 Ran Duan
Ran Duan Hui Xie
Hui Xie Zheng-Zhao Liu
Zheng-Zhao Liu- 1Movement System Injury and Repair Research Center, Xiangya Hospital, Central South University, Changsha, China
- 2Department of Sports Medicine, Xiangya Hospital, Central South University, Changsha, China
- 3Department of Orthopedics, Xiangya Hospital, Central South University, Changsha, China
- 4Hunan Key Laboratory of Organ Injury, Aging and Regenerative Medicine, Changsha, China
- 5Hunan Key Laboratory of Bone Joint Degeneration and Injury, Changsha, China
- 6National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Changsha, China
Chondrocytes are the only cell type in normal cartilage. The pathological changes of osteoarthritis (OA) mostly revolve around the apoptosis and dysfunction of chondrocytes. Autophagy, as an intracellular degradation system that maintains the steady state of energy metabolism in cells, has been shown to restore the function of damaged chondrocytes, alleviating the occurrence and progression of OA. In this review, we explored the relationship between autophagy and OA and the key molecules of autophagy pathway that regulate the progression of OA, providing new ideas for OA treatment by targeting autophagy.
Introduction
OA is a chronic and high prevalent arthropathy and the most common cause of pain and disability worldwide characterized by progressive cartilage degradation, synovitis, and conversion of joint peripheral sclerotin including osteophyte formation and subchondral bone sclerosis (Harrell et al., 2019). A variety of risk factors contribute to the development of OA, such as age, obesity, hereditary factors, and preceding joint injuries (Vina and Kwoh, 2018). Senility is the greatest risk factor of OA (Jeon et al., 2017). The incidence of OA is rising due to the increasing obesity and a rapidly aging population. Over 50% OA patients are over 65 years old (Loeser, 2017), and even 80% of OA patients are over 75 years old (O'Neill et al., 2018). Articular cartilage is a kind of connective tissue composed of chondrocytes and extracellular matrix (ECM) that is synthesized and secreted by chondrocytes. The matrix that encapsulates chondrocytes plays a role of lubrication and mechanical support for cartilage joint. Although chondrocytes account for only 1% of the total cartilage volume, it plays an indelible role in maintaining the integrity of the matrix (Charlier et al., 2019; Rim et al., 2020). In fact, when certain factors act on the cartilage matrix and change its structure, chondrocytes can respond accordingly. However, the ability of articular chondrocytes to maintain the structure and integrity of normal cartilage matrix is limited and declines with aging (Martel-Pelletier et al., 2016). The existing treatment for OA is still relatively conservative, mostly limited to control the pain. However, in terms of reducing and controlling joint inflammation and promoting the recovery of damaged chondrocyte function, effective interventions that proven to alter the natural course of OA are still lacking (Cutolo et al., 2015; Hunter and Bierma-Zeinstra, 2019). Therefore, an in-depth investigation of the pathogenesis of OA is essential to explore new therapeutic targets.
Macroautophagy/Autophagy (hereafter referred to as autophagy), as a highly conserved degradation system, plays a vital role in the regulation of energy and nutrition, maintaining energy metabolism in the body (Nakamura and Yoshimori, 2017; Yu et al., 2018). Autophagy plays a protective role on cells under abnormal physiological conditions, including external pressure, hypoalimentation, hypoxia, endoplasmic reticulum stress (ERS), and so on. Malfunctioning cytoplasmic macromolecules, membranes and organelles are transported to lysosomes for degradation and reusing through autophagy pathway (Nakamura and Yoshimori, 2017). The occurrence of autophagy can be artificially divided into the following steps, including initiation, nucleation, elongation, maturation, and degradation. With the formation of these autophagosomes, degradation of chelate products provides amino acids, nucleotides, saccharides and aliphatic acids for maintaining the steady state of the entire tissue and even the body (Glick et al., 2010).
As mentioned above, since autophagy can degrade and remove long-lived or impaired organelles and proteins, the reduction of autophagy with aging is related to various aging diseases (Ren and Zhang, 2018). With the growth of age, the basic autophagic activity of cells decreases, following a down-regulated clearance efficiency. Subsequently, the aggregation of various macromolecular proteins increases, leading to the eventual cell degeneration and functional defect, or even apoptosis. In recent years, the inhibition of chondrocytes apoptosis by autophagy activation has attracted much attention (Shapiro et al., 2014). Recent studies have shown that the level of autophagy in OA cartilage is reduced (Feng et al., 2020), and autophagy can protect chondrocytes from degradation (Caramés et al., 2012). With the progress of OA, mammalian target of rapamycin (mTOR), the main negative regulator of autophagy, is up-regulated and mediates the inhibition of autophagy signal transduction in articular cartilage, and the protective effect of autophagy on cartilage decreases, eventually promoting cartilage degeneration (Vasheghani et al., 2015). Study by Zhang et al. demonstrated that the surgical destabilization of the medial meniscus (DMM) OA mouse model has higher level of autophagy, lower level of apoptosis when knocked out of mTOR (Zhang Y. et al., 2015). Activation of autophagy in chondrocytes by intra-articular injection of resveratrol, an autophagy inducer, can significantly delay articular cartilage degeneration of DMM OA mouse model (Qin et al., 2017). In recent years, studies on pharmacological suppression and gene deletion of mTOR to reduce the severity of OA have also repeatedly demonstrated the protective effect of autophagy on chondrocytes (Takayama et al., 2014; Ribeiro et al., 2016).
Mechanistic Studies of Autophagy and OA
Association between aging and OA has been demonstrated in clinic and epidemiology. Risk factors related to aging, such as the limited ability of tissue and cell regeneration, increased expression of inflammatory mediators, oxidative stress, etc., cause damage to the cartilage matrix and cells, and promote the occurrence and development of OA (Rahmati et al., 2017). In recent years, autophagy, as a protective mechanism in cells, has attracted much attention because of its role in regulating the aging process. Autophagy will be activated when oxidative stress occurs, but excessive oxidative stress will exceed the tolerance of autophagy and impair its activity, eventually leading to cellular senescence and apoptosis (Roca-Agujetas et al., 2019). However, definitive studies on the relationship between autophagy and senescence are still lacking since some authors suggest a direct connection between autophagy and senescence and others indicative of an inverse relationship. Indeed, autophagy may promote or counteract senescence depending on the cellular context and stress stimuli (White and Lowe, 2009; Gewirtz, 2014). Study by Capasso et al. showed that in acute senescence, but low radiation, the autophagy flux is heavily impaired suggesting autophagy counteracts deteriorative processes, and its decline triggers senescence. This did not occur in replicative senescence. The authors hypothesized to reconcile these opposite events, that cells try to contend with stress by activating autophagy that eliminates damaged components. In that context, autophagy protects from senescence, and impairment of its function may promote senescence. On the other hand, if autophagy cannot counteract stress-induced damage, it may induce senescence (Capasso et al., 2015). In the state of excessive oxidative stress, chondrocytes can enhance autophagic activity and inhibit aging process by activating AMP-activated protein kinase (AMPK) or inhibiting mTOR (Han et al., 2016; Tai et al., 2017). Another important sign of aging is mitochondrial dysfunction associated with an increase of superoxide accumulation. It is noteworthy that mitochondrial phagocytosis can reduce phenotypes of cell senescence (Tai et al., 2017).
