
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 14 December 2020
Sec. Signaling
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.595849
This article is part of the Research Topic Editor's Pick 2021: Highlights in Signaling View all 12 articles
 Amanda Krajnik1†Joseph A. Brazzo III1†Kalyanaraman Vaidyanathan1Tuhin Das2Javier Redondo-Muñoz3,4Yongho Bae1*
Amanda Krajnik1†Joseph A. Brazzo III1†Kalyanaraman Vaidyanathan1Tuhin Das2Javier Redondo-Muñoz3,4Yongho Bae1*Phosphoinositides, which are membrane-bound phospholipids, are critical signaling molecules located at the interface between the extracellular matrix, cell membrane, and cytoskeleton. Phosphoinositides are essential regulators of many biological and cellular processes, including but not limited to cell migration, proliferation, survival, and differentiation, as well as cytoskeletal rearrangements and actin dynamics. Over the years, a multitude of studies have uniquely implicated phosphoinositide signaling as being crucial in cardiovascular biology and a dominant force in the development of cardiovascular disease and its progression. Independently, the cellular transduction of mechanical forces or mechanotransduction in cardiovascular cells is widely accepted to be critical to their homeostasis and can drive aberrant cellular phenotypes and resultant cardiovascular disease. Given the versatility and diversity of phosphoinositide signaling in the cardiovascular system and the dominant regulation of cardiovascular cell functions by mechanotransduction, the molecular mechanistic overlap and extent to which these two major signaling modalities converge in cardiovascular cells remain unclear. In this review, we discuss and synthesize recent findings that rightfully connect phosphoinositide signaling to cellular mechanotransduction in the context of cardiovascular biology and disease, and we specifically focus on phosphatidylinositol-4,5-phosphate, phosphatidylinositol-4-phosphate 5-kinase, phosphatidylinositol-3,4,5-phosphate, and phosphatidylinositol 3-kinase. Throughout the review, we discuss how specific phosphoinositide subspecies have been shown to mediate biomechanically sensitive cytoskeletal remodeling in cardiovascular cells. Additionally, we discuss the direct interaction of phosphoinositides with mechanically sensitive membrane-bound ion channels in response to mechanical stimuli. Furthermore, we explore the role of phosphoinositide subspecies in association with critical downstream effectors of mechanical signaling in cardiovascular biology and disease.
Phosphoinositides (PPIs) constitute less than five percent of all cell membrane phospholipids (Hammond and Hong, 2018) but are essential to the integrity of all living cells (Dickson and Hille, 2019). Numerous studies have shown that PPIs are critical to cellular functions, including but not limited to cell proliferation, survival, motility, differentiation, and cytoskeletal dynamics (Di Paolo et al., 2004; Huang et al., 2004; Tsujita and Itoh, 2015; De Craene et al., 2017; Senju et al., 2017; Hao et al., 2018; Ramos et al., 2018; Bilanges et al., 2019; Li H. et al., 2019; Hirsch et al., 2020). The generation of PPIs is mediated by phosphorylation and dephosphorylation of phosphatidylinositol, the membrane lipid precursor (De Craene et al., 2017). More specifically, the inositol head of phosphatidylinositol can be phosphorylated at the 3-, 4-, and 5-hydroxyl positions of the inositol ring. The attachment of phosphate(s) can occur at any of these positions singularly or in combination to generate seven biologically active PPI subspecies: PI(3)P, PI(4)P, PI(5)P, PI(3,4)P2, PI(4,5)P2, PI(3,5)P2, and PI(3,4,5)P3 (Di Paolo and De Camilli, 2006; De Craene et al., 2017). All seven PPI subspecies naturally occur in the cell membrane of eukaryotes to varying degrees and are chemically interconverted by cell-specific kinases (purple-colored text in Figure 1) and phosphatases (red-colored text in Figure 1). PPI subspecies are shown as the black-colored text in Figure 1. Once biochemically active, PPIs modulate a tremendous breadth of horizontal and vertical cell signaling crosstalk spanning the cell membrane and cytoplasm, respectively, in which high-affinity interactions occur among various pleckstrin homology (PH) domain-containing membrane-based and cytosolic effector proteins, including protein kinase B (PKB)/Akt, protein kinase C (PKC), phosphoinositide phospholipase C (PLC), 3-phosphoinositide-dependent protein kinase-1 (PDK1), and small G proteins (Prestwich, 2004; Ghigo and Li, 2015; Manna and Jain, 2015; De Craene et al., 2017). In the cardiovascular system, activated PPI signaling mediates enzymatic organic modification of secondary messenger proteins because PPIs are crucial scaffolding proteins to complex signalosomes of cardiac and vascular cellular functions, and their aberration is a prominent driving force in cardiovascular pathology (Falkenburger et al., 2010; Ghigo and Li, 2015; Schink et al., 2016).
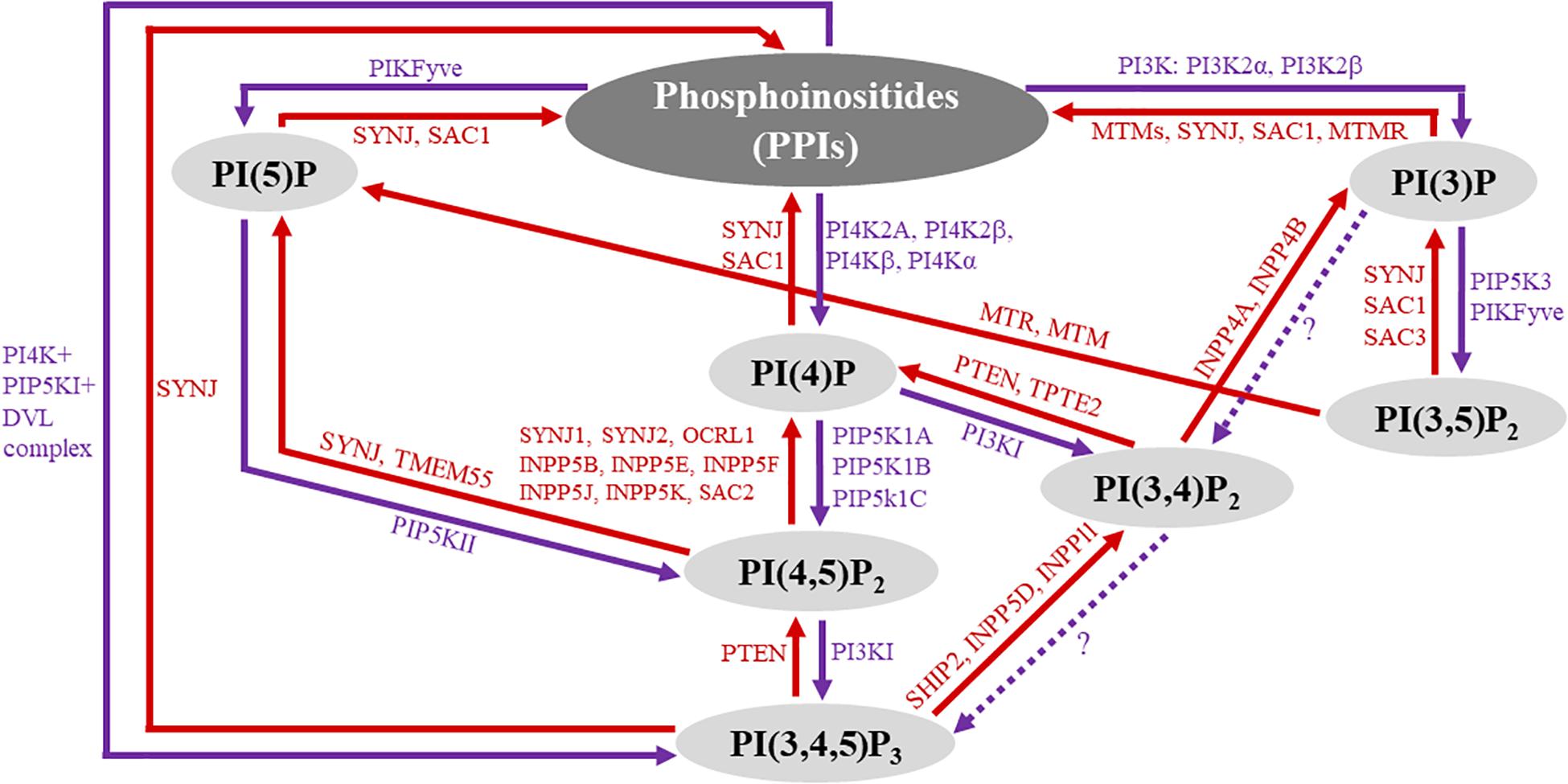
Figure 1. Overview of phosphoinositide (PPI) signaling. The diagram represents an overview of PPI subspecies and their biochemical interconversion. The seven biologically active PPI subspecies are highlighted in gray circles. PPI phosphatases are labeled red, and their associated reactions are represented by red arrows, indicating the direction of the reaction. Similarly, kinase reactions are represented by purple arrows with kinase enzymes labeled purple. Question marks along the dotted arrows represent areas that have yet to be explored.
Mechanotransduction describes the physiological process by which cells perceive and respond to mechanical stimuli, including tensile stretch and compression, shear stress, and extracellular matrix (ECM) stiffness. Moreover, mechanical cues are converted into intracellular biochemical signals in which the resultant cytoskeletal and nuclear remodeling modulates cellular functions (Tschumperlin, 2011; Maurer and Lammerding, 2019). Mechanotransduction is vital to cardiovascular tissue development, growth, and homeostasis because cells are continuously under mechanical stress (Garoffolo and Pesce, 2019). Dysregulation of the mechanical harmony between the cell and ECM can drive the development and progression of pathology, including but not limited to cardiac ischemia and fibrosis, hypertension, and atherosclerosis (Gimbrone and Garcia-Cardena, 2013; Yue et al., 2015; Schafer et al., 2017; Ochoa et al., 2018; Russo et al., 2018). Only recently have we begun to understand the cellular mechanisms that mediate the signal transduction of mechanical stimuli, which greatly overlap with canonical biochemical cellular signaling pathways.
Today, cardiovascular disease (CVD) remains the leading cause of death and morbidity worldwide. A great majority of biomedical research in CVD centers around the known mechanisms of biochemical and molecular biology modalities. With the recent emergence of novel biomechanical and cell biological technologies and techniques, there has been a new integrative movement toward understanding the mechanical regulation of cellular biochemistry and molecular biology inside the cell. This review will span the most recent findings in phosphoinositide biology as it relates to mechanically sensitive cellular processes in cardiovascular cells in both homeostasis and disease. We will discuss how specific PPI subspecies mediate cytoskeletal remodeling processes known to be dominantly regulated by mechanotransduction and the direct interaction of PPIs with membrane-bound channels in response to mechanical stress in cardiovascular cells. Furthermore, we will explore the role of PPI subspecies in association with the essential effectors of mechanical signaling in cardiovascular biology and disease.
