 Aitor Rodriguez-Casanova1,2†
Aitor Rodriguez-Casanova1,2† Nicolás Costa-Fraga1†
Nicolás Costa-Fraga1† Aida Bao-Caamano1†
Aida Bao-Caamano1† Rafael López-López2,3,4
Rafael López-López2,3,4 Laura Muinelo-Romay4,5*
Laura Muinelo-Romay4,5* Angel Diaz-Lagares1,4*
Angel Diaz-Lagares1,4*- 1Cancer Epigenomics Laboratory, Translational Medical Oncology Group (Oncomet), Health Research Institute of Santiago (IDIS), University Clinical Hospital of Santiago (CHUS/SERGAS), Santiago de Compostela, Spain
- 2Roche-Chus Joint Unit, Translational Medical Oncology Group (Oncomet), Health Research Institute of Santiago (IDIS), Santiago de Compostela, Spain
- 3Translational Medical Oncology Group (Oncomet), Health Research Institute of Santiago (IDIS), University Clinical Hospital of Santiago (CHUS/SERGAS), Santiago de Compostela, Spain
- 4Centro de Investigación Biomédica en Red Cáncer (CIBERONC), Madrid, Spain
- 5Liquid Biopsy Analysis Unit, Translational Medical Oncology Group (Oncomet), Health Research Institute of Santiago (IDIS), University Clinical Hospital of Santiago (CHUS/SERGAS), Santiago de Compostela, Spain
Colorectal cancer (CRC) is one of the most common malignancies and is a major cause of cancer-related deaths worldwide. Thus, there is a clinical need to improve early detection of CRC and personalize therapy for patients with this disease. In the era of precision oncology, liquid biopsy has emerged as a major approach to characterize the circulating tumor elements present in body fluids, including cell-free DNA and RNA, circulating tumor cells, and extracellular vesicles. This non-invasive tool has allowed the identification of relevant molecular alterations in CRC patients, including some indicating the disruption of epigenetic mechanisms. Epigenetic alterations found in solid and liquid biopsies have shown great utility as biomarkers for early detection, prognosis, monitoring, and evaluation of therapeutic response in CRC patients. Here, we summarize current knowledge of the most relevant epigenetic mechanisms associated with cancer development and progression, and the implications of their deregulation in cancer cells and liquid biopsy of CRC patients. In particular, we describe the methodologies used to analyze these epigenetic alterations in circulating tumor material, and we focus on the clinical utility of epigenetic marks in liquid biopsy as tumor biomarkers for CRC patients. We also discuss the great challenges and emerging opportunities of this field for the diagnosis and personalized management of CRC patients.
Introduction
Colorectal cancer (CRC) is the third most frequently detected cancer in both sexes worldwide. In particular, this tumor type accounts for ~10% of all diagnosed cancer cases, with ~1.8 million new cases estimated in 2018. Importantly, CRC is the second leading cause of cancer mortality in the world, being responsible for ~9% of all cancer deaths (Bray et al., 2018). Surgery remains the most common treatment for non-metastatic CRC, while the administration of adjuvant chemotherapy is mainly restricted to stage III tumors. Importantly, most CRC patients are diagnosed at an advanced stage because symptoms normally appear after disease progression (John et al., 2011). The 5-year survival rate after surgery of localized CRC patients is over 90%, while patients affected by stage III and IV tumors, exhibiting local lymph node invasion or distant metastases, respectively, normally show poor overall survival rates (Mattiuzzi et al., 2019). The high incidence and mortality of CRC highlight the clinical need for novel strategies to improve early CRC detection and personalize the management of patients with this type of tumor.
Currently, various screening assays are being used to detect CRC at an early stage. These detection strategies include the fecal immunochemical test (FIT), a non-invasive and cost-effective assay for detecting the presence of fecal hemoglobin (Song and Li, 2016). A positive result of this test implies the recommendation of a colonoscopy, which is the gold standard diagnostic technique for CRC detection. However, this method is clearly invasive, requires considerable patient preparation, and can eventually lead to serious complications (Triantafillidis et al., 2015).
Importantly, a refined understanding of the molecular aspects of CRC has recently been achieved owing to the application of next generation sequencing (NGS)-based approaches, which revealed a wide intratumor heterogeneity and general genomic instability (Molinari et al., 2018). Based on the advances in the molecular characterization of CRC, new biological drugs targeting vascular endothelial growth factors (VEGFRs), such as bevacizumab (Rosen et al., 2017), or epidermal growth factors (EGFRs), including cetuximab and panitumumab (Qin et al., 2018; Taniguchi et al., 2020), have improved the survival of patients, mainly in the context of metastatic CRC (mCRC). In addition, promising targeted therapies for mCRC are being evaluated in pre-clinical and clinical studies, including new drugs directed against different components of EGF/EGFR, VEGF/VEGFR, and HGF/c-MET pathways (Xie Y. H. et al., 2020). On the other hand, several recent pre-clinical studies have shown that blocking the PD-1/PD-L1 interaction or CTLA-4 with immune checkpoint inhibitors prevents colorectal tumor cells to escape from immune surveillance (Fiegle et al., 2019; Zhang Y. et al., 2020). In this sense, the use of immune checkpoint inhibitors has provided good outcomes in the treatment of mCRC with microsatellite instability (MSI) (Overman et al., 2018), which can be detected in ~15% of CRC patients (Ward et al., 2001; Le et al., 2015). Importantly, several molecular biomarkers, such as the presence of KRAS/NRAS/BRAF mutations and MSI markers, have been approved for supporting the selection of targeted therapies (Lo Nigro et al., 2016). However, these predictive biomarkers are currently analyzed in tumor tissue samples, which are not always available during the disease evolution and can provide partial information of the molecular profile of colorectal tumors, mainly in the metastatic setting.
Liquid biopsy has emerged in recent years as an important approach to address and overcome such limitations. Indeed, this non-invasive strategy allows to observe the molecular landscape of circulating tumor elements in body fluids to obtain diagnostic, prognostic, and therapy response biomarkers that improve the management of cancer patients (Siravegna et al., 2017). The analysis of these liquid biopsy components in several body fluids of CRC patients has highlighted relevant molecular alterations, such as those depending on epigenetic mechanisms (Lofton-Day et al., 2008; Maminezhad et al., 2020). Importantly, the combination of liquid biopsy and detection of epigenetic alterations represents a great opportunity in cancer research for the identification of new non-invasive clinical biomarkers to improve the detection of CRC and personalize the management of this disease.
In this review, we provide an overview of the epigenetic landscape of liquid biopsies in CRC. We describe the concept and clinical application of liquid biopsy, the most relevant epigenetic mechanisms (DNA modifications, histone modifications and nucleosome positioning, and non-coding RNAs), and the implications of their deregulation in cancer cells and liquid biopsy. Furthermore, we summarize the methodologies used for detecting these epigenetic modifications in liquid biopsy, and describe the clinical utility of epigenetic marks in liquid biopsy as tumor biomarkers for CRC patients. Finally, we discuss the great challenges and opportunities of liquid biopsy epigenetics for the detection of CRC and management of CRC patients.
Liquid Biopsy
In the era of precision oncology, liquid biopsies represent a key element for cancer detection, to guide treatment selection and monitor tumor evolution in real time. A liquid biopsy consists of any body fluid that contains tumor material suitable for molecular characterization. Therefore, this term includes blood, the most used human liquid sample, but also other fluids such as urine, ascitic fluid, pleural effusion, cerebrospinal fluid, and saliva. Both primary tumors and metastases can release tumor material into these body fluids, mainly consisting of circulating tumor cells (CTCs), nucleic acids (cNAs), and extracellular vesicles (cEVs). These circulating elements constitute a valuable source of non-invasive biomarkers and information about the molecular mechanisms underpinning tumor dissemination and evolution (Siravegna et al., 2017).
Although Thomas Ashworth described the presence of tumor cells in the blood of breast cancer patients for the first time in 1869 (Ashworth, 1869), it was not until recent years that the scientific community focused their attention on the study of blood CTCs. Like most circulating tumor elements, CTCs are poorly concentrated in blood, and thus require the implementation of highly sensitive and specific strategies for their enrichment and subsequent detection. Initially, the most common strategy to isolate CTCs was immune enrichment, based on the presence of cell surface markers such as the epithelial cell adhesion molecule (EpCAM) (Allard et al., 2004). For example, the Food and Drug Administration (FDA)-approved CellSearch System can isolate EpCAM-positive cells and determine the number of CTCs in a sample after an immunofluorescence assay to detect epithelial and hematopoietic markers (Cristofanilli et al., 2004). This platform has been employed in numerous studies to quantify CTCs in CRC patients, demonstrating the clinical value of CTC enumeration as a prognostic and follow-up biomarker (Cohen et al., 2008; Sastre et al., 2012; Bork et al., 2015). However, this strategy has not been widely adopted as a clinical tool, mainly due to the lack of clear benefits in terms of accuracy of treatment decisions. Nevertheless, the technology for isolating and characterizing CTCs has continued to improve during the last decade, mainly through the application of antigen-independent technologies allowing to isolate a broader and more molecularly heterogeneous CTC population (Barbazan et al., 2017). Despite the difficulty of translating CTCs into the clinical context, their molecular characterization has provided valuable information for understanding how colorectal tumor cells are able to disseminate, implant at distant locations, and generate metastasis (Barbazan et al., 2012, 2017). Such molecular characterization, including epigenetic mechanisms, is vital for unraveling the biological aspects of CRC and identifying new clinical and therapeutic biomarkers to manage this disease.
In contrast to the limited translation of CTCs into routine clinical practice, the analysis of cNAs has started to be applied to oncologic therapy selection. These analyses mainly focus on circulating cell-free DNA (cfDNA), released as a consequence of cell death and characterized by a high fragmentation in biological fluids (size, 160–180 bp). cfDNA generally contains small fractions of circulating tumor DNA (ctDNA), in a range as low as 0.01–1% in patients with advanced tumors (Thierry et al., 2016). Of note, several genetic alterations, such as point mutations, copy number variations, small indels, and translocations, together with epigenetic modifications, can be studied in cfDNA from different body fluids in a non-invasive and comprehensive way (Bardelli and Pantel, 2017). Furthermore, in 2014 Bettegowda et al. demonstrated that ctDNA is detectable in most patients with mCRC and in a considerable percentage of non-metastatic patients (Bettegowda et al., 2014). These results provided the basis for numerous studies that have demonstrated that it is possible to characterize the molecular alterations of CRC with high accuracy, thereby predicting the information obtained by standard image-based follow-up. For example, there is clear scientific evidence of the robustness of ctDNA analyses to monitor RAS mutational status without the need for invasive tissue biopsies. This approach is also interesting for improving the selection of anti-EGFR therapy at different time points of disease evolution (Siravegna et al., 2015; Vidal et al., 2017). Moreover, recent technological improvements have favored the application of ctDNA analysis in patients with early CRC as well as for the detection of minimal residual disease (MRD) after surgery (Cohen et al., 2018).
Among cNAs, numerous non-coding RNAs (ncRNAs) can be detected in liquid biopsy. Several studies have highlighted the role of ncRNAs in cell-to-cell communication through the promotion of differential gene expression in tumor cells and stroma, which has a relevant impact on cancer progression and therapy resistance (Anfossi et al., 2018; Pardini et al., 2019). Although cNA-based applications for CRC are still in the clinical validation phase, current genetic and epigenetic data are promising and will soon support a more extensive use of cNA analysis to manage this type of cancer.
Other circulating elements of great interest found in liquid biopsy are cEVs. These vesicles are a complex population of cell-derived membranous structures released by cells through different mechanisms, which can be grouped into exosomes (ranging from 30 to 100 nm), microvesicles (50–2,000 nm), and apoptotic bodies (500–4,000 nm) (Akers et al., 2013; van Niel et al., 2018). These vesicles play an important role in cancer, mediating the interaction between tumor and stromal cells, promoting cell proliferation and invasion, and significantly contributing to the establishment of pre-metastatic niches (de la Fuente et al., 2015; Becker et al., 2016). Such functions are mediated by their membrane components, but also their molecular cargo, composed of proteins, messenger RNAs (mRNAs), ncRNAs, and single- or double-stranded DNA (van Niel et al., 2018). Importantly, cEVs can be found in different body fluids at high concentrations. Their isolation is mainly achieved by ultracentrifugation, immunoaffinity, or precipitation strategies (Bu et al., 2019; Jayaseelan, 2020), and the selection of the optimal isolation strategy is a crucial point, significantly influencing the characteristics of the isolated vesicles. The presence of relevant molecular alterations has been described in exosomal DNA from cancer patients (Hao et al., 2017; Castellanos-Rizaldos et al., 2018). Furthermore, exosomal ncRNAs obtained from serum and plasma have also been explored in connection with CRC, with promising results (Matsumura et al., 2015).
In summary, the study of cNAs, CTCs, and cEVs has shown significant potential for early CRC diagnosis, therapy selection, and disease monitoring, through the analysis of several molecular alterations such as those depending on various epigenetic mechanisms, which are examined in detail in the present review.
The Epigenetic Machinery and Cancer
The concept of epigenetics was postulated for the first time in 1942 by Waddington (2012), and can now be defined as the study of hereditary changes in the activity and expression of genes that take place without alterations of the DNA sequence (Holliday, 1987; Berger et al., 2009). The epigenetic machinery has several mechanisms (Figure 1), including DNA methylation and hydroxymethylation, histone modifications and nucleosomes positioning, and ncRNAs (Rodriguez-Paredes and Esteller, 2011). These mechanisms play an important role in regulating gene expression during many biological processes, such as embryonic development, imprinting, and tissue differentiation (Sharma et al., 2010). However, the deregulation of all these epigenetic layers has important implications for cancer development and progression (Mari-Alexandre et al., 2017).
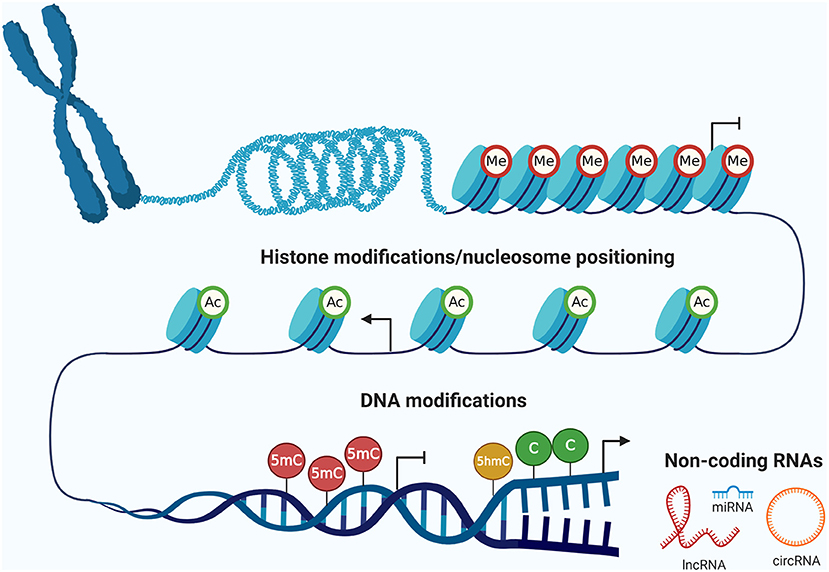
Figure 1. The epigenetic machinery. There are three fundamental epigenetic mechanisms, represented by (i) DNA modifications (methylation and hydroxymethylation), (ii) post-translational modifications of histones and nucleosome positioning, and (iii) non-coding RNAs. These epigenetic layers are highly interrelated among them and regulate gene expression of relevant biological processes in normal cells. However, these mechanisms can be deregulated in tumor cells leading to cancer development and progression. Me, methylation of histones; Ac, acetylation of histones; 5mC, 5-methylcytosine; 5hmC, 5-hydroxymethylcytosine; C, cytosine; lncRNA, long non-coding RNA; miRNA, microRNA; circRNA, circular RNA. Created with BioRender.com.
DNA Modifications: Methylation and Hydroxymethylation
DNA methylation is the most widely described epigenetic mechanism. This covalent modification of DNA consists in the incorporation of a methyl group (CH3) to the 5′ carbon of cytosines in cytosine-phosphate-guanine (CpG) dinucleotides to produce 5-methylcytosine (5mC) (Portela and Esteller, 2010). Such incorporation is regulated by DNA methyltransferases (DNMTs), including DNMT1, DNMT3A, and DNMT3B, which catalyze the transfer of methyl groups from S-adenosyl-L-methionine (SAM) to cytosines. In particular, DNMT3A and DNMT3B participate in producing de novo methylation patterns in cells, while DNMT1 maintains the parental methylation profile in each cell division (Gowher and Jeltsch, 2002; Jaenisch and Bird, 2003). DNA methylation usually occurs in specific regions of the genome with high density of CpG dinucleotides, called CpG islands (CGIs). These CpG-enriched sequences are usually located in the promoter regions of genes but can also be present in intragenic regions, including gene bodies (Diaz-Lagares et al., 2016a; Arechederra et al., 2018). According to their distance from CGIs, genomic sequences can be defined as CGI shores (up to 2 kb from CGIs), shelves (2–4 kb from CGIs), and open sea (>4 kb from CGIs) (Qu et al., 2014). In addition, DNA methylation can affect not only the intragenic but also the intergenic regions of the genome, increasing the complexity of this layer of epigenetic regulation (Zhao S. G. et al., 2020).
DNA methylation plays an important role in regulating gene expression as well as in maintaining the integrity and conformation of DNA, thereby protecting it from the potential damage by mobile genetic elements (Herceg and Vaissiere, 2011). This epigenetic modification can be enhanced (hypermethylation) or suppressed (hypomethylation) in different regions of the genome (Portela and Esteller, 2010). In cancer, hypermethylation of CGIs in promoters is usually linked to silencing of both coding and non-coding tumor suppressors (Diaz-Lagares et al., 2016a,b). However, hypomethylation of CpG-poor regions has been associated with proto-oncogene expression, genomic instability, and malignant transformation of tumors (Esteller, 2008; Sheaffer et al., 2016).
In human cells, DNA methylation can be reversed by TET (ten-eleven translocation) enzymes, which induce the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC) in a process defined as DNA hydroxymethylation (Tahiliani et al., 2009). This demethylation mechanism can modulate gene expression by adjusting the methylation levels (Xu and Gao, 2020). In cancer, deregulation of TET enzymes can alter the balance of genomic 5mC/5hmC levels, inducing cancer transformation (Chen et al., 2017). In addition, DNA methylation can be reversed through epigenetic-based drugs (epidrugs), which can decrease the methylation levels of hypermethylated genes (Berdasco and Esteller, 2019). For example, 5-azacytidine (5-AZA-CR) and 5-aza-2′-deoxycytidine (5-AZA-CdR, decitabine) are nucleoside analogs approved by the FDA, acting as DNA methyltransferase inhibitors (DNMTi) (Quintas-Cardama et al., 2010).
Histone Modifications and Nucleosome Positioning
Nucleosomes were first described in 1974 by Kornberg (1974) and represent the basic functional units of chromatin. They are composed of 147 bp of DNA wrapped around an octamer consisting of two copies of four core histone proteins (H2A, H2B, H3, and H4) (Kouzarides, 2007). This core of histones is organized into two H2A–H2B dimers and one H3–H4 tetramer. In addition, nucleosomes bind the linker histone H1, which protects the ~20–50 bp of free DNA (linker DNA) located between neighboring nucleosomal particles (Portela and Esteller, 2010). Nucleosomal histones can undergo different types of reversible post-translational modifications (PTMs), such as acetylation, methylation, phosphorylation, ubiquitylation, and sumoylation, that mainly occur in the histone tail and are enzymatically regulated (Kouzarides, 2007). Importantly, histone PTMs have demonstrated to play a relevant role in normal development and pathogenesis associated with transcriptional regulation, DNA repair and replication, or chromatin condensation (Bannister and Kouzarides, 2011; Bates, 2020). Specifically, multiple histone PTMs related to gene activation or silencing have been described, in particular histone acetylation and methylation. The balance of these types of PTMs is regulated by histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), and histone demethylases (HDMs). The possible combinations of PTMs provide various histone modification patterns that have been proposed to constitute a “histone code” associated with open/closed states of chromatin and gene regulation (Strahl and Allis, 2000; Rando, 2012).