Inflammatory cytokines, mechanical stress and senescence can cause elevated levels of reactive oxygen species (ROS) in chondrocytes. Known ROS include superoxide, hydrogen peroxide, peroxyl radicals, the reactive nitrogen species (including nitric oxide and peroxynitrite derived from the nitric oxide) (Su et al., 2019). Kongara and Karantza have shown that autophagy is responsible for eliminating intracellular sources of ROS, including mitochondria and peroxisomes (Kongara and Karantza, 2012), thus, autophagy defect leads to accumulation of ROS (Scherz-Shouval and Elazar, 2007). Under pathological conditions, the phenomenon of cartilage degradation can be attributed to excessive ROS acting as second messengers, which subsequently mediate the inhibition of matrix synthesis, affect cell migration and the biological activity of growth factors, which in turn regulate the degradation of matrix components, activation of matrix metalloproteinases (MMP) and cell death. Excess ROS can cause inhibition of the mitochondrial respiratory chain, decrease of ATP, mitochondrial DNA (mtDNA) mutations, and disorder of redox regulated cell signaling pathways such as protein kinase B (PKB/AKT) and mitogen-activated protein kinase (MAPK) signaling pathways. ROS-induced mitochondrial damage and activation of endoplasmic reticulum (ER) stress play a key role in OA, and may eventually trigger the cascade of chondrocytes apoptosis (Collins et al., 2018; Bolduc et al., 2019). Previous studies have shown that both autophagy defect and mitochondrial dysfunction were found in age-related and surgically induced OA mouse models, and mitochondrial function and autophagy in chondrocytes are directly mediated by the AKT-mTOR signaling pathway (Barranco, 2015). Inhibition of autophagy related 5 (Atg5) in chondrocytes increases the production of ROS, and induces mitochondrial dysfunction (Lopez de Figueroa et al., 2015). These results indicated that autophagy participates in the occurrence and development of OA by mediating apoptosis and ROS production (Charlier et al., 2016). When autophagy is activated, the damaged mitochondria can be removed and intracellular ROS reduces, protecting chondrocytes from the negative effects of OA. Therefore, we have every reason to believe that autophagy plays an irreplaceable role in protecting chondrocytes from oxidative stress (Ansari et al., 2018).
Key Molecules Involved in Autophagy and OA
mTOR kinase is a key molecule in the process of autophagy. It combines with various proteins to form two different multiprotein complexes known as mTOR complex 1 (mTORC1) and 2 (mTORC2). mTORC1, as a complex containing mTOR, plays a vital role in the regulation of autophagy (Laplante and Sabatini, 2012). Studies have shown that mTORC1 mainly acts as a negative regulator in autophagy and can be regulated by a variety of signaling molecules to affect autophagic activity (Choi et al., 2013). Thus, mTOR activation pathways, such as AKT and MAPK signaling pathways, inhibit autophagy, while mTOR negative regulation pathways, such as AMPK and p53 signaling pathways, promote autophagy. AMPK is an energy-sensing kinase, a highly conserved protein molecule during evolution. When metabolic stress or ATP consumption occurs, AMPK can be activated to promote catabolism. Once the AMPK pathway activated, AMPK phosphorylates tuberous sclerosis 2 (TSC2), which then inhibits mTOR and eventually promotes autophagy. In addition, AMPK can also directly regulate autophagy by acting on downstream signaling molecules of mTOR (Alers et al., 2012). In summary, both mTOR and AMPK are integral parts of autophagy initiation (Figure 1).
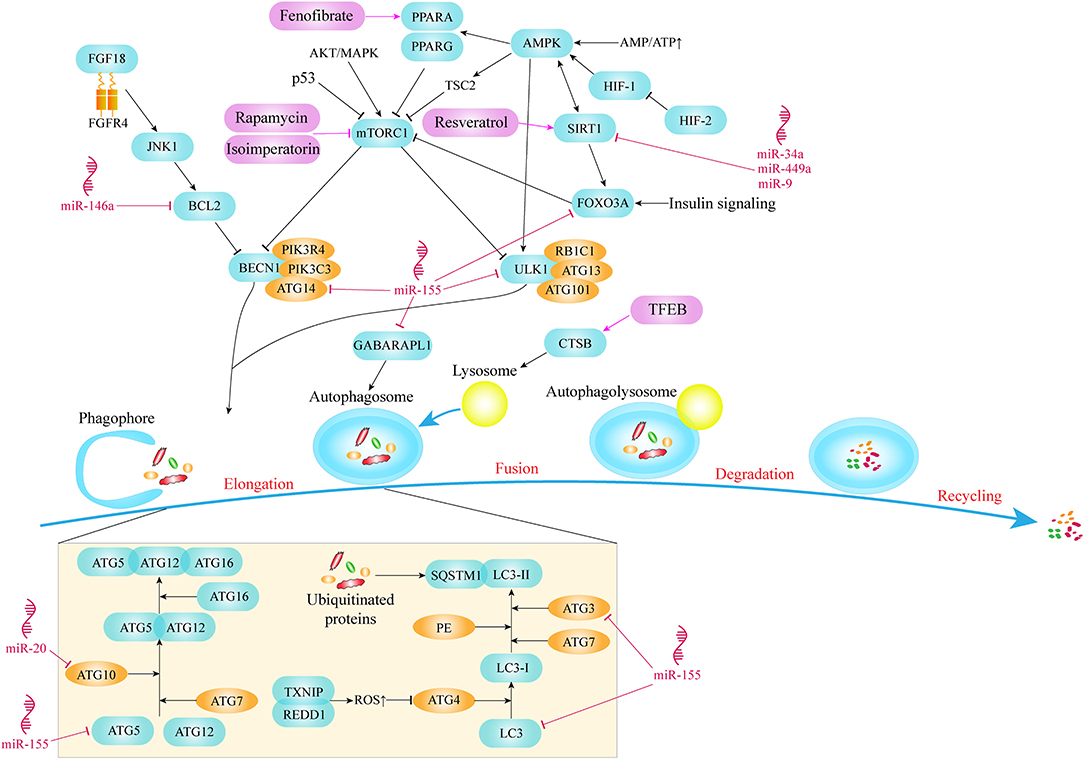
Figure 1. Key molecules and pathways of autophagy in OA.
