 Marte Sneeggen
Marte Sneeggen Noemi Antonella Guadagno
Noemi Antonella Guadagno Cinzia Progida
Cinzia Progida- Department of Biosciences, University of Oslo, Oslo, Norway
Tumor progression is a complex process consisting of several steps characterized by alterations in cellular behavior and morphology. These steps include uncontrolled cell division and proliferation, invasiveness and metastatic ability. Throughout these phases, cancer cells encounter a changing environment and a variety of metabolic stress. To meet their needs for energy while they proliferate and survive in their new environment, tumor cells need to continuously fine-tune their metabolism. The connection between intracellular transport and metabolic reprogramming during cancer progression is emerging as a central process of cellular adaptation to these changes. The trafficking of proteolytic enzymes, surface receptors, but also the regulation of downstream pathways, are all central to cancer progression. In this review, we summarize different hallmarks of cancer with a special focus on the role of intracellular trafficking in cell proliferation, epithelial to mesenchymal transition as well as invasion. We will further emphasize how intracellular trafficking contributes to the regulation of energy consumption and metabolism during these steps of cancer progression.
Introduction
During cancer progression, tumor cells go through different stages, which are defined as hallmarks of cancer. One of the main hallmarks is the ability to sustain proliferation. Misregulation of growth-promoting signals stimulates cell survival and energy metabolism, resulting in tumor growth (Hanahan and Weinberg Robert, 2011). As cancer further develops, cells may become able to disseminate from the primary site of origin. This is usually induced by loss of epithelial markers such E-cadherin, and characterized by the transition from an epithelial phenotype to a mesenchymal phenotype, a process known as epithelial to mesenchymal transition (EMT). Genes that in normal tissues express molecules involved in cell-to-cell adhesion and cell-to-extracellular matrix adhesions are altered in highly aggressive carcinomas, typically downregulated (Hanahan and Weinberg Robert, 2011). After losing cell-cell adhesions, cancer cells acquire migratory ability, leading eventually to invasion into neighboring tissues and forming metastatic sites in distant organs (Son and Moon, 2010).
During these transitions, cancer cells undergo metabolic changes, which allow them to satisfy their increased need of energy. The reprogramming of energy metabolism is now recognized as one of the hallmarks of cancer. One of the most known metabolic adaptation in malignant cells is the Warburg effect, that is characterized by increased glucose uptake and lactate production in the presence of oxygen (Potter et al., 2016). To satisfy their high nutritional and energetic requirements, cancer cells exploit intracellular trafficking pathways such as macropinocytosis and autophagy to scavenge the tumor microenvironment for nutrients and macromolecules. This fuels the cells to sustain proliferation, undergo EMT, as well as drive invasion. In this review, we will highlight the contribution of the intracellular transport for the metabolic adaptations required during the different stages of tumor progression.
Intracellular Trafficking in Cancer Cell Proliferation
One of the first challenges that cancer cells overcome during cancer transformation is the ability to sustain chronic proliferation (Hanahan and Weinberg Robert, 2011). Keeping up with a sustained proliferation signal has a cost in term of energy requirement. To be able to proliferate, cells must duplicate their mass. Therefore, they need to reprogram their metabolism to meet the need for larger amount of nutrients to support the synthesis of new macromolecules (Davidson and Vander Heiden, 2017).
Cells that are proliferating rapidly have different metabolic needs than those that are in a resting state. Even though glucose and glutamine have been believed to be the major source of energy, it is now clear that the cells also use nutrients and amino acids available in the environment rather than synthesizing them de novo. This is a more convenient strategy as de novo synthesis requires more energy compared to reusing already existing nutrients and amino acids (Hosios et al., 2016). Pre-existing nutrients necessitate to be transported either from the extracellular environment or from other cellular compartments inside the cells to lysosomes for their degradation into recyclable building blocks (Davidson and Vander Heiden, 2017; Figure 1).
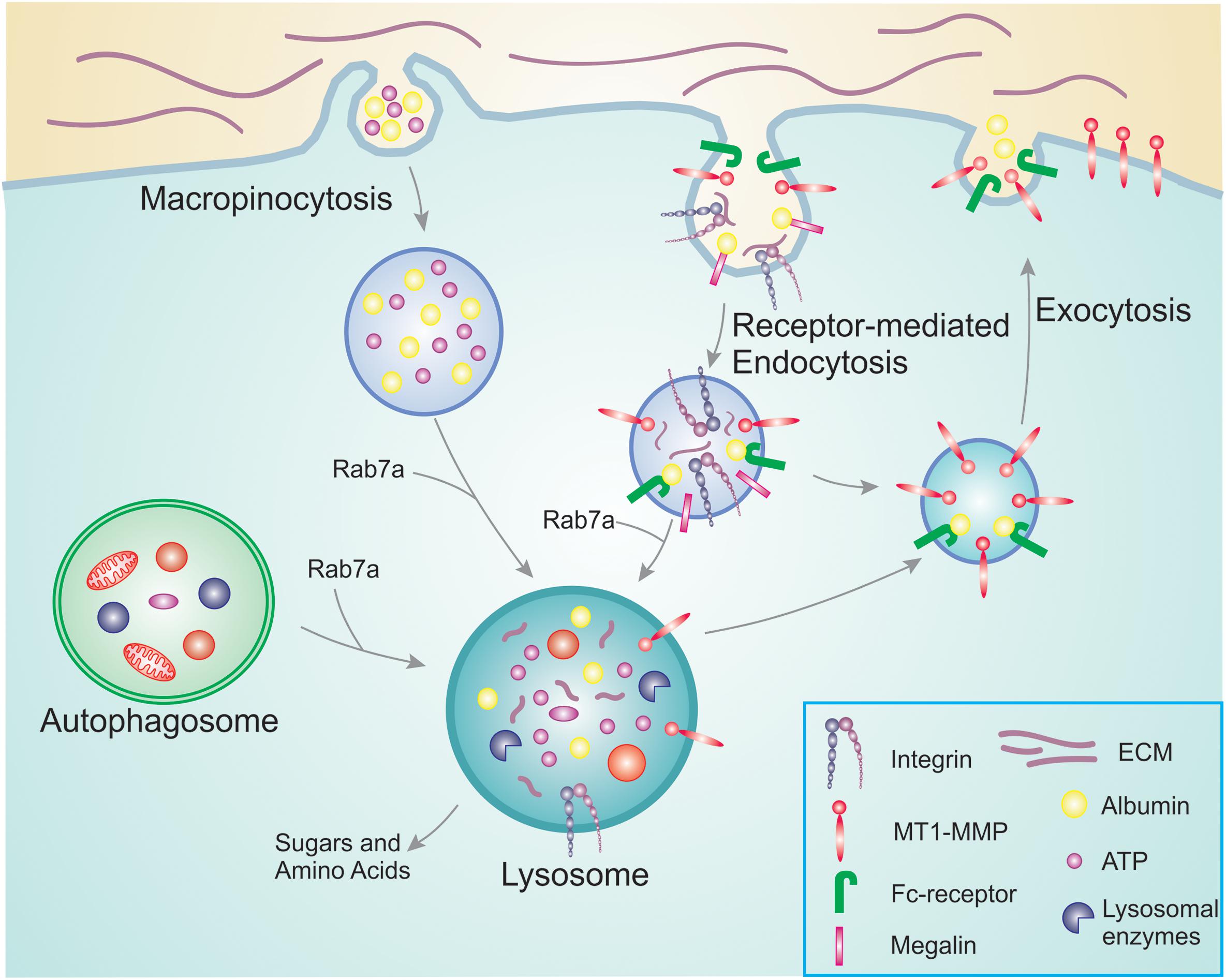
Figure 1. Overview of nutrient scavenging pathways in cancer cells. During cancer progression, cancer cells increase their energy and nutrient requirement to meet the demand of constant proliferation and sustain processes such as migration and invasion. For this, they use membrane trafficking pathways to scavenge nutrients already available. Macropinocytosis allows the bulk internalization of extracellular ATP as well as albumin and other nutrients. In a similar manner, receptor-mediated endocytosis is responsible for the uptake of receptors and their ligands that will be degraded in the lysosomes providing new building blocks to be reused. Examples are integrins that bind to ECM components, and megalin which binds to albumin in the extracellular environment. The internalized albumin can either bind to neonatal Fc receptor inside the endosomes and be recycled back to the plasma membrane, or degraded in the lysosomes. To access the pool of nutrients already available in the cell, cancer cells can hijack autophagy. Engulfed damaged organelles and protein aggregates can thus be broken down and degraded in the lysosomes for reuse. The altered metabolism during cancer progression results in increased MT1-MMP recycling, thus promoting cell invasion.