In addition to PTMs, the positioning of nucleosomes has shown to be importantly involved in the regulation of chromatin accessibility. Indeed, variations in the position of nucleosomes modulate the binding of RNA polymerase or transcription factors to regulatory elements that control gene expression (Kurup et al., 2019; Huertas et al., 2020). Nucleosome positioning is controlled by chromatin-remodeling proteins (called movers) that shift nucleosomes and allow gene expression (Bates, 2020). Active genes usually present nucleosome-depleted regions (NDR) around their transcription start sites (TSS) that facilitate the accessibility of transcription regulatory proteins (Mavrich et al., 2008). The organization of nucleosomes is dynamically regulated and their turnover in active promoters and enhancers is higher than in inactive regions (Deal et al., 2010; Klemm et al., 2019). Interestingly, nucleosome positioning not only regulates gene expression but also influences the type of DNA fragmentation that can occur in different cellular processes, such as apoptosis, resulting in a non-random fragmentation of DNA (Matassov et al., 2004; Ivanov et al., 2015).
In cancer, epigenetic mechanisms such as nucleosome positioning and PTMs of histones, and/or the enzymes that regulate these modifications, are often deregulated (Hesson et al., 2014; Yang et al., 2014). For example, mutations in histone-modifying enzymes can lead to cancer development due to alterations in the PTM balance of histones (Morin et al., 2010). However, the alteration of histone modifications can be reversed in some types of tumors through the administration of certain epidrugs approved by the FDA (Hoy, 2020).
Non-coding RNAs
Non-coding transcripts are important regulatory molecules that represent the vast majority of the transcriptome (~98%) (Kapranov et al., 2007). The number of identified ncRNAs has been rapidly increasing in recent years, and currently multiple types of ncRNAs build up the non-coding transcriptome. Similar to other epigenetic mechanisms, ncRNAs have relevant functions in controlling gene expression (Dragomir et al., 2018). Non-coding transcripts are usually classified based on their nucleotide length. Using this criterion, ncRNAs can be divided into: (i) small ncRNAs (sncRNAs), which are shorter than 200 nt and include microRNAs (miRNAs), small interfering RNAs (siRNAs), and piwi-interacting RNAs (piRNAs); and (ii) long ncRNAs (lncRNAs), which are longer than 200 nt and encompass long intergenic ncRNAs (lincRNAs) and long intronic ncRNAs (intronic lncRNAs) (Taft et al., 2010; Esteller, 2011; Memczak et al., 2013). Of note, some ncRNAs have variable lengths and might be attributed to both classes at the same time. This is the case of enhancer RNAs (eRNAs), which originate from transcriptional enhancers, and circular RNAs (circRNAs), which are circularization products arising from splicing events (Leveille et al., 2015; Zhang et al., 2019).
Among sncRNAs, miRNAs have been the most widely described ncRNAs. miRNAs are single-stranded molecules of 18–25 nt that bind to specific regions of target mRNAs, mediating post-transcriptional gene silencing by two possible mechanisms: blocking transcription or triggering mRNA degradation (Lee and Calin, 2011). As a consequence, a single miRNA can control the expression of hundreds of genes, regulating key pathways for cancer tumorigenesis and progression (Garzon et al., 2009). Importantly, miRNAs can have a dual effect in cancer, functioning either as tumor suppressors or oncogenes (oncomiRs) (Volinia et al., 2006; Bayraktar et al., 2017). In addition, different types of cancers have shown specific miRNA signatures, defining the molecular characteristics of tumors (Lu et al., 2005).
Although miRNAs are the most studied ncRNAs, lncRNAs have recently been shown to represent the vast majority of non-coding transcripts (Hon et al., 2017). LncRNAs do not have the potential to encode proteins, but they may exhibit some mRNA-like properties, such as multiexonic gene structures, polyadenylation, presence of 5′ caps, and transcription by RNA polymerase II (Guttman et al., 2009; Derrien et al., 2012). LncRNAs have relevant regulatory functions in the process of gene expression, for example during transcriptional regulation and splicing (Kotake et al., 2011; Leveille et al., 2015). Nevertheless, currently few lncRNAs have been well-characterized, although several studies have shown their implications in cancer, where they act by suppressing (Leveille et al., 2015; Diaz-Lagares et al., 2016a) or promoting the tumoral process (Gupta et al., 2010; Gutschner et al., 2013).
Interplay Between Epigenetic Mechanisms
In normal mammalian cells, epigenetic modifications can regulate each other through the interplay among distinct epigenetic players, forming a complex regulatory network (Caley et al., 2010; Rose and Klose, 2014). However, this crosstalk is also evident in cancer, where specific methylation and histone modification patterns regulate the expression of different types of ncRNAs, including miRNAs and lncRNAs (Shin et al., 2009; Leveille et al., 2015). Conversely, the disruption of ncRNA expression in cancer cells may alter histone PTMs and DNA methylation levels (Gupta et al., 2010). These are only some examples of the important crosstalk that usually occurs among the different components of the epigenetic machinery.
Methods for Detecting Epigenetic Marks in Liquid Biopsy
Multiple methodologies have been described at genome-wide or locus-specific level for the analysis of the status of different types of epigenetic marks (Bao-Caamano et al., 2020). However, it is important to consider the advantages and limitations of each method in order to choose the most appropriate approach depending on the type of epigenetic mechanism under investigation and on the conditions of the assay (Kurdyukov and Bullock, 2016). Table 1 shows the most representative methods used for detecting epigenetic marks in liquid biopsy.
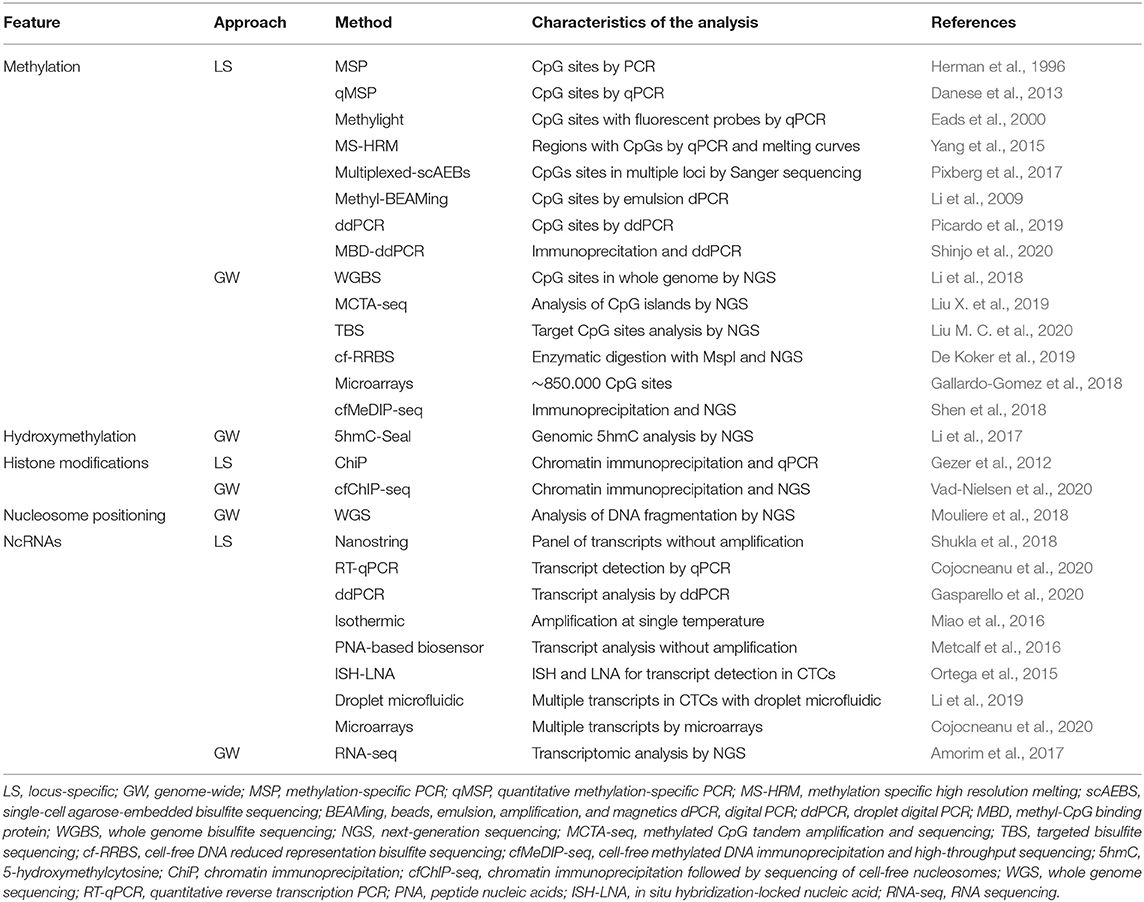
Table 1. Methods used for the analysis of epigenetic marks in liquid biopsy.
DNA Modifications: Methylation and Hydroxymethylation
The detection of DNA methylation patterns is based on methods that (i) depend on sodium bisulfite conversion or (ii) are independent of sodium bisulfite, such as approaches based on immunoprecipitation and methyl-sensitive restriction enzymes (MSRE) (Huang and Wang, 2019). Methodologies that use bisulfite conversion are considered the gold standard for methylation analyses. These approaches are based on the principle that after sodium bisulfite treatment, methylcytosines do not undergo any change, whereas cytosine residues are converted into uracils (Frommer et al., 1992).
At the locus-specific scale, common bisulfite-based methods used in liquid biopsy include methylation-specific PCR (MSP), quantitative methylation-specific PCR (qMSP), MethyLight assay, methylation-sensitive high-resolution melting (MS-HRM), and more recently, digital PCR (dPCR)-based approaches (Li et al., 2009; Bao-Caamano et al., 2020). In particular, the detection of methylation by dPCR represents a quantitative and highly sensitive method that allows the analysis of very low amounts of DNA. Therefore, several dPCR-based methods, including methyl-BEAMing (Barault et al., 2015) and droplet digital PCR (ddPCR) (Boeckx et al., 2018), have been used for methylation analysis in cfDNA. Regarding CTCs, the first studies describing DNA methylation alterations in these cells were reported using MSP (Chimonidou et al., 2011). Recently, a new method has been described for methylation analysis of CTCs, based on agarose-embedded bisulfite treatment (AEBS), allowing to analyze the DNA methylation status of multiple loci of single CTCs by multiplex PCR (multiplexed-scAEBS) (Pixberg et al., 2017).
Moreover, genome-wide bisulfite-based approaches based on NGS allow to evaluate the whole methylome in liquid biopsy. For instance, whole-genome bisulfite sequencing (WGBS) has proved useful for the inspection of the whole methylation landscape of not only cfDNA from cancer patients (Li et al., 2018), but also of single CTCs and CTC clusters (Gkountela et al., 2019). Although WGBS is highly informative, the high cost of this approach is a limitation for its general implementation in a clinical setting (Legendre et al., 2015; Zhang et al., 2020). Therefore, other high-throughput bisulfite-based approaches, not allowing to analyze the complete methylome but enabling to assay a great number of CpGs in a genome-wide scale, have been proposed. For instance, by MCTA-seq (methylated CpG tandem amplification and sequencing) it is possible to analyze the methylation status of CGIs in cfDNA (Liu et al., 2019). Similarly, CpG-targeted bisulfite sequencing methods have proved useful for the analysis of methylation in cfDNA. In this regard, Liu et al. introduced a novel approach by which, after bisulfite treatment of plasma cfDNA, regions of interest are pulled down and sequenced, and the results are analyzed in combination with machine learning (Liu M. C. et al., 2020). Recently, cfDNA reduced representation bisulfite sequencing (cf-RRBS) has been developed as an alternative genome-wide bisulfite sequencing method to analyze the cfDNA methylome (De Koker et al., 2019). This new method is based on the reduced representation bisulfite sequencing (RRBS) technology, which was first described by Meissner et al. (2005). In cf-RRBS, cfDNA is dephosphorylated prior to enzymatic digestion by the methylation-insensitive restriction enzyme MspI and sequencing. This approach represents a cost-effective method that allows methylation profiling of cfDNA in liquid biopsy (Van Paemel et al., 2020). In addition to NGS methods, DNA methylation can also be analyzed at the genome-wide level in liquid biopsy using methylation microarrays. This methodology implies the use of bisulfite-converted DNA and has been applied to the study of cfDNA and CTCs from cancer patients (Friedlander et al., 2014; Gallardo-Gomez et al., 2018). These genome-wide approaches based on NGS and microarrays are very promising, but have still been used in a small number of works. Therefore, more studies are necessary to validate their application for the analysis of epigenetic alterations in liquid biopsy.
One of the advantages of bisulfite conversion-based approaches is that they allow methylome profiling at base resolution (Sun et al., 2015). However, the use of bisulfite treatment for methylation analysis has some limitations, such as a potentially high degradation of DNA or an incomplete bisulfite conversion (Grunau et al., 2001; Rand et al., 2002). To overcome these limitations, Shen et al. recently developed a genome-wide method based on cell-free methylated DNA immunoprecipitation and high-throughput sequencing (cfMeDIP-seq) (Shen et al., 2018). In contrast to bisulfite single-base resolution technologies, cfMeDIP-seq is a region-based method that unravels the methylation status of genomic regions of at least 100 bp in length (Shen et al., 2019). This new bisulfite-free method was adapted from a previous methylated DNA immunoprecipitation (MeDIP) protocol based on the use of anti-methylcytosine antibodies (Taiwo et al., 2012). cfMeDIP-seq is a low-input (requiring 1 to 10 ng of DNA) and sensitive (with a detection limit down to 0.001%) approach that can be used for both early- and late-stage detection of multiple types of tumors (Shen et al., 2018, 2019). Recently, another enrichment method based on immunoprecipitation was developed to detect cfDNA methylation. This new locus-specific method is based on the immunoprecipitation of methyl-CpG binding (MBD) proteins coupled with ddPCR (MBD–ddPCR) (Shinjo et al., 2020). This highly sensitive technique allows the detection of methylation sites in cfDNA.
Similar to DNA methylation, genome-wide hydroxymethylation profiles can be obtained from cfDNA of cancer patients. This can be achieved by 5hmC-Seal, a robust and efficient sequencing method that has proved useful for detecting 5hmC in cfDNA with high sensitivity and specificity (Li et al., 2017).
Histone Modifications and Nucleosome Positioning
The global distribution of specific PTMs of histones in cells can be assayed by chromatin immunoprecipitation with massively parallel DNA sequencing (ChIP-seq) (Barski et al., 2007). Recently, Sadeh et al. developed a method to perform ChIP-seq of cell-free nucleosomes (cfChIP-seq). This method enables the capture of circulating nucleosomes with different active chromatin marks that maintain the cell-of-origin genomic distribution of modifications and expression patterns (Sadeh et al., 2019). Other similar approaches have been used to quantify the level of histone marks associated with circulating cell-free nucleosomes in plasma of cancer patients (Vad-Nielsen et al., 2020). In addition to these high-throughput technologies, circulating nucleosomes can also be analyzed by chromatin immunoprecipitation (ChIP) followed by quantitative PCR to detect histone modifications in individual genes (Gezer et al., 2012). Other methods, such as enzyme-linked immunosorbent assay (ELISA), are used to quantify the occurrence of specific histone marks based on their levels in circulating nucleosomes (Rahier et al., 2017).
In addition, the fragmentation patterns of cfDNA depending on nucleosome positioning can be analyzed using genome-wide fragmentation methods, based on the combination of NGS and machine learning. These approaches require a very low input of cfDNA from different types of fluids, allowing even early detection of cancer (Mouliere et al., 2018). Other genome-wide fragmentation methods, such as DNA evaluation of fragments for early interception (DELFI), have been recently proposed (Cristiano et al., 2019). These kinds of fragmentation approaches are based on prior knowledge on the different lengths of cfDNA fragments originating from tumor and non-tumor cells (Jiang et al., 2015). Moreover, the landscape of cfDNA fragmentation is associated with nucleosome occupancy and epigenetic regulation (Ivanov et al., 2015). In turn, nucleosome occupancy of cfDNA correlates well with the nuclear architecture, gene structure, and gene expression observed in cells (Snyder et al., 2016), providing relevant information about nucleosome organization and the tissue of origin (Ivanov et al., 2015; Mouliere et al., 2018; Cristiano et al., 2019).
Non-coding RNAs
Genome-wide expression analyses, based on microarrays or NGS technologies, enable the detection of a large number of ncRNAs in a high-throughput manner. Therefore, microarrays and NGS have been used in liquid biopsy for the detection of several types of ncRNAs, including miRNAs, lncRNAs, and circRNAs (Amorim et al., 2017; Cojocneanu et al., 2020; Gasparello et al., 2020). Although both methods allow a comprehensive analysis of ncRNA transcripts, NGS displays higher sensitivity than microarrays and does not require previous knowledge of the target transcripts (Hurd and Nelson, 2009; Wang et al., 2019). On the other hand, several methods and instruments are based on targeted detection of transcripts in liquid biopsy. This is the case of the NanoString nCounter, a platform that directly detects the expression levels of a wide panel of ncRNAs without any enzymatic amplification (Shukla et al., 2018). Other technologies, such as reverse transcription quantitative PCR (RT-qPCR) and ddPCR, despite requiring enzyme-assisted amplification, allow to quantify the expression levels of specific ncRNA transcripts in liquid biopsy with high sensitivity. Importantly, RT-qPCR and ddPCR are usually used as gold standard methods to validate the results obtained by genome-wide approaches (Sole et al., 2019; Cojocneanu et al., 2020; Gasparello et al., 2020). However, since these PCR-based methods require several cycles of temperature variations, isothermal amplification approaches, which use a single temperature, have been developed for ncRNA analysis in liquid biopsy (Miao et al., 2016). In contrast, other methods for miRNA detection, such as the use of peptide nucleic acids (PNAs)-based fluorogenic biosensors, do not require amplification (Metcalf et al., 2016). Some targeted approaches have also been shown to be specifically useful for the analysis of ncRNAs, especially miRNAs, in CTCs. Among these, in situ hybridization (ISH) with locked-nucleic-acid (LNA) probes has been successfully applied for the detection of miRNAs in CTCs (Ortega et al., 2015), since the use of LNA probes increases the efficiency of ISH for the detection of miRNAs (Kubota et al., 2006). Other methods for the analysis of CTCs have been recently developed, based on signal amplification in microfluidic droplets for single-cell analysis of multiple miRNAs (Li et al., 2019).
Deregulation of Epigenetic Mechanisms in Colorectal Cancer and Liquid Biopsy
CRC originates from an accumulation of both genetic and epigenetic alterations in normal colon epithelial cells, leading to their transformation, first into adenomas and then into adenocarcinomas (Fearon and Vogelstein, 1990; Lao and Grady, 2011). Epigenetic alterations are involved in all the steps of the adenoma-carcinoma sequence, participating in the initiation, progression, and metastasis of CRC (Kim et al., 2006; Wendt et al., 2006; Silva-Fisher et al., 2020). Importantly, the disruption of the epigenetic machinery has been proposed as a hallmark of cancer by several authors (Esteller, 2007; Bates, 2020).
Promoter hypermethylation of single TSGs has been widely described in colorectal tumor cells. In particular, the hypermethylation of CGIs in promoters of relevant genes (p16/CDKN2A, MLH1, MGMT, APC, and TIMP3, among others) is associated with CRC pathogenesis (Gonzalez-Zulueta et al., 1995; Hiltunen et al., 1997; Kane et al., 1997; Esteller et al., 1999; Lee et al., 2004). In addition, the hypermethylation of specific groups of genes has shown implications for this disease. In this sense, Toyota et al. described for the first time the CpG island methylator phenotype (CIMP), characterized by the hypermethylation of CGIs in the promoter regions of a panel of TSGs from a subset of CRC and adenoma tissues (Toyota et al., 1999). CIMP-High (CIMP-H) is considered a molecular subtype of sporadic CRC characterized by a high degree of methylation in CIMP-specific loci. Most studies have defined CIMP using a classic methylated-in-tumor (MINT) marker panel: p16/CDKN2A, MLH1, MINT1, MINT2, and MINT31 (Nazemalhosseini Mojarad et al., 2013). However, subsequent studies have used other types and number of genes to define CIMP, thereby increasing the difficulty of its implementation in the clinic (Weisenberger et al., 2006; Ogino et al., 2007). On the other hand, recent advances in the characterization of genome-wide DNA methylation patterns have allowed the discovery of methylation signatures based on multiple CpG sites associated with CRC (Sandoval et al., 2011; Luo et al., 2014). This type of epigenomic methodology has revealed that the disruption of methylation in colorectal tumor cells can affect both coding and non-coding genes, such as those producing miRNAs and lncRNAs (Mori et al., 2011; Diaz-Lagares et al., 2016a). In addition to the hypermethylation of TSGs, colorectal tumors are also characterized by global hypomethylation, presenting a low methylation status in LINE-1 repetitive sequences (Suter et al., 2004). On the other hand, total 5hmC levels are reduced in colon tumor cells compared to normal cells (Gilat et al., 2017). However, tumor cells can also show high levels of 5hmC in transcriptionally active regions, associated with the expression of particular coding and non-coding genes; this phenomenon contributes to CRC pathogenesis, even in early stages (Hu et al., 2017; Gao et al., 2019).
