 Kihwan Lee
Kihwan Lee Youn Yi Jo2†
Youn Yi Jo2† Yong Ho Kim
Yong Ho Kim Chul-Kyu Park
Chul-Kyu Park- 1Gachon Pain Center and Department of Physiology, Gachon University College of Medicine, Incheon, South Korea
- 2Department of Anesthesiology and Pain Medicine, Gil Medical Center, Gachon University, Incheon, South Korea
- 3Department of Oral Physiology and Program in Neurobiology, School of Dentistry, Seoul National University, Seoul, South Korea
- 4Program in Neurosciences and Mental Health, The Hospital for Sick Children, Toronto, ON, Canada
Transient receptor potential (TRP) channels are transmembrane protein complexes that play important roles in the physiology and pathophysiology of both the central nervous system (CNS) and the peripheral nerve system (PNS). TRP channels function as non-selective cation channels that are activated by several chemical, mechanical, and thermal stimuli as well as by pH, osmolarity, and several endogenous or exogenous ligands, second messengers, and signaling molecules. On the pathophysiological side, these channels have been shown to play essential roles in the reproductive system, kidney, pancreas, lung, bone, intestine, as well as in neuropathic pain in both the CNS and PNS. In this context, TRP channels have been implicated in several neurological disorders, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and epilepsy. Herein, we focus on the latest involvement of TRP channels, with a special emphasis on the recently identified functional roles of TRP channels in neurological disorders related to the disruption in calcium ion homeostasis.
Introduction
TRP Channel Subfamily
Transient receptor potential (TRP) channels are classified into 28 members that function as a group of unique non-selective cation channels in mammals. TRP channels are conserved in yeast, invertebrates, and vertebrates and share a common three-dimensional structure with six transmembrane helical segments (S1–S6), two variable and intracellular amino (-NH2) and a carboxy (-COOH) terminal cytosolic domain, and the channel pore formed by S5 and S6, which allow transport of various ions including sodium (Na+), potassium (K+), calcium (Ca2+), and magnesium ions (Mg2+). Based on significant sequence homology and a common structure, TRP channels are divided into six subfamilies: TRPC (canonical), TRPM (melastatin), TRPV (vanilloid), TRPA (ankyrin), TRPP (polycystin), and TRPML (mucolipin). TRP subfamilies are divided into Group 1 (TRPC, TRPM, TRPV, and TRPA) and Group 2 (TRPP and TRPML) according to differences in their sequence and topology. Subfamilies of TRP channels are divided into groups and subtypes as represented in the phylogenetic tree in Supplementary Figure 1.
Transient receptor potential channels are ubiquitously expressed in many cell types (especially neurons and non-neuronal cells in the central nervous system, CNS) and tissues, including brain, kidney, pancreas, lung, bone, intestine, reproductive system as well as dorsal root ganglia (DRG) neurons in the peripheral nervous system (PNS). In addition, TRP channels are primarily expressed in plasma membranes that play critical roles in stimulus perception (i.e., thermosensation, mechanosensation, and chemosensation) and ion homeostasis (Nishida et al., 2006; Nilius and Owsianik, 2011).
Initially, TRP channels were shown to regulate cellular Ca2+ influx through the so-called store-operated channels (Nilius, 2004; Ramsey et al., 2006; Yazgan and Naziroglu, 2017). Several studies have shown that TRP channels regulate neuronal excitability, intracellular Ca2+ and Mg2+ homeostasis, as well as cell proliferation and differentiation (Nilius, 2004).
In addition to their physiological functions, TRP channels are known to contribute to various pathophysiological roles in neurological disorders of the CNS (Nilius, 2007; Colsoul et al., 2013; Takada et al., 2013).
Neurological Disorders
Neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) and epilepsy, collectively known as “neurological disorders,” have distinct pathologies and represent a significant medical burden in the modern world.
Alzheimer’s disease is the most common neurodegenerative disease in the world and is characterized by the accumulation of beta-amyloid (Aβ) plaques from amyloid precursor protein (APP) and hyperphosphorylated tau protein (Iqbal et al., 2010; Murphy and Levine, 2010). PD is also a common brain disorder, primarily characterized by a resting tremor, postural instability, rigidity, and bradykinesia caused by dopaminergic (DA) neuronal loss in the substantia nigra (SN) pars compacta (SNpc) (Michel et al., 2013; Kalia and Lang, 2015). HD is an inherited neurodegenerative disorder that causes cognitive deficits, emotional imbalance, and uncontrolled excessive motor movements caused by a CAG trinucleotide repeat expansion within the Huntingtin gene that leads to the synthesis of polyglutamine tracts (Kremer et al., 1994). ALS, also known as Lou Gehrig’s disease, is another fatal type of neurodegenerative motor disease characterized by the deterioration of motor neurons in the motor cortex, brainstem, and spinal cord that leads to impairments in voluntary movement (Guatteo et al., 2007). Finally, epilepsy is a neurological disorder characterized by recurrent epileptic seizures, abnormal brain activity, and unusual behavior.
Over the past few decades, enormous efforts have been made to unveil the pathogenesis of neurological disorders. For example, endoplasmic reticulum (ER) stress, also known as oxidative stress, which is caused by misfolded proteins and abnormal Ca2+ homeostasis, neuroinflammation, and mitochondrial dysfunction have been shown to lead to neuronal cell death. Most notably, Ca2+ regulation, which is involved in normal physiological functions such as neuronal survival, proliferation, differentiation, gene transcription, and exocytosis at synapses, has been shown to be dysregulated in various neurological disorders (Bojarski et al., 2008; Bezprozvanny, 2009; Grosskreutz et al., 2010; Surmeier et al., 2010; Wu et al., 2011; Nikoletopoulou and Tavernarakis, 2012).
Interestingly, several studies have reported a correlation between intracellular Ca2+ concentrations ([Ca2+]i) and other pathogenic mechanisms, including the imbalance between antioxidant function and reactive oxygen species (ROS) production (Gorlach et al., 2015) as well as mitochondrial dysfunction (Contreras et al., 2010; Pivovarova and Andrews, 2010).
In fact, exposure of neuronal cells to Aβ peptides, induces an elevation of [Ca2+]i that leads to cell death as observed in in vitro experiments (Adhya and Sharma, 2019). Aggregation of α-synuclein, which is associated with the pathology of PD, can also induce neuronal cell death via the disruption of cellular Ca2+ homeostasis (Fonfria et al., 2005; Danzer et al., 2007). Furthermore, the polyglutamine-expanded huntingtin protein and mutant superoxide dismutase-1 (SOD1), which are implicated in the pathogenesis of HD and ALS, respectively, also disrupt cellular Ca2+ homeostasis (Giacomello et al., 2013; Barrett et al., 2014). The disruption of intracellular Ca2+ concentration in epilepsy induces ROS production, apoptosis, and caspase activation through mitochondrial oxidative stress (Yilmaz et al., 2011; Naziroglu and Ovey, 2015). Therefore, alleviating disturbances in Ca2+ homeostasis may represent a potential therapeutic target for the treatment of neurological disorders (Nilius, 2007; Colsoul et al., 2013; Takada et al., 2013).
