 Wen Li
Wen Li YuHong Fu1,3
YuHong Fu1,3 Glenda M. Halliday
Glenda M. Halliday Carolyn M. Sue
Carolyn M. Sue- 1Brain and Mind Centre, University of Sydney, Sydney, NSW, Australia
- 2Kolling Institute of Medical Research, Faculty of Medicine and Health, University of Sydney, Royal North Shore Hospital, St Leonards, NSW, Australia
- 3School of Medical Science, Faculty of Medicine and Health, University of Sydney, Sydney, NSW, Australia
Parkinson’s disease (PD) is an age-related neurodegenerative disorder affecting millions of people worldwide. The disease is characterized by the progressive loss of dopaminergic neurons and spread of Lewy pathology (α-synuclein aggregates) in the brain but the pathogenesis remains elusive. PD presents substantial clinical and genetic variability. Although its complex etiology and pathogenesis has hampered the breakthrough in targeting disease modification, recent genetic tools advanced our approaches. As such, mitochondrial dysfunction has been identified as a major pathogenic hub for both familial and sporadic PD. In this review, we summarize the effect of mutations in 11 PARK genes (SNCA, PRKN, PINK1, DJ-1, LRRK2, ATP13A2, PLA2G6, FBXO7, VPS35, CHCHD2, and VPS13C) on mitochondrial function as well as their relevance in the formation of Lewy pathology. Overall, these genes play key roles in mitochondrial homeostatic control (biogenesis and mitophagy) and functions (e.g., energy production and oxidative stress), which may crosstalk with the autophagy pathway, induce proinflammatory immune responses, and increase oxidative stress that facilitate the aggregation of α-synuclein. Thus, rectifying mitochondrial dysregulation represents a promising therapeutic approach for neuroprotection in PD.
Introduction
Parkinson’s disease (PD) is an age-related neurodegenerative disorder with an insidious onset and a substantial preclinical phase (estimated as >20 years). The progressive aggregation of α-synuclein (Lewy pathology) through the brain and the loss of midbrain dopamine neurons are the pathological landmarks of PD (Surmeier et al., 2017; Fares et al., 2021). Although the etiology of PD is multifactorial, mitochondrial dysfunction has been recognized as a main neuropathogenic mechanism that can affect energy provision and biological pathways (e.g., autophagy, proinflammatory, and antioxidant) to potentially facilitate the Lewy pathology and neuronal loss (Poewe et al., 2017; Park et al., 2018; Grunewald et al., 2019; Pang et al., 2019; Fenton et al., 2021; Onishi et al., 2021).
High-throughput next-generation sequencing and genome-wide association studies have revealed PD risk-associated genes, including 23 PARK genes (Schulte and Gasser, 2011; Marras et al., 2017; Poewe et al., 2017) and others (e.g., HLA-DRA, EIF4GI, GBA, MAPT, BSTI, TMEM230, APOE, and POLG). Most PARK genes have been revealed as low prevalence (Tran et al., 2020), perhaps a reason of limited research on their involvement in sporadic PD. We reviewed 11 PARK genes (Table 1) relevant to mitochondrial function, aiming to highlight their potential roles in the etiology of sporadic PD.
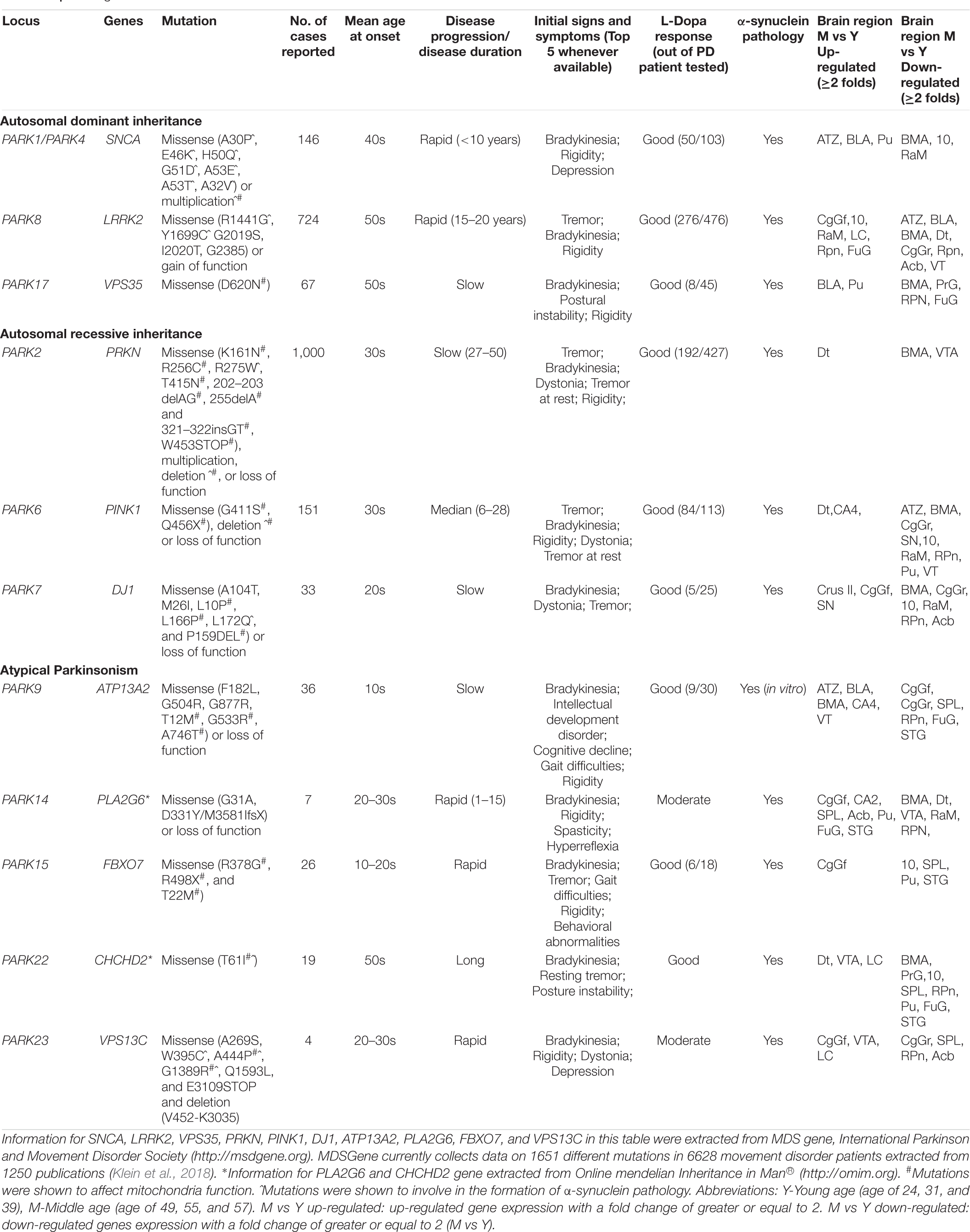
Table 1. PARK genes that are linked with mitochondrial function.
Park Genes in Mitochondrial Function and Homeostatic Control
PARK1/4 (SNCA): α-Synuclein
SNCA was the first PARK gene discovered to cause PD (Polymeropoulos et al., 1996). The prevalence of SNCA mutations is estimated as ∼0.05% in a cohort size of more than 2,000 sporadic PD patients (Tan et al., 2019). SNCA mutations can cause early-onset PD of variable clinical phenotypes and diverse Lewy pathologies (Campelo and Silva, 2017). Duplications or triplications of SNCA cause gene dosage effect on PD severity (Ibanez et al., 2004; Miller et al., 2004).
The physiological function of α-synuclein is not fully understood. Consistent with its many functions, this native disordered protein locates in multiple cellular organelles and sites: mitochondria, nucleus, synapse, endoplasmic reticulum (ER), Golgi, and lysosomes (Bernal-Conde et al., 2019; Shahmoradian et al., 2019).
α-Synuclein directly and indirectly interacts with mitochondria (Ganguly et al., 2018; Grunewald et al., 2019). It maintains mitochondrial membrane composition and structure and its deposition in neurons alters mitochondrial morphology and fragmentation, membrane potential, respiratory chain complex I function, and increases oxidative stress (Di Maio et al., 2016; Zambon et al., 2019; Hannestad et al., 2020). Overexpression of mitochondria-targeted α-synuclein results in mitochondrial reactive oxygen species (ROS) formation, reduced ATP levels, and neuronal death (Ganjam et al., 2019). Vice versa, mitochondrial dysfunction causes α-synuclein pathology as shown in traditional PD models induced by paraquat and rotenone. In addition, α-synuclein interacts with a number of critical mitochondrial proteins, including voltage-dependent anion-selective channel 1, PINK1, Parkin, and DJ-1 proteins (Bernal-Conde et al., 2019). It is also associated with mitochondrial Sirtuin 3, a nicotinamide adenine dinucleotide (NAD+)-dependent enzyme critical in mitochondrial quality control and the prevention of oxidative stress (Park et al., 2020).
