 Young-Sun Lee
Young-Sun Lee Gyun Jee Song2,3
Gyun Jee Song2,3 Hee-Sook Jun
Hee-Sook Jun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 21 January 2021
Sec. Cell Growth and Division
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.605110
Betacellulin (BTC), an epidermal growth factor family, is known to promote β-cell regeneration. Recently, pancreatic α-cells have been highlighted as a source of new β-cells. We investigated the effect of BTC on α-cells. Insulin+glucagon+ double stained bihormonal cell levels and pancreatic and duodenal homeobox-1 expression were increased in mice treated with recombinant adenovirus-expressing BTC (rAd-BTC) and β-cell-ablated islet cells treated with BTC. In the islets of rAd-BTC-treated mice, both BrdU+glucagon+ and BrdU+insulin+ cell levels were significantly increased, with BrdU+glucagon+ cells showing the greater increase. Treatment of αTC1-9 cells with BTC significantly increased proliferation and cyclin D2 expression. BTC induced phosphorylation of ErbB receptors in αTC1-9 cells. The proliferative effect of BTC was mediated by ErbB-3 or ErbB-4 receptor kinase. BTC increased phosphorylation of ERK1/2, AKT, and mTOR and PC1/3 expression and GLP-1 production in α-cells, but BTC-induced proliferation was not changed by the GLP-1 receptor antagonist, exendin-9. We suggest that BTC has a direct role in α-cell proliferation via interaction with ErbB-3 and ErbB-4 receptors, and these increased α-cells might be a source of new β-cells.
Diabetes mellitus (DM) is characterized by prolonged high blood glucose levels due to absolute or relative deficiency of insulin. One of the beneficial strategies postulated as a possible therapy for diabetes is to increase functional insulin-producing β-cells via endogenous pancreatic β-cell regeneration. The pancreatic β-cell regeneration could occur through promotion of existing β-cell replication or conversion of other cells into β-cell (Thorel et al., 2010; Aguayo-Mazzucato and Bonner-Weir, 2018).
Pancreatic islets are organized into glucagon-producing α-cells, insulin-secreting β-cells, somatostatin-releasing δ-cells, pancreatic polypeptide-producing PP-cells, and ghrelin-secreting ε-cells (Baetens et al., 1977; Berts et al., 1997; Wierup et al., 2014; Briant et al., 2016). The pancreatic β-cells produce insulin, which stimulates glucose uptake by muscle, adipose tissue, and liver, contributing to the lowering of blood glucose levels. The pancreatic α-cells are known to play an important role in blood glucose homeostasis through production of glucagon, which stimulates hepatic glucose production. However, recent reports indicated that the possible conversion of non-β-cells in the islets to β-cells to replenish the reduced β-cell mass is possible (Thorel et al., 2010; Aguayo-Mazzucato and Bonner-Weir, 2018; Gromada et al., 2018). In this regard, α-cells have been highlighted in the islets as a source of new β-cells and a direct progenitor of β-cells under conditions of extreme destruction of β-cells (Thorel et al., 2010).
Betacellulin (BTC), a member of the epidermal growth factor (EGF) family, was originally isolated from mouse beta cell tumor (betaTC-3) as a growth–promoting factor and is known to be expressed in pancreatic α-cells, β-cells, and duct cells in adult humans (Miyagawa et al., 1999). BTC acts through binding to four ErbB tyrosine kinase receptors (EGF-R/ErbB-1, neu/ErbB-2, ErbB-3, and ErbB-4) (Wieduwilt and Moasser, 2008). It was reported that EGF-R-deficiency in mice impairs pancreatic islet development (Miettinen et al., 2000). In addition, BTC is known to promote β-cell proliferation and insulin secretion and BTC induces neogenesis of β-cells and increases the number of islet-like cell clusters, consisting primarily of β-cells (Yamamoto et al., 2000). Several studies have also shown the regenerative effects of BTC on β-cells to ameliorate diabetes (Li et al., 2003; Song et al., 2014).
We have previously found that administration of recombinant adenovirus expressing BTC improved hyperglycemia by promoting regeneration of β-cells in streptozotocin-induced diabetic mice and that β-cell proliferation was one of the mechanisms for the regeneration of β-cells by BTC (Shin et al., 2008). However, the effects of BTC on α-cells remains uncharacterized. In this study, we investigated the possible contribution of BTC on β-cell regeneration via α-cell proliferation and the mechanisms involved in α-cell proliferation by BTC.
C57BL/6 mice were obtained from the Korea Research Institute of Bioscience and Biotechnology (Daejeon, Korea). These mice were maintained at the facility at Gachon University under a 12 h light:12 h dark photoperiod. Animals were fed ad libitum on a standard rodent diet. All animal experiments were carried out under a protocol approved by the Institutional Animal Care and Use Committee at Lee Gil Ya Cancer and Diabetes Institute, Gachon University.
Recombinant adenovirus (rAd) producing BTC (rAd-BTC) and the control, rAd producing β-galactosidase (rAd-βgal) were constructed and produced, as previously reported (Shin et al., 2008). The recombinant adenoviruses were amplified in a human embryonic kidney cell line (HEK-293). After purification of virus by CsCl-gradient ultracentrifugation, the viral titer was determined by 50% tissue culture infectious dose (TCID50).
β-cell destruction was achieved in C57BL/6 mice by i.p. injection of STZ (Sigma, 150 mg/kg), a β-cell specific toxin. The mice were monitored for the development of hyperglycemia using a glucometer. STZ-induced diabetic mice (blood glucose levels > 300 mg/dl for 3 consecutive days) were injected via the tail vein with rAd-BTC or rAd-βgal with 3 × 109 plaque-forming units (pfu). After viral injection, C57BL/6 mice were injected with 5-bromodeoxyuridine (BrdU; Sigma, 100 mg/kg) every day for 4 weeks.
Mice were not fed for 4 h and a 20% glucose solution was injected intraperitoneally (2 g/kg body weight). Blood glucose levels were measured at 0, 30, 60, 90, and 120 min following glucose injection.
C57BL/6 mice were sacrificed at 4 weeks after rAd-BTC or rAd-βgal injection. Pancreata were removed, fixed in 10% formalin, and embedded in paraffin. More than 200 serial sections (4 μm thick) were prepared from each pancreas, and every 20–25th section was used for immunohistochemical analysis. The tissue sections were boiled (100°C for 10 min, 10 mM sodium citrate, pH 6.0) for antigen retrieval, and blocked with blocking solution (DAKO, Carpinteria, CA, USA). The sections were then incubated with primary antibody solution: guinea-pig anti-insulin (DAKO, 1:100), rabbit anti-glucagon (DAKO, 1:100), mouse anti-PDX-1 (DSHB, Iowa, IA, 1:100) or mouse anti-BrdU (DAKO, 1:50). Fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology, 1:200), Texas Red (TR)-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology, 1:200) or Alexa-Fluor-633-conjugated goat anti-guinea-pig IgG (Thermo Fisher Scientific, Rockford, IL, 1:200) were used as secondary antibodies. Fluorescence was imaged using a laser scanning confocal fluorescent microscope (LSM 700, Carl Zeiss MicroImaging, Jena, Germany) and colocalization was analyzed using the ZEN 2009 Analysis Program.