TRP Channels in Neurological Disorders
TRP Channels in AD
Importantly, a strong correlation between the pathological hallmarks of AD (Aβ accumulation and neurofibrillary tangles) and perturbed cellular Ca2+ homeostasis have been reported in AD patients as well as in animal and cell culture models of AD (Mattson and Chan, 2001). To date, TRPC1, TRPC3, TRPC6, TRPM2, TRPM7, TRPV1, TRPV4, TRPA1, and TRPML1 have been shown to be involved in AD (Figure 1A). TRPC1 is a member of the most prevalent TRPC channels in the brain and is linked to the store-operated Ca2+ channel-mediated Ca2+ entry (SOCE) channels. Interestingly, SOCE was reduced by downregulation of TRPC1 in astrocytes in APP knockout (KO) mice (Linde et al., 2011). Additionally, the alteration of the brain-derived neurotrophic factor-tropomyosin receptor kinase B-TRPC3 (BDNF-TrkB-TRPC3) signaling pathway led to hyperphosphorylation of tau protein caused by increased [Ca2+]i levels in AD (Elliott and Ginzburg, 2006). Moreover, it has been reported that TRPC1 and TRPC3 are associated with caveolin-1, which is the main component of the plasma membrane caveolae that interacts with APP (Ikezu et al., 1998). Several studies have reported that TRPC6 in neurons promotes neuronal survival (Jia et al., 2007), synaptogenesis, and learning and memory (Zhou J. et al., 2008). Furthermore, in pharmacological studies using hyperforin, one of the main natural compounds of the medicinal plant Saint John’s wort that acts as an antidepressant drug (Zanoli, 2004) and TRPC6 activator (Tu et al., 2010), or tetrahydrohyperforin, a stable semisynthetic compound derived from hyperforin (Rozio et al., 2005), TRPC6 was shown to play a potential role in AD through the reduction of Aβ accumulation due to increased cerebrovascular P-glycoprotein (Brenn et al., 2014) and increased adult hippocampal neurogenesis and long-term spatial memory (Abbott et al., 2013), respectively. In contrast, AD-linked presenilin (PS)-2 mutants influenced TRPC6-mediated neurotoxic Ca2+ entry (Lessard et al., 2005), whereas TRPC6 was shown to be neuroprotective against AD through interaction with the cleavage of APP (Wang et al., 2015). In fact, a recent study observed that hyperforin induced activation of TRPC6, reduced Aβ levels, and improved mild cognitive impairment in AD models and that TRPC6 mRNA levels in the blood cells were reduced in AD patients (Lu et al., 2018).
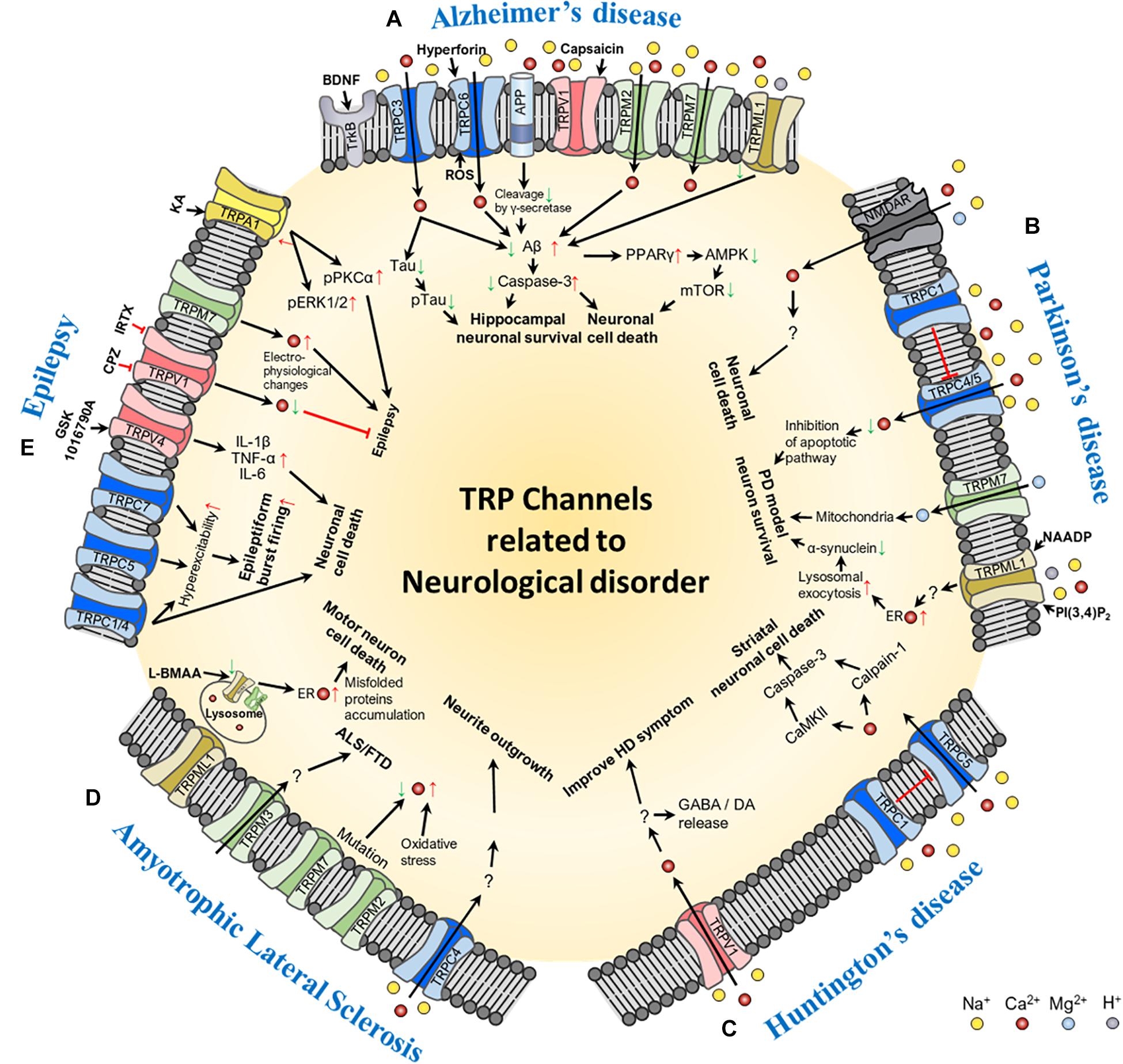
Figure 1. Schematic of the molecular mechanism of TRP channel-mediated pathogenesis of neurological disorders: (A) Alzheimer’s disease, (B) Parkinson’s disease, (C) Huntington’s disease, (D) Amyotrophic lateral sclerosis, and (E) epilepsy. In the figure, red arrows represent increase or up-regulation; green arrows, decrease or down-regulation; red thick bar, inhibition of the signaling pathway; and black arrows, activation of the signaling pathway. Ion channels involved in each of the neurological disorders and their transporting ions, channel agonists, and antagonists are also shown in the figure. Abbreviations: BDNF, brain-derived neurotrophic factor; ROS, reactive oxygen species; Aβ, beta-amyloid; PPAR, peroxisome proliferator-activated receptor gamma; AMPK, 5′ adenosine monophosphate-activated protein kinase; mTOR, mechanistic target of rapamycin; NMDAR, N-methyl-D-aspartate receptor; NAADP, Nicotinic acid adenine dinucleotide phosphate; PI(3,4)P2, phosphatidylinositol (3,4)-bisphosphate; PD, Parkinson’s disease; ER, endoplasmic reticulum; GABA, gamma-aminobutyric acid; DA, dopamine; ALS/FTD, amyotrophic lateral sclerosis patients with frontotemporal dementia; L-BMAA, L-beta-methylamino-L-alanine; IL, interleukin; TNF-α, tumor necrosis factor alpha; CPZ, capsazepine; IRTX, 5′-Iodoresiniferatoxin; KA, kainic acid; pPKCα, phospho-protein kinase C alpha; pERK1/2, phospho-extracellular signal-regulated kinase 1/2.