The chondrocytes in the growth plate survive in a microenvironment with almost no blood vessels and hypoxia, and have evolved a different way of energy generation, generating metabolic energy through anaerobic glycolysis. The hypoxia-inducible factors, HIF-1 and HIF-2, mediate the acclimatization of chondrocytes to this avascular environment (Zhang F.-J. et al., 2015). HIF-1α is highly expressed in hypertrophic chondrocytes (Taheem et al., 2018). Activation of HIF-1 enhances the autophagic activity of chondrocytes in hypoxia condition. Reduction of energy charge activates HIF-1, which leads to the phosphorylation and activation of AMPK. AMPK enhances autophagy flux by inhibiting mTOR and promoting other downstream signaling molecules (Bohensky et al., 2010). In contrast with HIF-1, HIF-2 is a potent negative regulator of autophagy in maturing chondrocytes which is elevated in OA cartilage and drives catabolic metalloproteinases, such as MMP-13 which is a major enzyme that mediates the degradation of type II collagen in cartilage (van den Berg, 2011; Wang et al., 2013) (Figure 1).
As a core protein with serine/threonine kinase activity in autophagy signaling pathway, unc-51 like autophagy activating kinase 1/2 (ULK1/2) can form the ULK complex with RB1 inducible coiled-coil 1 (RB1CC1/FIP200), autophagy related 13 (ATG13) and autophagy related 101 (ATG101) and mediate the activation of autophagy signaling pathway (Mizushima, 2010). ULK1 in a phosphorylation status has always been identified as a pivotal sign of autophagy initiation. Phosphorylation of ULK1 was mediated by AMPK and mTOR signaling pathway (Figure 1). A large number of studies have shown that with the change in the nutritional status of tissues or cells, the phosphorylation status of the ULK1/2-ATG13-RB1CC1 complex will also undergo corresponding changes. Specifically, in the context of glucose starvation, AMPK directly phosphorylates ULK1 at Ser 317 and Ser 777 to promote autophagy (Shang et al., 2011). However, in the case of sufficient nutrition, the high activity state of mTOR phosphorylates ULK1 at Ser 757, destroys the mutual effect between ULK1 and AMPK, and finally prevents the activation of ULK1 (Kim et al., 2011).
One of the initial steps in assembling pre-phagocytic structures into autophagosomes is to recruit and activate the class III phosphatidylinositol 3-kinase complex which is composed of Beclin1 (BECN1), phosphoinositide-3-kinase regulatory subunit 4 (PIK3R4/VPS15), phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3/VPS34), and autophagy related 14 (ATG14/ATG14L) proteins (Menon and Dhamija, 2018) (Figure 1). The pivotal role of ATG14 in autophagy is the recruitment of autophagy specific PI3 kinase complex in endoplasmic reticulum (Matsunaga et al., 2010). Meanwhile, PIK3C3, as a kind of catalytic phosphatidylinositol 3 kinase, catalyzes the phosphorylation of phosphatidylinositol to form phosphatidylinositol 3-phosphate (PI3P). PI3P on the autophagosome membrane has the ability to recruit the membrane-bound protein ATG18 and bind to the bilayer membrane, which is crucial for the elongation and formation of autophagosomes (Noda et al., 2010).
In addition to the above autophagy pathway signaling molecules, two protein conjugation systems are also involved in the formation and amplification of autophagosomes, among which ATG proteins microtubule associated protein light chain 3 (LC3) and ATG12 are important components of the related protein coupling system. LC3 is usually used as a marker to evaluate the degree of autophagy. During the process of autophagy, ATG4 can remove the C-terminus of LC3 to generate LC3-I in the cytoplasm, and LC3-I is connected with phosphatidylethanolamine (PE) in the way of ubiquitin like reaction, which requires the participation of E1-like enzyme ATG7 and E2-like enzyme ATG3. LC3-II is transformed from LC3-I through the lipidation of the ubiquitin-like reaction described above and binds to autophagic vesicles (Li et al., 2020). Therefore, the amount of LC3-II is related to the degree of autophagosome formation, and the ratio of LC3-II/LC3-I is usually used to assess the level of autophagy (Martin-Rincon et al., 2018). In selective autophagy, cytoplasmic components that are selectively degraded are labeled with ubiquitin in order to be recognized by means of autophagy receptors before being sequestered into an autophagosome (Lamark et al., 2017). As a multifunctional adaptor protein, sequestosome 1 (SQSTM1/p62) is able to combine with mono- or poly-ubiquitinated proteins. Through its LC3-interacting region (LIR) motif, SQSTM1 directly binds to LC3 and becomes a selective autophagy receptor which aggregates ubiquitinated proteins and brings them into the emerging autophagosomes (Shaid et al., 2013). SQSTM1 also acts as cargo receptor and is degraded by autophagy together with ubiquitinated substrate proteins (Figure 1).
Conjugation of ATG5 and ATG12 is an essential link in autophagy (Figure 1). The genetic ablation of Atg5 leads to the development of OA with age and the apoptosis of chondrocytes mediated by caspases (Vuppalapati et al., 2015; Bouderlique et al., 2016). As a ubiquitin activating enzyme, ATG7 plays an important role in autophagy formation and LC3 activation. Knocking out Atg7 in chondrocytes leads to growth retardation of chondrocytes, which is also related to the reduction of chondrocyte proliferation and the upregulation of apoptosis (Kang et al., 2017; Horigome et al., 2020).
The defects of autophagy in OA cartilage include the decrease of the number and size of autophagosomes, which is related to the decreased expression of ULK1, BECN1, LC3 and the overexpression of mTOR (D'Adamo et al., 2017b). At the same time, SQSTM1 increased in OA cartilage, indicating that autophagy was inhibited (Zheng et al., 2019).
Key Molecules Regulated the Autophagy Pathway in OA
MicroRNAs Regulated the Autophagy Pathway in OA
MicroRNAs (miRNAs) are a type of non-coding small RNAs that promote specific mRNA degradation and/or translation inhibition, and play a pivotal role in biological processes as new gene expression regulators, whose regulatory mechanism is thought to be through sequentially specific interactions with the 3' untranslated regions (UTRs) of specific mRNA targets (Bartel, 2004). miRNAs play a vital role in cartilage homeostasis as well (Miyaki and Asahara, 2012). There are a lot of evidences revealing that miRNAs targeting autophagy pathway act as key regulators in the occurrence and development of OA (Al-Modawi et al., 2019; Yu and Zhao, 2019).
miR-155 is one of the most upregulated miRNAs in human OA cartilage. Study by Adamo et al., revealed that miR-155 plays an inhibitory role in autophagy of chondrocytes and explored its targets in autophagy pathway, including ULK1 participated in the startup phase of autophagy, ATG14 responsible for recruiting autophagy-specific PI3 kinase complex, ATG5, ATG3, LC3, and GABA type A receptor associated protein like 1 (GABARAPL1/ATG8L) involved in the formation of autophagosomes, and forkhead box O3 (FOXO3) which is the key transcription factor of autophagy-related genes (D'Adamo et al., 2016) (Figure 1).