Pancreatic islets were isolated from 8 to 10-week-old male C57BL/6 mice as previously described (Jun et al., 1999). Intact islets were dissociated at 37°C in Accutase (Millipore), given STZ (1 mM) for 15 h, washed with fresh media, and then cultured with BTC (1 nM). The islet cells or the αTC1-9 cells were fixed in 4% paraformaldehyde, permeabilized in permeabilization buffer (Thermo Fisher Scientific), blocked in blocking solution (Thermo Fisher Scientific), and then incubated with mouse anti-glucagon (Sigma, 1:100), rabbit anti-glucagon (DAKO, 1:100), mouse anti-PDX-1 (DSHB, Iowa, IA, 1:100) or mouse anti-BrdU (DAKO, 1:50) antibodies. FITC-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology, 1:200) or TR-conjugated goat anti-mouse IgG or anti-rabbit IgG (Santa Cruz Biotechnology, 1:200) were used as secondary antibodies. Fluorescence was imaged using a laser scanning confocal fluorescent microscope (LSM 700).
The αTC1 clone 9 (αTC1-9) was obtained from American Type Culture Collection (ATCC, CRL-2350). This is a pancreatic α-cell line that produces glucagon, but not preproinsulin mRNA. αTC1-9 were cultured in Dulbecco's modified Eagle medium (DMEM) containing 16.7 mM glucose supplemented with 10% heat-inactivated dialyzed fetal bovine serum, 15 mM HEPES, 0.1 mM non-essential amino acids, and 0.02% BSA under an atmosphere of 95% humidified air-5% CO2 at 37°C.
RT-PCR was performed under the following conditions: ErbB-1 and ErbB-2: 35 cycles of 1 min at 94°C, 1 min at 60°C and 1 min at 72°C. ErbB-3 and ErbB-4: 35 cycles of 1 min at 94°C, 1 min at 58°C and 1 min at 72°C. Real-time quantitative PCR was performed using SYBR master mix (Applied Biosystems, Carlsbad, CA, USA) and carried out in a 7900HT fast real-time PCR system (Applied Biosystems). The specific PCR primers are given in Supplementary Table 1. The relative copy number was calculated using the threshold crossing point (Ct) as calculated by the 7900HT fast real time PCR software combined with the delta-delta Ct calculations.
αTC1-9 cells were seeded in 96-well plates at a density of 2 × 104 cells (100 μL) /well and cultured with or without BTC (0, 0.25, 0.5, 1 nM) for 24 h or 48 h (BTC was added every 24 h). The cells were pulsed with [3H]-thymidine (1 μCi/well). Eight hours after [3H]-thymidine addition, the cells were analyzed for [3H]-thymidine incorporation using a scintillation β-counter, 1450 LSC & Luminescence Counter MicroBeta TriLux (Perkin Elmer). αTC1-9 cells were seeded in 96-well plates at a density of 2 × 104 cells (100 μL) /well and cultured with BTC (1 nM) and with or without ErbB-1 inhibitor: AG1478 (4 nM) (Calbiochem, La Jolla, CA, USA), ErbB-2 inhibitor: AG825 (4 nM) (Calbiochem), ErbB-1,-2 and−3 inhibitor: AZD8931 (8 nM) (Selleckchem, Houston, TX, USA, IC50 = 4, 3, and 4 nM), ErbB-1,-2 and−4 inhibitor: AST 1306 tosylate (1.6 nM) (Selleckchem, IC50 = 0.5, 3.0, and 0.8 nM) for 24 or 48 h. Cell proliferation was measured by incubating with 10 μl of D-Plus TM CCK reagent (Dongjin LS, Seoul, Korea) for 2 h at 37°C. The optical density at 450 nm was determined and control cells were considered to represent 100%.
Whole lysates of cells were prepared as previously described. Western blotting was performed with rabbit-anti-cyclin D2 (Cell Signaling Technology), mouse-anti-cyclin E (Cell Signaling Technology), rabbit-cyclin A2 (NOVUS Biologicals), mouse-cyclin D3 (Cell Signaling Technology), mouse-anti-β actin (Santa Cruz Biotechnology), rabbit-anti-ErbB-1 (Santa Cruz Biotechnology), rabbit-anti-ErbB-2 (Santa Cruz Biotechnology), rabbit-anti-ErbB-3 (Santa Cruz Biotechnology), mouse-anti-ErbB-4 (Santa Cruz Biotechnology), mouse-anti-phospho-Tyr (Millipore), rabbit-anti-phospho-Erk1/2 (Cell Signaling Technology), rabbit-anti-Erk1/2 (Cell Signaling Technology), rabbit-anti-phospho-Akt (Ser473) (Cell Signaling Technology), rabbit-anti-Akt (Cell Signaling Technology), rabbit-anti-phospho-mTOR (Cell Signaling Technology), rabbit-anti-mTOR (Cell Signaling Technology), or mouse-anti-GAPDH (Santa Cruz Biotechnology).
αTC1-9 cells were treated with BTC (0, 0.5, 1, or 2 nM) for 24 h. Active GLP-1 (7-36) secretion was analyzed with cultured media. Rat islets were isolated and STZ (1 mM) was added for 15 h to ablate the β-cells. The STZ-treated islets were given new media and treated with BTC (1 nM added for 24 h) for 48 h. Active GLP-1 (7–36) secretion was measured and expressed as a percentage of the STZ-treated islet value. GLP-1 (7–36) secretion was analyzed by enzyme-linked immunosorbent assay (ELISA) (R&D System), with values normalized to protein.
Data are presented as means ± SD or ± SE. Statistical analysis was performed using an unpaired parametric Student's t-test for two groups or ANOVA followed by Fisher's protected least significant difference test for multiple groups. P < 0.05 was accepted as significant.
To confirm that rAd-BTC treatment efficiently regulates blood glucose levels, we injected rAd-BTC or rAd-βgal (4 × 109 pfu) into STZ-induced diabetic C57BL/6 male mice and blood glucose levels were monitored for 4 weeks. Blood glucose levels in rAd-BTC-treated mice were significantly decreased, compared with those in rAd-βgal-treated mice (Supplementary Figure 1A). Intraperitoneal glucose tolerance tests at 2 weeks after rAd-BTC treatment showed that blood glucose levels in rAd-BTC-treated mice were significantly lower at the 60, 90, and 120 min points following glucose injection compared with the rAd-βgal-treated mice (Supplementary Figure 1B), indicating that rAd-BTC treatment showed the glucose-lowering effects, as we previously found (Shin et al., 2008).
Recent studies show plasticity between pancreatic α- and β-cells. α-cells can be converted into new β-cells via a bihormonal insulin+glucagon+ transitional state in animals (Parker et al., 2002; Thorel et al., 2010; Habener and Stanojevic, 2012). We therefore examined the population of bihormonal insulin+glucagon+ cells in rAd-BTC-treated mice. Immunostaining of pancreatic sections from rAd-BTC- or rAd-βgal-treated C57BL/6 mice with anti-glucagon and anti-insulin antibodies revealed that levels of insulin+glucagon+ double stained bihormonal cells were higher in rAd-BTC-treated mice than in rAd-βgal-treated mice (Figures 1A,B). To investigate whether BTC induces the expression of β-cell transcription factors such as PDX-1 in α-cells, isolated islets from C57BL/6 mice were treated with STZ to ablate β-cells, which were incubated with BTC for 24 h, and then PDX-1 mRNA expression was analyzed. We found that PDX-1 mRNA expression was significantly increased by BTC treatment (Figure 1C). In addition, immunocytochemical analysis showed that glucagon+ PDX-1+ double positive cells were observed after BTC treatment. The intensity of PDX-1 staining in glucagon-producing α-cells was significantly increased in BTC-treated cells compared with cells without BTC treatment (Figures 1D,E). To investigate whether BTC induces the expression of PDX-1 in α-cells of rAd-BTC-treated mice, the pancreatic sections from rAd-BTC- or rAd-βgal-treated C57BL/6 mice were immunostained with anti-PDX-1 and anti-glucagon antibodies. The levels of glucagon+PDX-1+ double stained cells in glucagon-producing α-cells were higher in rAd-BTC-treated mice than in rAd-βgal-treated mice (Figures 1F,G).