While APP/PS1 transgenic (Tg) mice were observed to have age-dependent spatial memory deficits through TRPM2 channel activation by toxic oligomeric Aβ, genetic elimination of TRPM2 in APP/PS1 Tg mice ameliorated the synaptic loss and spatial memory deficits (Ostapchenko et al., 2015). It has been suggested that the TRPM2 channel may be a possible therapeutic agent of neuronal toxicity and memory impairment in AD. In contrast, TRPM7, which is another TRPM channel, plays an important role in not only inducing anoxic neuronal cell death by disrupting Ca2+ and Mg2+ homeostasis but also increasing susceptibility to degenerative processes (Aarts et al., 2003).
In recent studies of TRPV channels, the activity of TRPV1 was reported to reduce oxidative/nitrosative stress (Hofrichter et al., 2013; Jayant et al., 2016), rescue Aβ-induced degradation of hippocampal neurons (Balleza-Tapia et al., 2018), and increase levels of presynaptic synapsin I and postsynaptic density 93 (PSD93). In contrast, increased TRPV4 expression has been observed in the brain of aged rats (Lee and Choe, 2014), which is known to increase neuronal cell death via increased Ca2+ influx and ROS production (Hong et al., 2016). In APP/PS1 Tg/TRPA1 KO mice, the genetic loss of TRPA1 has been shown to exacerbate spatial learning and memory deficits, increase Aβ deposition, promote the release of pro-inflammatory cytokines such as interleukin (IL)-1β, IL-4, IL-6, and IL-10, and inhibit the activities of transcriptional factors NF-κB and nuclear factor of activated T cells (Lee et al., 2016).
Lastly, it has been shown that overexpression of TRPML1 is sufficient to rescue memory and cognitive deficits by diminishing neuronal apoptosis in APP/PS1 Tg mice (Zhang et al., 2017).
TRP Channels in PD
TRPC1, TRPC3, TRPM2, TRPM7, and TRPV1 have been shown to be involved in PD (Figure 1B). The activation of TRPC1 channels has been reported to induce neuroprotection against apoptosis in SH-SY5Y neuroblastoma cells (Bollimuntha et al., 2006), as well as to regulate SOCE channels and reduce DA neuronal cell death in the SN of TRPC1 KO mice (Selvaraj et al., 2009, 2012). Consistent with these reports, decreased levels of TRPC1 have been detected in brain lysates from the SNpc of PD patients (Sun et al., 2017). In contrast to TRPC1 levels, TRPC3 levels are not altered in SNpc DA neurons in PD patients (Sun et al., 2017) although their levels are increased by the compensatory effect of decreased TRPC1 in 1-methyl-4-phenyl-1,2,3,6-tetrahyrdropyridine (MPTP)-induced PD-like conditions (Selvaraj et al., 2009). Constitutively active TRPC3 channels are known to express and regulate firing intensity and pattern in GABAergic neurons of the SN pars reticulata (SNpr) (Zhou F. W. et al., 2008). Therefore, this channel may also be involved in different brain subregions or pathophysiological mechanisms of PD.
Additionally, 1-methyl-4-phenylpyridinium ion (MPP+)-induced oxidative stress has been shown to increase intracellular Ca2+ influx via TRPM2 channel activity and promote DA neuronal cell death in the SNpc (Sun et al., 2018). Importantly, TRPM7 channels regulate Mg2+ homeostasis in cells, and increased concentrations of Mg2+ significantly inhibit MPP + -induced neurotoxicity by reducing the number of DA neurons and ameliorating the length of DA neurites (Hashimoto et al., 2008; Paravicini et al., 2012).
The activation of TRPV1 has also been shown to induce cell death in DA neurons, increase Ca2+ influx, and mediate mitochondrial disruption (Kim et al., 2005; Nam et al., 2015).
Furthermore, activation of TRPML1 evokes global ER Ca2+ release and Ca2+ influx (Kilpatrick et al., 2016) and results in upregulated lysosomal exocytosis, thus preventing α-synuclein accumulation in DA neurons (Tsunemi et al., 2019).
TRP Channels in HD
TRPC1, TRPC5, and TRPV1 have been shown to be involved in HD (Figure 1C). Recently, it was shown that the expression level of endogenous TRPC1 was decreased in Q111 HD striatal cells compared to wild-type (Q7) cells (Hong et al., 2015). Furthermore, increased glutathionylation of TRPC5, activated by oxidants, leads to Ca2+-induced apoptosis of the striatal neurons in HD Tg mice (Hong et al., 2015). However, knockdown by siTRPC5 and inhibition of TRPC5 with ML204, a selective TRPC4 blocker produced by the Molecular Libraries Probe, produces a protective effect against oxidative stress in Q111 HD striatal cells and improves motor behavior in HD Tg mice (Miller et al., 2011; Hong et al., 2015, 2020a).
Moreover, it has been shown that administration of N-arachidonoylphenolamine (AM404), an inhibitor of endocannabinoid reuptake, has potential antihyperkinetic effects via the TRPV1 receptor using the 3-nitropropionic acid-induced HD model, suggesting that the activity of the TRPV1 channel may contribute to the motor dysfunction in HD patients (Lastres-Becker et al., 2003).
TRP Channels in ALS
TRPC4, TRPM2, TRPM3, TRPM7, and TRPML1 have been shown to be involved in ALS (Figure 1D). The clinical symptoms of ALS overlap with those of Parkinsonism dementia complex (PDC), a neurodegenerative disorder characterized by symptoms of PD and dementia (Garruto, 2006; Hermosura and Garruto, 2007). While the pathogenesis of ALS/PDC is not fully understood, it is thought to be caused by two potential scenarios: (i) low levels of Ca2+ and Mg2+, which cause excess ROS production and cell death, and (ii) excess exposure to putative neurotoxin β-methylamino-L-alanine (L-BMAA), derived from the cycad plant, which causes an increase of [Ca2+]i (Brownson et al., 2002).
A previous study demonstrated that the expression of TRPC4 is increased by nerve growth factor and dibutyryl-cAMP treatment in cultured DRG neurons. Conversely, inhibition of TRPC4 using a selective siRNA approach reduces neurite outgrowth in cultured DRG neurons (Wu et al., 2008). In ALS, reactive astrocytes accelerate nerve growth factor production (Pehar et al., 2004). In addition, a recent study demonstrated that ALS-resistant motor neurons from mutant SOD1 ALS models upregulate axonal outgrowth and dendritic branching (Osking et al., 2019). Taken together, these findings suggest that TRPC4 regulates DRG differentiation and plays a pivotal role in ALS.