Macropinocytosis and Cancer Cell Proliferation
Cancer cells have an increased need for nutrients and therefore adopt different strategies to access macromolecules from the tumor microenvironment. Macropinocytosis is an effective and rapid way to internalize macromolecules from the environment. It is an actin-dependent endocytic mechanism consisting of non-specific uptake of large amounts of extracellular fluid and nutrients into large vesicles (King and Kay, 2019). This process is crucial for nutrient uptake to support tumor cell fitness and it is associated with cell growth. Indeed, pharmacological inhibition of macropinocytosis suppresses tumor growth and it has been suggested that this process could be a possible target for anticancer therapies (Commisso et al., 2013). Macropinocytosis also boosts intracellular Adenosine triphosphate (ATP) concentration by directly ingesting ATP molecules when they are available in the extracellular environment (Figure 1; Qian et al., 2014). Extracellular ATP concentration in tumors is up to 104 times higher than in normal tissues (Pellegatti et al., 2008; Falzoni et al., 2013; Qian et al., 2014). This extracellular ATP increases the survival of cancer cells during metabolic stress by protecting against tumor inhibition drugs. It has been suggested that following ATP internalization, the increased intracellular ATP interferes with tumor inhibition drugs that compete with ATP for their anticancer activity (Qian et al., 2014). In line with this, extracellular ATP reduced the function of the cancer drug sunitinib that works as an ATP competitor targeting receptor tyrosine kinases (Papaetis and Syrigos, 2009; Qian et al., 2014).
After internalization, macropinosomes deliver their content to the lysosomes where the internalized macromolecules are broken down. The obtained amino acids provide a carbon source to the central metabolism, and serve as building blocks for protein synthesis during proliferation in conditions lacking free amino acids (Palm, 2019). The small GTPase Rab7a, which regulates the fusion between endosomes and autophagosomes with lysosomes, is enriched in melanoma cells where it also is important for sustaining cell proliferation and cancer progression (Alonso-Curbelo et al., 2014, 2015). In particular, at early stages of melanoma development, Rab7a is upregulated and sustains melanoma cell proliferation controlled by the lineage-specific transcription factor SOX10 and the oncogenic transcription factor MYC, which is activated at early stages of melanoma development (Alonso-Curbelo et al., 2014). Rab7a upregulation at these early stages of melanocyte transformation hyperactivates Rab7a-mediated lysosomal degradation to counteract the enhanced macropinocytic influx associated with oncogene-induced senescence (Alonso-Curbelo et al., 2015). During melanoma progression, Rab7a expression is then downregulated. In highly invasive melanoma cells, this favors invasive phenotypes supporting Rab7a as a risk factor for melanoma metastasis and poor survival (Alonso-Curbelo et al., 2014).
Nutrient scavenging consists in the uptake of macromolecules from the extracellular environment and their degradation to produce ATP or to be used in anabolism (Finicle et al., 2018). Scavenging is controlled by the mechanistic target of rapamycin complex-1 (mTORC1) and AMP kinase (AMPK), which are involved in the regulation of macropinocytosis (Swanson and King, 2019). AMPK activates all forms of scavenging, while mTORC1 represses the effect of scavenging by interfering with the catabolism happening in the lysosomes (Finicle et al., 2018). AMPK regulates diverse metabolic cellular processes, and also endocytic traffic during metabolic stress. When it is activated, it inhibits energy demanding processes and enhances catabolic reaction to generate ATP (Rahmani et al., 2019). AMPK can both suppress but also promote tumor growth (Hardie, 2015). It has been suggested that this depends on the timing of modification, mutation and overexpression of AMPK or of the upstream kinase Liver kinase B1 (LKB1). In the initial steps of cancer, inactivation of this pathway may help cell growth by utilizing anabolic pathways. In later stages, activation of the LKB1–AMPK pathway could protect the tumor cells against oxidative stress by facilitating metabolic adaptations (Jeon, 2016).
mTORC1 is a signaling hub that coordinates nutrient status and cell growth. Activated mTORC1 regulates cellular metabolism and growth by stimulating protein synthesis (Kim and Guan, 2019). The internalization of amino acids in macropinosomes and their delivery to the lysosomes is essential for mTORC1 growth factor-dependent activation (Yoshida et al., 2015). The control of mTOR signaling is critical for the cells and its dysregulation leads to several diseases such as cancer, diabetes, and metabolic diseases (Yoshida et al., 2015).
Amino acid depletion stimulates macropinocytosis and the scarcity of glutamine drives this process (Lee et al., 2019). The nutrient stress triggered from amino acid depletion enhances epidermal growth factor (EGF) receptor signaling that in turn increases macropinocytosis by regulating membrane ruffling and cytoskeleton dynamics (Lee et al., 2019). The activation of the actin cytoskeleton occurs through the small GTPase Ras (Bloomfield and Kay, 2016; Recouvreux and Commisso, 2017). Ras is frequently mutated in cancer and activated in almost 33% of all human cancer (Bloomfield and Kay, 2016; Lanfredini et al., 2019). Ras-driven cancer cells have a higher rate of macropinocytosis. Over-activation of Ras promotes metabolic rewiring and cell proliferation not only by activation of macropinocytosis to internalize extracellular nutrients and enhancing uptake of glucose, contributing to the Warburg effect, but also by inducing autophagy (Recouvreux and Commisso, 2017; Palm, 2019).
Ras-transformed cancer cells are able to take up albumin through macropinocytosis. Degradation of albumin is a source of glutamine, one of the most deprived nutrients in cancer environments. Hence, the macropinocytic uptake of albumin could serve to sustain the proliferation of oncogenic Ras cells by constituting a source of amino acid supply (Commisso et al., 2013; Ha et al., 2016; Palm, 2019). Glutamine serves indeed as important source of carbon, which in different tumors is utilized for TCA cycle anaplerosis. In proliferating cells, glutamine-dependent anaplerosis is critical for mitochondrial metabolism and essential for cell growth (Cluntun et al., 2017). The internalization of albumin through macropinocytosis and the downstream use of albumin-derived amino acids as a source of energy seems to be a unique property of cancer cells, since normal cells adjacent to a tumor lack this ability (Davidson et al., 2017). The stimulation of macropinocytosis in cancer cells is, however, not limited to Ras-transformed cells, as activating mutations in Src kinases also drive macropinocytosis (Amyere et al., 2000; Finicle et al., 2018).
Intriguingly, it has been recently demonstrated that under nutrient-limited conditions, cancer cells within pancreatic ductal adenocarcinoma are able to internalize collagen fragments through macropinocytosis. This extracellular matrix protein represents a proline reservoir that is used as a nutrient source in the absence of other fuels. In this way, the collagen-derived proline contributes to promoting cancer cell survival as well as cell proliferation (Olivares et al., 2017). Therefore, it seems that cancer cells have developed an efficient strategy to obtain nutrients from alternative sources through macropinocytosis followed by lysosomal degradation of extracellular proteins. This allows to furnish the energy and nutrient demand for sustained proliferation.
Receptor-Mediated Internalization for Nutrient Scavenging
Nutrient scavenging does not only occur by macropinocytosis. Cancer cells can also utilize receptor-mediated scavenging to sustain their growth and proliferation (Figure 1). An example of receptor-mediated scavenging is represented by the integrin-mediated endocytosis of extracellular matrix (ECM) components (Finicle et al., 2018). Integrins are cell surface receptors for ECM components that link the actin cytoskeleton to the ECM. During cancer progression, the trafficking of integrins is often upregulated resulting in the internalization of the receptors but also of the ECM components bound to the integrins. The ECM consists of collagen, laminin, and fibronectin. These extracellular proteins are also heavily glycosylated, thus ECM scavenging yields amino acids and sugars to sustain cell proliferation. Dietary restriction and nutrient deprivation induces laminin scavenging by integrin α6β4-mediated endocytosis. Laminin degradation in the lysosomes enhances mTORC1 signaling, preventing cell death and promoting cell survival (Muranen et al., 2017). Similarly, in ovarian cancer cells, integrin α5β1 binds fibronectin, which is then internalized and degraded in the lysosomes, and the resulting amino acids activate mTORC1 (Rainero et al., 2015). However, the mechanisms for integrin-mediated nutrient scavenging in tumors are still poorly characterized and further studies are required to better understand this process.