In addition, other studies explored the regulatory effect of miRNAs on sirtuin1 (SIRT1) which is another crucial signaling molecule of autophagy pathway. MiR-34a, miR-449a and miR-9 have been demonstrated to significantly reduce the expression of SIRT1 in chondrocytes (Figure 1). Inhibition of miR-34a, or miR-449a, or miR-9 can alleviate cartilage damage by upregulating the expression of SIRT1, indicating an improved prognosis of OA (Park et al., 2016; Yan et al., 2016; D'Adamo et al., 2017a).
miR-140-5p and miR-149 could target fucosyltransferase1 (FUT1). An in vitro experiment proved that overexpression of miR-140-5p or miR-149 can down-regulate the intracellular level of FUT1, promoting survival of chondrocytes by activating autophagy and inhibiting the apoptosis (Wang et al., 2018). Moreover, there are many other miRNAs that affect chondrocyte function by targeting autophagy pathway, such as miR-20 affects chondrocyte function by targeting ATG10 (He and Cheng, 2018), miR-146a enhances autophagic activity of chondrocyte by decreasing the expression of BCL2 which is an autophagy inhibitor (Chen et al., 2017) (Figure 1).
Although the autophagy control mechanism has been extensively studied, the changes of miRNA in chondrocytes are still in sore need of further research to explore the autophagy control network. So far, interfering the gene expression with miRNA in chondrocytes has been proved a promising strategy for the treatment of OA.
Other Key Molecules Regulated the Autophagy Pathway in OA
Based on the results of cell experiments, animal models of OA induced by various drugs or surgeries have been developed to study the causes and pathogenesis and to test some new therapeutic interventions. To explore the role of autophagy in cartilage of OA, mice specifically knocked out autophagy-related genes in cartilage were generated.
Cinque et al. discovered that the underlying mechanism of mild growth retardation after Atg7 gene ablation involves impaired type II collagen formation and secretion in cartilage. And they demonstrated that induction of chondral autophagy after birth is mediated by the fibroblast growth factor 18 (FGF18) through fibroblast growth factor 4 (FGFR4) and the autophagy initiation complex VPS34-BECN1 (Cinque et al., 2015) (Figure 1). This suggests that FGF18 may be a key molecule that regulates autophagy signaling pathways during OA generation and development.
Gene ablation of mTOR, an effective negative regulator of autophagy in cartilage can enhance autophagic activity, significantly reduce the degradation, apoptosis and synovium fibrosis of articular cartilage in DMM induced OA model, and effectively maintain joint homeostasis (Zhang Y. et al., 2015). Knocking out peroxisome proliferator activated receptor gamma (Pparg) in mouse cartilage accelerates the development of OA. As a ligand-activated transcription factor, the lack of PPARG in articular cartilage leads to an up-regulation of mTOR in articular cartilage, which subsequently suppresses the level of autophagy and is associated with increased apoptosis of chondrocytes (Vasheghani et al., 2015) (Figure 1).
DNA damage inducible transcript 4 (DDIT4/REDD1) can be induced by hypoxia and other stresses. The specific knockout of Ddit4 in a mouse model showed dramatically reduced LC3 and ATG5 expression levels, significant decrease of mtDNA, an increase apoptosis of chondrocytes in articular cartilage (Alvarez-Garcia et al., 2016, 2017). REDD1 has been shown to combine with thioredoxin interacting protein (TXNIP, a pro-oxidant protein) to form a protein complex in chondrocytes to induce the production of ROS, which is crucial for the induction of autophagy (Qiao et al., 2015). When the expression level of REDD1 in chondrocytes is reduced, ROS in the cytoplasm is down-regulated, which subsequently leads to the over-activation of cysteine endopeptidase ATG4, leading to delipidation of LC3 and defective autophagosome assembly (Figure 1). Therefore, we believe that REDD1 is the main molecule mediating articular cartilage homeostasis through autophagy pathway.
Deletion of Foxo transcription factors also produced OA-like changes (Matsuzaki et al., 2018). FOXO transcription factors could enhance the strength of chondrocytes to resist oxidative stress. Down-regulation of FOXO reduces the levels of autophagy-related proteins, such as LC3-II and BECN1 (Akasaki et al., 2014). Study also shows that the positive effect of FOXO3A in protecting chondrocytes by activating AMPK and autophagy pathway (Zhao et al., 2014) (Figure 1). FOXO transcription factors are also demonstrated as downstream effectors of insulin signaling under low nutrient situations, activating the transcription of glutamine synthetase and inhibiting mTOR to enhance autophagic activity (Webb and Brunet, 2014). Sirt1 haploid deficiency mouse model showed a slow-growing phenotype and was accompanied by spontaneous OA at 9 months, which may be related to the change in the number of chondrocytes (Gabay et al., 2012). Mice knocking out Sirt1 in chondrocytes develop OA in a time-dependent manner (Matsuzaki et al., 2014). Previous study also revealed that SIRT1 regulates autophagy and plays a protective role on chondrocytes through its effect on FOXO, as downstream of SIRT1 which is a conserved family of NAD+-dependent deacetylases, are the FOXO family members (Almeida and Porter, 2019) (Figure 1).
It has been shown that mouse models of OA are essential to improve our understanding of the potential molecular mechanisms of OA.
Status of OA Therapeutics Targeting Autophagy Signaling Pathway
To delay the progress of OA clinically, various treatments have been developed, including medication, non-drugs therapy and physical therapy. Medical treatment includes some conventional drugs such as non-steroidal anti-inflammatory drugs, but these drugs have serious side effects, including damage to the gastrointestinal tract and increased susceptibility to cardiovascular and kidney diseases. Non-pharmacologic treatments consist of weight loss, biomechanical intervention, electromagnetic stimulation, and shock wave therapy (Cooper et al., 2019). Surgical treatment is a routine treatment for advanced OA, usually total joint replacement (Nelson, 2018; Kloppenburg and Berenbaum, 2020).
The current treatment methods are limited to symptomatic treatment to delay the course of OA. Various new drugs developed have no obvious benefit or have serious side effects and do not applicable in clinic. As an important role in the pathogenesis of OA, autophagy has been attracted intensive attention to be a target for new OA treatment. The development of safe and effective drugs that can enhance autophagic activity or restore autophagy flux is a promising strategy for the treatment of OA.
mTOR, as a key signaling molecule in autophagy pathway, has become an important target of drugs targeting autophagy pathway. Rapamycin has been proved to reduce the expression of mTOR and delay the degradation of articular cartilage. Local application of rapamycin by intra-articular injection may be a potential therapy for OA (Takayama et al., 2014). Other drugs such as isoimperatorin and glucosamine can also activate autophagy and improve cell homeostasis by inhibiting mTOR pathway (Ouyang et al., 2017) (Figure 1). In addition, isoimperatorin improve the pathological changes induced by OA by reducing the expression of matrix metallopeptidase 13 (MMP13), RUNX family transcription factor 2 (RUNX2), collagen type X alpha 1 chain (COL10A1) and vascular endothelial growth factor A (VEGFA). However, Glucosamine shows a dual role in dose-dependent and time-dependent manner in the regulation of chondrocyte survival and apoptosis. This is because when chondrocytes are exposed to glucosamine for a short period of time, autophagic responses will be activated (called a short-term response), and long-term exposure will cause the suppression of the level of autophagy, especially pexophagy and peroxidative damage (called a long-term response) (Kang et al., 2015). Resveratrol can activate SIRT1 in order to inhibit OA disease progression (Deng et al., 2019) (Figure 1).
