 Guo Li
Guo Li Yuan Tian
Yuan Tian Wei-Guo Zhu
Wei-Guo Zhu- 1Guangdong Key Laboratory for Genome Stability and Human Disease Prevention, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Shenzhen University Health Science Center, Shenzhen, China
- 2Shenzhen Bay Laboratory, Shenzhen, China
Genetic mutations and abnormal gene regulation are key mechanisms underlying tumorigenesis. Nucleosomes, which consist of DNA wrapped around histone cores, represent the basic units of chromatin. The fifth amino group (Nε) of histone lysine residues is a common site for post-translational modifications (PTMs), and of these, acetylation is the second most common. Histone acetylation is modulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), and is involved in the regulation of gene expression. Over the past two decades, numerous studies characterizing HDACs and HDAC inhibitors (HDACi) have provided novel and exciting insights concerning their underlying biological mechanisms and potential anti-cancer treatments. In this review, we detail the diverse structures of HDACs and their underlying biological functions, including transcriptional regulation, metabolism, angiogenesis, DNA damage response, cell cycle, apoptosis, protein degradation, immunity and other several physiological processes. We also highlight potential avenues to use HDACi as novel, precision cancer treatments.
Introduction
In the nuclei of eukaryotic cells, the entire genome of an organism is condensed into chromatin. The nucleosome is the basic unit of chromatin: it contains 147 DNA base pairs coiled around a core histone octamer, which includes histones H2A, H2B, H3 and H4 (Luger et al., 1997; Tessarz and Kouzarides, 2014). The additional linker histone H1 interacts with chromatin outside of the core octamer to regulate higher order chromatin structure (Fyodorov et al., 2018). There are two major higher order structures: heterochromatin refers to condensed chromatin, and euchromatin refers to loosely packed chromatin that is more accessible to transcriptional regulators and RNA polymerase complexes (Allfrey et al., 1964). Thus, alteration and regulation of chromatin structure impacts gene expression by making certain genes more or less available for transcription.
The epigenome is comprised of modifications to chromatin, including DNA methylation and histone modifications. For example, DNA accessibility is regulated by nucleosome sliding or post-translational modifications (PTMs), which include phosphorylation, methylation and acetylation. These covalent modifications control the structure and function of chromatin through a number of regulators. These regulators can be broadly divided into “readers” (enzymes that bind to modifications and facilitate epigenetic activities), “writers”(enzymes that establish DNA methylation or histone modifications), and “erasers”(enzymes that remove these markers) (Taverna et al., 2007; Kutateladze, 2011; Musselman et al., 2012; Huang et al., 2014; Xu et al., 2017). As an example, acetylation occurs at the fifth NH2 (Nε) of histone lysine residues (Strahl and Allis, 2000; Jenuwein and Allis, 2001), and is read by the bromodomain-containing protein (BRD), written by histone acetyltransferases (HATs), and erased by histone deacetylases (HDACs) (Dawson and Kouzarides, 2012; Filippakopoulos and Knapp, 2014; Zaware and Zhou, 2019).
The acetylation of lysine residues (Kac) on histone tails generates positive charges, which neutralize negatively charged DNA and the unwinding of tightly coiled heterochromatin (Shogren-Knaak et al., 2006). Histone acetylation can increase the inner pore space of chromatin from 20 nm to 60−100 nm, altering spatial distance and accessibility during interphase (Gorisch et al., 2005); it also ensures sufficient space for local transcriptional events, including initiation and elongation (Wang et al., 2009). Acetylation is of particular importance because the interaction between histones and chromatin is generally very stable, and interruption of this interaction requires a high concentration of NaCl or acetate (Von Holt et al., 1989; Shechter et al., 2007). Notably, acetylation is often a necessary precursor to other modifications, such as phosphorylation, methylation and ubiquitylation (Yang and Gregoire, 2007; Yang and Seto, 2008a).
Acetylation is controlled by two antagonistic enzyme families: HATs and HDACs. HDACs are expressed by various tumors, and are involved in vital chromosomal translocation-mediated oncogenic protein fusion and carcinogenic events (Falkenberg and Johnstone, 2014; West and Johnstone, 2014). These enzymes were first revealed to remove acetyl groups from histones by Vincent Allfrey (Inoue and Fujimoto, 1969). The first HDAC that was discovered, HDAC1, was originally isolated by utilizing a microbe-derived cyclic tetrapeptide, Trapoxin, which inhibits histone deacetylation and induces cell-cycle arrest (Taunton et al., 1996). Sequence homology-dependent HDACs were subsequently identified, and shown to be involved in major biological functions such as transcription, metastasis, autophagy, cell cycle, DNA damage repair, angiogenesis, stress responses and senescence (Yang and Seto, 2008b; Li and Zhu, 2014).
Histone deacetylase inhibitors (HDACi) might be able to reverse the activation of tumor suppressor genes (TSG), and in this way inhibit the viability and malignant proliferation of tumor cells (Glozak and Seto, 2007). The efficacy of HDACi treatment has been demonstrated in numerous clinical studies. This review discusses HDACs and their inhibitors in the context of potential cancer treatments.
Classifications, Enzymatic Activities and Cellular Distributions of HDACs
Classifications of HDACs
According to their sequence similarities with yeast HDACs, 18 human HDACs have been identified and grouped into four classes (Yang and Seto, 2008b; Seto and Yoshida, 2014). Class I HDACs include HDAC1, -2, -3, and -8 (Rundlett et al., 1996; Taunton et al., 1996; Yang et al., 1996, 1997; Emiliani et al., 1998; Buggy et al., 2000; Hu et al., 2000; Van Den Wyngaert et al., 2000). Class II HDACs are further divided into two subgroups: class IIa and class IIb. Class IIa includes HDAC4, -5, -7, and -9 and class IIb includes HDAC6 and -10 (Grozinger et al., 1999; Miska et al., 1999; Wang et al., 1999; Kao et al., 2000, 2002; Zhou et al., 2001; Fischer et al., 2002; Guardiola and Yao, 2002; Tong et al., 2002). Class III, also known as the sirtuins (SIRTs), include SIRT1-7 (Imai et al., 2000; Lin et al., 2000), and class IV contains only HDAC11 (Gao et al., 2002). SIRTs are nicotinamide adenine dinucleotide (NAD+)-dependent enzymes, while the other three classes are Zinc cation (or Zn2+ ion)-dependent HDACs. Besides the deacetylase activity, a number of diverse enzymatic activities of HDACs are presented in Table 1 and the sequence characteristics of HDACs are presented in Figure 1.
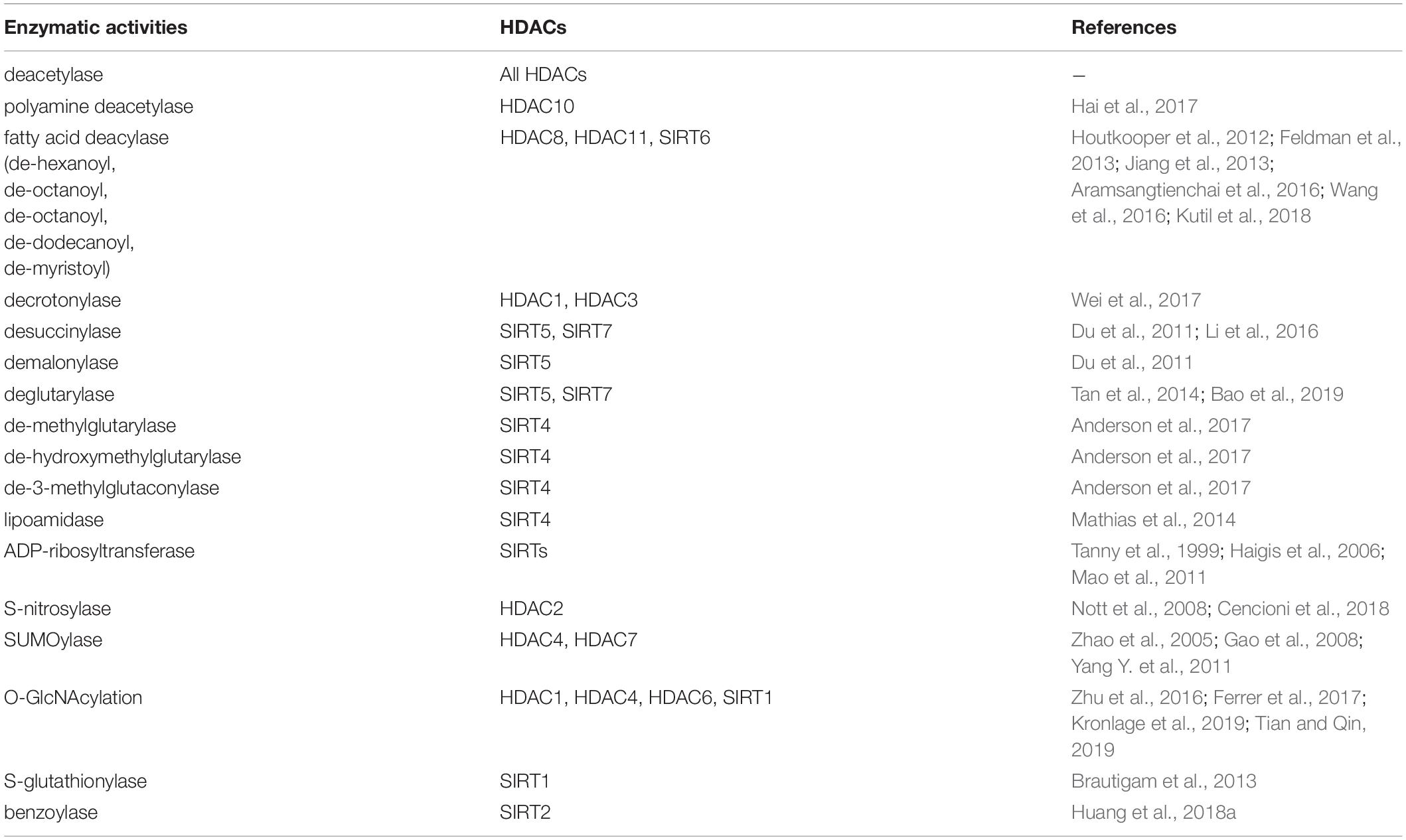
Table 1. The multifaceted catalytic functions of HDACs.
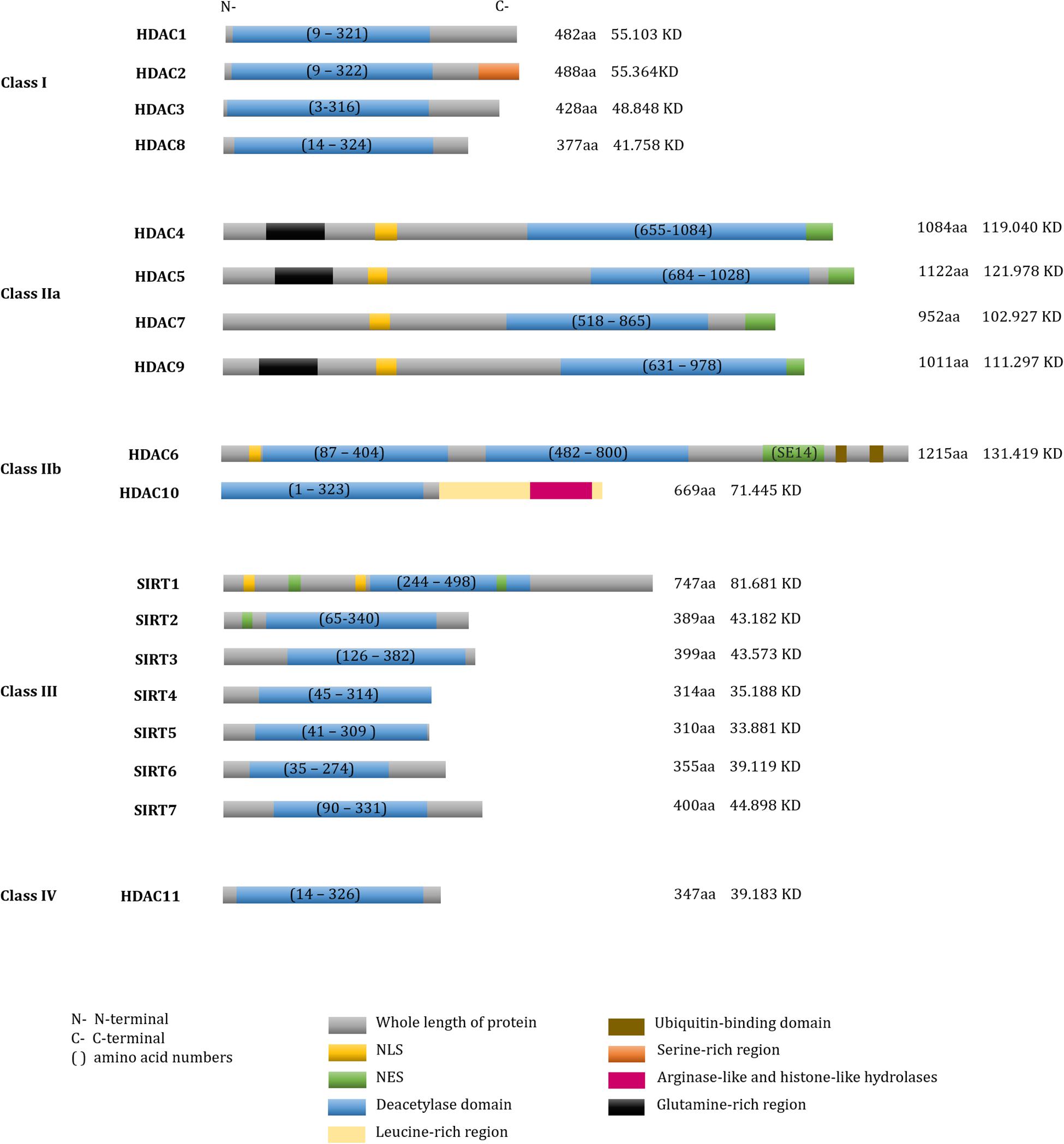
Figure 1. Domain structure of human HDACs. The fundamental structure of all deacetylases. Total number of amino acid residues and molecular weights in each HDAC were shown on the right of each protein.
Compositions, Sequence Characteristics, and Cellular Distributions of HDACs
Class I HDACs
Of class I HDACs, HDAC1, -2, and -3 catalytic activities depend on their respective co-repressor complexes. Based on the conserved structures and dimerization domains, HDAC1 and HDAC2 are often recruited to the same co-repressor complexes, including Mi-2/nucleosome remodeling deacetylase (NuRD), repressor element-1 silencing transcription co-repressor (RCOR1/CoREST), SWI-independent-3A (Sin3A) and mitotic deacetylase complex (MiDAC) (Hassig et al., 1997; Laherty et al., 1997; Nagy et al., 1997; Ayer, 1999; You et al., 2001; Bantscheff et al., 2011; Itoh et al., 2015; Turnbull et al., 2020). HDAC3 associates with the nuclear receptor co-repressor (NCoR) and silencing mediator for retinoid or thyroid-hormone receptors (SMRT) to form co-repressors. The NCoR/SMRT complex provides a platform for the recruitment and activation of HDAC3 in the deacetylase-activating domain (DAD) of SMRT (Wen et al., 2000; Oberoi et al., 2011; Emmett and Lazar, 2019). Inositol phosphate, an intermolecular “glue”; binds to the interface between the co-repressors and HDAC catalytic domains, improving the catalytic activity of the HDACs in NuRD and NCoR/SMRT complexes (Watson et al., 2012, 2016; Millard et al., 2013). Particularly, the HDAC8 monomer accommodates substrates with a unique flexible L1 loop in its N-terminal region, which is absent in other HDACs (Somoza et al., 2004). Therefore, this motif is likely to be conducive to the development of HDAC8-specific inhibitors (Ingham et al., 2016). Furthermore, its crystal structure indicates that dimerization occurs at the binding interface between HDAC8 and its substrate (Castaneda et al., 2017).
Class II HDACs
All Class IIa HDACs include an extended N-terminal domain that contains conserved serine (Ser) residues and other motifs for localization and function (Yang and Gregoire, 2005). Based on these Ser residues, several kinases such as calcium/calmodulin-dependent protein kinase (CaMK), salt-inducible kinase (SIK) and members of microtubule affinity-regulating kinase (MARK/hPar-1) phosphorylate Class IIa HDACs (Mckinsey et al., 2000, 2001; Dequiedt et al., 2006; Walkinshaw et al., 2013), which facilitates HDACs nuclear export through chromosomal region maintenance 1 protein (CRM1) [also called exportin 1 (XPO1)]- or ankyrin repeat family A protein 2 (ANKRA2)-recognized nuclear export sequence (NES) (Wang and Yang, 2001; Mckinsey et al., 2006; Xu et al., 2012). For nuclear localization, all class IIa HDACs contain nuclear localization sequence (NLS) (Zhang et al., 2002b). 14-3-3 protein inhibits the nuclear localization of these HDACs by blocking their interaction with importin α. The absence of 14-3-3 promotes HDAC4/5 nuclear localization, which also facilitates transcription repression by binding to HDAC3 (Grozinger and Schreiber, 2000; Wang et al., 2000). Of note, Class IIa HDACs contains myocyte enhancer factor 2 (MEF2) binding sites. The phosphorylated kinases-induced exported HDACs dissociate with nuclear MEF2 family proteins that are response for differentiated gene expression (Sparrow et al., 1999; Wang et al., 1999; Mckinsey et al., 2000; Walkinshaw et al., 2013). Nevertheless, Class IIa HDACs have significant weaker deacetylase activity compared to Class I. X-ray crystallography data have revealed that the catalytic pocket of histone deacetylase-like protein (HDLP) contains several key catalytic sites, such as histidine (His), aspartic acid (Asp) and tyrosine (Tyr). Class IIa HDACs have relatively low catalytic ability due to a substitution of asparagine to Asp on the Asp-His charge relay (Finnin et al., 1999). Moreover, the catalytic Tyr is conserved in other HDACs except for class IIa enzymes, where the Tyr residue is replaced by His. Substitution of His back to Tyr at 976 recovers the enzymatic activity of class IIa HDACs (Lahm et al., 2007).
Of the class IIb HDACs, HDAC6 is a microtubule-associated deacetylase that is predominantly localized in the cytoplasm. HDAC6 contains a microtubule-binding domain that promotes chemotactic cell motility (Hubbert et al., 2002; Ustinova et al., 2020); it also includes a double-tandem deacetylase domain and a serine-glutamine containing tetradecapeptide (SE14) repeats domain that is important for cytoplasmic anchoring (Bertos et al., 2004). HDAC6 undergoes nuclear export via leucine-rich motifs that are recognized by CRM1/exportin1 (Verdel et al., 2000), and contains NLS at adjacent Kac sites in the N-terminal (Liu et al., 2012). HDAC6 also contains zinc-finger ubiquitin binding domains (ZnF-UBP, also called PAZ domain) that negatively regulate polyubiquitin chain turnover (Seigneurin-Berny et al., 2001; Hook et al., 2002; Boyault et al., 2006). Recently, two deacetylase domains of HDAC6 have been re-classified as catalytic domain 1 (CD1) and CD2: these domains confer differential substrate recognition (Hai and Christianson, 2016).
HDAC10 is a polyamine deacetylase that preferentially catalyzes N8-acetylspermidine hydrolysis to generate acetate (Hai et al., 2017; Shinsky and Christianson, 2018). It contains a leucine-rich domain, a deacetylase domain and an inactivity domain (Guardiola and Yao, 2002; Kao et al., 2002). Similar to class IIa HDACs, HDAC10 associates with HDAC2, HDAC3, SMRT, and NCOR2 to enhance transcriptional repression (Fischer et al., 2002; Tong et al., 2002; Jin et al., 2018).
Class III HDACs
A total of seven NAD+-dependent class III HDACs, or SIRTs, have been identified in the cytoplasm, nucleus and mitochondria (Yao et al., 2014; Chalkiadaki and Guarente, 2015; Zhao and Zhou, 2019). SIRT1, which is distributed in the cytoplasm, mitochondria and the nucleus, has two CRM1-mediated NES and two NLS (Tanno et al., 2007): it undergoes conformational shifts in response to adenosine triphosphate (ATP), which impedes its ability to interact with substrates in the C-terminal domain (Kang et al., 2017). SIRT2 is even more widely distributed than SIRT1, being found in the plasma membrane and cytoskeleton-associated organelles in addition to the cytoplasm, nucleus and mitochondria: it contains a CRM1-dependent NES and a putative leucine-rich NES (Wilson et al., 2006). By contrast, SIRT3, SIRT4, and SIRT5 are primarily found in the mitochondria. SIRT3 has the capability to shuttle from the nucleus to the mitochondria via its mitochondrial localization sequence, which is also responsible for its mitochondrial deacetylation activity (Onyango et al., 2002; Schwer et al., 2002; Lombard et al., 2007; Bao et al., 2010). Of note, these mitochondrial regulators transfer to the nucleus in response to DNA damage induced by etoposide treatment or ultraviolet (UV) irradiation (Scher et al., 2007). SIRT6 is widely distributed, being found in the nuclear plasma, the heterochromatin, the nucleolus, as well as the cytoplasm (Michishita et al., 2005; Ardestani and Liang, 2012; Jedrusik-Bode et al., 2013). SIRT6 has a slower catalytic rate than other active SIRTs on substrates because SIRT6 lacks the conserved, highly flexible NAD+-binding loop, and instead contains a stable single helix (Pan et al., 2011). Finally, SIRT7, predominantly locates in the nucleolus but also exists in the cytoplasm (Nahalkova, 2015; Zhang et al., 2016) and is involved in mitochondrial function (Ryu et al., 2014; Mohrin et al., 2015). Two sequences in the N-terminal and C-terminal regions of SIRT7 permit nuclear and nucleolar localization, respectively (Kiran et al., 2013).
Class IV HDACs
HDAC11 is the exclusive member of the class IV HDACs. Recent studies have indicated that HDAC11 might predominantly be more involved in the fatty acylation of proteins compared to its weak deacetylation (Kutil et al., 2018; Cao et al., 2019).
Here, we recapitulate the detailed distributions of all 18 HDACs in Table 2.

Table 2. Some phenotypes observed after some manipulations of HDACs in different models.
Biological Functions of HDACs
Histone deacetylases are expressed in different tumors: class I and II HDACs are considered to be general oncoproteins that interact with substrates and regulate gene expression to promote tumorigenesis and cancer development either individually or alongside with co-repressors (Falkenberg and Johnstone, 2014; West and Johnstone, 2014). Paradoxically, SIRTs can serve as both oncoproteins and tumor suppressors (Kugel et al., 2016; Costa-Machado et al., 2018; Funato et al., 2018). We list the demonstrated knockout (KO) or knockdown models of 18 HDACs (Table 2). Because of the diverse biological function of HDACs, it is not surprising that HDACi regimens influence many cellular processes, including those that contribute to cancer progression.
Transcriptional Regulation
Transcription Modulators
Transcription factors (TFs) can either directly target DNA or undergo various PTMs to alter gene expression. In this manner, HDACs negatively modulate transcription through forming a complex with TFs or by directly regulating TF transcription (Grunstein, 1997). For instance, the v-myc avian myelocytomatosis viral oncogene homolog (Myc) is a well-characterized proto-oncogene that promotes tumorigenesis by directly recruiting and interacting with HDACs to regulate gene expression (Liu et al., 2007; Zhang et al., 2012a). Meanwhile, Myc acetylation is also modulated by HDACs either directly or indirectly. For example, SIRT2 stabilizes N-Myc and c-Myc proteins by deacetylating and repressing neuronal precursor cell-expressed developmentally downregulated 4 (NEDD4), which mediates Myc ubiquitination and degradation (Liu et al., 2013). Consequently, the SIRT2-specific inhibitor thiomyristoyl (TM) promotes Myc ubiquitination and degradation (Jing et al., 2016). HDACi suberoylanilide hydroxamic acid (SAHA) and entinostat (also called MS-275) induce Myc acetylation at K323, downregulating Myc and accompanying with tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) activation (Nebbioso et al., 2017). Therapeutic regimens that target Myc suppression using HDACi combined with DNA demethylation reagents seems to have a notable effect on non-small cell lung cancer (NSCLC) through activating immune system (Topper et al., 2017).
p53 is a well-known TSG that is crucial for mediating gene expression (Gu and Zhu, 2012; Zhu, 2017): its activity is modulated by various PTMs. HDACs and SIRTs downregulate p53 activity to promote cancer cell survival in response to oxidative stress (Juan et al., 2000; Luo et al., 2000, 2001; Vaziri et al., 2001). Specifically, HDAC1, -2, and -3 all can induce p53 deacetylation that represses p53-mediated apoptosis (Juan et al., 2000). In addition, HDAC2 modulates p53 transcriptional activity through direct p53-DNA binding (Harms and Chen, 2007). p53 binds to DNA depending on its acetylation state at K373/K382 by p300 (Gu and Roeder, 1997). HDACi depsipeptide induces acetylation at K373/K382 by recruiting p300. This in turn promotes the expression of p21Cip1/Waf1 (encoded by cyclin dependent kinase inhibitor 1A, CDKN1A) (Zhao et al., 2006). Compared to wild type p53, HDAC deficiency reduces mutant p53 (mtp53) expression both at the mRNA and protein level (Yan et al., 2013; Stojanovic et al., 2017). Besides the transcriptional regulation of mtp53, HDACs also modulate mtp53 protein stability. By inhibiting HDAC6, SAHA promotes the preferential degradation of mtp53 by downregulating heat shock protein 90 (HSP90) that suppresses p53 degradation via E3 murine double minute (MDM2) or carboxy terminus of HSP70-interacting protein (CHIP) (Li et al., 2011). Therefore, inducing mtp53 degradation by blocking HDAC6-HSP90 might represent a novel strategy to suppress oncogenesis in the future (Alexandrova et al., 2015).
In addition to TFs, HDACs also modulate the activity of super enhancers (SEs) (Gryder et al., 2019). Enhancer RNAs (eRNAs) are short, non-coding RNA molecules that alter the transcription of target genes in cooperation with promoters (Melo et al., 2013; Hsieh et al., 2014; Danko et al., 2015; Mao et al., 2019). Trichostatin A (TSA) and SAHA reduce eRNA synthesis by inhibiting HSP90 (Greer et al., 2015). MEF2D and HDAC4/9 form a corepressor to recognize intergenic regions. HDAC4/9 depleted cells show increased H3K27ac level around the gene transcriptional start sites where show the features of active enhancers within corresponded topologically associated domains (TAD) (Di Giorgio et al., 2020). Class I-specific HDACi 4SC-202 globally increases both of H3K27ac and H3K4me3 levels around the TSS of genes, but notably decreases occupancy at proximal regions of TSS of genes such as SMAD family member 6 (SMAD6) and E2F transcription factor 8 (E2F8), which are associated with enhancer deactivation (Mishra et al., 2017). Panobinostat and romidepsin alter the acetylation status of H3K27 by disrupting the SE topology in paired box 8 (PAX8) (Shi et al., 2019). Largazole (a cyclic peptides similar to depsipeptide) preferentially disturbs SE-driven transcripts that are frequently associated with oncogenic activities (Sanchez et al., 2018).
Transcriptional Activation
Although HDACs generally function as gene silencers, they can also activate transcription (Kurdistani et al., 2002; Wang et al., 2002). Besides the regulation of enhancers, a potential mechanism underlying this role includes the modulation of RNA polymerase II (RNAP2) by HDACs. HDACs participate in the crosstalk between RNAP2 C-terminal domain acetylation and phosphorylation (Wang et al., 2009; Blank et al., 2017; Ali et al., 2019). SIRT6 recruits and mono-ADP-ribosylates switch/sucrose non-fermenting (SWI/SNF) related, matrix associated, actin dependent regulator of chromatin subfamily c member 2 (SMARCC2/BAF170) to form active chromatin at the enhancer of heme oxygenase-1, which subsequently recruits RNAP2 (Rezazadeh et al., 2019). SIRT6 can also bind p53 to effectively recruit RNAP2 to local promoters (Li et al., 2018). SIRT6 deficiency mediates the activation of cyclin-dependent kinase 9 (CDK9) that can phosphorylate negative elongation factor (NELF) and mediate NELF release from RNAP2, facilitating the enrichment of TFs and RNAP2-related elongation factors to promote elongation of specific gene sets (Etchegaray et al., 2019). Consistently, TSA and SAHA disturb RNAP2-mediated transcriptional elongation by promoting the association between RNAP2 and NELF (Greer et al., 2015). Moreover, high doses of largazole can cause RNAP2-mediated transcriptional pausing and cell death (Sanchez et al., 2018).