It is not only integrins and ECM that are involved in nutrient scavenging by receptor-mediated endocytosis. Also albumin is endocytosed upon binding to megalin or other cell surface scavenger receptors. The internalized albumin can either bind to neonatal Fc receptor (FcRn) in endosomes and be recycled back to the plasma membrane, or degraded in the lysosomes. Degradation of albumin results in increased amino acid and possibly also lipid availability (Finicle et al., 2018).
Autophagy and Cancer Cell Proliferation
Autophagy is another process that provides nutrients and energy to the cell. It is used by cells to recycle their own compartments after sequestering them in a double membrane organelle, the autophagosome. When nutrients are running low, autophagosome formation is initiated to engulf macromolecules, protein aggregates and damaged organelles from the cytosol (Davidson and Vander Heiden, 2017). The autophagosomes then fuse with the lysosomes. Degradation inside the lysosomes provides the cells with new building blocks for protein synthesis (Kimmelman and White, 2017).
Upregulation of autophagy can occur in response to hypoxia or metabolic stress to ensure survival. Mice that lack essential autophagy genes such as Atg5 and Atg7, die from nutrient starvation underlying how essential autophagy is to provide nutrients during metabolic stress (Kuma et al., 2004). Similar to macropinocytosis, autophagy allows the cells to tap into a pool of macromolecules. Both pathways are exploited by cancer cells to obtain nutrients for survival and growth. The main difference is that macropinocytosis internalizes nutrients from the extracellular environment, while autophagy utilizes what is already available inside the cells (Palm, 2019). The importance of autophagy in cancer cell proliferation, therefore, seems to be connected to its role in supporting tumor metabolism. In line with this, mutations in the Ras pathway are often associated with high levels of autophagy required to maintain cancer cell metabolism (Guo et al., 2011; Lock et al., 2011; Yang et al., 2011).
Essential autophagy genes such as beclin-1, which is important in the formation of the autophagosome, are upregulated in several types of cancers, including colorectal and gastric cancer (Ahn et al., 2007). Furthermore, Rab escort protein 1 (REP1) is associated with cancer progression by contributing to cell growth and survival through the regulation of mTOR signaling and its downstream pathways (Choi et al., 2017). REP1 is involved in the recruitment of Rab proteins to membranes as well as in the regulation of autophagy (Alexandrov et al., 1994; Choi et al., 2017). Knockdown of REP1 suppresses mTOR activity, blocking autophagy and increasing macropinocytosis. Even though the exact mechanism used by REP1 to regulate autophagy is not known, it is suggested that REP1 modulates the localization of lysosomes and mTOR thereby affecting their activity. It is also reasonable to think that REP1 controls the recruitment of Rab proteins necessary for this process, such as Rab7a (Choi et al., 2017).
The role of autophagy in cancer is quite complex and not fully elucidated yet. It seems to be dependent on several factors such as the type of tumor or the cancer stage. In the initial stages of cancer, autophagy acts as a tumor suppressor through quality control of proteins and removing damaged organelles and protein aggregates (Mathew et al., 2009; Li et al., 2020). By controlling these events, it can prevent sustained proliferation and therefore tumor initiation. However, in the later stages, when a tumor has formed, autophagy can protect cancer cells by helping them cope with cellular stress using the same strategies as in the early phases (Li et al., 2020). Autophagy seems also to contribute to the ability of cancer cells to develop resistance to chemotherapy by protecting them from the stress inflicted by the therapy (Sui et al., 2013; Ma et al., 2014).
Epithelial to Mesenchymal Transition
The ability of cells to change their morphology and phenotype is crucial during embryonic development but also in tissue repair in adults. Transitions from epithelial to mesenchymal cells and back again are known as cellular plasticity (Corallino et al., 2015). Several cancers derive from epithelial cells. These cells are the building blocks of most organs and are organized in tissues by establishing contacts with their neighboring cells. When epithelial cells transition to cancer cells, they lose their epithelial phenotype and acquire a mesenchymal phenotype during a process called epithelial to mesenchymal transition (EMT) (Sciacovelli and Frezza, 2017). During this process, epithelial cells lose their junctions and apical-basal polarity, re-organize their cytoskeleton, change signaling programs and alter gene expression. This results in the loss of contacts with the neighboring cells leading to increased motility of individual cells and in the development of an invasive phenotype. EMT is indeed an essential step in cancer cell progression, which leads to invasion and metastasis (Figure 2).
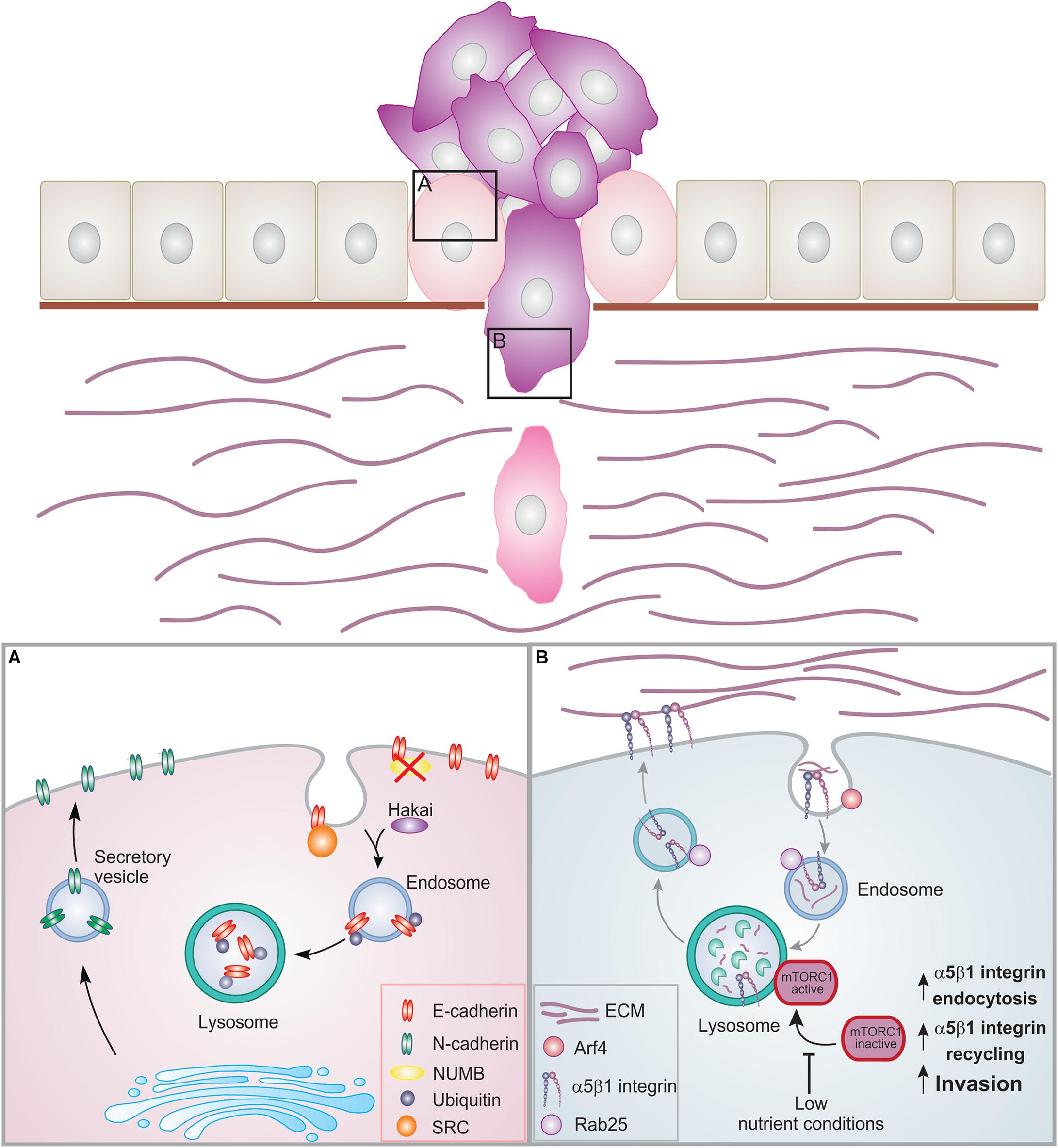
Figure 2. Membrane trafficking events in EMT and cell invasion. Epithelial cells are organized in layers by establishing contacts with neighboring cells as well as the basement membrane. During cancer progression, the cells can lose these contacts leading to EMT and increased proliferation. Cancer cells can then acquire migratory ability, breaching through the basement membrane and invading into the ECM. (A) During EMT, E-cadherin is internalized and degraded. NUMB regulates the internalization and recycling of E-cadherin. When NUMB is lost, E-cadherin relocalizes to the apical side. Upon internalization, E-cadherin can be phosphorylated by src leading to ubiquitination by the E3 ligase Hakai. Ubiquitinated E-cadherin is delivered to lysosomes for degradation. This results in the loss of adherens junctions. N-cadherin is then transported to the surface promoting migration. (B) Trafficking of α5β1 integrin under low nutrient conditions. Inhibition of the mTORC1 activation and of its recruitment to lysosomes promotes Arf4-dependent endocytosis, α5β1 integrin recycling, and cell invasion.