In contrast, mutations in both TRPM2P1018L and TRPM7T1482I have been found in Guamanian ALS/PDC patients. Importantly, the TRPM2P1018L variant was shown to attenuate oxidative stress-induced Ca2+ influx through inactivation of the channel (Hermosura et al., 2008). In contrast, the TRPM7T1482I variant promotes an imbalance in Ca2+ and Mg2+ homeostasis (Hermosura and Garruto, 2007). TRPM3, which is in the same group as TRPM2, has also been considered as a possible candidate gene involved in the pathogenesis of ALS with frontotemporal dementia (Lee et al., 2003).
TRPML1 was expressed in cell lysosomes or in the endosomal membrane and in the main Ca2+-releasing channel used as a regulator of lysosomal storage via phosphatidylinositol 3,5-biphosphate (PI(3,5)P2) (Cheng et al., 2010; Li et al., 2013). In addition, TRPML1 also regulates the maintenance of lysosome homeostasis and accumulation of autophagy (Cheng et al., 2010; Curcio-Morelli et al., 2010; Morgan et al., 2011). Interestingly, PI(3,5)P2 levels are significantly impaired in some forms of ALS (Chow et al., 2009; Osmanovic et al., 2017). Moreover, in an experimental model of ALS/PDC, the expression of TRPML1 was shown to be reduced through autophagy leading to the loss of motor neurons. Additionally, with an L-BMAA-induced ALS mouse model, it was discovered that TRPML1 is downregulated, and autophagy is impaired in primary motor neurons, which leads to ER stress and neuronal cell death (Tedeschi et al., 2019).
TRP Channels in Epilepsy
TRPC1, TRPC3-7, TRPM2, TRPM7, TRPV1, TRPV4, and TRPA1 have been shown to be involved in epilepsy (Figure 1E). As mentioned above, TRPC channels are not only known to play an important role in neuronal outgrowth and survival during brain development but are also believed to play a pivotal role in various epileptogenic processes. For example, the expression of TRPC1 is increased in cortical lesions of epilepsy patients and regulated by the mediation of astrocyte-induced epilepsy (Zang et al., 2015). Using a pilocarpine (muscarinic agonist)-induced status epilepticus (PISE) model, studies have shown that the genetic elimination of TRPC3 reduces the susceptibility of seizures to pilocarpine, while enhancing the expression of TRPC3 induces hyperexcitability and increases susceptibility to epileptiform activity in the cortex (Zhou and Roper, 2014; Phelan et al., 2017). In contrast, the expression of TRPC6 has been shown to be down-regulated in chronic epileptic rats, whereas the genetic ablation by siTRPC6 increases seizure susceptibility and seizure-induced neuronal damage in the dentate gyrus but not in CA1 and CA3 neurons of the hippocampus (Kim and Kang, 2015). In addition, in other members of the TRPC family, the genetic deletion of TRPC1/4 reduces seizure-induced neuronal cell death. Furthermore, TRPC5 KO mice exhibit significantly reduced seizures as well as minimal seizure-induced neuronal cell death in the CA1 and CA3 areas of the hippocampus (Phelan et al., 2013). Conversely, the TRPC7 channel plays an important role in spontaneous epileptiform bursting in the CA3, the reduction of which is correlated with a reduction in PISE in TRPC7 KO mice (Phelan et al., 2014).
The TRPM family is also involved in the pathogenesis of epilepsy. For example, TRPM2 channels are co-expressed with the EF-hand domain-containing protein 1 gene, which is related to an increased susceptibility to juvenile myoclonic epilepsy, and are regulated in the hippocampal neurons (Katano et al., 2012). Moreover, TRPM7 has been shown to be activated during epilepsy (Aarts and Tymianski, 2005). Importantly, genetic ablation of TRPM7 blocks the activation of a cation current, which is produced by oxygen-glucose deprivation (IOGD), and prevents ROS-mediated IOGD activation (Aarts et al., 2003).
Although the TRPV1 channel is believed to play an essential role in the development of neurogenic pain and inflammation in the sensory neurons (Caterina and Julius, 2001; Julius and Basbaum, 2001), it is also expressed in other brain regions, including the cerebral cortex, hippocampus, cerebellum, thalamus, hypothalamus, striatum, midbrain, and amygdala (Cristino et al., 2006). Furthermore, increased expression of TRPV1 has been found in the hippocampus of rats and the dentate gyrus of mice with temporal lobe epilepsy as well as in the cortex of patients with temporal lobe epilepsy (Bhaskaran and Smith, 2010; Sun et al., 2013; Saffarzadeh et al., 2015). In fact, a recent study suggested that the activation of the TRPV1 channel may play a key role in the development of epilepsy (Naziroglu and Ovey, 2015). Using capsazepine, 5′-iodoresiniferatoxin, and resolvins, the authors showed that inhibition of the TRPV1 channel induced protective effects against epilepsy and epilepsy-induced Ca2+ entry in the hippocampal and DRG neurons (Naziroglu and Ovey, 2015).
Activation of TRPV3 by eugenol was shown to suppress epileptiform field potentials and decrease the amplitude of field postsynaptic potentials evoked in CA1 neurons of the hippocampus and the third layer of the neocortex (Muller et al., 2006). Another study using the PISE model of epilepsy found that activation of TRPV4 by the specific agonist GSK1016790A increased pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6), while the inhibition of TRPV4 by HC-067047, a selective TRPV4 antagonist, significantly increased cell survival post status epilepticus (Wang et al., 2019).
Using a kainic acid-induced seizure model, TRPA1 was found to be upregulated while TRPV4 was not, which is contradictory to the earlier findings concerning TRPV4 (Hunt et al., 2012; Wang et al., 2019).
Conclusion and Future Perspectives
In this review, we described the functional importance of TRP channels in the regulation of Ca2+ and oxidative stress responses as well as their contributions to neurological disorders, including AD, PD, HD, ALS, and epilepsy.
Overall, in addition to playing a broad range of physiological roles throughout the CNS and PNS, TRP channels also contribute to pathophysiology across a wide range of diseases and disorders through abnormalities in Ca2+ homeostasis. In the CNS, TRP channels are expressed in several brain regions (including the spinal cord) and have been shown to be key regulatory proteins involved in lipid metabolism, glucose homeostasis (Liu et al., 2009; Zhu et al., 2011), and the pathobiology of aforementioned neurological disorders.
Apart from their important role in neurological disorders of the CNS, TRP channels are also expressed in the neurons of the DRG, trigeminal ganglion, and sympathetic ganglion, and contribute to both normal and pathological sensory processing in the PNS (Lee et al., 2019). For example, TRP channels are known to be involved in diabetic peripheral neuropathy, chemotherapy-induced peripheral neuropathy, and autonomic neuropathy.
In light of the physiological and pathophysiological functions of TRP channels in both the CNS and PNS, we believe that they represent potential therapeutic targets for treating neurological disorders of the CNS as well as neuropathic pain in the PNS.