The actin cytoskeleton is a complex and dynamic intracellular structure that gives mechanical rigor to the cell while simultaneously mediating the transduction of mechanical stress into biochemical signals. Given the unique contractile properties of cardiac and vascular cells, the actin cytoskeleton is most essential to their integrity (Allahverdian et al., 2018; Zhang et al., 2018). Alteration of cytoskeletal organization, specifically actin filament dynamics, can result in gene expression and cell proliferation modification with the subsequent adaptation of and changes to intracellular biochemical responses and cellular functions, respectively. Thus, cytoskeletal remodeling can mediate mechanical stress-induced gene expression, cell proliferation, and pathological processes (Ohashi et al., 2017; Martino et al., 2018). Recent studies have shown that PIPs regulate cytoskeletal arrangement and signaling dynamics (Di Paolo and De Camilli, 2006; Saarikangas et al., 2010; Senju and Lappalainen, 2019). Specifically, PIP2 is involved in cytoskeletal reorganizational events, including vesicle trafficking, cell migration, phagocytosis, and membrane cytoskeletal adhesion (Saarikangas et al., 2010; Shewan et al., 2011; Dickson and Hille, 2019; Phan et al., 2019). PIP2 binds to and affects actin-binding proteins, such as myristoylated alanine-rich C kinase substrate (MARCKS), cofilin, gelsolin, α-actinin, Wiskott-Aldrich syndrome protein (WASP), and the Rho family of small GTPases (Janmey et al., 2018; Figure 2). MARCKS is an actin-binding protein found in mammalian tissues and, upon phosphorylation, it binds reversibly to structural and regulatory molecules in the cell in which there is an associated decrease in PIP2 binding (Nairn and Aderem, 1992; Sheetz et al., 2006). Additionally, in vascular endothelial cells (ECs), MARCKS directly modulates PIP2-mediated insulin signaling. The treatment of vascular ECs with insulin increases the levels of PIP2, which is also released into lipid rafts (caveolar and non-caveolar fractions) to bind to the cytoskeletal protein, N-WASP. Subsequently, N-WASP phosphorylation and interaction with actin-related proteins 2/3 (Arp2/3) cause cytoskeletal remodeling to induce cell migration (Kalwa and Michel, 2011). Furthermore, MARCKS mediates a PIP2-dependent actin rearrangement process. In this process, when low levels of MARCKS are present, actin filaments form an actin gel. Conversely, actin filaments aggregate upon increased levels of MARCKS. In the plasma membrane, the overall PIP2 levels are relatively constant; however, changes in PIP2 levels can be observed locally in the membrane directly overlying actin protrusions and membrane ruffles (Sheetz et al., 2006).
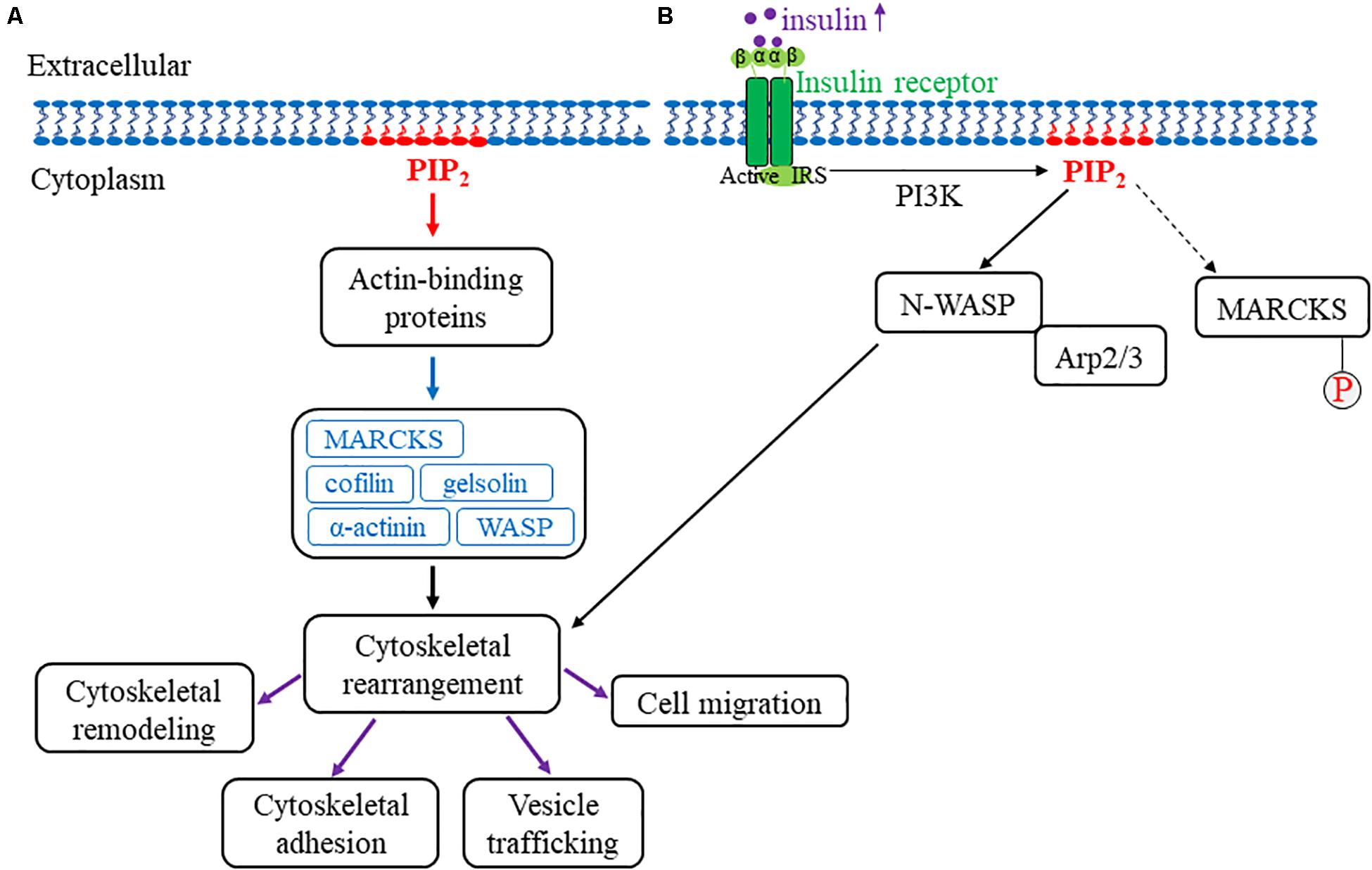
Figure 2. Model for PIP2 association with the actin cytoskeleton. (A) An overview of PIP2’s role in actin cytoskeletal remodeling. The red arrow represents PIP2 intracellular association with actin-binding proteins. The blue arrow represents the various PIP2-associated actin-binding proteins, including MARCKS, cofilin, WASP, gelsolin, and α-actinin. Upon interaction with PIP2, these proteins collectively mediate cellular cytoskeletal rearrangement (indicated by the black arrow). More specifically, actin cytoskeletal remodeling, cytoskeletal adhesion, vesicle trafficking, and cell migration are associated with cytoskeletal rearrangements that are mediated by PIP2 interaction with actin-binding proteins and are indicated by the purple arrows. (B) This figure depicts how PIP2 regulates cytoskeletal rearrangement through N-WASP and Arp2/3 with MARCKS phosphorylation. Once the insulin receptor is activated, insulin increases intracellularly and subsequently mediates the dissociation of MARCKS from PIP2 (as indicated by the dotted arrow) in which MARCKS is subsequently phosphorylated and activated. In addition, PIP2 separately binds to and activates both N-WASP and Arp2/3, which drives actin cytoskeletal rearrangement. Together, these findings highlight the role of PIP2 in mechanosensitive actin cytoskeletal rearrangement and remodeling.
Rac, a downstream small GTPase effector, is a regulator of membrane ruffles (Ridley, 1994). In addition, Rac is instrumental to the transduction of external mechanical stimuli (Labouesse, 2011; Lawson and Burridge, 2014; McGowan and McCoy, 2017), including the mechanotransduction of the FAK-Cas-Rac axis, which transmits ECM stiffness into intracellular stiffness and mechanosensitive cell cycling (Bae et al., 2014). Moreover, PIP2 levels fluctuate in membrane ruffles in a Rac-dependent manner. Although the immediate relationship of PIP2 and Rac has not been explored in the context of cardiovascular biology and disease specifically, acknowledging their possible connection could better aid the understanding of their effects in different cellular pathways and help implicate PIP2 as a Rac-mediated effector in vascular pathology (Polacheck et al., 2017; Narumiya and Thumkeo, 2018).
In striated muscle, capping protein Z (CapZ), an actin-capping protein, regulates cytoskeletal remodeling (Edwards et al., 2014). CapZ’s relationship with PIP2 (Figure 3) has been recently observed in relation to mechanical stiffness and cytoskeletal remodeling, and the PIP2-mediated interaction with CapZ has been shown to regulate cardiac myocyte hypertrophy (Li et al., 2016) and actin dynamics (Li and Russell, 2013). More specifically, ECM stiffening induces cardiac myocyte hypertrophy by increased PIP2 localization at the sarcomere Z-discs in cardiac myocytes (Li et al., 2016). The Z-disc is a critical site for mechanotransduction and the location of the β1-isoform of CapZ (CapZβ1) (Russell et al., 2010). Moreover, the localization of PIP2 to the sarcomere Z-disc is crucial to ventricular cardiac myocyte mechanotransduction and associated with pathological hypertrophic remodeling. Dysregulation of PIP2 signaling alters sarcomere integrity by modulating the function of CapZβ1 and actin dynamics. Taken together, PIP2 is vital to cardiac cell physiology, where it regulates CapZβ1 and actin dynamics in response to mechanical stimuli. Additionally, mechanical stimulation causes the production of PIP2, specifically through the RhoA/Rho-associated kinase (ROCK) pathway (Li and Russell, 2013). Solís et al. explored PIP2 signaling effects on CapZ through neomycin, a PIP2 sequestering agent, in neonatal ventricular cardiomyocytes cultured on varying substrate stiffnesses. Further studies have assessed the molecular mechanisms by which different mechanotransduction signaling pathways mediate the capping and uncapping of CapZ from actin filaments via PIP2. The results showed that interactions between PIP2 and the β-tentacle of CapZ after molecular stimulation become considerably modified by phosphorylation. Moreover, CapZ is bound tightly to actin when inactive; however, upon phosphorylation and activation in growth states of hypertrophy, the binding is loosened. This is triggered by external stimuli, including mechanical flexing, loading, a stiffer substrate, angiotensin II, and phenylephrine. CapZ is modified by the stimuli’s signaling pathways through phosphorylation, acetylation or PIP2 binding. Thus, an actin assembly mechanism can be presented where phosphorylation, acetylation or PIP2 anchorage causes CapZ to act as a nodal terminus for the integration of various signaling pathways (Solis and Russell, 2019). This mechanism implicates PIP2 as a critical mediator of mechanotransduction in cardiac myocytes by directly affecting CapZ in response to mechanical stiffness.