DNA Methylation and Deacetylation
DNA methylation and histone modification modulate transcription, either alone or cooperatively, by altering chromatin status. HDACs and their complexes are recruited to hyper-methylated DNA through methyl-CpG binding domain containing (MBD) protein (MeCP), which has transcriptional repression roles (Jones et al., 1998; Nan et al., 1998; Ng et al., 1999; Zhang et al., 1999; Zhu et al., 2003). To maintain DNA methylation, DNA (cytosine-5-)-methyltransferase 1 (DNMT1) binds to HDAC1 and HDAC2 to establish heritable transcriptional silencing (Robertson et al., 2000; Rountree et al., 2000). A number of major breakthroughs involving combinations of HDACi and DNA demethylation reagents [DNA methyltransferase inhibitors (DNMTi)] have occurred in the past two decades (Cameron et al., 1999; Zhu et al., 2001b; Topper et al., 2017). The rationale underlying combined DNMTi and HDACi therapy lies in their synergistic effects on compacted chromatin. Dense methylation of CpG islands (CGI) is responsible for silencing genes, which can be reactivated by HDACi. In this scenario, TSA loosens the structure of chromatin and induces the expression of previously silenced genes in the presence of DNMTi (Jones et al., 1998, 2016; Cameron et al., 1999). The combination of 5-aza-2’-deoxycytidine (5-Aza-CdR, decitabine, Dacogen, Otsuka) and either depsipeptide or TSA induces the expression of p21Cip/Waf1, p15 (CDKN2B/INK4B), p16 (CDKN2A/INK4B), and p19 (CDKN2D/INK4D) (Cameron et al., 1999; Zhu et al., 2001a). Depsipeptide and apicidin induce demethylation and re-activate silenced genes such as p16, GATA binding protein 4 (GATA4) and sal-like protein 3 (SALL3) by inhibiting DNMT1 binding to these gene promoters (Wu et al., 2008). In terms of the direct anti-tumor effects, DNMTi in combination with HDACi can provoke a durable, powerful clinical response in patients (Jones et al., 2016). Several combinational therapies present in regimen of reversing tumor immune evasion in NSCLC. Azacytidine plus ITF-2357 (givinostat) seems to be the most efficient strategy that augments antigen presentation machinery and interferon α/β (IFNα/β)-related immune gene activation, and mainly focuses on suppression of Myc-driven tumorigenesis (Topper et al., 2017). With their broad range of physiological functions in various tissues, the combined effects of HDACi and DMNTi hold substantial therapeutic promise going forward.
Synergetic Regulation of HDACs and Other Histone Modifiers
In addition to DNA methylation, HDACs also cooperate with other epigenetic modifiers. For example, lysine-specific demethylase 1 [LSD1, lysine demethylase 1A (KDM1A)] is responsible for removing mono- or di-methylation of H3K4, and represses transcription via the CoREST-HDACs complex (Humphrey et al., 2001; Lee et al., 2005; Shi et al., 2005). A dual inhibitor of HDAC and LSD1, corin, has been developed to suppress CoREST-HDACs and to coordinately increase H3K4me1, H3K27ac and H3K27me3 (Kalin et al., 2018; Anastas et al., 2019). KDM2B induces H3K79 demethylation and transcriptional repression in a SIRT1-dependent manner (Kang et al., 2018). KDM4A modulates gene repression though a physiological interaction with the NCoR-HDAC3 complex (Zhang et al., 2005). KDM5A directly associates with HDAC complexes to regulate H3K4me2/3 (Nishibuchi et al., 2014). The H3K36me2 demethylase KDM8 increases H3/H4 acetylation and Cyclin A1 transcriptional activation by impeding HDAC1 recruitment (Hsia et al., 2010).
Regarding histone lysine methyltransferases, HDAC3 modulates the H3K9ac/H3K9me3 transition in a suppressor of variegation 3-9 homolog 1 (SUV39H1, also called KMT1A)-dependent manner during the DNA damage response (DDR) (Ji et al., 2019). SIRT1 regulates H3K9 methylation by deacetylating K266 in the Su(var)3-9, enhancer-of-zeste and trithorax (SET) domain of SUV39H1, thus increasing its activity during heterochromatin formation (Shankaranarayana et al., 2003; Vaquero et al., 2007). While SIRT6 induces the monoubiquitination of cysteines (Cys) in the pre-SET domain of SUV39H1, removing SUV39H1 from IκBα negatively regulates the nuclear factor-kappaB (NF-κB) pathway (Santos-Barriopedro et al., 2018). Depsipeptide decreases H3K9me2/3 expression by reducing the expression of SUV39H1 and G9A (also called KMT1C) (Wu et al., 2008). SIRT2 binds and deacetylates PR-Set7/SET8/KMT5A at K90, and increases the H4K20me1 level (Serrano et al., 2013). HDACs also interact with polycomb-group (PcG) proteins to reset chromatin remodeling and transcriptional repression (Van Der Vlag and Otte, 1999; Kuzmichev et al., 2005; Zhang et al., 2012b; Fukumoto et al., 2018). SIRT1 interacts with Set7/9 (also called KMT7), with several sites being methylated by Set7/9. In response to DNA damage, SIRT1-p53 binding is significantly enhanced in the presence of Set7/9 and this binding coincide with increased p53 acetylation at K382 (Liu et al., 2011).
The BRD family proteins are readers of Kac (Dhalluin et al., 1999; Fujisawa and Filippakopoulos, 2017). Class I HDACi 4SC-202, mocetinostat and entinostat induce increase of hundreds of gene expression, which are mostly enriched upon BRD4- and MYC-targeted TSS-proximal regions. p21 is activated by 4SC-202 to inhibit cell proliferation (Mishra et al., 2017). Similarly, JQ1 cooperates with SAHA to inhibit the growth of pancreatic ductal adenocarcinoma (PDAC) by upregulating p57 (CDKN1C) that usually blocked by Myc (Mazur et al., 2015). Besides, HDACs also interact with protein arginine methyltransferases (PRMTs) to regulate gene transcription (Qi et al., 2018; Yan W. W. et al., 2018). These findings all highlight the competition among the “readers”, “writers” and “erasers” at acetylated histones and non-histones, and may provide additional, novel and combination epigenetic approaches for cancer therapy in the future (Figure 2).
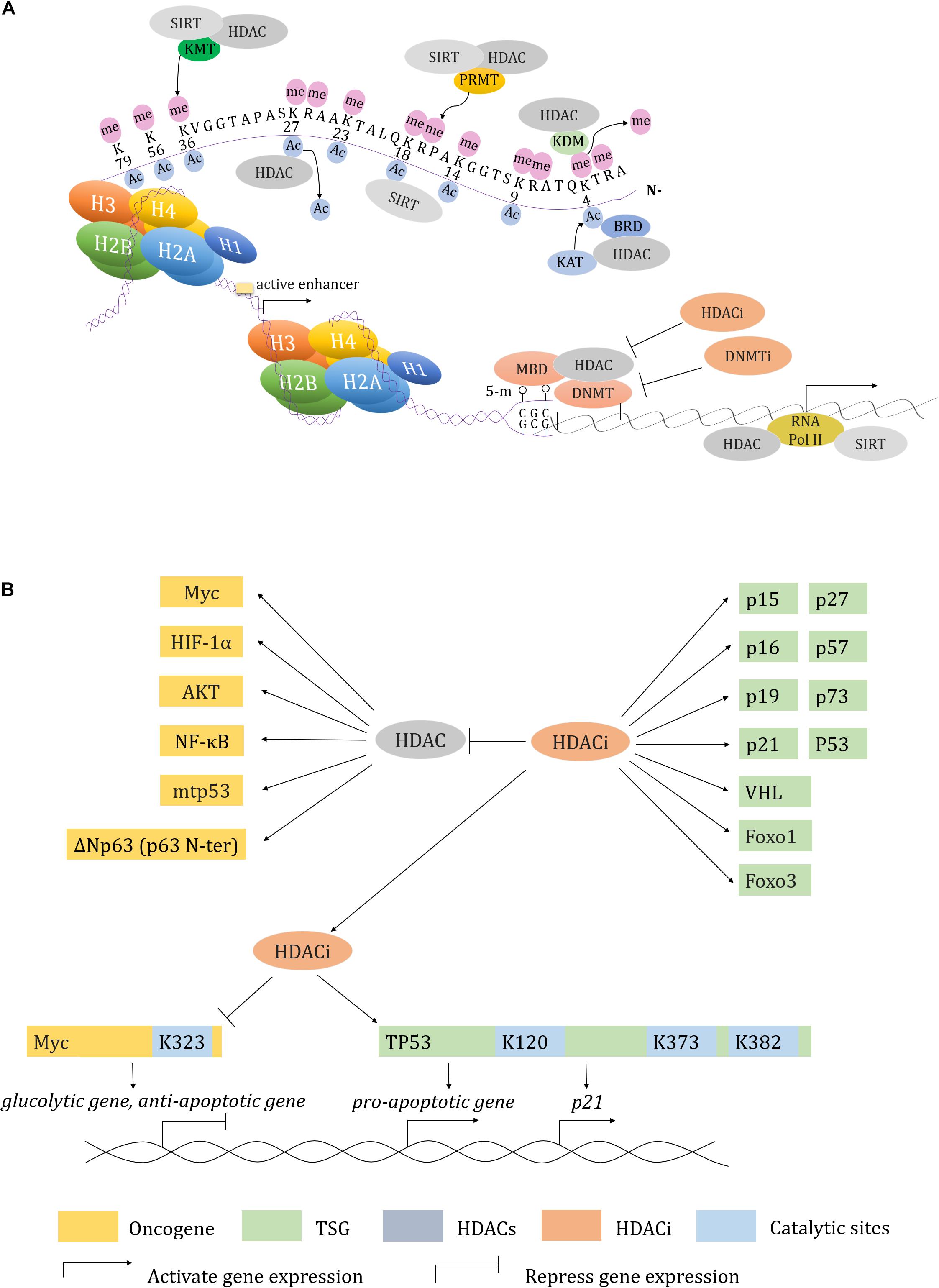
Figure 2. Transcription regulation in HDACs and HDACi. (A) HDAC and HDACi involved transcription regulation in concert with other epigenetic modifiers. (B) A working model described how HDAC and HDACi regulate both of oncogenes and tumor suppressor genes expression.
Metabolism
Various kinases and metabolic pathways form a complex network with epigenetic co-repressors to dynamically regulate metabolic flux and enzyme activity; aberrations in these processes can result in tumorigenesis and cancer progression. Metabolism can affect protein acetylation by altering the concentration of NAD+ and acetyl-CoA. In turn, HDACs also mediate metabolic reprogramming in cancer cells (Verdin and Ott, 2015).
Cancer cells are often characterized by their strong glycolytic activity, with aerobic glycolytic activity being preferred for tumor energy metabolism (Weinhouse, 1956). Increased glycolysis is associated with the abnormal regulation of glycolytic enzymes and other glucose metabolism pathways. Class II HDACs induce trans-repression of gluconeogenic enzymes from the cytoplasm to the nucleus in an HDAC3-dependent manner, and mediate the deacetylation and activation of the forkhead box class O (FoxO) family in the nucleus (Mihaylova et al., 2011). SIRT2 deacetylated isocitrate dehydrogenase 1 (IDH1) at K224 and promotes IDH1 enzymatic activity. The hypoacetylated IDH1 converts isocitrate into α-ketoglutarate in the tricarboxylic acid (TCA) cycle to inhibit liver metastases of colorectal cancer (CRC) (Wang B. et al., 2020). Pyruvate kinase (PKM2) promotes tumorigenesis by regulating oncogene expression and proliferation pathway activation in HDAC3-dependent way (Yang W. et al., 2011; Yang et al., 2012). SIRT6 can directly interact with and deacetylate PKM2, resulting in its nuclear export via exportin 4 and suppression of PKM2-related oncogenic functions (Bhardwaj and Das, 2016). Conversely, SIRT3 and SIRT6 also act as tumor suppressors, restricting aerobic glycolysis in cancer cells through destabilization of hypoxia inducible factor 1 subunit alpha (HIF-1α) and inhibition of glycolytic kinases, respectively (Finley et al., 2011; Sebastian et al., 2012). p53 directly binds and activates SIRT6 to regulate gluconeogenesis by mediating the nuclear exclusion and deacetylation of FoxO1 (Zhang et al., 2014).
The fatty acylation of proteins has a vital role in membrane synthesis, vesicle transport, protein-membrane interaction, cell signaling and localization (Resh, 2006). HDACs regulate fatty acylation during cancer progression. For example, HDAC8 performs lysine de-fatty-acylation functions. The HDAC8-selective inhibitor PCI-34051 also increases overall fatty acylation levels in Jurkat cells (Aramsangtienchai et al., 2016). HDAC11 has a relatively low effect on acetyl groups, but efficiently catalyzes dodecanoylated and myristoylated peptides (Kutil et al., 2018). Compared with acetyl peptides, some HDACs have higher catalytic efficiency on acyl groups (Houtkooper et al., 2012; Feldman et al., 2013; Jiang et al., 2013; Aramsangtienchai et al., 2016; Wang et al., 2016; Kutil et al., 2018) (see Table 1). HDAC11 efficiently removes acyl groups on the surface of serine hydroxymethyltransferase 2α (SHMT2α), causing SHMT2α dissociation from the late endosome/lysosome. This effect leads to type I interferon receptor chain 1 (IFNαR1) polyubiquitylation and degradation, as well as downregulation of IFN signaling (Cao et al., 2019). HDAC11-specific inhibitors, such as elevenostat, FT895, and SIS17, might represent promising future treatments that target lipid metabolic dysregulation in cancers (Martin et al., 2018; Kutil et al., 2019; Son et al., 2019). SIRT3 has a role in mitochondrial fatty-acid β-oxidation by regulating long-chain acyl-CoA dehydrogenase (LCAD) (Hirschey et al., 2010). SIRT6 is indispensable for hepatic β-oxidation by deacetylating the peroxisome proliferator-activated receptor α (PPARα) coactivator nuclear receptor coactivator 2 (NCOA2) at K780 (Naiman et al., 2019). Following palmitic acid treatment, SIRT6 interacts with p53 to regulate de novo cardiolipin biosynthesis and maintain lipid homeostasis (Li et al., 2018).
Amino acids are also involved in tumorigenesis. SIRT3 depletion suppresses glutamate dehydrogenase (GDH/GLUD), which impairs glutamine flux to the TCA cycle and causes reduction of acetyl-CoA pools (Li M. et al., 2019). SIRT4 is a lipoamidase that diminishes the activity of the pyruvate dehydrogenase complex (PDH) by hydrolyzing the lipoamide cofactor dihydrolipoyllysine acetyltransferase (DLAT) (Mathias et al., 2014). Furthermore, SIRT4 represses GDH activity through its ADP-ribosyltransferase function. SIRT4 deficiency activates GDH, stimulating amino acid-mediated insulin secretion in insulinoma cells (Haigis et al., 2006). SIRT4 also mediates other PTMs, including methylglutarylation, hydroxymethylglutarylation and 3-methylglutaconylation, and intermediates of these PTMs contribute to leucine oxidation. Indeed, SIRT4-KO induces leucine disordered metabolism and leads to glucose intolerance and insulin resistance (Anderson et al., 2017). Meanwhile, elevated SIRT5 expression in breast cancer mediates glutaminase desuccinylation and protects glutaminase from ubiquitin-mediated degradation; this effect has been associated with a poor prognosis in breast cancers (Greene et al., 2019). SIRT3 and SIRT5 also mediate desuccinylation and deacetylation of SHMT2, respectively, suggesting that suppression of serine catabolism might represent a novel strategy to restrain tumor growth (Wei et al., 2018; Yang et al., 2018).
Hypoxia and Angiogenesis
Activated HIFs (HIF-1α, HIF-2α, HIF-3α, and HIF-1β) have vital roles in adaptive responses, with HIF-1α and HIF-2α in particular being associated with tumorigenesis and angiogenesis in response to hypoxia (Gonzalez et al., 2018). Notably, SAHA specifically induces the accumulation of HIF-2α rather than HIF-1α in soft tissue sarcomas (Nakazawa et al., 2016). HIF-1α is ubiquitinated by von Hippel-Lindau (VHL) or by binding to p53-MDM2, inducing proteasomal dependent degradation (Vriend and Reiter, 2016; Gonzalez et al., 2018). HDAC1 downregulates p53 and VHL expression, and stimulates HIF-1α-dependent angiogenesis. TSA inhibits this process by blocking HIF-1α and the vascular endothelial growth factor (VEGF) receptor (Kim et al., 2001). Besides, HDAC4 and HDAC6 directly bind to HIF-1α. HDACi LAQ824, valproic acid (VPA) and trapoxin induce dose-dependent HIF-1α depletion in an VHL-independent manner (Qian et al., 2006). The class IIa-selective HDACi TMP195 effectively establishes an anti-tumor microenvironment and induces normalization of tumor vasculature in breast cancers by eliciting recruitment and differentiation of macrophages. TMP195 in combination with chemotherapeutic regimens such as carboplatin or paclitaxel can significantly reduce breast cancer burden (Guerriero et al., 2017).
As for SIRTs, they continuously perform an inhibitory role to HIF-1α-relevant transcriptional and metabolic regulation. During hypoxia, SIRT1 activity is inhibited due to reduced NAD+ levels, which leads to the acetylation and activation of HIF-1α and HIF-2α. SIRT1 negatively regulates angiogenesis by deacetylating FoxO1 (Potente et al., 2007; Dioum et al., 2009; Lim et al., 2010). In human breast cancers, a SIRT3 deficiency can stabilize HIF-1α (Finley et al., 2011). Both SIRT6 and SIRT7 can negatively modulate the expression and activity of HIF-1α and HIF-2α (Zhong et al., 2010; Hubbi et al., 2013).
Redox and Oxidative Stress
Histone deacetylase inhibitors treatment is often accompanied by oxidative stress related DNA damage that is primarily caused by the generation of reactive oxygen species (ROS) (Xu et al., 2006). In mammalian cells, two redox systems respond to oxidative stress: the thioredoxin (Trx) system and the glutathione-glutaredoxin (Grx) system. In response to nitric oxide (NO), HDAC2 is S-nitrosylated at Cys 262 and Cys 274, which induces chromatin remodeling to promote gene expression (Nott et al., 2008). A pair of redox-sensitive cysteine residues (Cys-667/Cys-669) in HDAC4 are involved in oxidative stress via the formation of intramolecular disulfide bonds (Ago et al., 2008). Compared with normal cells, tumor cells are enriched with the antioxidant Trx reductase (TrxR), which might represent a novel therapeutic target (Lu and Holmgren, 2014; West and Johnstone, 2014). Depsipeptide causes robust DNA damage and apoptosis by inducing ROS generation, primarily through the suppression of TrxR (Wang et al., 2012). HDAC5 represses mitochondrial ROS generation, and depletion of HDAC5 provokes nuclear factor, erythroid 2 like 2 (NRF2)-associated transcription (Hu et al., 2019). The DNA and RNA binding protein Y-box binding protein 1 (YB-1) binds to NRF2 in response to oxidative stress. Entinostat induces YB-1 acetylation and blocks its binding to NRF2, reducing NRF2 synthesis and increasing ROS levels in sarcoma cells (El-Naggar et al., 2019).
Sirtuins primarily serve as antioxidants in redox signaling. SIRT1, -2, and -3 all prevent oxidative stress by inducing or modulating manganese superoxide dismutase (MnSOD) (Brunet et al., 2004; Wang et al., 2007). Glucose-6-phosphate dehydrogenase (G6PD) is key enzyme of the pentose phosphate pathway (PPP) that regulates nicotinamide adenine dinucleotide phosphate (NADP)/NADPH levels. NADPH maintains glutathione (GSH) at a reduced state, which serves as an antagonist to prevent ROS generation (Chen et al., 2019). In response to oxidative stress, SIRT2 and SIRT3 promote NADPH generation by deacetylating and activating G6PD and IDH2 in the PPP or in the TCA cycle, respectively. The PPP also produces ribose-5-P, which synthesizes nucleotides and generates NAD+, which in turn supports SIRTs activity (Schlicker et al., 2008; Wang Y. P. et al., 2014). SIRT3 also activates NADH quinone oxidoreductase (Complex I) and succinate dehydrogenas (Complex II) in the electron transport chain (Ahn et al., 2008; Cimen et al., 2010). Furthermore, in the mitochondrial inter-membrane space, SIRT5 deacetylates cytochrome c (Schlicker et al., 2008). SIRT5 is also present in peroxisomes, where it desuccinylates and inhibits peroxisomal acyl-CoA oxidase 1 (ACOX1). A SIRT5 deficiency in hepatocellular carcinoma (HCC) increases oxidative DNA damage by elevating ACOX1-mediated H2O2 production (Chen et al., 2018). By contrast, SIRTs also inhibit antioxidation; for example, SIRT2 deacetylates and suppresses peroxiredoxin (an antioxidant) in breast cancer cells (Fiskus et al., 2016; Figure 3).
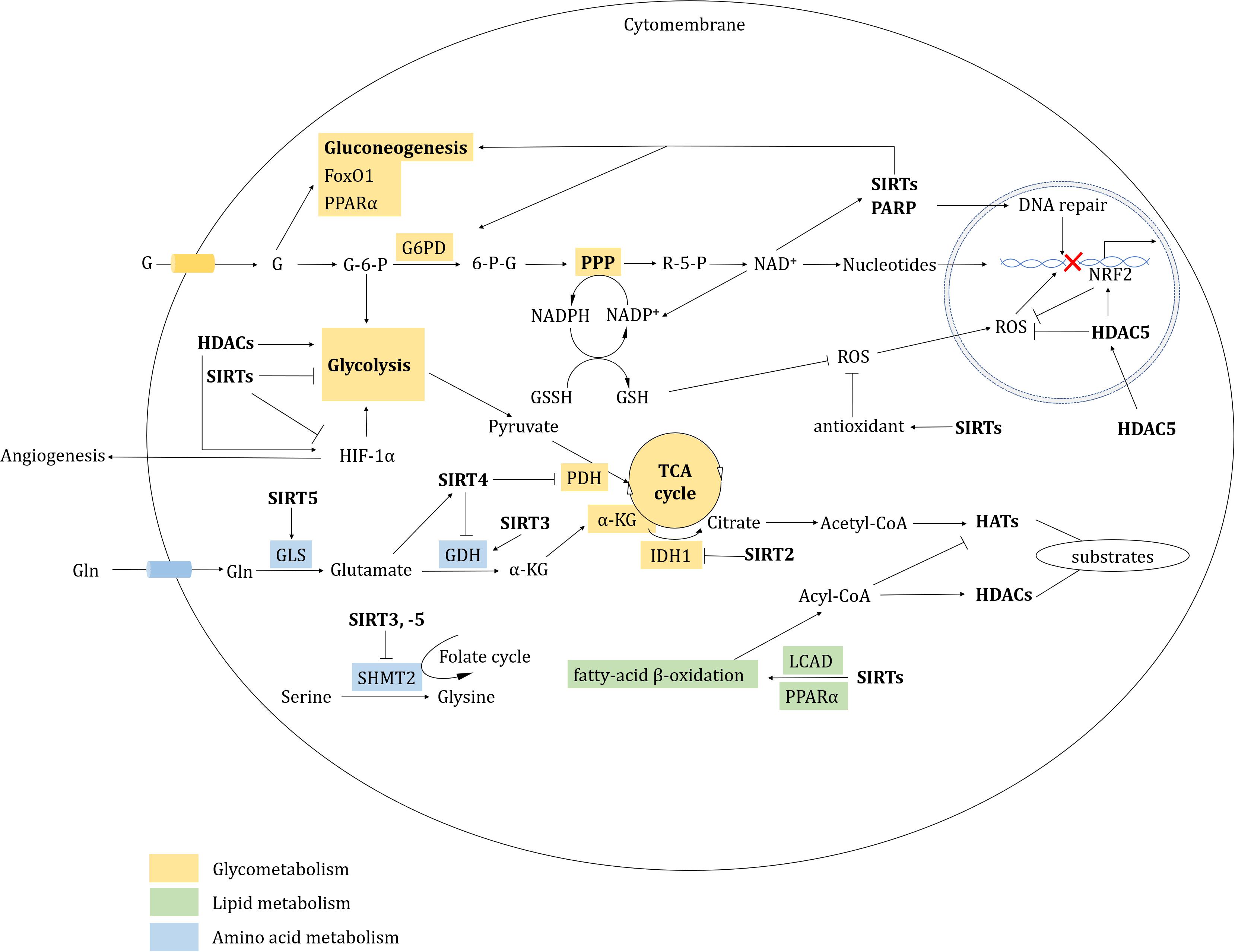
Figure 3. HDAC-involved metabolic regulation. HDACs regulate metabolism mainly including glycometabolism, lipid metabolism, amino acid metabolism and redox.
DNA Damage Response
The DDR is a vitally important regulatory mechanism that protects genomic DNA from damage induced by various stimuli (Jackson and Bartek, 2009). Different levels of DNA damage are inevitably caused by UV radiation and DNA adducts, which are produced by ROS and reactive nitrogen species (RNS), as well as exposure to chemical agents (Barker et al., 2015; Roos et al., 2016; Pouget et al., 2018; Guo et al., 2020). DNA double-strand breaks (DSBs) are the most severe form of DNA damage and are repaired via one of two pathways: homologous recombination (HR) or non-homologous end-joining (NHEJ) (Scully et al., 2019). Deacetylation of H3K56 and H4K16 by HDAC1/2 are involved in mediating dynamic chromatin regulation in response to NHEJ (Miller et al., 2010). Although H4K16 acetylation attenuates binding of p53 binding protein 1 (53BP1) to H4K20me2, euchromatic histone lysine methyltransferase 1 (EHMT1, also called GLP or KMT1D)-catalyzed H4K16 monomethylation could significantly enhance this binding (Lu X. et al., 2019). Meanwhile, SIRT1 redistributes to DSB foci to promote HR during oxidative stress (Oberdoerffer et al., 2008). NuRD complex subunit, chromodomain-helicase-DNA-binding protein (CHD4), was recently discovered to be recruited by SIRT6 to replace heterochromatin 1 (HP1) at H3K9me3 to ultimately promote chromatin relaxation through HR (Hou et al., 2020). SIRT6 also mono-ADP-ribosylates and displaces KDM2A (also called JmjC domain-containing histone demethylase 1A, JHDM1A) from chromatin, which leads to HP1α-dependent H3K9me3 deposition at DSBs and transient transcriptional repression, accompanying with the recruitment of NHEJ factors (Rezazadeh et al., 2020).
The DDR is controlled by three related kinases: ataxia telangiectasia mutated (ATM), ataxia telangiectasia mutated and Rad3 related (ATR), and DNA-dependent protein kinase catalytic subunits (DNA-PKcs) (Blackford and Jackson, 2017). Once a DSB occurs, the MRE11-RAD50-NBS1 (MRN) complex and the Ku family are rapidly recruited to DSB sites. As the sensor, ATM is recruited to DSB sites by the MRN complex (Blackford and Jackson, 2017). The interplay between HDAC1 and ATM increases chromatin condensation to prevent radio-sensitivity in response to ionizing radiation (Kim et al., 1999). SIRT1 binds to deleted in breast cancer 1 (DBC1), which induces p53 activation (Kim et al., 2008; Zhao et al., 2008). ATM also mediates DBC1 phosphorylation at threonine (Thr) 454 which contributes to DBC1-SIRT1 interactions during DNA damage (Yuan J. et al., 2012). Moreover, SIRT1 deacetylates and maintains the hypoacetylation of Nijmegen breakage syndrome protein 1 (NBS1), which is necessary for the ionizing radiation-induced phosphorylation of NBS1 and subsequent MRN complex recruitment to DSB sites (Yuan et al., 2007). SIRT7 directly binds and deacetylates ATM, which is prerequisite for ATM dephosphorylation and inactivation in the final stage of DNA repair (Tang et al., 2019). Panobinostat-induced downregulation of meiotic recombination 11 homolog (MRE11) enhances radio-sensitization of bladder cancer cells by promoting MRE11 ubiquitination that relies on the upregulated E3 inhibitor of apoptosis protein 2 (cIAP2) (Nicholson et al., 2017).
