 Camille Dejos
Camille Dejos Dimitra Gkika
Dimitra Gkika Anna Rita Cantelmo
Anna Rita Cantelmo- 1Univ. Lille, Inserm, U1003 - PHYCEL - Physiologie Cellulaire, Lille, France
- 2Univ. Lille, CNRS, INSERM, CHU Lille, Centre Oscar Lambret, UMR 9020-UMR 1277-Canther-Cancer Heterogeneity, Plasticity and Resistance to Therapies, Lille, France
- 3Institut Universitaire de France (IUF), Paris, France
Calcium ion (Ca2+) signaling is critical to many physiological processes, and its kinetics and subcellular localization are tightly regulated in all cell types. All Ca2+ flux perturbations impact cell function and may contribute to various diseases, including cancer. Several modulators of Ca2+ signaling are attractive pharmacological targets due to their accessibility at the plasma membrane. Despite this, the number of specific inhibitors is still limited, and to date there are no anticancer drugs in the clinic that target Ca2+ signaling. Ca2+ dynamics are impacted, in part, by modifications of cellular metabolic pathways. Conversely, it is well established that Ca2+ regulates cellular bioenergetics by allosterically activating key metabolic enzymes and metabolite shuttles or indirectly by modulating signaling cascades. A coordinated interplay between Ca2+ and metabolism is essential in maintaining cellular homeostasis. In this review, we provide a snapshot of the reciprocal interaction between Ca2+ and metabolism and discuss the potential consequences of this interplay in cancer cells. We highlight the contribution of Ca2+ to the metabolic reprogramming observed in cancer. We also describe how the metabolic adaptation of cancer cells influences this crosstalk to regulate protumorigenic signaling pathways. We suggest that the dual targeting of these processes might provide unprecedented opportunities for anticancer strategies. Interestingly, promising evidence for the synergistic effects of antimetabolites and Ca2+-modulating agents is emerging.
Introduction
Calcium ions (Ca2+) are second messengers involved in signaling in many physiological processes, including the regulation of metabolic pathways (Clapham, 2007). Collectively termed the “Ca2+ transportome,” a variety of Ca2+ channels and transporters ensure Ca2+ flux between the extracellular space and the cytosol and between organelles within the cell, such as the endoplasmic reticulum (ER) and mitochondria, which act as major Ca2+ storage sites (Cardenas et al., 2018). Once Ca2+ enters the cell, some of the intracellular Ca2+ moves into the ER through energy-dependent sarco/endoplasmic reticulum calcium ATPases (SERCAs). However, following stimulation by extracellular stimuli and intracellular signals, the activation of two receptors, the inositol 1,4,5-triphosphate receptor (IP3R) and ryanodine receptor (RYR), mobilizes Ca2+ from this store, creating cytosolic Ca2+ hotspots in close proximity to mitochondria (Clapham, 2007). Mitochondrial Ca2+ uptake via voltage-dependent anion-selective channel protein (VDAC) and mitochondrial calcium uniporter protein (MCU) is a major regulator of several mitochondrial processes, including energy production and cell death (Duchen, 2000; Finkel et al., 2015). An increase in cytosolic Ca2+ levels also activates several Ca2+-binding proteins that are major players in signaling pathways and are able to directly regulate a variety of enzymes, transportome function, and gene expression (Berridge et al., 2000).
In recent years, a growing number of studies have yielded significant insights into how Ca2+ integrates with multiple metabolic pathways within cells (Rossi et al., 2019). This interaction is primarily related to the ability of Ca2+ to allosterically regulate the activity of key metabolic enzymes, such as mitochondrial dehydrogenases, thus impacting biosynthetic and energy-generating pathways. Furthermore, Ca2+ signaling can influence the generation of reactive oxygen species (ROS) at different subcellular locations. In turn, many Ca2+ channels and transporters are regulated by redox modifications.
Most of our knowledge of the integration of Ca2+ signaling with metabolic processes has come from the cardiovascular system and organ physiology, with well-known examples of Ca2+ coupling workload and energy metabolism in cardiomyocytes and skeletal muscle. Spikes in intracellular Ca2+ levels during contractile activity lead to changes in glucose metabolism by enhancing glucose uptake and transport (Wright et al., 2004; Waller et al., 2015). In insulin-sensitive tissues, the posttranslational activation of glucokinase (one of the rate-limiting enzymes of glycolysis) requires an increase in cytoplasmic Ca2+ levels (Markwardt et al., 2016). Variations in Ca2+ oscillations also regulate the generation of glucose from non-carbohydrate precursors (gluconeogenesis) (Gaspers et al., 2019) by modulating the expression of genes related to glucose metabolism in the liver (Wang et al., 2012; Zou et al., 2018). Furthermore, in response to Ca2+-mobilizing hormones, Ca2+ controls the phosphorylation state, and hence the activity, of key enzymes regulating glucose storage (glycogen) (Amaya and Nathanson, 2013). The relevance of these observations is underscored by studies showing an increase in intracellular Ca2+ levels and Ca2+-dependent signaling in obese and diabetic mice (Ozcan et al., 2012; Wang et al., 2012). In these pathological conditions, high glucose concentrations increase cytosolic Ca2+ levels by stimulating extracellular Ca2+ influx, ultimately leading to cytosolic Ca2+ overload and cellular damage (Song et al., 2003).
Nevertheless, it is difficult to clearly separate the effects of Ca2+ from the metabolic changes associated with muscle contraction or insulin sensitivity, and the interpretation of this interaction is often hindered by the causality dilemma. Although the development of sensitive methods has allowed for the recording of subtle, transient and localized intracellular Ca2+ peaks in response to metabolic changes, much remains to be understood regarding the reciprocal regulatory role of metabolism.
The “chicken and egg” situation is particularly evident in cancer cells, where both Ca2+ signaling and metabolism are aberrant, and it is therefore difficult to define whether dysregulated Ca2+ homeostasis leads to metabolic reprogramming or vice versa. Moreover, since much of the research investigating Ca2+ signaling in cancer has focused on evaluating relevant functional processes (such as proliferation, migration, and survival) as a result of aberrant expression levels of channels, pumps and transporters, the contribution of this interplay to cancer pathogenesis has often been overlooked.