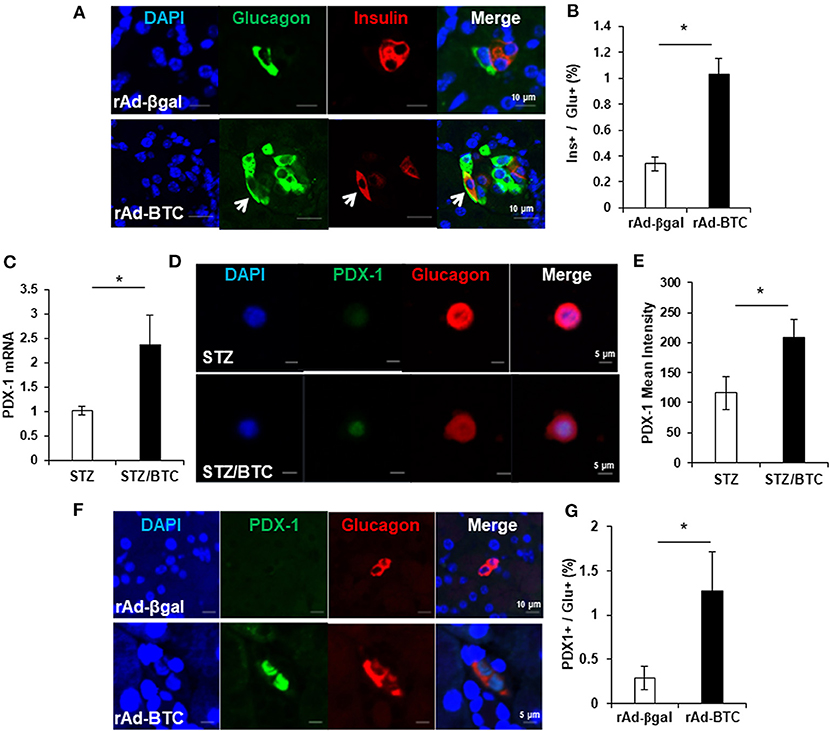
Figure 1. Increase of insulin+glucagon+ bihormonal cells in rAd-BTC-injected C57BL/6 mice. STZ-induced diabetic mice were treated with rAd-BTC (black squares, n = 5) or rAd-βgal (white squares, n = 4). (A) At 4 weeks after rAd-BTC or rAd-βgal virus injection, pancreatic sections were stained with anti-glucagon (Glu) and anti-insulin (Ins) antibodies. (B) The bihormonal (ins+glu+) cells were counted and expressed as a percentage of the number of glucagon+ cells in rAd-BTC or rAd-βgal injected mice (n = 92 or 40 islet). (C) Islet cells prepared form C57BL/6 mice (islet number = 400~450/group) were given STZ (1 mM) for 15 h followed by BTC (1 nM) for 24 or 48 h (add per 24 h). After 24 h, the expression of PDX-1 mRNA was analyzed by real-time quantitative PCR and normalized by cyclophilin expression. (D) After 48 h, the islet cells were double-stained with anti-PDX-1 and anti-glucagon antibodies. (E) The mean intensity was measured by confocal laser scanning microscope LSM 700 (Carl Zeiss) and analyzed with STZ- or STZ/BTC-treated islet cells (n = 13 or 17). (F) At 4 weeks after rAd-BTC virus injection, pancreatic sections were stained with anti-PDX-1 and anti-glucagon (Glu) antibodies. (G) The glucagon+ PDX-1+ cells were counted and expressed as a percentage of the number of glucagon+ cells in rAd-BTC or rAd-βgal injected mice (n = 54 or 101 islet). Data are represented as means ± SE. *P < 0.05 compared with rAd-βgal-treated mice or STZ treated cell.
Since insulin+glucagon+ bihormonal cells were increased by BTC, we hypothesized that BTC may increase the proliferation of α-cells and these increased α-cells may contribute to β-cells regeneration. To determine the validity of this, we first determined which cells are proliferated in rAd-BTC-treated mice by injecting BrdU (daily, i.p) for 4 weeks. BrdU-positive cells in the islets were significantly increased in rAd-BTC-treated mice compared with the rAd-βgal-treated mice. BrdU+insulin+ cells were significantly increased in the islets of rAd-BTC-treated mice compared with rAd-βgal-treated mice, as expected. Interestingly, BrdU+glucagon+ cells showed a greater increase than BrdU+insulin+ cells (Figures 2A,B). To investigate whether the cell numbers in an islet section was increased by BTC treatment, we counted the cell number in the islets. The average cell number in an islet was significantly increased in rAd-BTC-treated mice compared with rAd-βgal-treated mice (Figure 2C). In addition, we calculated the pancreatic α- or β-cell proportion. We found that the pancreatic α-cell proportion was similar between STZ-rAd-βgal-treated mice and STZ-rAd-BTC-treated mice. However, the pancreatic β-cell proportion was significantly increased in rAd-BTC-administered mice, even though the proliferation of α-cells was increased in STZ-rAd-BTC-treated mice (Figure 2D).
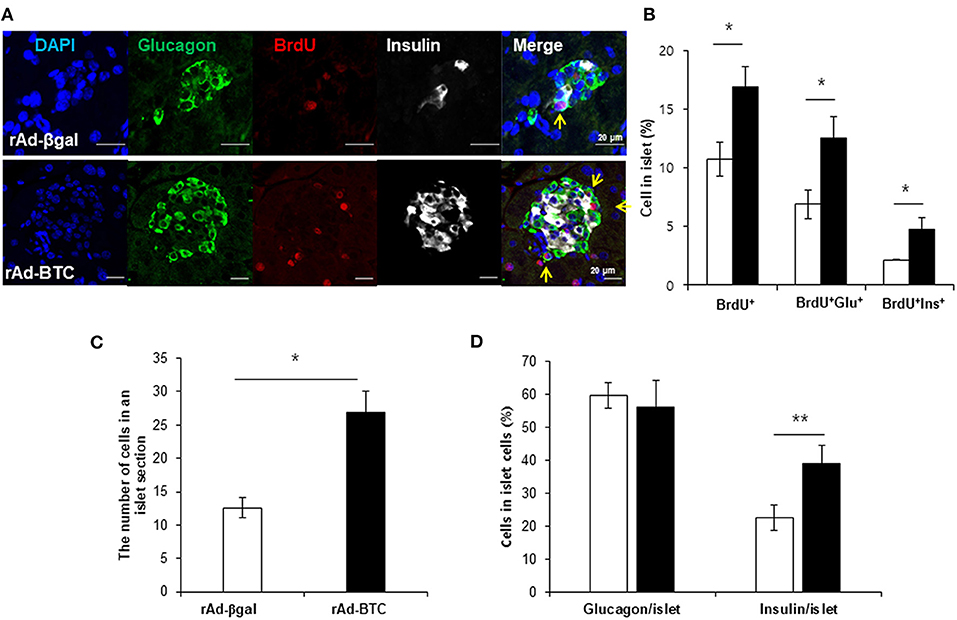
Figure 2. Proliferation of α-cells is increased in rAd-BTC-injected mice. STZ-induced diabetic C57BL/6 mice were injected with rAd-BTC (black, n = 3) or rAd-βgal (white, n = 3). Mice were then injected with BrdU (100 mg/kg, i.p) for 4 weeks, beginning on the first day following virus injection. (A) At 4 weeks after virus injection, pancreatic sections were stained with anti-glucagon (Glu), anti-insulin (Ins), and anti-BrdU antibodies. (B) The total BrdU+, BrdU+Glu+, and BrdU+Ins+ cell levels were calculated as a percentage of the number of total islet cells in rAd-BTC or rAd-βgal injected mice (n = 105 or 69 islet). (C) The cell number in an islet section were calculated as an average of the cell number per islet in rAd-BTC or rAd-βgal injected mice (n = 101 or 23 islet). (D) The Glu+ or Ins+ in islet cells were calculated as a percentage of the number of total islet cells in rAd-BTC or rAd-βgal injected mice (n = 105 or 56 islet). Data are means ± SE. *P < 0.05, **P < 0.01 compared with rAd-βgal-treated mice.