PARK2 (PRKN): Parkin RBR E3 Ubiquitin Protein Ligase (parkin)
PRKN, the second identified PD gene (Matsumine et al., 1997) is the most common autosomal recessive gene causing early onset PD (Klein and Lohmann-Hedrich, 2007; Jiang et al., 2020). There are over 100 known mutations in PRKN that lead to either a dysfunctional small Parkin protein being rapidly degraded or defective parkin without activity (Abbas et al., 1999). Some, but not all PRKN mutations cases have Lewy Pathology (Farrer et al., 2001; Shimura et al., 2001; Pramstaller et al., 2005; Miyakawa et al., 2013; Johansen et al., 2018).
Parkin is a HECT/RING hybrid ligase that receives ubiquitin on its catalytic center and passes ubiquitin onto its substrates (Trempe et al., 2013). It regulates mitochondrial quality control through mitophagy and mitochondrial biogenesis. A loss of Parkin function contributes to the pathogenesis of PD through affecting mitochondria, innate immunity, and interactions with α-synuclein. In human cell models, the lack of parkin altered mitochondrial respiratory chain function, oxidative stress levels, mitochondrial morphology and motility, mitophagy (Koentjoro et al., 2017; Bonello et al., 2019), and mitochondrial biogenesis [by upregulating PARIS and subsequently downregulating PGC-1α (Kumar et al., 2020)]. Notably, loss of Parkin alone is not sufficient to induce dopaminergic (DA) neuron loss or motor deficits in mouse models (AguiarJr., Tristao et al., 2013). However, in combination with a POLG mutation (a proofreading-defective mtDNA polymerase), Parkin-deficient mice have both DA neuron loss and motor defects (Sliter et al., 2018).
PARK6 (PINK1): PTEN Induced Kinase 1 (PINK1)
PINK1 is the second most common autosomal recessive gene for PD identified in 2004 (Valente et al., 2004). Heterozygous pathogenic mutations were found in both sporadic and familial PD (Klein et al., 2007). More than 70 mutations have been identified in PINK1 (Siuda et al., 2014; Puschmann et al., 2017). Heterozygous G411S mutation cells have normal PINK1 levels but reduced kinase activity, altered ubiquitin phosphorylation, parkin recruitment, and mitophagy, whereas heterozygous Q456X mutation cells have reduced levels of PINK1 with decreased kinase activity, but their mitochondrial response to damage remains intact (Puschmann et al., 2017). Most but not all PINK1 mutation cases have Lewy pathology, gliosis, and DA neuronal loss in the substantia nigra (Samaranch et al., 2010; Takanashi et al., 2016; Nybo et al., 2020).
PINK1 is a mitochondrial serine/threonine-protein kinase that recruits parkin to depolarized mitochondria for mitophagy (Matsuda et al., 2013). Pink1-deficient mice show significantly impaired mitochondrial respiration in the brain with aging, but no altered mitochondrial morphology, DA neuron loss, or Lewy pathology (Kitada et al., 2007; Gautier et al., 2008). Interestingly, Pink1-deficient rats exhibit DA neuron loss, altered neurotransmitters, and Lewy pathology at 12 months (Creed and Goldberg, 2018, 2020; Creed et al., 2019). Although PINK1/parkin are essential in the mitophagy pathway, they are not required in basal mitophagy (McWilliams et al., 2018). PINK1/parkin are significantly involved in regulating the basal inflammatory response (Sliter et al., 2018; Wang et al., 2019). Human cell models that lack PINK1 have altered mitochondrial respiratory chain function, morphology, motility, and mitophagy [reviewed in Grunewald et al. (2019)].
PARK7 (DJ-1): Parkinsonism Associated Deglycase (DJ-1)
Mutations in DJ-1 were identified as a rare cause of early onset recessive PD in 2003 (Bonifati et al., 2003). Around 20 pathogenic DJ-1 mutations have been identified with reduced protein due to rapid degradation (Ramsey and Giasson, 2010), and less dimerization into its functional form (Kumar et al., 2019). The autopsy of a patient with L172Q mutation showed severe DA neuronal loss in the substantia nigra with Lewy pathology (Taipa et al., 2016). About 57% of DJ-1 mutation carriers exhibit non-motor symptoms, a higher proportion than PRKN or PINK1 mutation carriers (Kasten et al., 2018).
DJ-1 is involved in cellular transformation, oxidative stress response, and mitochondrial function (Di Nottia et al., 2017; Raninga et al., 2017). DJ-1 responds to oxidative stress by accumulating on the outer mitochondrial membrane (OMM) in a PINK1/parkin dependent manner (Thomas et al., 2011; Joselin et al., 2012) which may be neuroprotective (Piston et al., 2017). Depletion of DJ-1 leads to increased ROS, decreased mitochondrial membrane potential, and accumulation of dysfunctional mitochondria, which can be rescued by increasing parkin (Andres-Mateos et al., 2007; Trempe and Fon, 2013; Ozawa et al., 2020). DJ-1 directly interacts with α-synuclein monomers and oligomers in mouse brains (Zondler et al., 2014) and DJ-1 deficiency increases α-synuclein aggregation in human and mouse models (Shendelman et al., 2004; Xu et al., 2017). Notably, the loss of DJ-1 does not induce nigral DA neuron demise in mice (Goldberg et al., 2005). Dj-1-deficient rats show DA neuron loss and evident motor abnormalities (Dave et al., 2014). Similar to SNCA, LRRK2, and UCHL1, mutations in DJ-1 block or reduce the activity of chaperone-mediated autophagy (Sala et al., 2016).
PARK8 (LRRK2): Leucine-Rich Repeat Kinase 2 (LRRK2)
LRRK2, discovered in 2004, is the most frequent autosomal dominant gene causing PD with more than 100 mutations (Paisan-Ruiz et al., 2004; Rui et al., 2018). Genome Aggregation Database predicts LOF in LRRK2 variants cause an 82.5% reduction in protein level, with no change in lifespan or clinical phenotype (Whiffin et al., 2020). LRRK2 mutations generally develop later in life and are clinically similar to sporadic PD, although up to 50% do not have Lewy pathology. Mouse models studying G2019S, R1441G, and Lrrk2-deficient failed to show correlation between loss of function (LOF) of LRRK2 and α-synuclein pathology (Daher et al., 2012; Xiong et al., 2017) but impaired parkin-mediated mitophagy is found in fibroblasts from patients with the G2019S mutation (Bonello et al., 2019).
LRRK2 has multiple domains including a kinase and GTPase enzyme. It is involved in a wide range of cellular processes (Berwick et al., 2019; Marchand et al., 2020) and interacts with Miro on OMM to promote its removal, stopping mitochondrial motility and initiating mitophagy (Hsieh et al., 2016). RAB10, a substrate of LRRK2 kinase activity, accumulates on depolarized mitochondria and interacts with the autophagy receptor OPTN (optineurin) to mediate mitophagy in a PINK1/parkin-dependent manner (Wauters et al., 2020). The toxic gain in function of LRRK2 kinase activity inhibits the accumulation of RAB10 on mitochondria (Wauters et al., 2020). The lack of LRRK2 in macrophages induces oxidative stress and dynamin-related protein 1 (DRP1)-dependent mitochondrial fragmentation (Weindel et al., 2020).
PARK9 (ATP13A2): ATPase Cation Transporting 13A2 (ATP13A2)
Loss of function of ATP13A2 was initially reported in Kufor-Rakeb syndrome (KRS; Ramirez et al., 2006) and in three other distinct neurodegenerative conditions: juvenile-onset neuronal ceroid lipofuscinosis (Bras et al., 2012), juvenile-onset hereditary spastic paraplegia (Estrada-Cuzcano et al., 2017), and amyotrophic lateral sclerosis-like phenotype (Spataro et al., 2019). More than 30 mutations have been identified in ATP13A2 and rare variants may contribute to PD risk (Cristina et al., 2020). Mutations in ATP13A2 cause decreased protein stability, increased proteasomal degradation, impaired polyamine transport and accumulation in lysosomes, and cell death (Podhajska et al., 2012; van Veen et al., 2020).