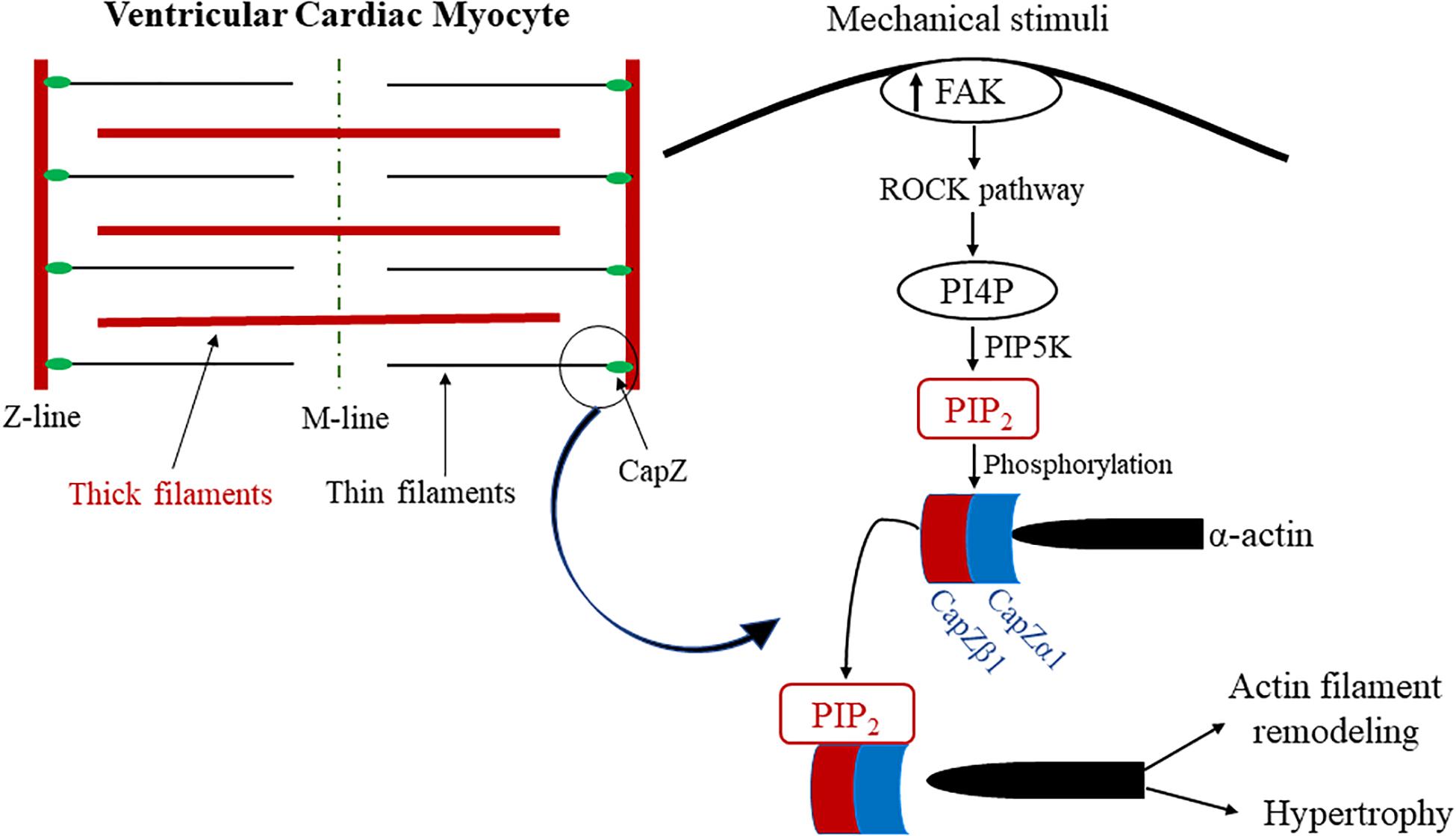
Figure 3. Proposed model of PIP2 and CapZ mechanotransduction in cardiac myocytes. Diagram of the proposed model for the mechanism of PIP2 association with CapZ because of mechanical stimuli. Mechanical strain activates the ROCK pathway when there is an increase in mechanosensitive FAK activation, which results in PIP5K phosphorylating PI4P into PIP2. PIP2 phosphorylates CapZ, which mediates its uncapping from α-actin. This results in pathological actin filament remodeling and cardiac hypertrophy. PIP2 is highlighted in and colored red. CapZα1 and CapZβ1 are colored blue to represent their association with PIP2 as well as their intricacy with biomechanically sensitive actin filament remodeling and pathological cardiac hypertrophy.
Phospholipase C (PLC) is a critical membrane-associated enzyme that, when stimulated by Gαq/11 subtype protein-coupled receptors (GαqPCRs), catalyzes the hydrolysis of PIP2 phosphodiester bonds to generate inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG), which further activates PKC. These secondary messengers and downstream effector proteins are important for cardiovascular cell functions because they orchestrate the regulation of intracellular calcium mobilization, which is critical not only to the contractile apparatus but also to cell survival, proliferation, and differentiation. Plasma membrane store-operated channels (SOCs), particularly transient receptor potential canonical channels 1, 3, and 6 (TRPC1/3/6) and calcium release-activated calcium (CRAC), also called Orai 1, 2, and 3, are activated as a consequence of PLC activation (Abdullaev et al., 2008; Baudel et al., 2020; Wang et al., 2020). Vascular smooth muscle cell (VSMC) contraction, proliferation, and migration are regulated by the stimulation of SOCs at the plasma membrane and their associated Ca2+ influx pathways (Baudel et al., 2020). Moreover, these cellular behaviors are associated with the development of diseases of the vasculature, such as hypertension and atherosclerosis (Baudel et al., 2020). In VSMCs, PKC activity and PIP2 are important in the activation pathway of SOCs, particularly transient receptor potential canonical channel 1 (TRPC1) (Saleh et al., 2009b; Shi et al., 2012; Baudel et al., 2020). Upon Ca2+ store depletion, TRPC1 is phosphorylated by PKC, which itself is stimulated by the PLC-PIP2-DAG pathway (Saleh et al., 2009a), thus establishing a potentially direct link between TRPC1 and PIP2. Additionally, TRPC1 is known to be an essential component of various mechanotransduction pathways, specifically in cells where TRPC1 is crucial for mechanosensitive cell migration (Formigli et al., 2009; Garrison et al., 2012; Canales et al., 2019; Li et al., 2019). Interestingly, TRPC1 is upregulated in pathological neointima remodeling in vessels induced by vascular injury, further suggesting that the induction of these channels is mechanosensitive (Kumar et al., 2006). Recent findings by Nikolaev et al. have suggested that the TRP ion channel superfamily is involved in a wide variety of mechanosensory processes, yet it has been shown that such channels are insensitive to tension induced by cell membrane stretching. Thus, although several TRP channels, including TRPC1, are essential components of mammalian stretch-activated mechanosensitive calcium-permeable cation channel heterologous systems, their true role in mechanotransduction remains unclear (Gottlieb et al., 2008). It is quite plausible that these ion channels are more likely to be activated by upstream components and consequently act as amplifiers of cellular mechanosensory signaling cascades, including PLC and PKC (Nikolaev et al., 2019). In addition to TRPC, the Orai channel, or CRAC, is another class of Ca2+-selective SOC activated as a consequence of PLC activation and subsequent PIP2 depletion (Abdullaev et al., 2008; Baudel et al., 2020; Wang et al., 2020). This channel is expressed in VSMCs and upregulated in such cells in vascular pathologies, including vascular injury and restenosis, which are known to be mechanically mediated (Wang et al., 2008; Spinelli and Trebak, 2016). Previous reports have shown that Orai interacts with TRPC channel subtypes, including TRPC3 and TRPC6 (Liao et al., 2007); however, TRPC 1 is independent of Orai function (DeHaven et al., 2009; Shi et al., 2017). Although these two proteins share great similarity in their functionality, it has yet to be explored how they may be coupled mechanically. Previous studies have assessed the mechanosensitivity of the Orai channel; however, it remains to be empirically determined (Dong et al., 2019). Furthermore, Piezo1 is a recently characterized putative mechanically activated calcium permeable cation channel that is ubiquitously expressed through the cardiovascular system (Beech and Kalli, 2019). It has been uniquely shown that Piezo1’s mechanosensitivity to membrane tension is regulated by PIP2 levels. Upon activation of TRPV1, PLC is activated and depletes the local levels of PIP2, which subsequently inhibits Piezo1’s mechanosensitive activity (Borbiro et al., 2015).
The capillary endothelial cell (cEC) inward rectifier K+ channel Kir2.1 is critical to sensing and translating neural activity and neurovascular coupling in brain cECs (Longden and Nelson, 2015). This process of capillary-to-arteriole signaling in cECs is regulated by PIP2. Moreover, PIP2 levels are required for sustained Kir2.1 activity, and such regulation of Kir2.1 channels mediates electrical signaling during neurovascular coupling (Harraz et al., 2018a). More specifically, GαqPCRs stimulate PLC to rapidly either deplete or decrease PIP2 levels and subsequently suppresses Kir2.1 channel signaling (Harraz et al., 2018a). This depletion of PIP2 also promotes the activation of transient receptor potential vanilloid 4 (TRPV4), a channel found in cECs that is inhibited by PIP2 under basal conditions and because of GαqPCR activation (Harraz et al., 2018b). Furthermore, PIP2 levels govern capillary-to-arteriole electrical signaling by modulating the activity of TRPV4 and Kir2.1, which regulate the cellular states of depolarization and hyperpolarization. Thus, the levels of PIP2 considerably modulate the magnitude of electrical signaling across cerebral capillaries, which ultimately affects cerebral microcirculatory blood flow in cECs (Harraz et al., 2018a,b). The relationship of TRPV4 to PIP2 is important given TRPV4’s involvement in shear stress mechanotransduction in endothelial cells and mesenchymal cells and its ability to act mechanosensitively (Kohler and Hoyer, 2007; Yin and Kuebler, 2010; Corrigan et al., 2018). Therefore, the role of PIP2 in this signaling pathway and its interaction with a known mechanotransducer, TRPV4, suggests that PIP2 itself acts in the mechanotransduction of capillary electrical signaling.