When cells undergo EMT, their metabolic needs become different (Kang et al., 2019). Shaul et al. (2014) found a mesenchymal metabolic signature consisting of 44 upregulated metabolic genes that are essential for EMT but not for cell proliferation. The reprogramming of gene expression has indeed a crucial role during EMT. However, also non-transcriptional changes, including alteration of intracellular trafficking, play a vital part in this process (Le Bras et al., 2012).
In epithelial cells, adherens junctions maintain cell-cell adhesion by connecting transmembrane proteins to the actin cytoskeleton. EMT is characterized by the loss of E-cadherin, one of the major structural components of these junctions. In normal cells, E-cadherin is rapidly internalized from the cell surface and then recycled back to form new cell-cell contacts (Lu et al., 2003; Palacios et al., 2005). However, the endocytic pathway is often dysregulated in cancer, with a shift in the balance between recycling and degradation. In the early phases of EMT, adherens junction dissociation often occurs as result of changes in E-cadherin transport, leading to the internalization of E-cadherin followed by its transportation and degradation into lysosomes (Janda et al., 2006; Ulrich and Heisenberg, 2009; Le Bras et al., 2012). The endocytosis and recycling of E-cadherin is regulated by its interactor NUMB. Loss of NUMB causes E-cadherin to relocate and accumulate at the apical side decreasing cell-cell adhesion, and promoting cell migration (Wang et al., 2009). In line with this, in triple-negative breast cancer, an aggressive type of cancer, reduced NUMB expression is often associated with elevated EMT (Zhang et al., 2016).
During EMT, activated v-Src, a kinase involved in oncogenesis, phosphorylates E-cadherin. After phosphorylation, the E3 ligase Hakai catalyzes the ubiquitination of E-cadherin leading to the trafficking and degradation of E-cadherin to lysosomes (Fujita et al., 2002). Even though the role of Hakai in E-cadherin ubiquitination in physiological conditions remains unclear (Niño et al., 2019), it is intriguing that the expression of this ligase is gradually increased during the different stages of colon cancer progression, which is in line with its suggested role in E-cadherin modulation in EMT (Castosa et al., 2018). Downregulation of E-cadherin facilitates a switch to N-cadherin, which is associated with enhanced migration and invasion (Figure 2A; Loh et al., 2019). Recently, it has been demonstrated that E-cadherin impacts cell metabolism as mechanical forces exerted on E-cadherin activates AMPK thereby stimulating actomyosin contractility, glucose uptake and ATP production (Bays et al., 2017).
One of the major EMT inducers is the transforming growth factor beta (TGF-β) (Katsuno et al., 2013; Corallino et al., 2015). Internalization of the TGF-β receptor triggers a series of downstream cascades, which eventually lead to exocytosis of ATP containing vesicles (Cao et al., 2019). The released ATP functions as an extracellular messenger. It binds to and activates the purinergic receptor P2X7, resulting in EMT induction by upregulating mesenchymal markers and downregulating epithelial markers (Cao et al., 2019). The extracellular ATP thus induces metalloproteinase expression, but it also serves as an energy source for cell detachment. This extracellular ATP is indeed internalized by macropinocytosis, providing the energy required to allow morphological changes and movement (Cao et al., 2019).
Intracellular Trafficking and Energy Requirement in Cell Invasion
Cancer cells that have undergone EMT experience changes that include not only the loss of adherens junctions and apical-basal polarity, but also the re-organization of their cytoskeleton and morphology. This leads to the acquisition of migratory ability that can develop in an invasive phenotype.
For efficient cell migration, adhesion molecules such as integrins are rapidly internalized and transported along the endosomal system. To evade lysosomal degradation, integrins are recycled back to the plasma membrane. This replenishes the plasma membrane pool of integrins and promotes their rapid turnover for cell migration (Mosesson et al., 2008; Dozynkiewicz et al., 2012; Sun et al., 2018; Moreno-Layseca et al., 2019). Therefore, it is not surprising that altered integrin trafficking is linked to invasive processes (Hamidi and Ivaska, 2018).
For example, gain-of-function mutant proteins of the tumor suppressor p53, which are often associate with cancer, increase α5β1 integrin recycling (Muller et al., 2009). α5β1, together with Rab-coupling protein (RCP; also known as Rab11-FIP1), recruits receptor tyrosine kinases, regulating their recycling and potentiating downstream signaling via protein kinase B (PKB)/Akt, thus resulting in invasive migration (Caswell et al., 2008; Muller et al., 2009; Jacquemet et al., 2013). RCP-driven endocytic recycling of α5β1 integrin enhances invasive migration of cancer cells by reprogramming the actin cytoskeleton to promote the formation of cell protrusions and actin-related protein 2/3 (Arp2/3) complex-independent cancer cell invasion in vivo (Jacquemet et al., 2013; Paul et al., 2015). Furthermore, mutant p53 increases the expression of the motor protein myosin X, which binds to β1 integrin to mediate its transport to filopodia (Arjonen et al., 2014). It has therefore been suggested that blocking α5β1-integrin might have therapeutic benefit in mutant p53-expressing cancers (Muller et al., 2009).
Gain-of-function p53 mutants, by promoting glucose transporter 1 (GLUT1) translocation to plasma membrane, stimulate glucose uptake, glycolysis and thus, the Warburg effect (Zhang et al., 2013). Hence, mutant p53, by affecting different intracellular transport pathways, coordinates cell metabolism and integrin recycling to promote cell invasion.
Small GTPases, along with their effectors, control integrin recycling with huge impact on cell invasion. Rab11- and Arf6- dependent recycling of integrins is associated with cancer invasion (Yoon et al., 2005; Das et al., 2018; Moreno-Layseca et al., 2019). A signaling pathway involving phosphorylation of Rab34 inhibits β3 integrin lysosomal degradation mediating its recycling back to the plasma membrane to promote cell migration (Sun et al., 2018). In triple-negative breast cancer cells, Rab5a stimulates Rab4-dependent fast recycling of α5β3 integrin, thus leading to cell invasion (Frittoli et al., 2014; Linder and Scita, 2015). Furthermore, Rab25, which directly associates with integrin α5β1, promotes integrin recycling from late endosomes/lysosomes at the cell front to drive invasion (Caswell et al., 2007; Dozynkiewicz et al., 2012).
Ligand-engaged α5β1 integrin are trafficked under control of Rab25 to late endosomes/lysosomes following Arf4-dependent internalization. This pathway is necessary to maintain lysosomal positioning at the perinuclear region and to recruit and activate the nutrient sensor mTORC1 on lysosomes. Interestingly, in response to low-nutrient status of cancer cells, the recruitment of mTOR to late endosomes/lysosomes is inhibited, further promoting ligand-bound α5β1 internalization and trafficking to lysosomes. This stimulates the degradation of ECM components in the lysosomes as well as Rab25-mediated α5β1 integrin recycling at the plasma membrane (Dozynkiewicz et al., 2012; Rainero et al., 2015). Consequently, this pathway connects nutrient sensing to ECM internalization and integrin recycling to promote cell invasion (Figure 2B).
Degradation of ECM components during cancer invasion occurs not only in the lysosomes but also outside the cell. Diverse polarized trafficking pathways converge at the invadopodia, plasma membrane protrusions responsible for ECM degradation and invasion, for the local delivery of proteolytic enzymes, which have pivotal role in defining the malignant features of cancer cells. Indeed, extracellular degradation-mediated cell invasion is carried by proteolytic enzymes, such as cathepsins and matrix metalloproteinases (MMPs), which can be trafficked either through the secretory pathway or via exocytosis of peripheral lysosomes (Bonnans et al., 2014).