ATP13A2 is a lysosomal protein, located in the ER, endosomal and lysosomal membranes of neurons (de Tezanos Pinto and Adamo, 2018; Spataro et al., 2019). Loss of ATP13A2 in mouse and human cell models increase mitochondrial fragmentation and increase ROS and cell death (Gusdon et al., 2012; Park et al., 2014). Cell models from KRS patients and cells with silenced ATP13A2 show α-synuclein oxidation and accumulation (Tsunemi and Krainc, 2014). Over-expression of ATP13A2 reduces intracellular α-synuclein via the release of exosomes (Kong et al., 2014).
PARK14 (PLA2G6): Phospholipase A2 Group VI (PLA2G6)
PLA2G6 mutations was discovered in a large family with neurodegeneration in 2006 (Morgan et al., 2006). Mutations in PLA2G6 can cause autosomal recessive PD with high clinical variability, but all show cerebral and cerebellar atrophy, iron accumulation in the basal ganglia, cognitive decline (Khateeb et al., 2006; Ferese et al., 2018) and marked Lewy pathology (Paisan-Ruiz et al., 2012). A total of 16 mutations have been reported. Although most mutations of this gene are homozygous, a heterozygous missense mutation (G31A) has been reported to increase the risk of PD (Ferese et al., 2018). In contrast, compound heterozygous mutations (D331Y/M358IfsX) cause dystonia-parkinsonism with a poor response to levodopa (Chu et al., 2020).
PLA2G6, a calcium-independent phospholipase A2, is involved in maintaining mitochondrial function (Chiu et al., 2017). Overexpression of PLA2G6 exerted neuroprotection in human cells by increasing the level of mitophagy proteins in response to rotenone (Chiu et al., 2017). The loss of PLA2G6 results in shortened acyl-chains in phospholipids, which affects ER homeostasis, neurotransmission, and promotes α-synuclein aggregation (Mori et al., 2019). Elevated expression of α-synuclein in neuronal mitochondria is observed in PLA2G6 deficiency (Sumi-Akamaru et al., 2016).
PARK15 (FBXO7): F-Box Protein 7 (FBXO7)
A homozygous mutation in FBXO7 was reported to cause autosomal recessive PD (Shojaee et al., 2008). Mutations in FBXO7 have not been detected in sporadic PD (Conedera et al., 2016). Mutations in FBXO7 promote the aggregation of the toxic form of this protein in mitochondria, resulting in impairment of mitophagy and the ubiquitin-proteasome system (Zhou et al., 2015). FBXO7 mutations and SNCA G51D mutation have been implicated in Parkinsonian-pyramidal syndrome with early onset and rapid progression (Joseph et al., 2018).
FBXO7 is an adaptor protein in Skp-Cullin-F-box (SCF) SCFFBXO7 ubiquitin E3 ligase complex, which recognizes substrates and mediates their ubiquitination and translocation to mitochondria following cellular stress (Winston et al., 1999; Joseph et al., 2018). FBXO7 recruits parkin into damaged mitochondria and facilitates its aggregation, but overexpression of FBXO7 can still rescue DA neuron degeneration in parkin null Drosophila (Burchell et al., 2013; Zhou et al., 2016) and restore PD phenotype in the absence of parkin, indicating FBXO7 mediates neuroprotective effects via a parkin-independent pathway (Burchell et al., 2013). Both soluble and insoluble FBXO7 are increased in PD (Zhou et al., 2015). FBXO7 immunoreactivity is detected in most α-synuclein aggregates in PD and in glial cytoplasmic inclusions of multiple system atrophy (Zhao et al., 2013). In contrast, only occasional tau-positive inclusions in Alzheimer’s disease and progressive supranuclear palsy contain FBXO7.
PARK17 (VPS35): Vacuolar Protein Sorting 35 Ortholog (VPS35)
Mutations in VPS35 were identified in 2008 (Wider et al., 2008) and are reported in patients with autosomal dominant PD. A heterozygous missense mutation D620N has been confirmed as pathogenic (Williams et al., 2017; Chen et al., 2019) and has been found in 0.056∼0.91% of the sporadic PD patients (Ando et al., 2012; Kumar et al., 2012). The D620N mutation did not affect the stability, assembly, or subcellular location of the retromer (Tian et al., 2015), instead it enhanced LRRK2 kinase activity (Mir et al., 2018). D620N mutant mice show no motor disorders but have increased mitochondrial fission and fragmentation (Wang et al., 2017; Cataldi et al., 2018).
VPS35 forms part of a retromer cargo-recognition complex involved in intracellular retrograde transport from endosomes to the trans-Golgi network (Hierro et al., 2007; Tabuchi et al., 2010). Loss of iPLA2-VIA (the Drosophila homolog of human PLAG2A) destabilizes VPS35 and impairs retromer function, resulting in ceramide accumulation and cell stress (Lin et al., 2018). VPS35 is implicated in the formation of mitochondria-derived vesicles directed to the peroxisome or lysosome for degradation of mitochondria proteins (Braschi et al., 2010; Wang et al., 2016). Lack of VPS35 in human cells with VPS35 mutations exhibit defective mitochondrial fusion and increased mitochondrial fragmentation (Tang et al., 2015). Mitochondrial dysfunction induced by VPS35 mutation can be restored by inhibition of mitochondrial fission (Wang et al., 2016). α-Synuclein is transported by the retromer complex (Miura et al., 2014). Heterozygous Vps35 KO mice show α-synuclein aggregation, DA neuron degeneration, impaired locomotor behavior, and altered lysosomal morphology (Tang et al., 2015). Overexpression of VPS35 reduces α-synuclein accumulation in mice overexpressing α-synuclein (Dhungel et al., 2015). Moreover, knockdown Vps35 in Drosophila results in α-synuclein accumulation (Miura et al., 2014).
PARK22 (CHCHD2): Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 2 (CHCHD2)
Mutations in CHCHD2 are a rare cause of autosomal dominant PD, originally found in 3/340 PD patients in 2015 (Funayama et al., 2015). To date, there is only one brain autopsy of a PD patient carrying CHCHD2 T61I mutation that revealed widespread Lewy pathology with additional amyloid plaques and neurofibrillary tangles in the brainstem, limbic regions, and neocortex (Ikeda et al., 2019). α-Synuclein aggregation was accelerated by CHCHD2 T61I in human cells and in Drosophila. Human cells from T61I patients show accumulated CHCHD2 in the mitochondrial intermembrane space (IMS), resulting in increased ROS and apoptosis (Cornelissen et al., 2020).
CHCHD2 (also called mitochondria nuclear retrograde regulator 1) contains at least one CHCH domain (Modjtahedi et al., 2016). The protein locates in the IMS (Aras et al., 2015). Loss of CHCHD2 function causes an abnormal mitochondrial matrix structure and impaired oxygen respiration in mitochondria resulting in oxidative stress, DA neuron loss, and motor dysfunction with aging (Meng et al., 2017). Importantly, overexpression of CHCHD2 rescues the phenotype of PD. In addition, CHCHD2 binds to cytochrome c and Bax inhibitor-1, suggesting the role CHCHD2 in regulating apoptosis and cell death (Liu et al., 2015). In Drosophila, CHCHD2 interacts with the mitochondrial protein P32 and indirectly regulates the level of mitochondrial fusion protein, Opa1, highlighting the role of CHCHD2 in regulating mitochondrial fusion and cristae morphology (Liu et al., 2015). Moreover, human cells lacking CHCHD2 have altered mitochondrial respiration (Harjuhaahto et al., 2020).
PARK23 (VPS13C): Vacuolar Protein Sorting-Associate Protein 13C
Mutations in VPS13C, identified in 2016 cause an autosomal recessive early onset PD, characterized by early cognitive decline and rapid disease progression (Lesage et al., 2016; Schormair et al., 2018). The post-mortem examination of the brain of the affected patient displayed reduced protein levels of VSP13C and the presence of α-synuclein pathology (Lesage et al., 2016; Smolders et al., 2021).
VPS13C acts at membrane contact sites on multiple organelles such ER, mitochondria, and late endosome and lysosome for lipid delivery, which is important for mitochondrial biogenesis and function (Kumar et al., 2018). VPS13C was localized to the OMM as shown in HEK293 cells (Lesage et al., 2016) and was found between lipid droplets and mitochondria (Ramseyer et al., 2018). LOF in VPS13C in COS-7 monkey cells resulted in abnormal mitochondrial morphology, increased vulnerability to stress and the activation of PINK1/parkin-dependent mitophagy (Lesage et al., 2016). Overexpression of W395C or A444P VPS13C in Hela or SH-SY5Y cells showed the ER-endosomal/lysosomal localization of VPS13C was lost, suggesting these mutants might affect the stability of the protein thereby influencing its localization (Smolders et al., 2021).