To examine whether BTC signaling directly increases proliferation in α-cells, αTC1-9 cells were cultured in the presence of BrdU. We found that BrdU incorporated α-cells were increased in BTC-treated αTC1-9 cells (Figure 3A). [3H]-thymidine incorporation assays and CCK8 proliferation assays also showed that BTC significantly increased proliferation of αTC1-9 cells in a dose dependent manner (Figures 3B,C). When we analyzed the expression of cyclins, which are important proteins that control the proliferation of cells through the cell cycle, the expression of cyclin D2 mRNA and protein was significantly increased; however, the expression of cyclin A2, cyclin E, and cyclin D3 was not changed in BTC-treated αTC1-9 cells (Figures 3D–H).
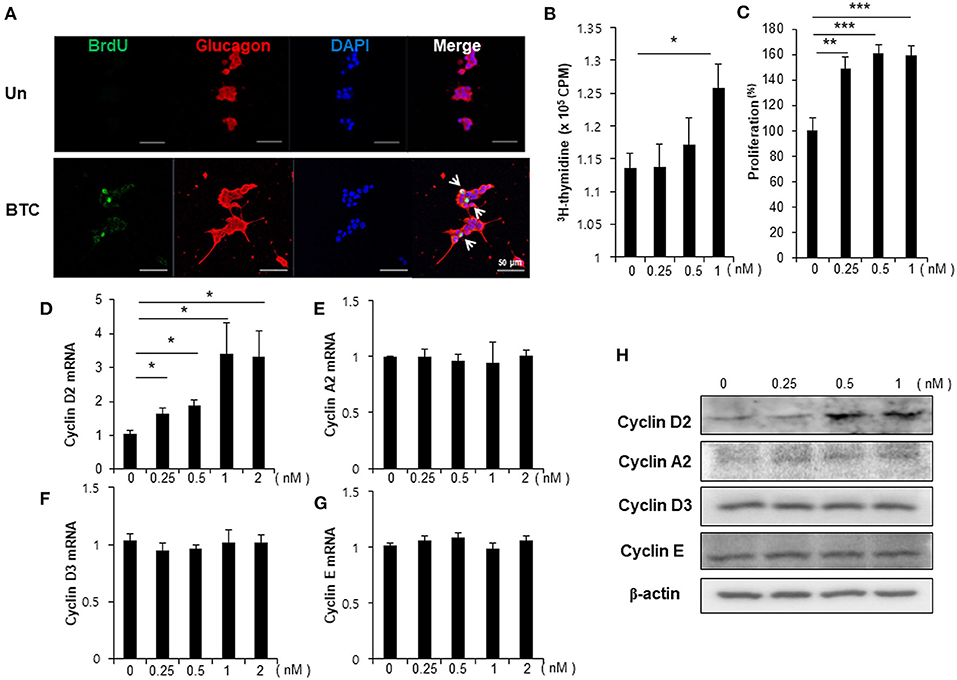
Figure 3. Proliferation of α-cells is increased in BTC-treated αTC1-9 cells. αTC1-9 cells were cultured without (Un) or with BTC (1 nM) for 2 days. (A) αTC1-9 cells were pulsed with BrdU for 24 h, and cells were then double-stained with anti-BrdU and anti-glucagon antibodies. (B) αTC1-9 cells were pulsed with [3H]-thymidine for 6 h and [3H]-thymidine incorporation was measured. (C) αTC1-9 cells were incubated with BTC (1 nM) for 24 h. Viable cells were measured by CCK8 assay. The data are presented as a percentage of the untreated control. The expression of (D) cyclin D2, (E) cyclin A2, (F) cyclin D3, (G) cyclin E mRNA was analyzed by real-time quantitative PCR, and (H) the proteins were analyzed by western blotting. Data are means ± SE from three to four independent experiments and are expressed as a ratio of the control (D–G). *P < 0.05, **P < 0.005, ***P < 0.0005 compared with untreated cells.
As BTC binds and activates ErbB receptors, we first examined the expression of ErbB receptors in αTC1-9 cells. The expression of ErbB-1, ErbB-2, ErbB-3, and ErbB-4 mRNA was detected in αTC1-9 cells, and islets from C57BL/6 mice also expressed all ErbB receptors (Figure 4A).
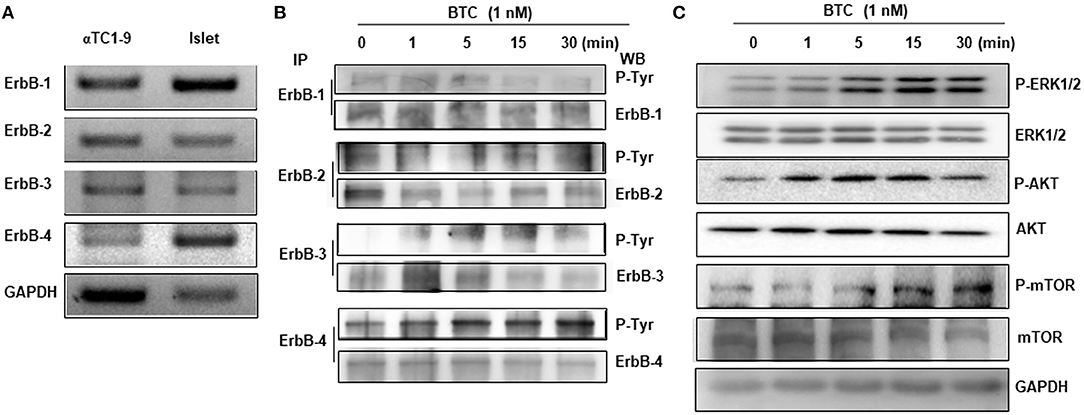
Figure 4. Expression and phosphorylation of ErbBs in BTC-treated αTC1-9 cells. (A) RT-PCR analysis of the expression of ErbB-1,−2,−3, and−4 in αTC1-9 cells and isolated C57BL/6 mouse islets. (B) αTC1-9 cells were treated with BTC for the indicated time. Cells were harvested and cell lysate was immunoprecipitated (IP) with anti-ErbB antibodies and then detected by phosphorylated ErbBs by western blot using phospho-Tyr antibody. (C) p-ERK1/2, p-AKT (Ser473), and p-mTOR, as well as ERK1/2, AKT, and mTOR were measured by western blot of total cell extracts. Results are representative of three independent experiments.
