 Jin-Fan Li1†
Jin-Fan Li1† Xue-ping Xiang
Xue-ping Xiang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 21 January 2021
Sec. Epigenomics and Epigenetics
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.622393
This article is part of the Research Topic Finding New Epigenomics and Epigenetics Biomarkers for Complex Diseases and Significant Developmental Events with Machine Learning Methods View all 36 articles
Acute lymphoblastic leukemia (ALL) as a common cancer is a heterogeneous disease which is mainly divided into BCP-ALL and T-ALL, accounting for 80–85% and 15–20%, respectively. There are many differences between BCP-ALL and T-ALL, including prognosis, treatment, drug screening, gene research and so on. In this study, starting with methylation and gene expression data, we analyzed the molecular differences between BCP-ALL and T-ALL and identified the multi-omics signatures using Boruta and Monte Carlo feature selection methods. There were 7 expression signature genes (CD3D, VPREB3, HLA-DRA, PAX5, BLNK, GALNT6, SLC4A8) and 168 methylation sites corresponding to 175 methylation signature genes. The overall accuracy, accuracy of BCP-ALL, accuracy of T-ALL of the RIPPER (Repeated Incremental Pruning to Produce Error Reduction) classifier using these signatures evaluated with 10-fold cross validation repeated 3 times were 0.973, 0.990, and 0.933, respectively. Two overlapped genes between 175 methylation signature genes and 7 expression signature genes were CD3D and VPREB3. The network analysis of the methylation and expression signature genes suggested that their common gene, CD3D, was not only different on both methylation and expression levels, but also played a key regulatory role as hub on the network. Our results provided insights of understanding the underlying molecular mechanisms of ALL and facilitated more precision diagnosis and treatment of ALL.
Acute lymphoblastic leukemia (ALL) as a common cancer is a heterogeneous disease that originates from lymphocyte progenitor cells of B-cells or T-cells. It is a childhood malignant tumor that comprises >25% of pediatric neoplasia in American (Jabbour et al., 2015; Pui et al., 2015). Among adults, the incidence of ALL is much lower, accounting for only 0.2% of all cancers. However, the prognosis of ALL remains worrying, with an estimated 5-year overall survival (OS) of between 20 and 40% (Sive et al., 2012; Wolach et al., 2017). According to the World Health Organization (WHO) classification, ALL can be divided into B-cell ALL (B-ALL) and T-cell ALL (T-ALL). B-cell precursor ALL (BCP-ALL) is one of the B-ALL (Herold et al., 2014; Jones et al., 2016). In children’s ALL, it is mainly divided into BCP-ALL and T-ALL, accounting for 80–85% and 15–20%, respectively (Graux, 2011). These different subtypes are characterized by structural chromosomal rearrangements and repeated copy number alterations, which with great clinical significance (Goldberg et al., 2003).
There are prognosis, treatment and genetics differences between BCP-ALL and T-ALL (Gutierrez et al., 2014; Pui et al., 2015): (1) The prognosis of T-ALL patients is always worse than BCP-ALL patients (Goldberg et al., 2003; Eckert et al., 2013); (2) Many targeted immunotherapies have been developed for BCP-ALL patients but not for T-ALL patients (Pui et al., 2015); (3) T-ALL is associated with a wide range of acquired genetic abnormalities, which leads to abnormal proliferation and development stagnation of malignant lymphoid progenitor cells (Van Vlierberghe et al., 2008; Teitell and Pandolfi, 2009). This poses a challenge to the development of targeted therapy with wide application value. In the studies of the gene expression profile of ALL, the high expression of CD45 in leukemia cells was not only related to the poor prognosis of BCP-ALL patients but also to the poor prognosis of T-ALL patients. However, the prognostic correlation of CD45 expression in T-ALL was much higher than that in BCP-ALL (Hermiston et al., 2003; Cario et al., 2014). Moreover, PR-104 has been shown to specifically target hypoxic regions of leukemia infiltration, and was effective in the treatment of T-ALL xenotransplantation, but not in the treatment of BCP-ALL xenograft (Benito et al., 2011).
In this study, starting with methylation and gene expression data, we analyzed the molecular differences between BCP-ALL and T-ALL, screened out the molecular characteristics, and explored the relationship between these characteristics and the two subtypes of ALL.
We downloaded the methylation and expression data of 69 BCP-ALL and 30 T-ALL patients from GEO (Gene Expression Omnibus) under accession number of GSE49031 and GSE47051 (Nordlund et al., 2013, 2015; Borssen et al., 2018), respectively. It was a large study performed by Uppsala University. There were originally 945 methylation samples and 108 expression samples. But the overlapped sample size between methylation data and expression data was 99 and within the 99 samples, there were 69 BCP-ALL and 30 T-ALL patients. Our goal was to systematically investigate the molecular differences between BCP-ALL and T-ALL and try to use these molecular differences to explain the clinical differences.
The methylation data were generated with Illumina HumanMethylation450 BeadChip and there were 485,577 methylation probes. Since there were missing values, we filtered the probes with missing values in at least 20% samples and kept 485,096 probes. Since the probes out of gene ranges were hard to explain, we kept the 317,845 probes that can be annotated onto genes and imputed the missing values using KNN (K = 10) method. Meanwhile, the expression data were generated with Affymetrix Human Genome U133 Plus 2.0 Array. The expression values of probes corresponding to the same gene were averaged. At last, the dataset was the expression levels of 15,888 genes and methylation levels of 317,845 probes in 69 BCP-ALL and 30 T-ALL patients.