One of the strategies adopted by cancer cells to sustain their high proliferation rate and survival is modulation of the Ca2+ transportome to control signaling and metabolism (Prevarskaya et al., 2018). Cell proliferation requires increased biomass and energy supply, which is provided by metabolic adaptation. Despite the presence of adequate oxygen (O2) levels to support mitochondrial respiration, cancer cells are highly glycolytic and, as such, demonstrate increased glucose consumption and lactate production, a phenomenon known as the Warburg effect (Vander Heiden et al., 2009). These cancer-related metabolic features are in part maintained by the activity of specific Ca2+ transporters, which limits mitochondrial Ca2+ influx and maintains glycolysis as a protective mechanism against oxidative stress. Moreover, by restricting mitochondrial Ca2+ entry, cancer cells reduce their vulnerability to apoptosis and cytotoxic agents (Lemeshko, 2015; Chakraborty et al., 2017). In turn, the metabolic shift toward glycolysis is a critical regulator of the Ca2+ supply in cancer cells. Thus, the dual pharmacological modulation of Ca2+ signaling and metabolism might have synergistic anticancer effects. Interestingly, preclinical evidence for the therapeutic potential of combined strategies is emerging (Park et al., 2018), yet it remains to be determined whether dual targeting will be effective in humans.
In this review, we examine the main findings regarding the interplay between Ca2+ and metabolism in cancer cells, with a focus on glycolysis and oxidative phosphorylation (OXPHOS). We discuss how Ca2+ flux between different cellular compartments affects these pivotal metabolic processes in the cell and highlight how metabolic enzymes and metabolites function as signaling molecules to influence Ca2+ homeostasis. We also describe how the cellular redox state defined by the metabolic features of cancer cells controls the activity of several Ca2+ transporters and pumps through posttranslational modifications.
The connection between Ca2+ and metabolism in cancer has only begun to be explored. Highlighting emerging findings on this crosstalk is important to provide further understanding of the mechanisms affecting cell function. This may yield great returns by opening up new areas of investigation and identifying new potential therapeutic opportunities.
Calcium – Glucose Metabolism Interplay
Ca2+ Regulation and Glycolysis
In all eukaryotic cells, intracellular Ca2+ levels are maintained at low resting concentrations (approximately 100 nM) by the activity of the major Ca2+ extrusion system, the plasma membrane Ca2+-ATPase (PMCA), which exchanges extracellular protons (H+) for cytosolic Ca2+ (Brini et al., 2013). The role played by PMCAs as housekeepers of low resting cytosolic Ca2+ levels is important for cell survival as it prevents Ca2+-dependent cell death (Brini et al., 2013). PMCAs also regulate dynamic Ca2+ signaling by modulating the frequency of Ca2+ oscillations (Caride et al., 2001).
It is well established that the activity of PMCAs is influenced by glycolysis, a cytosolic energy-conversion pathway that produces adenosine triphosphate (ATP) without the need for O2. In highly glycolytic cancer cells exhibiting the Warburg effect, the cytosolic ATP derived from glycolysis is crucial to maintaining PMCA function and cell survival (James et al., 2013, 2015). Pharmacological blockade of the glycolytic regulator 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) results in PMCA inhibition, toxic cytosolic Ca2+ overload, and cell death (Richardson et al., 2020). Recently, a functional coupling between PMCA and the ATP-generating enzyme pyruvate kinase M2 (PKM2) has been shown in pancreatic cancer cells (James et al., 2020; Figure 1A), further supporting the notion that the cytosolic ATP supply to PMCAs may represent a potential anticancer target. Interestingly, since the activity of PMCAs is influenced by extracellular acidification, it is tempting to speculate that the highly glycolytic phenotype of cancer cells would contribute to increased Ca2+ efflux by providing PMCAs with an abundant source of extracellular H+.