Colorectal tumors are also characterized by the disruption of histone modification patterns, such as methylation [e.g., methylation of lysine 9 on H3 (H3K9me)] and acetylation [e.g., acetylation of lysine 27 on H3 (H3K27ac)] (Liu et al., 2013; Karczmarski et al., 2014). In addition, the positioning of nucleosomes can also be modified in tumors, and such alteration has implications for the reorganization of chromatin and gene expression (Hesson et al., 2014). In summary, the positioning of nucleosomes over the TSS and differences in the occurrence of histone epigenetic marks can regulate gene expression in colorectal tumor cells (Gimeno-Valiente et al., 2019).
The expression of several types of ncRNAs, including miRNAs and lncRNAs, is usually dysregulated in CRC. For instance, the overexpression of particular miRNAs, such as miR-21, is associated with the activation of relevant specific pathways that promote pathogenesis of colorectal tumor cells (Xiong et al., 2013). In addition, recent transcriptomic analyses have revealed CRC-specific expression signatures of miRNAs, lncRNAs, and other types of ncRNAs (Di et al., 2020; Li et al., 2020; Song et al., 2020). One of the epigenetic mechanisms that may control the expression levels of these ncRNAs in colorectal tumor cells is DNA methylation. For instance, this is the case of the lncRNA TP53TG1, which exerts tumor suppressor activity in gastrointestinal tumors and participates in the p53 response to DNA damage (Diaz-Lagares et al., 2016a).
Of note, advances in our understanding of aberrant epigenetics have led to the identification of epigenetic alterations in liquid biopsy elements (cNAs, CTCs, and cEVs) associated with all steps of CRC progression (Gasch et al., 2015; Zeng et al., 2018; Jung et al., 2020). Disruption of epigenetic mechanisms has great relevance in cancer dissemination and metastasis of solid tumors, which remains the leading cause of cancer-related death (Gupta and Massague, 2006). For example, hypermethylation of TSGs (e.g., CDKN2A), in both tumor tissue and cfDNA, is broadly associated with distant metastasis in CRC patients (Mitomi et al., 2010). In addition, hypomethylation of LINE-1 in plasma cfDNA of CRC patients is also correlated with disease progression; this association is stronger in tumors with higher size, lymph node affectation, and distant metastasis (Nagai et al., 2017). More recently, the epigenetic markers of CTCs have been explored to better understand cancer progression (Chimonidou et al., 2011). In particular, Lyberopoulou et al. were the first to determine the DNA methylation profile of cancer-related genes in CTCs of CRC patients. This study revealed that CTCs from CRC patients are characterized by hypermethylation of the SFRP2 promoter and exon 1 of VIM (Lyberopoulou et al., 2017), which are genes related to epithelial-to-mesenchymal transition (EMT) and CRC metastasis (Shirahata et al., 2009; Loboda et al., 2011; Vincent and Postovit, 2017). In recent years, the analysis of single CTCs with genome-wide methylation (Gkountela et al., 2019) or multiplex approaches (Pixberg et al., 2017) has provided relevant information on cancer dissemination. However, the study of epigenetic alterations in CTCs from CRC patients remains limited.
In addition to CTCs, cancer-derived EVs play a major role in cancer progression as they participate in pre-metastatic niche formation (de la Fuente et al., 2015; Becker et al., 2016). For example, it has been shown that exosomal miR-25-3p from CRC cells regulates the expression of target genes, promoting vascular permeability, angiogenesis, and the formation of liver and lung metastasis in preclinical models (Zeng et al., 2018). Of note, plasma exosomal miR-25-3p levels are significantly higher in metastasic CRC patients than in those with no metastases, indicating a role for this miRNA in pre-metastatic niche formation (Zeng et al., 2018). On the other hand, the fact that serum exosomal miR-375 is less abundant in CRC patients with liver metastasis, together with functional studies, suggests its role as a tumor suppressor through the inhibition of the Bcl-2 pathway (Zaharie et al., 2015). Furthermore, recent results have indicated that decreased levels of serum exosomal miR-638 (Yan et al., 2017) and miR-548c-5p (Peng et al., 2018) are associated with liver metastasis in CRC patients.
Considering the relevance of epigenetic alterations in cancer dissemination and progression, there is a great interest in the development of epigenetic modifiers that can function as epidrugs. Importantly, some of these epigenetic modifiers have been tested preclinically or in early-phase clinical trials for CRC, representing a promising field for the treatment of this tumor (Baretti and Azad, 2018).
Clinical Utility of Epigenetic Marks in Liquid Biopsy as CRC Biomarkers
Alterations in epigenetic marks have shown great utility in both tissue and liquid biopsies as tumor biomarkers for early detection, prognosis, monitoring, and evaluation of therapeutic response in CRC (Jung et al., 2020). Of these epigenetic alterations, changes in DNA methylation patterns and ncRNA expression are among the most well-known epigenetic biomarkers of CRC in liquid biopsy. Epigenetic biomarkers have been detected in all the components of liquid biopsy as well as in multiple biological fluids (Figure 2). Although blood is the most explored biological fluid for the study of these types of biomarkers, other fluids (such as stool and saliva) have also been considered as a relevant source of epigenetic tumor biomarkers for CRC. Tables 2–7 include examples of representative circulating epigenetic biomarkers analyzed in blood and other fluids associated with clinical applications for CRC patients.
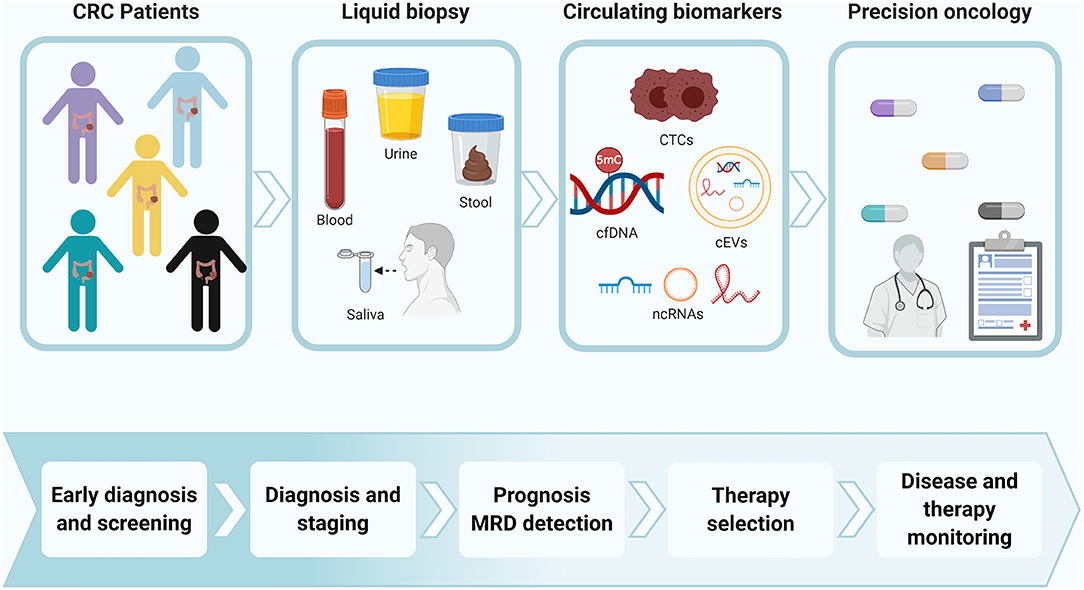
Figure 2. Epigenetic biomarkers in liquid biopsy for precision oncology of CRC patients. Colorectal primary tumors and metastasis can release epigenetic biomarkers into different types of biological fluids. The disruption of these epigenetic mechanisms can be detected in circulating tumor cells (CTCs), nucleic acids (cNAs) and extracellular vesicles (cEVs), showing clinical relevance as therapeutic targets and tumor biomarkers for early detection, prognosis, monitoring, therapy selection, and evaluation of therapeutic response in CRC. The detection of these epigenetic biomarkers in liquid biopsy have a great value to personalize the management of CRC patients. CRC, colorectal cancer; CTCs, circulating tumor cells; cfDNA, cell-free DNA; ncRNAs, non-coding RNAs; MRD, minimal residual disease. Created with BioRender.com.
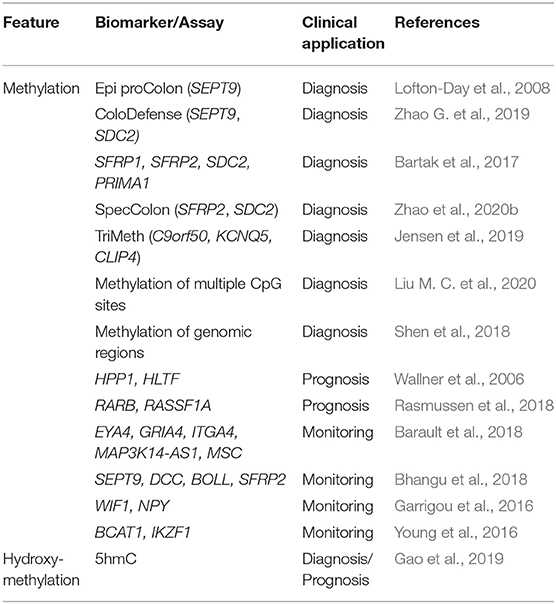
Table 2. DNA modifications in cfDNA of blood as epigenetic biomarkers of CRC.
Blood-Based Circulating Epigenetic Biomarkers
DNA Modifications: Methylation and Hydroxymethylation
A plethora of epigenetic biomarkers based on altered methylation have been evaluated in blood-derived cfDNA for the management of CRC (Table 2). Among them, plasma SEPTIN9 (SEPT9) methylation is one of the most studied epigenetic biomarkers for the screening and early detection of this tumor (Lofton-Day et al., 2008; Church et al., 2014). Epi proColon was the first commercially available test for the detection of plasma SEPT9 methylation, and Epi proColon 2.0 constitutes the improved second generation of this test. This qualitative assay is based on the detection of methylation in the promoter region of the SEPT9 gene from plasma cfDNA by real-time PCR. Importantly, the Epi proColon test was the first blood-based assay approved by the FDA for the screening of CRC (Pickhardt, 2016; Issa and Noureddine, 2017). According to several studies, assessing the promoter methylation status of SEPT9 allows to differentiate between CRC patients and healthy individuals with high overall sensitivity and specificity (Wang et al., 2018), although this assay has a limited capacity to predict precancerous lesions or adenomas (Church et al., 2014). In addition to this limitation, there are other aspects that have hampered the clinical implementation of this assay, including the high heterogeneity of the analytical characteristics among studies and its poor cost effectiveness in comparison with other methods (Wang et al., 2018). Recently, the combined analysis of SEPT9 and SDC2 methylation levels in blood has led to the development of a new test (ColoDefense) allowing significantly improved detection of CRC and adenomas, and thus representing a promising tool for tumor screening and early detection (Zhao G. et al., 2019). Other studies have described how the combined analysis of the methylation profiles of several genes in plasma allows for discrimination between healthy controls and patients with adenomas or CRC. For example, the analysis of a gene panel including SFRP1, SFRP2, SDC2, and PRIMA1 allowed to distinguish between CRC patients and healthy controls with high sensitivity and specificity (Bartak et al., 2017). Similarly, with the SpecColon test it was possible to analyze the methylation patterns of plasma SFRP2 and SDC2 simultaneously, thereby accurately detecting CRC and advanced adenomas (Zhao et al., 2020b). In another study, the analysis of methylation patterns in a three-gene panel (C9orf50, KCNQ5, and CLIP4) in plasma, through a test called TriMeth, enabled early detection of CRC with good sensitivity and specificity (Jensen et al., 2019).
Furthermore, approaches based on methylation microarrays and NGS have been used to identify epigenetic biomarkers in cfDNA for cancer detection. For instance, the analysis of ~850,000 CpGs in pooled cfDNA samples by MethylationEPIC array highlighted 1,384 differentially methylated CpG sites that discriminate CRC patients from healthy controls (Gallardo-Gomez et al., 2018). Moreover, the combination of NGS with machine learning has enabled the development of a test based on ~1 million CpG sites capable of detecting and localizing more than 50 tumor types, including CRC (Liu M. C. et al., 2020). Another research group designed a targeted NGS assay based on 9,223 hypermethylated CpG sites obtained from The Cancer Genome Atlas (TCGA), which proved useful for identifying advanced CRC as well as other tumor types (Liu et al., 2018). In addition, the PanSeer assay, which considers 10,613 CpG sites, allowed the detection of five cancer types, including CRC, regardless of the tissue of origin. Importantly, this assay enabled to detect the presence of cancer in asymptomatic individuals years before standard diagnosis (Chen et al., 2020). Another approach for the analysis of cfDNA methylation patterns, based on the combination of immunoprecipitation and NGS (cfMeDIP), was demonstrated to be effective for the detection of CRC and other tumor types (Shen et al., 2018).
At the prognosis level, several studies have revealed that the analysis of DNA methylation in liquid biopsy is useful to predict the outcome of patients. The potential prognostic biomarker for CRC that has been most studied is the hypermethylation of the p16 promoter in blood, which has been associated with worse overall survival (Xing et al., 2013). Similarly, several studies have analyzed the methylation status of other genes, such as HPP1 and HLTF, showing that hypermethylation of these genes in serum samples indicates worse prognosis and higher mortality of CRC patients (Wallner et al., 2006; Philipp et al., 2012). Another study demonstrated that the median number of hypermethylated promoter regions was higher in CRC patients with distant metastasis than in those without metastasis. These authors also found that the hypermethylation of RARB and RASSF1A was associated with the aggressiveness of the disease, representing an independent predictive factor of worse overall survival (Rasmussen et al., 2018).
Circulating DNA methylation can also be analyzed in series of fluid samples to monitor tumor burden and evaluate the therapeutic response of CRC patients to different types of treatments, including chemotherapy and anti-EGFR therapy (Barault et al., 2018; Bhangu et al., 2018). In this regard, the hypermethylation of two genes (WIF1 and NPY) in cfDNA has been described as a surrogate biomarker of tumor burden and applied to monitor patients without the need for mutational analysis in liquid biopsy (Garrigou et al., 2016). Of note, these types of circulating epigenetic alterations correlate with tumor volume and recurrence better than classical biomarkers, such as carcinoembryonic antigen (CEA) and carbohydrate antigen (CA) 19-9 (Young et al., 2016; Bhangu et al., 2018; Symonds et al., 2020). In line with these results, the analysis of circulating methylation markers has also shown consistency with the output of imaging tests in the assessment of response to therapy and surgery (Boeckx et al., 2018). All these findings indicate the potential of the analysis of circulating DNA methylation for improving the clinical evaluation of CRC patients and, if necessary, promptly redefining the treatment strategy.
The analysis of DNA methylation has also been explored in CTCs isolated from blood, although the number of studies focused on this circulating tumor population is still very low. Interestingly, when comparing the methylation levels of VIM and SFRP2 in CTCs of CRC patients with those of tumor tissues, researchers found a strong correlation between the methylation status of the SFRP2 promoter in CTCs and that of the corresponding tissue, but a weaker correlation in the case of VIM. Therefore, although the CTC population can show a different methylation pattern than tumor cells located in primary tissue and metastasis due to the gain of specific characteristics in the bloodstream, these data demonstrated that methylation profiling of CTCs in CRC patients represents a promising non-invasive approach for tumor detection (Lyberopoulou et al., 2017).
In addition to methylation, differential hydroxymethylation patterns of plasma cfDNA have been observed between patients with CRC and healthy controls, suggesting that markers of this epigenetic modification could also be used as a non-invasive tool for early detection and prognosis in CRC (Table 2). Importantly, the efficiency by which the presence of 5hmC in cfDNA points at CRC was shown to be similar to that in tumor tissues, and higher than that of well-known biomarkers, such as CEA, CA 19-9, and methylated SEPT9 (Li et al., 2017).
Histones and Nucleosomes
The study of histone modifications in blood-circulating nucleosomes (Table 3) has revealed that they can also contribute to CRC detection. For example, low levels of the circulating histone marks H3K9me3, H4K20me3, and H3K27me3 have been proposed as biomarkers for the diagnosis of CRC (Gezer et al., 2013, 2015). The combination of different PTMs detected in circulating nucleosomes has also proved useful for CRC screening (Rahier et al., 2017). In addition, high concentrations of circulating nucleosomes in CRC patients have been associated with disease progression, poor therapy response, and reduced survival (Fahmueller et al., 2012). Notably, the levels of nucleosomes in cancer patients are dynamic and thus can be useful to indicate the response to therapy in real time (Holdenrieder et al., 2001).
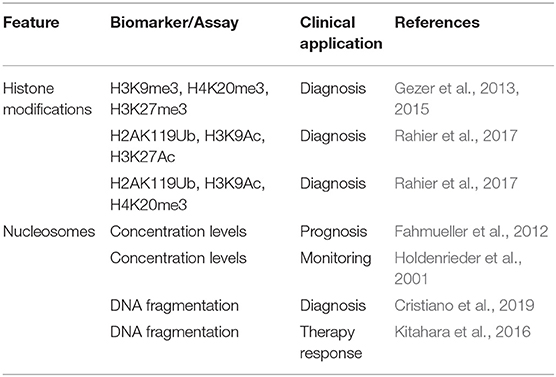
Table 3. Histone/nucleosomes in blood as epigenetic biomarkers of CRC.
Nucleosome occupancy is closely related to the fragmentation patterns of cfDNA (Ivanov et al., 2015). Notably, Mouliere et al. found that in multiple cancer types, including CRC, plasma cfDNA fragments exhibited different sizes between healthy individuals and cancer patients (Mouliere et al., 2018). Therefore, these authors proposed cfDNA fragmentation patterns as epigenetic biomarkers for early cancer detection. Consistently, fragmentation profiling of plasma cfDNA proved effective for CRC diagnosis, even when the tumor origin was initially unknown (Cristiano et al., 2019). The level of cfDNA fragmentation can also be associated with the prognosis of CRC patients, as patients displaying higher fragmentation showed worse prognosis (El Messaoudi et al., 2016). In addition, the analysis of cfDNA fragmentation patterns has proved useful for early detection of MRD after surgery and to predict the response of CRC patients to immunochemotherapy (Table 3).
Non-coding RNAs
Similar to other epigenetic alterations, the presence of ncRNAs in blood is a relevant source of biomarkers for CRC management (Table 4). For instance, a recent meta-analysis revealed that circulating miR-21 is a promising biomarker for CRC detection, and that its diagnostic properties can be improved by combining it with other biomarkers (Peng et al., 2017). Moreover, numerous studies have identified circulating miRNA signatures and applied them for non-invasive early detection of CRC; this enabled to distinguish healthy controls, patients with precancerous lesions (advanced adenomas), and CRC patients with high sensitivity and specificity (Wang et al., 2012; Kanaan et al., 2013; Herreros-Villanueva et al., 2019). The analysis of miRNAs in blood can also provide prognostic information. For instance, increased levels of circulating miR-210 and miR-141 are associated with shorter survival rates (Cheng et al., 2011; Wang et al., 2017). In contrast, high plasma levels of miR-23b are associated with longer survival (Kou et al., 2016). The levels of different miRNAs in blood can also provide information about the patient's disease stage (Sun et al., 2016), and represent a valuable tool for early detection of recurrence after surgery (Yuan et al., 2017) and the evaluation of response to therapy in CRC patients (Hansen et al., 2015; Schirripa et al., 2019).