ATR and its downstream kinase CHK1 are also involved in the response to DNA replication stress. SAHA slows down replication forks by restricting the ATR pathway (Conti et al., 2010). Following the conserved mechanism in yeast, VPA disrupts the formation of single-strand-DNA-RFA nucleofilaments and the activation of the Mec1 (ATR in human) and Rad53 (CHK2 in human) by suppressing the recruitment of replication factor A protein 1 [RFA1, replication protein A (RPA) in human] and DNA damage checkpoint protein Ddc2/LCD1 [ATR interacting protein (ATRIP) in human] to DNA damage sites (Robert et al., 2011). Entinostat represses checkpoint signaling during replication stress. Mechanically, HDAC1/2 suppress the cell cycle kinases WEE1 and CDK1 and induce the dephosphorylation of ATM and CHK2 by suppressing the expression of PP2A subunit. Entinostat also induces the incorrect incorporation of NTPs and metabolites during the induction of checkpoint kinase inactivation, which can result in mitosis catastrophe (Goder et al., 2018). Therefore, CHK inhibitors might be designed to prevent this event from occurring. Indeed, CHK1 inhibitor treatment combined with HDACi induces cell death via extensive mitotic disruption in a range of solid tumors (Lee et al., 2011).
DNA-PKcs is another sensor that is recruited to DSBs by Ku-bound DSB ends (Blackford and Jackson, 2017). Under conditions of fasting-induced oxidative stress, SIRTs act as protective factors in the DDR. Specifically, SIRT1 deacetylates Ku70, resulting in Ku70-Bcl-2 associated protein X (BAX) disassociation and the transport of BAX away from the mitochondria, leading to stress-induced resistance to apoptosis (Cohen et al., 2004). SIRT3 also physically interacts with and deacetylates Ku70 to impede BAX translocation to the mitochondria (Sundaresan et al., 2008).
SIRTs are highly important for DNA damage repair and genome stability (Tian et al., 2019; Ng and Huen, 2020). Recent data have shown that SIRT6 is a novel sensor for initiating the DDR (Onn et al., 2020). Deacetylated SIRT6 at K33 by SIRT1 results in SIRT6 polymerization and deposition at γH2AX foci. Moreover, a SIRT6 K33R hypoacetylation mimic can rescue DNA repair defects in SIRT1-deficient cancer cells (Meng et al., 2020). SIRT7 is also associated with NHEJ, as a Sirt7 deficiency impairs the recruitment of 53BP1 to DSB sites and inhibits NHEJ efficiency (Vazquez et al., 2016). SIRT7 also deacetylates ATM to mediate ATM inactivation in the final stage of DNA damage repair (Tang et al., 2019). SIRT7 acts as a deglutarylase to regulate H4K91 glutarylation (H4K91glu). This process is closely associated with chromatin remodeling in response to DNA damage (Bao et al., 2019). Treatment with 5-Fluorouracil (5-FU) induces SIRT7 degradation in the Tat-binding protein 1 (TBP1)-mediated proteasome-dependent pathway, increasing cell radiosensitivity in combination therapy (Tang M. et al., 2017).
Poly ADP-ribose polymerases (PARPs) are central to the activation of several downstream repair mechanisms, including single-strand DNA breaks (SSBs), base-excision repair (BER), HR and NHEJ (Pilie et al., 2019). SIRTs and PARPs all require NAD+ to elicit function. However, PARP1 consumes NAD+, and this affects NAD+-dependent SIRT activity. Thus, depleting PARP1 increases the catalytic function of SIRTs (Schreiber et al., 2006; Houtkooper et al., 2012; Imai and Guarente, 2014). Under conditions of oxidative stress, SIRT6 physically binds to and mono-ADP-ribosylates PARP1 at K521 to facilitate DNA repair (Mao et al., 2011). SIRT7 is recruited to DSB sites in a PARP1-dependent manner, and catalyzes H3K122 desuccinylation, which facilitates chromatin compaction and DNA repair (Li et al., 2016). Based on synthetic lethality, PARP inhibitors induce genomic instability in breast cancer susceptibility protein (BRCA1/2)-deficient cancer cells (Pilie et al., 2019). The combined use of HDAC and PARP inhibitors will likely be of great benefit for patients with BRCA1/2-deficient malignancies (Liszczak et al., 2018).
With the exception of DSBs, cancer cells can overcome DNA damage-induced cytotoxicity through BER, nucleotide excision repair (NER) and mismatch repair. Uracil-DNA N-glycosylase isoform 2 (UNG2) has a role in BER and can be deacetylated at K78 by HDAC, boosting disassociation from its E3 ubiquitin-like containing PHD and ring finger domain 1 (UHRF1) when stimulated by ROS. HDACi combined with genotoxic agents results in UNG2 degradation, resulting in a robust cell death effect (Bao et al., 2020). SIRT1 interacts with xeroderma pigmentosum group A (XPA) in NER by directly deacetylating XPA or mediating XPA binding to ATR to prevent UV irradiation (Fan and Luo, 2010; Jarrett et al., 2018). HDAC10 is mainly involved in DNA mismatch repair by deacetylating mutS homolog 2 (MSH2) at K73 (Radhakrishnan et al., 2015).
Besides, a number of other histone modifications are also actively involved in these DNA repair pathways, but are beyond the scope of this review (Cao et al., 2016; Kim J. J. et al., 2019; Li Z. et al., 2019). Suffice to say that the multiple sites of H3 and H4 acetylation are not absolutely related to checkpoint activation because the conversion of lysine to other amino acids can still activate checkpoints (Robert et al., 2011).
Cell Cycle
Cell cycle dysregulation is a central hallmark of oncogenesis; as such, cell cycle regulators are considered promising targets for cancer treatment. HDACs are often involved in cell cycle checkpoints. HDAC3-mediated deacetylation of cyclin A affects the progression of the S phase and G2/M transitions (Bhaskara et al., 2008, 2010). HDAC10 depletion induces G2-M transition arrest through the regulation of cyclin A2. Mechanically, HDAC10 depletion induces the downregulation of high mobility group AT hook 2 (HMGA2), which leads to enrichment of E4F transcription factor 1 (a cyclin A2 repressor) at the cyclin A2 promoter and G2-M arrest (Li et al., 2015). Regarding combination therapies, the CDK9 inhibitor dinaciclib and panobinostat together induce apoptosis over the short-term in MLL-AF9-driven acute myeloid leukemia (AML) (Baker et al., 2016). The emerging hybrid inhibitor Roxyl-zhc-84, which concordantly inhibits HDACs and CDKs, induces G1-phase arrest and apoptosis in ovarian and breast cancer cells (Huang et al., 2018c).
Spindle assembly checkpoint (SAC) is involved in regulating mitosis. Budding uninhibited by benzymidazol related-1 (BubR1), a component of the SAC, must be deacetylated by HDAC2/3 to initiate mitotic exit (Park et al., 2017). HDAC3 induces SAC activation and the dissociation of sister chromatids (Eot-Houllier et al., 2008). SIRT2 is strongly associated with mitosis exit (Dryden et al., 2003). SIRT2 regulates the anaphase-promoting complex/cyclosome (APC/C) by deacetylating its cofactors, cell-division cycle protein 20 (CDC20) and CDC20 homolog 1 (CDH1), which are both required for mitosis exit and chromosome segregation (Kim et al., 2011). SIRT2 also deacetylates α-tubulin at K40 to promote cell mobility (North et al., 2003). The SIRT2 inhibitor SirReal2 induces tubulin hyperacetylation and BubR1 destabilization (Rumpf et al., 2015). HDAC5 induces the transcription of the mitosis kinase Aurora A, by repressing the expression of the E3 ligase NEDD4 (Sun et al., 2014). Combination of the Aurora A kinase inhibitor alisertib with romidepsin causes dose-dependent cytotoxicity of lymphoma cells (Zullo et al., 2015).
p21Cip/Waf1 is a cyclin-dependent kinase inhibitor (CKI). HDACi induces p21 expression by re-activating hyperacetylation of H3 and H4 in its promoter region (Richon et al., 2000). Furthermore, depsipeptide induces p53 phosphorylation at Thr 18, which is a requirement for subsequent p53 acetylation at K373/382 and p21 activation (Wang et al., 2012). In liver cancer, the HDAC8-selective inhibitor PCI-34051 can induce p21 expression and G2-M phase cell cycle arrest (Tian et al., 2015). Nevertheless, p21 and p16 are activated by HDACi in a p53-independent manner (Yoshida and Horinouchi, 1999). Namely, SIRT7 indirectly modulates p21-mediated cell cycle arrest by elevating p53 activity. SIRT7 physically binds and deacetylates P300/CBP-associated factor (PCAF) at K720, and this interaction is enhanced under conditions of glucose deprivation. As a result, PCAF binding to MDM2 is promoted, resulting in a triggering of MDM2 degradation via the ubiquitin-dependent proteasome pathway (Lu Y. F. et al., 2020). Finally, in p21-KO lymphomas, p27Kip1 (CDKN1B) functions in a p21-independent manner to induce cell cycle arrest after SAHA treatment (Newbold et al., 2014).
Apoptosis
Apoptosis is a physiologically programmed cell death pathway that is essential for the maintenance of organismal homeostasis. Apoptosis is controlled by the B-cell lymphoma 2 (Bcl-2) family of proteins, which includes both pro-survival and pro-apoptotic proteins that control cell fate (Singh et al., 2019).
Regarding the intrinsic apoptotic pathway, the Bcl-2 interacting mediator of cell death (Bim), a Bcl-2 homology 3 (BH3)-only proapoptotic protein, is upregulated by depsipeptide via FoxO1 acetylation (Yang et al., 2009). Panobinostat elevates Sry-box transcription factor 7 (SOX7) expression and suppresses lung cancer cell proliferation. Mechanically, SOX7 triggers apoptosis by preventing Bim from proteasome-mediated degradation (Sun et al., 2019). The N-terminal truncated form of p63, ΔNp63, belongs to the p53 family, but acts as an oncoprotein. In squamous cell carcinoma, HDAC1 and HDAC2 form a complex with ΔNp63 to suppress the proapoptotic gene expression such as p53 upregulated modulator of apoptosis (PUMA) (Ramsey et al., 2011).
Histone deacetylase inhibitors treatment also affects the anti-apoptotic members of the Bcl-2 family. Specifically, depsipeptide induces apoptosis by decreasing the expression of pro-survival factors Bcl-2 and B-cell lymphoma-extra-large (Bcl-xL) (Adams and Eischen, 2016; Adams et al., 2016). The HDAC6-selective inhibitor ricolinostat exerts pronounced anti-lymphoma effects both alone and in combination with the alkylating agent bendamustine, by impairing the activation of caspase 8, -9, -3, and the Bcl-2 family (Cosenza et al., 2017). Myeloid cell leukaemia 1 (Mcl-1) is an E3-bound, anti-apoptotic protein that is involved in mitotic arrest (Senft et al., 2018). HDACi-induced Mcl-1 phosphorylation likely promotes apoptosis, whereas mutant phosphorylated Mcl-1 resists HDACi by binding to BH3-only proapoptotic proteins (Tong et al., 2018).
p53 is a crucial activator of apoptosis. HDAC1-3 all downregulate p53 activity, which represses p53-mediated activation of the pro-apoptotic gene BAX (Juan et al., 2000). Acetylation of p53 at K120 upregulates apoptotic peptidase activating factor 1 (Apaf-1) in the mitochondria (Yun et al., 2016). Under genotoxic stress, HDAC5 deacetylates p53 at K120, which activates pro-apoptotic target genes (Sen et al., 2013). Furthermore, HDAC6 directly deacetylates p53 at K120, which is required for p53-induced apoptosis in tumors with AT-rich interaction domain 1A (ARID1A) mutations (Bitler et al., 2017).
In summary, HDACi promote apoptosis via the intrinsic mitochondrial pathway, decreasing the expression of key anti-apoptotic factors (eg. Mcl-1, Bcl-2, and Bcl-xL), and/or increasing the expression of pro-apoptotic proteins (eg. BAX, Bim, Noxa and PUMA). HDACi also facilitate the activation of extrinsic apoptotic pathways, such as TRAIL (Nebbioso et al., 2017), driving mitochondrial outer membrane polarization (MOMP) and ultimately caspase-mediated cell death.
Degradation System
The modulation of protein degradation is of critical importance for cell function. Protein degradation occurs via two major pathways: the ubiquitin-dependent proteasome pathway and autophagy system.
Autophagy
Autophagy is a degradation process whereby autophagosomes engulf and recycle nutrient sources in response to energetic demands and organelle turnover (Mizushima et al., 2008; Mizushima, 2018). Autophagy can be effectively promoted by HDACs. For example, depletion of HDAC10 perturbs autophagy flux through increased LC3-II/I, and the accumulation of p62 and acidic vesicular organelles. HDAC10 inhibition results in increased sensitivity to cytotoxic reagents (Oehme et al., 2013). Sirt1 also forms complexes with autophagy related protein 5 (ATG5), ATG7 and ATG8 to promote autophagy, with organelles in Sirt1–/– mice being markedly damaged (Lee et al., 2008). The SIRT1 and -2 inhibitor tenovin-6 activates p53 and seems to be a specific regulator of mitochondrial acetylation (Lain et al., 2008; Scholz et al., 2015). Tenovin-6 suppresses Ewing sarcoma cells by regulating the NOTCH signaling pathway (Ban et al., 2014), and perturbs autophagic flux in chronic lymphocytic leukemia (CLL) cells and pediatric soft tissue sarcoma cells (Yuan et al., 2017). However, HDACs also interrupt autophagy, and various HDACi induce cancer cell death by promoting autophagy. In HDAC10-KO HeLa cells, chaperone-mediated autophagy (CMA) instead of macroautophagy is activated by the accumulation of lysosome-associated protein type 2A (LAMP2A)-positive lysosomes and the degradation of CMA substrate glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Obayashi et al., 2020). In response to serum starvation or oxidative stress, SIRT2 inhibition induces acetylated FoxO1 to locate in the cytoplasm, accelerating autophagy through interaction with ATG7 (Zhao et al., 2010). Under nutrient-rich conditions, FoxK1/2 bind to HDAC complex and restricts autophagic flux through the transcriptional repression of autophagy gene (Bowman et al., 2014). Under condition of nutrient deprivation, inhibition of the AKT serine/threonine kinase pathway facilitates nuclear import of FoxO3, which competitively replaces FoxK to bind the autophagy-associated gene promoters and upregulation of autophagy (Brunet et al., 1999; Bowman et al., 2014). Moreover, VPA activates autophagy by blocking HDAC1-mediated regulation of AKT pathway (Sun et al., 2020). The nutrient-sensor mammalian target of rapamycin (mTOR) negatively modulates downstream Unc-51-like autophagy activating kinase 1 (ULK1) that is involved in the non-transcriptional autophagic pathway. SAHA induces mTOR suppression, which ultimately activates autophagy by upregulating ULK1 (Gammoh et al., 2012). Of note, SAHA-induced autophagy seems to serve as a pro-survival mechanism to ameliorate SAHA-induced apoptosis by downregulating apoptotic factors (Gammoh et al., 2012). VPA also induces Sae2 [C-terminal-binding protein interacting protein (CtIP) in human] degradation in an autophagy related manner to impair HR-mediated DNA repair (Robert et al., 2011). As such, it seems that autophagy performs a dual role in DNA damage repair, depending on the cell states or DNA damage degree (Guo and Zhao, 2020).
Proteasome-Dependent Degradation
In addition to autophagy, proteasome-dependent degradation is also critical for cell function. HDACs target various E3s to affect basal cellular function. For example, panobinostat upregulates the E3 cIAP2 that causes the ubiquitination and proteasomal degradation of MRE11, elevating cellular sensitivity to chemoradiation (Nicholson et al., 2017). HDAC6 also modulates aggresome formation and the clearance of polyubiquitinated and misfolded proteins (Kawaguchi et al., 2003). In terms of therapeutic development, suppressing the aggresome pathway results in the accumulation of misfolded proteins, causing autophagy-associated DNA damage and apoptosis of cancer cells (Rodriguez-Gonzalez et al., 2008). Proteasome inhibitors (PIs), such as the United States Food and Drug Administration (FDA)-approved bortezomib (BTZ), have similar roles in preventing the degradation of polyubiquitin-misfolded proteins, which increases the production of ROS and disturbs DNA repair in tumor cells (Perez-Galan et al., 2006). However, long-term treatment with BTZ leads to drug-resistance in most patients. Low concentrations of HDACi combined with BTZ can downregulate anti-apoptotic proteins and upregulate pro-apoptotic proteins, thus accelerating cell death (Dai et al., 2008; Wang J. et al., 2019). Combining the HDAC6-selective inhibitor tubacin with BTZ induces significant anti-tumor activity triggering c-Jun NH2-terminal kinase (JNK)-caspase signaling and endoplasmic reticulum (ER) stress (Hideshima et al., 2005; Nawrocki et al., 2006). Another HDAC6 inhibitor, WT161, promotes the accumulation of acetylated tubulin and overcomes BTZ resistance to promote multiple myeloma (MM) cell death (Hideshima et al., 2016). RTS-V5, a dual inhibitor that targets HDAC6 and the 20S subunit of the proteasome, also possesses potent and selective anti-tumor activity in leukemia and MM cell lines (Bhatia et al., 2018).
Epithelial-Mesenchymal Transition, Cancer Stem Cells, and Senescence
Epithelial-mesenchymal transition (EMT) is characterized by the loss of the tight intercellular connections normally found in epithelial cells that then undergo cytoskeleton rearrangement and adopt the mesenchymal cell phenotype, which is associated with migration. Notably, cancer cell migration and invasion are promoted by a series of EMT-associated factors (such as SNAIL, ZEB, SLUG and TWIST) (Kim K. K. et al., 2018). These factors induce EMT-related stem cell properties and promote tumorigenesis via PTMs (Mani et al., 2008; Tam and Weinberg, 2013; Ye et al., 2015; Dai et al., 2020). S-nitrosylation of HDAC2 is regulated by endothelial nitric oxide synthase (eNOS) that is a crucial enzyme for NO synthesis, allowing ZEB1 re-activation (Cencioni et al., 2018). During hypoxia, HDAC3 is essential for the activation of mesenchymal gene expression by the interaction with WD repeat domain 5 (WDR5) (Wu et al., 2011). The transforming growth factor-β (TGF-β)-SMAD signaling pathway is the most important EMT stimulation pathway. HDAC6 also has an essential role in EMT by activating SMAD3 (Shan et al., 2008). SMAD3 and -4 induce SIRT7 transcriptional repression by forming a complex with HDAC8. HDAC8 inhibition significantly suppresses TGF-β signaling via SMAD-SIRT7 axis, and as a consequence, attenuates lung metastases of breast cancer (Tang et al., 2020). Class I HDACi 4SC-202 notably attenuates TGF-β-induced EMT (Mishra et al., 2017). By contrast, HDAC10 exhibits a potential TSG role by downregulating Sry-box transcription factor 9 (SOX9) in KRAS-driven lung adenocarcinoma. Furthermore, HDAC10 deficiency results in TGF-β pathway activation, leading to the induction of SOX9 and KRAS-expressing stem-like tumor growth (Li et al., 2020). Cancer-associated fibroblasts (CAFs) secrete extracellular matrix (ECM) that assists tumor progression and invasion. Scriptaid, a selective inhibitor of HDAC1, −3, and −8, represses TGF-β-mediated CAF by inhibiting ECM secretion and cell invasion (Kim D. J. et al., 2018). Of note, E-cadherin inhibits EMT, thus reduced E-cadherin expression indicates that “stemness” is increasing in cancer cells. HDAC and the 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) dual inhibitor JMF3086 restores E-cadherin expression and attenuates vimentin expression and stemness in NSCLC, which recovers sensitivity to gefitinib which is an epidermal growth factor receptor (EFGR) tyrosine kinase inhibitor (TKI) (Weng et al., 2019).
Cancer stem cells are hard to eradicate and prone to drug-resistance. HDAC3 interacts with p53 and forms complexes with tumor antigens melanoma antigen family A2 (MAGE-A2), establishing the resistance of melanoma cells to chemotherapeutic agents (Monte et al., 2006). In refractory and recurrent leukemia, HDAC8-selective inhibitor significantly restores acetylation and p53 activity, inducing apoptosis of AML cells but not of normal hematopoietic stem cells (Qi et al., 2015). In addition, SIRT1 inhibition increases the efficiency of BCR-ABL TKI imatinib mesylate to eliminate quiescent leukemia stem cells by reactivating p53 (Li et al., 2012).
Sirtuins are also closely involved in aging-related oncogene expression. Both SIRT6 and SIRT7 modulate long interspersed elements-1 (LINE-1, L1) expression and retrotransposition. SIRT6 mono-ADP-ribosylates the Krüppel-associated box domain-associated protein 1 (KAP1/TRIM28) and facilitates the KAP1 interaction with HP1α, resulting in the packaging of L1 elements into heterochromatin. SIRT7 directly binds L1 elements and promotes L1 sequences association with the nuclear lamina protein (Lamin A/C) by deacetylating H3K18 (Van Meter et al., 2014; Vazquez et al., 2019). These repressive functions of L1 highlight the protective roles of SIRTs on genome stability through preventing retrotransposition events. SIRT6 and SIRT7 deficiency result in aberrant heterochromatin and L1 activation especially in age-related diseases. Nucleoside reverse-transcriptase inhibitors can reverse a SIRT6 and SIRT7 deficiency by upregulating different immune signalings, such as the type I IFN pathway and cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway, respectively (Simon et al., 2019; Bi et al., 2020; Figure 4).
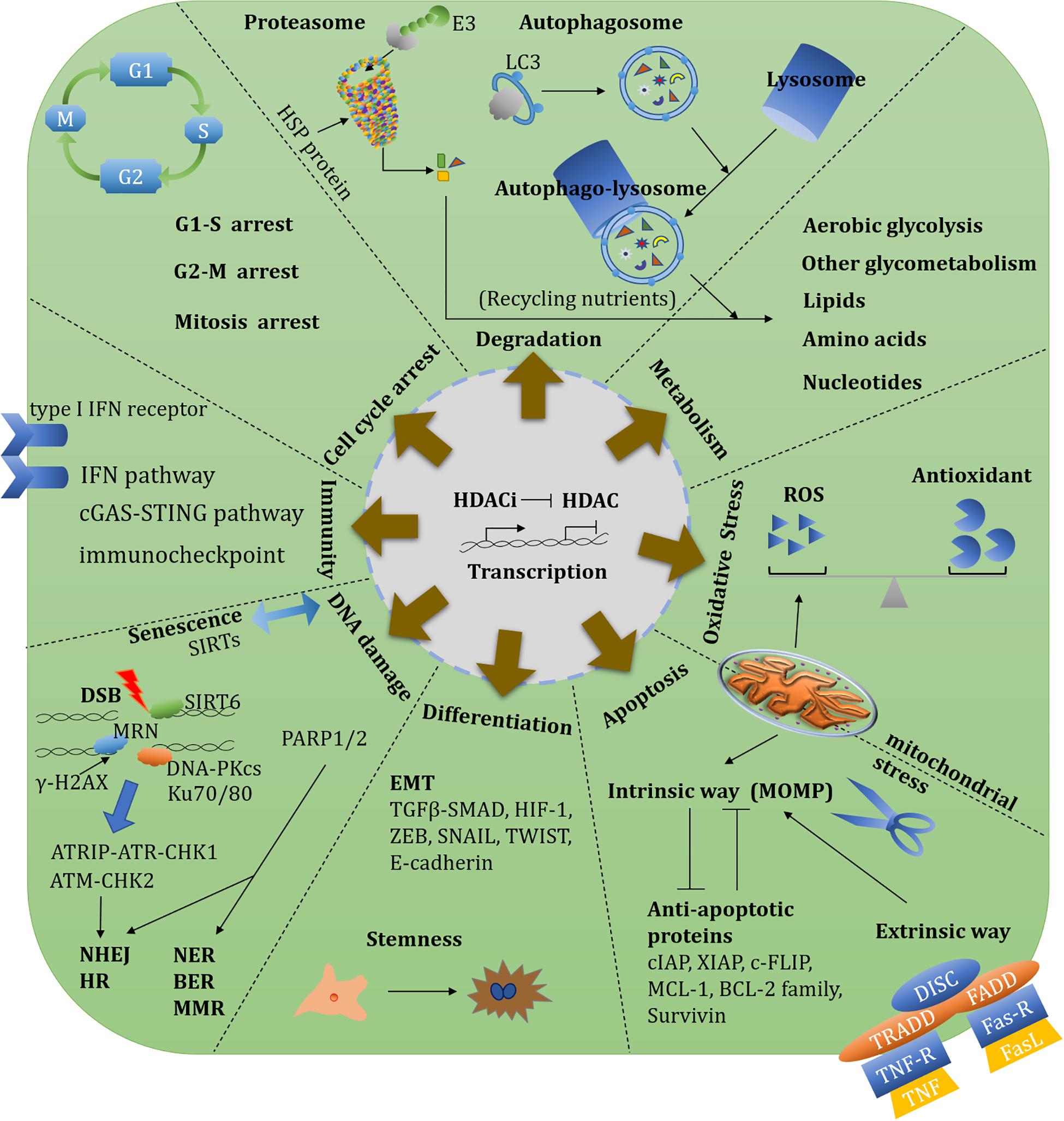
Figure 4. Overview of HDAC-involved biological functions and therapeutic targets. An overview of HDAC-involved biological functions including transcription, metabolism, oxidative stress, redox, protein degradation, cell cycle, DNA damage repair, apoptosis, angiogenesis, EMT, immunity, and stemness. There diverse functions could establish single or synergistic therapeutic targets.
HDAC Inhibitors in Cancer Therapy
In the 1970s, sodium butyrate was discovered to transform red leukemia cells into normal cells, and to resynthesize hemoglobin. This process was accompanied by strong histone hyperacetylation, and resulted in the discovery of the first HDACi (Ginsburg et al., 1973; Riggs et al., 1977; Vidali et al., 1978). In Tsuji et al. (1976) isolated the first natural HDACi, TSA, which was derived from Streptomyces hygroscopicus. Following the discovery of TSA, trapoxin was isolated from fungi and also found to act as an HDACi (Itazaki et al., 1990). A number of natural inhibitors have since been extracted from fungi, marine life, and plants that contain sulfur, polyphenol, flavonoid, terpenoid, selenium, and other organic molecules (Newkirk et al., 2009; Lascano et al., 2018; Singh et al., 2018). At present, HDACi are mainly divided into four categories following the FDA approval: (i) hydroxamic acids or hydroxamates, such as SAHA, panobinostat and belinostat; (ii) cyclic peptides, including depsipeptide; (iii) benzamides, such as chidamide; and (iv) short-chain fatty acids, including VPA (Li and Zhu, 2014; Seto and Yoshida, 2014; Li and Seto, 2016; Table 3).