We then investigated which ErbB receptors are activated by BTC treatment. αTC1-9 cells were treated with 1 nM BTC for 0, 1, 5, 15, and 30 min and then phosphorylation of ErbB receptors was determined by immunoprecipitation and western blotting. Phosphorylation was observed in ErbB-1, ErbB-2, ErbB-3, and ErbB-4 following BTC treatment in αTC1-9 cells (Figure 4B). Activation of ErbB induces the stimulation of intracellular pathways such as Ras/Raf/mitogen-activated protein kinase (MAPK)/ extracellular signal-regulated kinase (ERK) (MEK)/ERK, and phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) (Arteaga and Engelman, 2014). Thus, we examined the activation of ERK1/2, AKT, and mTOR and found that the phosphorylation of ERK, AKT, and mTOR was also increased after BTC treatment in αTC1-9 cells (Figure 4C).
Because all ErbBs (ErbB-1, ErbB-2, ErbB-3, and ErbB-4) were activated by BTC treatment in αTC1-9 cells, we investigated which ErbBs are involved in BTC-induced proliferation. αTC1-9 cells were stimulated with BTC (1 nM) for 24 h and 48 h in the presence of 4 nM of AG1478 or AG825 as specific ErbB-1 or ErbB-2 inhibitor. In BTC treated αTC1-9 cells, proliferation was significantly increased and specific ErbB-1 and ErbB-2 inhibitors did not show inhibitory effects on BTC-induced proliferation (Figures 5A,B).
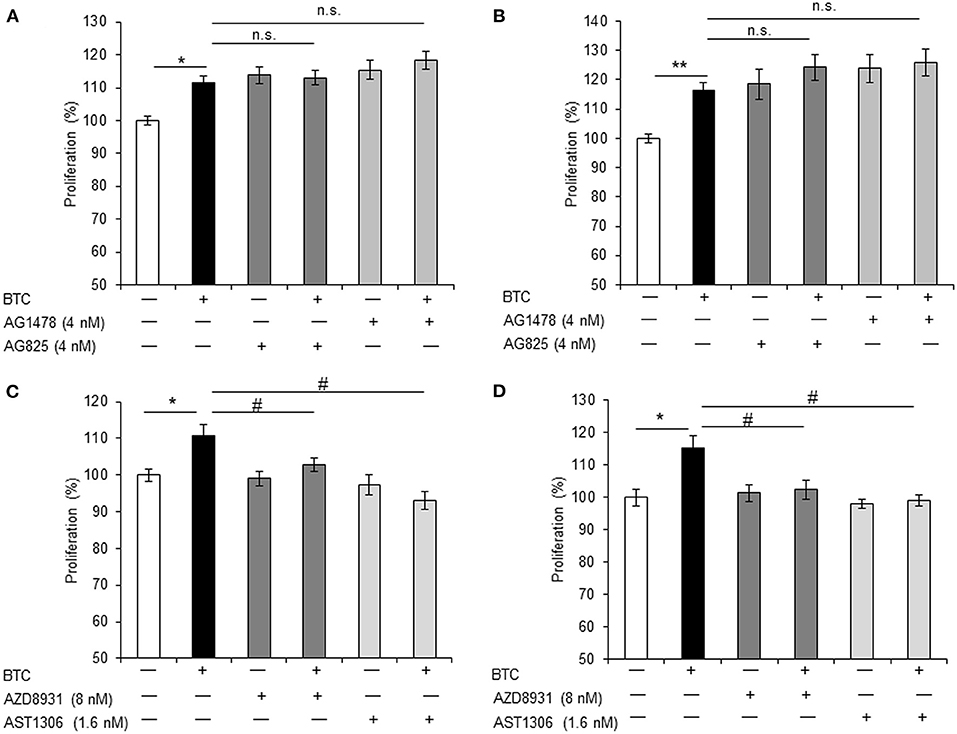
Figure 5. BTC-induced α-cell proliferation is inhibited by ErbB-3 and ErbB-4 inhibitors. αTC1-9 cells were pretreated with ErbB-1 inhibitor: AG1478, ErbB-2 inhibitor: AG825, ErbB-1,-2 and−3 inhibitor: AZD8931, or ErbB-1,-2 and−4 inhibitor: AST 1306 tosylate for 30 min and BTC (1 nM) was then added and incubated for 24 h (A,C) or 48 h (B,D). Viable cells were measured by CCK8 assay. The data are presented as a percentage of the untreated control. Data are means ± SE from three to four independent experiments. *P < 0.05, **P < 0.01 compared with untreated cells, #P < 0.05, n.s. (not significant) compared with BTC treated cells.
Treatment with either AZD8931 (inhibition of ErbB-1, ErbB-2, and ErbB3) or AST1306 tosylate (inhibition of ErbB-1, ErbB-2 and ErbB4), significantly suppressed BTC-induced proliferation of αTC1-9 cells (Figures 5C,D). These results suggest that BTC increases α-cell proliferation via activation of ErbB-3 and ErbB-4 receptors in αTC1-9 cells, not by ErbB-1 and ErbB-2 receptors.
Local secretion of GLP-1 from pancreatic α-cells may be beneficial for β-cell function and β-cell regeneration (Masur et al., 2005; Chen et al., 2012; Lee et al., 2018). Thus, we examined whether BTC induces GLP-1 secretion in α-cells. BTC induces GLP-1 secretion in αTC1-9 cells in a dose dependent manner at 24 h after BTC treatment (Figure 6A). GLP-1 is produced by PC1/3 in pancreatic α-cells (Masur et al., 2005; Chen et al., 2012). Thus, we determined the expression of PC1/3 mRNA in BTC treated αTC1-9 cells. We found that BTC treatment increased PC1/3 expression at 12 h after BTC treatment (Figure 6B). In addition, BTC treatment also significantly increased GLP-1 secretion in β-cell ablated islets following STZ treatment (mainly α-cells remain) (Figure 6C). To investigate whether BTC-induced proliferation in α-cells is mediated by the increased GLP-1 secretion by BTC, we pretreated αTC1-9 cells with a GLP-1 receptor antagonist, exendin-9, and then treated with BTC. However, BTC-induced proliferation was not changed by exendin-9 (Figure 6D) in αTC1-9 cells, suggesting that BTC increased pancreatic α-cell proliferation by itself, not mediated by GLP-1.