Drugs regulating other targets, such as Tougu Xiaotong Capsule, facilitate autophagy by balancing ATG12/LC3 coupling system (Li et al., 2014). Several autophagosome formation indexes including ATG3, ATG7, ATG12-5 and BECN1 in trehalose treated chondrocytes and the proportion of LC3-II:LC3-I increased as the dose of the drug increases, and the level of SQSTM1 decreased, indicating that trehalose can enhance autophagy flux (Tang et al., 2017). Fenofibrate (FN) as a PPARA agonist is commonly used to treat abnormal blood lipid levels in humans. Studies have found that FN can reduce the number of senescent cells, increase autophagy level and prevent cartilage degradation. The abnormal appearance of cartilage after Ppara knockdown also verified the role of FN (Nogueira-Recalde et al., 2019) (Figure 1). Transcription factor EB (TFEB) is known as a member of melanocyte inducing transcription factor (MITF)/transcription factor E (TFE) family. TFEB has indeed been proved to be the main regulator of autophagy level. It can induce the occurrence of lysosomes, promote the formation of autophagosomes and mediate their fusion (Settembre et al., 2011). The mechanism may be that on the one hand, increase the formation of LC3-II to promote the process of autophagy, on the other hand, up-regulate the expression of cathepsin B (CTSB) and increase the acidity of lysosomes to restore the function of lysosomes, ultimately rescuing the destruction of autophagy flux (Zheng et al., 2018) (Figure 1).
Conclusion
As an age-related disease, the incidence of OA increases with aging. In recent years, the basic researches of OA from various countries have centered around the age-related changes of chondrocytes. These changes include not only damaged proteins and abnormal accumulation of lipids, but also changes in ROS levels caused by impaired mitochondrial function and changes in autophagy and energy metabolism caused by oxidative stress. These subcellular disorders, on the one hand, destroy the relevant signaling pathways and normal function of cells, on the other hand they also promote catabolic activity and eventually lead to apoptosis of chondrocytes.
In the pathogenesis of OA, apoptosis and autophagy are considered to be two important links. At present, it is generally believed that autophagy as an adaptive response can reduce cell death in the early stage of OA, but with the development of OA, excessive autophagy may also cause cell death. Even so, we believe that the activation of autophagy has a positive significance for the survival of chondrocytes in the early development of OA. Although the role of autophagy disorder in the pathogenesis of OA has not been fully elucidated, autophagy as a therapeutic target for OA still shows broad clinical prospects. The targeted application of small molecule drugs to regulate the level of chondrocyte autophagy is expected to provide more options for the clinical treatment of OA.
Author Contributions
RD wrote the manuscript. HX and Z-ZL guided the planning, writing of the review manuscript, and critically amended the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the grants from National Natural Science Foundation of China (81974127, 81701383, 81871822), the Natural Science Foundation of Guangdong Province (2016A030306051, 2017A030310005), the Innovation-Driven Project of Central South University (2018CX029, 2019CX014), the Science and Technology Plan Project of Hunan Province (2018RS3029, 2017XK2039), the Shenzhen Foundation of Science and Technology (JCYJ20170306092009689), and the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (Grant Nos. 2019-RC-HL-024).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akasaki, Y., Alvarez-Garcia, O., Saito, M., Caramés, B., Iwamoto, Y., and Lotz, M. K. (2014). FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol. 66, 3349–3358. doi: 10.1002/art.38868
Alers, S., Löffler, A. S., Wesselborg, S., and Stork, B. (2012). Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 32, 2–11. doi: 10.1128/MCB.06159-11
Almeida, M., and Porter, R. M. (2019). Sirtuins and FoxOs in osteoporosis and osteoarthritis. Bone 121, 284–292. doi: 10.1016/j.bone.2019.01.018
Al-Modawi, R. N., Brinchmann, J. E., and Karlsen, T. A. (2019). Multi-pathway protective effects of microRNAs on human chondrocytes in an in vitro model of osteoarthritis. Mol. Ther. Nucleic Acids 17, 776–790. doi: 10.1016/j.omtn.2019.07.011
Alvarez-Garcia, O., Matsuzaki, T., Olmer, M., Plate, L., Kelly, J. W., and Lotz, M. K. (2017). Regulated in development and DNA damage response 1 deficiency impairs autophagy and mitochondrial biogenesis in articular cartilage and increases the severity of experimental osteoarthritis. Arthritis Rheumatol. 69, 1418–1428. doi: 10.1002/art.40104
Alvarez-Garcia, O., Olmer, M., Akagi, R., Akasaki, Y., Fisch, K. M., Shen, T., et al. (2016). Suppression of REDD1 in osteoarthritis cartilage, a novel mechanism for dysregulated mTOR signaling and defective autophagy. Osteoarthr. Cartil. 24, 1639–1647. doi: 10.1016/j.joca.2016.04.015
Ansari, M. Y., Khan, N. M., Ahmad, I., and Haqqi, T. M. (2018). Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthr. Cartil. 26, 1087–1097. doi: 10.1016/j.joca.2017.07.020
Barranco, C. (2015). Osteoarthritis: activate autophagy to prevent cartilage degeneration? Nat. Rev. Rheumatol. 11:127. doi: 10.1038/nrrheum.2015.12
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/s0092-8674(04)00045-5
Bohensky, J., Leshinsky, S., Srinivas, V., and Shapiro, I. M. (2010). Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr. Nephrol. 25, 633–642. doi: 10.1007/s00467-009-1310-y
Bolduc, J. A., Collins, J. A., and Loeser, R. F. (2019). Reactive oxygen species, aging and articular cartilage homeostasis. Free Radical Biol Med. 132, 73–82. doi: 10.1016/j.freeradbiomed.2018.08.038
Bouderlique, T., Vuppalapati, K. K., Newton, P. T., Li, L., Barenius, B., and Chagin, A. S. (2016). Targeted deletion of Atg5 in chondrocytes promotes age-related osteoarthritis. Ann. Rheum. Dis. 75, 627–631. doi: 10.1136/annrheumdis-2015-207742
Capasso, S., Alessio, N., Squillaro, T., Di Bernardo, G., Melone, M. A., Cipollaro, M., et al. (2015). Changes in autophagy, proteasome activity and metabolism to determine a specific signature for acute and chronic senescent mesenchymal stromal cells. Oncotarget 6, 39457–39468. doi: 10.18632/oncotarget.6277