As we mentioned before, there were 15,888+317,845 = 333,733 features for each ALL sample. The number of features was much larger than the sample size. If we directly analyze all these 333,733 features, there will be too much noise and too many random feature combinations that can classify the samples. Therefore, we filtered the irrelevant features using Boruta method (Kursa and Rudnicki, 2010). The Boruta method can find out the relevant features and significantly reduce the number of features based on ensemble learning of random forest classifiers. Boruta is a widely used method and has been proven to be an effective method to find all relevant features (Pan et al., 2020; Yuan et al., 2020; Zhang et al., 2020).
Although Boruta method can filter irrelevant features and keep the relevant features, usually the number of features was still too large and the importance of features were still unknown. We need more sophisticated feature selection method to calculate the importance of features and rank the features. In this study, we applied MCFS (Monte Carlo Feature Selection) (Draminski et al., 2008). The MCFS has been widely used for feature selection (Chen et al., 2018, 2019; Pan et al., 2018, 2019a,b; Li et al., 2020). It divided the whole dataset into many small subsets. The subsets had much less features and the data structure of these subsets were relatively simple. Decision trees can be easily constructed. Based on all the trees on all the subsets, the importance of each feature can be calculated. The basic idea was that if a feature appeared in many trees, it was important and if a feature can classify many samples correctly, it was important. Based on these two rules, the importance of each feature was calculated. What’s more, the data was shuffled to generate random importance of each feature, the significance of each feature can be estimated by comparing the random importance and actual importance. At last, the significant features with importance much greater than permutated importance can be selected. Meanwhile, the RIPPER (Repeated Incremental Pruning to Produce Error Reduction) rules within the trees can be cross-validated and their accuracy can be estimated.
As we mentioned there were 333,733 features (15,888 expression feature and 317,845 methylation features) for each ALL sample. The number of features were much larger than the sample size (99 in this study). Most of the features were not relevant to ALL. Keeping such features in the dataset will introduce noise and make the analysis inaccurate. Therefore, we adopted Boruta method (Kursa and Rudnicki, 2010) to remove irrelevant features. After running Boruta, 1,398 features were kept. Within these 1,398 features, there were 1,374 methylation features and 24 expression features.
The number of features filtered by Boruta (1,398) was still too large to be biomarkers. Therefore, we further reduced the number of features with MCFS method and finally identified 175 significant features. Within the 175 features, there were 168 methylation features (probe IDs starting with “cg”) and 7 expression features (CD3D, VPREB3, HLA-DRA, PAX5, BLNK, GALNT6, SLC4A8). These 175 features were given in Table 1. The annotations of the 168 methylation probes of in Supplementary Table 1.
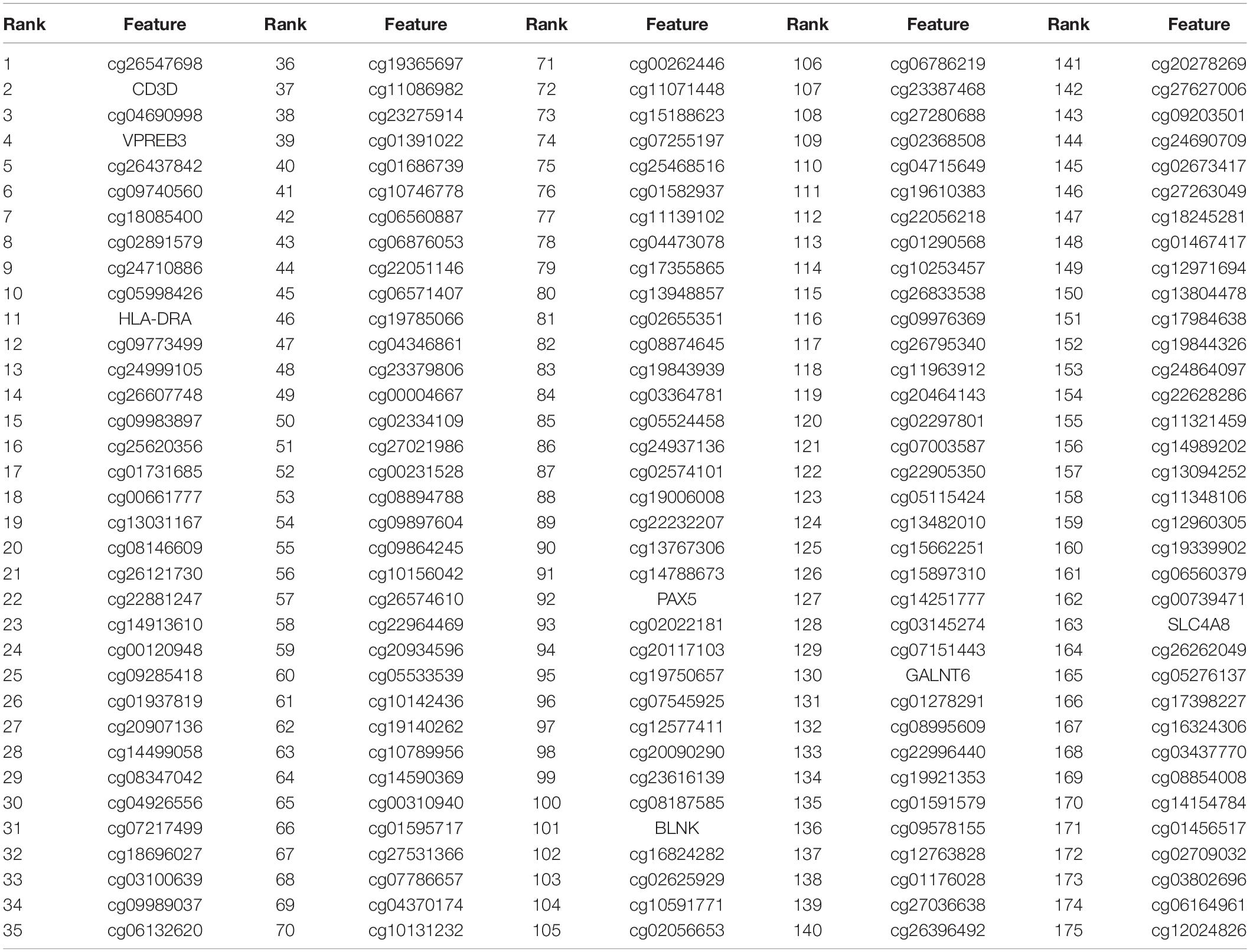
Table 1. The 175 important features identified by MCFS.