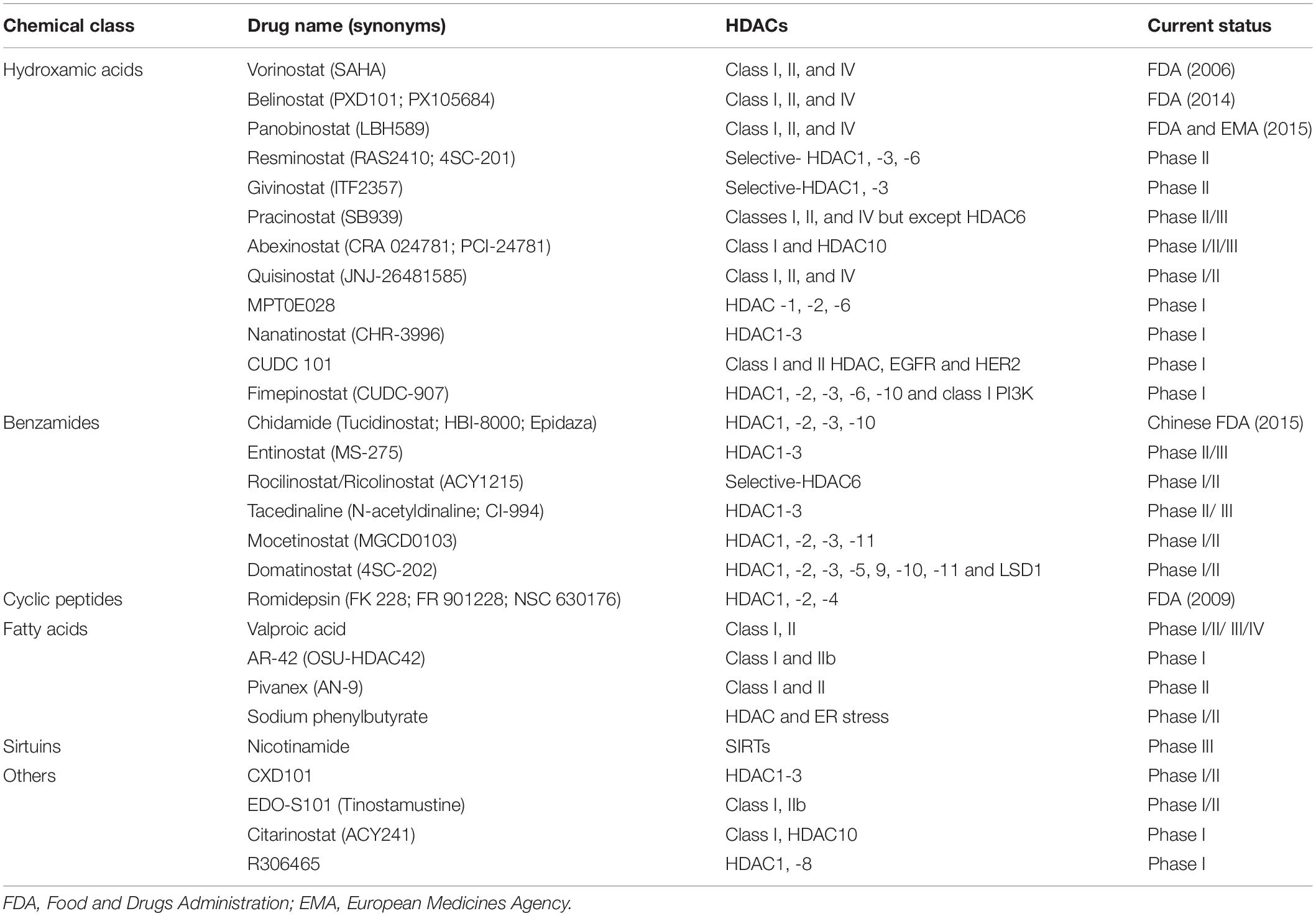
Table 3. Current clinical trials involving the use of HDAC inhibitors to treat cancer.
Efficiency of HDACi
From preclinical studies to clinical trials, HDACi have demonstrated powerful therapeutic effects in various cancers. HDACi can significantly attenuate tumor burden by limiting tumor growth and restraining aberrantly proliferated vessels (Guerriero et al., 2017). HDACi can also induce DNA damage, cell cycle arrest, apoptosis and autophagy to promote cancer cell death mentioned above. Some novel SIRT inhibitors, such as MC2494, MHY2245, MHY2256, tenovin-6, and YC8-02, also perform diverse anti-tumor activities through mediating apoptosis or autophagy (Carafa et al., 2018, 2019; De et al., 2018; Tae et al., 2018, 2020; Li M. et al., 2019; Igase et al., 2020).
Activation of the immune response by HDACi could also be an effective innate method to prevent cancer relapse when administered in a regimen with immunotherapeutic (Guerriero et al., 2017; Wang X. et al., 2020). The class IIa HDACi TMP195 efficiently improves the durability of tumor reduction in breast cancer by strengthening the phagocytic role of macrophages that are involved in the IFNγ axis; it also activates the adaptive anti-tumor immune response. Upon immune checkpoint blockade, TMP195 combined with an anti-programmed cell death-1 (PD-1) regimen could significantly reduce the tumor volume and induce a durable response in breast cancer (Guerriero et al., 2017). Myeloid-derived suppressor cells (MDSCs) suppress T-cell functions and promote tumor metastasis via the formation of an immunosuppressive tumor microenvironment (Azzaoui et al., 2016; Betsch et al., 2018). Entinostat exhibits remarkable curative effects when combined with PD-1 and cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) blockade; it has been shown to significantly reduce the number of MDSCs (Kim et al., 2014). 5-azacytidine combined with entinostat can also suppress MDSCs by downregulating C-C chemokine receptor type 2 (CCR2) and C-X-C chemokine receptor type 2 (CXCR2) expressions that ultimately, stimulates MDSC differentiation into a macrophage-like phenotype (Lu Z. et al., 2020). In human epidermal growth-factor receptor 2-positive (HER2+) breast cancer, panobinostat combined with trastuzumab (anti-HER2) stimulates the release of CXCR3-reactive chemokines and enhances the recruitment of tumor-associated natural killer (NK) cells to achieve eradication of tumors (Medon et al., 2017).
The first FDA-approved HDAC inhibitor, SAHA, belongs to the hydroxamic acid class, approved to treat patients with cutaneous T cell lymphoma (CTCL). In many clinical trials, SAHA has proven effective against advanced and refractory tumors, alone or in combination with other inhibitors. Subsequently, the cyclic peptide romidepsin was approved by the FDA in 2009 to treat CTCL. Panobinostat and belinostat were both approved by the FDA in 2014 to treat peripheral T cell lymphoma (PTCL), with belinostat gaining additional approval from the European Medicines Agency (EMA). Both panobinostat and belinostat are classified as hydroxamic acids. Meanwhile, chidamide has become the first benzamide HDACi to be approved by the China Food and Drug Administration(CFDA) in 2015, for the treatment of relapsed and refractory PTCL1,2,3,4. Most SIRT inhibitors still remain in the preclinical stages. So far, only nicotinamide (vitamin B3) has been used to treat cancer in clinical trials (e.g., NCT02416739 and NCT00033436). Nicotinamide has shown a potential role in inhibiting non-melanoma skin cancers that are principally generated by UV (Chen et al., 2015). Compared to other HDACi, nicotinamide exhibits the most catalytic sites: it is predominantly sensitive to acetylation sites in nuclear proteins that are involved in diverse biological processes (Scholz et al., 2015).
Application of Selective Inhibitors and Combination Therapy
With the development of HDACi, numerous clinical trials are ongoing or completed currently for cancer therapy. Many HDACi have already been approved for hematological malignancies and lymphomas, while clinical studies are ongoing for refractory, advanced and recurrent solid tumors (Table 4). For instance, the use of the HDAC6-selective inhibitor ACY1215 (also named rocilinostat or ricolinostat) as a regimen for relapsed or refractory lymphoma and MM is currently in phase I/II clinical trials [Lymphoma (NCT02091063), MM (NCT01323751, NCT01583283, NCT01997840)]. HDACi with multiple targets have also been developed and tested in clinical trials, such as the dual HDAC and phosphoinositide-3 kinase (PI3K) inhibitor CUDC-907 (also called fimepinostat), which has been reported to inhibit Myc transcriptional expression and reduce Myc-mediated proliferation of multiple cancer cell lines (Pei et al., 2016; Kotian et al., 2017; Pal et al., 2018; Guo et al., 2019). The safety, tolerability and efficacy of CUDC-907 has been assessed in phase I/II trials (Younes et al., 2016). CUDC-101 is another multiple-target inhibitor that blocks HDACs, the epidermal growth factor receptor (EGFR) and HER2 in head and neck squamous cell cancer (Galloway et al., 2015).
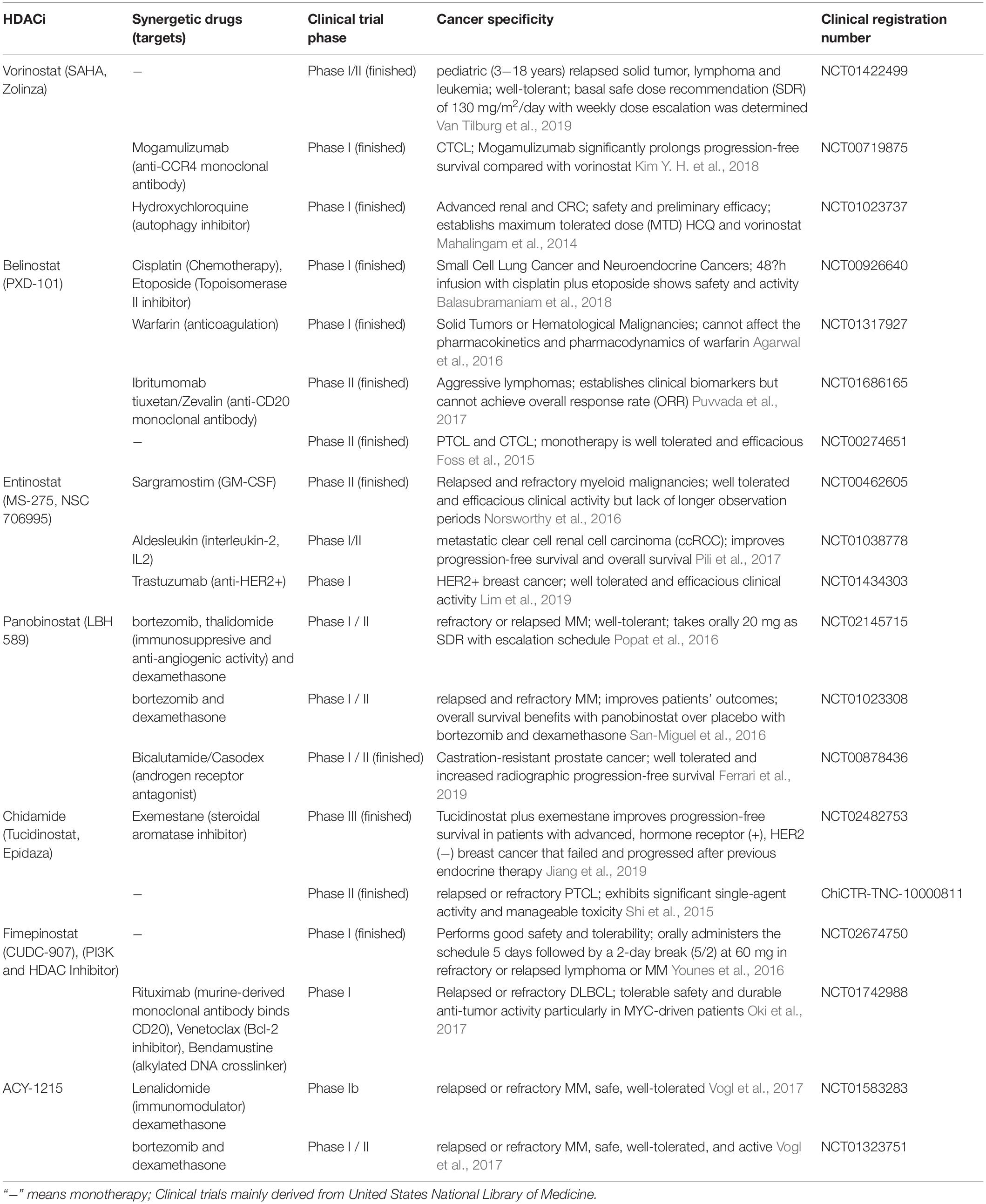
Table 4. Clinical trials investigating the single-agents or combined therapies in HDACi and other anti-neoplastic drugs.
Combination drugs can inhibit tumorigenesis from different aspects. A clinical phase I trial of SAHA combined with the autophagy inhibitor MLN9708 shows potential for this regimen in advanced p53-mutant malignancies (NCT02042989). Hydroxychloroquine (HCQ) is a common autophagy-targeting reagent that has been used in clinical research (Mahalingam et al., 2014). Several phase I/II clinical studies (e.g., NCT02316340 and NCT01023737) have thus tested the safety, tolerability and pharmacological efficacy of SAHA combined with HCQ in solid tumors. A phase I/II trial targeting BTZ-resistant MM cancer has found that a synergistic regimen of using ricolinostat and dexamethasone could be safe and well-tolerated in affected patients (Vogl et al., 2017). A recent phase III clinical trial found that a combination of chidamide and exemestane has therapeutic potential for postmenopausal patients with advanced, hormone receptor-positive (HR+), HER2– breast cancer and who have failed to respond to endocrine therapy (Jiang et al., 2019). Combining the SIRT1 inhibitor Ex527 with the WEE1 inhibitor MK-1775 produces efficient effects in lung cancers by impairing HR repair and mitotic catastrophe-associated apoptosis (Chen et al., 2017).
The overall survival (OS) and progression-free survival (PFS) are common and quite important indicators in the clinical trials. A phase I/II clinical study targets patients with metastatic clear cell renal cell carcinoma (ccRCC) by treatment with entinostat and high-dose interleukin-2 (IL2) that downregulates forkhead box P3 (Foxp3) expression and function of regulatory T cells (Treg). The median PFS reaches 13.8 months, and the median OS is 65.3 months (Pili et al., 2017) [NCT01038778]. A phase III trial enrolled 768 patients with relapsed MM exhibits that panobinostat shows the median OS of panobinostat (40.3 months) versus that of placebo (35.8 months) based on the existing treatments of bortezomib and dexamethasone. And patients who had received previous regimens such as immunomodulatory drug and bortezomib, median OS was only 25.5 months when received panobinostat, bortezomib, and dexamethasone versus that was merely 19.5 months who received placebo (San-Miguel et al., 2016) [NCT01023308]. These HDACi significantly exhibit potential median OS and PFS for patients with advanced tumors.
Limitations of HDACi
However, in a randomized, double-blind and placebo-controlled phase III trial, vorinostat did not improve OS and could not be recommended as a therapy as a second-line or third-line drug for patients with advanced malignant pleural mesothelioma (Krug et al., 2015). Moreover, a phase III study that recruited 370 patients with Sézary syndrome or relapsed or refractory mycosis fungoides in CTCL from different countries of the world showed that the overall response rate to vorinostat was less efficient than that to mogamulizumab, a novel monoclonal antibody against CCR4 that significantly prolongs PFS (Kim Y. H. et al., 2018). The overall response to vorinostat in this study was significantly lower than reported in a previous study (Olsen et al., 2007). Besides, in a phase II trial, mocetinostat did not reverse chemoresistance in patients with previous gemcitabine-resistant leiomyosarcoma and could not significantly prolong the median PFS of patients (Choy et al., 2019).
Sirtuins constitute a relatively unique class of HDACs; as such, SIRT modulators are attracting great interest in the research community (Yang et al., 2017; Dai et al., 2018; Wang Y. et al., 2019). A novel SIRT7 inhibitor has been identified to inhibit tumor growth by blockade of the direct interaction of SIRT7 and p53 (Vakhrusheva et al., 2008; Kim J. H. et al., 2019). However, there are also several reports demonstrating an indirect interaction between SIRT7 and p53 (Barber et al., 2012; Lu Y. F. et al., 2020; Yu et al., 2020). Thus, it is urgent to develop specific SIRT inhibitors for cancer therapy according to reasonable mechanisms. It is worth noting that SIRT activators and sirtuin-activating compounds (STACs) have been developed and studied in clinical trials to investigate their anti-aging, anti-inflammatory and metabolic regulatory effects (Howitz et al., 2003; Hubbard et al., 2013; Sinclair and Guarente, 2014; Bonkowski and Sinclair, 2016; Carafa et al., 2016; Huang et al., 2018b). Nevertheless, the SIRT1 activator SRT2183 has also been shown to inhibit growth and to promote cell death by causing ER stress in glioma cells (Ye et al., 2019).
Besides, most of clinical trials of HDACi have reported many adverse effects, including bleeding caused by different grades of thrombocytopenia, susceptibility to infection caused by neutropenia, anemia caused by hemoglobin reduction, arrhythmia, myocardial hypertrophy, neurotoxicity, and gastrointestinal toxicity such as nausea, vomiting, fatigue, diarrhea as well as electrolyte disturbance such as hypophosphatemia and hyponatremia. The most adverse effect reported is cell death caused by continuous cytotoxicity (Medina et al., 1997; Zhu et al., 2001b), as these agents also kill immortalized and normal cells from different tissues (Lee et al., 1996; Burgess et al., 2004).
Discussion and Perspectives
To date, only five HDACi have been approved by health authorities globally. Numerous clinical trials are currently evaluating the safety, application, and therapeutic benefits of HDAC inhibition when treating cancers, neurological disorders and other human diseases. Thus far, it is clear that FDA-approved HDACi have both beneficial and adverse effects. These effects might be accompanied by genomic instability, abnormal gene transcription, interference with chaperone protein function and free radical generation, which currently limit the therapeutic potential of HDACi.
Analyses of HDAC structural conformations and molecular mechanisms are necessary to improve treatment development. Complicating the application of HDACi is that the modification of the substrates by HDAC has spatio-temporal and tissue specificity. Consequently, dose-dependent and time-dependent treatments have different effects on gene expression regulation, various protein PTMs and chromatin remodeling. For now, combining different drugs to inhibit pathways such as tumor proliferation or angiogenesis, or to stimulate apoptosis requires more consideration.
Isolating the anti-cancer effects of HDACi and then synthesizing molecules with highly specific targets could be a promising avenue for cancer treatment in the future. Further investigation into combination treatments involving oncoprotein inhibitors and specific HDACi is also warranted. Overall, it seems that combination therapies have the advantage of reducing drug toxicity and lowering dose demand. SIRT protective factors should also be considered. Pending additional work to clarify HDACi that target specific HDACs or can be combined with other treatments, such as DNA methylation inhibitors or autophagy inhibitors, could be of great benefit to patients with cancers that have failed to respond to conventional treatments.
Author Contributions
GL wrote the primary manuscript and YT revised the manuscript. W-GZ conceived and designed the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funidng
This work was supported by the National Key R&D Program of China (Grant No. 2017YFA0503900), Science and Technology Program of Guangdong Province in China (2017B030301016), Shenzhen Municipal Commission of Science and Technology Innovation (JCYJ20170818092450901), and Discipline Construction Funding of Shenzhen [(2016)1452].
Acknowledgments
We are grateful to Dr. Jessica Tamanini (Shenzhen University) for proofreading the manuscript. We apologize to those whose works are not cited here due to limited space.
Footnotes
- ^ https://www.accessdata.fda.gov/scripts/cder/daf/
- ^ https://www.cancer.gov/about-cancer/treatment/drugs
- ^ https://www.ema.europa.eu/en
- ^ https://www.nmpa.gov.cn/
References
Adams, C. M., and Eischen, C. M. (2016). Histone deacetylase inhibition reveals a tumor-suppressive function of MYC-regulated miRNA in breast and lung carcinoma. Cell Death Differ. 23, 1312–1321. doi: 10.1038/cdd.2016.9
Adams, C. M., Hiebert, S. W., and Eischen, C. M. (2016). Myc induces miRNA-mediated apoptosis in response to HDAC inhibition in hematologic malignancies. Cancer Res. 76, 736–748. doi: 10.1158/0008-5472.can-15-1751
Agarwal, N., Mcpherson, J. P., Bailey, H., Gupta, S., Werner, T. L., Reddy, G., et al. (2016). A phase I clinical trial of the effect of belinostat on the pharmacokinetics and pharmacodynamics of warfarin. Cancer Chemother. Pharmacol. 77, 299–308. doi: 10.1007/s00280-015-2934-1
Ago, T., Liu, T., Zhai, P., Chen, W., Li, H., Molkentin, J. D., et al. (2008). A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 133, 978–993. doi: 10.1016/j.cell.2008.04.041
Ahn, B. H., Kim, H. S., Song, S., Lee, I. H., Liu, J., Vassilopoulos, A., et al. (2008). A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 14447–14452. doi: 10.1073/pnas.0803790105
Alexandrova, E. M., Yallowitz, A. R., Li, D., Xu, S., Schulz, R., Proia, D. A., et al. (2015). Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 523, 352–356. doi: 10.1038/nature14430
Ali, I., Ruiz, D. G., Ni, Z., Johnson, J. R., Zhang, H., Li, P. C., et al. (2019). Crosstalk between RNA Pol II C-terminal domain acetylation and phosphorylation via RPRD Proteins. Mol. Cell 74, 1164–1174.e4.
Allfrey, V. G., Faulkner, R., and Mirsky, A. E. (1964). Acetylation and methylation of histones and their possible role in the regulation of Rna synthesis. Proc. Natl. Acad. Sci. U.S.A. 51, 786–794. doi: 10.1073/pnas.51.5.786
Amano, H., Chaudhury, A., Rodriguez-Aguayo, C., Lu, L., Akhanov, V., Catic, A., et al. (2019). Telomere dysfunction induces sirtuin repression that drives telomere-dependent disease. Cell Metab. 29, 1274–1290.e9.
Anastas, J. N., Zee, B. M., Kalin, J. H., Kim, M., Guo, R., Alexandrescu, S., et al. (2019). Re-programing Chromatin with a Bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG. Cancer Cell 36, 528–544.e10.
Anderson, K. A., Huynh, F. K., Fisher-Wellman, K., Stuart, J. D., Peterson, B. S., Douros, J. D., et al. (2017). SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion. Cell Metab. 25, 838–855.e15.
Aramsangtienchai, P., Spiegelman, N. A., He, B., Miller, S. P., Dai, L., Zhao, Y., et al. (2016). HDAC8 Catalyzes the Hydrolysis of Long Chain Fatty Acyl Lysine. ACS Chem. Biol. 11, 2685–2692. doi: 10.1021/acschembio.6b00396
Ardestani, P. M., and Liang, F. (2012). Sub-cellular localization, expression and functions of Sirt6 during the cell cycle in HeLa cells. Nucleus 3, 442–451. doi: 10.4161/nucl.21134
Ayer, D. E. (1999). Histone deacetylases: transcriptional repression with SINers and NuRDs. Trends Cell Biol. 9, 193–198. doi: 10.1016/s0962-8924(99)01536-6
Azagra, A., Roman-Gonzalez, L., Collazo, O., Rodriguez-Ubreva, J., De Yebenes, V. G., Barneda-Zahonero, B., et al. (2016). In vivo conditional deletion of HDAC7 reveals its requirement to establish proper B lymphocyte identity and development. J. Exp. Med. 213, 2591–2601. doi: 10.1084/jem.20150821
Azzaoui, I., Uhel, F., Rossille, D., Pangault, C., Dulong, J., Le Priol, J., et al. (2016). T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 128, 1081–1092. doi: 10.1182/blood-2015-08-662783
Bagchi, R. A., Ferguson, B. S., Stratton, M. S., Hu, T., Cavasin, M. A., Sun, L., et al. (2018). HDAC11 suppresses the thermogenic program of adipose tissue via BRD2. JCI Insight 3:e120159.
Baker, A., Gregory, G. P., Verbrugge, I., Kats, L., Hilton, J. J., Vidacs, E., et al. (2016). The CDK9 inhibitor dinaciclib exerts potent apoptotic and antitumor effects in preclinical models of mll-rearranged acute myeloid leukemia. Cancer Res. 76, 1158–1169. doi: 10.1158/0008-5472.can-15-1070
Balasubramaniam, S., Redon, C. E., Peer, C. J., Bryla, C., Lee, M. J., Trepel, J. B., et al. (2018). Phase I trial of belinostat with cisplatin and etoposide in advanced solid tumors, with a focus on neuroendocrine and small cell cancers of the lung. Anticancer Drugs 29, 457–465.
Ban, J., Aryee, D. N., Fourtouna, A., Van Der Ent, W., Kauer, M., Niedan, S., et al. (2014). Suppression of deacetylase SIRT1 mediates tumor-suppressive NOTCH response and offers a novel treatment option in metastatic Ewing sarcoma. Cancer Res. 74, 6578–6588. doi: 10.1158/0008-5472.can-14-1736
Bantscheff, M., Hopf, C., Savitski, M. M., Dittmann, A., Grandi, P., Michon, A. M., et al. (2011). Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29, 255–265. doi: 10.1038/nbt.1759
Bao, J., Lu, Z., Joseph, J. J., Carabenciov, D., Dimond, C. C., Pang, L., et al. (2010). Characterization of the murine SIRT3 mitochondrial localization sequence and comparison of mitochondrial enrichment and deacetylase activity of long and short SIRT3 isoforms. J. Cell. Biochem. 110, 238–247.
Bao, X., Liu, Z., Zhang, W., Gladysz, K., Fung, Y. M. E., Tian, G., et al. (2019). Glutarylation of histone H4 Lysine 91 regulates chromatin dynamics. Mol. Cell 76, 660–675.e9.
Bao, Y., Tong, L., Song, B., Liu, G., Zhu, Q., Lu, X., et al. (2020). UNG2 deacetylation confers cancer cell resistance to hydrogen peroxide-induced cytotoxicity. Free Radic. Biol. Med. 160, 403–417. doi: 10.1016/j.freeradbiomed.2020.06.010
Barber, M. F., Michishita-Kioi, E., Xi, Y., Tasselli, L., Kioi, M., Moqtaderi, Z., et al. (2012). SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 487, 114–118. doi: 10.1038/nature11043
Barker, H. E., Paget, J. T., Khan, A. A., and Harrington, K. J. (2015). The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer 15, 409–425. doi: 10.1038/nrc3958
Bertos, N. R., Gilquin, B., Chan, G. K., Yen, T. J., Khochbin, S., and Yang, X. J. (2004). Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J. Biol. Chem. 279, 48246–48254. doi: 10.1074/jbc.m408583200
Betsch, A., Rutgeerts, O., Fevery, S., Sprangers, B., Verhoef, G., Dierickx, D., et al. (2018). Myeloid-derived suppressor cells in lymphoma: the good, the bad and the ugly. Blood Rev. 32, 490–498. doi: 10.1016/j.blre.2018.04.006
Bhardwaj, A., and Das, S. (2016). SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc. Natl. Acad. Sci. U.S.A. 113, E538–E547.