Discussion
Over the past 20 years, great progress has been in our understanding of PD with the identification of 23 PARK genes. No doubt there will be more that await discovery. The 11 PARK genes highlighted here collectively emphasize the mechanistic importance of mitochondrial function underlying that pathobiology of PD. These genes are involved in multiple pathways affecting mitochondrial morphology, quality control, respiratory chain function, release of ROS, and biogenesis (fission/fragmentation). More importantly, proteins encoded by five genes (PRKN, PINK1, DJ-1, LRRK2, and FBXO7) closely interact with α-synuclein. Mutations in LRRK2, ATP13A2, PLA2G6, VPS35, CHCHD2, and VPS13C lead to increased α-synuclein accumulation, and mutations in SNCA, PRKN, PINK1, DJ-1, LRRK2, and VPS35 are responsible for the loss of DA neurons.
Although there is a lack of topographical mapping of these 11 gene coding proteins, the heatmap of their RNA expression is available for the human brain (Figures 1A,B; © 2010 Allen Institute for Brain Science. Allen Human Brain Atlas. Available from: human.brain-map.org), suggesting PARK gene expression is both age and brain region related, which further highlight regional vulnerability in the profiling of these proteins. Assessment of pathways affected by these 11 PARK genes using Ingenuity® Pathway Analysis software (Ingenuity Systems Inc., Redwood city, CA, United States) reveals links to DA neuron survival, mitochondrial function, formation of Lewy body pathology, and their mutual protein interactions (Figures 1C,D). Hitherto, cell type specific expression of these gene coding proteins remains unknown. This review suggests the knowledge gap in the field and highlights the importance of studying these genes in sporadic PD, which is essential before targeting these mitochondrial pathways for disease modification.
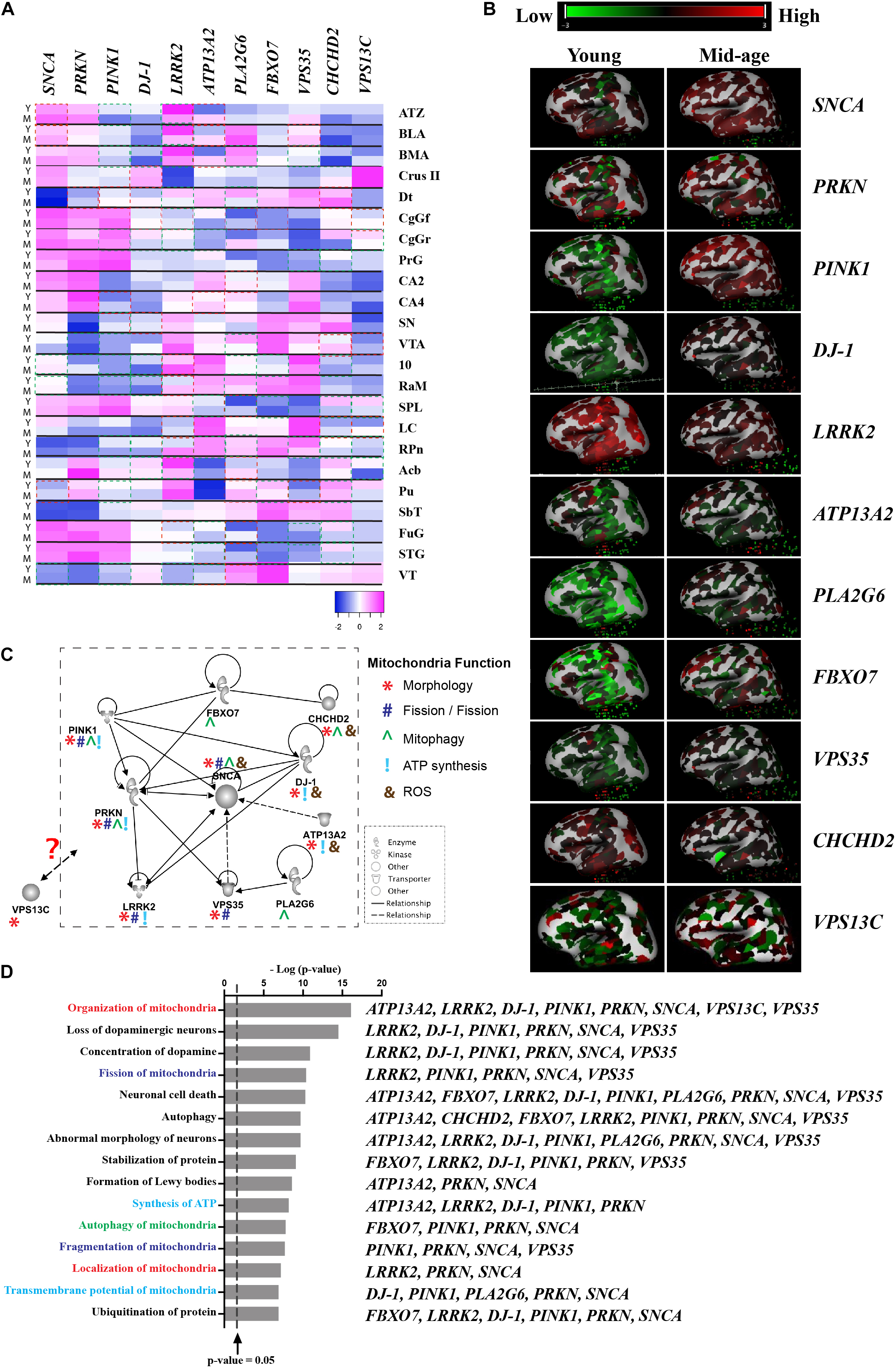
Figure 1. Interaction of proteins encoded by mitochondrial-related PARK genes. (A) Gene expression data was obtained from Allen Brain Atlas, Allen Institute. n = 2–3/group. Y-Young (age of 24, 31, and 39), M-Middle age (age of 49, 55, and 57). Heatmap showing the z-score was generated using online Heatmap program (Babicki et al., 2016). Red boxes indicates up-regulated gene expression with a fold change of greater or equal to 2 (M vs Y) and green boxes indicates down-regulated genes expression with a fold change of greater or equal to 2 (M vs Y). (B) 3D heatmap of the 11 genes from Allen Brain Atlas, Allen Institute. Young (age of 24) and mid-age (age of 57); (C) The interactive pathways of proteins encoded by the 11 PARK genes generated from IPA (Ingenuity Systems Inc., Redwood city, CA, United States). Symbols are used to show mitochondrial function: *Morphology; #Fission/Fusion; ^Mitophagy; !ATP synthesis; and &ROS. (D) Top 35 biological functions identified to be related to mitochondrial function and DA neuron survival and relevant PARK genes listed next to functions. The biological functions were plotted against the negative log10 p-value [–log(p-value)] as measured by Fischer’s exact test determined by IPA. Line represented p-value = 0.05. Abbreviations: ATZ, amygdalohippocampal transition zone; BLA, basolateral nucleus; BMA, basomedial nucleus; Cb-Crus II, crus II; Dt, dentate nucleus; CgGf, cingulate gyrus; frontal part; CgGr, cingulate gyrus, retrosplenial part; PrG, precentral gyrus; CA2, CA2 field; CA4, CA4 field; SN, substantia nigra; VTA, ventral tegmental area; 10, dorsal motor nucleus of the vagus; RaM, raphe nuclei of medulla; SPL, superior parietal lobule; LC, locus ceruleus; RPn, pontine raphe nucleus; Acb, nucleus accumbens; Pu, putamen; SbT, subthalamus; FuG, fusiform gyrus; STG, superior temporal gyrus; and VT, ventral thalamus.
Author Contributions
WL and YF conceived of the presented data. WL and YF wrote the manuscript in consultation with GH and CS. GH and CS were in charge of overall direction and planning. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Health and Medical Research Council (NHMRC) to CS and YF (#1141064) and funding to ForeFront, a collaborative research group at the Brain and Mind Centre University of Sydney, from NHMRC program (#1132524) and Dementia Team (#1095127) grants. GH is an NHMRC Leadership Fellow (#1176607). CS is an MRFF NHMRC Practitioner Fellow (#1136800).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Allen Institute for Brain Science for providing transcriptomic data from donors, MDS gene, International Parkinson and Movement Disorder Society and Online Mendelian Inheritance in Man® for genetic data and relevant clinical data.