As we mentioned in section “Methods,” the MCFS method can also extract the classification rules. The confusion matrix of these RIPPER classification rules evaluated with 10-fold cross validation repeated 3 times was given in Table 2. The overall accuracy, accuracy of BCP-ALL, accuracy of T-ALL were 0.973, 0.990, and 0.933, respectively. These results meant that these features can classify the BCP-ALL and T-ALL very well.

Table 2. The confusion matrix of the RIPPER rules evaluated with 10-fold cross validation repeated 3 times.
Based on the annotations in Supplementary Table 1, we mapped the 168 methylation probes onto 175 genes. There were two overlapped genes (CD3D and VPREB3) between the 175 methylation signature genes and the 7 expression signature genes. We combined the 175 methylation signature genes and the 7 expression signature genes. Since there were two overlapped genes between them, there were 180 selected genes. We enriched the 180 selected genes onto KEGG pathways using WebGestalt1 (Wang et al., 2017). The KEGG enrichment results were shown in Figure 1. The x axis was log2 of enrichment ratio while the y axis was the -Log10 of FDR. The pathways on the top right corner were the significantly enrich pathways. It can be seen that hsa04640 Hematopoietic cell lineage was the enriched KEGG pathway. The were 11 selected genes on hsa04640 Hematopoietic cell lineage pathway: CD3D, CD3E, CD3G, CD59, FCER2, GP9, HLA-DMA, HLA-DPA1, HLA-DPB1, HLA-DRA and IL1B. The enrichment p value and FDR were 3.28e-9 and 5.35e-7, respectively. Its enrichment ratio was 11. As CD3D was dysfunctional on both methylation and gene expression levels, HLA-DRA was dysfunctional on gene expression levels and other genes were dysfunctional on methylation levels, the hsa04640 Hematopoietic cell lineage pathway was dysfunctional on both methylation and gene expression levels.
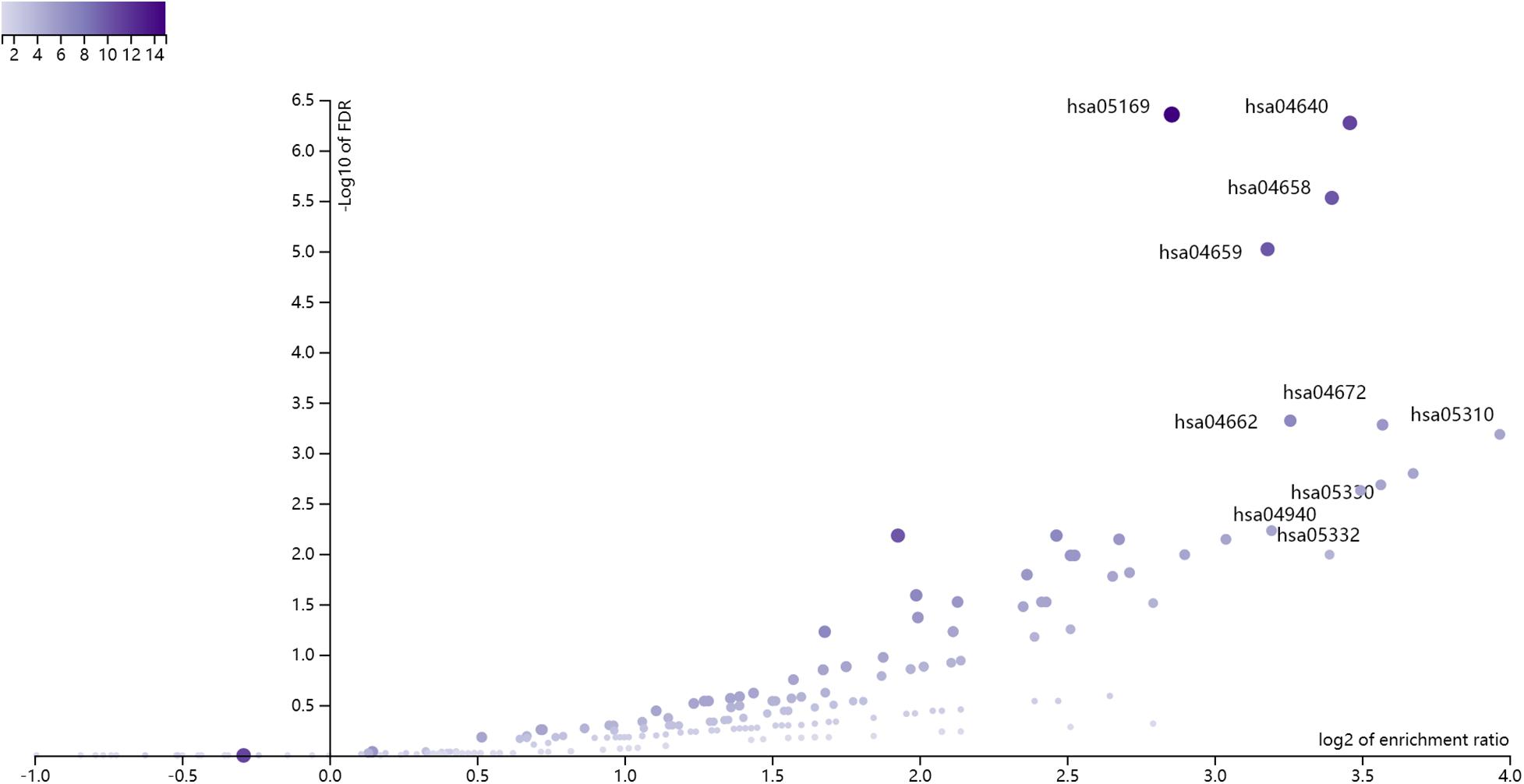
Figure 1. The enrichment results of the 180 selected genes using WebGestalt. The x axis was log2 of enrichment ratio while the y axis was the -Log10 of FDR. The pathways on the top right corner were the significantly enrich pathways. It can be seen that hsa04640 Hematopoietic cell lineage was the enriched KEGG pathway.