Caramés, B., Hasegawa, A., Taniguchi, N., Miyaki, S., Blanco, F. J., and Lotz, M. (2012). Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 71, 575–581. doi: 10.1136/annrheumdis-2011-200557
Charlier, E., Deroyer, C., Ciregia, F., Malaise, O., Neuville, S., Plener, Z., et al. (2019). Chondrocyte dedifferentiation and osteoarthritis (OA). Biochem. Pharmacol. 165, 49–65. doi: 10.1016/j.bcp.2019.02.036
Charlier, E., Relic, B., Deroyer, C., Malaise, O., Neuville, S., Collée, J., et al. (2016). Insights on molecular mechanisms of chondrocytes death in osteoarthritis. Int. J. Mol. Sci. 17:2146. doi: 10.3390/ijms17122146
Chen, G., Gao, X., Wang, J., Yang, C., Wang, Y., Liu, Y., et al. (2017). Hypoxia-induced microRNA-146a represses Bcl-2 through Traf6/IRAK1 but not Smad4 to promote chondrocyte autophagy. Biol. Chem. 398, 499–507. doi: 10.1515/hsz-2016-0211
Choi, A. M. K., Ryter, S. W., and Levine, B. (2013). Autophagy in human health and disease. N. Engl. J. Med. 368, 1845–1846. doi: 10.1056/NEJMc1303158
Cinque, L., Forrester, A., Bartolomeo, R., Svelto, M., Venditti, R., Montefusco, S., et al. (2015). FGF signalling regulates bone growth through autophagy. Nature 528, 272–275. doi: 10.1038/nature16063
Collins, J. A., Diekman, B. O., and Loeser, R. F. (2018). Targeting aging for disease modification in osteoarthritis. Curr. Opin. Rheumatol. 30, 101–107. doi: 10.1097/BOR.0000000000000456
Cooper, C., Chapurlat, R., Al-Daghri, N., Herrero-Beaumont, G., Bruyere, O., Rannou, F., et al. (2019). Safety of oral non-selective non-steroidal anti-inflammatory drugs in osteoarthritis: what does the literature say? Drugs Aging 36(Suppl. 1), 15–24. doi: 10.1007/s40266-019-00660-1
Cutolo, M., Berenbaum, F., Hochberg, M., Punzi, L., and Reginster, J. Y. (2015). Commentary on recent therapeutic guidelines for osteoarthritis. Semin. Arthritis Rheum. 44, 611–617. doi: 10.1016/j.semarthrit.2014.12.003
D'Adamo, S., Alvarez-Garcia, O., Muramatsu, Y., Flamigni, F., and Lotz, M. K. (2016). MicroRNA-155 suppresses autophagy in chondrocytes by modulating expression of autophagy proteins. Osteoarthr. Cartil. 24, 1082–1091. doi: 10.1016/j.joca.2016.01.005
D'Adamo, S., Cetrullo, S., Guidotti, S., Borz,ì, R. M., and Flamigni, F. (2017a). Hydroxytyrosol modulates the levels of microRNA-9 and its target sirtuin-1 thereby counteracting oxidative stress-induced chondrocyte death. Osteoarthr. Cartil. 25, 600–610. doi: 10.1016/j.joca.2016.11.014
D'Adamo, S., Cetrullo, S., Minguzzi, M., Silvestri, Y., Borzi, R. M., and Flamigni, F. (2017b). MicroRNAs and autophagy: fine players in the control of chondrocyte homeostatic activities in osteoarthritis. Oxid. Med. Cell. Longev. 2017:3720128. doi: 10.1155/2017/3720128
Deng, Z., Li, Y., Liu, H., Xiao, S., Li, L., Tian, J., et al. (2019). The role of sirtuin 1 and its activator, resveratrol in osteoarthritis. Biosci. Rep. 39:BSR20190189. doi: 10.1042/BSR20190189
Feng, L., Feng, C., Wang, C.-X., Xu, D.-Y., Chen, J.-J., Huang, J.-F., et al. (2020). Circulating microRNA let-7e is decreased in knee osteoarthritis, accompanied by elevated apoptosis and reduced autophagy. Int. J. Mol. Med. 45, 1464–1476. doi: 10.3892/ijmm.2020.4534
Gabay, O., Oppenhiemer, H., Meir, H., Zaal, K., Sanchez, C., and Dvir-Ginzberg, M. (2012). Increased apoptotic chondrocytes in articular cartilage from adult heterozygous SirT1 mice. Ann. Rheum. Dis. 71, 613–616. doi: 10.1136/ard.2011.200504
Gewirtz, D. A. (2014). The four faces of autophagy: implications for cancer therapy. Cancer Res. 74, 647–651. doi: 10.1158/0008-5472.CAN-13-2966
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Han, X., Tai, H., Wang, X., Wang, Z., Zhou, J., Wei, X., et al. (2016). AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell. 15, 416–427. doi: 10.1111/acel.12446
Harrell, C. R., Markovic, B. S., Fellabaum, C., Arsenijevic, A., and Volarevic, V. (2019). Mesenchymal stem cell-based therapy of osteoarthritis: current knowledge and future perspectives. Biomed. Pharmacother. 109, 2318–2326. doi: 10.1016/j.biopha.2018.11.099
He, W., and Cheng, Y. (2018). Inhibition of miR-20 promotes proliferation and autophagy in articular chondrocytes by PI3K/AKT/mTOR signaling pathway. Biomed. Pharmacother. 97, 607–615. doi: 10.1016/j.biopha.2017.10.152
Horigome, Y., Ida-Yonemochi, H., Waguri, S., Shibata, S., Endo, N., and Komatsu, M. (2020). Loss of autophagy in chondrocytes causes severe growth retardation. Autophagy 16, 501–511. doi: 10.1080/15548627.2019.1628541
Hunter, D. J., and Bierma-Zeinstra, S. (2019). Osteoarthritis. Lancet 393, 1745–1759. doi: 10.1016/s0140-6736(19)30417-9
Jeon, O. H., Kim, C., Laberge, R. M., Demaria, M., Rathod, S., Vasserot, A. P., et al. (2017). Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781. doi: 10.1038/nm.4324
Kang, X., Yang, W., Feng, D., Jin, X., Ma, Z., Qian, Z., et al. (2017). Cartilage-specific autophagy deficiency promotes ER stress and impairs chondrogenesis in PERK-ATF4-CHOP-dependent manner. J. Bone Min. Res. 32, 2128–2141. doi: 10.1002/jbmr.3134
Kang, Y.-H., Park, S., Ahn, C., Song, J., Kim, D., and Jin, E.-J. (2015). Beneficial reward-to-risk action of glucosamine during pathogenesis of osteoarthritis. Eur. J. Med. Res. 20:89. doi: 10.1186/s40001-015-0176-7
Kim, J., Kundu, M., Viollet, B., and Guan, K.-L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kloppenburg, M., and Berenbaum, F. (2020). Osteoarthritis year in review 2019: epidemiology and therapy. Osteoarthr. Cartil. 28, 242–248. doi: 10.1016/j.joca.2020.01.002
Kongara, S., and Karantza, V. (2012). The interplay between autophagy and ROS in tumorigenesis. Front. Oncol. 2:171. doi: 10.3389/fonc.2012.00171
Lamark, T., Svenning, S., and Johansen, T. (2017). Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 61, 609–624. doi: 10.1042/EBC20170035
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Li, X., He, S., and Ma, B. (2020). Autophagy and autophagy-related proteins in cancer. Mol. Cancer 19:12. doi: 10.1186/s12943-020-1138-4
Li, X., Liu, F., Liang, W., Ye, H., Li, H., Yu, F., et al. (2014). Tougu Xiaotong capsule promotes chondrocyte autophagy by regulating the Atg12/LC3 conjugation systems. Int. J. Mol. Med. 34, 545–552. doi: 10.3892/ijmm.2014.1794
Loeser, R. F. (2017). The role of aging in the development of osteoarthritis. Trans. Am. Clin. Climatol. Assoc. 128, 44–54.