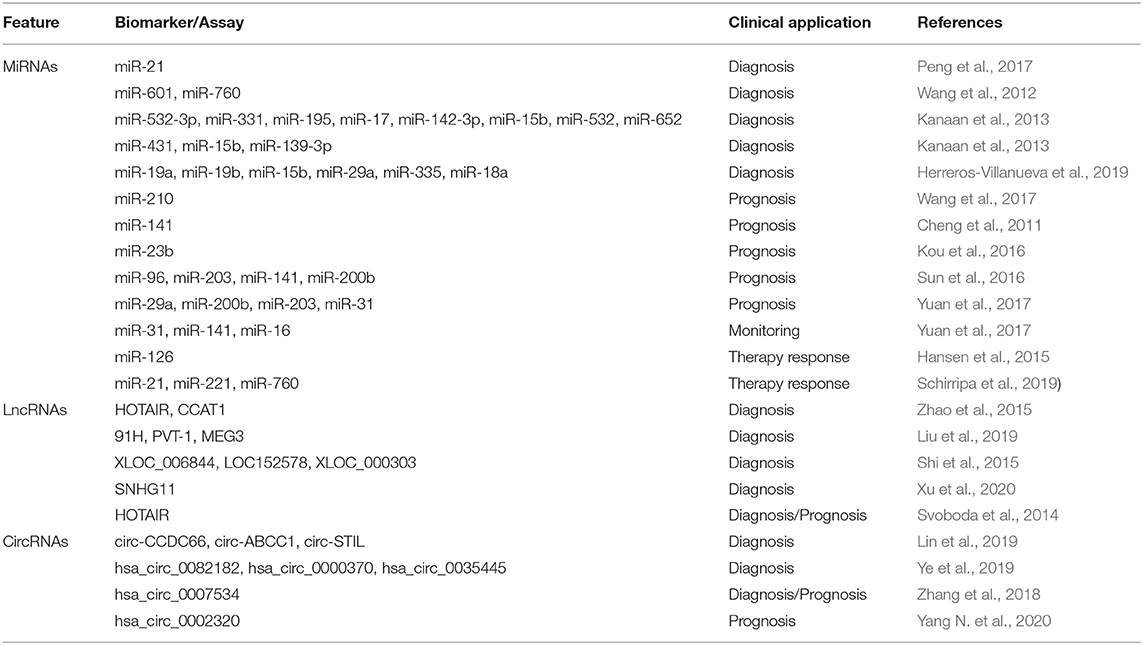
Table 4. Non-coding cfRNAs in blood as epigenetic biomarkers of CRC.
Other ncRNAs, such as lncRNAs, are also deregulated in CRC and can provide relevant clinical information (Table 4). For example, elevated plasma levels of the lncRNA HOTAIR, alone or in combination with other lncRNAs, have proved useful not only for CRC screening, but also as a biomarker associated with worse prognosis and higher mortality (Svoboda et al., 2014; Zhao et al., 2015). Of note, other studies have proposed different combinations of circulating lncRNAs as diagnostic biomarkers for the detection of CRC and precancerous lesions (Shi et al., 2015; Liu et al., 2019; Xu et al., 2020). In addition to lncRNAs, the analysis of circRNAs in blood, as biomarkers with relevant clinical properties for CRC diagnosis and prognosis, has raised considerable interest (Lin et al., 2019; Yang N. et al., 2020).
Finally, ncRNA levels have also been analyzed in cEVs, especially in exosomes, of CRC patients (Table 5). Exosomal miRNAs (e.g., miR-21 and miR-139-3p) can be more or less abundant than normal in the blood of CRC patients and their analysis represents a valuable tool for CRC diagnosis and prognosis (Fu et al., 2018; Liu W. et al., 2020). Furthermore, the analysis of miRNAs in circulating exosomes has revealed important strategies for the identification of treatment-resistant patients (Jin et al., 2019). For example, the exosomal levels of the lncRNAs HOTTIP, LINC02418, and RPPH1 were found deregulated in CRC and thus proposed as potential markers for the diagnosis of CRC, as well as in other clinical contexts (Liang et al., 2019; Oehme et al., 2019; Zhao Y. et al., 2019). Finally, recent studies have focused on exosomal circRNAs (e.g., circ-133 and circ-PNN), suggesting a relevant role of these ncRNAs as biomarkers for CRC (Xie Y. et al., 2020; Yang et al., 2020).
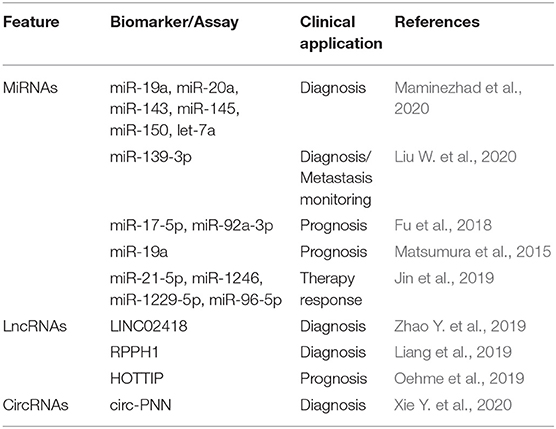
Table 5. Non-coding RNAs in EVs of blood as epigenetic biomarkers of CRC.
Circulating Epigenetic Biomarkers in Alternative Fluids
DNA Modifications: Methylation and Hydroxymethylation
Numerous studies have reported the application of DNA methylation patterning in stool (Table 6) for the detection of CRC or precancerous lesions (Raut et al., 2020). In particular, methylation of the promoter region of FBN1, SDC2, or VIM has been reported as a biomarker for early CRC diagnosis (Chen et al., 2005; Guo et al., 2013; Han et al., 2019). In addition, the hypermethylation of a three-gene panel (HPP1, SFRP2, and MGMT) in stool showed clinical value for detecting CRC and precancerous colorectal lesions (Huang et al., 2007). Other studies have also highlighted the hypermethylation of the promoters of numerous genes in stool samples from patients affected by CRC or adenomas, such as HIC1, COL4A1, COL4A2, GATA4, ITGA4, OSMR, and TLX2 (Lenhard et al., 2005; Kim et al., 2009; Liu et al., 2020). In addition, the methylation status of various miRNAs (e.g., miR-34a and miR-34b/c) was found to be altered in stool of CRC patients, and studied as a source of potential biomarkers (Kalimutho et al., 2011; Wu et al., 2014).
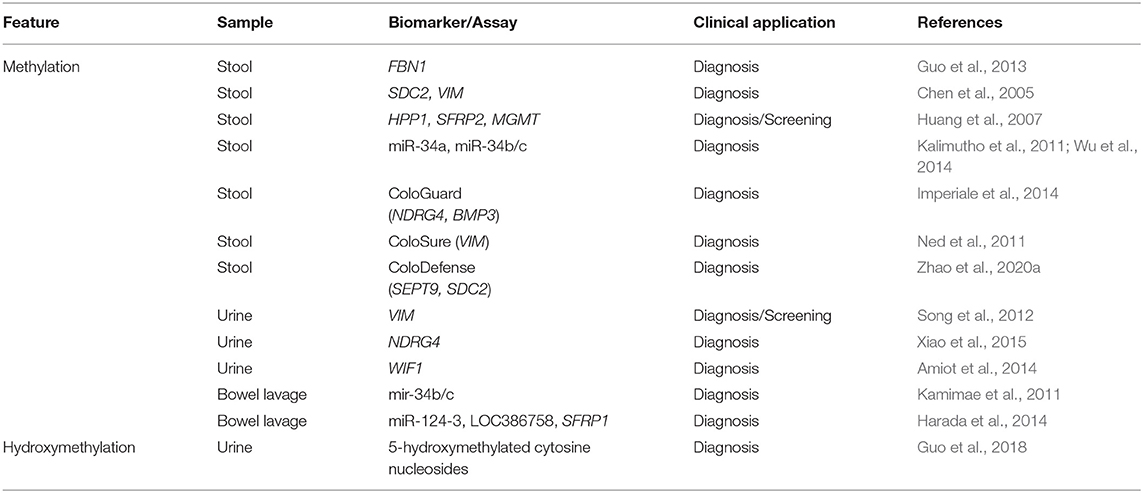
Table 6. DNA modifications in non-blood fluids as epigenetic biomarkers of CRC.
Importantly, some of the methylated biomarkers detected at an increased concentration in stool samples from CRC patients have been translated into clinical tests. This is the case of Cologuard, which is the first multi-target stool panel approved by the FDA for CRC screening (Pickhardt, 2016). The Cologuard assay combines the analysis of NDRG4 and BMP3 methylation, KRAS mutation, β-actin levels, and hemoglobin levels by immunochemistry. This multiparameter stool assay displays a high sensitivity in CRC detection, higher than that of FIT; however, Cologuard yields lower specificity in the case of asymptomatic patients, which represents a limit for its clinical implementation (Imperiale et al., 2014). Another similar FDA-approved test, ColoSure, allows to analyze VIM methylation in stool samples to identify the presence of precancerous adenomas or malignant colorectal tumors (Ned et al., 2011). Another strategy for addressing the methylation of known promoters is the application of the ColoDefense test to stool samples, which enables to detect the methylation status of SEPT9 and SDC2 with high sensitivity and specificity (Zhao et al., 2020a).
Similar to other biofluids, urine also contains cfDNA suitable for epigenetic analyses (Table 6). For instance, the VIM promoter in cfDNA from urine samples was found to display higher methylation in CRC patients than in controls, and thus methylation profiling of this promoter was proposed as a useful test for CRC screening (Song et al., 2012). The methylation status of NDRG4 has also been explored in urine from CRC patients, showing higher diagnostic properties than that in blood and stool (Xiao et al., 2015). The easier manipulation of urine samples with respect to stool supports the value of this approach as a potential clinical method for CRC detection. The methylation levels of WIF1, ALX4, and VIM were also assessed in either urine or serum samples of CRC patients and controls with promising results for CRC diagnosis in the case of WIF1 (Amiot et al., 2014). In addition, several epigenetic markers, including aberrant levels of methylated and hydroxymethylated cytosine nucleosides, have been analyzed in urine samples with promising results, and may be applied as potential biomarkers of CRC (Guo et al., 2018).
Interestingly, there are also other types of fluids that can be obtained from CRC patients. For example, mucosal wash fluid, in which DNA methylation is also detectable, can be collected during colonoscopy (Kamimae et al., 2011). Moreover, the release of cancer cells by the most aggressive tumors might impact the levels of methylated biomarkers in wash samples. To verify this hypothesis, the methylation levels of 15 genes were assessed in bowel lavage fluid (BLF) samples from a large cohort of individuals with CRC, adenomas, and small polyps, as well as from healthy individuals. The methylation levels of three gene promoters (mir-124-3, LOC386758, and SFRP1) showed good correlation with CRC, confirming that methylation studies in BLF samples represent a source of potential biomarkers for CRC detection (Table 6) (Harada et al., 2014).
Histones and Nucleosomes
As previously mentioned, nucleosome occupancy is closely related to the fragmentation patterns of cfDNA (Ivanov et al., 2015). In this regard, the different length of the DNA fragments present in stool has also been proposed as a good tool to discriminate CRC patients from healthy donors. Within this line of research, the integrity of stool DNA from CRC patients was analyzed using an oligonucleotide-based hybrid capture strategy to quantify DNA fragments of 200, 400, 800 bp, 1.3, 1.8, and 24 kb, unraveling the association between high-molecular-weight fragments and this tumor type (Boynton et al., 2003). The presence of long DNA fragments was also found to be increased in CRC patients with respect to healthy controls through the use of fluorescent primers and capillary electrophoresis (Calistri et al., 2004). Such presence of long DNA fragments in stool samples has been reported in several studies as a potential biomarker, either alone (Zou et al., 2006) or in combination with the presence of oncogenic mutations in different genes (KRAS, APC, or p53) (Ahlquist et al., 2000) or with altered methylation patterns (Ahlquist et al., 2012).
Non-coding RNAs
NcRNAs (mainly miRNAs) are stable in stool samples, and thus represent a relevant source of non-coding biomarkers (Table 7). For instance, the analysis of miR-451a and miR-144-5p levels enables to differentiate between patients with CRC and healthy donors with high sensitivity and specificity (Wu et al., 2017), while that of miR-20a levels is more suitable to discriminate CRC patients from both adenoma patients and healthy controls (Yau et al., 2016). In addition, the combined investigation of miR-421 and miR-27a-3p levels with hemoglobin quantification in stool was described as a valuable tool to improve the sensitivity of current screening strategies (Duran-Sanchon et al., 2020). Similarly, increased accumulation of several miRNAs (e.g., miR-21) has also been detected in stool from CRC patients with respect to that from healthy individuals or adenoma patients, and has thus been proposed as a potential screening tool (Wu et al., 2012).

Table 7. Non-coding cfRNAs in non-blood fluids as epigenetic biomarkers of CRC.
On the other hand, several studies have demonstrated the potential of saliva as a source of biomarkers for non-oral cancer detection (Table 7). In particular, two studies have validated the feasibility of detecting increased levels of miRNAs in the saliva of CRC patients. In one of these studies, miR-21 levels were found to be increased in both plasma and saliva of patients with stage II–IV CRC compared to those of healthy individuals. Importantly, the sensitivity and specificity of CRC identification were higher when analyzing saliva samples (Sazanov et al., 2017). On the other hand, Rapado-González et al. performed a unique massive profiling of miRNAs in saliva samples from both CRC patients and healthy donors, and found a total of 22 miRNAs whose accumulation was specifically altered in CRC patients. The levels of most of these miRNAs had been previously described as altered in tissue or plasma samples from CRC patients. Moreover, five of these altered miRNAs (miR-186-5p, miR-29a-3p, miR-29c-3p, miR-766-3p, and miR-491-5p) showed potential as diagnostic tools to detect CRC. Overall, these two studies demonstrated that salivary miRNA analysis represents a novel approach to detect cancer-associated epigenetic alterations with potential clinical value (Rapado-Gonzalez et al., 2019).
MiRNA content was also investigated in cEVs isolated from peritoneal lavage fluid and ascites of CRC patients and control individuals. Such global characterization of cEV-associated miRNAs provided a list of 210 miRNAs whose abundance is significantly altered in CRC patients, most of which were less abundant. From this altered pattern of miRNA accumulation, the authors could identify a 10-miRNA signature with clinical value for CRC detection (Roman-Canal et al., 2019).
Circulating Epigenetic Biomarkers in Blood vs. Alternative Fluids
As we have already described, nowadays there are several body fluids that can serve as liquid biopsies to interrogate epigenetic biomarkers in CRC. Beyond the great value of blood samples as a source of tumor material, stool, and other alternative liquid biopsies showed great potential to analyze clinically relevant biomarkers. Although liquid biopsy has the advantage of being non-invasive and accessible, the diagnostic utility of the different epigenetic biomarkers can be conditioned by the type of body fluid and biomarker analyzed. For example, the detection of some epigenetic biomarkers can be more sensitive in stool than plasma samples. Thus, some studies compared the performance of methylation biomarkers in plasma and stool in parallel, finding relevant differences. This is the case of SEPT9 methylation, which was evaluated in stool and plasma from patients with adenomas and malignant CRC tumors. Both strategies showed similar sensitivity and specificity for the detection of CRC at all stages, however, the methylation levels of SEPT9 in stool showed higher sensitivity for detecting adenomas and early CRC tumors, indicating that the methylation analysis of this gene in stool can improve CRC screening (Liu Y. et al., 2020). The higher diagnostic accuracy observed in stool respect to blood samples could be explained due to biomarkers can be released directly from tumor cells to the intestinal lumen (Osborn and Ahlquist, 2005). Similar results were obtained for the methylation analysis of VIM. Although in advanced CRC this biomarker showed similar diagnostic utility in both types of samples, in early stages the methylation of VIM offered higher diagnostic accuracy in stool than in plasma (Chen et al., 2005; Itzkowitz et al., 2007; Li et al., 2009). On the other hand, in independent studies the levels of methylated SFRP2 in stool and plasma showed similar results in terms of sensitivity and specificity to detect adenomas (Zhang et al., 2014; Bartak et al., 2017). MiRNA biomarkers can also show differences between stool and blood samples. Thus, although the analysis of miR-92a expression was described as a good diagnostic tool in both types of fluids, the sensitivity of miR-92a for adenomas detection was higher in blood than stool (Ng et al., 2009; Wu et al., 2012). In addition, miR-21 was found to have a similar value for discriminating CRC patients from advanced adenomas in blood and stool, but in stool samples the data variability was higher (Peng et al., 2017). In this sense, it is important to mention that stool samples represent a very heterogeneous biological material difficult to normalize among individuals for the quantitative analysis of miRNAs. Standardization of procedures from stool collection and amount of starting material to RNA extraction and detection methods, have been proposed for the detection of CRC with fecal miRNAs (Marcuello et al., 2019).
Similar to stool samples, urine represents a completely non-invasive body fluid that can be collected without pain or risk for the patient, making the sample very suitable for mass-screening of epigenetic biomarkers in CRC. This fluid has shown promising results to analyze the methylation of some genes, such as NDRG4, which provided higher diagnostic power to detect CRC than the same biomarker analyzed in blood and stool (Xiao et al., 2015). Additionally, saliva also has some advantages as a diagnostic tool in comparison with blood samples. Its collection is even less invasive than blood and without causing any discomfort for the patient. Recently, miR-21 expression was assessed in peripheral blood and saliva samples obtained from patients with CRC at different stages and healthy controls. Although miR-21 levels in both saliva and plasma, showed diagnostic value for CRC screening, the analysis of saliva demonstrated higher sensitivity and specificity than blood (Sazanov et al., 2017).
Overall, the selection of the most appropriated body fluid source to analyze the different tumor epigenetic marks will depend on the specific biomarker, the clinical context and the level of clinical validation, which is nowadays higher in blood and stool samples than in other alternative body fluids.
Conclusions and Future Perspectives
The interest in epigenetic alterations associated with CRC development and progression as potential clinical biomarkers or therapeutic targets has increased significantly in recent years. In the present review, we provide an overview of the different epigenetic mechanisms that regulate key steps of CRC development. Because of the clear advantage of epigenetic analysis in liquid biopsies as a non-invasive method for the dynamic characterization of CRC, we also summarize the techniques currently applied to characterize epigenetic modifications in liquid biopsy, and describe the circulating epigenetic biomarkers identified in different body fluids and their clinical potential for the personalized management of CRC.
There is a wide range of epigenetic mechanisms that are altered in CRC, including DNA methylation and hydroxymethylation, nucleosome positioning, histone modifications, and the expression of different ncRNAs (miRNAs, lncRNAs, and circRNAs). All these alterations can be explored in different fluid samples such as blood, stool, urine, or saliva, and represent a valuable source of clinical biomarkers. In fact, as detailed in this review, the differential methylation status of several gene promoters in CRC constitutes the rationale of several commercialized screening tests, such as Cologuard or Epi ProColon. However, substantial optimization is required prior to the general implementation of circulating epigenetic markers in the clinic to guide the management of CRC patients.
Indeed, some technical factors clearly hamper the potential translation of circulating biomarkers into the clinical routine. For example, CTCs, cNAs, and cEVs released by tumor and metastases into body fluids are usually poorly concentrated with respect to circulating non-tumor elements. Therefore, the methodology employed for their detection must be very sensitive in both localized and advanced CRC. On the other hand, new NGS-based strategies, together with ddPCR-based approaches, display improved sensitivity for the detection of epigenetic signatures in cfDNA, opening new avenues for early diagnosis of CRC. However, these approaches usually require sophisticated laboratory equipment, and are too laborious and expensive to be generically implemented into the clinical routine. Epigenetic patterns are starting to be explored also in CTCs, but mainly with the aim of characterizing the specific signatures associated with the disseminative behavior of these tumor cells, due to the difficulty of isolating CTCs even in metastatic patients. Together with their sensitivity, the specificity of epigenetic biomarkers represents a critical factor affecting their clinical utility. In fact, poor specificity of screening tests would result in unnecessary invasive evaluations and undesired side effects.
Molecular intratumoral heterogeneity also represents a clinical and technical challenge for the translation of epigenetic biomarkers in the clinical setting. It is well-acknowledged that CRC tumors are composed of multiple tumor clones with different biological properties. This heterogeneity also has an impact on the variability of several epigenetic marks, with possible clinical implications. Therefore, technical improvement is required for the study of epigenetic changes in single CTCs, which will be of great value to achieve a better understanding of CRC biology.