We searched the methylation and expression signature genes in STRING database2 (Szklarczyk et al., 2019) and their network with highest confidence (confidence score >0.900) was shown in Figure 2. The confidence score integrated the information from multiple sources including text mining, experiments, databases, co-expression, neighborhood, gene fusion and co-occurrence. It ranged from 0 to 1. The higher the confidence score was, the more reliable the interaction was. The cutoff of confidence score was set to be 0.900 since 0.900 was considered to be highest confidence in the STRING database. It can be seen that CD3D was the hub of the whole network. CD3D and another neighbor gene on the network, HLA-DRA, both belonged to hsa04640 Hematopoietic cell lineage pathway. The protein encoded by CD3D is part of the T cell receptor / CD3 complex (TCR/CD3 complex) and is involved in T cell development and signal transduction (Shi et al., 2019). CD3D has been shown to work with PKRCQ as a model to distinguish between B-ALL and T-ALL (Ma et al., 2016).
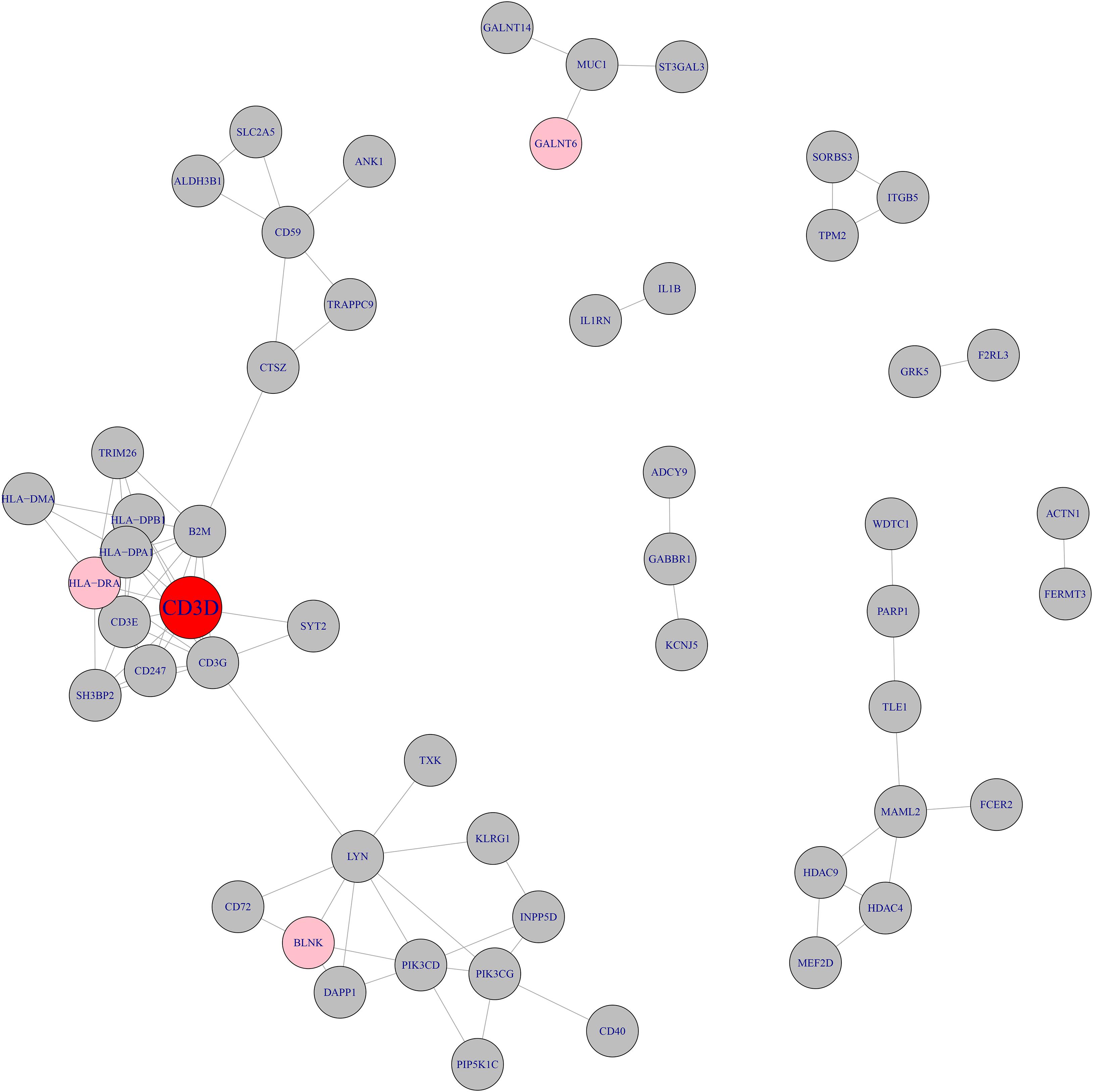
Figure 2. The network of methylation and expression signature genes. The methylation and expression signature genes were colored in gray and pink respectively. The red node, CD3D, was both methylation signature gene and expression signature gene. CD3D was the hub node of the network.