Phosphatidylinositol-4-phosphate 5-kinase (PIP5K) phosphorylates the fifth position of the inositol head of phosphatidylinositol-4-phosphate. The type 1 PIP5K subfamily comprises three isoforms, Iα, Iβ, and Iγ, and is critical to many cytoskeletal processes. It has been reported that the overexpression of these isoforms induces the formation of stress fibers, membrane ruffles, and microvilli and regulates actin cytoskeletal dynamics, suggesting that this enzyme and its PIP2 products are mechanosensitive (Chatah and Abrams, 2001). Weernink et al. reported that RhoA and its kinase, ROCK, which are both dominant effectors of mechanotransduction, are essential regulators of PIP5K in HEK-293 cells. The overexpression of ROCK enhances the PIP5K activity and subsequently elevates PIP2 formation. Conversely, the chemical inhibition of ROCK decreases PIP5K activity and PIP2 formation (Oude Weernink et al., 2000). Furthermore, Weernink et al. examined Type 1 PIP5K through other Rho family small GTPases, including Rac1 and Cdc42. Rho GTPases, RhoA, Rac1, and Cdc42 all mediate the PIP5K levels and lead to an increase in PIP2 levels (Weernink et al., 2004). Therefore, PIP5K activity is RhoA-dependent in which signals from RhoA to the actin cytoskeleton are mediated, and synthesis of PIP2 is enhanced (Oude Weernink et al., 2000).
The PIP2 synthesis pathway in platelets through the isoform PIP5K Iα was more closely studied by Chatah and Abrams (2001) and Trepat et al. (2005). Thrombin, a known mediator of actin cytoskeleton remodeling (Chatah and Abrams, 2001; Trepat et al., 2005), promotes PIP2 synthesis by PIP5K from PI4P in response to G protein-coupled receptor stimulation. PIP5K Iα localizes in the Golgi under basal conditions. Following stimulation of PAR1, a thrombin receptor, or overexpression of the active variant of Gαq, PIP5K Iα relocates to the plasma membrane. This translocation of PIP5K Iα is dependent on Rac1 and RhoA. Rac1 has been suggested to affect PIP5K indirectly, and activation is required by Rho (Chatah and Abrams, 2001).
Although these studies independently identified Rho GTPases in mediating PIP5K activity, the mechanisms by which Rho GTPase is suggested to activate PIP5K are separate. Taken together, these findings indicate that members of the Rho GTPase family, RhoA, Rac, and Cdc42, are vital in mediating PIP5K activation and, consequently, PIP2 synthesis, regardless of their interconversional crosstalk (Oude Weernink et al., 2000; Chatah and Abrams, 2001; Weernink et al., 2004). Furthermore, these GTPases act as a dynamic molecular switch between various cells, which play a key role in vascular pathology (Cai et al., 2015; Karoor et al., 2018; Barlow and Cleaver, 2019) and are involved in mechanosensing and mechanotransduction pathways (Verma et al., 2011; Chaterji et al., 2014; Zegers and Friedl, 2014; Ohashi et al., 2017). The relationship of PIP5K with small downstream GTPases in vascular pathology and mechanotransduction has not yet been explored. Due to the relevance of the small GTPases Rho, Rac, and Cdc42 for PIP5K activity and PIP2 synthesis, this pathway may be vitally important for better understanding vascular disease and may be potentially significant in the overall study of mechanotransduction in the context of vascular pathology. Therefore, the relationship of these PIP5Ks and these downstream GTPases should be explored in relation to mechanotransduction and vascular disease.
Phosphoinositide 3-kinase (PI3K) is a family of evolutionarily conserved lipid kinases that mediate many cellular responses to physiological and pathophysiological stimuli. The PI3K family is divided into three subgroups (classes I, II, and III), which together include eight isoforms. The class I isoforms, PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ, convert PIP2 to phosphatidylinositol-3,4,5-triphosphate (PIP3) (Vanhaesebroeck et al., 2010; Miller et al., 2019). Activated PI3K produces PIP3, which further recruits 3-phosphoinositide-dependent kinase 1 (PDK1) to the plasma membrane (Hagiwara et al., 2012). PIP3 activates PDK1 through its PH domain. PDK1 subsequently phosphorylates and activates Akt at threonine residue 308 (T308) (Ghigo and Li, 2015; Manning and Toker, 2017). More importantly, the phosphorylation of serine residue 473 (S473) by the mechanistic target of the mammalian target of rapamycin complex 2 (mTORC2) stabilizes not only T308 phosphorylation but also AKT in its active state (Manning and Toker, 2017). Together, Akt and PI3K create a unique signaling pathway (Akt/PI3K) that is instrumental in cardiomyocyte mechanotransduction (Li C.J. et al., 2019). Moreover, the Akt/PI3K signaling pathway regulates intracellular and extracellular activities in response to mechanical stress and molecular effectors, leading to a robust cellular mechanotransduction signaling cascade in cardiac myocytes. These cellular responses include modulation of cell metabolism, growth, proliferation, angiogenesis, and cardiac adaptation (Aoyagi and Matsui, 2011; Markowska et al., 2014; Yang et al., 2018). In a disease model, chronic activation of the Akt/PI3K pathway dysregulates cell contractility, which induces compensatory cardiac hypertrophy with preserved contractility and ultimately advances to chronic dilated cardiomyopathy (Shiojima et al., 2005; Li C.J. et al., 2019). Furthermore, alterations in the function and structure of titin, a giant sarcomeric filament protein, have been observed in similar cardiomyopathies, including cardiac remodeling, hypertrophy, and heart failure (Linke, 2008; Kruger and Linke, 2009; Lyon et al., 2015). In cardiac sarcomeres, titin isoforms exhibit varying properties of mechanical elasticity and are differentially expressed throughout cardiac development and during disease in which isoform switching is dynamically regulated by the Akt/PI3K signaling pathway (Kruger and Linke, 2009). Moreover, it is believed that these properties of titin are uniquely positioned to serve as a molecular sensor of mechanical stress in cardiac myocytes, including oscillatory changes in cell stretching known to induce PI3K activation through molecular mechanisms that remain unclear (Miller et al., 2004; Linke, 2008; Leychenko et al., 2011; Voelkel and Linke, 2011).
In cardiac myocytes, mechanotransduction critically mediates remodeling of the cytoskeleton, and dysregulation of this process can drive heart disease in response to aberrant biomechanical stress. Biomedical research on patients with hypertension has revealed how critical cardiac mechanotransduction plays in this response (Patel et al., 2013). One study by Patel et al. demonstrated that PI3Kα, a major PI3K isoform in the heart, negatively regulates gelsolin activity and suppresses pathological cytoskeletal remodeling in response to biomechanical stress-induced cardiac mechanotransduction and the resulting dilated cardiomyopathy (Guo et al., 2010). Similarly, a separate study showed that loss of PTEN in ventricular cardiac myocytes increases PI3Kα activity, which attenuates pressure overload-induced heart failure but loss of myocardial contractility (Oudit et al., 2008). Conversely, however, other studies have shown that constitutively activated PI3K drives the growth and hypertrophy of such cells, greatly increasing the heart size in mice, while knocking down PI3Kα results in mice with smaller hearts (Shioi et al., 2000). In response to mechanical stress, PI3Kα translocates to the plasma membrane to convert PIP2 to PIP3, which subsequently recruits gelsolin to the plasma membrane (Patel et al., 2018). A resulting spatial colocalization occurs between p110α, the catalytic subunit of PI3Kα, and gelsolin in which p110α-catalyzed PIP3 negatively regulates gelsolin activity and thus diminishes unfavorable remodeling of the actin cytoskeleton while conserving the cytoskeletal integrity. Consequently, PI3Kα-generated PIP3 plays a critical role in the mechanotransduction of cardiomyocytes by negatively regulating gelsolin, which subsequently inhibits actin remodeling (Patel et al., 2018).
In cardiac myocytes, GPCRs activate PI3Kγ in response to pressure overload or biomechanical stress, which mediates the adaptive role in cardiac mechanotransduction by negatively regulating cyclic adenosine monophosphate (cAMP) levels (Guo et al., 2010). It was first shown that complete deletion of PI3Kγ in cardiac myocytes alters heart function by inducing cell hypercontractility as a result of cAMP accumulation but does not alter the cell structure or growth (Crackower et al., 2002; Patrucco et al., 2004); however, a separate study has shown that deletion of PI3Kγ accelerates the development of pathological hypertrophy (Guo et al., 2010). Intriguingly, the regulation of cell contractility by PI3Kγ in response to mechanical stress is independent of its activity or functional kinase domain (Patrucco et al., 2004). More specifically, cardiac myocytes lacking PI3Kγ activity with preserved expression exhibit normal levels of cAMP that are believed to be the result of phosphodiesterase 3B positive regulation by a PI3Kγ-associated multifunctional protein complex (Patrucco et al., 2004). Critical to this complex is the anchoring of PKA to PI3Kγ and downstream activation of phosphodiesterases, type 3 and 4 (PDE3/4), and subsequently reducing the cAMP levels; upon its anchoring, PKA also phosphorylates and inhibits PI3Kγ lipid kinase activity, resulting in a reduction in PIP3 (Perino et al., 2011; Ghigo et al., 2017). In pressure overload-mediated sympathetic overdrive of cardiac myocytes, the beta2 adrenergic receptor is desensitized and internalized as a result of PKA-escaped PI3Kγ kinase activity and ultimately induces hypokinetic dilated heart failure (Prasad et al., 2005; Perino et al., 2011; Ghigo and Li, 2015; Ghigo et al., 2017). Despite enhanced calcium dynamics and contractility upon the loss of PI3Kγ in cardiac myocytes, decompensation ensues because of dysregulated cellular-ECM interactions (Guo et al., 2010). Furthermore, a more direct relationship between PI3Kγ and cardiac mechanotransduction is observed upon the loss of PI3Kγ, in which elevated cAMP levels mediate extracellular matrix remodeling and interactions (Guo et al., 2010). In this particular instance, inhibiting the beta2 adrenergic receptor protects N-cadherin adhesion complexes from degradation (Guo et al., 2010), whereas the loss of p110γ function, the catalytic subunit of PI3Kγ, leads to heart failure by the deterioration of N-cadherin and an increase in cAMP levels (Patel et al., 2018). Furthermore, N-cadherin complexes actively perceive biomechanical stress, and through the regulation of gelsolin, actin polymerization is promoted, therefore expressing a collaborative relationship between PI3Kγ and PI3Kα in cardiac mechanotransduction (Chan et al., 2004).