Bhaskara, S., Chyla, B. J., Amann, J. M., Knutson, S. K., Cortez, D., Sun, Z. W., et al. (2008). Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 30, 61–72. doi: 10.1016/j.molcel.2008.02.030
Bhaskara, S., Knutson, S. K., Jiang, G., Chandrasekharan, M. B., Wilson, A. J., Zheng, S., et al. (2010). Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 18, 436–447. doi: 10.1016/j.ccr.2010.10.022
Bhatia, S., Krieger, V., Groll, M., Osko, J. D., Ressing, N., Ahlert, H., et al. (2018). Discovery of the first-in-class dual histone deacetylase-proteasome inhibitor. J. Med. Chem. 61, 10299–10309. doi: 10.1021/acs.jmedchem.8b01487
Bi, S., Liu, Z., Wu, Z., Wang, Z., Liu, X., Wang, S., et al. (2020). SIRT7 antagonizes human stem cell aging as a heterochromatin stabilizer. Protein Cell 11, 483–504. doi: 10.1007/s13238-020-00728-4
Bitler, B. G., Wu, S., Park, P. H., Hai, Y., Aird, K. M., Wang, Y., et al. (2017). ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 19, 962–973. doi: 10.1038/ncb3582
Blackford, A. N., and Jackson, S. P. (2017). ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817. doi: 10.1016/j.molcel.2017.05.015
Blank, M. F., Chen, S., Poetz, F., Schnolzer, M., Voit, R., and Grummt, I. (2017). SIRT7-dependent deacetylation of CDK9 activates RNA polymerase II transcription. Nucleic Acids Res. 45, 2675–2686. doi: 10.1093/nar/gkx053
Bonkowski, M. S., and Sinclair, D. A. (2016). Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 17, 679–690. doi: 10.1038/nrm.2016.93
Boucheron, N., Tschismarov, R., Goeschl, L., Moser, M. A., Lagger, S., Sakaguchi, S., et al. (2014). CD4(+) T cell lineage integrity is controlled by the histone deacetylases HDAC1 and HDAC2. Nat. Immunol. 15, 439–448. doi: 10.1038/ni.2864
Bowman, C. J., Ayer, D. E., and Dynlacht, B. D. (2014). Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nat. Cell Biol. 16, 1202–1214. doi: 10.1038/ncb3062
Boyault, C., Gilquin, B., Zhang, Y., Rybin, V., Garman, E., Meyer-Klaucke, W., et al. (2006). HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 25, 3357–3366. doi: 10.1038/sj.emboj.7601210
Bradley, E. W., Carpio, L. R., Olson, E. N., and Westendorf, J. J. (2015). Histone deacetylase 7 (Hdac7) suppresses chondrocyte proliferation and beta-catenin activity during endochondral ossification. J. Biol. Chem. 290, 118–126. doi: 10.1074/jbc.m114.596247
Brautigam, L., Jensen, L. D., Poschmann, G., Nystrom, S., Bannenberg, S., Dreij, K., et al. (2013). Glutaredoxin regulates vascular development by reversible glutathionylation of sirtuin 1. Proc. Natl. Acad. Sci. U.S.A. 110, 20057–20062. doi: 10.1073/pnas.1313753110
Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., et al. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. doi: 10.1016/s0092-8674(00)80595-4
Brunet, A., Sweeney, L. B., Sturgill, J. F., Chua, K. F., Greer, P. L., Lin, Y., et al. (2004). Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015. doi: 10.1126/science.1094637
Buggy, J. J., Sideris, M. L., Mak, P., Lorimer, D. D., Mcintosh, B., and Clark, J. M. (2000). Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem. J. 350(Pt 1), 199–205. doi: 10.1042/bj3500199
Burgess, A., Ruefli, A., Beamish, H., Warrener, R., Saunders, N., Johnstone, R., et al. (2004). Histone deacetylase inhibitors specifically kill nonproliferating tumour cells. Oncogene 23, 6693–6701. doi: 10.1038/sj.onc.1207893
Cadot, B., Brunetti, M., Coppari, S., Fedeli, S., De Rinaldis, E., Dello Russo, C., et al. (2009). Loss of histone deacetylase 4 causes segregation defects during mitosis of p53-deficient human tumor cells. Cancer Res. 69, 6074–6082. doi: 10.1158/0008-5472.can-08-2796
Cameron, E. E., Bachman, K. E., Myohanen, S., Herman, J. G., and Baylin, S. B. (1999). Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 21, 103–107. doi: 10.1038/5047
Cao, J., Sun, L., Aramsangtienchai, P., Spiegelman, N. A., Zhang, X., Huang, W., et al. (2019). HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc. Natl. Acad. Sci. U.S.A. 116, 5487–5492. doi: 10.1073/pnas.1815365116
Cao, L. L., Shen, C., and Zhu, W. G. (2016). Histone modifications in DNA damage response. Sci. China Life Sci. 59, 257–270. doi: 10.1007/s11427-016-5011-z
Carafa, V., Nebbioso, A., Cuomo, F., Rotili, D., Cobellis, G., Bontempo, P., et al. (2018). RIP1-HAT1-SIRT complex identification and targeting in treatment and prevention of cancer. Clin. Cancer Res. 24, 2886–2900. doi: 10.1158/1078-0432.ccr-17-3081
Carafa, V., Poziello, A., Della Torre, L., Giovannelli, P., Di Donato, M., Safadeh, E., et al. (2019). Enzymatic and biological characterization of novel sirtuin modulators against cancer. Int. J. Mol. Sci. 20:5654. doi: 10.3390/ijms20225654
Carafa, V., Rotili, D., Forgione, M., Cuomo, F., Serretiello, E., Hailu, G. S., et al. (2016). Sirtuin functions and modulation: from chemistry to the clinic. Clin. Epigenetics 8:61.
Castaneda, C. A., Wolfson, N. A., Leng, K. R., Kuo, Y. M., Andrews, A. J., and Fierke, C. A. (2017). HDAC8 substrate selectivity is determined by long- and short-range interactions leading to enhanced reactivity for full-length histone substrates compared with peptides. J. Biol. Chem. 292, 21568–21577. doi: 10.1074/jbc.m117.811026
Cencioni, C., Spallotta, F., Savoia, M., Kuenne, C., Guenther, S., Re, A., et al. (2018). Zeb1-Hdac2-eNOS circuitry identifies early cardiovascular precursors in naive mouse embryonic stem cells. Nat. Commun. 9:1281.
Chalkiadaki, A., and Guarente, L. (2015). The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 15, 608–624. doi: 10.1038/nrc3985
Chang, S., Young, B. D., Li, S., Qi, X., Richardson, J. A., and Olson, E. N. (2006). Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 126, 321–334. doi: 10.1016/j.cell.2006.05.040
Chatterjee, T. K., Idelman, G., Blanco, V., Blomkalns, A. L., Piegore, M. G. Jr., Weintraub, D. S., et al. (2011). Histone deacetylase 9 is a negative regulator of adipogenic differentiation. J. Biol. Chem. 286, 27836–27847. doi: 10.1074/jbc.m111.262964
Chen, A. C., Martin, A. J., Choy, B., Fernandez-Penas, P., Dalziell, R. A., Mckenzie, C. A., et al. (2015). A Phase 3 randomized trial of nicotinamide for skin-cancer chemoprevention. N. Engl. J. Med. 373, 1618–1626.
Chen, G., Zhang, B., Xu, H., Sun, Y., Shi, Y., Luo, Y., et al. (2017). Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis. Oncogene 36, 6863–6872. doi: 10.1038/onc.2017.297
Chen, L., Zhang, Z., Hoshino, A., Zheng, H. D., Morley, M., Arany, Z., et al. (2019). NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 1, 404–415. doi: 10.1038/s42255-019-0043-x
Chen, X., Barozzi, I., Termanini, A., Prosperini, E., Recchiuti, A., Dalli, J., et al. (2012). Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. U.S.A. 109, E2865–E2874.
Chen, X. F., Tian, M. X., Sun, R. Q., Zhang, M. L., Zhou, L. S., Jin, L., et al. (2018). SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 19:e45124.
Choi, S. J., Lee, H. C., Kim, J. H., Park, S. Y., Kim, T. H., Lee, W. K., et al. (2016). HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I. EMBO J. 35, 429–442. doi: 10.15252/embj.201592586
Choy, E., Ballman, K., Chen, J., Dickson, M. A., Chugh, R., George, S., et al. (2019). Corrigendum to “SARC018_SPORE02: phase II Study of Mocetinostat Administered with Gemcitabine for Patients with Metastatic Leiomyosarcoma with Progression or Relapse following Prior Treatment with Gemcitabine-Containing Therapy”. Sarcoma 2019:7608743.
Cimen, H., Han, M. J., Yang, Y., Tong, Q., Koc, H., and Koc, E. C. (2010). Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 49, 304–311. doi: 10.1021/bi901627u
Cohen, H. Y., Miller, C., Bitterman, K. J., Wall, N. R., Hekking, B., Kessler, B., et al. (2004). Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305, 390–392. doi: 10.1126/science.1099196
Conti, C., Leo, E., Eichler, G. S., Sordet, O., Martin, M. M., Fan, A., et al. (2010). Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 70, 4470–4480. doi: 10.1158/0008-5472.can-09-3028
Cosenza, M., Civallero, M., Marcheselli, L., Sacchi, S., and Pozzi, S. (2017). Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine. Apoptosis 22, 827–840. doi: 10.1007/s10495-017-1364-4
Costa-Machado, L. F., Martin-Hernandez, R., Sanchez-Luengo, M. A., Hess, K., Vales-Villamarin, C., Barradas, M., et al. (2018). Sirt1 protects from K-Ras-driven lung carcinogenesis. EMBO Rep. 19:e43879.
Daenthanasanmak, A., Iamsawat, S., Chakraborty, P., Nguyen, H. D., Bastian, D., Liu, C., et al. (2019). Targeting Sirt-1 controls GVHD by inhibiting T-cell allo-response and promoting Treg stability in mice. Blood 133, 266–279. doi: 10.1182/blood-2018-07-863233
Dai, H., Sinclair, D. A., Ellis, J. L., and Steegborn, C. (2018). Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharmacol. Ther. 188, 140–154. doi: 10.1016/j.pharmthera.2018.03.004
Dai, X., Xin, Y., Xu, W., Tian, X., Wei, X., and Zhang, H. (2020). CBP-mediated Slug acetylation stabilizes Slug and promotes EMT and migration of breast cancer cells. Sci. China Life Sci. doi: 10.1007/s11427-020-1736-5 [Epub ahead of print].
Dai, Y., Chen, S., Kramer, L. B., Funk, V. L., Dent, P., and Grant, S. (2008). Interactions between bortezomib and romidepsin and belinostat in chronic lymphocytic leukemia cells. Clin. Cancer Res. 14, 549–558. doi: 10.1158/1078-0432.ccr-07-1934
Danko, C. G., Hyland, S. L., Core, L. J., Martins, A. L., Waters, C. T., Lee, H. W., et al. (2015). Identification of active transcriptional regulatory elements from GRO-seq data. Nat. Methods 12, 433–438. doi: 10.1038/nmeth.3329
Dawson, M. A., and Kouzarides, T. (2012). Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27. doi: 10.1016/j.cell.2012.06.013
De, U., Son, J. Y., Sachan, R., Park, Y. J., Kang, D., Yoon, K., et al. (2018). A New synthetic histone deacetylase inhibitor, MHY2256, induces apoptosis and autophagy cell death in endometrial cancer cells via p53 acetylation. Int. J. Mol. Sci. 19:2743. doi: 10.3390/ijms19092743
Dequiedt, F., Martin, M., Von Blume, J., Vertommen, D., Lecomte, E., Mari, N., et al. (2006). New role for hPar-1 kinases EMK and C-TAK1 in regulating localization and activity of class IIa histone deacetylases. Mol. Cell. Biol. 26, 7086–7102. doi: 10.1128/mcb.00231-06
Dhalluin, C., Carlson, J. E., Zeng, L., He, C., Aggarwal, A. K., and Zhou, M. M. (1999). Structure and ligand of a histone acetyltransferase bromodomain. Nature 399, 491–496. doi: 10.1038/20974
Di Giorgio, E., Dalla, E., Franforte, E., Paluvai, H., Minisini, M., Trevisanut, M., et al. (2020). Different class IIa HDACs repressive complexes regulate specific epigenetic responses related to cell survival in leiomyosarcoma cells. Nucleic Acids Res. 48, 646–664. doi: 10.1093/nar/gkz1120
Dioum, E. M., Chen, R., Alexander, M. S., Zhang, Q., Hogg, R. T., Gerard, R. D., et al. (2009). Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 324, 1289–1293. doi: 10.1126/science.1169956
Dovey, O. M., Foster, C. T., Conte, N., Edwards, S. A., Edwards, J. M., Singh, R., et al. (2013). Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 121, 1335–1344. doi: 10.1182/blood-2012-07-441949
Dryden, S. C., Nahhas, F. A., Nowak, J. E., Goustin, A. S., and Tainsky, M. A. (2003). Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol. 23, 3173–3185. doi: 10.1128/mcb.23.9.3173-3185.2003
Du, J., Zhou, Y., Su, X., Yu, J. J., Khan, S., Jiang, H., et al. (2011). Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809. doi: 10.1126/science.1207861
El-Naggar, A. M., Somasekharan, S. P., Wang, Y., Cheng, H., Negri, G. L., Pan, M., et al. (2019). Class I HDAC inhibitors enhance YB-1 acetylation and oxidative stress to block sarcoma metastasis. EMBO Rep. 20:e48375.
Emiliani, S., Fischle, W., Van Lint, C., Al-Abed, Y., and Verdin, E. (1998). Characterization of a human RPD3 ortholog, HDAC3. Proc. Natl. Acad. Sci. U.S.A. 95, 2795–2800. doi: 10.1073/pnas.95.6.2795
Emmett, M. J., and Lazar, M. A. (2019). Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell Biol. 20, 102–115. doi: 10.1038/s41580-018-0076-0
Eom, G. H., Nam, Y. S., Oh, J. G., Choe, N., Min, H. K., Yoo, E. K., et al. (2014). Regulation of acetylation of histone deacetylase 2 by p300/CBP-associated factor/histone deacetylase 5 in the development of cardiac hypertrophy. Circ. Res. 114, 1133–1143. doi: 10.1161/circresaha.114.303429
Eot-Houllier, G., Fulcrand, G., Watanabe, Y., Magnaghi-Jaulin, L., and Jaulin, C. (2008). Histone deacetylase 3 is required for centromeric H3K4 deacetylation and sister chromatid cohesion. Genes Dev. 22, 2639–2644. doi: 10.1101/gad.484108
Etchegaray, J. P., Chavez, L., Huang, Y., Ross, K. N., Choi, J., Martinez-Pastor, B., et al. (2015). The histone deacetylase SIRT6 controls embryonic stem cell fate via TET-mediated production of 5-hydroxymethylcytosine. Nat. Cell Biol. 17, 545–557. doi: 10.1038/ncb3147
Etchegaray, J. P., Zhong, L., Li, C., Henriques, T., Ablondi, E., Nakadai, T., et al. (2019). The histone deacetylase SIRT6 restrains transcription elongation via promoter-proximal pausing. Mol. Cell 75, 683–699.e7.
Falkenberg, K. J., and Johnstone, R. W. (2014). Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 13, 673–691. doi: 10.1038/nrd4360
Fan, W., and Luo, J. (2010). SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol. Cell 39, 247–258. doi: 10.1016/j.molcel.2010.07.006
Fang, J., Ianni, A., Smolka, C., Vakhrusheva, O., Nolte, H., Kruger, M., et al. (2017). Sirt7 promotes adipogenesis in the mouse by inhibiting autocatalytic activation of Sirt1. Proc. Natl. Acad. Sci. U.S.A. 114, E8352–E8361.
Feldman, J. L., Baeza, J., and Denu, J. M. (2013). Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 288, 31350–31356. doi: 10.1074/jbc.c113.511261
Ferrari, A. C., Alumkal, J. J., Stein, M. N., Taplin, M. E., Babb, J., Barnett, E. S., et al. (2019). Epigenetic therapy with panobinostat combined with bicalutamide rechallenge in castration-resistant prostate cancer. Clin. Cancer Res. 25, 52–63. doi: 10.1158/1078-0432.ccr-18-1589
Ferrer, C. M., Lu, T. Y., Bacigalupa, Z. A., Katsetos, C. D., Sinclair, D. A., and Reginato, M. J. (2017). O-GlcNAcylation regulates breast cancer metastasis via SIRT1 modulation of FOXM1 pathway. Oncogene 36, 559–569. doi: 10.1038/onc.2016.228
Filippakopoulos, P., and Knapp, S. (2014). Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 13, 337–356. doi: 10.1038/nrd4286
Finley, L. W., Carracedo, A., Lee, J., Souza, A., Egia, A., Zhang, J., et al. (2011). SIRT3 opposes reprogramming of cancer Cell Metab.olism through HIF1alpha destabilization. Cancer Cell 19, 416–428. doi: 10.1016/j.ccr.2011.02.014
Finnin, M. S., Donigian, J. R., Cohen, A., Richon, V. M., Rifkind, R. A., Marks, P. A., et al. (1999). Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193. doi: 10.1038/43710
Fischer, D. D., Cai, R., Bhatia, U., Asselbergs, F. A., Song, C., Terry, R., et al. (2002). Isolation and characterization of a novel class II histone deacetylase, HDAC10. J. Biol. Chem. 277, 6656–6666. doi: 10.1074/jbc.m108055200
Fiskus, W., Coothankandaswamy, V., Chen, J., Ma, H., Ha, K., Saenz, D. T., et al. (2016). SIRT2 deacetylates and inhibits the peroxidase activity of peroxiredoxin-1 to sensitize breast cancer cells to oxidant stress-inducing agents. Cancer Res. 76, 5467–5478. doi: 10.1158/0008-5472.can-16-0126
Foss, F., Advani, R., Duvic, M., Hymes, K. B., Intragumtornchai, T., Lekhakula, A., et al. (2015). A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 168, 811–819. doi: 10.1111/bjh.13222
Fujisawa, T., and Filippakopoulos, P. (2017). Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 18, 246–262. doi: 10.1038/nrm.2016.143
Fukumoto, T., Park, P. H., Wu, S., Fatkhutdinov, N., Karakashev, S., Nacarelli, T., et al. (2018). Repurposing Pan-HDAC inhibitors for ARID1A-mutated ovarian cancer. Cell Rep. 22, 3393–3400. doi: 10.1016/j.celrep.2018.03.019
Funato, K., Hayashi, T., Echizen, K., Negishi, L., Shimizu, N., Koyama-Nasu, R., et al. (2018). SIRT2-mediated inactivation of p73 is required for glioblastoma tumorigenicity. EMBO Rep. 19:e4558.
Fyodorov, D. V., Zhou, B. R., Skoultchi, A. I., and Bai, Y. (2018). Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 19, 192–206. doi: 10.1038/nrm.2017.94
Galloway, T. J., Wirth, L. J., Colevas, A. D., Gilbert, J., Bauman, J. E., Saba, N. F., et al. (2015). A Phase I Study of CUDC-101, a Multitarget Inhibitor of HDACs, EGFR, and HER2, in Combination with Chemoradiation in Patients with Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 21, 1566–1573. doi: 10.1158/1078-0432.ccr-14-2820
Gammoh, N., Lam, D., Puente, C., Ganley, I., Marks, P. A., and Jiang, X. (2012). Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. U.S.A. 109, 6561–6565. doi: 10.1073/pnas.1204429109
Gao, C., Ho, C. C., Reineke, E., Lam, M., Cheng, X., Stanya, K. J., et al. (2008). Histone deacetylase 7 promotes PML sumoylation and is essential for PML nuclear body formation. Mol. Cell. Biol. 28, 5658–5667. doi: 10.1128/mcb.00874-08
Gao, L., Cueto, M. A., Asselbergs, F., and Atadja, P. (2002). Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 277, 25748–25755. doi: 10.1074/jbc.m111871200
Gao, Y. S., Hubbert, C. C., Lu, J., Lee, Y. S., Lee, J. Y., and Yao, T. P. (2007). Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol. Cell. Biol. 27, 8637–8647. doi: 10.1128/mcb.00393-07
Ginsburg, E., Salomon, D., Sreevalsan, T., and Freese, E. (1973). Growth inhibition and morphological changes caused by lipophilic acids in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 70, 2457–2461. doi: 10.1073/pnas.70.8.2457
Goder, A., Emmerich, C., Nikolova, T., Kiweler, N., Schreiber, M., Kuhl, T., et al. (2018). HDAC1 and HDAC2 integrate checkpoint kinase phosphorylation and cell fate through the phosphatase-2A subunit PR130. Nat. Commun. 9:764.
Gonzalez, F. J., Xie, C., and Jiang, C. (2018). The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 15, 21–32. doi: 10.1038/s41574-018-0096-z
Gorisch, S. M., Wachsmuth, M., Toth, K. F., Lichter, P., and Rippe, K. (2005). Histone acetylation increases chromatin accessibility. J. Cell Sci. 118, 5825–5834. doi: 10.1242/jcs.02689
Greene, K. S., Lukey, M. J., Wang, X., Blank, B., Druso, J. E., Lin, M. J., et al. (2019). SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 116, 26625–26632. doi: 10.1073/pnas.1911954116
Greer, C. B., Tanaka, Y., Kim, Y. J., Xie, P., Zhang, M. Q., Park, I. H., et al. (2015). Histone deacetylases positively regulate transcription through the elongation machinery. Cell Rep. 13, 1444–1455. doi: 10.1016/j.celrep.2015.10.013
Grozinger, C. M., Hassig, C. A., and Schreiber, S. L. (1999). Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. U.S.A. 96, 4868–4873. doi: 10.1073/pnas.96.9.4868
Grozinger, C. M., and Schreiber, S. L. (2000). Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. U.S.A. 97, 7835–7840. doi: 10.1073/pnas.140199597
Grunstein, M. (1997). Histone acetylation in chromatin structure and transcription. Nature 389, 349–352. doi: 10.1038/38664
Gryder, B. E., Wu, L., Woldemichael, G. M., Pomella, S., Quinn, T. R., Park, P. M. C., et al. (2019). Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 10:3004.
Gu, B., and Zhu, W. G. (2012). Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci. 8, 672–684. doi: 10.7150/ijbs.4283
Gu, W., and Roeder, R. G. (1997). Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606. doi: 10.1016/s0092-8674(00)80521-8
Guardiola, A. R., and Yao, T. P. (2002). Molecular cloning and characterization of a novel histone deacetylase HDAC10. J. Biol. Chem. 277, 3350–3356. doi: 10.1074/jbc.m109861200
Guerriero, J. L., Sotayo, A., Ponichtera, H. E., Castrillon, J. A., Pourzia, A. L., Schad, S., et al. (2017). Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543, 428–432. doi: 10.1038/nature21409
Guo, C., and Zhao, Y. (2020). Autophagy and DNA damage repair. Genome Instabil. Dis. 1, 172–183. doi: 10.1007/s42764-020-00016-9
Guo, H., Zeng, D., Zhang, H., Bell, T., Yao, J., Liu, Y., et al. (2019). Dual inhibition of PI3K signaling and histone deacetylation halts proliferation and induces lethality in mantle cell lymphoma. Oncogene 38, 1802–1814. doi: 10.1038/s41388-018-0550-3
Guo, L., Zhou, S. R., Wei, X. B., Liu, Y., Chang, X. X., Liu, Y., et al. (2016). Acetylation of mitochondrial trifunctional protein alpha-subunit enhances its stability to promote fatty acid oxidation and is decreased in nonalcoholic fatty liver disease. Mol. Cell. Biol. 36, 2553–2567. doi: 10.1128/mcb.00227-16
Guo, Q., Li, F., Duan, Y., Wen, C., Wang, W., Zhang, L., et al. (2020). Oxidative stress, nutritional antioxidants and beyond. Sci. China Life Sci. 63, 866–874. doi: 10.1007/s11427-019-9591-5
Haberland, M., Johnson, A., Mokalled, M. H., Montgomery, R. L., and Olson, E. N. (2009). Genetic dissection of histone deacetylase requirement in tumor cells. Proc. Natl. Acad. Sci. U.S.A. 106, 7751–7755. doi: 10.1073/pnas.0903139106
Hai, Y., and Christianson, D. W. (2016). Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 12, 741–747. doi: 10.1038/nchembio.2134
Hai, Y., Shinsky, S. A., Porter, N. J., and Christianson, D. W. (2017). Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun. 8:15368.
Haigis, M. C., Mostoslavsky, R., Haigis, K. M., Fahie, K., Christodoulou, D. C., Murphy, A. J., et al. (2006). SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126, 941–954. doi: 10.1016/j.cell.2006.06.057
Harms, K. L., and Chen, X. (2007). Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 67, 3145–3152. doi: 10.1158/0008-5472.can-06-4397
Hassig, C. A., Fleischer, T. C., Billin, A. N., Schreiber, S. L., and Ayer, D. E. (1997). Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 89, 341–347. doi: 10.1016/s0092-8674(00)80214-7
Heideman, M. R., Wilting, R. H., Yanover, E., Velds, A., De Jong, J., Kerkhoven, R. M., et al. (2013). Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood 121, 2038–2050. doi: 10.1182/blood-2012-08-450916
Hershberger, K. A., Abraham, D. M., Liu, J., Locasale, J. W., Grimsrud, P. A., and Hirschey, M. D. (2018). Ablation of Sirtuin5 in the postnatal mouse heart results in protein succinylation and normal survival in response to chronic pressure overload. J. Biol. Chem. 293, 10630–10645. doi: 10.1074/jbc.ra118.002187
Hideshima, T., Bradner, J. E., Wong, J., Chauhan, D., Richardson, P., Schreiber, S. L., et al. (2005). Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. U.S.A. 102, 8567–8572. doi: 10.1073/pnas.0503221102
Hideshima, T., Qi, J., Paranal, R. M., Tang, W., Greenberg, E., West, N., et al. (2016). Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. U.S.A. 113, 13162–13167. doi: 10.1073/pnas.1608067113
Hirschey, M. D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D. B., et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. doi: 10.1038/nature08778
Ho, M., Chen, T., Liu, J., Dowling, P., Hideshima, T., Zhang, L., et al. (2020). Targeting histone deacetylase 3 (HDAC3) in the bone marrow microenvironment inhibits multiple myeloma proliferation by modulating exosomes and IL-6 trans-signaling. Leukemia 34, 196–209. doi: 10.1038/s41375-019-0493-x
Hook, S. S., Orian, A., Cowley, S. M., and Eisenman, R. N. (2002). Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc. Natl. Acad. Sci. U.S.A. 99, 13425–13430. doi: 10.1073/pnas.172511699
Hou, T., Cao, Z., Zhang, J., Tang, M., Tian, Y., Li, Y., et al. (2020). SIRT6 coordinates with CHD4 to promote chromatin relaxation and DNA repair. Nucleic Acids Res. 48, 2982–3000. doi: 10.1093/nar/gkaa006
Houtkooper, R. H., Pirinen, E., and Auwerx, J. (2012). Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238. doi: 10.1038/nrm3293
Howitz, K. T., Bitterman, K. J., Cohen, H. Y., Lamming, D. W., Lavu, S., Wood, J. G., et al. (2003). Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196. doi: 10.1038/nature01960
Hsia, D. A., Tepper, C. G., Pochampalli, M. R., Hsia, E. Y., Izumiya, C., Huerta, S. B., et al. (2010). KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 107, 9671–9676. doi: 10.1073/pnas.1000401107
Hsieh, C. L., Fei, T., Chen, Y., Li, T., Gao, Y., Wang, X., et al. (2014). Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. U.S.A. 111, 7319–7324. doi: 10.1073/pnas.1324151111
Hsu, F. C., Belmonte, P. J., Constans, M. M., Chen, M. W., Mcwilliams, D. C., Hiebert, S. W., et al. (2015). Histone Deacetylase 3 Is Required for T Cell Maturation. J. Immunol. 195, 1578–1590.
Hu, E., Chen, Z., Fredrickson, T., Zhu, Y., Kirkpatrick, R., Zhang, G. F., et al. (2000). Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 275, 15254–15264. doi: 10.1074/jbc.m908988199
Hu, F., Sun, X., Li, G., Wu, Q., Chen, Y., Yang, X., et al. (2018). Inhibition of SIRT2 limits tumour angiogenesis via inactivation of the STAT3/VEGFA signalling pathway. Cell Death Dis. 10:9.
Hu, T., Schreiter, F. C., Bagchi, R. A., Tatman, P. D., Hannink, M., and Mckinsey, T. A. (2019). HDAC5 catalytic activity suppresses cardiomyocyte oxidative stress and NRF2 target gene expression. J. Biol. Chem. 294, 8640–8652. doi: 10.1074/jbc.ra118.007006
Hua, W. K., Qi, J., Cai, Q., Carnahan, E., Ayala Ramirez, M., Li, L., et al. (2017). HDAC8 regulates long-term hematopoietic stem-cell maintenance under stress by modulating p53 activity. Blood 130, 2619–2630. doi: 10.1182/blood-2017-03-771386
Huang, F., Sun, J., Chen, W., He, X., Zhu, Y., Dong, H., et al. (2020). HDAC4 inhibition disrupts TET2 function in high-risk MDS and AML. Aging. doi: 10.18632/aging.103605 [Epub ahead of print].
Huang, H., Sabari, B. R., Garcia, B. A., Allis, C. D., and Zhao, Y. (2014). SnapShot: histone modifications. Cell 159, 458–458.e1.
Huang, H., Zhang, D., Wang, Y., Perez-Neut, M., Han, Z., Zheng, Y. G., et al. (2018a). Lysine benzoylation is a histone mark regulated by SIRT2. Nat. Commun. 9:3374.
Huang, Z., Zhao, J., Deng, W., Chen, Y., Shang, J., Song, K., et al. (2018b). Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol. 14, 1118–1126. doi: 10.1038/s41589-018-0150-0
Huang, Z., Zhou, W., Li, Y., Cao, M., Wang, T., Ma, Y., et al. (2018c). Novel hybrid molecule overcomes the limited response of solid tumours to HDAC inhibitors via suppressing JAK1-STAT3-BCL2 signalling. Theranostics 8, 4995–5011. doi: 10.7150/thno.26627
Hubbard, B. P., Gomes, A. P., Dai, H., Li, J., Case, A. W., Considine, T., et al. (2013). Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339, 1216–1219.