Within the 7 expression signature genes, beside CD3D which was discussed above, VPREB3 and HLA-DRA also looked promising.
VPREB3 is the B-cell receptor component and its overexpression can activate the pro-survival PI3K pathway (Soldini et al., 2014). It has been reported as a biomarker for B-cell lymphoma by many studies (Heerema-McKenney et al., 2010; Rodig et al., 2010; Soldini et al., 2014).
HLA-DRA is related to the antigen presentation steps of the immune system (Hotchkiss et al., 2013). In the study of Morrison et al. (2010), women and children with multiple sclerosis (MS) had a fourfold increased risk of developing ALL. And, there was a certain correlation between MS and HLA-DRA single nucleotide polymorphism (SNP) (Morrison et al., 2010). Moreover, HLA genes are candidate genetic susceptibility loci for childhood ALL, HLA-DP1 was significantly correlated with ALL in children (Urayama et al., 2012). According to Ross et al. (2019), the ablation of POZ domain of ZBTB17 (Miz-1) interferes with its interaction with c-MYC and delays the occurrence of T-ALL and B-ALL.
Within the 175 methylation signature genes, there were many great candidates, such as HDAC4, HDAC9, LMO2, MEF2D, CD40, PAX5, BLNK and TLE1.
HDAC4 and HDAC9 are Histone deacetylases (HDACs) which may be a potential target for cancer treatment, including hematological malignancies. Moreno et al. (2010) detected the expression profile of HDAC gene in ALL samples by PCR. It was found that HDAC1 and HDAC4 were highly expressed in T-ALL and HDAC5 was highly expressed in B-ALL. Moreover, the expression of HDAC9 was correlated with B-ALL patients (Moreno et al., 2010).
LMO2 plays an essential role during early hematopoiesis and is frequently activated in T-ALL patients (Morishima et al., 2019). Wu et al. have deeply studied the mechanism of LMO2 in T-ALL and found that LMO2 can induce the transcriptional inhibition of ZEB1, while ZEB1 plays an important role in promoting T cell differentiation and may play an anti-cancer role in T-ALL (Wu et al., 2018). Several studies have also confirmed that LMO2 plays an important role in T-ALL (Curtis and McCormack, 2010; Homminga et al., 2012; Rahman et al., 2017).
MEF2D has been reported as a biomarker for a B-ALL subtype with a low survival rate. According to Zhang M et al., MEF2D-SS18 fusion gene blocks the differentiation of B cells, which plays an important role in the pathogenesis and prognosis of B-ALL (Zhang et al., 2018). Besides, Suzuki et al. (2016) confirmed that MEF2D-BCL9 fusion gene is associated with juvenile acute BCP-ALL.
CD40 is the member of the tumor necrosis factor receptor (TNFR) family, are critical regulators of lymphocyte growth and differentiation. Troeger et al. (2008) confirmed that the high expression of CD40 in BCP-ALL cells is an independent prognostic indicator, which indicates a better recurrence-free survival.
PAX5 is a haplotype tumor suppressor gene in human B-All, which is involved in a variety of chromosome translocation (Jamrog et al., 2018). In the investigation and analysis of Bastian et al. (2019), it was found that the army of patients with BCP-ALL subgroup carried PAX5 mutation.
BLNK is an adapter molecule essential to the development of normal B cells and is associated with increased pro-B/pre-B-cell expansion in mice. It was reported that BLNK deficiency was one of the main causes of B-ALL (Imai et al., 2004). The results of Nakayama et al. suggested that somatic loss of BLNK and concomitant mutations leading to constitutive activation of Jak/STAT5 pathway result in the generation of BCP-ALL (Nakayama et al., 2009).
TLE1 can be used as an indicator of poor prognosis of T-ALL (Brassesco et al., 2018) and the expression of ATP10A was up-regulated in BCP-ALL (Olsson et al., 2014).
Although there have been studies on the clinical differences between BCP-ALL and T-ALL, there has been no in-depth study of their underlying mechanism. In our study, the multi-omics profiles in BCP-ALL and T-ALL were analyzed. The discovered epigenetic changes of ALL and their possible effects on gene expression can help us understand the molecular mechanisms of the development, progression and recurrence of ALL. In ALL, those molecular characteristics have the function of differential diagnosis, targeted therapy and so on. At the same time, our research not only provides new information about the methylation and gene expression pattern of ALL, but also provides a selective reference for the study of ALL genes and methylation sites.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
J-FL, X-pX, and X-JM contributed to the study design. L-LY conducted the literature search. Y-hT, J-FL, and X-JM acquired the data. J-FL and X-pX wrote the article. X-JM performed data analysis. J-FL and L-LY revised the article and gave the final approval of the version to be submitted. All authors read and approved the final manuscript.