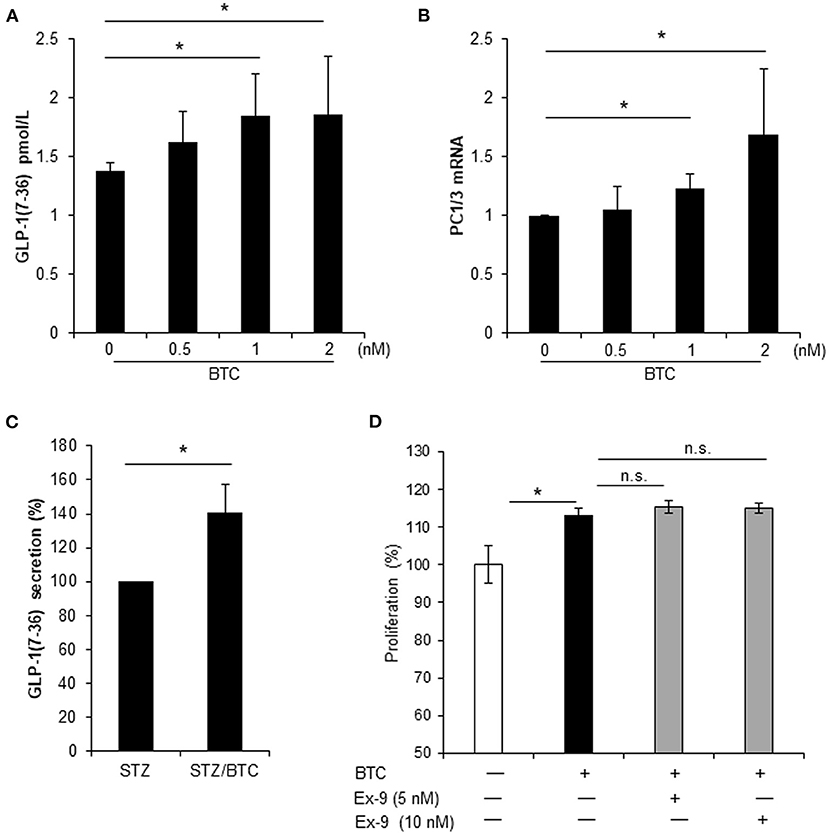
Figure 6. BTC increased GLP-1(7-36) secretion in αTC1-9 cells and rat islets but BTC-induced GLP-1 did not affect cell proliferation. (A) αTC1-9 cells were treated with the indicated concentration of BTC for 24 h and GLP-1 (active 7-36) secretion in media was measured. (B) αTC1-9 cells were treated with the indicated concentration of BTC for 6 h and the expression of PC1/3 mRNA was measured by RT-qPCR. (C) Rat islets were isolated and treated with STZ (1 mM) for 15 h and then with or without BTC (1 nM) for 48 h (add per 24 h). Active GLP-1 secretion in media was measured and expressed as a percentage of the value of STZ-treated islet. (D) αTC1-9 cells were pretreated with exendin-9 (5 nM or 10 nM) for 30 min and BTC was then added, incubated for 24 h, and a CCK8 assay was performed (n = 6~12). Data are expressed as a percentage of the value of untreated cells. Data are means ± SE from three to four independent experiments. *P < 0.05 compared with untreated or STZ only-treated islets and n.s. (not significant) compared with BTC treated cells.
Pancreatic α-cells are endocrine cells in the islets of the pancreas. The α-cells synthesize proglucagon which is cleaved by prohormone convertase 2 (PC2) to produce active glucagon. Glucagon is secreted by α-cells in response to hypoglycemia. Glucagon is a counter-regulatory hormone to insulin which together maintain homeostasis of glucose metabolism, which is the classical role of pancreatic α-cells. However, recent reports indicate an emerging role for pancreatic α-cells, as a source of newly generated β-cells (Gromada et al., 2018; Lee et al., 2018). Transdifferentiation of α-cells to β-cells was observed under conditions of extreme physiological demand for insulin (Thorel et al., 2010). The number of pancreatic α-cells increased under conditions of insufficient insulin, due to damage to β-cells, as found in patients of recent-onset type 1 diabetes (Yabe et al., 2013) and STZ-induced diabetic animals (Dusaulcy et al., 2016). In addition, the number of pancreatic α-cells also increased under the insulin resistance condition of high fat diet-induced obesity (Ellingsgaard et al., 2008). However, the reasons for compensatory increase of pancreatic α-cells are not yet clear.
We previously found that rAd-BTC treatment regenerated pancreatic β-cells and lowered blood glucose levels in STZ-induced diabetic mice, and BTC-induced β-cell proliferation was one of the mechanisms for β-cell regeneration (Shin et al., 2008). In this study, we investigated the effect of BTC on the proliferation of α-cells and found that α-cell proliferation increased in the pancreas of rAd-BTC-treated mice compared with rAd-βgal-treated mice after destruction of β-cells with STZ. In addition, we found that the proliferation of αTC1-9, a pancreatic α-cell line, increased with BTC treatment.
An increase of α-cell proliferation might increase glucagon production, which can increase blood glucose levels through hepatic glucose output, and deteriorates the diabetic condition. However, rAd-BTC did not increase blood glucagon levels (Supplementary Figure 2). We found that BTC induces GLP-1 secretion in αTC1-9 and β-cell-ablated islets (Figures 6A–C). GLP-1 has glucose-lowering effects as it inhibits glucagon secretion and stimulates insulin secretion in islets (Kieffer and Habener, 1999; Drucker, 2006; Holst et al., 2011). Thus, we speculate that BTC-induced GLP-1 secretion might inhibit glucagon secretion, contributing to the attenuation of hyperglycemia, even despite the increase in proliferation of α-cells.
Recent studies show that insulin + glucagon + bihormonal cells can become new-β cells in animals with almost total destruction of β-cells (Thorel et al., 2010). In this study, BTC increased not only pancreatic α-cells, but also insulin+glucagon+ bihormonal cells following rAd-BTC treatment in STZ-induced diabetic mice (Figures 1A,B). In addition, BTC increased the expression of PDX-1, an important transcription factor for the differentiation of progenitor cells into the β-cell phenotype, in α-cells and BTC has been known to have effects in regulation of β-cell mass (Shin et al., 2008; Oh et al., 2015). Therefore, it is possible to generate new β-cells via insulin+glucagon+ bihormonal cells and induction of PDX-1. The expression of PDX-1 mRNA was significantly increased at 24 h after BTC treatment in β-cell ablated islet cells (Figure 1C). In mice, PDX-1 is expressed during E7.5 −11.5 and re-expressed after E13.5 (Zhu et al., 2017) and forced PDX-1 expression induces α-cells to β-cells conversion (Yang et al., 2011). In this study, the levels of glucagon+PDX-1+ double stained cells in glucagon-producing cells were higher in rAd-BTC-treated mice than in rAd-βgal-treated mice (Figures 1F,G). Therefore, BTC induced PDX-1 might contribute to transdifferentiation of α-cells to β-cells. However, we need to further study for lineage tracing experiment, using for example gcg-rtTA;TetOcre;R-26YFP mice, where α-cells are tagged with YFP. In addition, the somatostatin producing δ-cells were significantly increased in rAd-BTC-treated mice compared with the rAd-βgal-treated group (Supplementary Figure 3), however we couldn't observe somatostatin + insulin + bihormonal cells. We need to further study for the effect of BTC on δ-cells.
BTC is a member of the EGF family and a ligand for the four tyrosine kinase receptors (ErbB-1/EGFR, neu/ErbB-2/HER2, ErbB-3/HER3 and ErbB-4/HER4). These receptors can form homo- and heterodimers with one another (Graus-Porta et al., 1997; Wieduwilt and Moasser, 2008). It was reported that BTC binds ErbB-1 and ErbB-3 heterodimers (Rush et al., 2018) or ErbB-1 and ErbB-4 heterodimers (Gomez-Gaviro et al., 2012). BTC was shown to induce proliferation of pancreatic β-cells through the activation of ErbB-1 and ErbB-2 receptors (Oh et al., 2011). We found that all ErbBs (ErbB-1, ErbB-2, ErbB-3, and ErbB-4) were expressed in both αTC1-9 cells and in islets of C57BL/6 mice. BTC-induced α-cell proliferation was not affected by specific inhibitors of ErbB-1 or ErbB2, but inhibited by pan-ErbB inhibitors AZD8931 (inhibition of ErbB-1, ErbB-2 and ErbB3) or AST 1306 tosylate (inhibition of ErbB-1, ErbB-2 and ErbB4), suggesting that BTC may induce proliferation of pancreatic α-cells via the ErbB-3 and ErbB-4 receptor. However, the role of each ErbB receptors in BTC-induced α-cell proliferation needs further study.