Another important aspect is the need for standardized protocols for sample collection, processing, and storage to improve the reproducibility of studies. In particular, sample collection and pretreatment are key steps for methylation studies, and a source of heterogeneity (Pharo et al., 2016; Merker et al., 2018; Zavridou et al., 2018). Therefore, efforts should be made to harmonize pre-analytical and analytical protocols according to the epigenetic alteration, circulating element, or body fluid under investigation. Hopefully, this standardization will be possible in the near future thanks to the commitment of international initiatives and scientific societies.
A major consideration for the clinical translation of epigenetic biomarkers is the cost effectiveness of the analyses. Nowadays, most technologies consist in time-demanding, mainly NGS-based approaches, which include library preparation and bioinformatic interpretation. However, in the future, these strategies should be associated with more adjusted costs and user-friendly bioinformatic solutions. To this purpose, it would be convenient to stimulate the technological development and commercial interest of epigenetic-based tests, thereby creating a competitive environment that will lead to significant benefits in terms of pricing and effectiveness.
Of note, the scientific community have focused their attention on the great potential of epigenomic approaches in the analysis of liquid biopsy for diagnostic purposes. For instance, one of the emerging trends is the use of broad panels of methylated biomarkers in cfDNA for pan-cancer detection. Moreover, recent studies on cfDNA have revealed the importance of NGS-based approaches for the analysis of methylation signatures, nucleosome positioning, or fragmentation footprints, opening a new scenario for epigenetics in liquid biopsy (Snyder et al., 2016; Liu et al., 2018; Cristiano et al., 2019; Liu M. C. et al., 2020). In addition, there is great interest in increasing the knowledge of the epigenetic characteristics of CTCs and cEVs also in terms of other epigenetic layers, such as 5hmC levels, differential presence of various types of ncRNAs, and epitranscriptomic modifications.
The methylation analysis of SEPT9 (Epi proColon test) was the first blood-based assay approved by the FDA (Pickhardt, 2016; Issa and Noureddine, 2017), opening great opportunities for the clinical applications of circulating epigenetic biomarkers. One of the most challenging goals to be addressed is the clinical validation of the numerous epigenetic biomarkers that have provided promising results but have not yet reached a routine use, in order to assess their prognostic value for the outcome of patients with CRC. Indeed, the translation of the results obtained with epigenetic biomarkers into diagnostic tools is still limited, probably due to the lack of standardization and the limited number of large independent cohorts that have been analyzed. Therefore, more clinical trials, including the study of the dynamics of these epigenetics biomarkers, are warranted to validate their impact in terms of survival benefit. Similarly, numerous studies have shown that epigenetic alterations can be reversed through pharmacological intervention. However, evidence supporting the benefit of epigenetic modifiers in patients with CRC is still faint (Rezapour et al., 2019).
In summary, epigenetic circulating biomarkers have demonstrated a great potential for diagnosis of CRC, as well as for monitoring the progression and therapy response of CRC patients in a non-invasive and dynamic way. However, although significant progress has been made in the clinical translation of these biomarkers in recent years, further research is required to overcome some technologic and clinical barriers. Indeed, the translation of circulating epigenetic biomarkers into the clinical setting will require large multicenter studies to demonstrate the clinical benefit of their use. Such studies should be carried out in the near future and are expected to yield valuable results, toward a more personalized management of CRC patients.
Author Contributions
RL-L, LM-R, and AD-L designed the work. AR-C, NC-F, AB-C, LM-R, and AD-L wrote the original manuscript and prepared the tables. NC-F, LM-R, and AD-L designed all the figures. AR-C, NC-F, AB-C, LM-R, RL-L, and AD-L reviewed the manuscript. LM-R and AD-L supervised the work. All the authors contributed to the article and approved the submitted version.
Funding
This work was co-funded by the ISCIII (PI18/00307) and the European Regional Development Fund (FEDER). AD-L was funded by a contract Juan Rodés from ISCIII (JR17/00016). AB-C was funded by a predoctoral contract PFIS from ISCIII (FI19/00240) co-funded by Fondo Social Europeo (FSE). AR-C was supported by Roche-Chus Joint Unit (IN853B 2018/03) funded by GAIN, Consellería de Economía, Emprego e Industria. NC-F was funded by a predoctoral contract from Xunta de Galicia (IN606A-2020/004). LM-R was supported by AECC.
Conflict of Interest
RL-L has received honoraria for participation in Advisory Boards from Roche, AstraZeneca, Merck, MSD, Bayer, BMS, Novartis, Janssen, Lilly, Pfizer and Leo; travel, accommodations and expenses from Pharmamar, Roche, BMS and Pierre Fabre; research funding from Roche and Merck; and is co-founder and shareholder in Nasasbiotech, S.L., Mtrap Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahlquist, D. A., Skoletsky, J. E., Boynton, K. A., Harrington, J. J., Mahoney, D. W., Pierceall, W. E., et al. (2000). Colorectal cancer screening by detection of altered human DNA in stool: feasibility of a multitarget assay panel. Gastroenterology 119, 1219–1227. doi: 10.1053/gast.2000.19580
Ahlquist, D. A., Zou, H., Domanico, M., Mahoney, D. W., Yab, T. C., Taylor, W. R., et al. (2012). Next-generation stool DNA test accurately detects colorectal cancer and large adenomas. Gastroenterology 142, 248–256; quiz e225–246. doi: 10.1053/j.gastro.2011.10.031
Akers, J. C., Gonda, D., Kim, R., Carter, B. S., and Chen, C. C. (2013). Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 113, 1–11. doi: 10.1007/s11060-013-1084-8
Allard, W. J., Matera, J., Miller, M. C., Repollet, M., Connelly, M. C., Rao, C., et al. (2004). Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 10, 6897–6904. doi: 10.1158/1078-0432.CCR-04-0378
Amiot, A., Mansour, H., Baumgaertner, I., Delchier, J. C., Tournigand, C., Furet, J. P., et al. (2014). The detection of the methylated Wif-1 gene is more accurate than a fecal occult blood test for colorectal cancer screening. PLoS ONE 9:e99233. doi: 10.1371/journal.pone.0099233
Amorim, M. G., Valieris, R., Drummond, R. D., Pizzi, M. P., Freitas, V. M., Sinigaglia-Coimbra, R., et al. (2017). A total transcriptome profiling method for plasma-derived extracellular vesicles: applications for liquid biopsies. Sci. Rep. 7:14395. doi: 10.1038/s41598-017-14264-5
Anfossi, S., Babayan, A., Pantel, K., and Calin, G. A. (2018). Clinical utility of circulating non-coding RNAs - an update. Nat. Rev. Clin. Oncol. 15, 541–563. doi: 10.1038/s41571-018-0035-x
Arechederra, M., Daian, F., Yim, A., Bazai, S. K., Richelme, S., Dono, R., et al. (2018). Hypermethylation of gene body CpG islands predicts high dosage of functional oncogenes in liver cancer. Nat. Commun. 9:3164. doi: 10.1038/s41467-018-05550-5
Ashworth, T. (1869). A case of cancer in which cells similar to those in the tumors were seen in the blood after death. Med. J. Aust. 14, 146–147.
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Bao-Caamano, A., Rodriguez-Casanova, A., and Diaz-Lagares, A. (2020). Epigenetics of circulating tumor cells in breast cancer. Adv. Exp. Med. Biol. 1220, 117–134. doi: 10.1007/978-3-030-35805-1_8
Barault, L., Amatu, A., Bleeker, F. E., Moutinho, C., Falcomata, C., Fiano, V., et al. (2015). Digital PCR quantification of MGMT methylation refines prediction of clinical benefit from alkylating agents in glioblastoma and metastatic colorectal cancer. Ann. Oncol. 26, 1994–1999. doi: 10.1093/annonc/mdv272
Barault, L., Amatu, A., Siravegna, G., Ponzetti, A., Moran, S., Cassingena, A., et al. (2018). Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer. Gut 67, 1995–2005. doi: 10.1136/gutjnl-2016-313372
Barbazan, J., Alonso-Alconada, L., Elkhatib, N., Geraldo, S., Gurchenkov, V., Glentis, A., et al. (2017). Liver metastasis is facilitated by the adherence of circulating tumor cells to vascular fibronectin deposits. Cancer Res. 77, 3431–3441. doi: 10.1158/0008-5472.CAN-16-1917
Barbazan, J., Alonso-Alconada, L., Muinelo-Romay, L., Vieito, M., Abalo, A., Alonso-Nocelo, M., et al. (2012). Molecular characterization of circulating tumor cells in human metastatic colorectal cancer. PLoS ONE 7:e40476. doi: 10.1371/journal.pone.0040476
Bardelli, A., and Pantel, K. (2017). Liquid biopsies, what we do not know (Yet). Cancer Cell 31, 172–179. doi: 10.1016/j.ccell.2017.01.002
Baretti, M., and Azad, N. S. (2018). The role of epigenetic therapies in colorectal cancer. Curr. Probl. Cancer 42, 530–547. doi: 10.1016/j.currproblcancer.2018.03.001
Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. doi: 10.1016/j.cell.2007.05.009
Bartak, B. K., Kalmar, A., Peterfia, B., Patai, A. V., Galamb, O., Valcz, G., et al. (2017). Colorectal adenoma and cancer detection based on altered methylation pattern of SFRP1, SFRP2, SDC2, and PRIMA1 in plasma samples. Epigenetics 12, 751–763. doi: 10.1080/15592294.2017.1356957
Bates, S. E. (2020). Epigenetic therapies for cancer. N. Engl. J. Med 383, 650–663. doi: 10.1056/NEJMra1805035
Bayraktar, R., Pichler, M., Kanlikilicer, P., Ivan, C., Bayraktar, E., Kahraman, N., et al. (2017). MicroRNA 603 acts as a tumor suppressor and inhibits triple-negative breast cancer tumorigenesis by targeting elongation factor 2 kinase. Oncotarget 8, 11641–11658. doi: 10.18632/oncotarget.14264
Becker, A., Thakur, B. K., Weiss, J. M., Kim, H. S., Peinado, H., and Lyden, D. (2016). Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell 30, 836–848. doi: 10.1016/j.ccell.2016.10.009
Berdasco, M., and Esteller, M. (2019). Clinical epigenetics: seizing opportunities for translation. Nat. Rev. Genet. 20, 109–127. doi: 10.1038/s41576-018-0074-2
Berger, S. L., Kouzarides, T., Shiekhattar, R., and Shilatifard, A. (2009). An operational definition of epigenetics. Genes Dev. 23, 781–783. doi: 10.1101/gad.1787609
Bettegowda, C., Sausen, M., Leary, R. J., Kinde, I., Wang, Y., Agrawal, N., et al. (2014). Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 6:ra224. doi: 10.1126/scitranslmed.3007094
Bhangu, J. S., Beer, A., Mittlbock, M., Tamandl, D., Pulverer, W., Schonthaler, S., et al. (2018). Circulating free methylated tumor DNA markers for sensitive assessment of tumor burden and early response monitoring in patients receiving systemic chemotherapy for colorectal cancer liver metastasis. Ann. Surg. 268, 894–902. doi: 10.1097/SLA.0000000000002901
Boeckx, N., Op de Beeck, K., Beyens, M., Deschoolmeester, V., Hermans, C., De Clercq, P., et al. (2018). Mutation and methylation analysis of circulating tumor DNA can be used for follow-up of metastatic colorectal cancer patients. Clin. Colorectal Cancer 17, e369–e379. doi: 10.1016/j.clcc.2018.02.006
Bork, U., Rahbari, N. N., Scholch, S., Reissfelder, C., Kahlert, C., Buchler, M. W., et al. (2015). Circulating tumour cells and outcome in non-metastatic colorectal cancer: a prospective study. Br. J. Cancer 112, 1306–1313. doi: 10.1038/bjc.2015.88
Boynton, K. A., Summerhayes, I. C., Ahlquist, D. A., and Shuber, A. P. (2003). DNA integrity as a potential marker for stool-based detection of colorectal cancer. Clin. Chem. 49, 1058–1065. doi: 10.1373/49.7.1058
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Bu, H., He, D., He, X., and Wang, K. (2019). Exosomes: isolation, analysis, and applications in cancer detection and therapy. Chembiochem 20, 451–461. doi: 10.1002/cbic.201800470
Caley, D. P., Pink, R. C., Trujillano, D., and Carter, D. R. (2010). Long noncoding RNAs, chromatin, and development. Sci. World J. 10, 90–102. doi: 10.1100/tsw.2010.7
Calistri, D., Rengucci, C., Lattuneddu, A., Francioni, G., Polifemo, A. M., Nanni, O., et al. (2004). Detection of colorectal cancer by a quantitative fluorescence determination of DNA amplification in stool. Neoplasia 6, 536–540. doi: 10.1593/neo.04190
Castellanos-Rizaldos, E., Grimm, D. G., Tadigotla, V., Hurley, J., Healy, J., Neal, P. L., et al. (2018). Exosome-based detection of EGFR T790M in plasma from non-small cell lung cancer patients. Clin. Cancer Res. 24, 2944–2950. doi: 10.1158/1078-0432.CCR-17-3369
Chen, J. Y., Luo, C. W., Lai, Y. S., Wu, C. C., and Hung, W. C. (2017). Lysine demethylase KDM2A inhibits TET2 to promote DNA methylation and silencing of tumor suppressor genes in breast cancer. Oncogenesis 6:e369. doi: 10.1038/oncsis.2017.71
Chen, W. D., Han, Z. J., Skoletsky, J., Olson, J., Sah, J., Myeroff, L., et al. (2005). Detection in fecal DNA of colon cancer-specific methylation of the nonexpressed vimentin gene. J. Natl. Cancer Inst. 97, 1124–1132. doi: 10.1093/jnci/dji204
Chen, X., Gole, J., Gore, A., He, Q., Lu, M., Min, J., et al. (2020). Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 11:3475. doi: 10.1038/s41467-020-17316-z
Cheng, H., Zhang, L., Cogdell, D. E., Zheng, H., Schetter, A. J., Nykter, M., et al. (2011). Circulating plasma MiR-141 is a novel biomarker for metastatic colon cancer and predicts poor prognosis. PLoS ONE 6:e17745. doi: 10.1371/journal.pone.0017745
Chimonidou, M., Strati, A., Tzitzira, A., Sotiropoulou, G., Malamos, N., Georgoulias, V., et al. (2011). DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin. Chem. 57, 1169–1177. doi: 10.1373/clinchem.2011.165902
Church, T. R., Wandell, M., Lofton-Day, C., Mongin, S. J., Burger, M., Payne, S. R., et al. (2014). Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut 63, 317–325. doi: 10.1136/gutjnl-2012-304149
Cohen, J. D., Li, L., Wang, Y., Thoburn, C., Afsari, B., Danilova, L., et al. (2018). Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930. doi: 10.1126/science.aar3247
Cohen, S. J., Punt, C. J., Iannotti, N., Saidman, B. H., Sabbath, K. D., Gabrail, N. Y., et al. (2008). Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 26, 3213–3221. doi: 10.1200/JCO.2007.15.8923
Cojocneanu, R., Braicu, C., Raduly, L., Jurj, A., Zanoaga, O., Magdo, L., et al. (2020). Plasma and tissue specific miRNA expression pattern and functional analysis associated to colorectal cancer patients. Cancers 12:843. doi: 10.3390/cancers12040843
Cristiano, S., Leal, A., Phallen, J., Fiksel, J., Adleff, V., Bruhm, D. C., et al. (2019). Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 570, 385–389. doi: 10.1038/s41586-019-1272-6
Cristofanilli, M., Budd, G. T., Ellis, M. J., Stopeck, A., Matera, J., Miller, M. C., et al. (2004). Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med 351, 781–791. doi: 10.1056/NEJMoa040766
Danese, E., Minicozzi, A. M., Benati, M., Montagnana, M., Paviati, E., Salvagno, G. L., et al. (2013). Epigenetic alteration: new insights moving from tissue to plasma - the example of PCDH10 promoter methylation in colorectal cancer. Br. J. Cancer 109, 807–813. doi: 10.1038/bjc.2013.351
De Koker, A., Van Paemel, R., De Wilde, B., De Preter, K., and Callewaert, N. (2019). A versatile method for circulating cell-free DNA methylome profiling by reduced representation bisulfite sequencing. bioRxiv 663195. doi: 10.1101/663195
de la Fuente, A., Alonso-Alconada, L., Costa, C., Cueva, J., Garcia-Caballero, T., Lopez-Lopez, R., et al. (2015). M-Trap: exosome-based capture of tumor cells as a new technology in peritoneal metastasis. J. Natl. Cancer Inst. 107:djv184. doi: 10.1093/jnci/djv184
Deal, R. B., Henikoff, J. G., and Henikoff, S. (2010). Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328, 1161–1164. doi: 10.1126/science.1186777
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H., et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. doi: 10.1101/gr.132159.111
Di, Z., Di, M., Fu, W., Tang, Q., Liu, Y., Lei, P., et al. (2020). Integrated analysis identifies a nine-microrna signature biomarker for diagnosis and prognosis in colorectal cancer. Front. Genet. 11:192. doi: 10.3389/fgene.2020.00192
Diaz-Lagares, A., Crujeiras, A. B., Lopez-Serra, P., Soler, M., Setien, F., Goyal, A., et al. (2016a). Epigenetic inactivation of the p53-induced long noncoding RNA TP53 target 1 in human cancer. Proc. Natl. Acad. Sci. U.S.A. 113, E7535–E7544. doi: 10.1073/pnas.1608585113
Diaz-Lagares, A., Mendez-Gonzalez, J., Hervas, D., Saigi, M., Pajares, M. J., Garcia, D., et al. (2016b). A novel epigenetic signature for early diagnosis in lung cancer. Clin. Cancer Res. 22, 3361–3371. doi: 10.1158/1078-0432.CCR-15-2346
Dragomir, M., Mafra, A. C. P., Dias, S. M. G., Vasilescu, C., and Calin, G. A. (2018). Using microRNA Networks to Understand Cancer. Int. J. Mol. Sci. 19:1871. doi: 10.3390/ijms19071871
Duran-Sanchon, S., Moreno, L., Auge, J. M., Serra-Burriel, M., Cuatrecasas, M., Moreira, L., et al. (2020). Identification and validation of microRNA profiles in fecal samples for detection of colorectal cancer. Gastroenterology 158, 947–957 e944. doi: 10.1053/j.gastro.2019.10.005
Eads, C. A., Danenberg, K. D., Kawakami, K., Saltz, L. B., Blake, C., Shibata, D., et al. (2000). MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 28:E32. doi: 10.1093/nar/28.8.e32
El Messaoudi, S., Mouliere, F., Du Manoir, S., Bascoul-Mollevi, C., Gillet, B., Nouaille, M., et al. (2016). Circulating DNA as a strong multimarker prognostic tool for metastatic colorectal cancer patient management care. Clin. Cancer Res. 22, 3067–3077. doi: 10.1158/1078-0432.CCR-15-0297
Esteller, M. (2007). Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum. Mol. Genet. 16, R50–R59. doi: 10.1093/hmg/ddm018
Esteller, M. (2008). Epigenetics in cancer. N. Engl. J. Med. 358, 1148–1159. doi: 10.1056/NEJMra072067
Esteller, M. (2011). Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861–874. doi: 10.1038/nrg3074
Esteller, M., Hamilton, S. R., Burger, P. C., Baylin, S. B., and Herman, J. G. (1999). Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 59, 793–797.