This research was supported by the National Natural Science Foundation of China under Grant No. 81803549 and Zhejiang Provincial Natural Science Foundation of China under Grant No. LQ18H310002.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.622393/full#supplementary-material
Supplementary Table 1 | The annotations of the 168 methylation features.
Bastian, L., Schroeder, M. P., Eckert, C., Schlee, C., Tanchez, J. O., Kämpf, S., et al. (2019). PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 33, 1895–1909. doi: 10.1038/s41375-019-0430-z
Benito, J., Shi, Y., Szymanska, B., Carol, H., Boehm, I., Lu, H., et al. (2011). Pronounced hypoxia in models of murine and human leukemia: high efficacy of hypoxia-activated prodrug PR-104. PLoS One 6:e23108. doi: 10.1371/journal.pone.0023108
Borssen, M., Nordlund, J., Haider, Z., Landfors, M., Larsson, P., Kanerva, J., et al. (2018). DNA methylation holds prognostic information in relapsed precursor B-cell acute lymphoblastic leukemia. Clin. Epigenetics 10:31. doi: 10.1186/s13148-018-0466-3
Brassesco, M. S., Pezuk, J. A., Cortez, M. A., Bezerra Salomão, K., Scrideli, C. A., and Tone, L. G. (2018). TLE1 as an indicator of adverse prognosis in pediatric acute lymphoblastic leukemia. Leuk. Res. 74, 42–46. doi: 10.1016/j.leukres.2018.09.010
Cario, G., Rhein, P., Mitlöhner, R., Zimmermann, M., Bandapalli, O. R., Romey, R., et al. (2014). High CD45 surface expression determines relapse risk in children with precursor B-cell and T-cell acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 99, 103–110. doi: 10.3324/haematol.2013.090225
Chen, L., Li, J., Zhang, Y. H., Feng, K., Wang, S., Zhang, Y., et al. (2018). Identification of gene expression signatures across different types of neural stem cells with the Monte-Carlo feature selection method. J. Cell. Biochem. 119, 3394–3403. doi: 10.1002/jcb.26507
Chen, L., Pan, X., Zhang, Y.-H., Kong, X., Huang, T., and Cai, Y.-D. (2019). Tissue differences revealed by gene expression profiles of various cell lines. J. Cell. Biochem. 120, 7068–7081. doi: 10.1002/jcb.27977
Curtis, D. J., and McCormack, M. P. (2010). The molecular basis of Lmo2-induced T-cell acute lymphoblastic leukemia. Clin. Cancer Res. 16, 5618–5623.
Draminski, M., Rada-Iglesias, A., Enroth, S., Wadelius, C., Koronacki, J., and Komorowski, J. (2008). Monte Carlo feature selection for supervised classification. Bioinformatics 24, 110–117. doi: 10.1093/bioinformatics/btm486
Eckert, C., von Stackelberg, A., Seeger, K., Groeneveld, T. W., Peters, C., Klingebiel, T., et al. (2013). Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk relapsed acute lymphoblastic leukaemia - long-term results of trial ALL-REZ BFM P95/96. Eur. J. Cancer 49, 1346–1355. doi: 10.1016/j.ejca.2012.11.010
Goldberg, J. M., Silverman, L. B., Levy, D. E., Dalton, V. K., Gelber, R. D., Lehmann, L., et al. (2003). Childhood T-cell acute lymphoblastic leukemia: the Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J. Clin. Oncol. 21, 3616–3622. doi: 10.1200/jco.2003.10.116
Graux, C. (2011). Biology of acute lymphoblastic leukemia (ALL): clinical and therapeutic relevance. Transfus. Apher. Sci. 44, 183–189. doi: 10.1016/j.transci.2011.01.009
Gutierrez, A., Feng, H., Stevenson, K., Neuberg, D. S., Calzada, O., Zhou, Y., et al. (2014). Loss of function tp53 mutations do not accelerate the onset of myc-induced T-cell acute lymphoblastic leukaemia in the zebrafish. Br. J. Haematol. 166, 84–90. doi: 10.1111/bjh.12851
Heerema-McKenney, A., Waldron, J., Hughes, S., Zhan, F., Sawyer, J., Barlogie, B., et al. (2010). Clinical, immunophenotypic, and genetic characterization of small lymphocyte-like plasma cell myeloma: a potential mimic of mature B-cell lymphoma. Am. J. Clin. Pathol. 133, 265–270. doi: 10.1309/ajcpus3prrt5zxvs
Hermiston, M. L., Xu, Z., and Weiss, A. (2003). CD45: a critical regulator of signaling thresholds in immune cells. Annu. Rev. Immunol. 21, 107–137. doi: 10.1146/annurev.immunol.21.120601.140946
Herold, T., Baldus, C. D., and Gökbuget, N. (2014). Ph-like acute lymphoblastic leukemia in older adults. N. Engl. J. Med. 371:2235.