Lopez de Figueroa, P., Lotz, M. K., Blanco, F. J., and Carames, B. (2015). Autophagy activation and protection from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol. 67, 966–976. doi: 10.1002/art.39025
Martel-Pelletier, J., Barr, A. J., Cicuttini, F. M., Conaghan, P. G., Cooper, C., Goldring, M. B., et al. (2016). Osteoarthritis. Nat. Rev. Dis. Prim. 2:16072. doi: 10.1038/nrdp.2016.72
Martin-Rincon, M., Morales-Alamo, D., and Calbet, J. A. L. (2018). Exercise-mediated modulation of autophagy in skeletal muscle. Scand. J. Med. Sci. Sports 28, 772–781. doi: 10.1111/sms.12945
Matsunaga, K., Morita, E., Saitoh, T., Akira, S., Ktistakis, N. T., Izumi, T., et al. (2010). Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 190, 511–521. doi: 10.1083/jcb.200911141
Matsuzaki, T., Alvarez-Garcia, O., Mokuda, S., Nagira, K., Olmer, M., Gamini, R., et al. (2018). FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci. Transl. Med. 10:eaan0746. doi: 10.1126/scitranslmed.aan0746
Matsuzaki, T., Matsushita, T., Takayama, K., Matsumoto, T., Nishida, K., Kuroda, R., et al. (2014). Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann. Rheum. Dis. 73, 1397–1404. doi: 10.1136/annrheumdis-2012-202620
Menon, M. B., and Dhamija, S. (2018). Beclin 1 phosphorylation - at the center of autophagy regulation. Front. Cell Dev. Biol. 6:137. doi: 10.3389/fcell.2018.00137
Miyaki, S., and Asahara, H. (2012). Macro view of microRNA function in osteoarthritis. Nat. Rev. Rheumatol. 8, 543–552. doi: 10.1038/nrrheum.2012.128
Mizushima, N. (2010). The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 22, 132–139. doi: 10.1016/j.ceb.2009.12.004
Nakamura, S., and Yoshimori, T. (2017). New insights into autophagosome-lysosome fusion. J. Cell. Sci. 130, 1209–1216. doi: 10.1242/jcs.196352
Nelson, A. E. (2018). Osteoarthritis year in review 2017: clinical. Osteoarthr. Cartil. 26, 319–325. doi: 10.1016/j.joca.2017.11.014
Noda, T., Matsunaga, K., Taguchi-Atarashi, N., and Yoshimori, T. (2010). Regulation of membrane biogenesis in autophagy via PI3P dynamics. Semin. Cell Dev. Biol. 21, 671–676. doi: 10.1016/j.semcdb.2010.04.002
Nogueira-Recalde, U., Lorenzo-Gómez, I., Blanco, F. J., Loza, M. I., Grassi, D., Shirinsky, V., et al. (2019). Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 45, 588–605. doi: 10.1016/j.ebiom.2019.06.049
O'Neill, T. W., McCabe, P. S., and McBeth, J. (2018). Update on the epidemiology, risk factors and disease outcomes of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 32, 312–326. doi: 10.1016/j.berh.2018.10.007
Ouyang, J., Jiang, H., Fang, H., Cui, W., and Cai, D. (2017). Isoimperatorin ameliorates osteoarthritis by downregulating the mammalian target of rapamycin C1 signaling pathway. Mol. Med. Rep. 16, 9636–9644. doi: 10.3892/mmr.2017.7777
Park, K. W., Lee, K. M., Yoon, D. S., Park, K. H., Choi, W. J., Lee, J. W., et al. (2016). Inhibition of microRNA-449a prevents IL-1β-induced cartilage destruction via SIRT1. Osteoarthr. Cartil. 24, 2153–2161. doi: 10.1016/j.joca.2016.07.002
Qiao, S., Dennis, M., Song, X., Vadysirisack, D. D., Salunke, D., Nash, Z., et al. (2015). A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat. Commun. 6:7014. doi: 10.1038/ncomms8014
Qin, N., Wei, L., Li, W., Yang, W., Cai, L., Qian, Z., et al. (2017). Local intra-articular injection of resveratrol delays cartilage degeneration in C57BL/6 mice by inducing autophagy via AMPK/mTOR pathway. J. Pharmacol. Sci. 134, 166–174. doi: 10.1016/j.jphs.2017.06.002
Rahmati, M., Nalesso, G., Mobasheri, A., and Mozafari, M. (2017). Aging and osteoarthritis: central role of the extracellular matrix. Ageing Res. Rev. 40, 20–30. doi: 10.1016/j.arr.2017.07.004
Ren, J., and Zhang, Y. (2018). Targeting autophagy in aging and aging-related cardiovascular diseases. Trends Pharmacol. Sci. 39, 1064–1076. doi: 10.1016/j.tips.2018.10.005
Ribeiro, M., López de Figueroa, P., Nogueira-Recalde, U., Centeno, A., Mendes, A. F., Blanco, F. J., et al. (2016). Diabetes-accelerated experimental osteoarthritis is prevented by autophagy activation. Osteoarthr. Cartil. 24, 2116–2125. doi: 10.1016/j.joca.2016.06.019
Rim, Y. A., Nam, Y., and Ju, J. H. (2020). The role of chondrocyte hypertrophy and senescence in osteoarthritis initiation and progression. Int. J. Mol. Sci. 21:2358. doi: 10.3390/ijms21072358
Roca-Agujetas, V., de Dios, C., Lestón, L., Marí, M., Morales, A., and Colell, A. (2019). Recent insights into the mitochondrial role in autophagy and its regulation by oxidative stress. Oxid. Med. Cell. Longev. 2019:3809308. doi: 10.1155/2019/3809308
Scherz-Shouval, R., and Elazar, Z. (2007). ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 17, 422–427. doi: 10.1016/j.tcb.2007.07.009
Settembre, C., Di Malta, C., Polito, V. A., Garcia Arencibia, M., Vetrini, F., Erdin, S., et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. doi: 10.1126/science.1204592
Shaid, S., Brandts, C. H., Serve, H., and Dikic, I. (2013). Ubiquitination and selective autophagy. Cell Death Different. 20, 21–30. doi: 10.1038/cdd.2012.72