Author Contributions
KL, YYJ, YHK, and C-KP contributed to the conception and design. KL and YYJ drafted the manuscript. KL, GC, JHJ, YHK, and C-KP revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Research Foundation of Korea (NRF-2020R1I1A1A01068037 for KL and NRF-2020R1A2C1008084 for YYJ), which is funded by the Korean Government.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.611773/full#supplementary-material
Supplementary Figure 1 | Summary of TRP subfamilies-mediated studies related to neurological disorders [modified from Nilius (2007), Hong et al. (2020b), Thapak et al. (2020)]. Different colors are used in the table for better classification and the gray boxes in the “Related Disease” column represent the roles of specific TRP subfamilies in each neurological disorders as reported in the published literature. Abbreviations: AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; AMG, amygdala; APP, amyloid precursor protein; BBB, blood-brain barrier; BG, basal ganglia; Bs, brainstem; CB, cerebellum; CC, corpus callosum; Cd, caudate; Cg, cingulate gyrus; CNS, central nervous system; CTX, cortex; DA, dopamine; DG, dentate gyrus; DRG, dorsal root ganglion; ER, endoplasmic reticulum; FB, forebrain; fCTX, frontal cortex; GABA, gamma-aminobutyric acid; GP, globus pallidus; HD, Huntington’s disease; HPC, hippocampus; HT, hypothalamus; IC, inferior colliculus; JME, juvenile myoclonic epilepsy; KA, kainic acid; LS, lateral septum; MB, midbrain; mFBb, medial forebrain bundle; mPFC, medial prefrontal cortex; mTOR, mechanistic target of rapamycin; n.d., not determined; OB, olfactory bulb; PD, Parkinson’s disease; PISE, pilocarpine-induced status epilepticus; PNS, peripheral nervous system; Pm, putamen; PS1, presenilin 1; RCh, retrochiasmatic area; SC, spinal cord; Sep, septum; SN, substantia nigra; SNpc, substantia nigra pars compacta; SNpr, substantia nigra pars reticulata; SOCE, store-operated calcium entry; Str, striatum; TH, thalamus; vS, ventral subiculum.
References
Aarts, M., Iihara, K., Wei, W. L., Xiong, Z. G., Arundine, M., Cerwinski, W., et al. (2003). A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877. doi: 10.1016/s0092-8674(03)01017-1
Aarts, M. M., and Tymianski, M. (2005). TRsPMs and neuronal cell death. Pflugers Arch. 451, 243–249.
Abbott, A. C., Calderon Toledo, C., Aranguiz, F. C., Inestrosa, N. C., and Varela-Nallar, L. (2013). Tetrahydrohyperforin increases adult hippocampal neurogenesis in wild-type and APPswe/PS1DeltaE9 mice. J. Alzheimers Dis. 34, 873–885. doi: 10.3233/jad-121714
Adhya, P., and Sharma, S. S. (2019). Redox TRPs in diabetes and diabetic complications: mechanisms and pharmacological modulation. Pharmacol. Res. 146:104271. doi: 10.1016/j.phrs.2019.104271
Balleza-Tapia, H., Crux, S., Andrade-Talavera, Y., Dolz-Gaiton, P., Papadia, D., Chen, G., et al. (2018). TrpV1 receptor activation rescues neuronal function and network gamma oscillations from Abeta-induced impairment in mouse hippocampus in vitro. Elife 7:e37703.
Barrett, E. F., Barrett, J. N., and David, G. (2014). Dysfunctional mitochondrial Ca(2+) handling in mutant SOD1 mouse models of fALS: integration of findings from motor neuron somata and motor terminals. Front. Cell. Neurosci. 8:184.
Bezprozvanny, I. (2009). Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 15, 89–100. doi: 10.1016/j.molmed.2009.01.001
Bhaskaran, M. D., and Smith, B. N. (2010). Effects of TRPV1 activation on synaptic excitation in the dentate gyrus of a mouse model of temporal lobe epilepsy. Exp. Neurol. 223, 529–536. doi: 10.1016/j.expneurol.2010.01.021
Bojarski, L., Herms, J., and Kuznicki, J. (2008). Calcium dysregulation in Alzheimer’s disease. Neurochem. Int. 52, 621–633.
Bollimuntha, S., Ebadi, M., and Singh, B. B. (2006). TRPC1 protects human SH-SY5Y cells against salsolinol-induced cytotoxicity by inhibiting apoptosis. Brain Res. 1099, 141–149. doi: 10.1016/j.brainres.2006.04.104
Brenn, A., Grube, M., Jedlitschky, G., Fischer, A., Strohmeier, B., Eiden, M., et al. (2014). St. John’s Wort reduces beta-amyloid accumulation in a double transgenic Alzheimer’s disease mouse model-role of P-glycoprotein. Brain Pathol. 24, 18–24. doi: 10.1111/bpa.12069
Brownson, D. M., Mabry, T. J., and Leslie, S. W. (2002). The cycad neurotoxic amino acid, beta-N-methylamino-L-alanine (BMAA), elevates intracellular calcium levels in dissociated rat brain cells. J. Ethnopharmacol. 82, 159–167. doi: 10.1016/s0378-8741(02)00170-8
Caterina, M. J., and Julius, D. (2001). The vanilloid receptor: a molecular gateway to the pain pathway. Annu. Rev. Neurosci. 24, 487–517. doi: 10.1146/annurev.neuro.24.1.487
Cheng, X., Shen, D., Samie, M., and Xu, H. (2010). Mucolipins: intracellular TRPML1-3 channels. FEBS Lett. 584, 2013–2021. doi: 10.1016/j.febslet.2009.12.056
Chow, C. Y., Landers, J. E., Bergren, S. K., Sapp, P. C., Grant, A. E., Jones, J. M., et al. (2009). Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am. J. Hum. Genet. 84, 85–88. doi: 10.1016/j.ajhg.2008.12.010
Colsoul, B., Nilius, B., and Vennekens, R. (2013). Transient receptor potential (TRP) cation channels in diabetes. Curr. Top. Med. Chem. 13, 258–269. doi: 10.2174/1568026611313030004
Contreras, L., Drago, I., Zampese, E., and Pozzan, T. (2010). Mitochondria: the calcium connection. Biochim. Biophys. Acta 1797, 607–618. doi: 10.1016/j.bbabio.2010.05.005
Cristino, L., De Petrocellis, L., Pryce, G., Baker, D., Guglielmotti, V., and Di Marzo, V. (2006). Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 139, 1405–1415. doi: 10.1016/j.neuroscience.2006.02.074
Curcio-Morelli, C., Charles, F. A., Micsenyi, M. C., Cao, Y., Venugopal, B., Browning, M. F., et al. (2010). Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol. Dis. 40, 370–377. doi: 10.1016/j.nbd.2010.06.010
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 27, 9220–9232.
Elliott, E., and Ginzburg, I. (2006). The role of neurotrophins and insulin on tau pathology in Alzheimer’s disease. Rev. Neurosci. 17, 635–642.