Homminga, I., Vuerhard, M. J., Langerak, A. W., Buijs-Gladdines, J., Pieters, R., and Meijerink, J. P. (2012). Characterization of a pediatric T-cell acute lymphoblastic leukemia patient with simultaneous LYL1 and LMO2 rearrangements. Haematologica 97, 258–261. doi: 10.3324/haematol.2011.051722
Hotchkiss, R. S., Monneret, G., and Payen, D. (2013). Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 13, 862–874. doi: 10.1038/nri3552
Imai, C., Ross, M. E., Reid, G., Coustan-Smith, E., Schultz, K. R., Pui, C. H., et al. (2004). Expression of the adaptor protein BLNK/SLP-65 in childhood acute lymphoblastic leukemia. Leukemia 18, 922–925. doi: 10.1038/sj.leu.2403349
Jabbour, E., O’Brien, S., Konopleva, M., and Kantarjian, H. (2015). New insights into the pathophysiology and therapy of adult acute lymphoblastic leukemia. Cancer 121, 2517–2528. doi: 10.1002/cncr.29383
Jamrog, L., Chemin, G., Fregona, V., Coster, L., Pasquet, M., Oudinet, C., et al. (2018). PAX5-ELN oncoprotein promotes multistep B-cell acute lymphoblastic leukemia in mice. Proc. Natl. Acad. Sci. U.S.A. 115, 10357–10362. doi: 10.1073/pnas.1721678115
Jones, L., Carol, H., Evans, K., Richmond, J., Houghton, P. J., Smith, M. A., et al. (2016). A review of new agents evaluated against pediatric acute lymphoblastic leukemia by the Pediatric Preclinical Testing Program. Leukemia 30, 2133–2141. doi: 10.1038/leu.2016.192
Kursa, M., and Rudnicki, W. (2010). Feature Selection with the Boruta Package. J. Stat. Softw. 36, 1–13. doi: 10.18637/jss.v036.i11
Li, J., Lu, L., Zhang, Y. H., Xu, Y., Liu, M., Feng, K., et al. (2020). Identification of leukemia stem cell expression signatures through Monte Carlo feature selection strategy and support vector machine. Cancer Gene Ther. 27(1-2), 56–69. doi: 10.1038/s41417-019-0105-y
Ma, D., Zhong, S., Liu, X., Mai, H., Mai, G., Xu, C., et al. (2016). CD3D and PRKCQ work together to discriminate between B-cell and T-cell acute lymphoblastic leukemia. Comput. Biol. Med. 77, 16–22. doi: 10.1016/j.compbiomed.2016.07.004
Moreno, D. A., Scrideli, C. A., Cortez, M. A., de Paula Queiroz, R., Valera, E. T., da Silva Silveira, V., et al. (2010). Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 150, 665–673. doi: 10.1111/j.1365-2141.2010.08301.x
Morishima, T., Krahl, A. C., Nasri, M., Xu, Y., Aghaallaei, N., Findik, B., et al. (2019). LMO2 activation by deacetylation is indispensable for hematopoiesis and T-ALL leukemogenesis. Blood 134, 1159–1175. doi: 10.1182/blood.2019000095
Morrison, B. A., Ucisik-Akkaya, E., Flores, H., Alaez, C., Gorodezky, C., and Dorak, M. T. (2010). Multiple sclerosis risk markers in HLA-DRA, HLA-C, and IFNG genes are associated with sex-specific childhood leukemia risk. Autoimmunity 43, 690–697. doi: 10.3109/08916930903567492
Nakayama, J., Yamamoto, M., Hayashi, K., Satoh, H., Bundo, K., Kubo, M., et al. (2009). BLNK suppresses pre-B-cell leukemogenesis through inhibition of JAK3. Blood 113, 1483–1492.
Nordlund, J., Backlin, C. L., Wahlberg, P., Busche, S., Berglund, E. C., Eloranta, M. L., et al. (2013). Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 14:r105. doi: 10.1186/gb-2013-14-9-r105
Nordlund, J., Backlin, C. L., Zachariadis, V., Cavelier, L., Dahlberg, J., Ofverholm, I., et al. (2015). DNA methylation-based subtype prediction for pediatric acute lymphoblastic leukemia. Clin. Epigenetics 7:11. doi: 10.1186/s13148-014-0039-z
Olsson, L., Castor, A., Behrendtz, M., Biloglav, A., Forestier, E., Paulsson, K., et al. (2014). Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011. Leukemia 28, 302–310. doi: 10.1038/leu.2013.206
Pan, X., Chen, L., Feng, K. Y., Hu, X. H., Zhang, Y. H., Kong, X. Y., et al. (2019a). Analysis of expression pattern of snoRNAs in different cancer types with machine learning algorithms. Int. J. Mol. Sci. 20:2185. doi: 10.3390/ijms20092185
Pan, X., Hu, X., Zhang, Y.-H., Chen, L., Zhu, L., Wan, S., et al. (2019b). Identification of the copy number variant biomarkers for breast cancer subtypes. Mol. Genet. Genomics 294, 95–110. doi: 10.1007/s00438-018-1488-4
Pan, X., Hu, X., Zhang, Y. H., Feng, K., Wang, S. P., Chen, L., et al. (2018). Identifying patients with atrioventricular septal defect in down syndrome populations by using self-normalizing neural networks and feature selection. Genes 9:208. doi: 10.3390/genes9040208
Pan, X., Zeng, T., Zhang, Y. H., Chen, L., Feng, K., Huang, T., et al. (2020). Investigation and prediction of human interactome based on quantitative features. Front. Bioeng. Biotechnol. 8:730. doi: 10.3389/fbioe.2020.00730
Pui, C. H., Yang, J. J., Hunger, S. P., Pieters, R., Schrappe, M., Biondi, A., et al. (2015). Childhood acute lymphoblastic leukemia: progress through collaboration. J. Clin. Oncol. 33, 2938–2948. doi: 10.1200/jco.2014.59.1636
Rahman, S., Magnussen, M., León, T. E., Farah, N., Li, Z., Abraham, B. J., et al. (2017). Activation of the oncogene through a somatically acquired neomorphic promoter in T-cell acute lymphoblastic leukemia. Blood 129, 3221–3226.