Fahmueller, Y. N., Nagel, D., Hoffmann, R. T., Tatsch, K., Jakobs, T., Stieber, P., et al. (2012). Predictive and prognostic value of circulating nucleosomes and serum biomarkers in patients with metastasized colorectal cancer undergoing Selective Internal Radiation Therapy. BMC Cancer 12:5. doi: 10.1186/1471-2407-12-5
Fearon, E. R., and Vogelstein, B. (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759–767. doi: 10.1016/0092-8674(90)90186-i
Fiegle, E., Doleschel, D., Koletnik, S., Rix, A., Weiskirchen, R., Borkham-Kamphorst, E., et al. (2019). Dual CTLA-4 and PD-L1 blockade inhibits tumor growth and liver metastasis in a highly aggressive orthotopic mouse model of colon cancer. Neoplasia 21, 932–944. doi: 10.1016/j.neo.2019.07.006
Friedlander, T. W., Ngo, V. T., Dong, H., Premasekharan, G., Weinberg, V., Doty, S., et al. (2014). Detection and characterization of invasive circulating tumor cells derived from men with metastatic castration-resistant prostate cancer. Int. J. Cancer 134, 2284–2293. doi: 10.1002/ijc.28561
Frommer, M., McDonald, L. E., Millar, D. S., Collis, C. M., Watt, F., Grigg, G. W., et al. (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. U.S.A. 89, 1827–1831. doi: 10.1073/pnas.89.5.1827
Fu, F., Jiang, W., Zhou, L., and Chen, Z. (2018). Circulating exosomal miR-17-5p and miR-92a-3p predict pathologic stage and grade of colorectal cancer. Transl. Oncol. 11, 221–232. doi: 10.1016/j.tranon.2017.12.012
Gallardo-Gomez, M., Moran, S., Paez de la Cadena, M., Martinez-Zorzano, V. S., Rodriguez-Berrocal, F. J., Rodriguez-Girondo, M., et al. (2018). A new approach to epigenome-wide discovery of non-invasive methylation biomarkers for colorectal cancer screening in circulating cell-free DNA using pooled samples. Clin. Epigenetics 10:53. doi: 10.1186/s13148-018-0487-y
Gao, P., Lin, S., Cai, M., Zhu, Y., Song, Y., Sui, Y., et al. (2019). 5-Hydroxymethylcytosine profiling from genomic and cell-free DNA for colorectal cancers patients. J. Cell. Mol. Med. 23, 3530–3537. doi: 10.1111/jcmm.14252
Garrigou, S., Perkins, G., Garlan, F., Normand, C., Didelot, A., Le Corre, D., et al. (2016). A study of hypermethylated circulating tumor DNA as a universal colorectal cancer biomarker. Clin. Chem. 62, 1129–1139. doi: 10.1373/clinchem.2015.253609
Garzon, R., Calin, G. A., and Croce, C. M. (2009). MicroRNAs in cancer. Annu. Rev. Med. 60, 167–179. doi: 10.1146/annurev.med.59.053006.104707
Gasch, C., Plummer, P. N., Jovanovic, L., McInnes, L. M., Wescott, D., Saunders, C. M., et al. (2015). Heterogeneity of miR-10b expression in circulating tumor cells. Sci. Rep. 5:15980. doi: 10.1038/srep15980
Gasparello, J., Papi, C., Allegretti, M., Giordani, E., Carboni, F., Zazza, S., et al. (2020). A Distinctive microRNA (miRNA) signature in the blood of colorectal cancer (CRC) patients at surgery. Cancers 12:2410. doi: 10.3390/cancers12092410
Gezer, U., Mert, U., Ozgur, E., Yoruker, E. E., Holdenrieder, S., and Dalay, N. (2012). Correlation of histone methyl marks with circulating nucleosomes in blood plasma of cancer patients. Oncol. Lett. 3, 1095–1098. doi: 10.3892/ol.2012.600
Gezer, U., Ustek, D., Yoruker, E. E., Cakiris, A., Abaci, N., Leszinski, G., et al. (2013). Characterization of H3K9me3- and H4K20me3-associated circulating nucleosomal DNA by high-throughput sequencing in colorectal cancer. Tumour Biol. 34, 329–336. doi: 10.1007/s13277-012-0554-5
Gezer, U., Yoruker, E. E., Keskin, M., Kulle, C. B., Dharuman, Y., and Holdenrieder, S. (2015). Histone methylation marks on circulating nucleosomes as novel blood-based biomarker in colorectal cancer. Int. J. Mol. Sci 16, 29654–29662. doi: 10.3390/ijms161226180
Gilat, N., Tabachnik, T., Shwartz, A., Shahal, T., Torchinsky, D., Michaeli, Y., et al. (2017). Single-molecule quantification of 5-hydroxymethylcytosine for diagnosis of blood and colon cancers. Clin. Epigenetics 9:70. doi: 10.1186/s13148-017-0368-9
Gimeno-Valiente, F., Riffo-Campos, A. L., Vallet-Sanchez, A., Siscar-Lewin, S., Gambardella, V., Tarazona, N., et al. (2019). ZNF518B gene up-regulation promotes dissemination of tumour cells and is governed by epigenetic mechanisms in colorectal cancer. Sci. Rep. 9:9339. doi: 10.1038/s41598-019-45411-9
Gkountela, S., Castro-Giner, F., Szczerba, B. M., Vetter, M., Landin, J., Scherrer, R., et al. (2019). Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 176, 98–112 e114. doi: 10.1016/j.cell.2018.11.046
Gonzalez-Zulueta, M., Bender, C. M., Yang, A. S., Nguyen, T., Beart, R. W., Van Tornout, J. M., et al. (1995). Methylation of the 5' CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res. 55, 4531–4535
Gowher, H., and Jeltsch, A. (2002). Molecular enzymology of the catalytic domains of the Dnmt3a and Dnmt3b DNA methyltransferases. J. Biol. Chem. 277, 20409–20414. doi: 10.1074/jbc.M202148200
Grunau, C., Clark, S. J., and Rosenthal, A. (2001). Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 29, E65–65. doi: 10.1093/nar/29.13.e65
Guo, C., Xie, C., Chen, Q., Cao, X., Guo, M., Zheng, S., et al. (2018). A novel malic acid-enhanced method for the analysis of 5-methyl-2'-deoxycytidine, 5-hydroxymethyl-2'-deoxycytidine, 5-methylcytidine and 5-hydroxymethylcytidine in human urine using hydrophilic interaction liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 1034, 110–118. doi: 10.1016/j.aca.2018.06.081
Guo, Q., Song, Y., Zhang, H., Wu, X., Xia, P., and Dang, C. (2013). Detection of hypermethylated fibrillin-1 in the stool samples of colorectal cancer patients. Med. Oncol. 30, 695. doi: 10.1007/s12032-013-0695-4
Gupta, G. P., and Massague, J. (2006). Cancer metastasis: building a framework. Cell 127, 679–695. doi: 10.1016/j.cell.2006.11.001
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., et al. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. doi: 10.1038/nature08975
Gutschner, T., Hammerle, M., Eissmann, M., Hsu, J., Kim, Y., Hung, G., et al. (2013). The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 73, 1180–1189. doi: 10.1158/0008-5472.CAN-12-2850
Guttman, M., Amit, I., Garber, M., French, C., Lin, M. F., Feldser, D., et al. (2009). Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227. doi: 10.1038/nature07672
Han, Y. D., Oh, T. J., Chung, T. H., Jang, H. W., Kim, Y. N., An, S., et al. (2019). Early detection of colorectal cancer based on presence of methylated syndecan-2 (SDC2) in stool DNA. Clin. Epigenetics 11:51. doi: 10.1186/s13148-019-0642-0
Hansen, T. F., Carlsen, A. L., Heegaard, N. H., Sorensen, F. B., and Jakobsen, A. (2015). Changes in circulating microRNA-126 during treatment with chemotherapy and bevacizumab predicts treatment response in patients with metastatic colorectal cancer. Br. J. Cancer 112, 624–629. doi: 10.1038/bjc.2014.652
Hao, Y. X., Li, Y. M., Ye, M., Guo, Y. Y., Li, Q. W., Peng, X. M., et al. (2017). KRAS and BRAF mutations in serum exosomes from patients with colorectal cancer in a Chinese population. Oncol. Lett. 13, 3608–3616. doi: 10.3892/ol.2017.5889
Harada, T., Yamamoto, E., Yamano, H. O., Nojima, M., Maruyama, R., Kumegawa, K., et al. (2014). Analysis of DNA methylation in bowel lavage fluid for detection of colorectal cancer. Cancer Prev. Res. 7, 1002–1010. doi: 10.1158/1940-6207.CAPR-14-0162
Herceg, Z., and Vaissiere, T. (2011). Epigenetic mechanisms and cancer: an interface between the environment and the genome. Epigenetics 6, 804–819. doi: 10.4161/epi.6.7.16262
Herman, J. G., Graff, J. R., Myohanen, S., Nelkin, B. D., and Baylin, S. B. (1996). Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U.S.A. 93, 9821–9826. doi: 10.1073/pnas.93.18.9821
Herreros-Villanueva, M., Duran-Sanchon, S., Martin, A. C., Perez-Palacios, R., Vila-Navarro, E., Marcuello, M., et al. (2019). Plasma microRNA signature validation for early detection of colorectal cancer. Clin. Transl. Gastroenterol. 10:e00003. doi: 10.14309/ctg.0000000000000003
Hesson, L. B., Sloane, M. A., Wong, J. W., Nunez, A. C., Srivastava, S., Ng, B., et al. (2014). Altered promoter nucleosome positioning is an early event in gene silencing. Epigenetics 9, 1422–1430. doi: 10.4161/15592294.2014.970077
Hiltunen, M. O., Alhonen, L., Koistinaho, J., Myohanen, S., Paakkonen, M., Marin, S., et al. (1997). Hypermethylation of the APC (adenomatous polyposis coli) gene promoter region in human colorectal carcinoma. Int. J. Cancer 70, 644–648. doi: 10.1002/(sici)1097-0215(19970317)70:6<644::aid-ijc3>3.0.co
Holdenrieder, S., Stieber, P., Bodenmuller, H., Busch, M., Fertig, G., Furst, H., et al. (2001). Nucleosomes in serum of patients with benign and malignant diseases. Int. J. Cancer 95, 114–120. doi: 10.1002/1097-0215(20010320)95:2<114::aid-ijc1020>3.0.co
Holliday, R. (1987). The inheritance of epigenetic defects. Science 238, 163–170. doi: 10.1126/science.3310230
Hon, C. C., Ramilowski, J. A., Harshbarger, J., Bertin, N., Rackham, O. J., Gough, J., et al. (2017). An atlas of human long non-coding RNAs with accurate 5' ends. Nature 543, 199–204. doi: 10.1038/nature21374
Hu, H., Shu, M., He, L., Yu, X., Liu, X., Lu, Y., et al. (2017). Epigenomic landscape of 5-hydroxymethylcytosine reveals its transcriptional regulation of lncRNAs in colorectal cancer. Br. J. Cancer 116, 658–668. doi: 10.1038/bjc.2016.457
Huang, J., and Wang, L. (2019). Cell-Free DNA methylation profiling analysis-technologies and bioinformatics. Cancers 11:1741. doi: 10.3390/cancers11111741
Huang, Z. H., Li, L. H., Yang, F., and Wang, J. F. (2007). Detection of aberrant methylation in fecal DNA as a molecular screening tool for colorectal cancer and precancerous lesions. World J. Gastroenterol. 13, 950–954. doi: 10.3748/wjg.v13.i6.950
Huertas, J., MacCarthy, C. M., Scholer, H. R., and Cojocaru, V. (2020). Nucleosomal DNA dynamics mediate Oct4 pioneer factor binding. Biophys. J. 118, 2280–2296. doi: 10.1016/j.bpj.2019.12.038
Hurd, P. J., and Nelson, C. J. (2009). Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief. Funct. Genomics Proteomics 8, 174–183. doi: 10.1093/bfgp/elp013
Imperiale, T. F., Ransohoff, D. F., Itzkowitz, S. H., Levin, T. R., Lavin, P., Lidgard, G. P., et al. (2014). Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 370, 1287–1297. doi: 10.1056/NEJMoa1311194
Issa, I. A., and Noureddine, M. (2017). Colorectal cancer screening: an updated review of the available options. World J. Gastroenterol. 23, 5086–5096. doi: 10.3748/wjg.v23.i28.5086
Itzkowitz, S. H., Jandorf, L., Brand, R., Rabeneck, L., Schroy, P. C. III., Sontag, S., et al. (2007). Improved fecal DNA test for colorectal cancer screening. Clin. Gastroenterol. Hepatol 5, 111–117. doi: 10.1016/j.cgh.2006.10.006
Ivanov, M., Baranova, A., Butler, T., Spellman, P., and Mileyko, V. (2015). Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genomics 16 (Suppl. 13):S1. doi: 10.1186/1471-2164-16-S13-S1
Jaenisch, R., and Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33(Suppl.), 245–254. doi: 10.1038/ng1089
Jayaseelan, V. P. (2020). Emerging role of exosomes as promising diagnostic tool for cancer. Cancer Gene Ther. 27, 395–398. doi: 10.1038/s41417-019-0136-4
Jensen, S. O., Ogaard, N., Orntoft, M. W., Rasmussen, M. H., Bramsen, J. B., Kristensen, H., et al. (2019). Novel DNA methylation biomarkers show high sensitivity and specificity for blood-based detection of colorectal cancer-a clinical biomarker discovery and validation study. Clin. Epigenetics 11:158. doi: 10.1186/s13148-019-0757-3
Jiang, P., Chan, C. W., Chan, K. C., Cheng, S. H., Wong, J., Wong, V. W., et al. (2015). Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 112, E1317–1325. doi: 10.1073/pnas.1500076112
Jin, G., Liu, Y., Zhang, J., Bian, Z., Yao, S., Fei, B., et al. (2019). A panel of serum exosomal microRNAs as predictive markers for chemoresistance in advanced colorectal cancer. Cancer Chemother. Pharmacol. 84, 315–325. doi: 10.1007/s00280-019-03867-6
John, S. K., George, S., Primrose, J. N., and Fozard, J. B. (2011). Symptoms and signs in patients with colorectal cancer. Colorectal Dis. 13, 17–25. doi: 10.1111/j.1463-1318.2010.02221.x
Jung, G., Hernandez-Illan, E., Moreira, L., Balaguer, F., and Goel, A. (2020). Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 17, 111–130. doi: 10.1038/s41575-019-0230-y
Kalimutho, M., Di Cecilia, S., Del Vecchio Blanco, G., Roviello, F., Sileri, P., Cretella, M., et al. (2011). Epigenetically silenced miR-34b/c as a novel faecal-based screening marker for colorectal cancer. Br. J. Cancer 104, 1770–1778. doi: 10.1038/bjc.2011.82
Kamimae, S., Yamamoto, E., Yamano, H. O., Nojima, M., Suzuki, H., Ashida, M., et al. (2011). Epigenetic alteration of DNA in mucosal wash fluid predicts invasiveness of colorectal tumors. Cancer Prev. Res. 4, 674–683. doi: 10.1158/1940-6207.CAPR-10-0214
Kanaan, Z., Roberts, H., Eichenberger, M. R., Billeter, A., Ocheretner, G., Pan, J., et al. (2013). A plasma microRNA panel for detection of colorectal adenomas: a step toward more precise screening for colorectal cancer. Ann. Surg. 258, 400–408. doi: 10.1097/SLA.0b013e3182a15bcc
Kane, M. F., Loda, M., Gaida, G. M., Lipman, J., Mishra, R., Goldman, H., et al. (1997). Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 57, 808–811
Kapranov, P., Cheng, J., Dike, S., Nix, D. A., Duttagupta, R., Willingham, A. T., et al. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488. doi: 10.1126/science.1138341
Karczmarski, J., Rubel, T., Paziewska, A., Mikula, M., Bujko, M., Kober, P., et al. (2014). Histone H3 lysine 27 acetylation is altered in colon cancer. Clin. Proteomics 11:24. doi: 10.1186/1559-0275-11-24
Kim, M. S., Louwagie, J., Carvalho, B., Terhaar Sive Droste, J. S., Park, H. L., Chae, Y. K., et al. (2009). Promoter DNA methylation of oncostatin m receptor-beta as a novel diagnostic and therapeutic marker in colon cancer. PLoS ONE 4:e6555. doi: 10.1371/journal.pone.0006555
Kim, Y. H., Petko, Z., Dzieciatkowski, S., Lin, L., Ghiassi, M., Stain, S., et al. (2006). CpG island methylation of genes accumulates during the adenoma progression step of the multistep pathogenesis of colorectal cancer. Genes Chromosomes Cancer 45, 781–789. doi: 10.1002/gcc.20341
Kitahara, M., Hazama, S., Tsunedomi, R., Takenouchi, H., Kanekiyo, S., Inoue, Y., et al. (2016). Prediction of the efficacy of immunotherapy by measuring the integrity of cell-free DNA in plasma in colorectal cancer. Cancer Sci. 107, 1825–1829. doi: 10.1111/cas.13085
Klemm, S. L., Shipony, Z., and Greenleaf, W. J. (2019). Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 20, 207–220. doi: 10.1038/s41576-018-0089-8
Kornberg, R. D. (1974). Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871. doi: 10.1126/science.184.4139.868
Kotake, Y., Nakagawa, T., Kitagawa, K., Suzuki, S., Liu, N., Kitagawa, M., et al. (2011). Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30, 1956–1962. doi: 10.1038/onc.2010.568
Kou, C. H., Zhou, T., Han, X. L., Zhuang, H. J., and Qian, H. X. (2016). Downregulation of mir-23b in plasma is associated with poor prognosis in patients with colorectal cancer. Oncol. Lett. 12, 4838–4844. doi: 10.3892/ol.2016.5265
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Kubota, K., Ohashi, A., Imachi, H., and Harada, H. (2006). Improved in situ hybridization efficiency with locked-nucleic-acid-incorporated DNA probes. Appl. Environ. Microbiol. 72, 5311–5317. doi: 10.1128/AEM.03039-05
Kurdyukov, S., and Bullock, M. (2016). DNA methylation analysis: choosing the right method. Biology 5:3. doi: 10.3390/biology5010003
Kurup, J. T., Campeanu, I. J., and Kidder, B. L. (2019). Contribution of H3K4 demethylase KDM5B to nucleosome organization in embryonic stem cells revealed by micrococcal nuclease sequencing. Epigenetics Chromatin 12:20. doi: 10.1186/s13072-019-0266-9
Lao, V. V., and Grady, W. M. (2011). Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 8, 686–700. doi: 10.1038/nrgastro.2011.173
Le, D. T., Uram, J. N., Wang, H., Bartlett, B. R., Kemberling, H., Eyring, A. D., et al. (2015). PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520. doi: 10.1056/NEJMoa1500596
Lee, S., Hwang, K. S., Lee, H. J., Kim, J. S., and Kang, G. H. (2004). Aberrant CpG island hypermethylation of multiple genes in colorectal neoplasia. Lab. Invest. 84, 884–893. doi: 10.1038/labinvest.3700108
Lee, S. K., and Calin, G. A. (2011). Non-coding RNAs and cancer: new paradigms in oncology. Discov. Med. 11, 245–254.