ErbB receptors contribute to biological effects through the activation of PI3K/AKT/mTOR and MAPK/ERK signaling pathways (Kainulainen et al., 2000; Mishra et al., 2018). mTOR regulates cell growth, cell proliferation, and cell survival (Lipton and Sahin, 2014) and its function is mediated through mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). The loss of mTORC1 signaling in α-cells reduced α-cell mass, which resulted from decreased proliferation and increased apoptosis during postnatal maintenance (Bozadjieva et al., 2017). In this study, BTC increased mTOR phosphorylation in αTC1-9 cells, which might regulate α-cell proliferation. In addition, BTC enhanced growth and migration of vascular smooth muscle cells through activation of ERK1/2, AKT, and MAPK (Mifune et al., 2004). We also found increased phosphorylation of ERK1/2 and AKT in BTC-treated αTC1-9 cells. These results indicate that activation of PI3K/AKT/mTOR and MAPK/ERK signaling pathways might be the mechanisms underlying BTC-induced α-cell proliferation.
Pancreatic α-cells produce GLP-1 through post-translational processing of proglucagon by PC1/3 (Piro et al., 2014; Knop, 2018). Interestingly, we found that BTC also induced GLP-1 secretion and PC1/3 expression in αTC1-9 cell and β-cell ablated islets. In a previous study, we reported that treatment with GLP-1 increases the proliferation in α-cell and transdifferentiation of α-cell to β-cells, as analyzed using lineage tracing transgenic mouse models (Lee et al., 2018). Thus, we consider that GLP-1-induced by BTC can affect proliferation of α-cells. Therefore, we investigated whether the increase in GLP-1 caused by BTC treatment increases proliferation of α-cells. We used the GLP-1 receptor antagonist, exendin-9, to inhibit the effect of GLP-1 in BTC-induced α-cell proliferation. We found that BTC-induced proliferation was not changed by exendin-9 in α-cells. These results suggest that the effect of BTC induced α-cell proliferation is a direct effect of BTC on α-cell, not an indirect effect mediated by BTC-induced GLP-1. In this study, BTC increased GLP-1 secretion in BTC-treated α-cells, however this may be not enough to increase α-cell proliferation. Further studies are required to identify the detailed mechanisms.
In conclusion, we investigated the effect of BTC treatment on proliferation of pancreatic α-cells and found that treatment of BTC increased α-cell proliferation via ErbB receptors and activation of Erk1/2, AKT, and mTOR signaling pathways in α-cells. The increased α-cells may contribute to β-cells regeneration.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee at Lee Gil Ya Cancer and Diabetes Institute, Gachon University.
Y-SL designed the study, performed experiments and data analysis, and wrote the manuscript. GS performed experiments and wrote the revised manuscript. H-SJ designed the study, analyzed data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF-2015R1D1A1A01060232, NRF-2018R1D1A1B07048223), and by Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI14C1135).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.605110/full#supplementary-material
Aguayo-Mazzucato, C., and Bonner-Weir, S. (2018). Pancreatic beta cell regeneration as a possible therapy for diabetes. Cell Metab. 27, 57–67. doi: 10.1016/j.cmet.2017.08.007
Arteaga, C. L., and Engelman, J. A. (2014). ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25, 282–303. doi: 10.1016/j.ccr.2014.02.025
Baetens, D., De Mey, J., and Gepts, W. (1977). Immunohistochemical and ultrastructural identification of the pancreatic polypeptide-producing cell (PP-cell) in the human pancreas. Cell Tissue Res. 185, 239–246. doi: 10.1007/BF00220668
Berts, A., Liu, Y. J., Gylfe, E., and Hellman, B. (1997). Oscillatory Ca2+ signaling in somatostatin-producing cells from the human pancreas. Metab. Clin. Exp. 46, 366–369. doi: 10.1016/S0026-0495(97)90048-2
Bozadjieva, N., Blandino-Rosano, M., Chase, J., Dai, X. Q., Cummings, K., Gimeno, J., et al. (2017). Loss of mTORC1 signaling alters pancreatic alpha cell mass and impairs glucagon secretion. J. Clin. Invest. 127, 4379–4393. doi: 10.1172/JCI90004
Briant, L., Salehi, A., Vergari, E., Zhang, Q., and Rorsman, P. (2016). Glucagon secretion from pancreatic alpha-cells. Ups. J. Med. Sci. 121, 113–119. doi: 10.3109/03009734.2016.1156789
Chen, X., Hermansen, K., and Jeppesen, P. B. (2012). Impact of glucagon-like peptide-1 (7-36) amide, isosteviol and 5-aminoimidazole-4-carboxamide 1-beta-d-ribofuranoside on leucine-mediated alpha-cell dysfunction. Diabetes Obes. Metab. 14, 1020–1031. doi: 10.1111/j.1463-1326.2012.01633.x
Drucker, D. J. (2006). The biology of incretin hormones. Cell Metab. 3, 153–165. doi: 10.1016/j.cmet.2006.01.004
Dusaulcy, R., Handgraaf, S., Heddad-Masson, M., Visentin, F., Vesin, C., Reimann, F., et al. (2016). alpha-cell dysfunctions and molecular alterations in male insulinopenic diabetic mice are not completely corrected by insulin. Endocrinology 157, 536–547. doi: 10.1210/en.2015-1725
Ellingsgaard, H., Ehses, J. A., Hammar, E. B., Van Lommel, L., Quintens, R., Martens, G., et al. (2008). Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc. Natl. Acad. Sci. U.S.A. 105, 13163–13168. doi: 10.1073/pnas.0801059105
Gomez-Gaviro, M. V., Scott, C. E., Sesay, A. K., Matheu, A., Booth, S., Galichet, C., et al. (2012). Betacellulin promotes cell proliferation in the neural stem cell niche and stimulates neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 1317–1322. doi: 10.1073/pnas.1016199109
Graus-Porta, D., Beerli, R. R., Daly, J. M., and Hynes, N. E. (1997). ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 16, 1647–1655. doi: 10.1093/emboj/16.7.1647
Gromada, J., Chabosseau, P., and Rutter, G. A. (2018). The alpha-cell in diabetes mellitus. Nature Rev. Endocrinol. 14, 694–704. doi: 10.1038/s41574-018-0097-y
Habener, J. F., and Stanojevic, V. (2012). alpha-cell role in beta-cell generation and regeneration. Islets 4, 188–198. doi: 10.4161/isl.20500
Holst, J. J., Christensen, M., Lund, A., de Heer, J., Svendsen, B., Kielgast, U., et al. (2011). Regulation of glucagon secretion by incretins. Diabetes Obes. Metab. 13(Suppl. 1), 89–94. doi: 10.1111/j.1463-1326.2011.01452.x