The Hippo signaling pathway, which was originally observed in Drosophila, mediates the VSMC stretch response that inhibits cell proliferation and participates in mechanotransduction pathways (Huang et al., 2005; Ota and Sasaki, 2008; Yu et al., 2015; Chakraborty et al., 2017; Fletcher et al., 2018). Inhibition of the Hippo pathway promotes tissue growth in epithelial cells through the PI3K-PDK1-Akt axis upon mechanical stimulation and growth factor signaling (Borreguero-Munoz et al., 2019). Yes-associated protein 1 (YAP) and transcriptional coactivator with the PDZ-binding motif (TAZ) are downstream transcriptional activators of the Hippo pathway (Halder et al., 2012). These effectors are regulated by mechanical cues, specifically, matrix stiffness, stretch, and cell density, which influence cell proliferation and differentiation (Halder et al., 2012; Codelia et al., 2014; Meng et al., 2016). Thus, YAP and TAZ function as essential effectors of mechanotransduction (Meng et al., 2018). YAP/TAZ-dependent glutaminolysis and anaplerosis are mechanoactivated by vascular stiffness to drive cell proliferation in pulmonary hypertension (Bertero et al., 2016). Additionally, mechanical stretching regulates YAP/TAZ activity via the PI3K-PDK1-mediated pathway in human umbilical arterial VSMCs (Wang et al., 2018). Furthermore, the PDK1 interaction with the Hippo complex is mediated through Sav1, where PDK1 directly controls the Hippo pathway (Wang et al., 2018). The consequential association of PI3K with the Hippo signaling pathway effectors YAP and TAZ in vascular cells further implicates PI3K in the mechanotransduction of the cardiovascular system.
Mechanical forces of a hemodynamic nature are uniquely fundamental for vascular homeostasis as well as pathological vascular remodeling that are commonly observed in CVD (Cahill and Redmond, 2016; Russo et al., 2018). In cells of the vasculature, harmony in cell proliferation, apoptosis, migration, and differentiation is integral to vascular wall homeostasis. Mechanical forces perceived by ECs and VSMCs generate a biological response, i.e., mechanotransduction to induce physiological vascular remodeling (Qi et al., 2018). Consequently, vascular remodeling involves a variety of cellular components to mediate these biophysical and biochemical events, including PI3K, which has previously been connected to the vascular remodeling pathway. During angiogenesis, vessel remodeling can help with cell proliferation and maturation (Wang and Khalil, 2018). Vascular remodeling in pericytes is regulated by PI3Kβ. Mature pericytes, which are mostly found in vessels undergoing remodeling, are quiescent and express low activation of the PI3K signaling. Inactivation of PI3Kβ in these cells generates early pericyte maturation, with an increase in PI3K signaling that obstructs pericyte maturation. Thus, pericytes in a sustained immature state will result in vascular hyperplasia and block vascular remodeling, whereas accurate PI3K signaling is necessary for pericyte maturation and correct vessel formation (Figueiredo et al., 2020).
One of the most prevalent cardiovascular diseases involving vascular remodeling is atherosclerosis. During atherosclerosis, vascular injury occurs, causing abnormal proliferation of VSMCs, which leads to neointima formation and vessel lumen narrowing and ultimately limits blood flow and oxygen supply (Yu et al., 2018). PI3K has been directly associated with the molecular pathways that mediate vascular remodeling and atherosclerosis. The catalytic subunit of PI3Kα, p110α, is important for receptor tyrosine kinase (RTK) signaling, which is upstream of class 1A PI3K isoforms, in VSMCs. Furthermore, p110α is critical to neointima formation after balloon angioplasty by mediating VSMC proliferation and migration, while the PI3Kα isoforms p100β and p110δ do not play a significant role (Vantler et al., 2015).
PI3Kγ functions in both leukocytes and cardiomyocytes and plays a role in atherosclerosis and heart disease. PI3Kγ controls leukocyte infiltration in the myocardium and arteries. PI3Kγ is involved in neuraminidase-1 (Neu-1) signaling, which governs atherosclerosis development (Gayral et al., 2014). Genetic and pharmacological inhibitory targeting of PI3Kγ in leukocytes reduces atherosclerosis in mouse models (Fougerat et al., 2008). Ghigo et al. (2017) recently reviewed PI3K and calcium signaling in cardiovascular disease. The PI3K pathway has recently been interconnected with Ca2+ signaling. PI3Kγ appears to be preferentially linked to Ca2+ signaling in smooth muscle cells (Lupieri et al., 2020), where Class I PI3Ks are highly expressed. This interconnection between the PI3Kγ pathway and Ca2+ signaling has been involved in smooth muscle cell proliferation and migration, atherosclerosis and arterial injury. The development of arterial lesions through various immune functions requires PI3Kγ activity with PI3Kγ playing an important role in arterial injury in T cells. For example, it has been found that PI3Kγ regulates T-cell function, and it has been proposed that PI3Kγ interacts with Ca2+ signaling, leading to Ca2+ influx downstream of T-cell receptor activation; thus, PI3Kγ interconnects with Ca2+, playing an important role in arterial injury (Smirnova et al., 2014; Lupieri et al., 2015; Ghigo et al., 2017). Taken together, PI3Kβ and PI3Kγ are paramount pathways that drive cardiovascular remodeling seen in heart failure as well as in atherosclerosis, and this strongly suggests that PI3K is critically involved in mechanotransduction-mediated cardiovascular disease.
This review summarizes the relationship between PPIs and mechanotransduction in regard to cardiovascular biology and disease (Table 1). PPIs are central mediators in multiple biological processes, although understanding the specific contribution of PPIs to cellular dynamics can be difficult, especially regarding mechanotransduction in cardiovascular disease. PIP2, PIP3, PI3K, and PIP5K all play important roles in different mechanotransduction pathways of the cardiovascular system. These PPI functions include cytoskeletal arrangements, association with actin-binding proteins and ion channels, and response to mechanical stimuli. Indeed, PPIs are critical modulators of mechanotransduction. Complete knowledge of these pathways is not yet fully known and should be further explored to address how these pathways influence cellular mechanotransduction in cardiovascular cells in both homeostasis and disease.

Table 1. Overview of phosphoinositide signaling and mechanotransduction in cardiovascular biology and pathology.
AK, JB, and YB conceptualized the review. AK, JB, KV, TD, and YB wrote the original draft. AK and JB prepared the figures and table. AK, JB, KV, TD, JR-M, and YB critically reviewed and edited the final manuscript version. All authors contributed to the article and approved the submitted version.
This work was supported by the American Heart Association Career Development Award (18CDA34080415) to YB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdullaev, I. F., Bisaillon, J. M., Potier, M., Gonzalez, J. C., Motiani, R. K., and Trebak, M. (2008). Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 103, 1289–1299. doi: 10.1161/01.RES.0000338496.95579.56
Allahverdian, S., Chaabane, C., Boukais, K., Francis, G. A., and Bochaton-Piallat, M. L. (2018). Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 114, 540–550. doi: 10.1093/cvr/cvy022
Aoyagi, T., and Matsui, T. (2011). Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr. Pharm. Des. 17, 1818–1824. doi: 10.2174/138161211796390976
Bae, Y. H., Mui, K. L., Hsu, B. Y., Liu, S. L., Cretu, A., Razinia, Z., et al. (2014). A FAK-Cas-Rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci. Signal. 7:ra57. doi: 10.1126/scisignal.2004838
Barlow, H. R., and Cleaver, O. (2019). Building Blood Vessels-One Rho GTPase at a Time. Cells 8:545. doi: 10.3390/cells8060545
Baudel, M., Shi, J., Large, W. A., and Albert, A. P. (2020). Insights into activation mechanisms of store-operated TRPC1 channels in vascular smooth muscle. Cells 9:179. doi: 10.3390/cells9010179
Beech, D. J., and Kalli, A. C. (2019). Force sensing by Piezo channels in cardiovascular health and disease. Arterioscler. Thromb. Vasc. Biol. 39, 2228–2239. doi: 10.1161/ATVBAHA.119.313348
Bertero, T., Oldham, W. M., Cottrill, K. A., Pisano, S., Vanderpool, R. R., Yu, Q., et al. (2016). Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Invest. 126, 3313–3335. doi: 10.1172/JCI86387
Bilanges, B., Posor, Y., and Vanhaesebroeck, B. (2019). PI3K isoforms in cell signalling and vesicle trafficking. Nat. Rev. Mol. Cell Biol. 20, 515–534. doi: 10.1038/s41580-019-0129-z
Borbiro, I., Badheka, D., and Rohacs, T. (2015). Activation of TRPV1 channels inhibits mechanosensitive Piezo channel activity by depleting membrane phosphoinositides. Sci. Signal. 8:ra15. doi: 10.1126/scisignal.2005667
Borreguero-Munoz, N., Fletcher, G. C., Aguilar-Aragon, M., Elbediwy, A., Vincent-Mistiaen, Z. I., and Thompson, B. J. (2019). The Hippo pathway integrates PI3K-Akt signals with mechanical and polarity cues to control tissue growth. PLoS Biol. 17:e3000509. doi: 10.1371/journal.pbio.3000509