Legendre, C., Gooden, G. C., Johnson, K., Martinez, R. A., Liang, W. S., and Salhia, B. (2015). Whole-genome bisulfite sequencing of cell-free DNA identifies signature associated with metastatic breast cancer. Clin. Epigenetics 7:100. doi: 10.1186/s13148-015-0135-8
Lenhard, K., Bommer, G. T., Asutay, S., Schauer, R., Brabletz, T., Goke, B., et al. (2005). Analysis of promoter methylation in stool: a novel method for the detection of colorectal cancer. Clin. Gastroenterol. Hepatol. 3, 142–149. doi: 10.1016/s1542-3565(04)00624-x
Leveille, N., Melo, C. A., Rooijers, K., Diaz-Lagares, A., Melo, S. A., Korkmaz, G., et al. (2015). Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat. Commun. 6:6520. doi: 10.1038/ncomms7520
Li, L., Lu, M., Fan, Y., Shui, L., Xie, S., Sheng, R., et al. (2019). High-throughput and ultra-sensitive single-cell profiling of multiple microRNAs and identification of human cancer. Chem. Commun. 55, 10404–10407. doi: 10.1039/c9cc05553c
Li, M., Chen, W. D., Papadopoulos, N., Goodman, S. N., Bjerregaard, N. C., Laurberg, S., et al. (2009). Sensitive digital quantification of DNA methylation in clinical samples. Nat. Biotechnol. 27, 858–863. doi: 10.1038/nbt.1559
Li, W., Li, Q., Kang, S., Same, M., Zhou, Y., Sun, C., et al. (2018). CancerDetector: ultrasensitive and non-invasive cancer detection at the resolution of individual reads using cell-free DNA methylation sequencing data. Nucleic Acids Res. 46:e89. doi: 10.1093/nar/gky423
Li, W., Yu, W., Jiang, X., Gao, X., Wang, G., Jin, X., et al. (2020). The construction and comprehensive prognostic analysis of the LncRNA-associated competitive endogenous RNAs network in colorectal cancer. Front. Genet. 11:583. doi: 10.3389/fgene.2020.00583
Li, W., Zhang, X., Lu, X., You, L., Song, Y., Luo, Z., et al. (2017). 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 27, 1243–1257. doi: 10.1038/cr.2017.121
Liang, Z. X., Liu, H. S., Wang, F. W., Xiong, L., Zhou, C., Hu, T., et al. (2019). LncRNA RPPH1 promotes colorectal cancer metastasis by interacting with TUBB3 and by promoting exosomes-mediated macrophage M2 polarization. Cell Death Dis. 10:829. doi: 10.1038/s41419-019-2077-0
Lin, J., Cai, D., Li, W., Yu, T., Mao, H., Jiang, S., et al. (2019). Plasma circular RNA panel acts as a novel diagnostic biomarker for colorectal cancer. Clin. Biochem. 74, 60–68. doi: 10.1016/j.clinbiochem.2019.10.012
Liu, H., Ye, D., Chen, A., Tan, D., Zhang, W., Jiang, W., et al. (2019). A pilot study of new promising non-coding RNA diagnostic biomarkers for early-stage colorectal cancers. Clin. Chem. Lab. Med. 57, 1073–1083. doi: 10.1515/cclm-2019-0052
Liu, L., Toung, J. M., Jassowicz, A. F., Vijayaraghavan, R., Kang, H., Zhang, R., et al. (2018). Targeted methylation sequencing of plasma cell-free DNA for cancer detection and classification. Ann. Oncol. 29, 1445–1453. doi: 10.1093/annonc/mdy119
Liu, M. C., Oxnard, G. R., Klein, E. A., Swanton, C., and Seiden, M. V. (2020). Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 31, 745–759. doi: 10.1016/j.annonc.2020.02.011
Liu, W., Yang, D., Chen, L., Liu, Q., Wang, W., Yang, Z., et al. (2020). Plasma exosomal miRNA-139-3p is a novel biomarker of colorectal cancer. J. Cancer 11, 4899–4906. doi: 10.7150/jca.45548
Liu, X., Ren, J., Luo, N., Guo, H., Zheng, Y., Li, J., et al. (2019). Comprehensive DNA methylation analysis of tissue of origin of plasma cell-free DNA by methylated CpG tandem amplification and sequencing (MCTA-Seq). Clin. Epigenetics 11:93. doi: 10.1186/s13148-019-0689-y
Liu, X., Wen, J., Li, C., Wang, H., Wang, J., and Zou, H. (2020). High-yield methylation markers for stool-based detection of colorectal cancer. Dig. Dis. Sci. 65, 1710–1719. doi: 10.1007/s10620-019-05908-9
Liu, Y., Zhao, G., Miao, J., Li, H., Ma, Y., Liu, X., et al. (2020). Performance comparison between plasma and stool methylated SEPT9 tests for detecting colorectal cancer. Front. Genet. 11:24. doi: 10.3389/fgene.2020.00324
Liu, Y., Zheng, P., Liu, Y., Ji, T., Liu, X., Yao, S., et al. (2013). An epigenetic role for PRL-3 as a regulator of H3K9 methylation in colorectal cancer. Gut 62, 571–581. doi: 10.1136/gutjnl-2011-301059
Lo Nigro, C., Ricci, V., Vivenza, D., Granetto, C., Fabozzi, T., Miraglio, E., et al. (2016). Prognostic and predictive biomarkers in metastatic colorectal cancer anti-EGFR therapy. World J. Gastroenterol. 22, 6944–6954. doi: 10.3748/wjg.v22.i30.6944
Loboda, A., Nebozhyn, M. V., Watters, J. W., Buser, C. A., Shaw, P. M., Huang, P. S., et al. (2011). EMT is the dominant program in human colon cancer. BMC Med. Genomics 4:9. doi: 10.1186/1755-8794-4-9
Lofton-Day, C., Model, F., Devos, T., Tetzner, R., Distler, J., Schuster, M., et al. (2008). DNA methylation biomarkers for blood-based colorectal cancer screening. Clin. Chem. 54, 414–423. doi: 10.1373/clinchem.2007.095992
Lu, J., Getz, G., Miska, E. A., Alvarez-Saavedra, E., Lamb, J., Peck, D., et al. (2005). MicroRNA expression profiles classify human cancers. Nature 435, 834–838. doi: 10.1038/nature03702
Luo, Y., Wong, C. J., Kaz, A. M., Dzieciatkowski, S., Carter, K. T., Morris, S. M., et al. (2014). Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology 147, 418–429 e418. doi: 10.1053/j.gastro.2014.04.039
Lyberopoulou, A., Galanopoulos, M., Aravantinos, G., Theodoropoulos, G. E., Marinos, E., Efstathopoulos, E. P., et al. (2017). Identification of methylation profiles of cancer-related genes in circulating tumor cells population. Anticancer Res. 37, 1105–1112. doi: 10.21873/anticanres.11423
Maminezhad, H., Ghanadian, S., Pakravan, K., Razmara, E., Rouhollah, F., Mossahebi-Mohammadi, M., et al. (2020). A panel of six-circulating miRNA signature in serum and its potential diagnostic value in colorectal cancer. Life Sci. 258:118226. doi: 10.1016/j.lfs.2020.118226
Marcuello, M., Vymetalkova, V., Neves, R. P. L., Duran-Sanchon, S., Vedeld, H. M., Tham, E., et al. (2019). Circulating biomarkers for early detection and clinical management of colorectal cancer. Mol. Aspects Med. 69, 107–122. doi: 10.1016/j.mam.2019.06.002
Mari-Alexandre, J., Diaz-Lagares, A., Villalba, M., Juan, O., Crujeiras, A. B., Calvo, A., et al. (2017). Translating cancer epigenomics into the clinic: focus on lung cancer. Transl. Res. 189, 76–92. doi: 10.1016/j.trsl.2017.05.008
Matassov, D., Kagan, T., Leblanc, J., Sikorska, M., and Zakeri, Z. (2004). Measurement of apoptosis by DNA fragmentation. Methods Mol. Biol. 282, 1–17. doi: 10.1385/1-59259-812-9:001
Matsumura, T., Sugimachi, K., Iinuma, H., Takahashi, Y., Kurashige, J., Sawada, G., et al. (2015). Exosomal microRNA in serum is a novel biomarker of recurrence in human colorectal cancer. Br. J. Cancer 113, 275–281. doi: 10.1038/bjc.2015.201
Mattiuzzi, C., Sanchis-Gomar, F., and Lippi, G. (2019). Concise update on colorectal cancer epidemiology. Ann. Transl. Med. 7:609. doi: 10.21037/atm.2019.07.91
Mavrich, T. N., Ioshikhes, I. P., Venters, B. J., Jiang, C., Tomsho, L. P., Qi, J., et al. (2008). A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 18, 1073–1083. doi: 10.1101/gr.078261.108
Meissner, A., Gnirke, A., Bell, G. W., Ramsahoye, B., Lander, E. S., and Jaenisch, R. (2005). Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877. doi: 10.1093/nar/gki901
Memczak, S., Jens, M., Elefsinioti, A., Torti, F., Krueger, J., Rybak, A., et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338. doi: 10.1038/nature11928
Merker, J. D., Oxnard, G. R., Compton, C., Diehn, M., Hurley, P., Lazar, A. J., et al. (2018). Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J. Clin. Oncol. 36, 1631–1641. doi: 10.1200/JCO.2017.76.8671
Metcalf, G. A., Shibakawa, A., Patel, H., Sita-Lumsden, A., Zivi, A., Rama, N., et al. (2016). Amplification-free detection of circulating microRNA biomarkers from body fluids based on fluorogenic oligonucleotide-templated reaction between engineered peptide nucleic acid probes: application to prostate cancer diagnosis. Anal. Chem. 88, 8091–8098. doi: 10.1021/acs.analchem.6b01594
Miao, X., Ning, X., Li, Z., and Cheng, Z. (2016). Sensitive detection of miRNA by using hybridization chain reaction coupled with positively charged gold nanoparticles. Sci. Rep. 6:32358. doi: 10.1038/srep32358
Mitomi, H., Fukui, N., Tanaka, N., Kanazawa, H., Saito, T., Matsuoka, T., et al. (2010). Aberrant p16((INK4a)) methylation is a frequent event in colorectal cancers: prognostic value and relation to mRNA expression and immunoreactivity. J. Cancer Res. Clin. Oncol. 136, 323–331. doi: 10.1007/s00432-009-0688-z
Molinari, C., Marisi, G., Passardi, A., Matteucci, L., De Maio, G., and Ulivi, P. (2018). Heterogeneity in colorectal cancer: a challenge for personalized medicine? Int. J. Mol. Sci. 19:3733. doi: 10.3390/ijms19123733
Mori, Y., Olaru, A. V., Cheng, Y., Agarwal, R., Yang, J., Luvsanjav, D., et al. (2011). Novel candidate colorectal cancer biomarkers identified by methylation microarray-based scanning. Endocr. Relat. Cancer 18, 465–478. doi: 10.1530/ERC-11-0083
Morin, R. D., Johnson, N. A., Severson, T. M., Mungall, A. J., An, J., Goya, R., et al. (2010). Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42, 181–185. doi: 10.1038/ng.518
Mouliere, F., Chandrananda, D., Piskorz, A. M., Moore, E. K., Morris, J., Ahlborn, L. B., et al. (2018). Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 10:eaat4921. doi: 10.1126/scitranslmed.aat4921
Nagai, Y., Sunami, E., Yamamoto, Y., Hata, K., Okada, S., Murono, K., et al. (2017). LINE-1 hypomethylation status of circulating cell-free DNA in plasma as a biomarker for colorectal cancer. Oncotarget 8, 11906–11916. doi: 10.18632/oncotarget.14439
Nazemalhosseini Mojarad, E., Kuppen, P. J., Aghdaei, H. A., and Zali, M. R. (2013). The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed Bench 6, 120–128.
Ned, R. M., Melillo, S., and Marrone, M. (2011). Fecal DNA testing for Colorectal Cancer Screening: the ColoSure test. PLoS Curr. 3:RRN1220. doi: 10.1371/currents.RRN1220
Ng, E. K., Chong, W. W., Jin, H., Lam, E. K., Shin, V. Y., Yu, J., et al. (2009). Differential expression of microRNAs in plasma of patients with colorectal cancer: a potential marker for colorectal cancer screening. Gut 58, 1375–1381. doi: 10.1136/gut.2008.167817
Oehme, F., Krahl, S., Gyorffy, B., Muessle, B., Rao, V., Greif, H., et al. (2019). Low level of exosomal long non-coding RNA HOTTIP is a prognostic biomarker in colorectal cancer. RNA Biol. 16, 1339–1345. doi: 10.1080/15476286.2019.1637697
Ogino, S., Kawasaki, T., Kirkner, G. J., Kraft, P., Loda, M., and Fuchs, C. S. (2007). Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J. Mol. Diagn. 9, 305–314. doi: 10.2353/jmoldx.2007.060170
Ortega, F. G., Lorente, J. A., Garcia Puche, J. L., Ruiz, M. P., Sanchez-Martin, R. M., de Miguel-Perez, D., et al. (2015). miRNA in situ hybridization in circulating tumor cells–MishCTC. Sci. Rep. 5:9207. doi: 10.1038/srep09207
Osborn, N. K., and Ahlquist, D. A. (2005). Stool screening for colorectal cancer: molecular approaches. Gastroenterology 128, 192–206. doi: 10.1053/j.gastro.2004.10.041
Overman, M. J., Lonardi, S., Wong, K. Y. M., Lenz, H. J., Gelsomino, F., Aglietta, M., et al. (2018). Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 36, 773–779. doi: 10.1200/JCO.2017.76.9901
Pardini, B., Sabo, A. A., Birolo, G., and Calin, G. A. (2019). Noncoding RNAs in extracellular fluids as cancer biomarkers: the new frontier of liquid biopsies. Cancers 11:1170. doi: 10.3390/cancers11081170
Peng, Q., Zhang, X., Min, M., Zou, L., Shen, P., and Zhu, Y. (2017). The clinical role of microRNA-21 as a promising biomarker in the diagnosis and prognosis of colorectal cancer: a systematic review and meta-analysis. Oncotarget 8, 44893–44909. doi: 10.18632/oncotarget.16488
Peng, Z. Y., Gu, R. H., and Yan, B. (2018). Downregulation of exosome-encapsulated miR-548c-5p is associated with poor prognosis in colorectal cancer. J. Cell. Biochem. doi: 10.1002/jcb.27291. [Epub ahead of print].
Pharo, H. D., Honne, H., Vedeld, H. M., Dahl, C., Andresen, K., Liestol, K., et al. (2016). Experimental factors affecting the robustness of DNA methylation analysis. Sci. Rep. 6:33936. doi: 10.1038/srep33936
Philipp, A. B., Stieber, P., Nagel, D., Neumann, J., Spelsberg, F., Jung, A., et al. (2012). Prognostic role of methylated free circulating DNA in colorectal cancer. Int. J. Cancer 131, 2308–2319. doi: 10.1002/ijc.27505
Picardo, F., Romanelli, A., Muinelo-Romay, L., Mazza, T., Fusilli, C., Parrella, P., et al. (2019). Diagnostic and Prognostic Value of B4GALT1 Hypermethylation and its clinical significance as a novel circulating cell-free DNA biomarker in colorectal cancer. Cancers 11:1598. doi: 10.3390/cancers11101598
Pickhardt, P. J. (2016). Emerging stool-based and blood-based non-invasive DNA tests for colorectal cancer screening: the importance of cancer prevention in addition to cancer detection. Abdom. Radiol. 41, 1441–1444. doi: 10.1007/s00261-016-0798-4
Pixberg, C. F., Raba, K., Muller, F., Behrens, B., Honisch, E., Niederacher, D., et al. (2017). Analysis of DNA methylation in single circulating tumor cells. Oncogene 36, 3223–3231. doi: 10.1038/onc.2016.480
Portela, A., and Esteller, M. (2010). Epigenetic modifications and human disease. Nat. Biotechnol. 28, 1057–1068. doi: 10.1038/nbt.1685
Qin, S., Li, J., Wang, L., Xu, J., Cheng, Y., Bai, Y., et al. (2018). Efficacy and tolerability of first-line cetuximab plus leucovorin, fluorouracil, and oxaliplatin (FOLFOX-4) versus FOLFOX-4 in patients with RAS wild-type metastatic colorectal cancer: the open-label, randomized, phase III TAILOR trial. J. Clin. Oncol. 36, 3031–3039. doi: 10.1200/JCO.2018.78.3183
Qu, Y., Lennartsson, A., Gaidzik, V. I., Deneberg, S., Karimi, M., Bengtzen, S., et al. (2014). Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics 9, 1108–1119. doi: 10.4161/epi.29315
Quintas-Cardama, A., Santos, F. P., and Garcia-Manero, G. (2010). Therapy with azanucleosides for myelodysplastic syndromes. Nat. Rev. Clin. Oncol. 7, 433–444. doi: 10.1038/nrclinonc.2010.87
Rahier, J. F., Druez, A., Faugeras, L., Martinet, J. P., Gehenot, M., Josseaux, E., et al. (2017). Circulating nucleosomes as new blood-based biomarkers for detection of colorectal cancer. Clin. Epigenetics 9:53. doi: 10.1186/s13148-017-0351-5
Rand, K., Qu, W., Ho, T., Clark, S. J., and Molloy, P. (2002). Conversion-specific detection of DNA methylation using real-time polymerase chain reaction (ConLight-MSP) to avoid false positives. Methods 27, 114–120. doi: 10.1016/s1046-2023(02)00062-2
Rando, O. J. (2012). Combinatorial complexity in chromatin structure and function: revisiting the histone code. Curr. Opin. Genet. Dev. 22, 148–155. doi: 10.1016/j.gde.2012.02.013
Rapado-Gonzalez, O., Majem, B., Alvarez-Castro, A., Diaz-Pena, R., Abalo, A., Suarez-Cabrera, L., et al. (2019). A novel saliva-based miRNA signature for colorectal cancer diagnosis. J. Clin. Med. 8:2029. doi: 10.3390/jcm8122029
Rasmussen, S. L., Krarup, H. B., Sunesen, K. G., Johansen, M. B., Stender, M. T., Pedersen, I. S., et al. (2018). The prognostic efficacy of cell-free DNA hypermethylation in colorectal cancer. Oncotarget 9, 7010–7022. doi: 10.18632/oncotarget.24097
Raut, J. R., Guan, Z., Schrotz-King, P., and Brenner, H. (2020). Fecal DNA methylation markers for detecting stages of colorectal cancer and its precursors: a systematic review. Clin. Epigenetics 12:122. doi: 10.1186/s13148-020-00904-7
Rezapour, S., Hosseinzadeh, E., Marofi, F., and Hassanzadeh, A. (2019). Epigenetic-based therapy for colorectal cancer: prospect and involved mechanisms. J. Cell. Physiol. 234, 19366–19383. doi: 10.1002/jcp.28658
Rodriguez-Paredes, M., and Esteller, M. (2011). Cancer epigenetics reaches mainstream oncology. Nat. Med. 17, 330–339. doi: 10.1038/nm.2305
Roman-Canal, B., Tarragona, J., Moiola, C. P., Gatius, S., Bonnin, S., Ruiz-Miro, M., et al. (2019). EV-associated miRNAs from peritoneal lavage as potential diagnostic biomarkers in colorectal cancer. J. Transl. Med. 17:208. doi: 10.1186/s12967-019-1954-8
Rose, N. R., and Klose, R. J. (2014). Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta 1839, 1362–1372. doi: 10.1016/j.bbagrm.2014.02.007
Rosen, L. S., Jacobs, I. A., and Burkes, R. L. (2017). Bevacizumab in colorectal cancer: current role in treatment and the potential of biosimilars. Target. Oncol. 12, 599–610. doi: 10.1007/s11523-017-0518-1
Sadeh, R., Sharkia, I., Fialkoff, G., Rahat, A., Gutin, J., Chappleboim, A., et al. (2019). ChIP-seq of plasma cell-free nucleosomes identifies cell-of-origin gene expression programs. bioRxiv 638643. doi: 10.1101/638643
Sandoval, J., Heyn, H., Moran, S., Serra-Musach, J., Pujana, M. A., Bibikova, M., et al. (2011). Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 6, 692–702. doi: 10.4161/epi.6.6.16196
Sastre, J., Maestro, M. L., Gomez-Espana, A., Rivera, F., Valladares, M., Massuti, B., et al. (2012). Circulating tumor cell count is a prognostic factor in metastatic colorectal cancer patients receiving first-line chemotherapy plus bevacizumab: a Spanish Cooperative Group for the Treatment of Digestive Tumors study. Oncologist 17, 947–955. doi: 10.1634/theoncologist.2012-0048
Sazanov, A. A., Kiselyova, E. V., Zakharenko, A. A., Romanov, M. N., and Zaraysky, M. I. (2017). Plasma and saliva miR-21 expression in colorectal cancer patients. J. Appl. Genet. 58, 231–237. doi: 10.1007/s13353-016-0379-9
Schirripa, M., Borelli, B., D'Aurizio, R., Lubrano, S., Cremolini, C., Zucchelli, G., et al. (2019). Early modifications of circulating microRNAs levels in metastatic colorectal cancer patients treated with regorafenib. Pharmacogenomics J. 19, 455–464. doi: 10.1038/s41397-019-0075-3
Sharma, S., Kelly, T. K., and Jones, P. A. (2010). Epigenetics in cancer. Carcinogenesis 31, 27–36. doi: 10.1093/carcin/bgp220
Sheaffer, K. L., Elliott, E. N., and Kaestner, K. H. (2016). DNA hypomethylation contributes to genomic instability and intestinal cancer initiation. Cancer Prev. Res. 9, 534–546. doi: 10.1158/1940-6207.CAPR-15-0349
Shen, S. Y., Burgener, J. M., Bratman, S. V., and De Carvalho, D. D. (2019). Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 14, 2749–2780. doi: 10.1038/s41596-019-0202-2
Shen, S. Y., Singhania, R., Fehringer, G., Chakravarthy, A., Roehrl, M. H. A., Chadwick, D., et al. (2018). Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563, 579–583. doi: 10.1038/s41586-018-0703-0
Shi, J., Li, X., Zhang, F., Zhang, C., Guan, Q., Cao, X., et al. (2015). Circulating lncRNAs associated with occurrence of colorectal cancer progression. Am. J. Cancer Res. 5, 2258–2265.
