., Palikuqi, B., Poulos, M. G., James, D., et al. (2013). Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev. Cell 26, 204–219. doi: 10.1016/j.devcel.2013.06.017
Paul, F., Arkin, Y., Giladi, A., Jaitin, D. A., Kenigsberg, E., Keren-Shaul, H., et al. (2015). Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell 163, 1663–1677. doi: 10.1016/j.cell.2015.11.013
Pellin, D., Loperfido, M., Baricordi, C., Wolock, S. L., Montepeloso, A., Weinberg, O. K., et al. (2019). A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun. 10:2395. doi: 10.1038/s41467-019-10291-0
Pietras, E. M., Reynaud, D., Kang, Y.-A., Carlin, D., Calero-Nieto, F. J., Leavitt, A. D., et al. (2015). Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell 17, 35–46. doi: 10.1016/j.stem.2015.05.003
Pinho, S., and Frenette, P. S. (2019). Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 20, 303–320. doi: 10.1038/s41580-019-0103-9
Poulos, M. G., Guo, P., Kofler, N. M., Pinho, S., Gutkin, M. C., Tikhonova, A., et al. (2013). Endothelial jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep. 4, 1022–1034. doi: 10.1016/j.celrep.2013.07.048
Qiu, X., Hill, A., Packer, J., Lin, D., Ma, Y.-A., and Trapnell, C. (2017). Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 14, 309–315. doi: 10.1038/nmeth.4150
Ramasamy, S. K., Kusumbe, A. P., Wang, L., and Adams, R. H. (2014). Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 507, 376–380. doi: 10.1038/nature13146
Reya, T., Morrison, S. J., Clarke, M. F., and Weissman, I. L. (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105–111. doi: 10.1038/35102167
Street, K., Risso, D., Fletcher, R. B., Das, D., Ngai, J., Yosef, N., et al. (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19:477. doi: 10.1186/s12864-018-4772-0
Stuart, T., Butler, A., Hoffman, P., Hafemeister, C., Papalexi, E., Mauck, W. M., et al. (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. doi: 10.1016/j.cell.2019.05.031
Sugiyama, T., Kohara, H., Noda, M., and Nagasawa, T. (2006). Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988. doi: 10.1016/j.immuni.2006.10.016
Tikhonova, A. N., Dolgalev, I., Hu, H., Sivaraj, K. K., Hoxha, E., Cuesta-Domínguez, Á, et al. (2019). The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228. doi: 10.1038/s41586-019-1104-8
Tikhonova, A. N., Lasry, A., Austin, R., and Aifantis, I. (2020). Cell-by-cell deconstruction of stem cell niches. Cell Stem Cell 27, 19–34. doi: 10.1016/j.stem.2020.06.013
Trapnell, C., Cacchiarelli, D., Grimsby, J., Pokharel, P., Li, S., Morse, M., et al. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. doi: 10.1038/nbt.2859
Tusi, B. K., Wolock, S. L., Weinreb, C., Hwang, Y., Hidalgo, D., Zilionis, R., et al. (2018). Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 555, 54–60. doi: 10.1038/nature25741
van der Maaten, L., and Hinton, G. (2008). Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605.
Velten, L., Haas, S. F., Raffel, S., Blaszkiewicz, S., Islam, S., Hennig, B. P., et al. (2017). Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 19, 271–281. doi: 10.1038/ncb3493
Watcham, S., Kucinski, I., and Gottgens, B. (2019). New insights into hematopoietic differentiation landscapes from single-cell RNA sequencing. Blood 133, 1415–1426. doi: 10.1182/blood-2018-08-835355
Weinreb, C., Wolock, S., and Klein, A. M. (2018a). SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246–1248. doi: 10.1093/bioinformatics/btx792
Weinreb, C., Wolock, S., Tusi, B. K., Socolovsky, M., and Klein, A. M. (2018b). Fundamental limits on dynamic inference from single-cell snapshots. Proc. Natl. Acad. Sci. U.S.A. 115, E2467–E2476. doi: 10.1073/pnas.1714723115
Weissman, I. L. (2000). Stem cells: units of development, units of regeneration, and units in evolution. Cell 100, 157–168. doi: 10.1016/S0092-8674(00)81692-X
Wolf, F. A., Hamey, F. K., Plass, M., Solana, J., Dahlin, J. S., Göttgens, B., et al. (2019). PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20:59. doi: 10.1186/s13059-019-1663-x
Wolock, S. L., Krishnan, I., Tenen, D. E., Matkins, V., Camacho, V., Patel, S., et al. (2019). Mapping distinct bone marrow niche populations and their differentiation paths. Cell Rep. 28, 302–311.e5. doi: 10.1016/j.celrep.2019.06.031
Xu, C., Gao, X., Wei, Q., Nakahara, F., Zimmerman, S. E., Mar, J., et al. (2018). Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat. Commun. 9:2449. doi: 10.1038/s41467-018-04726-3
Zhang, J., Wu, Q., Johnson, C. B., Pham, G., Kinder, J. M., Olsson, A., et al. (2021). In situ mapping identifies distinct vascular niches for myelopoiesis. Nature. 590, 457–462. doi: 10.1038/s41586-021-03201-2
Zheng, S., Papalexi, E., Butler, A., Stephenson, W., and Satija, R. (2018). Molecular transitions in early progenitors during human cord blood hematopoiesis. Mol. Syst. Biol. 14:e8041. doi: 10.15252/msb.20178041
Keywords: microenvironment, stromal – hematopoietic cells interactions, bone marrow, cell-to cell communication, hematopoiesis, single-cell RNA-seq (scRNA-seq), single-cell ‘omics
Citation: Dolgalev I and Tikhonova AN (2021) Connecting the Dots: Resolving the Bone Marrow Niche Heterogeneity. Front. Cell Dev. Biol. 9:622519. doi: 10.3389/fcell.2021.622519
Received: 28 October 2020; Accepted: 15 February 2021;
Published: 12 March 2021.
Edited by:
Tomer Itkin, Cornell University, United StatesReviewed by:
Simon Haas, Charité Medical University of Berlin, GermanyCopyright © 2021 Dolgalev and Tikhonova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Igor Dolgalev, SWdvci5Eb2xnYWxldkBueXVsYW5nb25lLm9yZw==; Anastasia N. Tikhonova, QW5hc3Rhc2lhLlRpa2hvbm92YUB1aG5yZXNlYXJjaC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.