References
Abbas, N., Lucking, C. B., Ricard, S., Durr, A., Bonifati, V., De Michele, G., et al. (1999). A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum. Mol. Genet. 8, 567–574. doi: 10.1093/hmg/8.4.567
Aguiar, A. S. Jr., Tristao, F. S., Amar, M., Chevarin, C., Lanfumey, L., Mongeau, R., et al. (2013). Parkin-knockout mice did not display increased vulnerability to intranasal administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Neurotox. Res. 24, 280–287. doi: 10.1007/s12640-013-9389-0
Ando, M., Funayama, M., Li, Y., Kashihara, K., Murakami, Y., Ishizu, N., et al. (2012). VPS35 mutation in Japanese patients with typical Parkinson’s disease. Mov. Disord. 27, 1413–1417. doi: 10.1002/mds.25145
Andres-Mateos, E., Perier, C., Zhang, L., Blanchard-Fillion, B., Greco, T. M., Thomas, B., et al. (2007). DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. U.S.A. 104, 14807–14812. doi: 10.1073/pnas.0703219104
Aras, S., Bai, M., Lee, I., Springett, R., Huttemann, M., and Grossman, L. I. (2015). MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 20, 43–51. doi: 10.1016/j.mito.2014.10.003
Babicki, S., Arndt, D., Marcu, A., Liang, Y., Grant, J. R., Maciejewski, A., et al. (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44, W147–W153.
Bernal-Conde, L. D., Ramos-Acevedo, R., Reyes-Hernandez, M. A., Balbuena-Olvera, A. J., Morales-Moreno, I. D., Arguero-Sanchez, R., et al. (2019). Alpha-Synuclein physiology and pathology: a perspective on cellular structures and organelles. Front. Neurosci. 13:1399. doi: 10.3389/fnins.2019.01399
Berwick, D. C., Heaton, G. R., Azeggagh, S., and Harvey, K. (2019). LRRK2 Biology from structure to dysfunction: research progresses, but the themes remain the same. Mol. Neurodegener. 14:49.
Bonello, F., Hassoun, S. M., Mouton-Liger, F., Shin, Y. S., Muscat, A., Tesson, C., et al. (2019). LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 28, 1645–1660. doi: 10.1093/hmg/ddz004
Bonifati, V., Rizzu, P., Van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Bras, J., Verloes, A., Schneider, S. A., Mole, S. E., and Guerreiro, R. J. (2012). Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum. Mol. Genet. 21, 2646–2650. doi: 10.1093/hmg/dds089
Braschi, E., Goyon, V., Zunino, R., Mohanty, A., Xu, L., and Mcbride, H. M. (2010). Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 20, 1310–1315. doi: 10.1016/j.cub.2010.05.066
Burchell, V. S., Nelson, D. E., Sanchez-Martinez, A., Delgado-Camprubi, M., Ivatt, R. M., Pogson, J. H., et al. (2013). The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 16, 1257–1265. doi: 10.1038/nn.3489
Campelo, C., and Silva, R. H. (2017). Genetic variants in SNCA and the risk of sporadic Parkinson’s disease and clinical outcomes: a review. Parkinsons Dis. 2017:4318416.
Cataldi, S., Follett, J., Fox, J. D., Tatarnikov, I., Kadgien, C., Gustavsson, E. K., et al. (2018). Altered dopamine release and monoamine transporters in Vps35 p.D620N knock-in mice. NPJ Parkinsons Dis. 4:27.
Chen, C., Turnbull, D. M., and Reeve, A. K. (2019). Mitochondrial dysfunction in parkinson’s disease-cause or consequence? Biology (Basel) 8:38. doi: 10.3390/biology8020038
Chiu, C. C., Yeh, T. H., Lu, C. S., Huang, Y. C., Cheng, Y. C., Huang, Y. Z., et al. (2017). PARK14 PLA2G6 mutants are defective in preventing rotenone-induced mitochondrial dysfunction, ROS generation and activation of mitochondrial apoptotic pathway. Oncotarget 8, 79046–79060. doi: 10.18632/oncotarget.20893
Chu, Y. T., Lin, H. Y., Chen, P. L., and Lin, C. H. (2020). Genotype-phenotype correlations of adult-onset PLA2G6-associated Neurodegeneration: case series and literature review. BMC Neurol. 20:101. doi: 10.1186/s12883-020-01684-6
Conedera, S., Apaydin, H., Li, Y., Yoshino, H., Ikeda, A., Matsushima, T., et al. (2016). FBXO7 mutations in Parkinson’s disease and multiple system atrophy. Neurobiol. Aging 40, 192.e191–192.e195.
Cornelissen, T., Spinazzi, M., Martin, S., Imberechts, D., Vangheluwe, P., Bird, M., et al. (2020). CHCHD2 harboring Parkinson’s disease-linked T61I mutation precipitates inside mitochondria and induces precipitation of wild-type CHCHD2. Hum. Mol. Genet. 29, 1096–1106. doi: 10.1093/hmg/ddaa028
Creed, R. B., and Goldberg, M. S. (2018). Analysis of alpha-Synuclein pathology in PINK1 knockout rat brains. Front. Neurosci. 12:1034. doi: 10.3389/fnins.2018.01034
Creed, R. B., and Goldberg, M. S. (2020). Enhanced susceptibility of PINK1 knockout rats to alpha-synuclein fibrils. Neuroscience 437, 64–75. doi: 10.1016/j.neuroscience.2020.04.032
Creed, R. B., Menalled, L., Casey, B., Dave, K. D., Janssens, H. B., Veinbergs, I., et al. (2019). Basal and evoked neurotransmitter levels in parkin, DJ-1, PINK1 and LRRK2 knockout rat striatum. Neuroscience 409, 169–179. doi: 10.1016/j.neuroscience.2019.04.033
Cristina, T. P., Pablo, M., Teresa, P. M., Lydia, V. D., Irene, A. R., Araceli, A. C., et al. (2020). A genetic analysis of a Spanish population with early onset Parkinson’s disease. PLoS One 15:e0238098. doi: 10.1371/journal.pone.0238098
Daher, J. P., Pletnikova, O., Biskup, S., Musso, A., Gellhaar, S., Galter, D., et al. (2012). Neurodegenerative phenotypes in an A53T alpha-synuclein transgenic mouse model are independent of LRRK2. Hum. Mol. Genet. 21, 2420–2431. doi: 10.1093/hmg/dds057
Dave, K. D., De Silva, S., Sheth, N. P., Ramboz, S., Beck, M. J., Quang, C., et al. (2014). Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol. Dis. 70, 190–203. doi: 10.1016/j.nbd.2014.06.009
de Tezanos Pinto, F., and Adamo, H. P. (2018). The strategic function of the P5-ATPase ATP13A2 in toxic waste disposal. Neurochem. Int. 112, 108–113. doi: 10.1016/j.neuint.2017.11.008
Dhungel, N., Eleuteri, S., Li, L. B., Kramer, N. J., Chartron, J. W., Spencer, B., et al. (2015). Parkinson’s disease genes VPS35 and EIF4G1 interact genetically and converge on alpha-synuclein. Neuron 85, 76–87. doi: 10.1016/j.neuron.2014.11.027
Di Maio, R., Barrett, P. J., Hoffman, E. K., Barrett, C. W., Zharikov, A., Borah, A., et al. (2016). alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 8:342ra378.
Di Nottia, M., Masciullo, M., Verrigni, D., Petrillo, S., Modoni, A., Rizzo, V., et al. (2017). DJ-1 modulates mitochondrial response to oxidative stress: clues from a novel diagnosis of PARK7. Clin. Genet. 92, 18–25. doi: 10.1111/cge.12841
Estrada-Cuzcano, A., Martin, S., Chamova, T., Synofzik, M., Timmann, D., Holemans, T., et al. (2017). Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 140, 287–305. doi: 10.1093/brain/aww307
Fares, M. B., Jagannath, S., and Lashuel, H. A. (2021). Reverse engineering Lewy bodies: how far have we come and how far can we go? Nat. Rev. Neurosci. 22, 111–131. doi: 10.1038/s41583-020-00416-6
Farrer, M., Chan, P., Chen, R., Tan, L., Lincoln, S., Hernandez, D., et al. (2001). Lewy bodies and parkinsonism in families with parkin mutations. Ann. Neurol. 50, 293–300.
Fenton, A. R., Jongens, T. A., and Holzbaur, E. L. F. (2021). Mitochondrial dynamics: shaping and remodeling an organelle network. Curr. Opin. Cell Biol. 68, 28–36. doi: 10.1016/j.ceb.2020.08.014
Ferese, R., Scala, S., Biagioni, F., Giardina, E., Zampatti, S., Modugno, N., et al. (2018). Heterozygous PLA2G6 mutation leads to iron accumulation within Basal Ganglia and Parkinson’s disease. Front. Neurol. 9:536. doi: 10.3389/fneur.2018.00536
Funayama, M., Ohe, K., Amo, T., Furuya, N., Yamaguchi, J., Saiki, S., et al. (2015). CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: a genome-wide linkage and sequencing study. Lancet Neurol. 14, 274–282. doi: 10.1016/s1474-4422(14)70266-2
Ganguly, U., Chakrabarti, S. S., Kaur, U., Mukherjee, A., and Chakrabarti, S. (2018). Alpha-synuclein, proteotoxicity and Parkinson’s disease: search for neuroprotective therapy. Curr. Neuropharmacol. 16, 1086–1097. doi: 10.2174/1570159x15666171129100944
Ganjam, G. K., Bolte, K., Matschke, L. A., Neitemeier, S., Dolga, A. M., Hollerhage, M., et al. (2019). Mitochondrial damage by alpha-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 10:865.