Shin, S., Lee, E. M., Cha, H. J., Bae, S., Jung, J. H., Lee, S. M., et al. (2009). MicroRNAs that respond to histone deacetylase inhibitor SAHA and p53 in HCT116 human colon carcinoma cells. Int. J. Oncol. 35, 1343–1352. doi: 10.3892/ijo_00000452
Shinjo, K., Hara, K., Nagae, G., Umeda, T., Katsushima, K., Suzuki, M., et al. (2020). A novel sensitive detection method for DNA methylation in circulating free DNA of pancreatic cancer. PLoS ONE 15:e0233782. doi: 10.1371/journal.pone.0233782
Shirahata, A., Sakata, M., Sakuraba, K., Goto, T., Mizukami, H., Saito, M., et al. (2009). Vimentin methylation as a marker for advanced colorectal carcinoma. Anticancer Res. 29, 279–281.
Shukla, N., Yan, I. K., and Patel, T. (2018). Multiplexed detection and quantitation of extracellular vesicle RNA expression using nanostring. Methods Mol. Biol 1740, 177–185. doi: 10.1007/978-1-4939-7652-2_14
Silva-Fisher, J. M., Dang, H. X., White, N. M., Strand, M. S., Krasnick, B. A., Rozycki, E. B., et al. (2020). Long non-coding RNA RAMS11 promotes metastatic colorectal cancer progression. Nat. Commun. 11:2156. doi: 10.1038/s41467-020-15547-8
Siravegna, G., Marsoni, S., Siena, S., and Bardelli, A. (2017). Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 14, 531–548. doi: 10.1038/nrclinonc.2017.14
Siravegna, G., Mussolin, B., Buscarino, M., Corti, G., Cassingena, A., Crisafulli, G., et al. (2015). Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 21, 795–801. doi: 10.1038/nm.3870
Snyder, M. W., Kircher, M., Hill, A. J., Daza, R. M., and Shendure, J. (2016). Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 164, 57–68. doi: 10.1016/j.cell.2015.11.050
Sole, C., Tramonti, D., Schramm, M., Goicoechea, I., Armesto, M., Hernandez, L. I., et al. (2019). The circulating transcriptome as a source of biomarkers for melanoma. Cancers 11:70. doi: 10.3390/cancers11010070
Song, B. P., Jain, S., Lin, S. Y., Chen, Q., Block, T. M., Song, W., et al. (2012). Detection of hypermethylated vimentin in urine of patients with colorectal cancer. J. Mol. Diagn. 14, 112–119. doi: 10.1016/j.jmoldx.2011.12.003
Song, L. L., and Li, Y. M. (2016). Current noninvasive tests for colorectal cancer screening: an overview of colorectal cancer screening tests. World J. Gastrointest. Oncol. 8, 793–800. doi: 10.4251/wjgo.v8.i11.793
Song, W., Ren, J., Wang, C., Ge, Y., and Fu, T. (2020). Analysis of circular RNA-related competing endogenous RNA identifies the immune-related risk signature for colorectal cancer. Front. Genet. 11:505. doi: 10.3389/fgene.2020.00505
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi: 10.1038/47412
Sun, Y., Liu, Y., Cogdell, D., Calin, G. A., Sun, B., Kopetz, S., et al. (2016). Examining plasma microRNA markers for colorectal cancer at different stages. Oncotarget 7, 11434–11449. doi: 10.18632/oncotarget.7196
Sun, Z., Cunningham, J., Slager, S., and Kocher, J. P. (2015). Base resolution methylome profiling: considerations in platform selection, data preprocessing and analysis. Epigenomics 7, 813–828. doi: 10.2217/epi.15.21
Suter, C. M., Martin, D. I., and Ward, R. L. (2004). Hypomethylation of L1 retrotransposons in colorectal cancer and adjacent normal tissue. Int. J. Colorectal Dis. 19, 95–101. doi: 10.1007/s00384-003-0539-3
Svoboda, M., Slyskova, J., Schneiderova, M., Makovicky, P., Bielik, L., Levy, M., et al. (2014). HOTAIR long non-coding RNA is a negative prognostic factor not only in primary tumors, but also in the blood of colorectal cancer patients. Carcinogenesis 35, 1510–1515. doi: 10.1093/carcin/bgu055
Symonds, E. L., Pedersen, S. K., Murray, D., Byrne, S. E., Roy, A., Karapetis, C., et al. (2020). Circulating epigenetic biomarkers for detection of recurrent colorectal cancer. Cancer 126, 1460–1469. doi: 10.1002/cncr.32695
Taft, R. J., Pang, K. C., Mercer, T. R., Dinger, M., and Mattick, J. S. (2010). Non-coding RNAs: regulators of disease. J. Pathol. 220, 126–139. doi: 10.1002/path.2638
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Taiwo, O., Wilson, G. A., Morris, T., Seisenberger, S., Reik, W., Pearce, D., et al. (2012). Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 7, 617–636. doi: 10.1038/nprot.2012.012
Taniguchi, H., Yamanaka, T., Sakai, D., Muro, K., Yamazaki, K., Nakata, S., et al. (2020). Efficacy of panitumumab and cetuximab in patients with colorectal cancer previously treated with bevacizumab; a combined analysis of individual patient data from ASPECCT and WJOG6510G. Cancers 12:1715. doi: 10.3390/cancers12071715
Thierry, A. R., El Messaoudi, S., Gahan, P. B., Anker, P., and Stroun, M. (2016). Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 35, 347–376. doi: 10.1007/s10555-016-9629-x
Toyota, M., Ahuja, N., Ohe-Toyota, M., Herman, J. G., Baylin, S. B., and Issa, J. P. (1999). CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 96, 8681–8686. doi: 10.1073/pnas.96.15.8681
Triantafillidis, J. K., Vagianos, C., and Malgarinos, G. (2015). Colonoscopy in colorectal cancer screening: current aspects. Indian J. Surg. Oncol. 6, 237–250. doi: 10.1007/s13193-015-0410-3
Vad-Nielsen, J., Meldgaard, P., Sorensen, B. S., and Nielsen, A. L. (2020). Cell-free Chromatin Immunoprecipitation (cfChIP) from blood plasma can determine gene-expression in tumors from non-small-cell lung cancer patients. Lung Cancer 147, 244–251. doi: 10.1016/j.lungcan.2020.07.023
van Niel, G., D'Angelo, G., and Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228. doi: 10.1038/nrm.2017.125
Van Paemel, R., De Koker, A., Vandeputte, C., van Zogchel, L., Lammens, T., Laureys, G., et al. (2020). Minimally invasive classification of paediatric solid tumours using reduced representation bisulphite sequencing of cell-free DNA: a proof-of-principle study. Epigenetics 1–13. doi: 10.1080/15592294.2020.1790950. [Epub ahead of print].
Vidal, J., Muinelo, L., Dalmases, A., Jones, F., Edelstein, D., Iglesias, M., et al. (2017). Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 28, 1325–1332. doi: 10.1093/annonc/mdx125
Vincent, K. M., and Postovit, L. M. (2017). A pan-cancer analysis of secreted Frizzled-related proteins: re-examining their proposed tumour suppressive function. Sci. Rep. 7:42719. doi: 10.1038/srep42719
Volinia, S., Calin, G. A., Liu, C. G., Ambs, S., Cimmino, A., Petrocca, F., et al. (2006). A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U.S.A. 103, 2257–2261. doi: 10.1073/pnas.0510565103
Waddington, C. H. (2012). The epigenotype. 1942. Int. J. Epidemiol. 41, 10–13. doi: 10.1093/ije/dyr184
Wallner, M., Herbst, A., Behrens, A., Crispin, A., Stieber, P., Goke, B., et al. (2006). Methylation of serum DNA is an independent prognostic marker in colorectal cancer. Clin. Cancer Res. 12, 7347–7352. doi: 10.1158/1078-0432.CCR-06-1264
Wang, Q., Huang, Z., Ni, S., Xiao, X., Xu, Q., Wang, L., et al. (2012). Plasma miR-601 and miR-760 are novel biomarkers for the early detection of colorectal cancer. PLoS ONE 7:e44398. doi: 10.1371/journal.pone.0044398
Wang, W., Qu, A., Liu, W., Liu, Y., Zheng, G., Du, L., et al. (2017). Circulating miR-210 as a diagnostic and prognostic biomarker for colorectal cancer. Eur. J. Cancer Care 26:e12448. doi: 10.1111/ecc.12448
Wang, Y., Chen, P. M., and Liu, R. B. (2018). Advance in plasma SEPT9 gene methylation assay for colorectal cancer early detection. World J. Gastrointest. Oncol. 10, 15–22. doi: 10.4251/wjgo.v10.i1.15
Wang, Y. M., Trinh, M. P., Zheng, Y., Guo, K., Jimenez, L. A., and Zhong, W. (2019). Analysis of circulating non-coding RNAs in a non-invasive and cost-effective manner. Trends Analyt. Chem. 117, 242–262. doi: 10.1016/j.trac.2019.07.001
Ward, R., Meagher, A., Tomlinson, I., O'Connor, T., Norrie, M., Wu, R., et al. (2001). Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut 48, 821–829. doi: 10.1136/gut.48.6.821
Weisenberger, D. J., Siegmund, K. D., Campan, M., Young, J., Long, T. I., Faasse, M. A., et al. (2006). CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 38, 787–793. doi: 10.1038/ng1834
Wendt, M. K., Johanesen, P. A., Kang-Decker, N., Binion, D. G., Shah, V., and Dwinell, M. B. (2006). Silencing of epithelial CXCL12 expression by DNA hypermethylation promotes colonic carcinoma metastasis. Oncogene 25, 4986–4997. doi: 10.1038/sj.onc.1209505
Wu, C. W., Cao, X., Berger, C. K., Foote, P. H., Mahoney, D. W., Simonson, J. A., et al. (2017). Novel approach to fecal occult blood testing by assay of erythrocyte-specific microRNA markers. Dig. Dis. Sci. 62, 1985–1994. doi: 10.1007/s10620-017-4627-6
Wu, C. W., Ng, S. S., Dong, Y. J., Ng, S. C., Leung, W. W., Lee, C. W., et al. (2012). Detection of miR-92a and miR-21 in stool samples as potential screening biomarkers for colorectal cancer and polyps. Gut 61, 739–745. doi: 10.1136/gut.2011.239236
Wu, X. D., Song, Y. C., Cao, P. L., Zhang, H., Guo, Q., Yan, R., et al. (2014). Detection of miR-34a and miR-34b/c in stool sample as potential screening biomarkers for noninvasive diagnosis of colorectal cancer. Med. Oncol 31, 894. doi: 10.1007/s12032-014-0894-7
Xiao, W., Zhao, H., Dong, W., Li, Q., Zhu, J., Li, G., et al. (2015). Quantitative detection of methylated NDRG4 gene as a candidate biomarker for diagnosis of colorectal cancer. Oncol. Lett. 9, 1383–1387. doi: 10.3892/ol.2014.2815
Xie, Y., Li, J., Li, P., Li, N., Zhang, Y., Binang, H., et al. (2020). RNA-seq profiling of serum exosomal circular RNAs reveals Circ-PNN as a potential biomarker for human colorectal cancer. Front. Oncol. 10:982. doi: 10.3389/fonc.2020.00982
Xie, Y. H., Chen, Y. X., and Fang, J. Y. (2020). Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target Ther. 5:22. doi: 10.1038/s41392-020-0116-z
Xing, X. B., Cai, W. B., Luo, L., Liu, L. S., Shi, H. J., and Chen, M. H. (2013). The prognostic value of p16 hypermethylation in cancer: a meta-analysis. PLoS ONE 8:e66587. doi: 10.1371/journal.pone.0066587
Xiong, B., Cheng, Y., Ma, L., and Zhang, C. (2013). MiR-21 regulates biological behavior through the PTEN/PI-3 K/Akt signaling pathway in human colorectal cancer cells. Int. J. Oncol 42, 219–228. doi: 10.3892/ijo.2012.1707
Xu, T., and Gao, H. (2020). Hydroxymethylation and tumors: can 5-hydroxymethylation be used as a marker for tumor diagnosis and treatment? Hum. Genomics 14:15. doi: 10.1186/s40246-020-00265-5
Xu, W., Zhou, G., Wang, H., Liu, Y., Chen, B., Chen, W., et al. (2020). Circulating lncRNA SNHG11 as a novel biomarker for early diagnosis and prognosis of colorectal cancer. Int. J. Cancer 146, 2901–2912. doi: 10.1002/ijc.32747
Yan, S., Han, B., Gao, S., Wang, X., Wang, Z., Wang, F., et al. (2017). Exosome-encapsulated microRNAs as circulating biomarkers for colorectal cancer. Oncotarget 8, 60149–60158. doi: 10.18632/oncotarget.18557
Yang, H., Salz, T., Zajac-Kaye, M., Liao, D., Huang, S., and Qiu, Y. (2014). Overexpression of histone deacetylases in cancer cells is controlled by interplay of transcription factors and epigenetic modulators. FASEB J. 28, 4265–4279. doi: 10.1096/fj.14-250654
Yang, H., Zhang, H., Yang, Y., Wang, X., Deng, T., Liu, R., et al. (2020). Hypoxia induced exosomal circRNA promotes metastasis of Colorectal Cancer via targeting GEF-H1/RhoA axis. Theranostics 10, 8211–8226. doi: 10.7150/thno.44419
Yang, N., Xu, B., Kong, P., Han, M., and Li, B. H. (2020). Hsa_circ_0002320: a novel clinical biomarker for colorectal cancer prognosis. Medicine 99:e21224. doi: 10.1097/MD.0000000000021224
Yang, X., Dai, W., Kwong, D. L., Szeto, C. Y., Wong, E. H., Ng, W. T., et al. (2015). Epigenetic markers for noninvasive early detection of nasopharyngeal carcinoma by methylation-sensitive high resolution melting. Int. J. Cancer 136, E127–135. doi: 10.1002/ijc.29192
Yau, T. O., Wu, C. W., Tang, C. M., Chen, Y., Fang, J., Dong, Y., et al. (2016). MicroRNA-20a in human faeces as a non-invasive biomarker for colorectal cancer. Oncotarget 7, 1559–1568. doi: 10.18632/oncotarget.6403
Ye, D. X., Wang, S. S., Huang, Y., and Chi, P. (2019). A 3-circular RNA signature as a noninvasive biomarker for diagnosis of colorectal cancer. Cancer Cell Int. 19:276. doi: 10.1186/s12935-019-0995-7
Young, G. P., Pedersen, S. K., Mansfield, S., Murray, D. H., Baker, R. T., Rabbitt, P., et al. (2016). A cross-sectional study comparing a blood test for methylated BCAT1 and IKZF1 tumor-derived DNA with CEA for detection of recurrent colorectal cancer. Cancer Med. 5, 2763–2772. doi: 10.1002/cam4.868
Yuan, Z., Baker, K., Redman, M. W., Wang, L., Adams, S. V., Yu, M., et al. (2017). Dynamic plasma microRNAs are biomarkers for prognosis and early detection of recurrence in colorectal cancer. Br. J. Cancer 117, 1202–1210. doi: 10.1038/bjc.2017.266
Zaharie, F., Muresan, M. S., Petrushev, B., Berce, C., Gafencu, G. A., Selicean, S., et al. (2015). Exosome-carried microRNA-375 inhibits cell progression and dissemination via Bcl-2 blocking in colon cancer. J. Gastrointestin. Liver Dis. 24, 435–443. doi: 10.15403/jgld.2014.1121.244.375
Zavridou, M., Mastoraki, S., Strati, A., Tzanikou, E., Chimonidou, M., and Lianidou, E. (2018). Evaluation of preanalytical conditions and implementation of quality control steps for reliable gene expression and DNA methylation analyses in liquid biopsies. Clin. Chem. 64, 1522–1533. doi: 10.1373/clinchem.2018.292318
Zeng, Z., Li, Y., Pan, Y., Lan, X., Song, F., Sun, J., et al. (2018). Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 9:5395. doi: 10.1038/s41467-018-07810-w
Zhang, H., Dong, P., Guo, S., Tao, C., Chen, W., Zhao, W., et al. (2020). Hypomethylation in HBV integration regions aids non-invasive surveillance to hepatocellular carcinoma by low-pass genome-wide bisulfite sequencing. BMC Med. 18:200. doi: 10.1186/s12916-020-01667-x
Zhang, H., Zhu, Y. Q., Wu, Y. Q., Zhang, P., and Qi, J. (2014). Detection of promoter hypermethylation of Wnt antagonist genes in fecal samples for diagnosis of early colorectal cancer. World J. Gastroenterol. 20, 6329–6335. doi: 10.3748/wjg.v20.i20.6329
Zhang, P., Wu, W., Chen, Q., and Chen, M. (2019). Non-coding RNAs and their integrated networks. J. Integr. Bioinform. 16:20190027. doi: 10.1515/jib-2019-0027
Zhang, W., Yang, S., Liu, Y., Wang, Y., Lin, T., Li, Y., et al. (2018). Hsa_circ_0007534 as a blood-based marker for the diagnosis of colorectal cancer and its prognostic value. Int. J. Clin. Exp. Pathol. 11, 1399–1406.
Zhang, Y., Dong, X., Bai, L., Shang, X., and Zeng, Y. (2020). MUC1-induced immunosuppression in colon cancer can be reversed by blocking the PD1/PDL1 signaling pathway. Oncol. Lett. 20:317. doi: 10.3892/ol.2020.12180
Zhao, G., Li, H., Yang, Z., Wang, Z., Xu, M., Xiong, S., et al. (2019). Multiplex methylated DNA testing in plasma with high sensitivity and specificity for colorectal cancer screening. Cancer Med. 8, 5619–5628. doi: 10.1002/cam4.2475
Zhao, G., Liu, X., Liu, Y., Li, H., Ma, Y., Li, S., et al. (2020a). Aberrant DNA methylation of SEPT9 and SDC2 in stool specimens as an integrated biomarker for colorectal cancer early detection. Front. Genet. 11:643. doi: 10.3389/fgene.2020.00643
Zhao, G., Ma, Y., Li, H., Li, S., Zhu, Y., Liu, X., et al. (2020b). A novel plasma based early colorectal cancer screening assay base on methylated SDC2 and SFRP2. Clin. Chim. Acta 503, 84–89. doi: 10.1016/j.cca.2020.01.010
Zhao, S. G., Chen, W. S., Li, H., Foye, A., Zhang, M., Sjostrom, M., et al. (2020). The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 52, 778–789. doi: 10.1038/s41588-020-0648-8
Zhao, W., Song, M., Zhang, J., Kuerban, M., and Wang, H. (2015). Combined identification of long non-coding RNA CCAT1 and HOTAIR in serum as an effective screening for colorectal carcinoma. Int. J. Clin. Exp. Pathol. 8, 14131–14140.
Zhao, Y., Du, T., Du, L., Li, P., Li, J., Duan, W., et al. (2019). Long noncoding RNA LINC02418 regulates MELK expression by acting as a ceRNA and may serve as a diagnostic marker for colorectal cancer. Cell Death Dis. 10:568. doi: 10.1038/s41419-019-1804-x
Keywords: epigenetics, liquid biopsy, biomarkers, colorectal cancer, precision oncology, circulating nucleic acids, CTCs, extracellular vesicles
Citation: Rodriguez-Casanova A, Costa-Fraga N, Bao-Caamano A, López-López R, Muinelo-Romay L and Diaz-Lagares A (2021) Epigenetic Landscape of Liquid Biopsy in Colorectal Cancer. Front. Cell Dev. Biol. 9:622459. doi: 10.3389/fcell.2021.622459
Received: 28 October 2020; Accepted: 05 January 2021;
Published: 05 February 2021.
Edited by:
Catherine Alix-Panabieres, Centre Hospitalier Universitaire de Montpellier, FranceReviewed by:
Shiaw-Yih Lin, University of Texas MD Anderson Cancer Center, United StatesEvi Lianidou, National and Kapodistrian University of Athens, Greece
Copyright © 2021 Rodriguez-Casanova, Costa-Fraga, Bao-Caamano, López-López, Muinelo-Romay and Diaz-Lagares. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Muinelo-Romay, bG11aXJvbUBnbWFpbC5jb20=; Angel Diaz-Lagares, YW5nZWwuZGlhei5sYWdhcmVzQHNlcmdhcy5lcw==
†These authors share first authorship