Rodig, S. J., Kutok, J. L., Paterson, J. C., Nitta, H., Zhang, W., Chapuy, B., et al. (2010). The pre-B-cell receptor associated protein VpreB3 is a useful diagnostic marker for identifying c-MYC translocated lymphomas. Haematologica 95, 2056–2062. doi: 10.3324/haematol.2010.025767
Ross, J., Rashkovan, M., Fraszczak, J., Joly-Beauparlant, C., Vadnais, C., Winkler, R., et al. (2019). Deletion of the Miz-1 POZ domain increases efficacy of cytarabine treatment in T- and B-ALL/lymphoma mouse models. Cancer Res. 79, 4184–4195.
Shi, M. J., Meng, X. Y., Wu, Q. J., and Zhou, X. H. (2019). High CD3D/CD4 ratio predicts better survival in muscle-invasive bladder cancer. Cancer Manag. Res. 11, 2987–2995. doi: 10.2147/cmar.S191105
Sive, J. I., Buck, G., Fielding, A., Lazarus, H. M., Litzow, M. R., Luger, S., et al. (2012). Outcomes in older adults with acute lymphoblastic leukaemia (ALL): results from the international MRC UKALL XII/ECOG2993 trial. Br. J. Haematol. 157, 463–471. doi: 10.1111/j.1365-2141.2012.09095.x
Soldini, D., Georgis, A., Montagna, C., Schüffler, P. J., Martin, V., Curioni-Fontecedro, A., et al. (2014). The combined expression of VPREB3 and ID3 represents a new helpful tool for the routine diagnosis of mature aggressive B-cell lymphomas. Hematol. Oncol. 32, 120–125. doi: 10.1002/hon.2094
Suzuki, K., Okuno, Y., Kawashima, N., Muramatsu, H., Okuno, T., Wang, X., et al. (2016). MEF2D-BCL9 fusion gene is associated with high-risk acute B-cell precursor lymphoblastic leukemia in adolescents. J. Clin. Oncol. 34, 3451–3459. doi: 10.1200/jco.2016.66.5547
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47(D1), D607–D613. doi: 10.1093/nar/gky1131
Teitell, M. A., and Pandolfi, P. P. (2009). Molecular genetics of acute lymphoblastic leukemia. Annu. Rev. Pathol. 4, 175–198. doi: 10.1146/annurev.pathol.4.110807.092227
Troeger, A., Glouchkova, L., Ackermann, B., Escherich, G., Meisel, R., Hanenberg, H., et al. (2008). High expression of CD40 on B-cell precursor acute lymphoblastic leukemia blasts is an independent risk factor associated with improved survival and enhanced capacity to up-regulate the death receptor CD95. Blood 112, 1028–1034. doi: 10.1182/blood-2007-11-123315
Urayama, K. Y., Chokkalingam, A. P., Metayer, C., Ma, X., Selvin, S., Barcellos, L. F., et al. (2012). HLA-DP genetic variation, proxies for early life immune modulation and childhood acute lymphoblastic leukemia risk. Blood 120, 3039–3047. doi: 10.1182/blood-2012-01-404723
Van Vlierberghe, P., Pieters, R., Beverloo, H. B., and Meijerink, J. P. (2008). Molecular-genetic insights in paediatric T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 143, 153–168. doi: 10.1111/j.1365-2141.2008.07314.x
Wang, J., Vasaikar, S., Shi, Z., Greer, M., and Zhang, B. (2017). WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 45, W130–W137.
Wolach, O., Amitai, I., and DeAngelo, D. J. (2017). Current challenges and opportunities in treating adult patients with Philadelphia-negative acute lymphoblastic leukaemia. Br. J. Haematol. 179, 705–723. doi: 10.1111/bjh.14916
Wu, C., Li, J., Tian, C., Shi, W., Jiang, H., Zhang, Z., et al. (2018). Epigenetic dysregulation of ZEB1 is involved in LMO2-promoted T-cell acute lymphoblastic leukaemia leukaemogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2511–2525. doi: 10.1016/j.bbadis.2018.05.013
Yuan, F., Pan, X., Zeng, T., Zhang, Y.-H., Chen, L., Gan, Z., et al. (2020). Identifying cell-type specific genes and expression rules based on single-cell transcriptomic atlas data. Front. Bioeng. Biotechnol. 8:350. doi: 10.3389/fbioe.2020.00350
Zhang, M., Mao, D., and Zhang, W. (2018). The pathogenic role of MEF2D-SS18 fusion gene in B-cell acute lymphoblastic leukemia. Biochem. Biophys. Res. Commun. 496, 1331–1336. doi: 10.1016/j.bbrc.2018.02.013
Keywords: acute lymphoblastic leukemia, Boruta, Monte Carlo feature selection, network analysis, hub, multi-omics, expression, methylation
Citation: Li J-F, Ma X-J, Ying L-L, Tong Y-h and Xiang X-p (2021) Multi-Omics Analysis of Acute Lymphoblastic Leukemia Identified the Methylation and Expression Differences Between BCP-ALL and T-ALL. Front. Cell Dev. Biol. 8:622393. doi: 10.3389/fcell.2020.622393
Received: 28 October 2020; Accepted: 15 December 2020;
Published: 21 January 2021.
Edited by:
Tao Huang, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Zhangsen Huang, Sun Yat-sen University, ChinaCopyright © 2021 Li, Ma, Ying, Tong and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue-ping Xiang, eGlhbmd4dWVwaW5nQHpqdS5lZHUuY24=; Ying-hui Tong, dG9uZ3loQHpqY2Mub3JnLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.