Rab5-mediated endocytosis regulates the internalization and delivery of the membrane-associated MT1-MMP (membrane-type 1 matrix metalloproteinase), an important invasion-promoting enzyme, to non-degrading Rab7a-positive endosomal reservoirs before being exocytosed at invadopodia for ECM degradation (Planchon et al., 2018). A further regulation of MT1-MMP recycling to the plasma membrane has been described to be dependent on WDFY2 and Rab4 following a VAMP3-dependent mechanism (Sneeggen et al., 2019). However, while recycling is considered the major route for fast delivery of proteases to the plasma membrane for ECM degradation, MT1-MMP can additionally be delivered to the plasma membrane following Rab8-dependent polarized exocytosis (Bravo-Cordero et al., 2007) or Rab27-dependent exosomal release (Hoshino et al., 2013). High levels of glutamine consumption contribute to cancer aggressiveness by generating a source of extracellular glutamate. This extracellular glutamate activates its receptor GRM3 on the plasma membrane, stimulating Rab27-mediated recycling of MT1-MMP to promote invasiveness (Dornier et al., 2017). This highlights how changes of tumor environment such as the increased extracellular glutamate and low-nutrient status of cancer cells alter cellular metabolism leading to aberrant endosomal recycling to drive cell invasion.
In addition to have a role in catabolic and metabolic signaling, lysosomes can also function as secretory compartments releasing their luminal content in the extracellular space in a calcium-dependent process (Blott and Griffiths, 2002; Xu et al., 2012; Kimmelman and White, 2017; Buratta et al., 2020). The lysosomal calcium-channel TRPML1 is activated by the metabolic stress conditions typical of cancer cells. Its activation promotes mTORC1 activity and ATP release via lysosomal exocytosis (Liu et al., 2012; Takai et al., 2012; Machado et al., 2015; Naegeli et al., 2017; Xu et al., 2019). Extracellular ATP interacts with purinergic receptors on the plasma membrane, and acts as cancer invasion stimulator by activating Rho GTPase-dependent pathways and upregulating the expression of MMPs (Zhang et al., 2010; Li et al., 2013).
Recent evidence demonstrates that the presence of mitochondria at cell protrusions stimulates ATP-driven actin polymerization to drive cell motility and invasion during Caenorhabditis elegans development, even in absence of MMPs (Kelley et al., 2019). This indicates special energy requirement in protruding regions. In line with that, a connection between intracellular mitochondrial trafficking and energy gradients has been described, where ATP:ADP ratio changes depending on positioning and density of mitochondria (Altieri, 2017; Schuler et al., 2017). Long-range mitochondrial trafficking relies on microtubule-associated molecular motors kinesins and dyneins as well as on the mitochondrial Rho-GTPase Miro1. In Miro1-deficient mouse embryonic fibroblasts (MEFs), mitochondria reposition to the perinuclear area, which correlates with high ATP production in this region. This inhibits energy-demanding processes such as protrusion formation and focal adhesion dynamics at the cell periphery, resulting in decreased cell migration and invasion (Schuler et al., 2017). Conversely, in migrating and invasive cancer cells, mitochondria accumulate at the leading edge (Arismendi-Morillo et al., 2012). Thus, the traffic and dynamics of mitochondria are coupled to the localized energy demand at the protruding cell front for focal adhesion dynamics, cell membrane dynamics and invasion.
ATP consumption at the leading edge promotes mitochondria trafficking with a positive feedback mechanism that depends on the energy sensor AMPK (Cunniff et al., 2016; Furnish and Caino, 2020). This is in agreement with evidence showing that some key glycolytic enzymes are located at the plasma membrane of invasive cells (Attanasio et al., 2011; Havrylov and Park, 2015; James et al., 2020) where the actomyosin machinery used to displace the ECM relies on the readily available supply of ATP (Oser et al., 2009; van Horssen et al., 2009; Kelley et al., 2019).
Interestingly, tumor exposure to inhibitors of the therapeutic target phosphatidylinositol-3-kinase (PI3K) has shown a unique repositioning of energetically active mitochondria in proximity to focal complexes, which supports membrane dynamics and cytoskeletal remodeling, resulting in increased cell motility and invasion (Caino et al., 2015). Although this response may possibly increase the risk of metastasis, it illustrates the feasibility of targeting mitochondrial reprogramming.
Concluding Remarks
It is now widely recognized that different metabolic needs are encountered during cancer progression. Therefore, it is of high importance understanding the underlying molecular mechanisms behind this metabolic cancer plasticity for the development of target therapies and also to prevent therapy resistance.
The contribution of intracellular membrane transport to the metabolic rewiring in disease progression is, however, still poorly characterized. Many questions remain unanswered due to the limitations of studying cancer cells in their complex tumor environment. However, studies where nutrient access was restricted by pharmacologically altering membrane trafficking have shown positive results, simultaneously blocking lysosomal degradation of autophagosomes and macropinosomes, and starving cancer cells to death (Kim et al., 2016). In line with this, ongoing studies with agents that target scavenging, macropinocytosis, autophagy or lysosomes seem to be promising (Towers and Thorburn, 2017; Towers et al., 2020). For example, recently developed lysosomal inhibitors for cancer therapy can inhibit multiple lysosomal activities needed for tumor cell survival and growth (Rebecca et al., 2017). Therefore, future research should further explore the molecular mechanisms of intracellular trafficking characterizing tumor initiation, progression and metastasis in relation to the different cellular metabolic needs as these aspects could help to identify new ways or targets for therapy.
Dormant cancer cells are one of the most threatening aspects of cancer and can lead to reoccurrence of metastatic tumors after a long period of latency. Tumor cell dormancy can be induced by nutrient deprivation (Jahanban-Esfahlan et al., 2019) but the mechanism behind the revival of the dormant cells remains mainly elusive. Therefore, further investigation is required to understand whether and how changes in nutrient availability as well as the metabolic adaptation influence this process. Moreover, the role of intracellular trafficking in the re-activation of the dormant cells is still unknown and its characterization may further improve our understanding of tumor dormancy with impact on tumor relapse.