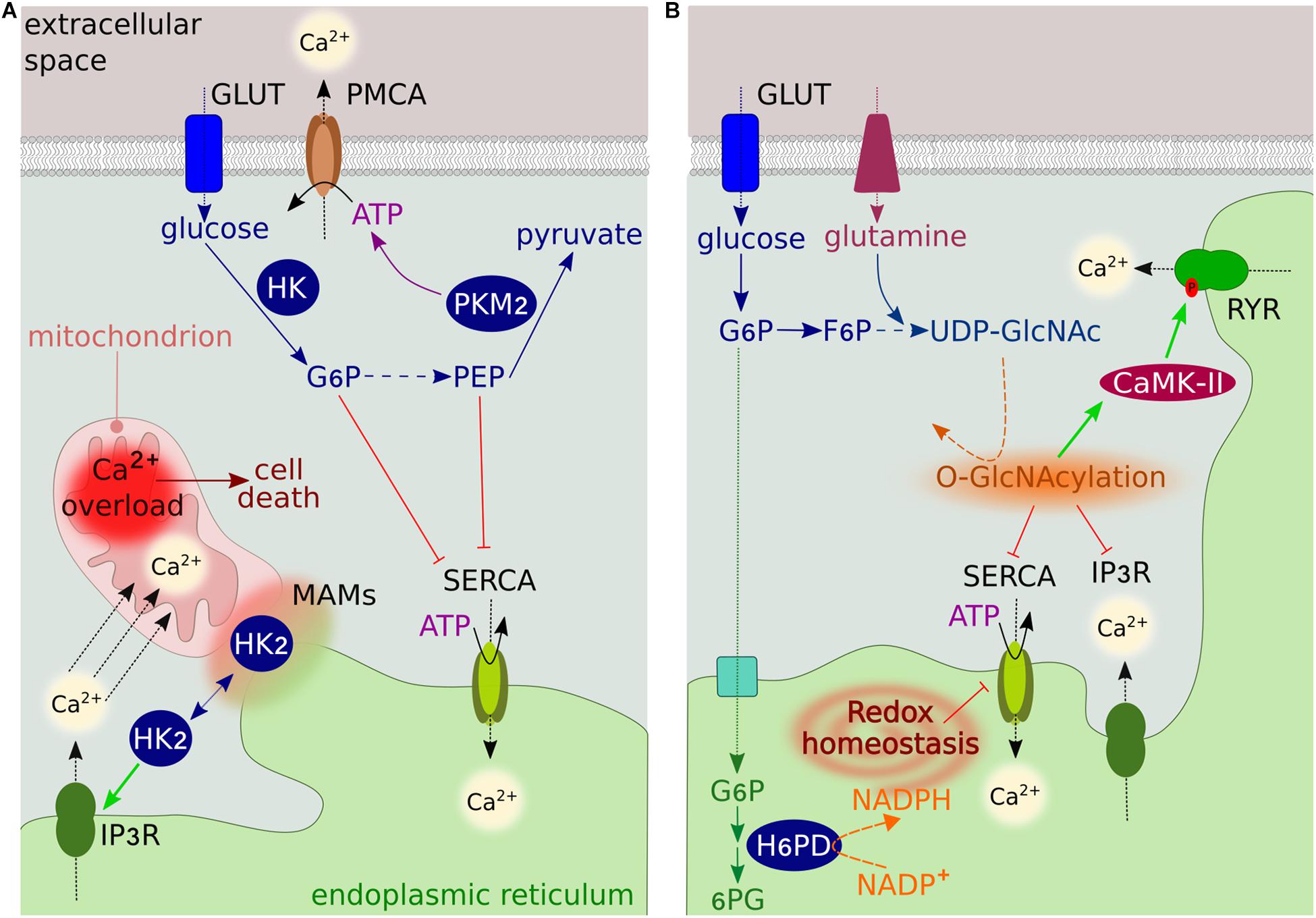
Figure 1. Overview of glucose metabolism-Ca2+ interplay. (A) The GLUT family transporters transfer glucose into cells, where it is converted to pyruvate though the glycolytic pathway. The rate-limiting glycolytic enzyme HK generates G6P, which inhibits ER Ca2+ uptake by binding SERCA. In the final step of glycolysis, PKM2 converts PEP into pyruvate, generating ATP. PEP inhibits SERCA, while ATP regulates the activity of PMCA. HK2 at MAMs modulates Ca2+ flux from the ER into mitochondria through IP3R to prevent mitochondrial Ca2+ overload-induced cell death. (B) The ER luminal enzyme H6PD catalyzes the first two reactions of the oxidative branch of the PPP. The conversion of G6P into 6-phosphogluconate (6PG) generates NADPH, which is used as a cofactor by several ER-reducing enzymes. By regulating the ER redox state, H6PD modulates the activity of several proteins, including SERCA and IP3R. The glycolytic intermediate fructose 6-phosphate (F6P) can enter the HBP to generate UDP-GlcNAc through a series of reactions requiring glutamine. UDP-GlcNAc modifies the activity of several Ca2+ transporters and regulatory proteins by O-GlcNAcylating serine and threonine residues.
In normal cells and under physiological conditions, cytosolic ATP derived from glycolysis saturates PMCA function. However, in the case of impaired mitochondrial metabolism, ATP derived from glycolysis becomes a critical regulator of PMCA activity. Acute metabolic stress inhibits the PMCA, but this effect can be attenuated by a metabolic switch from mitochondrial metabolism toward glycolysis, which restores the ATP supply to the PMCA (Mankad et al., 2012; Samad et al., 2014).
While the amount of ATP derived from glycolysis regulates PMCA activity, there is no significant evidence of regulatory feedback by the PMCA on metabolism. Indeed, silencing of PMCA4 in pancreatic ductal adenocarcinoma cells has only minimal effects on metabolic functions (Sritangos et al., 2020).
The mechanisms regulating the interplay between glycolysis and Ca2+ homeostasis are much more complex than simple changes in energy supply.
A first example of this complexity is related to the subcellular localization of glycolytic enzymes, which is crucial for supporting PMCA activity, and is in turn modulated by Ca2+. ATP is provided to the PMCA by glycolytic enzymes located in close proximity to the plasma membrane (Hardin et al., 1992; Chu et al., 2012). Studies using inside-out smooth muscle plasma membrane vesicles have demonstrated that the localized glycolytic pool of ATP regulates PMCA activity regardless of the overall cellular ATP levels (Paul et al., 1989; Hardin et al., 1993). On the other hand, cytosolic Ca2+ accumulation following treatment of melanoma cells with the Ca2+-ionophore A23187 alters the cytoskeletal compartmentalization of phosphofructokinase, thereby inhibiting glycolysis (Glass-Marmor et al., 1999). The exact mechanism is currently unknown, however, it may be related to an indirect effect of Ca2+ on the cytoskeletal structure.
In several cancer cells, the specific localization of the glycolytic enzyme hexokinase-2 (HK2) at mitochondria-ER contact sites [called mitochondria-associated ER membranes (MAMs)] prevents mitochondrial Ca2+ overload, permeability transition pore (mPTP) opening and cell death by preventing ER Ca2+ release through IP3R (Ciscato et al., 2020; Figure 1A). HK2 can also bind VDAC in the outer mitochondrial membrane (OMM) (Azoulay-Zohar et al., 2004; Shoshan-Barmatz et al., 2009). This association with VDAC is regulated by channel switching between an open and closed state, which is controlled by Ca2+ (Bathori et al., 2006). The binding reduces the channel conductance while providing HK direct access to mitochondrial ATP for enzymatic activity (Azoulay-Zohar et al., 2004), resulting in a high glycolytic rate and increased cancer cell proliferation and survival (Azoulay-Zohar et al., 2004; Abu-Hamad et al., 2008). These observations led to the hypothesis that the closure of VDAC contributes to the Warburg effect (Lemeshko, 2015).