Fonfria, E., Marshall, I. C., Boyfield, I., Skaper, S. D., Hughes, J. P., Owen, D. E., et al. (2005). Amyloid beta-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J. Neurochem. 95, 715–723. doi: 10.1111/j.1471-4159.2005.03396.x
Garruto, R. M. (2006). A commentary on neuronal degeneration and cell death in Guam ALS and PD: an evolutionary process of understanding. Curr. Alzheimer Res. 3, 397–401. doi: 10.2174/156720506778249425
Giacomello, M., Oliveros, J. C., Naranjo, J. R., and Carafoli, E. (2013). Neuronal Ca(2+) dyshomeostasis in Huntington disease. Prion 7, 76–84. doi: 10.4161/pri.23581
Gorlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271. doi: 10.1016/j.redox.2015.08.010
Grosskreutz, J., Van Den Bosch, L., and Keller, B. U. (2010). Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47, 165–174. doi: 10.1016/j.ceca.2009.12.002
Guatteo, E., Carunchio, I., Pieri, M., Albo, F., Canu, N., Mercuri, N. B., et al. (2007). Altered calcium homeostasis in motor neurons following AMPA receptor but not voltage-dependent calcium channels’ activation in a genetic model of amyotrophic lateral sclerosis. Neurobiol. Dis. 28, 90–100. doi: 10.1016/j.nbd.2007.07.002
Hashimoto, T., Nishi, K., Nagasao, J., Tsuji, S., and Oyanagi, K. (2008). Magnesium exerts both preventive and ameliorating effects in an in vitro rat Parkinson disease model involving 1-methyl-4-phenylpyridinium (MPP+) toxicity in dopaminergic neurons. Brain Res. 1197, 143–151. doi: 10.1016/j.brainres.2007.12.033
Hermosura, M. C., Cui, A. M., Go, R. C., Davenport, B., Shetler, C. M., Heizer, J. W., et al. (2008). Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc. Natl. Acad. Sci. U.S.A. 105, 18029–18034. doi: 10.1073/pnas.0808218105
Hermosura, M. C., and Garruto, R. M. (2007). TRPM7 and TRPM2-Candidate susceptibility genes for Western Pacific ALS and PD? Biochim. Biophys. Acta 1772, 822–835. doi: 10.1016/j.bbadis.2007.02.008
Hofrichter, J., Krohn, M., Schumacher, T., Lange, C., Feistel, B., Walbroel, B., et al. (2013). Reduced Alzheimer’s disease pathology by St. John’s Wort treatment is independent of hyperforin and facilitated by ABCC1 and microglia activation in mice. Curr. Alzheimer Res. 10, 1057–1069. doi: 10.2174/15672050113106660171
Hong, C., Choi, S. H., Kwak, M., Jeong, B., Ko, J., Park, H. J., et al. (2020a). TRPC5 channel instability induced by depalmitoylation protects striatal neurons against oxidative stress in Huntington’s disease. Biochim. Biophys. Acta Mol. Cell Res. 1867:118620. doi: 10.1016/j.bbamcr.2019.118620
Hong, C., Jeong, B., Park, H. J., Chung, J. Y., Lee, J. E., Kim, J., et al. (2020b). TRP channels as emerging therapeutic targets for neurodegenerative diseases. Front. Physiol. 11:238. doi: 10.3389/fphys.2020.00238
Hong, C., Seo, H., Kwak, M., Jeon, J., Jang, J., Jeong, E. M., et al. (2015). Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington’s disease. Brain 138, 3030–3047. doi: 10.1093/brain/awv188
Hong, Z., Tian, Y., Yuan, Y., Qi, M., Li, Y., Du, Y., et al. (2016). Enhanced oxidative stress is responsible for TRPV4-induced neurotoxicity. Front. Cell. Neurosci. 10:232. doi: 10.3389/fncel.2016.00232
Hunt, R. F., Hortopan, G. A., Gillespie, A., and Baraban, S. C. (2012). A novel zebrafish model of hyperthermia-induced seizures reveals a role for TRPV4 channels and NMDA-type glutamate receptors. Exp. Neurol. 237, 199–206. doi: 10.1016/j.expneurol.2012.06.013
Ikezu, T., Trapp, B. D., Song, K. S., Schlegel, A., Lisanti, M. P., and Okamoto, T. (1998). Caveolae, plasma membrane microdomains for alpha-secretase-mediated processing of the amyloid precursor protein. J. Biol. Chem. 273, 10485–10495. doi: 10.1074/jbc.273.17.10485
Iqbal, K., Liu, F., Gong, C. X., and Grundke-Iqbal, I. (2010). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664. doi: 10.2174/156720510793611592
Jayant, S., Sharma, B. M., and Sharma, B. (2016). Protective effect of transient receptor potential vanilloid subtype 1 (TRPV1) modulator, against behavioral, biochemical and structural damage in experimental models of Alzheimer’s disease. Brain Res. 1642, 397–408. doi: 10.1016/j.brainres.2016.04.022
Jia, Y., Zhou, J., Tai, Y., and Wang, Y. (2007). TRPC channels promote cerebellar granule neuron survival. Nat. Neurosci. 10, 559–567. doi: 10.1038/nn1870
Julius, D., and Basbaum, A. I. (2001). Molecular mechanisms of nociception. Nature 413, 203–210. doi: 10.1038/35093019
Katano, M., Numata, T., Aguan, K., Hara, Y., Kiyonaka, S., Yamamoto, S., et al. (2012). The juvenile myoclonic epilepsy-related protein EFHC1 interacts with the redox-sensitive TRPM2 channel linked to cell death. Cell Calcium 51, 179–185. doi: 10.1016/j.ceca.2011.12.011
Kilpatrick, B. S., Yates, E., Grimm, C., Schapira, A. H., and Patel, S. (2016). Endo-lysosomal TRP mucolipin-1 channels trigger global ER Ca2+ release and Ca2+ influx. J. Cell Sci. 129, 3859–3867. doi: 10.1242/jcs.190322
Kim, S. R., Lee, D. Y., Chung, E. S., Oh, U. T., Kim, S. U., and Jin, B. K. (2005). Transient receptor potential vanilloid subtype 1 mediates cell death of mesencephalic dopaminergic neurons in vivo and in vitro. J. Neurosci. 25, 662–671. doi: 10.1523/jneurosci.4166-04.2005
Kim, Y. J., and Kang, T. C. (2015). The role of TRPC6 in seizure susceptibility and seizure-related neuronal damage in the rat dentate gyrus. Neuroscience 307, 215–230. doi: 10.1016/j.neuroscience.2015.08.054
Kremer, B., Goldberg, P., Andrew, S. E., Theilmann, J., Telenius, H., Zeisler, J., et al. (1994). A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 330, 1401–1406. doi: 10.1056/nejm199405193302001
Lastres-Becker, I., De Miguel, R., De Petrocellis, L., Makriyannis, A., Di Marzo, V., and Fernandez-Ruiz, J. (2003). Compounds acting at the endocannabinoid and/or endovanilloid systems reduce hyperkinesia in a rat model of Huntington’s disease. J. Neurochem. 84, 1097–1109. doi: 10.1046/j.1471-4159.2003.01595.x
Lee, J. C., and Choe, S. Y. (2014). Age-related changes in the distribution of transient receptor potential vanilloid 4 channel (TRPV4) in the central nervous system of rats. J. Mol. Histol. 45, 497–505. doi: 10.1007/s10735-014-9578-z
Lee, K., Lee, B. M., Park, C. K., Kim, Y. H., and Chung, G. (2019). Ion channels involved in tooth pain. Int. J. Mol. Sci. 20:2266. doi: 10.3390/ijms20092266
Lee, K. I., Lee, H. T., Lin, H. C., Tsay, H. J., Tsai, F. C., Shyue, S. K., et al. (2016). Role of transient receptor potential ankyrin 1 channels in Alzheimer’s disease. J. Neuroinflammation 13:92.