Thus, the tight connection between intracellular trafficking and cell metabolism should be taken into account in the search of novel therapeutic targets for a more integrated cancer therapy.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The work in the authors’ laboratory is supported by grants from the Norwegian Cancer Society (grant 198094), the Research Council of Norway (grant 287560), the Anders Jahre Foundation, and UNIFOR-FRIMED 2020.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahn, C. H., Jeong, E. G., Lee, J. W., Kim, M. S., Kim, S. H., Kim, S. S., et al. (2007). Expression of beclin-1., an autophagy-related protein., in gastric and colorectal cancers. Apmis 115, 1344–1349. doi: 10.1111/j.1600-0463.2007.00858.x
Alexandrov, K., Horiuchi, H., Steele-Mortimer, O., Seabra, M. C., and Zerial, M. (1994). Rab escort protein-1 is a multifunctional protein that accompanies newly prenylated rab proteins to their target membranes. EMBO J. 13, 5262–5273. doi: 10.1002/j.1460-2075.1994.tb06860.x
Alonso-Curbelo, D., Osterloh, L., Cañón, E., Calvo, T. G., Martínez-Herranz, R., Karras, P., et al. (2015). RAB7 counteracts PI3K-driven macropinocytosis activated at early stages of melanoma development. Oncotarget 6, 11848–11862. doi: 10.18632/oncotarget.4055
Alonso-Curbelo, D., Riveiro-Falkenbach, E., Pérez-Guijarro, E., Cifdaloz, M., Karras, P., Osterloh, L., et al. (2014). RAB7 controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell 26, 61–76. doi: 10.1016/j.ccr.2014.04.030
Altieri, D. C. (2017). Mitochondria on the move: emerging paradigms of organelle trafficking in tumour plasticity and metastasis. Br. J. Cancer 117, 301–305. doi: 10.1038/bjc.2017.201
Amyere, M., Payrastre, B., Krause, U., Van Der Smissen, P., Veithen, A., and Courtoy, P. J. (2000). Constitutive macropinocytosis in oncogene-transformed fibroblasts depends on sequential permanent activation of phosphoinositide 3-kinase and phospholipase C. Mol. Biol. Cell 11, 3453–3467. doi: 10.1091/mbc.11.10.3453
Arismendi-Morillo, G., Hoa, N. T., Ge, L., and Jadus, M. R. (2012). Mitochondrial network in glioma’s invadopodia displays an activated state both in situ and in vitro: potential functional implications. Ultrastruct Pathol. 36, 409–414. doi: 10.3109/01913123.2012.694582
Arjonen, A., Kaukonen, R., Mattila, E., Rouhi, P., Högnäs, G., Sihto, H., et al. (2014). Mutant p53-associated myosin-X upregulation promotes breast cancer invasion and metastasis. J. Clin. Invest. 124, 1069–1082. doi: 10.1172/jci67280
Attanasio, F., Caldieri, G., Giacchetti, G., van Horssen, R., Wieringa, B., and Buccione, R. (2011). Novel invadopodia components revealed by differential proteomic analysis. Eur. J. Cell Biol. 90, 115–127. doi: 10.1016/j.ejcb.2010.05.004
Bays, J. L., Campbell, H. K., Heidema, C., Sebbagh, M., and DeMali, K. A. (2017). Linking E-cadherin mechanotransduction to cell metabolism through force-mediated activation of AMPK. Nat. Cell Biol. 19, 724–731. doi: 10.1038/ncb3537
Bloomfield, G., and Kay, R. R. (2016). Uses and abuses of macropinocytosis. J. Cell Sci. 129, 2697. doi: 10.1242/jcs.176149
Blott, E. J., and Griffiths, G. M. (2002). Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 3, 122–131. doi: 10.1038/nrm732
Bonnans, C., Chou, J., and Werb, Z. (2014). Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 15, 786–801. doi: 10.1038/nrm3904
Bravo-Cordero, J. J., Marrero-Diaz, R., Megías, D., Genís, L., García-Grande, A., García, M. A., et al. (2007). MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. EMBO J. 26, 1499–1510. doi: 10.1038/sj.emboj.7601606
Buratta, S., Tancini, B., Sagini, K., Delo, F., Chiaradia, E., Urbanelli, L., et al. (2020). Lysosomal Exocytosis., exosome release and secretory autophagy: the autophagic- and endo-lysosomal systems go extracellular. Int. J. Mol. Sci. 21:2576. doi: 10.3390/ijms21072576
Caino, M. C., Ghosh, J. C., Chae, Y. C., Vaira, V., Rivadeneira, D. B., Faversani, A., et al. (2015). PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc. Natl. Acad. Sci. U.S.A. 112, 8638–8643. doi: 10.1073/pnas.1500722112
Cao, Y., Wang, X., Li, Y., Evers, M., Zhang, H., and Chen, X. (2019). Extracellular and macropinocytosis internalized ATP work together to induce epithelial–mesenchymal transition and other early metastatic activities in lung cancer. Cancer Cell Int. 19:254. doi: 10.1186/s12935-019-0973-0
Castosa, R., Martinez-Iglesias, O., Roca-Lema, D., Casas-Pais, A., Díaz-Díaz, A., Iglesias, P., et al. (2018). Hakai overexpression effectively induces tumour progression and metastasis in vivo. Sci. Rep. 8:3466. doi: 10.1038/s41598-018-21808-w
Caswell, P. T., Chan, M., Lindsay, A. J., McCaffrey, M. W., Boettiger, D., and Norman, J. C. (2008). Rab-coupling protein coordinates recycling of alpha5beta1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J. Cell Biol. 183, 143–155. doi: 10.1083/jcb.200804140
Caswell, P. T., Spence, H. J., Parsons, M., White, D. P., Clark, K., Cheng, K. W., et al. (2007). Rab25 associates with alpha5beta1 integrin to promote invasive migration in 3D microenvironments. Dev. Cell 13, 496–510. doi: 10.1016/j.devcel.2007.08.012
Choi, J., Kim, H., Bae, Y. K., and Cheong, H. (2017). REP1 modulates autophagy and macropinocytosis to enhance cancer cell survival. Int. J. Mol. Sci. 18:1866. doi: 10.3390/ijms18091866
Cluntun, A. A., Lukey, M. J., Cerione, R. A., and Locasale, J. W. (2017). Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer 3, 169–180. doi: 10.1016/j.trecan.2017.01.005
Commisso, C., Davidson, S. M., Soydaner-Azeloglu, R. G., Parker, S. J., Kamphorst, J. J., Hackett, S., et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. doi: 10.1038/nature12138
Corallino, S., Malabarba, M. G., Zobel, M., Di Fiore, P. P., and Scita, G. (2015). Epithelial-to-Mesenchymal plasticity harnesses endocytic circuitries. Front. Oncol. 5:45. doi: 10.3389/fonc.2015.00045
Cunniff, B., McKenzie, A. J., Heintz, N. H., and Howe, A. K. (2016). AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell 27, 2662–2674. doi: 10.1091/mbc.e16-05-0286
Das, L., Gard, J. M. C., Prekeris, R., Nagle, R. B., Morrissey, C., Knudsen, B. S., et al. (2018). Novel regulation of integrin trafficking by Rab11-FIP5 in aggressive prostate cancer. Mol. Cancer Res. 16, 1319–1331. doi: 10.1158/1541-7786.mcr-17-0589
Davidson, S. M., Jonas, O., Keibler, M. A., Hou, H. W., Luengo, A., Mayers, J. R., et al. (2017). Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 23, 235–241. doi: 10.1038/nm.4256
Davidson, S. M., and Vander Heiden, M. G. (2017). Critical functions of the lysosome in cancer biology. Annu. Rev. Pharmacol. Toxicol. 57, 481–507. doi: 10.1146/annurev-pharmtox-010715-103101
Dornier, E., Rabas, N., Mitchell, L., Novo, D., Dhayade, S., Marco, S., et al. (2017). Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat. Commun. 8:2255. doi: 10.1038/s41467-017-02101-2
Dozynkiewicz, M. A., Jamieson, N. B., Macpherson, I., Grindlay, J., van den Berghe, P. V. E., von Thun, A., et al. (2012). Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell 22, 131–145. doi: 10.1016/j.devcel.2011.11.008
Falzoni, S., Donvito, G., and Virgilio, F. D. (2013). Detecting adenosine triphosphate in the pericellular space. Interf. Focus 3:20120101. doi: 10.1098/rsfs.2012.0101
Finicle, B. T., Jayashankar, V., and Edinger, A. L. (2018). Nutrient scavenging in cancer. Nat. Rev. Cancer 18, 619–633. doi: 10.1038/s41568-018-0048-x
Frittoli, E., Palamidessi, A., Marighetti, P., Confalonieri, S., Bianchi, F., Malinverno, C., et al. (2014). A RAB5/RAB4 recycling circuitry induces a proteolytic invasive program and promotes tumor dissemination. J. Cell Biol. 206, 307–328. doi: 10.1083/jcb.201403127