An additional regulatory mechanism involves glycolytic metabolites. The metabolite glucose 6-phosphate (G6P) has recently been identified as a potent regulator of SERCA pumps, which transfer Ca2+ from the cytosol to the ER at the expense of ATP hydrolysis (Figure 1A). G6P reduces ER Ca2+ uptake and accumulation by directly binding SERCAs, resulting in loss of ER Ca2+ compartmentalization and cell death (Cole et al., 2012). This ATP-independent mechanism of regulation resembles that of the chemical SERCA inhibitor thapsigargin and is specific to G6P, as the addition of other glycolytic intermediates had no effect on ER Ca2+ accumulation (Cole et al., 2012).
Sarcoplasmic/endoplasmic reticulum calcium ATPase pumps are also regulated by the metabolic intermediate phosphoenolpyruvate (PEP) (Figure 1A). In cytotoxic T cells, PEP regulates the amplitude of Ca2+ flux and activation of the downstream nuclear factor of activated T-cell (NFAT) signaling pathway, which is critical for anti-tumor T cell functions (Ho et al., 2015). Insufficient PEP production leads to defects in Ca2+-NFAT signaling by increasing SERCA-mediated ER Ca2+ storage. Interestingly, metabolic reprogramming of T cells to generate PEP in low-glucose conditions improves anti-tumor responses (Ho et al., 2015). Although the precise molecular mechanism(s) by which this occurs remains unknown, PEP may inhibit SERCA activity by increasing its oxidative state (Ho et al., 2015).
Recently, the regulation of Ca2+ homeostasis by PEP has been described in two human colon carcinoma cell lines, in which increased cytosolic Ca2+ due to SERCA modulation stabilizes the master regulator of glycolytic metabolism, the Myc proto-oncogene protein, ultimately supporting cancer cell proliferation (Moreno-Felici et al., 2019).
While further studies are required to unravel the reciprocal regulation of Ca2+ levels and glycolysis in cancer cells, these examples shed light on the potential benefits of targeting those metabolic pathways that are associated with malignant transformation and regulate Ca2+ signaling. Most cancer cells preferentially express the M2 isoform of the pyruvate kinase enzyme catalyzing the final ATP-generating step of glycolysis in which PEP is converted to pyruvate. This provides cancer cells the advantage to adapt their metabolism depending on nutrient availability, as PKM2 switches between an active and inactive state (Dayton et al., 2016). Targeting this glycolytic enzyme may inhibit PMCA pumps specifically in cancer cells. Furthermore, the therapeutic potential of the dual inhibition of glycolysis and Ca2+ signaling should be considered. In support of this, combined treatment with a SERCA inhibitor (thapsigargin) and the glycolytic inhibitor 2-deoxy-d-glucose has recently shown significant anti-tumor effects in preclinical studies (Park et al., 2018).
Contribution of Glycolytic Side Pathways
There is interesting evidence for the indirect regulation of Ca2+ levels by two glycolytic side branches, the pentose phosphate pathway (PPP) and the hexosamine biosynthetic pathway (HBP).
The metabolite G6P can enter the PPP, resulting in the generation of nicotinamide adenine dinucleotide phosphate (NADPH), which maintains antioxidant capacity, and ribose, the backbone of nucleotides. Recent studies in human breast cancer cells have shown that the production of NADPH by hexose-6-phosphate dehydrogenase (H6PD) in the ER lumen is indispensable for the activity of SERCAs (Figure 1B). Following ER oxidation, H6PD depletion causes an increase in SERCA expression, leading to increased ER Ca2+ levels and impairing cancer cell proliferation and migration (Tsachaki et al., 2018). Dysregulated SERCA expression has also been observed in H6pd knockout mice (Lavery et al., 2008).
These findings define, for the first time, a direct link between a metabolic pathway and ER stress, paving the way for further investigations into the ER-related metabolic pathways that sustain tumor growth by affecting Ca2+ homeostasis.
The activity of several proteins involved in Ca2+ signaling [such as calcium/calmodulin-dependent protein kinase type II (CaMK-II)], as well as Ca2+ pumps and channels (such as SERCA, RYR, and IP3R), is influenced by O-linked-beta-N-acetylglucosamine (O-GlcNAc) posttranslational modification (Bidasee et al., 2004; Rengifo et al., 2007; Erickson et al., 2013; Figure 1B). The amount of O-GlcNAc modification is dependent on the concentration of uridine-diphosphate-N-acetylglucosamine (UDP-GlcNAc), the end product of the HBP, which uses glucose as a substrate. Excess glucose uptake, as occurs in cancer cells, contributes to increased flux through the HBP and thus increased levels of O-GlcNAc (Jozwiak et al., 2014). The impact of this modification on channel function and activity has not been fully elucidated in cancer. Similar to other pathological conditions, increased O-GlcNAc modification is likely associated with low channel activity (Bidasee et al., 2004; Rengifo et al., 2007).
Mitochondrial Bioenergetics and Ca2+ Dynamics
Mitochondrial Membrane Potential
In vertebrates, mitochondria play an important role in ATP production and act as Ca2+ storage sites. The main driving force for mitochondrial Ca2+ uptake is the mitochondrial electrical gradient (or mitochondrial membrane potential, ΔΨm) generated by the activity of the respiratory electron transport chain (ETC) (Santo-Domingo and Demaurex, 2010). The ETC consists of four enzymatic complexes embedded in the inner mitochondrial membrane (IMM) that transport electrons delivered by reducing equivalents [nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2)] to O2 while pumping H+ into the intermembrane space (IMS) (Marreiros et al., 2016). Together with the ΔΨm, the H+ gradient provides the energy to generate ATP through the F1F0 ATP synthase (or complex V) (Saraste, 1999), a process referred as OXPHOS. Furthermore, H+ power allows the transportation of metabolites, proteins and ions into and out of the mitochondrial matrix (Poburko and Demaurex, 2012).
Since maintenance of the ΔΨm is essential for cellular Ca2+ homeostasis (Contreras, 2010), it has been assumed that mitochondrial Ca2+ homeostasis remains normal as long as ETC dysfunction does not affect the ΔΨM. However, in recent work, Jana et al. (2019) demonstrated that mitochondrial Ca2+ homeostasis can be altered without changes in the ΔΨm. The authors generated two stable knockdown breast cancer cell lines for NDUFAF3 and SDHA genes, encoding proteins essential for the assembly of ETC complex I and complex II, respectively. In this model, altered ETC activity was associated with reduced mitochondrial Ca2+ accumulation, despite no significant ΔΨm changes, suggesting that the modulation of mitochondrial Ca2+ may occur independently of the ΔΨm in cancer cells (Jana et al., 2019).