Gautier, C. A., Kitada, T., and Shen, J. (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 105, 11364–11369. doi: 10.1073/pnas.0802076105
Goldberg, M. S., Pisani, A., Haburcak, M., Vortherms, T. A., Kitada, T., Costa, C., et al. (2005). Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45, 489–496. doi: 10.1016/j.neuron.2005.01.041
Grunewald, A., Kumar, K. R., and Sue, C. M. (2019). New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 177, 73–93. doi: 10.1016/j.pneurobio.2018.09.003
Gusdon, A. M., Zhu, J., Van Houten, B., and Chu, C. T. (2012). ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol. Dis. 45, 962–972. doi: 10.1016/j.nbd.2011.12.015
Hannestad, J. K., Rocha, S., Agnarsson, B., Zhdanov, V. P., Wittung-Stafshede, P., and Hook, F. (2020). Single-vesicle imaging reveals lipid-selective and stepwise membrane disruption by monomeric alpha-synuclein. Proc. Natl. Acad. Sci. U.S.A. 117, 14178–14186.
Harjuhaahto, S., Rasila, T. S., Molchanova, S. M., Woldegebriel, R., Kvist, J., Konovalova, S., et al. (2020). ALS and Parkinson’s disease genes CHCHD10 and CHCHD2 modify synaptic transcriptomes in human iPSC-derived motor neurons. Neurobiol. Dis. 141:104940. doi: 10.1016/j.nbd.2020.104940
Hierro, A., Rojas, A. L., Rojas, R., Murthy, N., Effantin, G., Kajava, A. V., et al. (2007). Functional architecture of the retromer cargo-recognition complex. Nature 449, 1063–1067. doi: 10.1038/nature06216
Hsieh, C. H., Shaltouki, A., Gonzalez, A. E., Bettencourt Da Cruz, A., Burbulla, L. F., St Lawrence, E., et al. (2016). Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 19, 709–724. doi: 10.1016/j.stem.2016.08.002
Ibanez, P., Bonnet, A. M., Debarges, B., Lohmann, E., Tison, F., Pollak, P., et al. (2004). Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171. doi: 10.1016/s0140-6736(04)17104-3
Ikeda, A., Nishioka, K., Meng, H., Takanashi, M., Hasegawa, I., Inoshita, T., et al. (2019). Mutations in CHCHD2 cause alpha-synuclein aggregation. Hum. Mol. Genet. 28, 3895–3911.
Jiang, Y., Yu, M., Chen, J., Zhou, H., Sun, W., Sun, Y., et al. (2020). Parkin is the most common causative gene in a cohort of mainland Chinese patients with sporadic early-onset Parkinson’s disease. Brain Behav. e01765. doi: 10.1002/brb3.1765
Johansen, K. K., Torp, S. H., Farrer, M. J., Gustavsson, E. K., and Aasly, J. O. (2018). A case of Parkinson’s disease with no lewy body pathology due to a homozygous exon deletion in parkin. Case Rep. Neurol. Med. 2018:6838965.
Joselin, A. P., Hewitt, S. J., Callaghan, S. M., Kim, R. H., Chung, Y. H., Mak, T. W., et al. (2012). ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 21, 4888–4903. doi: 10.1093/hmg/dds325
Joseph, S., Schulz, J. B., and Stegmuller, J. (2018). Mechanistic contributions of FBXO7 to Parkinson disease. J. Neurochem. 144, 118–127. doi: 10.1111/jnc.14253
Kasten, M., Hartmann, C., Hampf, J., Schaake, S., Westenberger, A., Vollstedt, E. J., et al. (2018). Genotype-Phenotype relations for the Parkinson’s disease genes parkin, PINK1, DJ1: MDSGene systematic review. Mov. Disord. 33, 730–741. doi: 10.1002/mds.27352
Khateeb, S., Flusser, H., Ofir, R., Shelef, I., Narkis, G., Vardi, G., et al. (2006). PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am. J. Hum. Genet. 79, 942–948. doi: 10.1086/508572
Kitada, T., Pisani, A., Porter, D. R., Yamaguchi, H., Tscherter, A., Martella, G., et al. (2007). Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 104, 11441–11446. doi: 10.1073/pnas.0702717104
Klein, C., Hattori, N., and Marras, C. (2018). MDSGene: closing data gaps in genotype-phenotype correlations of monogenic Parkinson’s disease. J. Parkinsons Dis. 8, S25–S30. doi: 10.3233/JPD-181505
Klein, C., and Lohmann-Hedrich, K. (2007). Impact of recent genetic findings in Parkinson’s disease. Curr. Opin. Neurol. 20, 453–464. doi: 10.1097/WCO.0b013e3281e6692b
Klein, C., Lohmann-Hedrich, K., Rogaeva, E., Schlossmacher, M. G., and Lang, A. E. (2007). Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 6, 652–662. doi: 10.1016/s1474-4422(07)70174-6
Koentjoro, B., Park, J. S., and Sue, C. M. (2017). Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 7:44373.
Kong, S. M., Chan, B. K., Park, J. S., Hill, K. J., Aitken, J. B., Cottle, L., et al. (2014). Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum. Mol. Genet. 23, 2816–2833. doi: 10.1093/hmg/ddu099
Kumar, A., Mukherjee, D., and Satpati, P. (2019). Mutations in Parkinson’s disease associated protein DJ-1 alter the energetics of DJ-1 dimerization. J. Chem. Inf. Model 59, 1497–1507. doi: 10.1021/acs.jcim.8b00687
Kumar, K. R., Weissbach, A., Heldmann, M., Kasten, M., Tunc, S., Sue, C. M., et al. (2012). Frequency of the D620N mutation in VPS35 in Parkinson disease. Arch. Neurol. 69, 1360–1364. doi: 10.1001/archneurol.2011.3367
Kumar, M., Acevedo-Cintron, J., Jhaldiyal, A., Wang, H., Andrabi, S. A., Eacker, S., et al. (2020). Defects in mitochondrial biogenesis drive mitochondrial alterations in PARKIN-Deficient human dopamine neurons. Stem Cell Rep. 15, 629–645. doi: 10.1016/j.stemcr.2020.07.013
Kumar, N., Leonzino, M., Hancock-Cerutti, W., Horenkamp, F. A., Li, P., Lees, J. A., et al. (2018). VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 217, 3625–3639. doi: 10.1083/jcb.201807019
Lesage, S., Drouet, V., Majounie, E., Deramecourt, V., Jacoupy, M., Nicolas, A., et al. (2016). Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am. J. Hum. Genet. 98, 500–513.
Lin, G., Lee, P. T., Chen, K., Mao, D., Tan, K. L., Zuo, Z., et al. (2018). Phospholipase PLA2G6, a Parkinsonism-associated gene, affects Vps26 and Vps35, retromer function, and ceramide levels, similar to alpha-synuclein gain. Cell Metab. 28, 605–618.e606.
Liu, Y., Clegg, H. V., Leslie, P. L., Di, J., Tollini, L. A., He, Y., et al. (2015). CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate Bax activation. Cell Death Differ. 22, 1035–1046. doi: 10.1038/cdd.2014.194
Marchand, A., Drouyer, M., Sarchione, A., Chartier-Harlin, M. C., and Taymans, J. M. (2020). LRRK2 phosphorylation, more than an epiphenomenon. Front. Neurosci. 14:527. doi: 10.3389/fnins.2020.00527
Marras, C., Lang, A., Van De Warrenburg, B. P., Sue, C. M., Tabrizi, S. J., Bertram, L., et al. (2017). Nomenclature of genetic movement disorders: recommendations of the International Parkinson and Movement Disorder Society task force. Mov. Disord. 32, 724–725. doi: 10.1002/mds.27045
Matsuda, S., Kitagishi, Y., and Kobayashi, M. (2013). Function and characteristics of PINK1 in mitochondria. Oxid. Med. Cell. Longev. 2013:601587.
Matsumine, H., Saito, M., Shimoda-Matsubayashi, S., Tanaka, H., Ishikawa, A., Nakagawa-Hattori, Y., et al. (1997). Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am. J. Hum. Genet. 60, 588–596.