Fujita, Y., Krause, G., Scheffner, M., Zechner, D., Leddy, H. E. M., Behrens, J., et al. (2002). Hakai., a c-Cbl-like protein., ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 4, 222–231. doi: 10.1038/ncb758
Furnish, M., and Caino, M. C. (2020). Altered mitochondrial trafficking as a novel mechanism of cancer metastasis. Cancer Rep. 3:e1157. doi: 10.1002/cnr2.1157
Guo, J. Y., Chen, H. Y., Mathew, R., Fan, J., Strohecker, A. M., Karsli-Uzunbas, G., et al. (2011). Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470. doi: 10.1101/gad.2016311
Ha, K. D., Bidlingmaier, S. M., and Liu, B. (2016). Macropinocytosis exploitation by cancers and cancer therapeutics. Front. Physiol. 7:381. doi: 10.3389/fphys.2016.00381
Hamidi, H., and Ivaska, J. (2018). Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer 18, 533–548. doi: 10.1038/s41568-018-0038-z
Hanahan, D., and Weinberg Robert, A. (2011). Hallmarks of Cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hardie, D. G. (2015). Molecular pathways: is AMPK a friend or a foe in cancer? Clin. Cancer Res. 21, 3836–3840. doi: 10.1158/1078-0432.ccr-14-3300
Havrylov, S., and Park, M. (2015). MS/MS-based strategies for proteomic profiling of invasive cell structures. Proteomics 15, 272–286. doi: 10.1002/pmic.201400220
Hoshino, D., Kirkbride, K. C., Costello, K., Clark, E. S., Sinha, S., Grega-Larson, N., et al. (2013). Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 5, 1159–1168. doi: 10.1016/j.celrep.2013.10.050
Hosios, A. M., Hecht, V. C., Danai, L. V., Johnson, M. O., Rathmell, J. C., Steinhauser, M. L., et al. (2016). Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell 36, 540–549. doi: 10.1016/j.devcel.2016.02.012
Jacquemet, G., Green, D. M., Bridgewater, R. E., von Kriegsheim, A., Humphries, M. J., Norman, J. C., et al. (2013). RCP-driven α5β1 recycling suppresses Rac and promotes RhoA activity via the RacGAP1-IQGAP1 complex. J. Cell Biol. 202, 917–935. doi: 10.1083/jcb.201302041
Jahanban-Esfahlan, R., Seidi, K., Manjili, M. H., Jahanban-Esfahlan, A., Javaheri, T., and Zare, P. (2019). Tumor cell dormancy: threat or opportunity in the fight against cancer. Cancers 11:1207. doi: 10.3390/cancers11081207
James, A. D., Richardson, D. A., Oh, I.-W., Sritangos, P., Attard, T., Barrett, L., et al. (2020). Cutting off the fuel supply to calcium pumps in pancreatic cancer cells: role of pyruvate kinase-M2 (PKM2). Br. J. Cancer 122, 266–278. doi: 10.1038/s41416-019-0675-3
Janda, E., Nevolo, M., Lehmann, K., Downward, J., Beug, H., and Grieco, M. (2006). Raf plus TGFβ-dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene 25, 7117–7130. doi: 10.1038/sj.onc.1209701
Jeon, S.-M. (2016). Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 48:e245. doi: 10.1038/emm.2016.81
Kang, H., Kim, H., Lee, S., Youn, H., and Youn, B. (2019). Role of metabolic reprogramming in epithelial–mesenchymal transition (EMT). Int. J. Mol. Sci. 20:2042. doi: 10.3390/ijms20082042
Katsuno, Y., Lamouille, S., and Derynck, R. (2013). TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 25, 76–84. doi: 10.1097/cco.0b013e32835b6371
Kelley, L. C., Chi, Q., Cáceres, R., Hastie, E., Schindler, A. J., Jiang, Y., et al. (2019). Adaptive F-actin polymerization and localized ATP production drive basement membrane invasion in the absence of MMPs. Dev. Cell 48, 313.e8–328.e8. doi: 10.1016/j.devcel.2018.12.018
Kim, J., and Guan, K.-L. (2019). mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 21, 63–71. doi: 10.1038/s41556-018-0205-1
Kim, S. M., Roy, S. G., Chen, B., Nguyen, T. M., McMonigle, R. J., McCracken, A. N., et al. (2016). Targeting cancer metabolism by simultaneously disrupting parallel nutrient access pathways. J. Clin. Invest. 126, 4088–4102. doi: 10.1172/jci87148
Kimmelman, A. C., and White, E. (2017). Autophagy and tumor metabolism. Cell Metab. 25, 1037–1043. doi: 10.1016/j.cmet.2017.04.004
King, J. S., and Kay, R. R. (2019). The origins and evolution of macropinocytosis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374:20180158. doi: 10.1098/rstb.2018.0158
Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., et al. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036. doi: 10.1038/nature03029
Lanfredini, S., Thapa, A., and O’Neill, E. (2019). RAS in pancreatic cancer. Biochem. Soc. Trans. 47, 961–972. doi: 10.1042/BST20170521
Le Bras, G. F., Taubenslag, K. J., and Andl, C. D. (2012). The regulation of cell-cell adhesion during epithelial-mesenchymal transition., motility and tumor progression. Cell Adh. Migr. 6, 365–373. doi: 10.4161/cam.21326
Lee, S.-W., Zhang, Y., Jung, M., Cruz, N., Alas, B., and Commisso, C. (2019). EGFR-Pak signaling selectively regulates glutamine deprivation-induced macropinocytosis. Dev. Cell 50, 381.e–392.e.
Li, W. H., Qiu, Y., Zhang, H. Q., Liu, Y., You, J. F., Tian, X. X., et al. (2013). P2Y2 receptor promotes cell invasion and metastasis in prostate cancer cells. Br. J. Cancer 109, 1666–1675. doi: 10.1038/bjc.2013.484
Li, X., He, S., and Ma, B. (2020). Autophagy and autophagy-related proteins in cancer. Mol. Cancer 19, 12.
Linder, S., and Scita, G. (2015). RABGTPases in MT1-MMP trafficking and cell invasion: physiology versus pathology. Small GTPases 6, 145–152. doi: 10.4161/21541248.2014.985484
Liu, Y., Zhou, Y., and Zhu, K. (2012). Inhibition of glioma cell lysosome exocytosis inhibits glioma invasion. PLoS One 7:e45910. doi: 10.1371/journal.pone.0045910
Lock, R., Roy, S., Kenific, C. M., Su, J. S., Salas, E., Ronen, S. M., et al. (2011). Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 22, 165–178. doi: 10.1091/mbc.e10-06-0500
Loh, C.-Y., Chai, J. Y., Tang, T. F., Wong, W. F., Sethi, G., Shanmugam, M. K., et al. (2019). The E-Cadherin and N-cadherin switch in epithelial-to-mesenchymal transition: signaling. Ther. Implicat. Challenges. Cells 8:1118. doi: 10.3390/cells8101118
Lu, Z., Ghosh, S., Wang, Z., and Hunter, T. (2003). Downregulation of caveolin-1 function by EGF leads to the loss of E-cadherin., increased transcriptional activity of β-catenin., and enhanced tumor cell invasion. Cancer Cell 4, 499–515. doi: 10.1016/s1535-6108(03)00304-0
Ma, X. H., Piao, S. F., Dey, S., McAfee, Q., Karakousis, G., Villanueva, J., et al. (2014). Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Invest. 124, 1406–1417. doi: 10.1172/jci70454
Machado, E., White-Gilbertson, S., van de Vlekkert, D., Janke, L., Moshiach, S., Campos, Y., et al. (2015). Regulated lysosomal exocytosis mediates cancer progression. Sci. Adv. 1:e1500603. doi: 10.1126/sciadv.1500603
Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G., Chen, H. Y., et al. (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell 137, 1062–1075. doi: 10.1016/j.cell.2009.03.048
Moreno-Layseca, P., Icha, J., Hamidi, H., and Ivaska, J. (2019). Integrin trafficking in cells and tissues. Nat. Cell Biol. 21, 122–132. doi: 10.1038/s41556-018-0223-z
Mosesson, Y., Mills, G. B., and Yarden, Y. (2008). Derailed endocytosis: an emerging feature of cancer. Nat. Rev. Cancer 8, 835–850. doi: 10.1038/nrc2521
Muller, P. A., Caswell, P. T., Doyle, B., Iwanicki, M. P., Tan, E. H., Karim, S., et al. (2009). Mutant p53 drives invasion by promoting integrin recycling. Cell 139, 1327–1341. doi: 10.1016/j.cell.2009.11.026
Muranen, T., Iwanicki, M. P., Curry, N. L., Hwang, J., DuBois, C. D., Coloff, J. L., et al. (2017). Starved epithelial cells uptake extracellular matrix for survival. Nat. Commun. 8:13989. doi: 10.1038/ncomms13989
Naegeli, K. M., Hastie, E., Garde, A., Wang, Z., Keeley, D. P., Gordon, K. L., et al. (2017). Cell invasion in vivo via rapid exocytosis of a transient lysosome-derived membrane domain. Dev. Cell 43, 403.e–417.e.