Cahill, P. A., and Redmond, E. M. (2016). Vascular endothelium - Gatekeeper of vessel health. Atherosclerosis 248, 97–109. doi: 10.1016/j.atherosclerosis.2016.03.007
Cai, A., Zhou, Y., and Li, L. (2015). Rho-GTPase and atherosclerosis: pleiotropic effects of statins. J. Am. Heart Assoc. 4:e002113. doi: 10.1161/JAHA.115.002113
Canales, J., Morales, D., Blanco, C., Rivas, J., Diaz, N., Angelopoulos, I., et al. (2019). A TR(i)P to cell migration: new roles of TRP channels in mechanotransduction and cancer. Front. Physiol. 10:757. doi: 10.3389/fphys.2019.00757
Chakraborty, S., Njah, K., Pobbati, A. V., Lim, Y. B., Raju, A., Lakshmanan, M., et al. (2017). Agrin as a mechanotransduction signal regulating YAP through the hippo pathway. Cell Rep. 18, 2464–2479. doi: 10.1016/j.celrep.2017.02.041
Chan, M. W., El Sayegh, T. Y., Arora, P. D., Laschinger, C. A., Overall, C. M., Morrison, C., et al. (2004). Regulation of intercellular adhesion strength in fibroblasts. J. Biol. Chem. 279, 41047–41057. doi: 10.1074/jbc.M406631200
Chatah, N. E., and Abrams, C. S. (2001). G-protein-coupled receptor activation induces the membrane translocation and activation of phosphatidylinositol-4-phosphate 5-kinase I alpha by a Rac- and Rho-dependent pathway. J. Biol. Chem. 276, 34059–34065. doi: 10.1074/jbc.M104917200
Chaterji, S., Kim, P., Choe, S. H., Tsui, J. H., Lam, C. H., Ho, D. S., et al. (2014). Synergistic effects of matrix nanotopography and stiffness on vascular smooth muscle cell function. Tissue Eng. Part A 20, 2115–2126. doi: 10.1089/ten.tea.2013.0455
Codelia, V. A., Sun, G., and Irvine, K. D. (2014). Regulation of YAP by mechanical strain through Jnk and Hippo signaling. Curr. Biol. 24, 2012–2017. doi: 10.1016/j.cub.2014.07.034
Corrigan, M. A., Johnson, G. P., Stavenschi, E., Riffault, M., Labour, M. N., and Hoey, D. A. (2018). TRPV4-mediates oscillatory fluid shear mechanotransduction in mesenchymal stem cells in part via the primary cilium. Sci. Rep. 8:3824. doi: 10.1038/s41598-018-22174-3
Crackower, M. A., Oudit, G. Y., Kozieradzki, I., Sarao, R., Sun, H., Sasaki, T., et al. (2002). Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell 110, 737–749. doi: 10.1016/s0092-8674(02)00969-8
De Craene, J. O., Bertazzi, D. L., Bar, S., and Friant, S. (2017). Phosphoinositides, major actors in membrane trafficking and lipid signaling pathways. Int. J. Mol. Sci. 18:634. doi: 10.3390/ijms18030634
DeHaven, W. I., Jones, B. F., Petranka, J. G., Smyth, J. T., Tomita, T., Bird, G. S., et al. (2009). TRPC channels function independently of STIM1 and Orai1. J. Physiol. 587(Pt 10), 2275–2298. doi: 10.1113/jphysiol.2009.170431
Di Paolo, G., and De Camilli, P. (2006). Phosphoinositides in cell regulation and membrane dynamics. Nature 443, 651–657. doi: 10.1038/nature05185
Di Paolo, G., Moskowitz, H. S., Gipson, K., Wenk, M. R., Voronov, S., Obayashi, M., et al. (2004). Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431, 415–422. doi: 10.1038/nature02896
Dickson, E. J., and Hille, B. (2019). Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem. J. 476, 1–23. doi: 10.1042/BCJ20180022
Dong, H., Zhang, Y., Song, R., Xu, J., Yuan, Y., Liu, J., et al. (2019). Toward a model for activation of Orai channel. iScience 16, 356–367. doi: 10.1016/j.isci.2019.05.041
Edwards, M., Zwolak, A., Schafer, D. A., Sept, D., Dominguez, R., and Cooper, J. A. (2014). Capping protein regulators fine-tune actin assembly dynamics. Nat. Rev. Mol. Cell Biol. 15, 677–689. doi: 10.1038/nrm3869
Falkenburger, B. H., Jensen, J. B., Dickson, E. J., Suh, B. C., and Hille, B. (2010). Phosphoinositides: lipid regulators of membrane proteins. J. Physiol. 588(Pt 17), 3179–3185. doi: 10.1113/jphysiol.2010.192153
Figueiredo, A. M., Villacampa, P., Dieguez-Hurtado, R., Lozano, J. J., Kobialka, P., Cortazar, A. R., et al. (2020). PI3Kbeta-regulated pericyte maturation governs vascular remodeling. Circulation 142, 688–704. doi: 10.1161/CIRCULATIONAHA.119.042354
Fletcher, G. C., Diaz-de-la-Loza, M. D., Borreguero-Munoz, N., Holder, M., Aguilar-Aragon, M., and Thompson, B. J. (2018). Mechanical strain regulates the Hippo pathway in Drosophila. Development 145:dev159467. doi: 10.1242/dev.159467
Formigli, L., Sassoli, C., Squecco, R., Bini, F., Martinesi, M., Chellini, F., et al. (2009). Regulation of transient receptor potential canonical channel 1 (TRPC1) by sphingosine 1-phosphate in C2C12 myoblasts and its relevance for a role of mechanotransduction in skeletal muscle differentiation. J. Cell Sci. 122(Pt 9), 1322–1333. doi: 10.1242/jcs.035402
Fougerat, A., Gayral, S., Gourdy, P., Schambourg, A., Ruckle, T., Schwarz, M. K., et al. (2008). Genetic and pharmacological targeting of phosphoinositide 3-kinase-gamma reduces atherosclerosis and favors plaque stability by modulating inflammatory processes. Circulation 117, 1310–1317. doi: 10.1161/circulationaha.107.720466
Garoffolo, G., and Pesce, M. (2019). Mechanotransduction in the cardiovascular system: from developmental origins to homeostasis and pathology. Cells 8:1607. doi: 10.3390/cells8121607
Garrison, S. R., Dietrich, A., and Stucky, C. L. (2012). TRPC1 contributes to light-touch sensation and mechanical responses in low-threshold cutaneous sensory neurons. J. Neurophysiol. 107, 913–922. doi: 10.1152/jn.00658.2011
Gayral, S., Garnotel, R., Castaing-Berthou, A., Blaise, S., Fougerat, A., Berge, E., et al. (2014). Elastin-derived peptides potentiate atherosclerosis through the immune Neu1-PI3Kgamma pathway. Cardiovasc. Res. 102, 118–127. doi: 10.1093/cvr/cvt336
Ghigo, A., Laffargue, M., Li, M., and Hirsch, E. (2017). PI3K and calcium signaling in cardiovascular disease. Circ. Res. 121, 282–292. doi: 10.1161/CIRCRESAHA.117.310183
Ghigo, A., and Li, M. (2015). Phosphoinositide 3-kinase: friend and foe in cardiovascular disease. Front. Pharmacol. 6:169. doi: 10.3389/fphar.2015.00169
Gimbrone, M. A. Jr., and Garcia-Cardena, G. (2013). Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 22, 9–15. doi: 10.1016/j.carpath.2012.06.006
Gottlieb, P., Folgering, J., Maroto, R., Raso, A., Wood, T. G., Kurosky, A., et al. (2008). Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch. 455, 1097–1103. doi: 10.1007/s00424-007-0359-3
Guo, D., Kassiri, Z., Basu, R., Chow, F. L., Kandalam, V., Damilano, F., et al. (2010). Loss of PI3Kgamma enhances cAMP-dependent MMP remodeling of the myocardial N-cadherin adhesion complexes and extracellular matrix in response to early biomechanical stress. Circ. Res. 107, 1275–1289. doi: 10.1161/CIRCRESAHA.110.229054
Hagiwara, A., Cornu, M., Cybulski, N., Polak, P., Betz, C., Trapani, F., et al. (2012). Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 15, 725–738. doi: 10.1016/j.cmet.2012.03.015
Halder, G., Dupont, S., and Piccolo, S. (2012). Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 13, 591–600. doi: 10.1038/nrm3416
Hammond, G. R., and Hong, Y. (2018). Phosphoinositides and membrane targeting in cell polarity. Cold Spring Harb. Perspect. Biol. 10:a027938. doi: 10.1101/cshperspect.a027938
Hao, X. J., Xu, C. Z., Wang, J. T., Li, X. J., Wang, M. M., Gu, Y. H., et al. (2018). miR-21 promotes proliferation and inhibits apoptosis of hepatic stellate cells through targeting PTEN/PI3K/AKT pathway. J. Recept. Signal. Transduct. Res. 38, 455–461. doi: 10.1080/10799893.2019.1585452
Harraz, O. F., Longden, T. A., Dabertrand, F., Hill-Eubanks, D., and Nelson, M. T. (2018a). Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc. Natl. Acad. Sci. U.S.A. 115, E3569–E3577. doi: 10.1073/pnas.1800201115
Harraz, O. F., Longden, T. A., Hill-Eubanks, D., and Nelson, M. T. (2018b). PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. eLife 7:e38689. doi: 10.7554/eLife.38689
Hirsch, E., Gulluni, F., and Martini, M. (2020). Phosphoinositides in cell proliferation and metabolism. Adv. Biol. Regul. 75:100693. doi: 10.1016/j.jbior.2020.100693
Huang, J., Wu, S., Barrera, J., Matthews, K., and Pan, D. (2005). The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122, 421–434. doi: 10.1016/j.cell.2005.06.007
Huang, S., Lifshitz, L., Patki-Kamath, V., Tuft, R., Fogarty, K., and Czech, M. P. (2004). Phosphatidylinositol-4,5-bisphosphate-rich plasma membrane patches organize active zones of endocytosis and ruffling in cultured adipocytes. Mol. Cell. Biol. 24, 9102–9123. doi: 10.1128/MCB.24.20.9102-9123.2004
Janmey, P. A., Bucki, R., and Radhakrishnan, R. (2018). Regulation of actin assembly by PI(4,5)P2 and other inositol phospholipids: an update on possible mechanisms. Biochem. Biophys. Res. Commun. 506, 307–314. doi: 10.1016/j.bbrc.2018.07.155
Kalwa, H., and Michel, T. (2011). The MARCKS protein plays a critical role in phosphatidylinositol 4,5-bisphosphate metabolism and directed cell movement in vascular endothelial cells. J. Biol. Chem. 286, 2320–2330. doi: 10.1074/jbc.M110.196022
Karoor, V., Fini, M. A., Loomis, Z., Sullivan, T., Hersh, L. B., Gerasimovskaya, E., et al. (2018). Sustained activation of Rho GTPases promotes a synthetic pulmonary artery smooth muscle cell phenotype in neprilysin null mice. Arterioscler. Thromb. Vasc. Biol. 38, 154–163. doi: 10.1161/ATVBAHA.117.310207
Kohler, R., and Hoyer, J. (2007). “Role of TRPV4 in the mechanotransduction of shear stress in endothelial cells,” in TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades, eds W. B. Liedtke and S. Heller (Boca Raton, FL: CRC Press).