Lee, N., Chen, J., Sun, L., Wu, S., Gray, K. R., Rich, A., et al. (2003). Expression and characterization of human transient receptor potential melastatin 3 (hTRPM3). J. Biol. Chem. 278, 20890–20897. doi: 10.1074/jbc.m211232200
Lessard, C. B., Lussier, M. P., Cayouette, S., Bourque, G., and Boulay, G. (2005). The overexpression of presenilin2 and Alzheimer’s-disease-linked presenilin2 variants influences TRPC6-enhanced Ca2+ entry into HEK293 cells. Cell. Signal. 17, 437–445. doi: 10.1016/j.cellsig.2004.09.005
Li, X., Garrity, A. G., and Xu, H. (2013). Regulation of membrane trafficking by signalling on endosomal and lysosomal membranes. J. Physiol. 591, 4389–4401. doi: 10.1113/jphysiol.2013.258301
Linde, C. I., Baryshnikov, S. G., Mazzocco-Spezzia, A., and Golovina, V. A. (2011). Dysregulation of Ca2+ signaling in astrocytes from mice lacking amyloid precursor protein. Am. J. Physiol. Cell Physiol. 300, C1502–C1512.
Liu, H., Dear, A. E., Knudsen, L. B., and Simpson, R. W. (2009). A long-acting glucagon-like peptide-1 analogue attenuates induction of plasminogen activator inhibitor type-1 and vascular adhesion molecules. J. Endocrinol. 201, 59–66. doi: 10.1677/joe-08-0468
Lu, R., Wang, J., Tao, R., Wang, J., Zhu, T., Guo, W., et al. (2018). Reduced TRPC6 mRNA levels in the blood cells of patients with Alzheimer’s disease and mild cognitive impairment. Mol. Psychiatry 23, 767–776. doi: 10.1038/mp.2017.136
Mattson, M. P., and Chan, S. L. (2001). Dysregulation of cellular calcium homeostasis in Alzheimer’s disease: bad genes and bad habits. J. Mol. Neurosci. 17, 205–224. doi: 10.1385/jmn:17:2:205
Michel, P. P., Toulorge, D., Guerreiro, S., and Hirsch, E. C. (2013). Specific needs of dopamine neurons for stimulation in order to survive: implication for Parkinson disease. FASEB J. 27, 3414–3423. doi: 10.1096/fj.12-220418
Miller, M., Shi, J., Zhu, Y., Kustov, M., Tian, J. B., Stevens, A., et al. (2011). Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J. Biol. Chem. 286, 33436–33446. doi: 10.1074/jbc.m111.274167
Morgan, A. J., Platt, F. M., Lloyd-Evans, E., and Galione, A. (2011). Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 439, 349–374. doi: 10.1042/bj20110949
Muller, M., Pape, H. C., Speckmann, E. J., and Gorji, A. (2006). Effect of eugenol on spreading depression and epileptiform discharges in rat neocortical and hippocampal tissues. Neuroscience 140, 743–751. doi: 10.1016/j.neuroscience.2006.02.036
Murphy, M. P., and Levine, H. III (2010). Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 19, 311–323.
Nam, J. H., Park, E. S., Won, S. Y., Lee, Y. A., Kim, K. I., Jeong, J. Y., et al. (2015). TRPV1 on astrocytes rescues nigral dopamine neurons in Parkinson’s disease via CNTF. Brain 138, 3610–3622. doi: 10.1093/brain/awv297
Naziroglu, M., and Ovey, I. S. (2015). Involvement of apoptosis and calcium accumulation through TRPV1 channels in neurobiology of epilepsy. Neuroscience 293, 55–66. doi: 10.1016/j.neuroscience.2015.02.041
Nikoletopoulou, V., and Tavernarakis, N. (2012). Calcium homeostasis in aging neurons. Front. Genet. 3:200. doi: 10.3389/fgene.2012.00200
Nilius, B., and Owsianik, G. (2011). The transient receptor potential family of ion channels. Genome Biol. 12:218. doi: 10.1186/gb-2011-12-3-218
Nishida, M., Hara, Y., Yoshida, T., Inoue, R., and Mori, Y. (2006). TRP channels: molecular diversity and physiological function. Microcirculation 13, 535–550. doi: 10.1080/10739680600885111
Osking, Z., Ayers, J. I., Hildebrandt, R., Skruber, K., Brown, H., Ryu, D., et al. (2019). ALS-Linked SOD1 mutants enhance neurite outgrowth and branching in adult motor neurons. iScience 19, 448–449. doi: 10.1016/j.isci.2019.08.004
Osmanovic, A., Rangnau, I., Kosfeld, A., Abdulla, S., Janssen, C., Auber, B., et al. (2017). FIG4 variants in central European patients with amyotrophic lateral sclerosis: a whole-exome and targeted sequencing study. Eur. J. Hum. Genet. 25, 324–331. doi: 10.1038/ejhg.2016.186
Ostapchenko, V. G., Chen, M., Guzman, M. S., Xie, Y. F., Lavine, N., Fan, J., et al. (2015). The transient receptor potential melastatin 2 (TRPM2) channel contributes to beta-amyloid oligomer-related neurotoxicity and memory impairment. J. Neurosci. 35, 15157–15169. doi: 10.1523/jneurosci.4081-14.2015
Paravicini, T. M., Chubanov, V., and Gudermann, T. (2012). TRPM7: a unique channel involved in magnesium homeostasis. Int. J. Biochem. Cell Biol. 44, 1381–1384. doi: 10.1016/j.biocel.2012.05.010
Pehar, M., Cassina, P., Vargas, M. R., Castellanos, R., Viera, L., Beckman, J. S., et al. (2004). Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J. Neurochem. 89, 464–473. doi: 10.1111/j.1471-4159.2004.02357.x
Phelan, K. D., Shwe, U. T., Abramowitz, J., Birnbaumer, L., and Zheng, F. (2014). Critical role of canonical transient receptor potential channel 7 in initiation of seizures. Proc. Natl. Acad. Sci. U.S.A. 111, 11533–11538. doi: 10.1073/pnas.1411442111
Phelan, K. D., Shwe, U. T., Abramowitz, J., Wu, H., Rhee, S. W., Howell, M. D., et al. (2013). Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol. Pharmacol. 83, 429–438. doi: 10.1124/mol.112.082271
Phelan, K. D., Shwe, U. T., Cozart, M. A., Wu, H., Mock, M. M., Abramowitz, J., et al. (2017). TRPC3 channels play a critical role in the theta component of pilocarpine-induced status epilepticus in mice. Epilepsia 58, 247–254. doi: 10.1111/epi.13648
Pivovarova, N. B., and Andrews, S. B. (2010). Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 277, 3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x
Ramsey, I. S., Delling, M., and Clapham, D. E. (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647.