McWilliams, T. G., Prescott, A. R., Montava-Garriga, L., Ball, G., Singh, F., Barini, E., et al. (2018). Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 27, 439–449.e435.
Meng, H., Yamashita, C., Shiba-Fukushima, K., Inoshita, T., Funayama, M., Sato, S., et al. (2017). Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 8:15500.
Miller, D. W., Hague, S. M., Clarimon, J., Baptista, M., Gwinn-Hardy, K., Cookson, M. R., et al. (2004). Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62, 1835–1838. doi: 10.1212/01.wnl.0000127517.33208.f4
Mir, R., Tonelli, F., Lis, P., Macartney, T., Polinski, N. K., Martinez, T. N., et al. (2018). The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem. J. 475, 1861–1883. doi: 10.1042/bcj20180248
Miura, E., Hasegawa, T., Konno, M., Suzuki, M., Sugeno, N., Fujikake, N., et al. (2014). VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 71, 1–13. doi: 10.1016/j.nbd.2014.07.014
Miyakawa, S., Ogino, M., Funabe, S., Uchino, A., Shimo, Y., Hattori, N., et al. (2013). Lewy body pathology in a patient with a homozygous parkin deletion. Mov. Disord. 28, 388–391.
Modjtahedi, N., Tokatlidis, K., Dessen, P., and Kroemer, G. (2016). Mitochondrial proteins containing coiled-coil-helix-coiled-coil-helix (CHCH) domains in health and disease. Trends Biochem. Sci. 41, 245–260. doi: 10.1016/j.tibs.2015.12.004
Morgan, N. V., Westaway, S. K., Morton, J. E., Gregory, A., Gissen, P., Sonek, S., et al. (2006). PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet. 38, 752–754. doi: 10.1038/ng1826
Mori, A., Hatano, T., Inoshita, T., Shiba-Fukushima, K., Koinuma, T., Meng, H., et al. (2019). Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and alpha-synuclein stability through membrane remodeling. Proc. Natl. Acad. Sci. U.S.A. 116, 20689–20699. doi: 10.1073/pnas.1902958116
Nybo, C. J., Gustavsson, E. K., Farrer, M. J., and Aasly, J. O. (2020). Neuropathological findings in PINK1-associated Parkinson’s disease. Parkinsonism Relat. Disord. 78, 105–108. doi: 10.1016/j.parkreldis.2020.07.023
Onishi, M., Yamano, K., Sato, M., Matsuda, N., and Okamoto, K. (2021). Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40: e104705.
Ozawa, K., Tsumoto, H., Miura, Y., Yamaguchi, J., Iguchi-Ariga, S. M. M., Sakuma, T., et al. (2020). DJ-1 is indispensable for the S-nitrosylation of Parkin, which maintains function of mitochondria. Sci. Rep. 10:4377.
Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., Van Der Brug, M., et al. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. doi: 10.1016/j.neuron.2004.10.023
Paisan-Ruiz, C., Li, A., Schneider, S. A., Holton, J. L., Johnson, R., Kidd, D., et al. (2012). Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol. Aging 33, 814–823. doi: 10.1016/j.neurobiolaging.2010.05.009
Pang, S. Y., Ho, P. W., Liu, H. F., Leung, C. T., Li, L., Chang, E. E. S., et al. (2019). The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 8:23.
Park, J. H., Burgess, J. D., Faroqi, A. H., Demeo, N. N., Fiesel, F. C., Springer, W., et al. (2020). Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 15:5.
Park, J. S., Koentjoro, B., Veivers, D., Mackay-Sim, A., and Sue, C. M. (2014). Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 23, 2802–2815. doi: 10.1093/hmg/ddt623
Park, J. T., Lee, Y. S., Cho, K. A., and Park, S. C. (2018). Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing Res. Rev. 47, 176–182. doi: 10.1016/j.arr.2018.08.003
Piston, D., Alvarez-Erviti, L., Bansal, V., Gargano, D., Yao, Z., Szabadkai, G., et al. (2017). DJ-1 is a redox sensitive adapter protein for high molecular weight complexes involved in regulation of catecholamine homeostasis. Hum. Mol. Genet. 26, 4028–4041. doi: 10.1093/hmg/ddx294
Podhajska, A., Musso, A., Trancikova, A., Stafa, K., Moser, R., Sonnay, S., et al. (2012). Common pathogenic effects of missense mutations in the P-type ATPase ATP13A2 (PARK9) associated with early-onset parkinsonism. PLoS One 7:e39942. doi: 10.1371/journal.pone.0039942
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:17013.
Polymeropoulos, M. H., Higgins, J. J., Golbe, L. I., Johnson, W. G., Ide, S. E., Di Iorio, G., et al. (1996). Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 274, 1197–1199. doi: 10.1126/science.274.5290.1197
Pramstaller, P. P., Schlossmacher, M. G., Jacques, T. S., Scaravilli, F., Eskelson, C., Pepivani, I., et al. (2005). Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann. Neurol. 58, 411–422. doi: 10.1002/ana.20587
Puschmann, A., Fiesel, F. C., Caulfield, T. R., Hudec, R., Ando, M., Truban, D., et al. (2017). Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 140, 98–117. doi: 10.1093/brain/aww261
Ramirez, A., Heimbach, A., Grundemann, J., Stiller, B., Hampshire, D., Cid, L. P., et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 38, 1184–1191. doi: 10.1038/ng1884
Ramsey, C. P., and Giasson, B. I. (2010). L10p and P158DEL DJ-1 mutations cause protein instability, aggregation, and dimerization impairments. J. Neurosci. Res. 88, 3111–3124. doi: 10.1002/jnr.22477
Ramseyer, V. D., Kimler, V. A., and Granneman, J. G. (2018). Vacuolar protein sorting 13C is a novel lipid droplet protein that inhibits lipolysis in brown adipocytes. Mol. Metab. 7, 57–70. doi: 10.1016/j.molmet.2017.10.014
Raninga, P. V., Di Trapani, G., and Tonissen, K. F. (2017). The multifaceted roles of DJ-1 as an antioxidant. Adv. Exp. Med. Biol. 1037, 67–87. doi: 10.1007/978-981-10-6583-5_6
Rui, Q., Ni, H., Li, D., Gao, R., and Chen, G. (2018). The role of LRRK2 in neurodegeneration of Parkinson disease. Curr. Neuropharmacol. 16, 1348–1357. doi: 10.2174/1570159x16666180222165418
Sala, G., Marinig, D., Arosio, A., and Ferrarese, C. (2016). Role of chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson’s disease. Front. Mol. Neurosci. 9:157. doi: 10.3389/fnmol.2016.00157
Samaranch, L., Lorenzo-Betancor, O., Arbelo, J. M., Ferrer, I., Lorenzo, E., Irigoyen, J., et al. (2010). PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133, 1128–1142. doi: 10.1093/brain/awq051
Schormair, B., Kemlink, D., Mollenhauer, B., Fiala, O., Machetanz, G., Roth, J., et al. (2018). Diagnostic exome sequencing in early-onset Parkinson’s disease confirms VPS13C as a rare cause of autosomal-recessive Parkinson’s disease. Clin. Genet. 93, 603–612. doi: 10.1111/cge.13124
Schulte, C., and Gasser, T. (2011). Genetic basis of Parkinson’s disease: inheritance, penetrance, and expression. Appl. Clin. Genet. 4, 67–80. doi: 10.2147/tacg.s11639
Shahmoradian, S. H., Lewis, A. J., Genoud, C., Hench, J., Moors, T. E., Navarro, P. P., et al. (2019). Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 22, 1099–1109. doi: 10.1038/s41593-019-0423-2
Shendelman, S., Jonason, A., Martinat, C., Leete, T., and Abeliovich, A. (2004). DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2:e362. doi: 10.1371/journal.pbio.0020362
Shimura, H., Schlossmacher, M. G., Hattori, N., Frosch, M. P., Trockenbacher, A., Schneider, R., et al. (2001). Ubiquitination of a new form of alpha-synuclein by parkin from human brain: implications for Parkinson’s disease. Science 293, 263–269. doi: 10.1126/science.1060627
Shojaee, S., Sina, F., Banihosseini, S. S., Kazemi, M. H., Kalhor, R., Shahidi, G. A., et al. (2008). Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am. J. Hum. Genet. 82, 1375–1384. doi: 10.1016/j.ajhg.2008.05.005
Siuda, J., Jasinska-Myga, B., Boczarska-Jedynak, M., Opala, G., Fiesel, F. C., Moussaud-Lamodiere, E. L., et al. (2014). Early-onset Parkinson’s disease due to PINK1 p.Q456X mutation–clinical and functional study. Parkinsonism Relat. Disord. 20, 1274–1278. doi: 10.1016/j.parkreldis.2014.08.019
Sliter, D. A., Martinez, J., Hao, L., Chen, X., Sun, N., Fischer, T. D., et al. (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. doi: 10.1038/s41586-018-0448-9
Smolders, S., Philtjens, S., Crosiers, D., Sieben, A., Hens, E., Heeman, B., et al. (2021). Contribution of rare homozygous and compound heterozygous VPS13C missense mutations to dementia with Lewy bodies and Parkinson’s disease. Acta Neuropathol. Commun. 9:25.