Many metabolic pathways, including glycolysis, the tricarboxylic acid (TCA) cycle, and fatty acid oxidation, produce the electron donors that fuel the ETC. Upon exposure to high extracellular glucose concentrations, glycolysis provides a significant amount of pyruvate substrate to the TCA, elevating mitochondrial respiration and driving hyperpolarization of the IMM (Park et al., 2008). Conversely, energy deprivation of mitochondria secondary to low glucose is linked to mild mitochondrial depolarization (Gerencser et al., 2017). These glucose-driven intracellular changes control mitochondrial Ca2+ flux primarily via allosteric regulation of the major mitochondrial Ca2+ efflux system, the mitochondrial sodium/calcium exchanger protein NCLX (Kostic et al., 2018). Mild depolarization significantly reduces NCLX-mediated efflux (Figure 2). In turn, the consequent increase in matrix Ca2+ concentration stimulates OXPHOS, leading to repolarization that overcomes NCLX allosteric inhibition (Kostic et al., 2018). Thus, NCLX regulation by the ΔΨm links mitochondrial metabolism and Ca2+ signaling. This feedback mechanism can be manipulated to induce mitochondrial Ca2+ accumulation and cancer cell apoptosis under stress conditions such as nutrient deprivation.
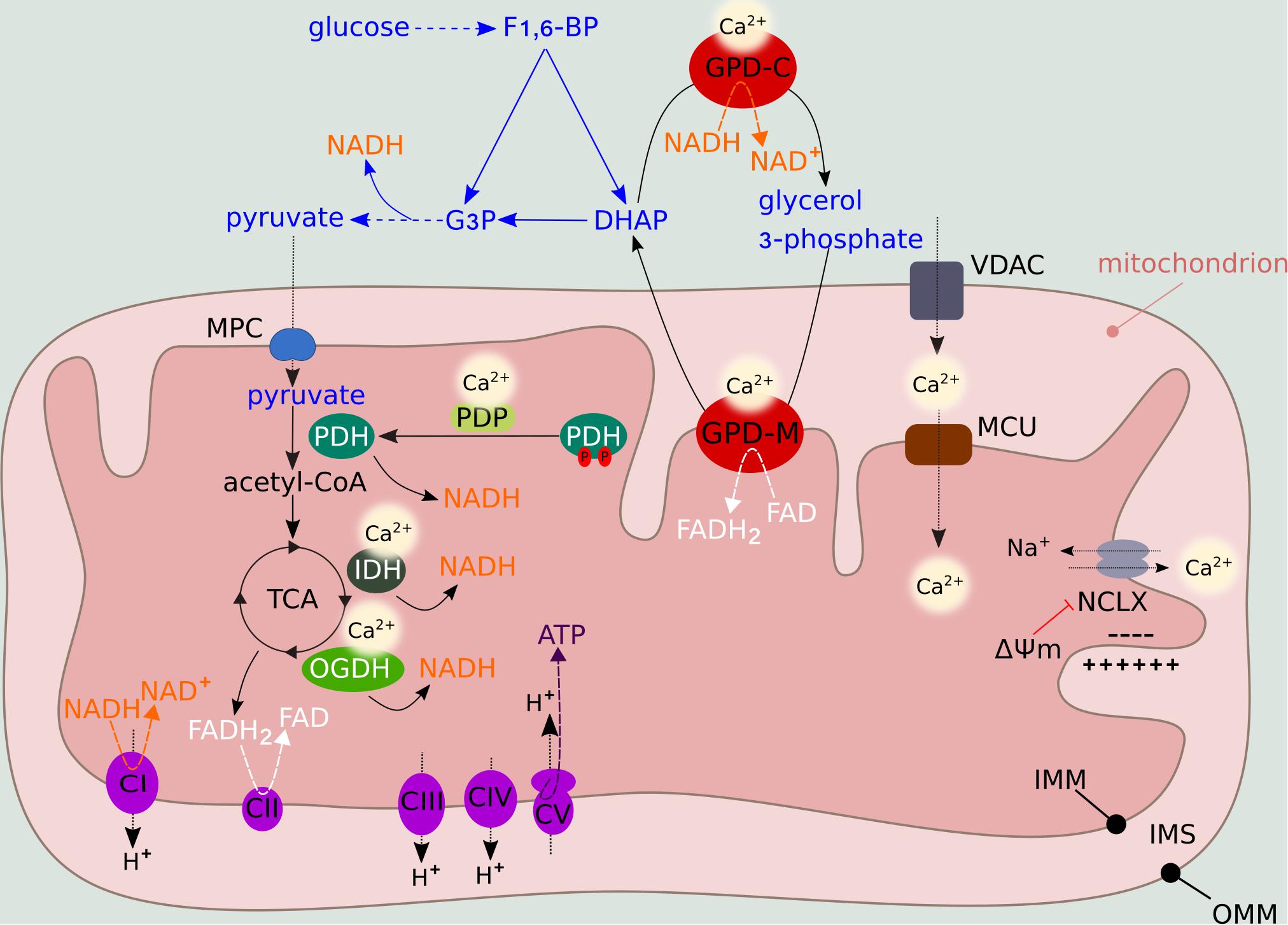
Figure 2. Metabolic regulation by mitochondrial Ca2+ levels. Mitochondrial Ca2+ entry through VDAC and MCU activates PDH, IDH and OGDH, providing NADH to the ETC. NADH and FADH2 are generated during the TCA feeding of complex I (NADH dehydrogenase) and complex II (succinate dehydrogenase), respectively. Complexes I, III, and IV (cytochrome c oxidase) function as H+ pumps by extruding H+ from the matrix to the IMS. Complex V (F1F0 ATP synthase) uses the H+ gradient to generate ATP in the presence of O2 (OXPHOS). ΔΨm fluctuations influence the activity of NCLX, the major mitochondrial Ca2+ efflux system. Ca2+ also regulates the activity of GPD-M, which constitutes the glycerol-phosphate shuttle, thus coupling mitochondrial metabolism to glycolysis. In the presence of O2, pyruvate derived from glycolysis enters the mitochondria via the mitochondria pyruvate carrier (MPC), where it is oxidized into acetyl-CoA. The glycolytic intermediate fructose 1,6-bisphosphate (F1,6-BP) is converted into dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate (G3P), which enters the subsequent glycolytic step, producing NADH. DHAP can be converted into glycerol-3-phosphate by GPD-C, regenerating NAD+ to sustain glycolysis. Subsequently, glycerol-3-phosphate is converted back into DHAP by GPD-M, converting FAD into FADH2.