Kruger, M., and Linke, W. A. (2009). Titin-based mechanical signalling in normal and failing myocardium. J. Mol. Cell. Cardiol. 46, 490–498. doi: 10.1016/j.yjmcc.2009.01.004
Kumar, B., Dreja, K., Shah, S. S., Cheong, A., Xu, S. Z., Sukumar, P., et al. (2006). Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ. Res. 98, 557–563. doi: 10.1161/01.RES.0000204724.29685.db
Labouesse, M. (2011). Rac GTPase signaling in mechanotransduction during embryonic morphogenesis. Small GTPases 2, 305–309. doi: 10.4161/sgtp.18035
Lawson, C. D., and Burridge, K. (2014). The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 5:e27958. doi: 10.4161/sgtp.27958
Leychenko, A., Konorev, E., Jijiwa, M., and Matter, M. L. (2011). Stretch-induced hypertrophy activates NFkB-mediated VEGF secretion in adult cardiomyocytes. PLoS One 6:e29055. doi: 10.1371/journal.pone.0029055
Li, C. J., Chen, C. S., Yiang, G. T., Tsai, A. P., Liao, W. T., and Wu, M. Y. (2019). Advanced evolution of pathogenesis concepts in cardiomyopathies. J. Clin. Med. 8:520. doi: 10.3390/jcm8040520
Li, H., Mao, Y., Bouaziz, M., Yu, H., Qu, X., Wang, F., et al. (2019). Lens differentiation is controlled by the balance between PDGF and FGF signaling. PLoS Biol. 17:e3000133. doi: 10.1371/journal.pbio.3000133
Li, J., Mkrtschjan, M. A., Lin, Y. H., and Russell, B. (2016). Variation in stiffness regulates cardiac myocyte hypertrophy via signaling pathways. Can. J. Physiol. Pharmacol. 94, 1178–1186. doi: 10.1139/cjpp-2015-0578
Li, J., and Russell, B. (2013). Phosphatidylinositol 4,5-bisphosphate regulates CapZbeta1 and actin dynamics in response to mechanical strain. Am. J. Physiol. Heart Circ. Physiol. 305, H1614–H1623. doi: 10.1152/ajpheart.00477.2013
Li, N., He, Y., Yang, G., Yu, Q., and Li, M. (2019). Role of TRPC1 channels in pressure-mediated activation of airway remodeling. Respir. Res. 20:91. doi: 10.1186/s12931-019-1050-x
Li, N., He, Y., Yang, G., Yu, Q., and Li, M. (2019). Role of TRPC1 channels in pressure-mediated activation of airway remodeling. Respir. Res. 20:91. doi: 10.1186/s12931-019-1050-x
Liao, Y., Erxleben, C., Yildirim, E., Abramowitz, J., Armstrong, D. L., and Birnbaumer, L. (2007). Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc. Natl. Acad. Sci. U.S.A. 104, 4682–4687. doi: 10.1073/pnas.0611692104
Linke, W. A. (2008). Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc. Res. 77, 637–648. doi: 10.1016/j.cardiores.2007.03.029
Longden, T. A., and Nelson, M. T. (2015). Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation 22, 183–196. doi: 10.1111/micc.12190
Luo, J., McMullen, J. R., Sobkiw, C. L., Zhang, L., Dorfman, A. L., Sherwood, M. C., et al. (2005). Class IA phosphoinositide 3-kinase regulates heart size and physiological cardiac hypertrophy. Mol. Cell. Biol. 25, 9491–9502. doi: 10.1128/MCB.25.21.9491-9502.2005
Lupieri, A., Smirnova, N., Malet, N., Gayral, S., and Laffargue, M. (2015). PI3K signaling in arterial diseases: non redundant functions of the PI3K isoforms. Adv. Biol. Regul. 59, 4–18. doi: 10.1016/j.jbior.2015.06.002
Lupieri, A., Smirnova, N. F., Solinhac, R., Malet, N., Benamar, M., Saoudi, A., et al. (2020). Smooth muscle cells-derived CXCL10 prevents endothelial healing through PI3Kgamma-dependent T cells response. Cardiovasc. Res. 116, 438–449. doi: 10.1093/cvr/cvz122
Lyon, R. C., Zanella, F., Omens, J. H., and Sheikh, F. (2015). Mechanotransduction in cardiac hypertrophy and failure. Circ. Res. 116, 1462–1476. doi: 10.1161/CIRCRESAHA.116.304937
Manna, P., and Jain, S. K. (2015). Phosphatidylinositol-3,4,5-triphosphate and cellular signaling: implications for obesity and diabetes. Cell. Physiol. Biochem. 35, 1253–1275. doi: 10.1159/000373949
Manning, B. D., and Toker, A. (2017). AKT/PKB signaling: navigating the network. Cell 169, 381–405. doi: 10.1016/j.cell.2017.04.001
Markowska, A., Pawalowska, M., Lubin, J., and Markowska, J. (2014). Signalling pathways in endometrial cancer. Contemp. Oncol. 18, 143–148. doi: 10.5114/wo.2014.43154
Martino, F., Perestrelo, A. R., Vinarsky, V., Pagliari, S., and Forte, G. (2018). Cellular mechanotransduction: from tension to function. Front. Physiol. 9:824. doi: 10.3389/fphys.2018.00824
Maurer, M., and Lammerding, J. (2019). The driving force: nuclear mechanotransduction in cellular function, fate, and disease. Annu. Rev. Biomed. Eng. 21, 443–468. doi: 10.1146/annurev-bioeng-060418-052139
McGowan, S. E., and McCoy, D. M. (2017). Platelet-derived growth factor receptor-alpha and Ras-related C3 botulinum toxin substrate-1 regulate mechano-responsiveness of lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L1174–L1187. doi: 10.1152/ajplung.00185.2017
Meng, Z., Moroishi, T., and Guan, K. L. (2016). Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17. doi: 10.1101/gad.274027.115
Meng, Z., Qiu, Y., Lin, K. C., Kumar, A., Placone, J. K., Fang, C., et al. (2018). RAP2 mediates mechanoresponses of the Hippo pathway. Nature 560, 655–660. doi: 10.1038/s41586-018-0444-0
Miller, M. K., Granzier, H., Ehler, E., and Gregorio, C. C. (2004). The sensitive giant: the role of titin-based stretch sensing complexes in the heart. Trends Cell Biol. 14, 119–126. doi: 10.1016/j.tcb.2004.01.003
Miller, M. S., Thompson, P. E., and Gabelli, S. B. (2019). Structural determinants of isoform selectivity in PI3K inhibitors. Biomolecules 9:82. doi: 10.3390/biom9030082
Nairn, A. C., and Aderem, A. (1992). Calmodulin and protein kinase C cross-talk: the MARCKS protein is an actin filament and plasma membrane cross-linking protein regulated by protein kinase C phosphorylation and by calmodulin. Ciba Found. Symp. 164, 145–154. doi: 10.1002/9780470514207.ch10
Narumiya, S., and Thumkeo, D. (2018). Rho signaling research: history, current status and future directions. FEBS Lett. 592, 1763–1776. doi: 10.1002/1873-3468.13087
Nikolaev, Y. A., Cox, C. D., Ridone, P., Rohde, P. R., Cordero-Morales, J. F., Vasquez, V., et al. (2019). Mammalian TRP ion channels are insensitive to membrane stretch. J. Cell Sci. 132:jcs238360. doi: 10.1242/jcs.238360
Ochoa, C. D., Wu, R. F., and Terada, L. S. (2018). ROS signaling and ER stress in cardiovascular disease. Mol. Aspects Med. 63, 18–29. doi: 10.1016/j.mam.2018.03.002
Ohashi, K., Fujiwara, S., and Mizuno, K. (2017). Roles of the cytoskeleton, cell adhesion and rho signalling in mechanosensing and mechanotransduction. J. Biochem. 161, 245–254. doi: 10.1093/jb/mvw082
Ota, M., and Sasaki, H. (2008). Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development 135, 4059–4069. doi: 10.1242/dev.027151
Oude Weernink, P. A., Schulte, P., Guo, Y., Wetzel, J., Amano, M., Kaibuchi, K., et al. (2000). Stimulation of phosphatidylinositol-4-phosphate 5-kinase by Rho-kinase. J. Biol. Chem. 275, 10168–10174. doi: 10.1074/jbc.275.14.10168
Oudit, G. Y., Kassiri, Z., Zhou, J., Liu, Q. C., Liu, P. P., Backx, P. H., et al. (2008). Loss of PTEN attenuates the development of pathological hypertrophy and heart failure in response to biomechanical stress. Cardiovasc. Res. 78, 505–514. doi: 10.1093/cvr/cvn041
Patel, V. B., Wang, Z., Fan, D., Zhabyeyev, P., Basu, R., Das, S. K., et al. (2013). Loss of p47phox subunit enhances susceptibility to biomechanical stress and heart failure because of dysregulation of cortactin and actin filaments. Circ. Res. 112, 1542–1556. doi: 10.1161/CIRCRESAHA.111.300299
Patel, V. B., Zhabyeyev, P., Chen, X., Wang, F., Paul, M., Fan, D., et al. (2018). PI3Kalpha-regulated gelsolin activity is a critical determinant of cardiac cytoskeletal remodeling and heart disease. Nat. Commun. 9:5390. doi: 10.1038/s41467-018-07812-8
Patrucco, E., Notte, A., Barberis, L., Selvetella, G., Maffei, A., Brancaccio, M., et al. (2004). PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 118, 375–387. doi: 10.1016/j.cell.2004.07.017
Perino, A., Ghigo, A., Ferrero, E., Morello, F., Santulli, G., Baillie, G. S., et al. (2011). Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110gamma. Mol. Cell 42, 84–95. doi: 10.1016/j.molcel.2011.01.030
Phan, T. K., Williams, S. A., Bindra, G. K., Lay, F. T., Poon, I. K. H., and Hulett, M. D. (2019). Phosphoinositides: multipurpose cellular lipids with emerging roles in cell death. Cell Death Differ. 26, 781–793. doi: 10.1038/s41418-018-0269-2
Polacheck, W. J., Kutys, M. L., Yang, J., Eyckmans, J., Wu, Y., Vasavada, H., et al. (2017). A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 552, 258–262. doi: 10.1038/nature24998
Prasad, S. V., Jayatilleke, A., Madamanchi, A., and Rockman, H. A. (2005). Protein kinase activity of phosphoinositide 3-kinase regulates β-adrenergic receptor endocytosis. Nat. Cell Biol. 7, 785–796. doi: 10.1038/ncb1278
Prestwich, G. D. (2004). Phosphoinositide signaling; from affinity probes to pharmaceutical targets. Chem. Biol. 11, 619–637. doi: 10.1016/j.chembiol.2004.03.025
Qi, Y. X., Han, Y., and Jiang, Z. L. (2018). Mechanobiology and vascular remodeling: from membrane to nucleus. Adv. Exp. Med. Biol. 1097, 69–82. doi: 10.1007/978-3-319-96445-4_4