Hubbert, C., Guardiola, A., Shao, R., Kawaguchi, Y., Ito, A., Nixon, A., et al. (2002). HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458. doi: 10.1038/417455a
Hubbi, M. E., Hu, H., Kshitiz, Gilkes, D. M., and Semenza, G. L. (2013). Sirtuin-7 inhibits the activity of hypoxia-inducible factors. J. Biol. Chem. 288, 20768–20775. doi: 10.1074/jbc.m113.476903
Humphrey, G. W., Wang, Y., Russanova, V. R., Hirai, T., Qin, J., Nakatani, Y., et al. (2001). Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J. Biol. Chem. 276, 6817–6824. doi: 10.1074/jbc.m007372200
Igase, M., Fujiki, N., Shibutani, S., Sakai, H., Noguchi, S., Nemoto, Y., et al. (2020). Tenovin-6 induces the SIRT-independent cell growth suppression and blocks autophagy flux in canine hemangiosarcoma cell lines. Exp. Cell Res. 388:111810. doi: 10.1016/j.yexcr.2019.111810
Imai, S., Armstrong, C. M., Kaeberlein, M., and Guarente, L. (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800. doi: 10.1038/35001622
Imai, S., and Guarente, L. (2014). NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471. doi: 10.1016/j.tcb.2014.04.002
Ingham, O. J., Paranal, R. M., Smith, W. B., Escobar, R. A., Yueh, H., Snyder, T., et al. (2016). Development of a potent and selective HDAC8 inhibitor. ACS Med. Chem. Lett. 7, 929–932.
Inoue, A., and Fujimoto, D. (1969). Enzymatic deacetylation of histone. Biochem. Biophys. Res. Commun. 36, 146–150. doi: 10.1016/0006-291x(69)90661-5
Itazaki, H., Nagashima, K., Sugita, K., Yoshida, H., Kawamura, Y., Yasuda, Y., et al. (1990). Isolation and structural elucidation of new cyclotetrapeptides, trapoxins A and B, having detransformation activities as antitumor agents. J. Antibiot. 43, 1524–1532. doi: 10.7164/antibiotics.43.1524
Itoh, T., Fairall, L., Muskett, F. W., Milano, C. P., Watson, P. J., Arnaudo, N., et al. (2015). Structural and functional characterization of a cell cycle associated HDAC1/2 complex reveals the structural basis for complex assembly and nucleosome targeting. Nucleic Acids Res. 43, 2033–2044. doi: 10.1093/nar/gkv068
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Jamaladdin, S., Kelly, R. D., O’regan, L., Dovey, O. M., Hodson, G. E., Millard, C. J., et al. (2014). Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, 9840–9845. doi: 10.1073/pnas.1321330111
Jarrett, S. G., Carter, K. M., Bautista, R. M., He, D., Wang, C., and D’orazio, J. A. (2018). Sirtuin 1-mediated deacetylation of XPA DNA repair protein enhances its interaction with ATR protein and promotes cAMP-induced DNA repair of UV damage. J. Biol. Chem. 293, 19025–19037. doi: 10.1074/jbc.ra118.003940
Jedrusik-Bode, M., Studencka, M., Smolka, C., Baumann, T., Schmidt, H., Kampf, J., et al. (2013). The sirtuin SIRT6 regulates stress granule formation in C. elegans and mammals. J. Cell Sci. 126, 5166–5177. doi: 10.1242/jcs.130708
Jenuwein, T., and Allis, C. D. (2001). Translating the histone code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Jeong, S. M., Lee, A., Lee, J., and Haigis, M. C. (2014). SIRT4 protein suppresses tumor formation in genetic models of Myc-induced B cell lymphoma. J. Biol. Chem. 289, 4135–4144. doi: 10.1074/jbc.m113.525949
Jeong, S. M., Xiao, C., Finley, L. W., Lahusen, T., Souza, A. L., Pierce, K., et al. (2013). SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell 23, 450–463. doi: 10.1016/j.ccr.2013.02.024
Ji, H., Zhou, Y., Zhuang, X., Zhu, Y., Wu, Z., Lu, Y., et al. (2019). HDAC3 deficiency promotes liver cancer through a defect in H3K9ac/H3K9me3 transition. Cancer Res. 79, 3676–3688.
Jiang, H., Khan, S., Wang, Y., Charron, G., He, B., Sebastian, C., et al. (2013). SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496, 110–113. doi: 10.1038/nature12038
Jiang, Y., and Hsieh, J. (2014). HDAC3 controls gap 2/mitosis progression in adult neural stem/progenitor cells by regulating CDK1 levels. Proc. Natl. Acad. Sci. U.S.A. 111, 13541–13546. doi: 10.1073/pnas.1411939111
Jiang, Z., Li, W., Hu, X., Zhang, Q., Sun, T., Cui, S., et al. (2019). Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 20, 806–815. doi: 10.1016/s1470-2045(19)30164-0
Jin, X., Yan, Y., Wang, D., Ding, D., Ma, T., Ye, Z., et al. (2018). DUB3 Promotes BET inhibitor resistance and cancer progression by deubiquitinating BRD4. Mol. Cell 71, 592–605.e4.
Jing, H., Hu, J., He, B., Negron Abril, Y. L., Stupinski, J., Weiser, K., et al. (2016). A SIRT2-selective inhibitor promotes c-Myc oncoprotein degradation and exhibits broad anticancer activity. Cancer Cell 29, 297–310. doi: 10.1016/j.ccell.2016.02.007
Jones, P. A., Issa, J. P., and Baylin, S. (2016). Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 17, 630–641.
Jones, P. L., Veenstra, G. J., Wade, P. A., Vermaak, D., Kass, S. U., Landsberger, N., et al. (1998). Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19, 187–191. doi: 10.1038/561
Juan, L. J., Shia, W. J., Chen, M. H., Yang, W. M., Seto, E., Lin, Y. S., et al. (2000). Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 275, 20436–20443. doi: 10.1074/jbc.m000202200
Kalin, J. H., Wu, M., Gomez, A. V., Song, Y., Das, J., Hayward, D., et al. (2018). Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 9:53.
Kaliszczak, M., Van Hechanova, E., Li, Y., Alsadah, H., Parzych, K., Auner, H. W., et al. (2018). The HDAC6 inhibitor C1A modulates autophagy substrates in diverse cancer cells and induces cell death. Br. J. Cancer 119, 1278–1287. doi: 10.1038/s41416-018-0232-5
Kaluza, D., Kroll, J., Gesierich, S., Yao, T. P., Boon, R. A., Hergenreider, E., et al. (2011). Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 30, 4142–4156. doi: 10.1038/emboj.2011.298
Kang, H., Oka, S., Lee, D. Y., Park, J., Aponte, A. M., Jung, Y. S., et al. (2017). Sirt1 carboxyl-domain is an ATP-repressible domain that is transferrable to other proteins. Nat. Commun. 8:15560.
Kang, J. Y., Kim, J. Y., Kim, K. B., Park, J. W., Cho, H., Hahm, J. Y., et al. (2018). KDM2B is a histone H3K79 demethylase and induces transcriptional repression via sirtuin-1-mediated chromatin silencing. FASEB J. 32, 5737–5750. doi: 10.1096/fj.201800242r
Kao, H. Y., Downes, M., Ordentlich, P., and Evans, R. M. (2000). Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 14, 55–66.
Kao, H. Y., Lee, C. H., Komarov, A., Han, C. C., and Evans, R. M. (2002). Isolation and characterization of mammalian HDAC10, a novel histone deacetylase. J. Biol. Chem. 277, 187–193. doi: 10.1074/jbc.m108931200
Kasler, H. G., Young, B. D., Mottet, D., Lim, H. W., Collins, A. M., Olson, E. N., et al. (2011). Histone deacetylase 7 regulates cell survival and TCR signaling in CD4/CD8 double-positive thymocytes. J. Immunol. 186, 4782–4793. doi: 10.4049/jimmunol.1001179
Kawaguchi, Y., Kovacs, J. J., Mclaurin, A., Vance, J. M., Ito, A., and Yao, T. P. (2003). The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738. doi: 10.1016/s0092-8674(03)00939-5
Kim, D. J., Dunleavey, J. M., Xiao, L., Ollila, D. W., Troester, M. A., Otey, C. A., et al. (2018). Suppression of TGFbeta-mediated conversion of endothelial cells and fibroblasts into cancer associated (myo)fibroblasts via HDAC inhibition. Br. J. Cancer 118, 1359–1368. doi: 10.1038/s41416-018-0072-3
Kim, G. D., Choi, Y. H., Dimtchev, A., Jeong, S. J., Dritschilo, A., and Jung, M. (1999). Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J. Biol. Chem. 274, 31127–31130. doi: 10.1074/jbc.274.44.31127
Kim, H. S., Patel, K., Muldoon-Jacobs, K., Bisht, K. S., Aykin-Burns, N., Pennington, J. D., et al. (2010). SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17, 41–52. doi: 10.1016/j.ccr.2009.11.023
Kim, H. S., Vassilopoulos, A., Wang, R. H., Lahusen, T., Xiao, Z., Xu, X., et al. (2011). SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 20, 487–499. doi: 10.1016/j.ccr.2011.09.004
Kim, J. E., Chen, J., and Lou, Z. (2008). DBC1 is a negative regulator of SIRT1. Nature 451, 583–586. doi: 10.1038/nature06500
Kim, J. H., Kim, D., Cho, S. J., Jung, K. Y., Kim, J. H., Lee, J. M., et al. (2019). Identification of a novel SIRT7 inhibitor as anticancer drug candidate. Biochem. Biophys. Res. Commun. 508, 451–457. doi: 10.1016/j.bbrc.2018.11.120
Kim, J. J., Lee, S. Y., and Miller, K. M. (2019). Preserving genome integrity and function: the DNA damage response and histone modifications. Crit. Rev. Biochem. Mol. Biol. 54, 208–241. doi: 10.1080/10409238.2019.1620676
Kim, K., Skora, A. D., Li, Z., Liu, Q., Tam, A. J., Blosser, R. L., et al. (2014). Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. U.S.A. 111, 11774–11779. doi: 10.1073/pnas.1410626111
Kim, K. K., Sheppard, D., and Chapman, H. A. (2018). TGF-beta1 signaling and tissue fibrosis. Cold Spring Harb. Perspect. Biol. 10:a022293.
Kim, M. S., Kwon, H. J., Lee, Y. M., Baek, J. H., Jang, J. E., Lee, S. W., et al. (2001). Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 7, 437–443. doi: 10.1038/86507
Kim, Y. H., Bagot, M., Pinter-Brown, L., Rook, A. H., Porcu, P., Horwitz, S. M., et al. (2018). Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 19, 1192–1204.
Kiran, S., Chatterjee, N., Singh, S., Kaul, S. C., Wadhwa, R., and Ramakrishna, G. (2013). Intracellular distribution of human SIRT7 and mapping of the nuclear/nucleolar localization signal. FEBS J. 280, 3451–3466. doi: 10.1111/febs.12346
Kosciuczuk, E. M., Mehrotra, S., Saleiro, D., Kroczynska, B., Majchrzak-Kita, B., Lisowski, P., et al. (2019). Sirtuin 2-mediated deacetylation of cyclin-dependent kinase 9 promotes STAT1 signaling in type I interferon responses. J. Biol. Chem. 294, 827–837. doi: 10.1074/jbc.ra118.005956
Kotian, S., Zhang, L., Boufraqech, M., Gaskins, K., Gara, S. K., Quezado, M., et al. (2017). Dual Inhibition of HDAC and tyrosine kinase signaling pathways with CUDC-907 inhibits thyroid cancer growth and metastases. Clin. Cancer Res. 23, 5044–5054. doi: 10.1158/1078-0432.ccr-17-1043
Kronlage, M., Dewenter, M., Grosso, J., Fleming, T., Oehl, U., Lehmann, L. H., et al. (2019). O-GlcNAcylation of histone deacetylase 4 protects the diabetic heart from failure. Circulation 140, 580–594. doi: 10.1161/circulationaha.117.031942
Krug, L. M., Kindler, H. L., Calvert, H., Manegold, C., Tsao, A. S., Fennell, D., et al. (2015). Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol. 16, 447–456.
Kugel, S., Sebastian, C., Fitamant, J., Ross, K. N., Saha, S. K., Jain, E., et al. (2016). SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell 165, 1401–1415. doi: 10.1016/j.cell.2016.04.033
Kurdistani, S. K., Robyr, D., Tavazoie, S., and Grunstein, M. (2002). Genome-wide binding map of the histone deacetylase Rpd3 in yeast. Nat. Genet. 31, 248–254. doi: 10.1038/ng907
Kutil, Z., Mikesova, J., Zessin, M., Meleshin, M., Novakova, Z., Alquicer, G., et al. (2019). Continuous activity assay for HDAC11 enabling reevaluation of HDAC inhibitors. ACS Omega 4, 19895–19904. doi: 10.1021/acsomega.9b02808
Kutil, Z., Novakova, Z., Meleshin, M., Mikesova, J., Schutkowski, M., and Barinka, C. (2018). Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem. Biol. 13, 685–693. doi: 10.1021/acschembio.7b00942
Kuzmichev, A., Margueron, R., Vaquero, A., Preissner, T. S., Scher, M., Kirmizis, A., et al. (2005). Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc. Natl. Acad. Sci. U.S.A. 102, 1859–1864. doi: 10.1073/pnas.0409875102
Laherty, C. D., Yang, W. M., Sun, J. M., Davie, J. R., Seto, E., and Eisenman, R. N. (1997). Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell 89, 349–356. doi: 10.1016/s0092-8674(00)80215-9
Lahm, A., Paolini, C., Pallaoro, M., Nardi, M. C., Jones, P., Neddermann, P., et al. (2007). Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 104, 17335–17340. doi: 10.1073/pnas.0706487104
Lain, S., Hollick, J. J., Campbell, J., Staples, O. D., Higgins, M., Aoubala, M., et al. (2008). Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 13, 454–463. doi: 10.1016/j.ccr.2008.03.004
Lascano, S., Lopez, M., and Arimondo, P. B. (2018). Natural products and chemical biology tools: alternatives to target epigenetic mechanisms in cancers. Chem. Rec. 18, 1854–1876. doi: 10.1002/tcr.201800133
Lee, E., Furukubo, T., Miyabe, T., Yamauchi, A., and Kariya, K. (1996). Involvement of histone hyperacetylation in triggering DNA fragmentation of rat thymocytes undergoing apoptosis. FEBS Lett. 395, 183–187. doi: 10.1016/0014-5793(96)01033-2
Lee, I. H., Cao, L., Mostoslavsky, R., Lombard, D. B., Liu, J., Bruns, N. E., et al. (2008). A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. U.S.A. 105, 3374–3379. doi: 10.1073/pnas.0712145105
Lee, J. H., Choy, M. L., Ngo, L., Venta-Perez, G., and Marks, P. A. (2011). Role of checkpoint kinase 1 (Chk1) in the mechanisms of resistance to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 108, 19629–19634. doi: 10.1073/pnas.1117544108
Lee, M. G., Wynder, C., Cooch, N., and Shiekhattar, R. (2005). An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437, 432–435. doi: 10.1038/nature04021
Lehmann, L. H., Jebessa, Z. H., Kreusser, M. M., Horsch, A., He, T., Kronlage, M., et al. (2018). A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat. Med. 24, 62–72. doi: 10.1038/nm.4452
Lei, Y., Liu, L., Zhang, S., Guo, S., Li, X., Wang, J., et al. (2017). Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation. Mol. Cancer 16:170.
Leslie, P. L., Chao, Y. L., Tsai, Y. H., Ghosh, S. K., Porrello, A., Van Swearingen, A. E. D., et al. (2019). Histone deacetylase 11 inhibition promotes breast cancer metastasis from lymph nodes. Nat. Commun. 10:4192.
Li, D., Marchenko, N. D., and Moll, U. M. (2011). SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 18, 1904–1913. doi: 10.1038/cdd.2011.71
Li, H., Tian, Z., Qu, Y., Yang, Q., Guan, H., Shi, B., et al. (2019). SIRT7 promotes thyroid tumorigenesis through phosphorylation and activation of Akt and p70S6K1 via DBC1/SIRT1 axis. Oncogene 38, 345–359. doi: 10.1038/s41388-018-0434-6
Li, L., Shi, L., Yang, S., Yan, R., Zhang, D., Yang, J., et al. (2016). SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 7:12235.
Li, L., Wang, L., Li, L., Wang, Z., Ho, Y., Mcdonald, T., et al. (2012). Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 21, 266–281. doi: 10.1016/j.ccr.2011.12.020
Li, M., Chiang, Y. L., Lyssiotis, C. A., Teater, M. R., Hong, J. Y., Shen, H., et al. (2019). Non-oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 35, 916–931.e9.
Li, M., Hou, T., Gao, T., Lu, X., Yang, Q., Zhu, Q., et al. (2018). p53 cooperates with SIRT6 to regulate cardiolipin de novo biosynthesis. Cell Death Dis. 9:941.
Li, Y., Peng, L., and Seto, E. (2015). Histone deacetylase 10 regulates the cell cycle G2/M phase transition via a novel Let-7-HMGA2-Cyclin A2 Pathway. Mol. Cell Biol 35, 3547–3565. doi: 10.1128/mcb.00400-15
Li, Y., and Seto, E. (2016). HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 6:a026831. doi: 10.1101/cshperspect.a026831
Li, Y., Zhang, X., Zhu, S., Dejene, E. A., Peng, W., Sepulveda, A., et al. (2020). HDAC10 Regulates Cancer Stem-like Cell Properties in KRAS-driven Lung Adenocarcinoma. Cancer Res. 80, 3265–3278. doi: 10.1158/0008-5472.can-19-3613
Li, Z., Chen, Y., Tang, M., Li, Y., and Zhu, W.-G. (2019). Regulation of DNA damage-induced ATM activation by histone modifications. Genome Instabil. Dis. 1, 20–33. doi: 10.1007/s42764-019-00004-8
Li, Z., and Zhu, W. G. (2014). Targeting histone deacetylases for cancer therapy: from molecular mechanisms to clinical implications. Int. J. Biol. Sci. 10, 757–770. doi: 10.7150/ijbs.9067
Lim, B., Murthy, R. K., Lee, J., Jackson, S. A., Iwase, T., Davis, D. W., et al. (2019). A phase Ib study of entinostat plus lapatinib with or without trastuzumab in patients with HER2-positive metastatic breast cancer that progressed during trastuzumab treatment. Br. J. Cancer 120, 1105–1112. doi: 10.1038/s41416-019-0473-y
Lim, J. H., Lee, Y. M., Chun, Y. S., Chen, J., Kim, J. E., and Park, J. W. (2010). Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 38, 864–878. doi: 10.1016/j.molcel.2010.05.023
Lin, C. L., Tsai, M. L., Lin, C. Y., Hsu, K. W., Hsieh, W. S., Chi, W. M., et al. (2019). HDAC1 and HDAC2 double knockout triggers cell apoptosis in advanced thyroid cancer. Int. J. Mol. Sci. 20:454. doi: 10.3390/ijms20020454
Lin, S. J., Defossez, P. A., and Guarente, L. (2000). Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128. doi: 10.1126/science.289.5487.2126
Liszczak, G., Diehl, K. L., Dann, G. P., and Muir, T. W. (2018). Acetylation blocks DNA damage-induced chromatin ADP-ribosylation. Nat. Chem. Biol. 14, 837–840. doi: 10.1038/s41589-018-0097-1
Liu, G., Bi, Y., Shen, B., Yang, H., Zhang, Y., Wang, X., et al. (2014). SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. 74, 727–737. doi: 10.1158/0008-5472.can-13-2584
Liu, P. Y., Xu, N., Malyukova, A., Scarlett, C. J., Sun, Y. T., Zhang, X. D., et al. (2013). The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ. 20, 503–514. doi: 10.1038/cdd.2012.147
Liu, T., Tee, A. E., Porro, A., Smith, S. A., Dwarte, T., Liu, P. Y., et al. (2007). Activation of tissue transglutaminase transcription by histone deacetylase inhibition as a therapeutic approach for Myc oncogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 18682–18687. doi: 10.1073/pnas.0705524104
Liu, X., Wang, D., Zhao, Y., Tu, B., Zheng, Z., Wang, L., et al. (2011). Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proc. Natl. Acad. Sci. U.S.A. 108, 1925–1930. doi: 10.1073/pnas.1019619108
Liu, Y., Peng, L., Seto, E., Huang, S., and Qiu, Y. (2012). Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J. Biol. Chem. 287, 29168–29174. doi: 10.1074/jbc.m112.371120
Lombard, D. B., Alt, F. W., Cheng, H. L., Bunkenborg, J., Streeper, R. S., Mostoslavsky, R., et al. (2007). Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814. doi: 10.1128/mcb.01636-07
Lu, J., and Holmgren, A. (2014). The thioredoxin antioxidant system. Free Radic. Biol. Med. 66, 75–87. doi: 10.1016/j.freeradbiomed.2013.07.036
Lu, X., Tang, M., Zhu, Q., Yang, Q., Li, Z., Bao, Y., et al. (2019). GLP-catalyzed H4K16me1 promotes 53BP1 recruitment to permit DNA damage repair and cell survival. Nucleic Acids Res. 47, 10977–10993. doi: 10.1093/nar/gkz897
Lu, X. F., Cao, X. Y., Zhu, Y. J., Wu, Z. R., Zhuang, X., Shao, M. Y., et al. (2018). Histone deacetylase 3 promotes liver regeneration and liver cancer cells proliferation through signal transducer and activator of transcription 3 signaling pathway. Cell Death Dis. 9:398.
Lu, Y., Stuart, J. H., Talbot-Cooper, C., Agrawal-Singh, S., Huntly, B., Smid, A. I., et al. (2019). Histone deacetylase 4 promotes type I interferon signaling, restricts DNA viruses, and is degraded via vaccinia virus protein C6. Proc. Natl. Acad. Sci. U.S.A. 116, 11997–12006.
Lu, Y. F., Xu, X. P., Lu, X. P., Zhu, Q., Liu, G., Bao, Y. T., et al. (2020). SIRT7 activates p53 by enhancing PCAF-mediated MDM2 degradation to arrest the cell cycle. Oncogene 39, 4650–4665. doi: 10.1038/s41388-020-1305-5
Lu, Z., Zou, J., Li, S., Topper, M. J., Tao, Y., Zhang, H., et al. (2020). Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 579, 284–290. doi: 10.1038/s41586-020-2054-x
Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., and Richmond, T. J. (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260. doi: 10.1038/38444
Luo, J., Nikolaev, A. Y., Imai, S., Chen, D., Su, F., Shiloh, A., et al. (2001). Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107, 137–148. doi: 10.1016/s0092-8674(01)00524-4
Luo, J., Su, F., Chen, D., Shiloh, A., and Gu, W. (2000). Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408, 377–381. doi: 10.1038/35042612
Mahalingam, D., Mita, M., Sarantopoulos, J., Wood, L., Amaravadi, R. K., Davis, L. E., et al. (2014). Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 10, 1403–1414. doi: 10.4161/auto.29231
Mani, S. A., Guo, W., Liao, M. J., Eaton, E. N., Ayyanan, A., Zhou, A. Y., et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715.
Mao, R., Wu, Y., Ming, Y., Xu, Y., Wang, S., Chen, X., et al. (2019). Enhancer RNAs: a missing regulatory layer in gene transcription. Sci. China Life Sci. 62, 905–912. doi: 10.1007/s11427-017-9370-9
Mao, Z., Hine, C., Tian, X., Van Meter, M., Au, M., Vaidya, A., et al. (2011). SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446. doi: 10.1126/science.1202723
Marquardt, J. U., Fischer, K., Baus, K., Kashyap, A., Ma, S., Krupp, M., et al. (2013). Sirtuin-6-dependent genetic and epigenetic alterations are associated with poor clinical outcome in hepatocellular carcinoma patients. Hepatology 58, 1054–1064. doi: 10.1002/hep.26413
Martin, M. W., Lee, J. Y., Lancia, D. R. Jr., Ng, P. Y., Han, B., Thomason, J. R., et al. (2018). Discovery of novel N-hydroxy-2-arylisoindoline-4-carboxamides as potent and selective inhibitors of HDAC11. Bioorg. Med. Chem. Lett. 28, 2143–2147. doi: 10.1016/j.bmcl.2018.05.021
Mathias, R. A., Greco, T. M., Oberstein, A., Budayeva, H. G., Chakrabarti, R., Rowland, E. A., et al. (2014). Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625. doi: 10.1016/j.cell.2014.11.046
Mazur, P. K., Herner, A., Mello, S. S., Wirth, M., Hausmann, S., Sanchez-Rivera, F. J., et al. (2015). Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 21, 1163–1171. doi: 10.1038/nm.3952
Mckinsey, T. A., Kuwahara, K., Bezprozvannaya, S., and Olson, E. N. (2006). Class II histone deacetylases confer signal responsiveness to the ankyrin-repeat proteins ANKRA2 and RFXANK. Mol. Biol. Cell 17, 438–447. doi: 10.1091/mbc.e05-07-0612
Mckinsey, T. A., Zhang, C. L., Lu, J., and Olson, E. N. (2000). Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111. doi: 10.1038/35040593
Mckinsey, T. A., Zhang, C. L., and Olson, E. N. (2001). Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol. Cell. Biol. 21, 6312–6321. doi: 10.1128/mcb.21.18.6312-6321.2001
Medina, V., Edmonds, B., Young, G. P., James, R., Appleton, S., and Zalewski, P. D. (1997). Induction of caspase-3 protease activity and apoptosis by butyrate and trichostatin A (inhibitors of histone deacetylase): dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res. 57, 3697–3707.
Medon, M., Vidacs, E., Vervoort, S. J., Li, J., Jenkins, M. R., Ramsbottom, K. M., et al. (2017). HDAC Inhibitor Panobinostat Engages Host Innate Immune Defenses to Promote the Tumoricidal Effects of Trastuzumab in HER2(+) Tumors. Cancer Res. 77, 2594–2606. doi: 10.1158/0008-5472.can-16-2247
Melo, C. A., Drost, J., Wijchers, P. J., Van De Werken, H., De Wit, E., Oude Vrielink, J. A., et al. (2013). eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cell 49, 524–535. doi: 10.1016/j.molcel.2012.11.021
Meng, F., Qian, M., Peng, B., Peng, L., Wang, X., Zheng, K., et al. (2020). Synergy between SIRT1 and SIRT6 helps recognize DNA breaks and potentiates the DNA damage response and repair in humans and mice. eLife 9:e55828.