Shang, L., Chen, S., Du, F., Li, S., Zhao, L., and Wang, X. (2011). Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U.S.A. 108, 4788–4793. doi: 10.1073/pnas.1100844108
Shapiro, I. M., Layfield, R., Lotz, M., Settembre, C., and Whitehouse, C. (2014). Boning up on autophagy: the role of autophagy in skeletal biology. Autophagy 10, 7–19. doi: 10.4161/auto.26679
Su, L.-J., Zhang, J.-H., Gomez, H., Murugan, R., Hong, X., Xu, D., et al. (2019). Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid. Med. Cell. Longev. 2019:5080843. doi: 10.1155/2019/5080843
Taheem, D. K., Foyt, D. A., Loaiza, S., Ferreira, S. A., Ilic, D., Auner, H. W., et al. (2018). Differential regulation of human bone marrow mesenchymal stromal cell chondrogenesis by hypoxia inducible factor-1α hydroxylase inhibitors. Stem Cells 36, 1380–1392. doi: 10.1002/stem.2844
Tai, H., Wang, Z., Gong, H., Han, X., Zhou, J., Wang, X., et al. (2017). Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 13, 99–113. doi: 10.1080/15548627.2016.1247143
Takayama, K., Kawakami, Y., Kobayashi, M., Greco, N., Cummins, J. H., Matsushita, T., et al. (2014). Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res. Ther. 16:482. doi: 10.1186/s13075-014-0482-4
Tang, Q., Zheng, G., Feng, Z., Chen, Y., Lou, Y., Wang, C., et al. (2017). Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis. 8:e3081. doi: 10.1038/cddis.2017.453
van den Berg, W. B. (2011). Osteoarthritis year 2010 in review: pathomechanisms. Osteoarthr. Cartil. 19, 338–341. doi: 10.1016/j.joca.2011.01.022
Vasheghani, F., Zhang, Y., Li, Y.-H., Blati, M., Fahmi, H., Lussier, B., et al. (2015). PPARγ deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann. Rheum. Dis. 74, 569–578. doi: 10.1136/annrheumdis-2014-205743
Vina, E. R., and Kwoh, C. K. (2018). Epidemiology of osteoarthritis: literature update. Curr. Opin. Rheumatol. 30, 160–167. doi: 10.1097/BOR.0000000000000479
Vuppalapati, K. K., Bouderlique, T., Newton, P. T., Kaminskyy, V. O., Wehtje, H., Ohlsson, C., et al. (2015). Targeted deletion of autophagy genes atg5 or atg7 in the chondrocytes promotes caspase-dependent cell death and leads to mild growth retardation. J. Bone Min. Res. 30, 2249–2261. doi: 10.1002/jbmr.2575
Wang, M., Sampson, E. R., Jin, H., Li, J., Ke, Q. H., Im, H.-J., et al. (2013). MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 15:R5. doi: 10.1186/ar4133
Wang, Z., Hu, J., Pan, Y., Shan, Y., Jiang, L., Qi, X., et al. (2018). miR-140-5p/miR-149 affects chondrocyte proliferation, apoptosis, and autophagy by targeting FUT1 in osteoarthritis. Inflammation 41, 959–971. doi: 10.1007/s10753-018-0750-6
Webb, A. E., and Brunet, A. (2014). FOXO transcription factors: key regulators of cellular quality control. Trends Biochem. Sci. 39, 159–169. doi: 10.1016/j.tibs.2014.02.003
White, E., and Lowe, S. W. (2009). Eating to exit: autophagy-enabled senescence revealed. Genes Dev. 23, 784–787. doi: 10.1101/gad.1795309
Yan, S., Wang, M., Zhao, J., Zhang, H., Zhou, C., Jin, L., et al. (2016). MicroRNA-34a affects chondrocyte apoptosis and proliferation by targeting the SIRT1/p53 signaling pathway during the pathogenesis of osteoarthritis. Int. J. Mol. Med. 38, 201–209. doi: 10.3892/ijmm.2016.2618
Yu, L., Chen, Y., and Tooze, S. A. (2018). Autophagy pathway: cellular and molecular mechanisms. Autophagy 14, 207–215. doi: 10.1080/15548627.2017.1378838
Yu, Y., and Zhao, J. (2019). Modulated autophagy by micrornas in osteoarthritis chondrocytes. BioMed. Res. Int. 2019:1484152. doi: 10.1155/2019/1484152
Zhang, F.-J., Luo, W., and Lei, G.-H. (2015). Role of HIF-1α and HIF-2α in osteoarthritis. Joint Bone Spine 82, 144–147. doi: 10.1016/j.jbspin.2014.10.003
Zhang, Y., Vasheghani, F., Li, Y. H., Blati, M., Simeone, K., Fahmi, H., et al. (2015). Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 74, 1432–1440. doi: 10.1136/annrheumdis-2013-204599
Zhao, X., Petursson, F., Viollet, B., Lotz, M., Terkeltaub, R., and Liu-Bryan, R. (2014). Peroxisome proliferator-activated receptor γ coactivator 1α and FoxO3A mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheumatol. 66, 3073–3082. doi: 10.1002/art.38791
Zheng, G., Zhan, Y., Li, X., Pan, Z., Zheng, F., Zhang, Z., et al. (2018). TFEB, a potential therapeutic target for osteoarthritis via autophagy regulation. Cell Death Dis. 9:858. doi: 10.1038/s41419-018-0909-y
Keywords: apoptosis, autophagy, cartilage, chondrocyte, osteoarthritis
Citation: Duan R, Xie H and Liu Z-Z (2020) The Role of Autophagy in Osteoarthritis. Front. Cell Dev. Biol. 8:608388. doi: 10.3389/fcell.2020.608388
Received: 20 September 2020; Accepted: 06 November 2020;
Published: 25 November 2020.
Edited by:
Hang Lin, University of Pittsburgh, United StatesReviewed by:
Louis Charles Penning, Utrecht University, NetherlandsUmberto Galderisi, University of Campania Luigi Vanvitelli, Italy
Copyright © 2020 Duan, Xie and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zheng-Zhao Liu, bGl1emhlbmd6aGFvQGNzdS5lZHUuY24=; Hui Xie, aHVpeGllQGNzdS5lZHUuY24=