Ramos, A. R., Elong Edimo, W., and Erneux, C. (2018). Phosphoinositide 5-phosphatase activities control cell motility in glioblastoma: two phosphoinositides PI(4,5)P2 and PI(3,4)P2 are involved. Adv. Biol. Regul. 67, 40–48. doi: 10.1016/j.jbior.2017.09.001
Ridley, A. J. (1994). Membrane ruffling and signal transduction. Bioessays 16, 321–327. doi: 10.1002/bies.950160506
Russell, B., Curtis, M. W., Koshman, Y. E., and Samarel, A. M. (2010). Mechanical stress-induced sarcomere assembly for cardiac muscle growth in length and width. J. Mol. Cell. Cardiol. 48, 817–823. doi: 10.1016/j.yjmcc.2010.02.016
Russo, T. A., Stoll, D., Nader, H. B., and Dreyfuss, J. L. (2018). Mechanical stretch implications for vascular endothelial cells: altered extracellular matrix synthesis and remodeling in pathological conditions. Life Sci. 213, 214–225. doi: 10.1016/j.lfs.2018.10.030
Saarikangas, J., Zhao, H., and Lappalainen, P. (2010). Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol. Rev. 90, 259–289. doi: 10.1152/physrev.00036.2009
Saleh, S. N., Albert, A. P., and Large, W. A. (2009a). Activation of native TRPC1/C5/C6 channels by endothelin-1 is mediated by both PIP3 and PIP2 in rabbit coronary artery myocytes. J. Physiol. 587(Pt 22), 5361–5375. doi: 10.1113/jphysiol.2009.180331
Saleh, S. N., Albert, A. P., and Large, W. A. (2009b). Obligatory role for phosphatidylinositol 4,5-bisphosphate in activation of native TRPC1 store-operated channels in vascular myocytes. J. Physiol. 587, 531–540. doi: 10.1113/jphysiol.2008.166678
Schafer, S., Viswanathan, S., Widjaja, A. A., Lim, W. W., Moreno-Moral, A., DeLaughter, D. M., et al. (2017). IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 552, 110–115. doi: 10.1038/nature24676
Schink, K. O., Tan, K. W., and Stenmark, H. (2016). Phosphoinositides in control of membrane dynamics. Annu. Rev. Cell Dev. Biol. 32, 143–171. doi: 10.1146/annurev-cellbio-111315-125349
Senju, Y., Kalimeri, M., Koskela, E. V., Somerharju, P., Zhao, H., Vattulainen, I., et al. (2017). Mechanistic principles underlying regulation of the actin cytoskeleton by phosphoinositides. Proc. Natl. Acad. Sci. U.S.A. 114, E8977–E8986. doi: 10.1073/pnas.1705032114
Senju, Y., and Lappalainen, P. (2019). Regulation of actin dynamics by PI(4,5)P2 in cell migration and endocytosis. Curr. Opin. Cell Biol. 56, 7–13. doi: 10.1016/j.ceb.2018.08.003
Sheetz, M. P., Sable, J. E., and Dobereiner, H. G. (2006). Continuous membrane-cytoskeleton adhesion requires continuous accommodation to lipid and cytoskeleton dynamics. Annu. Rev. Biophys. Biomol. Struct. 35, 417–434. doi: 10.1146/annurev.biophys.35.040405.102017
Shewan, A., Eastburn, D. J., and Mostov, K. (2011). Phosphoinositides in cell architecture. Cold Spring Harb. Perspect. Biol. 3:a004796. doi: 10.1101/cshperspect.a004796
Shi, J., Ju, M., Abramowitz, J., Large, W. A., Birnbaumer, L., and Albert, A. P. (2012). TRPC1 proteins confer PKC and phosphoinositol activation on native heteromeric TRPC1/C5 channels in vascular smooth muscle: comparative study of wild-type and TRPC1-/- mice. FASEB J. 26, 409–419. doi: 10.1096/fj.11-185611
Shi, J., Miralles, F., Kinet, J. P., Birnbaumer, L., Large, W. A., and Albert, A. P. (2017). Evidence that Orai1 does not contribute to store-operated TRPC1 channels in vascular smooth muscle cells. Channels 11, 329–339. doi: 10.1080/19336950.2017.1303025
Shioi, T., Kang, P. M., Douglas, P. S., Hampe, J., Yballe, C. M., Lawitts, J., et al. (2000). The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 19, 2537–2548. doi: 10.1093/emboj/19.11.2537
Shiojima, I., Sato, K., Izumiya, Y., Schiekofer, S., Ito, M., Liao, R., et al. (2005). Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest. 115, 2108–2118. doi: 10.1172/JCI24682
Smirnova, N. F., Gayral, S., Pedros, C., Loirand, G., Vaillant, N., Malet, N., et al. (2014). Targeting PI3Kgamma activity decreases vascular trauma-induced intimal hyperplasia through modulation of the Th1 response. J. Exp. Med. 211, 1779–1792. doi: 10.1084/jem.20131276
Solis, C., and Russell, B. (2019). CapZ integrates several signaling pathways in response to mechanical stiffness. J. Gen. Physiol. 151, 660–669. doi: 10.1085/jgp.201812199
Spinelli, A. M., and Trebak, M. (2016). Orai channel-mediated Ca2+ signals in vascular and airway smooth muscle. Am. J. Physiol. Cell Physiol. 310, C402–C413. doi: 10.1152/ajpcell.00355.2015
Trepat, X., Grabulosa, M., Buscemi, L., Rico, F., Farre, R., and Navajas, D. (2005). Thrombin and histamine induce stiffening of alveolar epithelial cells. J. Appl. Physiol. 98, 1567–1574. doi: 10.1152/japplphysiol.00925.2004
Tschumperlin, D. J. (2011). Mechanotransduction. Compr. Physiol. 1, 1057–1073. doi: 10.1002/cphy.c100016
Tsujita, K., and Itoh, T. (2015). Phosphoinositides in the regulation of actin cortex and cell migration. Biochim. Biophys. Acta 1851, 824–831. doi: 10.1016/j.bbalip.2014.10.011
Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M., and Bilanges, B. (2010). The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11, 329–341. doi: 10.1038/nrm2882
Vantler, M., Jesus, J., Leppanen, O., Scherner, M., Berghausen, E. M., Mustafov, L., et al. (2015). Class IA Phosphatidylinositol 3-Kinase isoform p110alpha mediates vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 35, 1434–1444. doi: 10.1161/ATVBAHA.114.304887
Verma, S. K., Lal, H., Golden, H. B., Gerilechaogetu, F., Smith, M., Guleria, R. S., et al. (2011). Rac1 and RhoA differentially regulate angiotensinogen gene expression in stretched cardiac fibroblasts. Cardiovasc. Res. 90, 88–96. doi: 10.1093/cvr/cvq385
Voelkel, T., and Linke, W. A. (2011). Conformation-regulated mechanosensory control via titin domains in cardiac muscle. Pflugers Arch. 462, 143–154. doi: 10.1007/s00424-011-0938-1
Wang, H., Cheng, X., Tian, J., Xiao, Y., Tian, T., Xu, F., et al. (2020). TRPC channels: Structure, function, regulation and recent advances in small molecular probes. Pharmacol. Ther. 209, 107497. doi: 10.1016/j.pharmthera.2020.107497
Wang, X., and Khalil, R. A. (2018). Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv. Pharmacol. 81, 241–330. doi: 10.1016/bs.apha.2017.08.002
Wang, Y., Cao, W., Cui, J., Yu, Y., Zhao, Y., Shi, J., et al. (2018). Arterial wall stress induces phenotypic switching of arterial smooth muscle cells in vascular remodeling by activating the YAP/TAZ signaling pathway. Cell. Physiol. Biochem. 51, 842–853. doi: 10.1159/000495376
Wang, Y., Deng, X., Hewavitharana, T., Soboloff, J., and Gill, D. L. (2008). Stim, ORAI and TRPC channels in the control of calcium entry signals in smooth muscle. Clin. Exp. Pharmacol. Physiol. 35, 1127–1133. doi: 10.1111/j.1440-1681.2008.05018.x
Weernink, P. A., Meletiadis, K., Hommeltenberg, S., Hinz, M., Ishihara, H., Schmidt, M., et al. (2004). Activation of type I phosphatidylinositol 4-phosphate 5-kinase isoforms by the Rho GTPases, RhoA, Rac1, and Cdc42. J. Biol. Chem. 279, 7840–7849. doi: 10.1074/jbc.M312737200
Yang, C. Y., Chen, C. S., Yiang, G. T., Cheng, Y. L., Yong, S. B., Wu, M. Y., et al. (2018). New insights into the immune molecular regulation of the pathogenesis of acute respiratory distress syndrome. Int. J. Mol. Sci. 19:588. doi: 10.3390/ijms19020588
Yin, J., and Kuebler, W. M. (2010). Mechanotransduction by TRP channels: general concepts and specific role in the vasculature. Cell Biochem. Biophys. 56, 1–18. doi: 10.1007/s12013-009-9067-2
Yu, B., Chen, Q., Le Bras, A., Zhang, L., and Xu, Q. (2018). Vascular stem/progenitor cell migration and differentiation in atherosclerosis. Antioxid. Redox Signal. 29, 219–235. doi: 10.1089/ars.2017.7171
Yu, F. X., Zhao, B., and Guan, K. L. (2015). Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 163, 811–828. doi: 10.1016/j.cell.2015.10.044
Yue, Z., Xie, J., Yu, A. S., Stock, J., Du, J., and Yue, L. (2015). Role of TRP channels in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 308, H157–H182. doi: 10.1152/ajpheart.00457.2014
Zegers, M. M., and Friedl, P. (2014). Rho GTPases in collective cell migration. Small GTPases 5:e28997. doi: 10.4161/sgtp.28997
Keywords: phosphoinositides, cardiovascular mechanotransduction, actin cytoskeleton, ion channel, focal adhesion, PIP2, PIP3, PI3K
Citation: Krajnik A, Brazzo JA III, Vaidyanathan K, Das T, Redondo-Muñoz J and Bae Y (2020) Phosphoinositide Signaling and Mechanotransduction in Cardiovascular Biology and Disease. Front. Cell Dev. Biol. 8:595849. doi: 10.3389/fcell.2020.595849
Received: 17 August 2020; Accepted: 25 November 2020;
Published: 14 December 2020.
Edited by:
Isabel Merida, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Tibor Rohacs, Rutgers New Jersey Medical School, United StatesCopyright © 2020 Krajnik, Brazzo, Vaidyanathan, Das, Redondo-Muñoz and Bae. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongho Bae, yonghoba@buffalo.edu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.