Jun, H. S., Yoon, C. S., Zbytnuik, L., van Rooijen, N., and Yoon, J. W. (1999). The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Exp. Med. 189, 347–358. doi: 10.1084/jem.189.2.347
Kainulainen, V., Sundvall, M., Maatta, J. A., Santiestevan, E., Klagsbrun, M., and Elenius, K. (2000). A natural ErbB4 isoform that does not activate phosphoinositide 3-kinase mediates proliferation but not survival or chemotaxis. J. Biol. Chem. 275, 8641–8649. doi: 10.1074/jbc.275.12.8641
Kieffer, T. J., and Habener, J. F. (1999). The glucagon-like peptides. Endocr. Rev. 20, 876–913. doi: 10.1210/edrv.20.6.0385
Knop, F. K. (2018). EJE PRIZE 2018: a gut feeling about glucagon. Eur. J. Endocrinol. 178, R267–R280. doi: 10.1530/EJE-18-0197
Lee, Y. S., Lee, C., Choung, J. S., Jung, H. S., and Jun, H. S. (2018). Glucagon-like peptide 1 increases β-cell regeneration by promoting α- to β-cell transdifferentiation. Diabetes 67, 2601–2614. doi: 10.2337/db18-0155
Li, L., Seno, M., Yamada, H., and Kojima, I. (2003). Betacellulin improves glucose metabolism by promoting conversion of intraislet precursor cells to beta-cells in streptozotocin-treated mice. Am. J. Physiol. Endocrinol. Metab. 285, E577–E583. doi: 10.1152/ajpendo.00120.2003
Lipton, J. O., and Sahin, M. (2014). The neurology of mTOR. Neuron 84, 275–291. doi: 10.1016/j.neuron.2014.09.034
Masur, K., Tibaduiza, E. C., Chen, C., Ligon, B., and Beinborn, M. (2005). Basal receptor activation by locally produced glucagon-like peptide-1 contributes to maintaining beta-cell function. Mol. Endocrinol. 19, 1373–1382. doi: 10.1210/me.2004-0350
Miettinen, P. J., Huotari, M., Koivisto, T., Ustinov, J., Palgi, J., Rasilainen, S., et al. (2000). Impaired migration and delayed differentiation of pancreatic islet cells in mice lacking EGF-receptors. Development 127, 2617–2627. Available online at: https://dev.biologists.org/content/127/12/2617.long
Mifune, M., Ohtsu, H., Suzuki, H., Frank, G. D., Inagami, T., Utsunomiya, H., et al. (2004). Signal transduction of betacellulin in growth and migration of vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 287, C807–C813. doi: 10.1152/ajpcell.00436.2003
Mishra, R., Patel, H., Alanazi, S., Yuan, L., and Garrett, J. T. (2018). HER3 signaling and targeted therapy in cancer. Oncol. Rev. 12:355. doi: 10.4081/oncol.2018.355
Miyagawa, J., Hanafusa, O., Sasada, R., Yamamoto, K., Igarashi, K., Yamamori, K., et al. (1999). Immunohistochemical localization of betacellulin, a new member of the EGF family, in normal human pancreas and islet tumor cells. Endocr. J. 46, 755–764. doi: 10.1507/endocrj.46.755
Oh, Y. S., Shin, S., Lee, Y. J., Kim, E. H., and Jun, H. S. (2011). Betacellulin-induced beta cell proliferation and regeneration is mediated by activation of ErbB-1 and ErbB-2 receptors. PLoS ONE 6:e23894. doi: 10.1371/journal.pone.0023894
Oh, Y. S., Shin, S., Li, H. Y., Park, E. Y., Lee, S. M., Choi, C. S., et al. (2015). Betacellulin ameliorates hyperglycemia in obese diabetic db/db mice. J. Mol. Med. 93, 1235–1245. doi: 10.1007/s00109-015-1303-1
Parker, J. C., Andrews, K. M., Allen, M. R., Stock, J. L., and McNeish, J. D. (2002). Glycemic control in mice with targeted disruption of the glucagon receptor gene. Biochem. Biophys. Res. Commun. 290, 839–843. doi: 10.1006/bbrc.2001.6265
Piro, S., Mascali, L. G., Urbano, F., Filippello, A., Malaguarnera, R., Calanna, S., et al. (2014). Chronic exposure to GLP-1 increases GLP-1 synthesis and release in a pancreatic alpha cell line (alpha-TC1): evidence of a direct effect of GLP-1 on pancreatic alpha cells. PLoS ONE 9:e90093. doi: 10.1371/journal.pone.0090093
Rush, J. S., Peterson, J. L., and Ceresa, B. P. (2018). Betacellulin (BTC) Biases the EGFR To Dimerize with ErbB3. Mol. Pharmacol. 94, 1382–1390. doi: 10.1124/mol.118.113399
Shin, S., Li, N., Kobayashi, N., Yoon, J. W., and Jun, H. S. (2008). Remission of diabetes by beta-cell regeneration in diabetic mice treated with a recombinant adenovirus expressing betacellulin. Mol. Ther. 16, 854–861. doi: 10.1038/mt.2008.22
Song, M. Y., Bae, U. J., Jang, K. Y., and Park, B. H. (2014). Transplantation of betacellulin-transduced islets improves glucose intolerance in diabetic mice. Exp. Mol. Med. 46:e98. doi: 10.1038/emm.2014.24
Thorel, F., Nepote, V., Avril, I., Kohno, K., Desgraz, R., Chera, S., et al. (2010). Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464, 1149–1154. doi: 10.1038/nature08894
Wieduwilt, M. J., and Moasser, M. M. (2008). The epidermal growth factor receptor family: biology driving targeted therapeutics. Cellular Mol. Life Sci. 65, 1566–1584. doi: 10.1007/s00018-008-7440-8
Wierup, N., Sundler, F., and Heller, R. S. (2014). The islet ghrelin cell. J. Mol. Endocrinol. 52, R35–R49. doi: 10.1530/JME-13-0122
Yabe, D., Rokutan, M., Miura, Y., Komoto, I., Usui, R., Kuwata, H., et al. (2013). Enhanced glucagon-like peptide-1 secretion in a patient with glucagonoma: implications for glucagon-like peptide-1 secretion from pancreatic alpha cells in vivo. Diabetes Res. Clin. Pract. 102, e1–4. doi: 10.1016/j.diabres.2013.08.005
Yamamoto, K., Miyagawa, J., Waguri, M., Sasada, R., Igarashi, K., Li, M., et al. (2000). Recombinant human betacellulin promotes the neogenesis of beta-cells and ameliorates glucose intolerance in mice with diabetes induced by selective alloxan perfusion. Diabetes 49, 2021–2027. doi: 10.2337/diabetes.49.12.2021
Yang, Y. P., Thorel, F., Boyer, D. F., Herrera, P. L., and Wright, C. V. (2011). Context-specific alpha- to-beta-cell reprogramming by forced Pdx1 expression. Genes Dev. 25, 1680–1685. doi: 10.1101/gad.16875711
Keywords: pancreatic α-cell, pancreatic β-cell, regeneration, betacellulin, ErbB receptor
Citation: Lee Y-S, Song GJ and Jun H-S (2021) Betacellulin-Induced α-Cell Proliferation Is Mediated by ErbB3 and ErbB4, and May Contribute to β-Cell Regeneration. Front. Cell Dev. Biol. 8:605110. doi: 10.3389/fcell.2020.605110
Received: 11 September 2020; Accepted: 15 December 2020;
Published: 21 January 2021.
Edited by:
Lisa Porter, University of Windsor, CanadaReviewed by:
Peter Flatt, Ulster University, United KingdomCopyright © 2021 Lee, Song and Jun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Young-Sun Lee, YmlvbHlzQGhhbm1haWwubmV0; Hee-Sook Jun, aHNqdW5AZ2FjaG9uLmFjLmty
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.