Spataro, R., Kousi, M., Farhan, S. M. K., Willer, J. R., Ross, J. P., Dion, P. A., et al. (2019). Mutations in ATP13A2 (PARK9) are associated with an amyotrophic lateral sclerosis-like phenotype, implicating this locus in further phenotypic expansion. Hum. Genomics 13:19.
Sumi-Akamaru, H., Beck, G., Shinzawa, K., Kato, S., Riku, Y., Yoshida, M., et al. (2016). High expression of alpha-synuclein in damaged mitochondria with PLA2G6 dysfunction. Acta Neuropathol. Commun. 4:27.
Surmeier, D. J., Obeso, J. A., and Halliday, G. M. (2017). Parkinson’s disease is not simply a prion disorder. J. Neurosci. 37, 9799–9807. doi: 10.1523/jneurosci.1787-16.2017
Tabuchi, M., Yanatori, I., Kawai, Y., and Kishi, F. (2010). Retromer-mediated direct sorting is required for proper endosomal recycling of the mammalian iron transporter DMT1. J. Cell Sci. 123, 756–766. doi: 10.1242/jcs.060574
Taipa, R., Pereira, C., Reis, I., Alonso, I., Bastos-Lima, A., Melo-Pires, M., et al. (2016). DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 139, 1680–1687. doi: 10.1093/brain/aww080
Takanashi, M., Li, Y., and Hattori, N. (2016). Absence of Lewy pathology associated with PINK1 homozygous mutation. Neurology 86, 2212–2213. doi: 10.1212/wnl.0000000000002744
Tan, M. M. X., Malek, N., Lawton, M. A., Hubbard, L., Pittman, A. M., Joseph, T., et al. (2019). Genetic analysis of Mendelian mutations in a large UK population-based Parkinson’s disease study. Brain 142, 2828–2844. doi: 10.1093/brain/awz191
Tang, F. L., Liu, W., Hu, J. X., Erion, J. R., Ye, J., Mei, L., et al. (2015). VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 12, 1631–1643. doi: 10.1016/j.celrep.2015.08.001
Thomas, K. J., Mccoy, M. K., Blackinton, J., Beilina, A., Van Der Brug, M., Sandebring, A., et al. (2011). DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 20, 40–50. doi: 10.1093/hmg/ddq430
Tian, Y., Tang, F. L., Sun, X., Wen, L., Mei, L., Tang, B. S., et al. (2015). VPS35-deficiency results in an impaired AMPA receptor trafficking and decreased dendritic spine maturation. Mol. Brain 8:70.
Tran, J., Anastacio, H., and Bardy, C. (2020). Genetic predispositions of Parkinson’s disease revealed in patient-derived brain cells. NPJ Parkinsons Dis. 6:8.
Trempe, J. F., and Fon, E. A. (2013). Structure and function of parkin, PINK1, and DJ-1, the three musketeers of neuroprotection. Front. Neurol. 4:38. doi: 10.3389/fneur.2013.00038
Trempe, J. F., Sauve, V., Grenier, K., Seirafi, M., Tang, M. Y., Menade, M., et al. (2013). Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455. doi: 10.1126/science.1237908
Tsunemi, T., and Krainc, D. (2014). Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 23, 2791–2801. doi: 10.1093/hmg/ddt572
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
van Veen, S., Martin, S., Van Den Haute, C., Benoy, V., Lyons, J., Vanhoutte, R., et al. (2020). ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 578, 419–424. doi: 10.1038/s41586-020-1968-7
Wang, H., Ni, H. M., Chao, X., Ma, X., Rodriguez, Y. A., Chavan, H., et al. (2019). Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol. 22:101148. doi: 10.1016/j.redox.2019.101148
Wang, W., Ma, X., Zhou, L., Liu, J., and Zhu, X. (2017). A conserved retromer sorting motif is essential for mitochondrial DLP1 recycling by VPS35 in Parkinson’s disease model. Hum. Mol. Genet. 26, 781–789.
Wang, W., Wang, X., Fujioka, H., Hoppel, C., Whone, A. L., Caldwell, M. A., et al. (2016). Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 22, 54–63. doi: 10.1038/nm.3983
Wauters, F., Cornelissen, T., Imberechts, D., Martin, S., Koentjoro, B., Sue, C., et al. (2020). LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 16, 203–222. doi: 10.1080/15548627.2019.1603548
Weindel, C. G., Bell, S. L., Vail, K. J., West, K. O., Patrick, K. L., and Watson, R. O. (2020). LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. Elife 9:e51071.
Whiffin, N., Armean, I. M., Kleinman, A., Marshall, J. L., Minikel, E. V., Goodrich, J. K., et al. (2020). The effect of LRRK2 loss-of-function variants in humans. Nat. Med. 26, 869–877.
Wider, C., Skipper, L., Solida, A., Brown, L., Farrer, M., Dickson, D., et al. (2008). Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat. Disord. 14, 465–470. doi: 10.1016/j.parkreldis.2007.11.013
Williams, E. T., Chen, X., and Moore, D. J. (2017). VPS35, the retromer complex and Parkinson’s disease. J. Parkinsons Dis. 7, 219–233.
Winston, J. T., Koepp, D. M., Zhu, C., Elledge, S. J., and Harper, J. W. (1999). A family of mammalian F-box proteins. Curr. Biol. 9, 1180–1182. doi: 10.1016/s0960-9822(00)80021-4
Xiong, Y., Neifert, S., Karuppagounder, S. S., Stankowski, J. N., Lee, B. D., Grima, J. C., et al. (2017). Overexpression of Parkinson’s disease-associated mutation LRRK2 G2019S in mouse forebrain induces behavioral deficits and alpha-synuclein pathology. eNeuro 4:e0004.
Xu, C. Y., Kang, W. Y., Chen, Y. M., Jiang, T. F., Zhang, J., Zhang, L. N., et al. (2017). DJ-1 inhibits alpha-synuclein aggregation by regulating chaperone-mediated autophagy. Front. Aging Neurosci. 9:308. doi: 10.3389/fnagi.2017.00308
Zambon, F., Cherubini, M., Fernandes, H. J. R., Lang, C., Ryan, B. J., Volpato, V., et al. (2019). Cellular alpha-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 28, 2001–2013. doi: 10.1093/hmg/ddz038
Zhao, T., Severijnen, L. A., Van Der Weiden, M., Zheng, P. P., Oostra, B. A., Hukema, R. K., et al. (2013). FBXO7 immunoreactivity in alpha-synuclein-containing inclusions in Parkinson disease and multiple system atrophy. J. Neuropathol. Exp. Neurol. 72, 482–488. doi: 10.1097/nen.0b013e318293c586
Zhou, Z. D., Sathiyamoorthy, S., Angeles, D. C., and Tan, E. K. (2016). Linking F-box protein 7 and parkin to neuronal degeneration in Parkinson’s disease (PD). Mol. Brain 9:41.
Zhou, Z. D., Xie, S. P., Sathiyamoorthy, S., Saw, W. T., Sing, T. Y., Ng, S. H., et al. (2015). F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum. Mol. Genet. 24, 6314–6330. doi: 10.1093/hmg/ddv340
Keywords: Parkinson’s disease, mitochondria, mitophagy, α-synuclein pathology, PARK genes
Citation: Li W, Fu Y, Halliday GM and Sue CM (2021) PARK Genes Link Mitochondrial Dysfunction and Alpha-Synuclein Pathology in Sporadic Parkinson’s Disease. Front. Cell Dev. Biol. 9:612476. doi: 10.3389/fcell.2021.612476
Received: 30 September 2020; Accepted: 10 June 2021;
Published: 06 July 2021.
Edited by:
Kiyoung Kim, Soonchunhyang University, South KoreaReviewed by:
Keiichi Inoue, Niigata University, JapanTongzheng Liu, Jinan University, China
Kenya Nishioka, Juntendo University, Japan
Seon-Heui Cha, Hanseo University, South Korea
Copyright © 2021 Li, Fu, Halliday and Sue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolyn M. Sue, Y2Fyb2x5bi5zdWVAc3lkbmV5LmVkdS5hdQ==