Ca2+ Entry in the Mitochondria and Regulation of Mitochondrial Energy Machinery
Mitochondrial Ca2+ uptake by the transporters VDAC (at the OMM) and MCU (at the IMM) is a key process in cell bioenergetics and functionality. In response to cellular demand, Ca2+ can directly modulate cellular metabolism by activating mitochondrial dehydrogenase enzymes and regulating ETC function (Denton, 2009).
By controlling the activity of Ca2+-sensitive pyruvate dehydrogenase phosphatase (PDP), Ca2+ regulates the dephosphorylation state, and hence activation, of pyruvate dehydrogenase (PDH) complex, thus feeding the TCA (Figure 2). Two TCA enzymes, NAD-isocitrate dehydrogenase (IDH) and mitochondrial 2-oxoglutarate dehydrogenase (OGDH, also known as alpha-ketoglutarate dehydrogenase), which generate NADH for ATP synthesis, are allosterically activated by mitochondrial Ca2+ levels (Figure 2). The fourth enzyme, mitochondrial glycerol-3-phosphate dehydrogenase (GPD-M), constitutes the glycerol-phosphate shuttle and transfers reducing equivalents from NADH (produced by glycolysis) to the ETC. As for IDH and OGDH, the effect of Ca2+ on GPD-M is to lower the dissociation constant of the substrate (Denton, 2009; Figure 2).
The stimulation of TCA dehydrogenases by Ca2+ triggers the activation of mitochondrial metabolic machinery, leading to increased ATP production to balance the energy demands of cancer cells during periods of increased proliferation. Inhibition of mitochondrial Ca2+ influx by genetic manipulation of the MCU complex in HeLa cells disrupts OXPHOS, lowers cellular ATP levels and activates 5′-adenosine monophosphate (AMP)-activated protein kinase catalytic subunit alpha-2 (AMPK)-dependent pro-survival autophagy (Mallilankaraman et al., 2012). A similar phenotype has been described following inhibition of IP3R-mediated ER Ca2+ release (Cardenas et al., 2010), suggesting that Ca2+ transfer from the ER to mitochondria is crucial for optimal cellular bioenergetics. Conversely, deprivation of mitochondrial TCA substrates alters mitochondrial Ca2+ flux by regulating the expression of the gatekeeper of the mitochondrial calcium uniporter MICU1, thereby protecting the cells from Ca2+ overload under conditions of limiting metabolite availability (Nemani et al., 2020). Many cancer cells are thought to have defective mitochondria, thus relying primarily on aerobic glycolysis. However, interestingly, a recent paper has shown that Ca2+ signaling in mitochondria remains a critical regulator of mitochondrial metabolism even in cells with impaired OXPHOS. In these cells, mitochondrial Ca2+ uptake is crucial to sustain OGDH-mediated metabolic reprogramming, and thus cancer cell survival (Cardenas et al., 2020). This evidence suggests that dual targeting of metabolism and Ca2+ shuttling to mitochondria may have synergistic effects in specific cancer subtypes.
Several other studies have shown that the close proximity of mitochondria to Ca2+ transporters in the ER is fundamental to determining mitochondrial Ca2+ handling properties (Rutter and Rizzuto, 2000). Increased ER-mitochondria coupling leads to cancer cell death through mitochondrial Ca2+ overload, while a reduction in contact sites shifts bioenergetics to glycolysis and contributes to therapy resistance (Cardenas et al., 2016; Madreiter-Sokolowski et al., 2016).
The direct link between mitochondrial Ca2+ levels and mitochondrial ATP production has been established by the seminal work of the Rizzuto group (Jouaville et al., 1999). By using mitochondrial aequorin as a targeted recombinant Ca2+ probe and an ATP-sensitive luciferase, the authors showed that stimulation of HeLa cells with histamine, which releases Ca2+ from ER stores, induces an increase in both mitochondrial Ca2+ and ATP levels. Interestingly, in the presence of pyruvate and lactate (oxidative substrates), ATP synthesis continued for several minutes after the mitochondrial Ca2+ signal returned to basal levels, suggesting the existence of a metabolic memory imposed by Ca2+ in cancer cells (Jouaville et al., 1999). This mechanism is currently unknown; however, it may involve persistent Ca2+ activation of mitochondrial dehydrogenases (Robb-Gaspers et al., 1998), prolonged changes in the ΔΨm (Jouaville et al., 1999) or activation of ATP synthase (see below). An alternative possibility is that of Ca2+-induced long-lasting events, such as activation of the ETC by posttranslational modifications (Glancy et al., 2013) or changes in mitochondrial volume (Halestrap, 1989).