Michishita, E., Park, J. Y., Burneskis, J. M., Barrett, J. C., and Horikawa, I. (2005). Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16, 4623–4635. doi: 10.1091/mbc.e05-01-0033
Mihaylova, M. M., Vasquez, D. S., Ravnskjaer, K., Denechaud, P. D., Yu, R. T., Alvarez, J. G., et al. (2011). Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145, 607–621. doi: 10.1016/j.cell.2011.03.043
Millard, C. J., Watson, P. J., Celardo, I., Gordiyenko, Y., Cowley, S. M., Robinson, C. V., et al. (2013). Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell 51, 57–67. doi: 10.1016/j.molcel.2013.05.020
Miller, K. M., Tjeertes, J. V., Coates, J., Legube, G., Polo, S. E., Britton, S., et al. (2010). Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 17, 1144–1151. doi: 10.1038/nsmb.1899
Ming, M., Han, W., Zhao, B., Sundaresan, N. R., Deng, C. X., Gupta, M. P., et al. (2014). SIRT6 promotes COX-2 expression and acts as an oncogene in skin cancer. Cancer Res. 74, 5925–5933. doi: 10.1158/0008-5472.can-14-1308
Ming, M., Shea, C. R., Guo, X., Li, X., Soltani, K., Han, W., et al. (2010). Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc. Natl. Acad. Sci. U.S.A. 107, 22623–22628. doi: 10.1073/pnas.1010377108
Mishra, V. K., Wegwitz, F., Kosinsky, R. L., Sen, M., Baumgartner, R., Wulff, T., et al. (2017). Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 45, 6334–6349. doi: 10.1093/nar/gkx212
Miska, E. A., Karlsson, C., Langley, E., Nielsen, S. J., Pines, J., and Kouzarides, T. (1999). HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 18, 5099–5107. doi: 10.1093/emboj/18.18.5099
Mizushima, N. (2018). A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 20, 521–527. doi: 10.1038/s41556-018-0092-5
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Mohrin, M., Shin, J., Liu, Y., Brown, K., Luo, H., Xi, Y., et al. (2015). Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347, 1374–1377. doi: 10.1126/science.aaa2361
Monte, M., Simonatto, M., Peche, L. Y., Bublik, D. R., Gobessi, S., Pierotti, M. A., et al. (2006). MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc. Natl. Acad. Sci. U.S.A. 103, 11160–11165. doi: 10.1073/pnas.0510834103
Montgomery, R. L., Davis, C. A., Potthoff, M. J., Haberland, M., Fielitz, J., Qi, X., et al. (2007). Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21, 1790–1802. doi: 10.1101/gad.1563807
Montgomery, R. L., Potthoff, M. J., Haberland, M., Qi, X., Matsuzaki, S., Humphries, K. M., et al. (2008). Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Invest. 118, 3588–3597. doi: 10.1172/jci35847
Moon, H., Zhu, J., and White, A. C. (2019). Sirt5 is dispensable for Braf(V600E) -mediated cutaneous melanoma development and growth in vivo. Exp. Dermatol. 28, 83–85. doi: 10.1111/exd.13845
Moresi, V., Carrer, M., Grueter, C. E., Rifki, O. F., Shelton, J. M., Richardson, J. A., et al. (2012). Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc. Natl. Acad. Sci. U.S.A. 109, 1649–1654. doi: 10.1073/pnas.1121159109
Moresi, V., Williams, A. H., Meadows, E., Flynn, J. M., Potthoff, M. J., Mcanally, J., et al. (2010). Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 143, 35–45. doi: 10.1016/j.cell.2010.09.004
Musselman, C. A., Lalonde, M. E., Cote, J., and Kutateladze, T. G. (2012). Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227. doi: 10.1038/nsmb.2436
Nagy, L., Kao, H. Y., Chakravarti, D., Lin, R. J., Hassig, C. A., Ayer, D. E., et al. (1997). Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell 89, 373–380. doi: 10.1016/s0092-8674(00)80218-4
Nahalkova, J. (2015). Novel protein-protein interactions of TPPII, p53, and SIRT7. Mol. Cell. Biochem. 409, 13–22. doi: 10.1007/s11010-015-2507-y
Naiman, S., Huynh, F. K., Gil, R., Glick, Y., Shahar, Y., Touitou, N., et al. (2019). SIRT6 promotes hepatic beta-oxidation via activation of PPARalpha. Cell Rep. 29, 4127–4143.e8.
Nakazawa, M. S., Eisinger-Mathason, T. S., Sadri, N., Ochocki, J. D., Gade, T. P., Amin, R. K., et al. (2016). Epigenetic re-expression of HIF-2alpha suppresses soft tissue sarcoma growth. Nat. Commun. 7:10539.
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389. doi: 10.1038/30764
Nawrocki, S. T., Carew, J. S., Pino, M. S., Highshaw, R. A., Andtbacka, R. H., Dunner, K., et al. (2006). Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 66, 3773–3781. doi: 10.1158/0008-5472.can-05-2961
Nebbioso, A., Carafa, V., Conte, M., Tambaro, F. P., Abbondanza, C., Martens, J., et al. (2017). c-Myc Modulation and Acetylation Is a Key HDAC inhibitor target in cancer. Clin. Cancer Res. 23, 2542–2555. doi: 10.1158/1078-0432.ccr-15-2388
Newbold, A., Salmon, J. M., Martin, B. P., Stanley, K., and Johnstone, R. W. (2014). The role of p21(waf1/cip1) and p27(Kip1) in HDACi-mediated tumor cell death and cell cycle arrest in the Emu-myc model of B-cell lymphoma. Oncogene 33, 5415–5423. doi: 10.1038/onc.2013.482
Newkirk, T. L., Bowers, A. A., and Williams, R. M. (2009). Discovery, biological activity, synthesis and potential therapeutic utility of naturally occurring histone deacetylase inhibitors. Nat. Prod. Rep. 26, 1293–1320. doi: 10.1039/b817886k
Ng, H. H., Zhang, Y., Hendrich, B., Johnson, C. A., Turner, B. M., Erdjument-Bromage, H., et al. (1999). MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 23, 58–61. doi: 10.1038/12659
Ng, R., and Huen, M. S. Y. (2020). Deacetylation of a deacetylase drives the DNA damage response. Genome Instabil. Dis. 1, 151–154. doi: 10.1007/s42764-020-00018-7
Nguyen, P., Lee, S., Lorang-Leins, D., Trepel, J., and Smart, D. K. (2014). SIRT2 interacts with beta-catenin to inhibit Wnt signaling output in response to radiation-induced stress. Mol. Cancer Res. 12, 1244–1253. doi: 10.1158/1541-7786.mcr-14-0223-t
Nguyen, P., Shukla, S., Liu, R., Abbineni, G., and Smart, D. K. (2019). Sirt2 regulates radiation-induced injury. Radiat. Res. 191, 398–412. doi: 10.1667/rr15282.1
Nicholson, J., Jevons, S. J., Groselj, B., Ellermann, S., Konietzny, R., Kerr, M., et al. (2017). E3 Ligase cIAP2 Mediates Downregulation of MRE11 and Radiosensitization in Response to HDAC Inhibition in Bladder Cancer. Cancer Res. 77, 3027–3039. doi: 10.1158/0008-5472.can-16-3232
Ning, Y., Ding, J., Sun, X., Xie, Y., Su, M., Ma, C., et al. (2020). HDAC9 deficiency promotes tumor progression by decreasing the CD8(+) dendritic cell infiltration of the tumor microenvironment. J. Immunother. Cancer 8:e000529. doi: 10.1136/jitc-2020-000529
Nishibuchi, G., Shibata, Y., Hayakawa, T., Hayakawa, N., Ohtani, Y., Sinmyozu, K., et al. (2014). Physical and functional interactions between the histone H3K4 demethylase KDM5A and the nucleosome remodeling and deacetylase (NuRD) complex. J. Biol. Chem. 289, 28956–28970. doi: 10.1074/jbc.m114.573725
Nishimori, S., Lai, F., Shiraishi, M., Kobayashi, T., Kozhemyakina, E., Yao, T. P., et al. (2019). PTHrP targets HDAC4 and HDAC5 to repress chondrocyte hypertrophy. JCI Insight 4:e97903.
Norsworthy, K. J., Cho, E., Arora, J., Kowalski, J., Tsai, H. L., Warlick, E., et al. (2016). Differentiation therapy in poor risk myeloid malignancies: results of companion phase II studies. Leuk. Res. 49, 90–97. doi: 10.1016/j.leukres.2016.09.003
North, B. J., Marshall, B. L., Borra, M. T., Denu, J. M., and Verdin, E. (2003). The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444. doi: 10.1016/s1097-2765(03)00038-8
Nott, A., Watson, P. M., Robinson, J. D., Crepaldi, L., and Riccio, A. (2008). S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 455, 411–415. doi: 10.1038/nature07238
Obayashi, H., Nagano, Y., Takahashi, T., Seki, T., Tanaka, S., Sakai, N., et al. (2020). Histone deacetylase 10 knockout activates chaperone-mediated autophagy and accelerates the decomposition of its substrate. Biochem. Biophys. Res. Commun. 523, 246–252. doi: 10.1016/j.bbrc.2019.12.048
Oberdoerffer, P., Michan, S., Mcvay, M., Mostoslavsky, R., Vann, J., Park, S. K., et al. (2008). SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918. doi: 10.1016/j.cell.2008.10.025
Oberoi, J., Fairall, L., Watson, P. J., Yang, J. C., Czimmerer, Z., Kampmann, T., et al. (2011). Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 18, 177–184. doi: 10.1038/nsmb.1983
Oehme, I., Linke, J. P., Bock, B. C., Milde, T., Lodrini, M., Hartenstein, B., et al. (2013). Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. U.S.A. 110, E2592–E2601.
Oki, Y., Kelly, K. R., Flinn, I., Patel, M. R., Gharavi, R., Ma, A., et al. (2017). CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: results from an expanded phase I trial. Haematologica 102, 1923–1930. doi: 10.3324/haematol.2017.172882
Olsen, E. A., Kim, Y. H., Kuzel, T. M., Pacheco, T. R., Foss, F. M., Parker, S., et al. (2007). Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 25, 3109–3115. doi: 10.1200/jco.2006.10.2434
Onn, L., Portillo, M., Ilic, S., Cleitman, G., Stein, D., Kaluski, S., et al. (2020). SIRT6 is a DNA double-strand break sensor. eLife 9:e51636.
Onyango, P., Celic, I., Mccaffery, J. M., Boeke, J. D., and Feinberg, A. P. (2002). SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 99, 13653–13658. doi: 10.1073/pnas.222538099
Pal, S., Kozono, D., Yang, X., Fendler, W., Fitts, W., Ni, J., et al. (2018). Dual HDAC and PI3K Inhibition Abrogates NFkappaB- and FOXM1-Mediated DNA Damage Response to Radiosensitize Pediatric High-Grade Gliomas. Cancer Res. 78, 4007–4021. doi: 10.1158/0008-5472.can-17-3691
Pan, P. W., Feldman, J. L., Devries, M. K., Dong, A., Edwards, A. M., and Denu, J. M. (2011). Structure and biochemical functions of SIRT6. J. Biol. Chem. 286, 14575–14587. doi: 10.1074/jbc.m111.218990
Park, I., Kwon, M. S., Paik, S., Kim, H., Lee, H. O., Choi, E., et al. (2017). HDAC2/3 binding and deacetylation of BubR1 initiates spindle assembly checkpoint silencing. FEBS J. 284, 4035–4050. doi: 10.1111/febs.14286
Park, S. H., Ozden, O., Liu, G., Song, H. Y., Zhu, Y., Yan, Y., et al. (2016). SIRT2-mediated deacetylation and tetramerization of pyruvate kinase directs glycolysis and tumor growth. Cancer Res. 76, 3802–3812. doi: 10.1158/0008-5472.can-15-2498
Pei, Y., Liu, K. W., Wang, J., Garancher, A., Tao, R., Esparza, L. A., et al. (2016). HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 29, 311–323. doi: 10.1016/j.ccell.2016.02.011
Perez-Galan, P., Roue, G., Villamor, N., Montserrat, E., Campo, E., and Colomer, D. (2006). The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood 107, 257–264. doi: 10.1182/blood-2005-05-2091
Phelps, M. P., Bailey, J. N., Vleeshouwer-Neumann, T., and Chen, E. Y. (2016). CRISPR screen identifies the NCOR/HDAC3 complex as a major suppressor of differentiation in rhabdomyosarcoma. Proc. Natl. Acad. Sci. U.S.A. 113, 15090–15095. doi: 10.1073/pnas.1610270114
Pili, R., Quinn, D. I., Hammers, H. J., Monk, P., George, S., Dorff, T. B., et al. (2017). Immunomodulation by entinostat in renal cell carcinoma patients receiving high-dose interleukin 2: a multicenter, single-Arm, Phase I/II Trial (NCI-CTEP#7870). Clin. Cancer Res. 23, 7199–7208. doi: 10.1158/1078-0432.ccr-17-1178
Pilie, P. G., Tang, C., Mills, G. B., and Yap, T. A. (2019). State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 16, 81–104. doi: 10.1038/s41571-018-0114-z
Popat, R., Brown, S. R., Flanagan, L., Hall, A., Gregory, W., Kishore, B., et al. (2016). Bortezomib, thalidomide, dexamethasone, and panobinostat for patients with relapsed multiple myeloma (MUK-six): a multicentre, open-label, phase 1/2 trial. Lancet Haematol. 3, e572–e580.
Potente, M., Ghaeni, L., Baldessari, D., Mostoslavsky, R., Rossig, L., Dequiedt, F., et al. (2007). SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 21, 2644–2658. doi: 10.1101/gad.435107
Pouget, J. P., Georgakilas, A. G., and Ravanat, J. L. (2018). Targeted and off-target (bystander and abscopal) effects of radiation therapy: redox mechanisms and risk/benefit analysis. Antioxid. Redox Signal. 29, 1447–1487. doi: 10.1089/ars.2017.7267
Preglej, T., Hamminger, P., Luu, M., Bulat, T., Andersen, L., Goschl, L., et al. (2020). Histone deacetylases 1 and 2 restrain CD4+ cytotoxic T lymphocyte differentiation. JCI Insight 5:e133393.
Puvvada, S. D., Guillen-Rodriguez, J. M., Rivera, X. I., Heard, K., Inclan, L., Schmelz, M., et al. (2017). A Phase II exploratory study of PXD-101 (Belinostat) followed by zevalin in patients with relapsed aggressive high-risk lymphoma. Oncology 93, 401–405.
Qi, H., Shi, X., Yu, M., Liu, B., Liu, M., Song, S., et al. (2018). Sirtuin 7-mediated deacetylation of WD repeat domain 77 (WDR77) suppresses cancer cell growth by reducing WDR77/PRMT5 transmethylase complex activity. J. Biol. Chem. 293, 17769–17779. doi: 10.1074/jbc.ra118.003629
Qi, J., Singh, S., Hua, W. K., Cai, Q., Chao, S. W., Li, L., et al. (2015). HDAC8 inhibition specifically targets Inv(16) acute myeloid leukemic stem cells by restoring p53 acetylation. Cell Stem Cell 17, 597–610. doi: 10.1016/j.stem.2015.08.004
Qian, D. Z., Kachhap, S. K., Collis, S. J., Verheul, H. M., Carducci, M. A., Atadja, P., et al. (2006). Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 66, 8814–8821. doi: 10.1158/0008-5472.can-05-4598
Radhakrishnan, R., Li, Y., Xiang, S., Yuan, F., Yuan, Z., Telles, E., et al. (2015). Histone deacetylase 10 regulates DNA mismatch repair and may involve the deacetylation of MutS homolog 2. J. Biol. Chem. 290, 22795–22804. doi: 10.1074/jbc.m114.612945
Ramsey, M. R., He, L., Forster, N., Ory, B., and Ellisen, L. W. (2011). Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res. 71, 4373–4379. doi: 10.1158/0008-5472.can-11-0046
Resh, M. D. (2006). Trafficking and signaling by fatty-acylated and prenylated proteins. Nat. Chem. Biol. 2, 584–590. doi: 10.1038/nchembio834
Rezazadeh, S., Yang, D., Biashad, S. A., Firsanov, D., Takasugi, M., Gilbert, M., et al. (2020). SIRT6 mono-ADP ribosylates KDM2A to locally increase H3K36me2 at DNA damage sites to inhibit transcription and promote repair. Aging 12, 11165–11184. doi: 10.18632/aging.103567
Rezazadeh, S., Yang, D., Tombline, G., Simon, M., Regan, S. P., Seluanov, A., et al. (2019). SIRT6 promotes transcription of a subset of NRF2 targets by mono-ADP-ribosylating BAF170. Nucleic Acids Res. 47, 7914–7928. doi: 10.1093/nar/gkz528
Richon, V. M., Sandhoff, T. W., Rifkind, R. A., and Marks, P. A. (2000). Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. U.S.A. 97, 10014–10019. doi: 10.1073/pnas.180316197
Riggs, M. G., Whittaker, R. G., Neumann, J. R., and Ingram, V. M. (1977). n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 268, 462–464. doi: 10.1038/268462a0
Robert, T., Vanoli, F., Chiolo, I., Shubassi, G., Bernstein, K. A., Rothstein, R., et al. (2011). HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 471, 74–79. doi: 10.1038/nature09803
Robertson, K. D., Ait-Si-Ali, S., Yokochi, T., Wade, P. A., Jones, P. L., and Wolffe, A. P. (2000). DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 25, 338–342. doi: 10.1038/77124
Rodriguez-Gonzalez, A., Lin, T., Ikeda, A. K., Simms-Waldrip, T., Fu, C., and Sakamoto, K. M. (2008). Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 68, 2557–2560. doi: 10.1158/0008-5472.can-07-5989
Roos, W. P., Thomas, A. D., and Kaina, B. (2016). DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 16, 20–33. doi: 10.1038/nrc.2015.2
Rountree, M. R., Bachman, K. E., and Baylin, S. B. (2000). DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 25, 269–277. doi: 10.1038/77023
Rumpf, T., Schiedel, M., Karaman, B., Roessler, C., North, B. J., Lehotzky, A., et al. (2015). Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 6:6263.
Rundlett, S. E., Carmen, A. A., Kobayashi, R., Bavykin, S., Turner, B. M., and Grunstein, M. (1996). HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc. Natl. Acad. Sci. U.S.A. 93, 14503–14508. doi: 10.1073/pnas.93.25.14503
Ryu, D., Jo, Y. S., Lo Sasso, G., Stein, S., Zhang, H., Perino, A., et al. (2014). A SIRT7-dependent acetylation switch of GABPbeta1 controls mitochondrial function. Cell Metab. 20, 856–869. doi: 10.1016/j.cmet.2014.08.001
Sanchez, G. J., Richmond, P. A., Bunker, E. N., Karman, S. S., Azofeifa, J., Garnett, A. T., et al. (2018). Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 46, 1756–1776. doi: 10.1093/nar/gkx1225
San-Miguel, J. F., Hungria, V. T., Yoon, S. S., Beksac, M., Dimopoulos, M. A., Elghandour, A., et al. (2016). Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): a randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 3, e506–e515.
Santos-Barriopedro, I., Bosch-Presegue, L., Marazuela-Duque, A., De La Torre, C., Colomer, C., Vazquez, B. N., et al. (2018). SIRT6-dependent cysteine monoubiquitination in the PRE-SET domain of Suv39h1 regulates the NF-kappaB pathway. Nat. Commun. 9:101.
Scher, M. B., Vaquero, A., and Reinberg, D. (2007). SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 21, 920–928. doi: 10.1101/gad.1527307
Schlicker, C., Gertz, M., Papatheodorou, P., Kachholz, B., Becker, C. F., and Steegborn, C. (2008). Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 382, 790–801. doi: 10.1016/j.jmb.2008.07.048
Scholz, C., Weinert, B. T., Wagner, S. A., Beli, P., Miyake, Y., Qi, J., et al. (2015). Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol. 33, 415–423. doi: 10.1038/nbt.3130
Schreiber, V., Dantzer, F., Ame, J. C., and De Murcia, G. (2006). Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 7, 517–528. doi: 10.1038/nrm1963
Schwer, B., North, B. J., Frye, R. A., Ott, M., and Verdin, E. (2002). The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 158, 647–657. doi: 10.1083/jcb.200205057
Scully, R., Panday, A., Elango, R., and Willis, N. A. (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 20, 698–714. doi: 10.1038/s41580-019-0152-0
Sebastian, C., Zwaans, B. M., Silberman, D. M., Gymrek, M., Goren, A., Zhong, L., et al. (2012). The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151, 1185–1199. doi: 10.1016/j.cell.2012.10.047
Seigneurin-Berny, D., Verdel, A., Curtet, S., Lemercier, C., Garin, J., Rousseaux, S., et al. (2001). Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol. Cell. Biol. 21, 8035–8044. doi: 10.1128/mcb.21.23.8035-8044.2001
Sen, N., Kumari, R., Singh, M. I., and Das, S. (2013). HDAC5, a key component in temporal regulation of p53-mediated transactivation in response to genotoxic stress. Mol. Cell 52, 406–420. doi: 10.1016/j.molcel.2013.09.003
Senft, D., Qi, J., and Ronai, Z. A. (2018). Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 18, 69–88. doi: 10.1038/nrc.2017.105
Serrano, L., Martinez-Redondo, P., Marazuela-Duque, A., Vazquez, B. N., Dooley, S. J., Voigt, P., et al. (2013). The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 27, 639–653. doi: 10.1101/gad.211342.112
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6:a018713. doi: 10.1101/cshperspect.a018713
Shan, B., Yao, T. P., Nguyen, H. T., Zhuo, Y., Levy, D. R., Klingsberg, R. C., et al. (2008). Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J. Biol. Chem. 283, 21065–21073. doi: 10.1074/jbc.m802786200
Shankaranarayana, G. D., Motamedi, M. R., Moazed, D., and Grewal, S. I. (2003). Sir2 regulates histone H3 lysine 9 methylation and heterochromatin assembly in fission yeast. Curr. Biol. 13, 1240–1246. doi: 10.1016/s0960-9822(03)00489-5
Shaw, E., Talwadekar, M., Rashida, Z., Mohan, N., Acharya, A., Khatri, S., et al. (2020). Anabolic SIRT4 exerts retrograde control over torc1 signaling by glutamine sparing in the mitochondria. Mol. Cell. Biol. 40:e00212-19.
Shechter, D., Dormann, H. L., Allis, C. D., and Hake, S. B. (2007). Extraction, purification and analysis of histones. Nat. Protoc. 2, 1445–1457. doi: 10.1038/nprot.2007.202
Shi, K., Yin, X., Cai, M. C., Yan, Y., Jia, C., Ma, P., et al. (2019). PAX8 regulon in human ovarian cancer links lineage dependency with epigenetic vulnerability to HDAC inhibitors. eLife 8:e44306.
Shi, Y., Dong, M., Hong, X., Zhang, W., Feng, J., Zhu, J., et al. (2015). Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann. Oncol. 26, 1766–1771. doi: 10.1093/annonc/mdv237
Shi, Y. J., Matson, C., Lan, F., Iwase, S., Baba, T., and Shi, Y. (2005). Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 19, 857–864. doi: 10.1016/j.molcel.2005.08.027
Shinsky, S. A., and Christianson, D. W. (2018). Polyamine deacetylase structure and catalysis: prokaryotic acetylpolyamine amidohydrolase and eukaryotic HDAC10. Biochemistry 57, 3105–3114. doi: 10.1021/acs.biochem.8b00079
Shogren-Knaak, M., Ishii, H., Sun, J. M., Pazin, M. J., Davie, J. R., and Peterson, C. L. (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847. doi: 10.1126/science.1124000
Simon, M., Van Meter, M., Ablaeva, J., Ke, Z., Gonzalez, R. S., Taguchi, T., et al. (2019). LINE1 Derepression in Aged Wild-Type and SIRT6-deficient mice drives inflammation. Cell Metab. 29, 871–885.e5.
Sinclair, D. A., and Guarente, L. (2014). Small-molecule allosteric activators of sirtuins. Annu. Rev. Pharmacol. Toxicol. 54, 363–380. doi: 10.1146/annurev-pharmtox-010611-134657
Singh, A. K., Bishayee, A., and Pandey, A. K. (2018). Targeting histone deacetylases with natural and synthetic agents: an emerging anticancer strategy. Nutrients 10:731. doi: 10.3390/nu10060731
Singh, R., Letai, A., and Sarosiek, K. (2019). Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 20, 175–193. doi: 10.1038/s41580-018-0089-8
Somoza, J. R., Skene, R. J., Katz, B. A., Mol, C., Ho, J. D., Jennings, A. J., et al. (2004). Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12, 1325–1334. doi: 10.1016/j.str.2004.04.012
Son, S. I., Cao, J., Zhu, C. L., Miller, S. P., and Lin, H. (2019). Activity-Guided Design of HDAC11-Specific Inhibitors. ACS Chem. Biol. 14, 1393–1397. doi: 10.1021/acschembio.9b00292
Sparrow, D. B., Miska, E. A., Langley, E., Reynaud-Deonauth, S., Kotecha, S., Towers, N., et al. (1999). MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 18, 5085–5098. doi: 10.1093/emboj/18.18.5085
Stojanovic, N., Hassan, Z., Wirth, M., Wenzel, P., Beyer, M., Schafer, C., et al. (2017). HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 36, 1804–1815. doi: 10.1038/onc.2016.344
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi: 10.1038/47412
Sun, J., Piao, J., Li, N., Yang, Y., Kim, K. Y., and Lin, Z. (2020). Valproic acid targets HDAC1/2 and HDAC1/PTEN/Akt signalling to inhibit cell proliferation via the induction of autophagy in gastric cancer. FEBS J. 287, 2118–2133. doi: 10.1111/febs.15122
Sun, L., Marin De Evsikova, C., Bian, K., Achille, A., Telles, E., Pei, H., et al. (2018). Programming and regulation of metabolic homeostasis by HDAC11. EBioMedicine 33, 157–168. doi: 10.1016/j.ebiom.2018.06.025
Sun, Q. Y., Ding, L. W., Johnson, K., Zhou, S., Tyner, J. W., Yang, H., et al. (2019). SOX7 regulates MAPK/ERK-BIM mediated apoptosis in cancer cells. Oncogene 38, 6196–6210. doi: 10.1038/s41388-019-0865-8
Sun, Y., Liu, P. Y., Scarlett, C. J., Malyukova, A., Liu, B., Marshall, G. M., et al. (2014). Histone deacetylase 5 blocks neuroblastoma cell differentiation by interacting with N-Myc. Oncogene 33, 2987–2994. doi: 10.1038/onc.2013.253
Sundaresan, N. R., Samant, S. A., Pillai, V. B., Rajamohan, S. B., and Gupta, M. P. (2008). SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 28, 6384–6401. doi: 10.1128/mcb.00426-08
Tae, I. H., Park, E. Y., Dey, P., Son, J. Y., Lee, S. Y., Jung, J. H., et al. (2018). Novel SIRT1 inhibitor 15-deoxy-Delta12,14-prostaglandin J2 and its derivatives exhibit anticancer activity through apoptotic or autophagic cell death pathways in SKOV3 cells. Int. J. Oncol. 53, 2518–2530.
Tae, I. H., Son, J. Y., Lee, S. H., Ahn, M. Y., Yoon, K., Yoon, S., et al. (2020). A new SIRT1 inhibitor, MHY2245, induces autophagy and inhibits energy metabolism via PKM2/mTOR pathway in human ovarian cancer cells. Int. J. Biol. Sci. 16, 1901–1916. doi: 10.7150/ijbs.44343
Tam, W. L., and Weinberg, R. A. (2013). The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 19, 1438–1449. doi: 10.1038/nm.3336
Tan, M., Peng, C., Anderson, K. A., Chhoy, P., Xie, Z., Dai, L., et al. (2014). Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 19, 605–617. doi: 10.1016/j.cmet.2014.03.014
Tang, M., Li, Z., Zhang, C., Lu, X., Tu, B., Cao, Z., et al. (2019). SIRT7-mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Sci. Adv. 5:eaav1118. doi: 10.1126/sciadv.aav1118
Tang, M., Lu, X., Zhang, C., Du, C., Cao, L., Hou, T., et al. (2017). Downregulation of SIRT7 by 5-fluorouracil induces radiosensitivity in human colorectal cancer. Theranostics 7, 1346–1359. doi: 10.7150/thno.18804
Tang, S., Fang, Y., Huang, G., Xu, X., Padilla-Banks, E., Fan, W., et al. (2017). Methionine metabolism is essential for SIRT1-regulated mouse embryonic stem cell maintenance and embryonic development. EMBO J. 36, 3175–3193. doi: 10.15252/embj.201796708
Tang, S., Huang, G., Fan, W., Chen, Y., Ward, J. M., Xu, X., et al. (2014). SIRT1-mediated deacetylation of CRABPII regulates cellular retinoic acid signaling and modulates embryonic stem cell differentiation. Mol. Cell 55, 843–855. doi: 10.1016/j.molcel.2014.07.011
Tang, X., Li, G., Su, F., Cai, Y., Shi, L., Meng, Y., et al. (2020). HDAC8 cooperates with SMAD3/4 complex to suppress SIRT7 and promote cell survival and migration. Nucleic Acids Res. 48, 2912–2923. doi: 10.1093/nar/gkaa039
Tang, X., Shi, L., Xie, N., Liu, Z., Qian, M., Meng, F., et al. (2017). SIRT7 antagonizes TGF-beta signaling and inhibits breast cancer metastasis. Nat. Commun. 8:318.