Rozio, M., Fracasso, C., Riva, A., Morazzoni, P., and Caccia, S. (2005). High-performance liquid chromatography measurement of hyperforin and its reduced derivatives in rodent plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 816, 21–27. doi: 10.1016/j.jchromb.2004.10.015
Saffarzadeh, F., Eslamizade, M. J., Ghadiri, T., Modarres Mousavi, S. M., Hadjighassem, M., and Gorji, A. (2015). Effects of TRPV1 on the hippocampal synaptic plasticity in the epileptic rat brain. Synapse 69, 375–383. doi: 10.1002/syn.21825
Selvaraj, S., Sun, Y., Watt, J. A., Wang, S., Lei, S., Birnbaumer, L., et al. (2012). Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Invest. 122, 1354–1367. doi: 10.1172/jci61332
Selvaraj, S., Watt, J. A., and Singh, B. B. (2009). TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP(+). Cell Calcium 46, 209–218. doi: 10.1016/j.ceca.2009.07.008
Sun, F. J., Guo, W., Zheng, D. H., Zhang, C. Q., Li, S., Liu, S. Y., et al. (2013). Increased expression of TRPV1 in the cortex and hippocampus from patients with mesial temporal lobe epilepsy. J. Mol. Neurosci. 49, 182–193. doi: 10.1007/s12031-012-9878-2
Sun, Y., Sukumaran, P., Selvaraj, S., Cilz, N. I., Schaar, A., Lei, S., et al. (2018). TRPM2 promotes neurotoxin MPP(+)/MPTP-induced cell death. Mol. Neurobiol. 55, 409–420. doi: 10.1007/s12035-016-0338-9
Sun, Y., Zhang, H., Selvaraj, S., Sukumaran, P., Lei, S., Birnbaumer, L., et al. (2017). Inhibition of L-type Ca(2+) channels by TRPC1-STIM1 complex is essential for the protection of dopaminergic neurons. J. Neurosci. 37, 3364–3377. doi: 10.1523/jneurosci.3010-16.2017
Surmeier, D. J., Guzman, J. N., and Sanchez-Padilla, J. (2010). Calcium, cellular aging, and selective neuronal vulnerability in Parkinson’s disease. Cell Calcium 47, 175–182. doi: 10.1016/j.ceca.2009.12.003
Takada, Y., Numata, T., and Mori, Y. (2013). Targeting TRPs in neurodegenerative disorders. Curr. Top. Med. Chem. 13, 322–334. doi: 10.2174/1568026611313030009
Tedeschi, V., Petrozziello, T., Sisalli, M. J., Boscia, F., Canzoniero, L. M. T., and Secondo, A. (2019). The activation of Mucolipin TRP channel 1 (TRPML1) protects motor neurons from L-BMAA neurotoxicity by promoting autophagic clearance. Sci. Rep. 9:10743.
Thapak, P., Vaidya, B., Joshi, H. C., Singh, J. N., and Sharma, S. S. (2020). Therapeutic potential of pharmacological agents targeting TRP channels in CNS disorders. Pharmacol. Res. 159:105026. doi: 10.1016/j.phrs.2020.105026
Tsunemi, T., Perez-Rosello, T., Ishiguro, Y., Yoroisaka, A., Jeon, S., Hamada, K., et al. (2019). Increased lysosomal exocytosis induced by lysosomal Ca(2+) channel agonists protects human dopaminergic neurons from alpha-synuclein toxicity. J. Neurosci. 39, 5760–5772. doi: 10.1523/jneurosci.3085-18.2019
Tu, P., Gibon, J., and Bouron, A. (2010). The TRPC6 channel activator hyperforin induces the release of zinc and calcium from mitochondria. J. Neurochem. 112, 204–213. doi: 10.1111/j.1471-4159.2009.06446.x
Wang, J., Lu, R., Yang, J., Li, H., He, Z., Jing, N., et al. (2015). TRPC6 specifically interacts with APP to inhibit its cleavage by gamma-secretase and reduce Abeta production. Nat. Commun. 6:8876.
Wang, Z., Zhou, L., An, D., Xu, W., Wu, C., Sha, S., et al. (2019). TRPV4-induced inflammatory response is involved in neuronal death in pilocarpine model of temporal lobe epilepsy in mice. Cell Death Dis. 10:491.
Wu, D., Huang, W., Richardson, P. M., Priestley, J. V., and Liu, M. (2008). TRPC4 in rat dorsal root ganglion neurons is increased after nerve injury and is necessary for neurite outgrowth. J. Biol. Chem. 283, 416–426. doi: 10.1074/jbc.m703177200
Wu, J., Shih, H. P., Vigont, V., Hrdlicka, L., Diggins, L., Singh, C., et al. (2011). Neuronal store-operated calcium entry pathway as a novel therapeutic target for Huntington’s disease treatment. Chem. Biol. 18, 777–793. doi: 10.1016/j.chembiol.2011.04.012
Yazgan, Y., and Naziroglu, M. (2017). Ovariectomy-induced mitochondrial oxidative stress, apoptosis, and calcium ion influx through TRPA1, TRPM2, and TRPV1 are prevented by 17beta-estradiol, tamoxifen, and raloxifene in the hippocampus and dorsal root ganglion of rats. Mol. Neurobiol. 54, 7620–7638. doi: 10.1007/s12035-016-0232-5
Yilmaz, M., Naziroglu, M., Kutluhan, S., Yilmaz, N., Yurekli, V. A., and Vural, H. (2011). Topiramate modulates hippocampus NMDA receptors via brain Ca(2+) homeostasis in pentylentetrazol-induced epilepsy of rats. J. Recept. Signal Transduct. Res. 31, 173–179. doi: 10.3109/10799893.2011.555914
Zang, Z., Li, S., Zhang, W., Chen, X., Zheng, D., Shu, H., et al. (2015). Expression patterns of TRPC1 in cortical lesions from patients with focal cortical dysplasia. J. Mol. Neurosci. 57, 265–272. doi: 10.1007/s12031-015-0615-5
Zanoli, P. (2004). Role of hyperforin in the pharmacological activities of St. John’s Wort. CNS Drug Rev. 10, 203–218. doi: 10.1111/j.1527-3458.2004.tb00022.x
Zhang, L., Fang, Y., Cheng, X., Lian, Y., Xu, H., Zeng, Z., et al. (2017). TRPML1 participates in the progression of Alzheimer’s disease by regulating the PPARgamma/AMPK/Mtor signalling pathway. Cell. Physiol. Biochem. 43, 2446–2456. doi: 10.1159/000484449
Zhou, F. W., Matta, S. G., and Zhou, F. M. (2008). Constitutively active TRPC3 channels regulate basal ganglia output neurons. J. Neurosci. 28, 473–482. doi: 10.1523/jneurosci.3978-07.2008
Zhou, F. W., and Roper, S. N. (2014). TRPC3 mediates hyperexcitability and epileptiform activity in immature cortex and experimental cortical dysplasia. J. Neurophysiol. 111, 1227–1237. doi: 10.1152/jn.00607.2013
Zhou, J., Du, W., Zhou, K., Tai, Y., Yao, H., Jia, Y., et al. (2008). Critical role of TRPC6 channels in the formation of excitatory synapses. Nat. Neurosci. 11, 741–743. doi: 10.1038/nn.2127
Keywords: TRP channels, neurological disorders, calcium homeostasis, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, epilepsy
Citation: Lee K, Jo YY, Chung G, Jung JH, Kim YH and Park C-K (2021) Functional Importance of Transient Receptor Potential (TRP) Channels in Neurological Disorders. Front. Cell Dev. Biol. 9:611773. doi: 10.3389/fcell.2021.611773
Received: 29 September 2020; Accepted: 09 February 2021;
Published: 04 March 2021.
Edited by:
Sandra Derouiche, National Institute for Physiological Sciences (NIPS), JapanReviewed by:
Peter Illes, Leipzig University, GermanyAntonio Orlacchio, Santa Lucia Foundation (IRCCS), Italy
Copyright © 2021 Lee, Jo, Chung, Jung, Kim and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Ho Kim, ZXVybzE2QGdhY2hvbi5hYy5rcg==; Chul-Kyu Park, cGNrMDcwOEBnYWNob24uYWMua3I=
†These authors have contributed equally to this work