Nevertheless, while Ca2+-mediated activation of dehydrogenases in mitochondria is well established, recent reports have questioned the importance of mitochondrial Ca2+ levels in controlling substrate supply for OXPHOS. Surprisingly, mitochondrial OXPHOS is not altered in MCU knockout mice, suggesting that additional mechanisms are involved in OXPHOS regulation (Szibor et al., 2020). Experimental findings have demonstrated that the malate/aspartate shuttle (MAS) acts as the “gas pedal” for mitochondrial metabolism by providing a supply of cytosolic pyruvate for OXPHOS (Szibor et al., 2020). An essential component of the MAS is the Ca2+-binding mitochondrial carrier protein Aralar1/2, an aspartate/glutamate transporter, which exchanges glutamate and one H+ (from the cytosol) for one aspartate (from the mitochondria) (Palmieri et al., 2001). Since the Ca2+-regulatory binding site of Aralar1/2 faces the mitochondrial IMS, MAS senses cytosolic, but not mitochondrial, Ca2+ levels (Palmieri et al., 2001). Thus, cytosolic Ca2+ controls the pyruvate supply to the mitochondria, while mitochondrial Ca2+ accounts for only 15% of the pyruvate-driven OXPHOS rate (Szibor et al., 2020). While mathematical models and additional experimental findings corroborate these observations, the debate is still ongoing (Gellerich et al., 2012; Korzeniewski, 2017), as findings are further complicated by compensatory metabolic mechanisms, as well as cell-type and tissue-specific effects (Pan et al., 2013; Boyman et al., 2020).
In addition to Aralar1/2, ATP-magnesium/phosphate carriers are regulated by Ca2+. This shuttle system catalyzes the exchange of ATP-magnesium ions (Mg2+) and adenosine diphosphate (ADP)-Mg2+ for one phosphate across the IMM, thus determining the levels of adenine nucleotides (AMP, ADP, ATP) in the mitochondria. The N-terminal region of these carriers contains Ca2+-binding domains that respond to high Ca2+ levels (Nosek et al., 1990). An unbalanced exchange capacity of these carriers can alter the concentration of the adenine nucleotide pool in the matrix, thus impacting cellular growth and energy metabolism.
Recently, it has been proposed that Ca2+ can directly modulate the enzymatic activity and velocity of F1F0 ATP synthase (Territo et al., 2000). Indeed, pharmacological blockade of mitochondrial Ca2+ entry leads to reduced ATP synthase activity (Das and Harris, 1990). However, the underlying mechanism is unknown, and evidence for Ca2+ binding or direct regulation of the ETC is still limited (Hubbard and McHugh, 1996; Wescott et al., 2019).
In general, as discussed above, increased mitochondrial Ca2+ levels result in increased mitochondrial metabolism. However, this is not always the case. When Ca2+ levels increase to just below the threshold sufficient to cause mPTP opening, the ability of mitochondria to synthesize ATP is compromised (Pandya et al., 2013). Most of the evidence to date has attributed this effect to inhibition of mitochondrial dehydrogenases, loss of purine nucleotides, and reduced ΔΨm (Fagian et al., 1986; Lai et al., 1988; Fink et al., 2017). Moreover, a recent study suggested that inhibition of ETC complex I is the most likely cause of the decrease in OXPHOS through accumulation of mitochondrial Ca2+ as phosphate precipitates (Malyala et al., 2019).
Ca2+ signals can also influence mitochondrial bioenergetics independent of ATP production. In response to repetitive cytosolic Ca2+ oscillations, NADH generated in the mitochondria is not used as a substrate for ATP synthesis, but it is transported (via MAS) to the cytosol. Once located in the cytosol, NADH influences the activity and expression of sirtuins (Marcu et al., 2014). This family of proteins comprises NAD-dependent deacetylases, thus regulating the posttranslational modifications of multiple targets, including metabolic genes (Zhao et al., 2010). Thus, mitochondrial Ca2+ accumulation impacts the regulation of metabolic adaptation by influencing protein acetylation and epigenetics.
Recent studies have provided evidence for this link. Alteration of mitochondrial metabolism upon VDAC1 depletion in glioblastoma cells limits the production of essential metabolic mediators of epigenetic processes, such as NADH, citrate, and acetyl-CoA (Arif et al., 2017; Amsalem et al., 2020). The associated changes in histone acetylation and methylation have profound effects on tumor growth, suggesting that this intricate link between Ca2+, metabolism and epigenetics may be exploited as an innovative therapeutic strategy for glioblastoma and other cancers (Arif et al., 2017; Amsalem et al., 2020).
Redox State and Ca2+ Balance
Oxidative Stress as a Modulator of Ca2+ Signaling
Mitochondria produce ROS as a natural byproduct of the activity of ETC. Due to electron leakage during respiration, O2 is reduced to form the radical superoxide anion (O2–), which is rapidly converted into non-radical hydrogen peroxide (H2O2), the major source of oxidative stress in cells (Schieber and Chandel, 2014). An increased metabolic rate results in more O2 consumption, resulting in increased ETC leakage and higher ROS levels (Zhao et al., 2019).
The interplay between Ca2+ and ROS is well established, with Ca2+ signaling influencing the cellular generation of ROS and oxidants regulating the activity of many Ca2+ channels and transporters.
Changes in the cellular redox state induce cytosolic Ca2+ overload by stimulating Ca2+ release from the ER (via IP3R), decreasing SERCA activity and inhibiting Ca2+ extrusion from the plasma membrane (Camello-Almaraz et al., 2006; Qin et al., 2013; Figure 3A). These events are associated with redox modifications that alter the sensitivity of the transporters to Ca2+ (Zaidi et al., 2003; Sharov et al., 2006; Joseph et al., 2018). The increase in cytosolic Ca2+ results in transient mPTP opening (called “flicker”) to prevent cell death by Ca2+ overload. mPTP Ca2+ desensitization and PMCA overexpression are escape mechanisms used by cancer cells to increase their resistance to death (Rasola and Bernardi, 2015; Peters et al., 2016). Recently, the redox-sensing transient receptor potential cation channel (TRP) subfamily A member 1 (TRPA1) has been shown to upregulate Ca2+-dependent anti-apoptotic signaling pathways in response to oxidative stress, allowing cancer cells to adapt to therapy by promoting oxidative stress tolerance (Takahashi et al., 2018; Figure 3B).