Tanno, M., Sakamoto, J., Miura, T., Shimamoto, K., and Horio, Y. (2007). Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 282, 6823–6832. doi: 10.1074/jbc.m609554200
Tanny, J. C., Dowd, G. J., Huang, J., Hilz, H., and Moazed, D. (1999). An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell 99, 735–745. doi: 10.1016/s0092-8674(00)81671-2
Taunton, J., Hassig, C. A., and Schreiber, S. L. (1996). A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 272, 408–411. doi: 10.1126/science.272.5260.408
Taverna, S. D., Li, H., Ruthenburg, A. J., Allis, C. D., and Patel, D. J. (2007). How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040. doi: 10.1038/nsmb1338
Tessarz, P., and Kouzarides, T. (2014). Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 15, 703–708. doi: 10.1038/nrm3890
Tian, J. L., and Qin, H. (2019). O-GlcNAcylation regulates primary ciliary length by promoting microtubule disassembly. iScience 12, 379–391. doi: 10.1016/j.isci.2019.01.031
Tian, X., Firsanov, D., Zhang, Z., Cheng, Y., Luo, L., Tombline, G., et al. (2019). SIRT6 is responsible for more efficient DNA double-strand break repair in long-lived species. Cell 177, 622–638.e22.
Tian, Y., Wong, V. W., Wong, G. L., Yang, W., Sun, H., Shen, J., et al. (2015). Histone Deacetylase HDAC8 promotes insulin resistance and beta-catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res. 75, 4803–4816. doi: 10.1158/0008-5472.can-14-3786
Toiber, D., Erdel, F., Bouazoune, K., Silberman, D. M., Zhong, L., Mulligan, P., et al. (2013). SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell 51, 454–468. doi: 10.1016/j.molcel.2013.06.018
Tong, J., Zheng, X., Tan, X., Fletcher, R., Nikolovska-Coleska, Z., Yu, J., et al. (2018). Mcl-1 phosphorylation without degradation mediates sensitivity to HDAC inhibitors by liberating BH3-only proteins. Cancer Res. 78, 4704–4715. doi: 10.1158/0008-5472.can-18-0399
Tong, J. J., Liu, J., Bertos, N. R., and Yang, X. J. (2002). Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res. 30, 1114–1123. doi: 10.1093/nar/30.5.1114
Topper, M. J., Vaz, M., Chiappinelli, K. B., Destefano Shields, C. E., Niknafs, N., Yen, R. C., et al. (2017). Epigenetic therapy Ties MYC depletion to reversing immune evasion and treating lung cancer. Cell 171, 1284–1300.e21.
Toubai, T., Tamaki, H., Peltier, D. C., Rossi, C., Oravecz-Wilson, K., Liu, C., et al. (2018). Mitochondrial Deacetylase SIRT3 plays an important role in donor T cell responses after experimental allogeneic hematopoietic transplantation. J. Immunol. 201, 3443–3455. doi: 10.4049/jimmunol.1800148
Tsuji, N., Kobayashi, M., Nagashima, K., Wakisaka, Y., and Koizumi, K. (1976). A new antifungal antibiotic, trichostatin. J. Antibiot. 29, 1–6. doi: 10.7164/antibiotics.29.1
Turnbull, R. E., Fairall, L., Saleh, A., Kelsall, E., Morris, K. L., Ragan, T. J., et al. (2020). The MiDAC histone deacetylase complex is essential for embryonic development and has a unique multivalent structure. Nat. Commun. 11:3252.
Ustinova, K., Novakova, Z., Saito, M., Meleshin, M., Mikesova, J., Kutil, Z., et al. (2020). The disordered N-terminus of HDAC6 is a microtubule-binding domain critical for efficient tubulin deacetylation. J. Biol. Chem. 295, 2614–2628. doi: 10.1074/jbc.ra119.011243
Vakhrusheva, O., Smolka, C., Gajawada, P., Kostin, S., Boettger, T., Kubin, T., et al. (2008). Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 102, 703–710. doi: 10.1161/circresaha.107.164558
Van Den Wyngaert, I., De Vries, W., Kremer, A., Neefs, J., Verhasselt, P., Luyten, W. H., et al. (2000). Cloning and characterization of human histone deacetylase 8. FEBS Lett. 478, 77–83. doi: 10.1016/s0014-5793(00)01813-5
Van Der Vlag, J., and Otte, A. P. (1999). Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet. 23, 474–478. doi: 10.1038/70602
Van Meter, M., Kashyap, M., Rezazadeh, S., Geneva, A. J., Morello, T. D., Seluanov, A., et al. (2014). SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 5:5011.
Van Tilburg, C. M., Milde, T., Witt, R., Ecker, J., Hielscher, T., Seitz, A., et al. (2019). Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma, or leukemia. Clin. Epigenetics 11:188.
Vaquero, A., Scher, M., Erdjument-Bromage, H., Tempst, P., Serrano, L., and Reinberg, D. (2007). SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature 450, 440–444. doi: 10.1038/nature06268
Vaziri, H., Dessain, S. K., Ng Eaton, E., Imai, S. I., Frye, R. A., Pandita, T. K., et al. (2001). hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159. doi: 10.1016/s0092-8674(01)00527-x
Vazquez, B. N., Thackray, J. K., Simonet, N. G., Chahar, S., Kane-Goldsmith, N., Newkirk, S. J., et al. (2019). SIRT7 mediates L1 elements transcriptional repression and their association with the nuclear lamina. Nucleic Acids Res. 47, 7870–7885. doi: 10.1093/nar/gkz519
Vazquez, B. N., Thackray, J. K., Simonet, N. G., Kane-Goldsmith, N., Martinez-Redondo, P., Nguyen, T., et al. (2016). SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 35, 1488–1503. doi: 10.15252/embj.201593499
Verdel, A., Curtet, S., Brocard, M. P., Rousseaux, S., Lemercier, C., Yoshida, M., et al. (2000). Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr. Biol. 10, 747–749. doi: 10.1016/s0960-9822(00)00542-x
Verdin, E., and Ott, M. (2015). 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 16, 258–264. doi: 10.1038/nrm3931
Vidali, G., Boffa, L. C., Bradbury, E. M., and Allfrey, V. G. (1978). Butyrate suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc. Natl. Acad. Sci. U.S.A. 75, 2239–2243. doi: 10.1073/pnas.75.5.2239
Vogl, D. T., Raje, N., Jagannath, S., Richardson, P., Hari, P., Orlowski, R., et al. (2017). Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin. Cancer Res. 23, 3307–3315. doi: 10.1158/1078-0432.ccr-16-2526
Von Holt, C., Brandt, W. F., Greyling, H. J., Lindsey, G. G., Retief, J. D., Rodrigues, J. D., et al. (1989). Isolation and characterization of histones. Methods Enzymol. 170, 431–523.
Vriend, J., and Reiter, R. J. (2016). Melatonin and the von Hippel-Lindau/HIF-1 oxygen sensing mechanism: a review. Biochim. Biophys. Acta 1865, 176–183. doi: 10.1016/j.bbcan.2016.02.004
Walkinshaw, D. R., Weist, R., Kim, G. W., You, L., Xiao, L., Nie, J., et al. (2013). The tumor suppressor kinase LKB1 activates the downstream kinases SIK2 and SIK3 to stimulate nuclear export of class IIa histone deacetylases. J. Biol. Chem. 288, 9345–9362. doi: 10.1074/jbc.m113.456996
Wang, A., Kurdistani, S. K., and Grunstein, M. (2002). Requirement of Hos2 histone deacetylase for gene activity in yeast. Science 298, 1412–1414. doi: 10.1126/science.1077790
Wang, A. H., Bertos, N. R., Vezmar, M., Pelletier, N., Crosato, M., Heng, H. H., et al. (1999). HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol. Cell. Biol. 19, 7816–7827. doi: 10.1128/mcb.19.11.7816
Wang, A. H., Kruhlak, M. J., Wu, J., Bertos, N. R., Vezmar, M., Posner, B. I., et al. (2000). Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol. Cell. Biol. 20, 6904–6912. doi: 10.1128/mcb.20.18.6904-6912.2000
Wang, A. H., and Yang, X. J. (2001). Histone deacetylase 4 possesses intrinsic nuclear import and export signals. Mol. Cell. Biol. 21, 5992–6005. doi: 10.1128/mcb.21.17.5992-6005.2001
Wang, B., Rao, Y. H., Inoue, M., Hao, R., Lai, C. H., Chen, D., et al. (2014). Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production. Nat. Commun. 5:3479.
Wang, B., Ye, Y., Yang, X., Liu, B., Wang, Z., Chen, S., et al. (2020). SIRT2-dependent IDH1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep. 21:e48183.
Wang, F., Nguyen, M., Qin, F. X., and Tong, Q. (2007). SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6, 505–514. doi: 10.1111/j.1474-9726.2007.00304.x
Wang, H., Zhou, W., Zheng, Z., Zhang, P., Tu, B., He, Q., et al. (2012). The HDAC inhibitor depsipeptide transactivates the p53/p21 pathway by inducing DNA damage. DNA Repair 11, 146–156. doi: 10.1016/j.dnarep.2011.10.014
Wang, J., Jiang, J., Chen, H., Wang, L., Guo, H., Yang, L., et al. (2019). FDA-approved drug screen identifies proteasome as a synthetic lethal target in MYC-driven neuroblastoma. Oncogene 38, 6737–6751. doi: 10.1038/s41388-019-0912-5
Wang, R. H., Sengupta, K., Li, C., Kim, H. S., Cao, L., Xiao, C., et al. (2008). Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14, 312–323. doi: 10.1016/j.ccr.2008.09.001
Wang, W. W., Zeng, Y., Wu, B., Deiters, A., and Liu, W. R. (2016). A chemical biology approach to reveal sirt6-targeted histone H3 sites in nucleosomes. ACS Chem. Biol. 11, 1973–1981. doi: 10.1021/acschembio.6b00243
Wang, X., Waschke, B. C., Woolaver, R. A., Chen, S. M. Y., Chen, Z., and Wang, J. H. (2020). HDAC inhibitors overcome immunotherapy resistance in B-cell lymphoma. Protein Cell 11, 472–482. doi: 10.1007/s13238-020-00694-x
Wang, Y., He, J., Liao, M., Hu, M., Li, W., Ouyang, H., et al. (2019). An overview of Sirtuins as potential therapeutic target: Structure, function and modulators. Eur. J. Med. Chem. 161, 48–77. doi: 10.1016/j.ejmech.2018.10.028
Wang, Y. P., Zhou, L. S., Zhao, Y. Z., Wang, S. W., Chen, L. L., Liu, L. X., et al. (2014). Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 33, 1304–1320.
Wang, Z., Zang, C., Cui, K., Schones, D. E., Barski, A., Peng, W., et al. (2009). Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031. doi: 10.1016/j.cell.2009.06.049
Watson, P. J., Fairall, L., Santos, G. M., and Schwabe, J. W. (2012). Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 481, 335–340. doi: 10.1038/nature10728
Watson, P. J., Millard, C. J., Riley, A. M., Robertson, N. S., Wright, L. C., Godage, H. Y., et al. (2016). Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat. Commun. 7:11262.
Wei, W., Liu, X., Chen, J., Gao, S., Lu, L., Zhang, H., et al. (2017). Class I histone deacetylases are major histone decrotonylases: evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 27, 898–915. doi: 10.1038/cr.2017.68
Wei, Z., Song, J., Wang, G., Cui, X., Zheng, J., Tang, Y., et al. (2018). Deacetylation of serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal carcinogenesis. Nat. Commun. 9:4468.
Weinhouse, S. (1956). On respiratory impairment in cancer cells. Science 124, 267–269. doi: 10.1126/science.124.3215.267
Wen, Y. D., Perissi, V., Staszewski, L. M., Yang, W. M., Krones, A., Glass, C. K., et al. (2000). The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. U.S.A. 97, 7202–7207. doi: 10.1073/pnas.97.13.7202
Weng, C. H., Chen, L. Y., Lin, Y. C., Shih, J. Y., Lin, Y. C., Tseng, R. Y., et al. (2019). Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 38, 455–468. doi: 10.1038/s41388-018-0454-2
West, A. C., and Johnstone, R. W. (2014). New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 124, 30–39. doi: 10.1172/jci69738
Wilson, J. M., Le, V. Q., Zimmerman, C., Marmorstein, R., and Pillus, L. (2006). Nuclear export modulates the cytoplasmic Sir2 homologue Hst2. EMBO Rep. 7, 1247–1251. doi: 10.1038/sj.embor.7400829
Winter, M., Moser, M. A., Meunier, D., Fischer, C., Machat, G., Mattes, K., et al. (2013). Divergent roles of HDAC1 and HDAC2 in the regulation of epidermal development and tumorigenesis. EMBO J. 32, 3176–3191. doi: 10.1038/emboj.2013.243
Woods, D. M., Woan, K. V., Cheng, F., Sodre, A. L., Wang, D., Wu, Y., et al. (2017). T cells lacking HDAC11 have increased effector functions and mediate enhanced alloreactivity in a murine model. Blood 130, 146–155. doi: 10.1182/blood-2016-08-731505
Wu, L. P., Wang, X., Li, L., Zhao, Y., Lu, S., Yu, Y., et al. (2008). Histone deacetylase inhibitor depsipeptide activates silenced genes through decreasing both CpG and H3K9 methylation on the promoter. Mol. Cell. Biol. 28, 3219–3235. doi: 10.1128/mcb.01516-07
Wu, M. Z., Tsai, Y. P., Yang, M. H., Huang, C. H., Chang, S. Y., Chang, C. C., et al. (2011). Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol. Cell 43, 811–822. doi: 10.1016/j.molcel.2011.07.012
Xiao, H., Jiao, J., Wang, L., O’brien, S., Newick, K., Wang, L. C., et al. (2016). HDAC5 controls the functions of Foxp3(+) T-regulatory and CD8(+) T cells. Int. J. Cancer 138, 2477–2486. doi: 10.1002/ijc.29979
Xu, C., Jin, J., Bian, C., Lam, R., Tian, R., Weist, R., et al. (2012). Sequence-specific recognition of a PxLPxI/L motif by an ankyrin repeat tumbler lock. Sci. Signal. 5:ra39. doi: 10.1126/scisignal.2002979
Xu, L., Li, Y., Zhou, L., Dorfman, R. G., Liu, L., Cai, R., et al. (2019). SIRT3 elicited an anti-Warburg effect through HIF1alpha/PDK1/PDHA1 to inhibit cholangiocarcinoma tumorigenesis. Cancer Med. 8, 2380–2391. doi: 10.1002/cam4.2089
Xu, W., Ngo, L., Perez, G., Dokmanovic, M., and Marks, P. A. (2006). Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc. Natl. Acad. Sci. U.S.A. 103, 15540–15545. doi: 10.1073/pnas.0607518103
Xu, Y., Zhang, S., Lin, S., Guo, Y., Deng, W., Zhang, Y., et al. (2017). WERAM: a database of writers, erasers and readers of histone acetylation and methylation in eukaryotes. Nucleic Acids Res. 45, D264–D270.
Yan, W., Liu, S., Xu, E., Zhang, J., Zhang, Y., Chen, X., et al. (2013). Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 32, 599–609. doi: 10.1038/onc.2012.81
Yan, W. W., Liang, Y. L., Zhang, Q. X., Wang, D., Lei, M. Z., Qu, J., et al. (2018). Arginine methylation of SIRT7 couples glucose sensing with mitochondria biogenesis. EMBO Rep. 19:e46377.
Yan, Y., An, J., Yang, Y., Wu, D., Bai, Y., Cao, W., et al. (2018). Dual inhibition of AKT-mTOR and AR signaling by targeting HDAC3 in PTEN- or SPOP-mutated prostate cancer. EMBO Mol. Med. 10:e8478.
Yang, L., Ma, X., He, Y., Yuan, C., Chen, Q., Li, G., et al. (2017). Sirtuin 5: a review of structure, known inhibitors and clues for developing new inhibitors. Sci. China Life Sci. 60, 249–256. doi: 10.1007/s11427-016-0060-7
Yang, W., Nagasawa, K., Munch, C., Xu, Y., Satterstrom, K., Jeong, S., et al. (2016). Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell 167, 985–1000.e21.
Yang, W., Xia, Y., Hawke, D., Li, X., Liang, J., Xing, D., et al. (2012). PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 150, 685–696. doi: 10.1016/j.cell.2012.07.018
Yang, W., Xia, Y., Ji, H., Zheng, Y., Liang, J., Huang, W., et al. (2011). Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 480, 118–122. doi: 10.1038/nature10598
Yang, W. M., Inouye, C., Zeng, Y., Bearss, D., and Seto, E. (1996). Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. U.S.A. 93, 12845–12850. doi: 10.1073/pnas.93.23.12845
Yang, W. M., Yao, Y. L., Sun, J. M., Davie, J. R., and Seto, E. (1997). Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 272, 28001–28007. doi: 10.1074/jbc.272.44.28001
Yang, X., Wang, Z., Li, X., Liu, B., Liu, M., Liu, L., et al. (2018). SHMT2 desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Res. 78, 372–386. doi: 10.1158/0008-5472.can-17-1912
Yang, X. J., and Gregoire, S. (2005). Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol. Cell. Biol. 25, 2873–2884. doi: 10.1128/mcb.25.8.2873-2884.2005
Yang, X. J., and Gregoire, S. (2007). Metabolism, cytoskeleton and cellular signalling in the grip of protein Nepsilon - and O-acetylation. EMBO Rep. 8, 556–562. doi: 10.1038/sj.embor.7400977
Yang, X. J., and Seto, E. (2008a). Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 31, 449–461. doi: 10.1016/j.molcel.2008.07.002
Yang, X. J., and Seto, E. (2008b). The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218. doi: 10.1038/nrm2346
Yang, Y., Tse, A. K., Li, P., Ma, Q., Xiang, S., Nicosia, S. V., et al. (2011). Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene 30, 2207–2218. doi: 10.1038/onc.2010.600
Yang, Y., Zhao, Y., Liao, W., Yang, J., Wu, L., Zheng, Z., et al. (2009). Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia 11, 313–324. doi: 10.1593/neo.81358
Yao, Y., Yang, Y., and Zhu, W. G. (2014). Sirtuins: nodes connecting aging, metabolism and tumorigenesis. Curr. Pharm. Des. 20, 1614–1624. doi: 10.2174/13816128113199990513
Ye, T., Wei, L., Shi, J., Jiang, K., Xu, H., Hu, L., et al. (2019). Sirtuin1 activator SRT2183 suppresses glioma cell growth involving activation of endoplasmic reticulum stress pathway. BMC Cancer 19:706. doi: 10.1186/s12885-019-5852-5
Ye, X., Tam, W. L., Shibue, T., Kaygusuz, Y., Reinhardt, F., Ng Eaton, E., et al. (2015). Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 525, 256–260. doi: 10.1038/nature14897
Yoshida, M., and Horinouchi, S. (1999). Trichostatin and leptomycin. Inhibition of histone deacetylation and signal-dependent nuclear export. Ann. N. Y. Acad. Sci. 886, 23–36. doi: 10.1111/j.1749-6632.1999.tb09397.x
You, A., Tong, J. K., Grozinger, C. M., and Schreiber, S. L. (2001). CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc. Natl. Acad. Sci. U.S.A. 98, 1454–1458. doi: 10.1073/pnas.98.4.1454
Younes, A., Berdeja, J. G., Patel, M. R., Flinn, I., Gerecitano, J. F., Neelapu, S. S., et al. (2016). Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol. 17, 622–631. doi: 10.1016/s1470-2045(15)00584-7
Yu, M., Shi, X., Ren, M., Liu, L., Qi, H., Zhang, C., et al. (2020). SIRT7 deacetylates STRAP to regulate p53 activity and stability. Int. J. Mol. Sci. 21:4122. doi: 10.3390/ijms21114122
Yuan, H., Tan, B., and Gao, S. J. (2017). Tenovin-6 impairs autophagy by inhibiting autophagic flux. Cell Death Dis. 8:e2608. doi: 10.1038/cddis.2017.25
Yuan, H., Wang, Z., Li, L., Zhang, H., Modi, H., Horne, D., et al. (2012). Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 119, 1904–1914. doi: 10.1182/blood-2011-06-361691
Yuan, J., Luo, K., Liu, T., and Lou, Z. (2012). Regulation of SIRT1 activity by genotoxic stress. Genes Dev. 26, 791–796. doi: 10.1101/gad.188482.112
Yuan, Z., Zhang, X., Sengupta, N., Lane, W. S., and Seto, E. (2007). SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 27, 149–162. doi: 10.1016/j.molcel.2007.05.029
Yue, L., Sharma, V., Horvat, N. P., Akuffo, A. A., Beatty, M. S., Murdun, C., et al. (2020). HDAC11 deficiency disrupts oncogene-induced hematopoiesis in myeloproliferative neoplasms. Blood 135, 191–207. doi: 10.1182/blood.2019895326
Yun, T., Yu, K., Yang, S., Cui, Y., Wang, Z., Ren, H., et al. (2016). Acetylation of p53 Protein at Lysine 120 Up-regulates Apaf-1 Protein and Sensitizes the Mitochondrial Apoptotic Pathway. J. Biol. Chem. 291, 7386–7395. doi: 10.1074/jbc.m115.706341
Zaware, N., and Zhou, M. M. (2019). Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 26, 870–879. doi: 10.1038/s41594-019-0309-8
Zhang, C. L., Mckinsey, T. A., Chang, S., Antos, C. L., Hill, J. A., and Olson, E. N. (2002a). Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110, 479–488. doi: 10.1016/s0092-8674(02)00861-9
Zhang, C. L., Mckinsey, T. A., and Olson, E. N. (2002b). Association of class II histone deacetylases with heterochromatin protein 1: potential role for histone methylation in control of muscle differentiation. Mol. Cell. Biol. 22, 7302–7312. doi: 10.1128/mcb.22.20.7302-7312.2002
Zhang, D., Yoon, H. G., and Wong, J. (2005). JMJD2A is a novel N-CoR-interacting protein and is involved in repression of the human transcription factor achaete scute-like homologue 2 (ASCL2/Hash2). Mol. Cell. Biol. 25, 6404–6414. doi: 10.1128/mcb.25.15.6404-6414.2005
Zhang, M., Wu, J., Sun, R., Tao, X., Wang, X., Kang, Q., et al. (2019). SIRT5 deficiency suppresses mitochondrial ATP production and promotes AMPK activation in response to energy stress. PLoS One 14:e0211796. doi: 10.1371/journal.pone.0211796
Zhang, P., Tu, B., Wang, H., Cao, Z., Tang, M., Zhang, C., et al. (2014). Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc. Natl. Acad. Sci. U.S.A. 111, 10684–10689. doi: 10.1073/pnas.1411026111
Zhang, P. Y., Li, G., Deng, Z. J., Liu, L. Y., Chen, L., Tang, J. Z., et al. (2016). Dicer interacts with SIRT7 and regulates H3K18 deacetylation in response to DNA damaging agents. Nucleic Acids Res. 44, 3629–3642. doi: 10.1093/nar/gkv1504
Zhang, X., Chen, X., Lin, J., Lwin, T., Wright, G., Moscinski, L. C., et al. (2012a). Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene 31, 3002–3008. doi: 10.1038/onc.2011.470
Zhang, X., Zhao, X., Fiskus, W., Lin, J., Lwin, T., Rao, R., et al. (2012b). Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell 22, 506–523. doi: 10.1016/j.ccr.2012.09.003
Zhang, Y., Kwon, S., Yamaguchi, T., Cubizolles, F., Rousseaux, S., Kneissel, M., et al. (2008). Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 28, 1688–1701. doi: 10.1128/mcb.01154-06
Zhang, Y., Ng, H. H., Erdjument-Bromage, H., Tempst, P., Bird, A., and Reinberg, D. (1999). Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13, 1924–1935. doi: 10.1101/gad.13.15.1924
Zhang, Y., Wang, X. L., Zhou, M., Kang, C., Lang, H. D., Chen, M. T., et al. (2018). Crosstalk between gut microbiota and Sirtuin-3 in colonic inflammation and tumorigenesis. Exp. Mol. Med. 50:21.
Zhao, K., and Zhou, Z. (2019). Post-translational modifications of nuclear sirtuins. Genome Instabil. Dis. 1, 34–45. doi: 10.1007/s42764-019-00001-x
Zhao, W., Kruse, J. P., Tang, Y., Jung, S. Y., Qin, J., and Gu, W. (2008). Negative regulation of the deacetylase SIRT1 by DBC1. Nature 451, 587–590. doi: 10.1038/nature06515
Zhao, X., Sternsdorf, T., Bolger, T. A., Evans, R. M., and Yao, T. P. (2005). Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol. Cell. Biol. 25, 8456–8464. doi: 10.1128/mcb.25.19.8456-8464.2005
Zhao, Y., Lu, S., Wu, L., Chai, G., Wang, H., Chen, Y., et al. (2006). Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol. Cell. Biol. 26, 2782–2790. doi: 10.1128/mcb.26.7.2782-2790.2006
Zhao, Y., Yang, J., Liao, W., Liu, X., Zhang, H., Wang, S., et al. (2010). Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 12, 665–675. doi: 10.1038/ncb2069
Zhong, L., D’urso, A., Toiber, D., Sebastian, C., Henry, R. E., Vadysirisack, D. D., et al. (2010). The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140, 280–293. doi: 10.1016/j.cell.2009.12.041
Zhou, L., Wang, F., Sun, R., Chen, X., Zhang, M., Xu, Q., et al. (2016). SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 17, 811–822. doi: 10.15252/embr.201541643
Zhou, X., Marks, P. A., Rifkind, R. A., and Richon, V. M. (2001). Cloning and characterization of a histone deacetylase, HDAC9. Proc. Natl. Acad. Sci. U.S.A. 98, 10572–10577. doi: 10.1073/pnas.191375098
Zhu, G., Tao, T., Zhang, D., Liu, X., Qiu, H., Han, L., et al. (2016). O-GlcNAcylation of histone deacetylases 1 in hepatocellular carcinoma promotes cancer progression. Glycobiology 26, 820–833. doi: 10.1093/glycob/cww025
Zhu, W. G. (2017). Regulation of p53 acetylation. Sci. China Life Sci. 60, 321–323. doi: 10.1007/s11427-016-0353-0
Zhu, W. G., Dai, Z., Ding, H., Srinivasan, K., Hall, J., Duan, W., et al. (2001a). Increased expression of unmethylated CDKN2D by 5-aza-2’-deoxycytidine in human lung cancer cells. Oncogene 20, 7787–7796. doi: 10.1038/sj.onc.1204970
Zhu, W. G., Lakshmanan, R. R., Beal, M. D., and Otterson, G. A. (2001b). DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Res. 61, 1327–1333.
Zhu, W. G., Srinivasan, K., Dai, Z., Duan, W., Druhan, L. J., Ding, H., et al. (2003). Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21(Cip1) promoter. Mol. Cell. Biol. 23, 4056–4065. doi: 10.1128/mcb.23.12.4056-4065.2003
Zou, X., Zhu, Y., Park, S. H., Liu, G., O’brien, J., Jiang, H., et al. (2017). SIRT3-mediated dimerization of IDH2 directs cancer cell metab.olism and tumor growth. Cancer Res. 77, 3990–3999. doi: 10.1158/0008-5472.can-16-2393
Zullo, K. M., Guo, Y., Cooke, L., Jirau-Serrano, X., Mangone, M., Scotto, L., et al. (2015). Aurora A kinase inhibition selectively synergizes with histone deacetylase inhibitor through cytokinesis failure in T-cell Lymphoma. Clin. Cancer Res. 21, 4097–4109. doi: 10.1158/1078-0432.ccr-15-0033
Keywords: cancer therapy, HDAC, HDAC inhibitors, HDAC sequence, histone modification
Citation: Li G, Tian Y and Zhu W-G (2020) The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 8:576946. doi: 10.3389/fcell.2020.576946
Received: 27 June 2020; Accepted: 04 September 2020;
Published: 29 September 2020.
Edited by:
Christiane Pienna Soares, São Paulo State University, BrazilReviewed by:
Claudio Brancolini, Università di Udine, ItalyAngela Nebbioso, Università degli Studi della Campania Luigi Vanvitelli Naples, Italy
Xiongbin Lu, Indiana University, United States
Copyright © 2020 Li, Tian and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Guo Zhu, emh1d2VpZ3VvQHN6dS5lZHUuY24=
†These authors have contributed equally to this work