Niño, C. A., Sala, S., and Polo, S. (2019). When ubiquitin meets E-cadherin: plasticity of the epithelial cellular barrier. Semin. Cell Dev. Biol. 93, 136–144. doi: 10.1016/j.semcdb.2018.12.005
Olivares, O., Mayers, J. R., Gouirand, V., Torrence, M. E., Gicquel, T., Borge, L., et al. (2017). Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 8:16031. doi: 10.1038/ncomms16031
Oser, M., Yamaguchi, H., Mader, C. C., Bravo-Cordero, J. J., Arias, M., Chen, X., et al. (2009). Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J. Cell Biol. 186, 571–587. doi: 10.1083/jcb.200812176
Palacios, F., Tushir, J. S., Fujita, Y., and Souza-Schorey, C. (2005). Lysosomal Targeting of E-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions. Mol. Cell. Biol. 25:389. doi: 10.1128/mcb.25.1.389-402.2005
Palm, W. (2019). Metabolic functions of macropinocytosis. Biol. Sci. 374:20180285. doi: 10.1098/rstb.2018.0285
Paul, N. R., Allen, J. L., Chapman, A., Morlan-Mairal, M., Zindy, E., Jacquemet, G., et al. (2015). α5β1 integrin recycling promotes Arp2/3-independent cancer cell invasion via the formin FHOD3. J. Cell Biol. 210, 1013–1031. doi: 10.1083/jcb.201502040
Pellegatti, P., Raffaghello, L., Bianchi, G., Piccardi, F., Pistoia, V., and Di Virgilio, F. (2008). Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One 3:e2599. doi: 10.1371/journal.pone.0002599
Planchon, D., Rios Morris, E., Genest, M., Comunale, F., Vacher, S., Bièche, I., et al. (2018). MT1-MMP targeting to endolysosomes is mediated by upregulation of flotillins. J. Cell Sci. 131:jcs218925. doi: 10.1242/jcs.218925
Potter, M., Newport, E., and Morten, K. J. (2016). The Warburg effect: 80 years on. Biochem. Soc. Trans. 44, 1499–1505. doi: 10.1042/bst20160094
Qian, Y., Wang, X., Liu, Y., Li, Y., Colvin, R. A., Tong, L., et al. (2014). Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett. 351, 242–251. doi: 10.1016/j.canlet.2014.06.008
Rahmani, S., Defferrari, M. S., Wakarchuk, W. W., and Antonescu, C. N. (2019). Energetic adaptations: metabolic control of endocytic membrane traffic. Traffic 20, 912–931. doi: 10.1111/tra.12705
Rainero, E., Howe, J. D., Caswell, P. T., Jamieson, N. B., Anderson, K., Critchley, D. R., et al. (2015). Ligand-occupied integrin internalization links nutrient signaling to invasive migration. Cell Rep. 10, 398–413. doi: 10.1016/j.celrep.2014.12.037
Rebecca, V. W., Nicastri, M. C., McLaughlin, N., Fennelly, C., McAfee, Q., Ronghe, A., et al. (2017). A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 7, 1266–1283. doi: 10.1158/2159-8290.cd-17-0741
Recouvreux, M. V., and Commisso, C. (2017). Macropinocytosis: a metabolic adaptation to nutrient stress in cancer. Front. Endocrinol. 8:261. doi: 10.3389/fendo.2017.00261
Schuler, M. H., Lewandowska, A., Caprio, G. D., Skillern, W., Upadhyayula, S., Kirchhausen, T., et al. (2017). Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol. Biol. Cell 28, 2159–2169. doi: 10.1091/mbc.e16-10-0741
Sciacovelli, M., and Frezza, C. (2017). Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. Febs J. 284, 3132–3144. doi: 10.1111/febs.14090
Shaul, Y. D., Freinkman, E., Comb, W. C., Cantor, J. R., Tam, W. L., Thiru, P., et al. (2014). Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell 158, 1094–1109. doi: 10.1016/j.cell.2014.07.032
Sneeggen, M., Pedersen, N. M., Campsteijn, C., Haugsten, E. M., Stenmark, H., and Schink, K. O. (2019). WDFY2 restrains matrix metalloproteinase secretion and cell invasion by controlling VAMP3-dependent recycling. Nat. Commun. 10:2850.
Son, H., and Moon, A. (2010). Epithelial-mesenchymal transition and cell invasion. Toxicol Res 26, 245–252. doi: 10.5487/tr.2010.26.4.245
Sui, X., Chen, R., Wang, Z., Huang, Z., Kong, N., Zhang, M., et al. (2013). Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 4:e838. doi: 10.1038/cddis.2013.350
Sun, L., Xu, X., Chen, Y., Zhou, Y., Tan, R., Qiu, H., et al. (2018). Rab34 regulates adhesion., migration., and invasion of breast cancer cells. Oncogene 37, 3698–3714. doi: 10.1038/s41388-018-0202-7
Swanson, J. A., and King, J. S. (2019). The breadth of macropinocytosis research. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374:20180146. doi: 10.1098/rstb.2018.0146
Takai, E., Tsukimoto, M., Harada, H., Sawada, K., Moriyama, Y., and Kojima, S. (2012). Autocrine regulation of TGF-β1-induced cell migration by exocytosis of ATP and activation of P2 receptors in human lung cancer cells. J. Cell Sci. 125, 5051–5060. doi: 10.1242/jcs.104976
Towers, C. G., and Thorburn, A. (2017). Targeting the lysosome for cancer therapy. Cancer Discov. 7, 1218–1220. doi: 10.1158/2159-8290.cd-17-0996
Towers, C. G., Wodetzki, D., and Thorburn, A. (2020). Autophagy and cancer: modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 219:e201909033.
Ulrich, F., and Heisenberg, C.-P. (2009). Trafficking and Cell Migration. Traffic 10, 811–818. doi: 10.1111/j.1600-0854.2009.00929.x
van Horssen, R., Janssen, E., Peters, W., van de Pasch, L., Lindert, M. M., van Dommelen, M. M., et al. (2009). Modulation of cell motility by spatial repositioning of enzymatic ATP/ADP exchange capacity. J. Biol. Chem. 284, 1620–1627. doi: 10.1074/jbc.m806974200
Wang, Z., Sandiford, S., Wu, C., and Li, S. S. C. (2009). Numb regulates cell-cell adhesion and polarity in response to tyrosine kinase signalling. EMBO J. 28, 2360–2373. doi: 10.1038/emboj.2009.190
Xu, J., Toops, K. A., Diaz, F., Carvajal-Gonzalez, J. M., Gravotta, D., Mazzoni, F., et al. (2012). Mechanism of polarized lysosome exocytosis in epithelial cells. J. Cell Sci. 125(Pt. 24), 5937–5943. doi: 10.1242/jcs.109421
Xu, M., Almasi, S., Yang, Y., Yan, C., Sterea, A. M., Rizvi Syeda, A. K., et al. (2019). The lysosomal TRPML1 channel regulates triple negative breast cancer development by promoting mTORC1 and purinergic signaling pathways. Cell Calc. 79, 80–88. doi: 10.1016/j.ceca.2019.02.010
Yang, S., Wang, X., Contino, G., Liesa, M., Sahin, E., Ying, H., et al. (2011). Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729. doi: 10.1101/gad.2016111
Yoon, S. O., Shin, S., and Mercurio, A. M. (2005). Hypoxia stimulates carcinoma invasion by stabilizing microtubules and promoting the Rab11 trafficking of the alpha6beta4 integrin. Cancer Res. 65, 2761–2769. doi: 10.1158/0008-5472.can-04-4122
Yoshida, S., Pacitto, R., Yao, Y., Inoki, K., and Swanson, J. A. (2015). Growth factor signaling to mTORC1 by amino acid-laden macropinosomes. J. Cell Biol. 211, 159–172. doi: 10.1083/jcb.201504097
Zhang, C., Liu, J., Liang, Y., Wu, R., Zhao, Y., Hong, X., et al. (2013). Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 4:2935. doi: 10.1038/ncomms3935(2013
Zhang, J., Shao, X., Sun, H., Liu, K., Ding, Z., Chen, J., et al. (2016). NUMB negatively regulates the epithelial-mesenchymal transition of triple-negative breast cancer by antagonizing Notch signaling. Oncotarget 7, 61036–61053. doi: 10.18632/oncotarget.11062
Keywords: membrane trafficking, cancer cell metabolism, cell proliferation, epithelial to mesenchymal transition, invasion
Citation: Sneeggen M, Guadagno NA and Progida C (2020) Intracellular Transport in Cancer Metabolic Reprogramming. Front. Cell Dev. Biol. 8:597608. doi: 10.3389/fcell.2020.597608
Received: 21 August 2020; Accepted: 12 October 2020;
Published: 30 October 2020.
Edited by:
Carlos M. Guardia, National Institutes of Health (NIH), United StatesReviewed by:
Chad Williamson, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), United StatesAntonino Colanzi, Institute of Biochemistry and Cell Biology, National Research Council (CNR), Italy
Copyright © 2020 Sneeggen, Guadagno and Progida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cinzia Progida, Yy5hLm0ucHJvZ2lkYUBpYnYudWlvLm5v