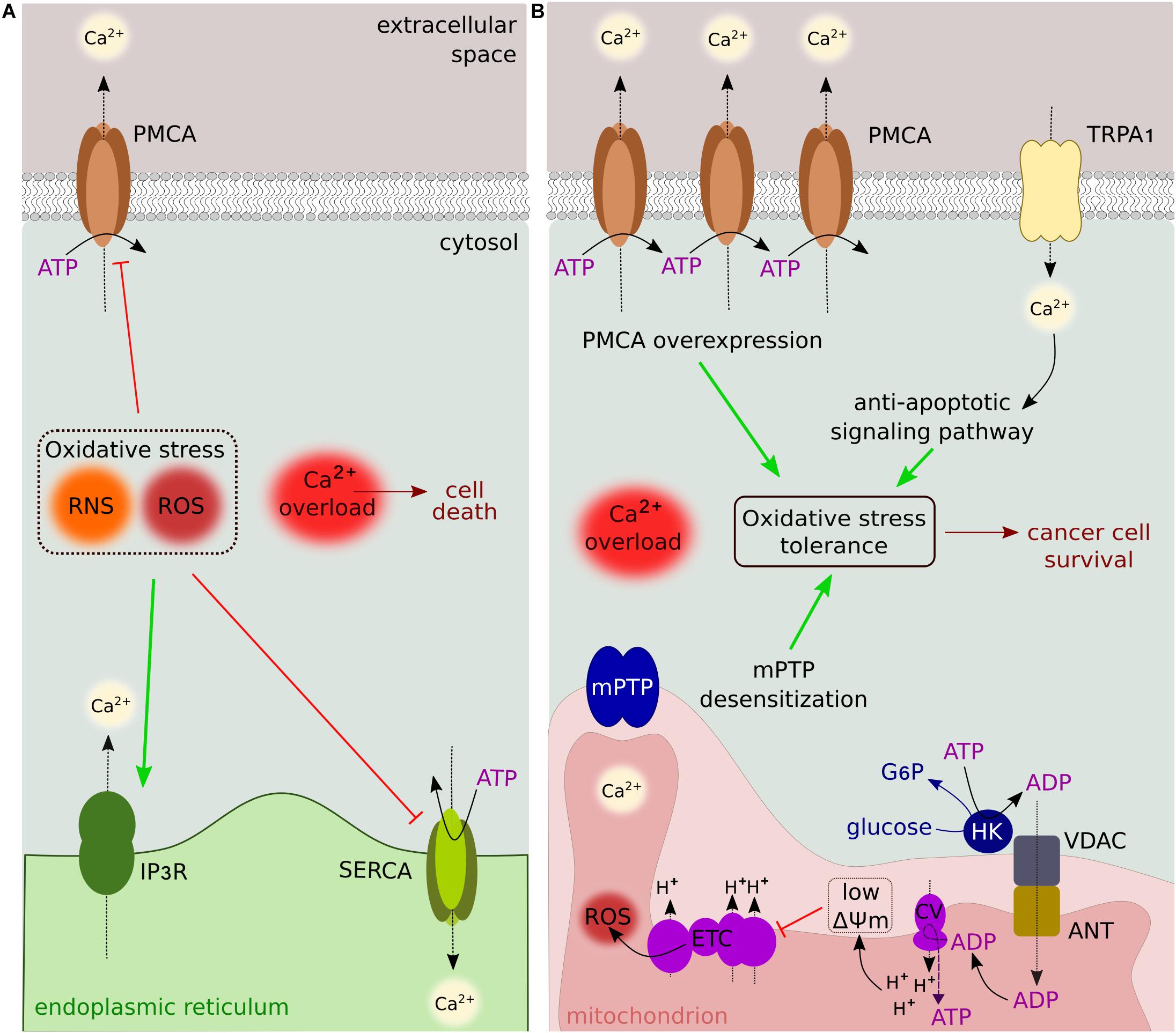
Figure 3. Oxidative stress and Ca2+ signaling. (A) Reactive nitrogen/oxygen species alter the sensitivity of transporters and pumps to Ca2+. Cytosolic Ca2+ overload is induced by IP3R stimulation, and SERCA/PMCA inhibition. (B) Oxidative stress tolerance is sustained by several Ca2+-dependent mechanisms, including PMCA overexpression, activation of anti-apoptotic signaling pathways, and mPTP desensitization. The binding of HK to VDAC contributes to reduced O2– leakage by lowering ΔΨm. These mechanisms are used by cancer cells to mediate resistance to ROS-producing anticancer drugs.
Mitochondrial ROS accumulation further augments ROS production through a positive feedback mechanism, overall increasing mitochondrial Ca2+ uptake by altering VDAC and MCU activity via redox modifications (De Pinto et al., 2016; Dong et al., 2017).
Another major endogenous source of ROS in cells is the NADPH oxidase isoform family (NOX1-5); these enzymes generate O2– and H2O2 in specific cell compartments, including the ER and plasma membrane (Ushio-Fukai, 2006). Although the mechanisms of NOX regulation of Ca2+ ion channels are not completely understood, the majority of NOX-derived ROS have been shown to induce extracellular Ca2+ entry. In cancer cells, modulation of NOX expression to manipulate ROS-dependent intracellular Ca2+ signaling could potentially be used to inhibit cancer progression (Song et al., 2014). However, current evidence is still mixed, as redox regulation can have different consequences depending on the specific NOX isoform involved and the cell context. For example, the interplay between NOX3 and transient receptor potential cation channel subfamily V member 1 (TRPV1) appears to be implicated in chemotherapy-induced toxicity through the augmentation of oxidative stress and regulation of cell death (Mukherjea et al., 2008).
In addition to channels, several other proteins involved in Ca2+ signaling are targeted by ROS-dependent oxidation. Oxidants generated by mitochondria can oxidize, among others, CaMK-II, thus enhancing its activity (Erickson et al., 2008). Following activation, CaMK-II regulates numerous Ca2+-regulatory proteins, including SERCA and RYR, leading to aberrant intracellular Ca2+ handling (Hook and Means, 2001). Considering the involvement of CaMK in several Ca2+-dependent cell processes, such as proliferation, apoptosis, and migration (Wayman et al., 2011), it is not surprising that its activity has been found to be aberrant in many cancer types (Brzozowski and Skelding, 2019).
Cells with high metabolic demand, such as cancer cells, also generate reactive nitrogen species (RNS). RNS are formed by peroxidase- and nitric oxide synthase (NOS)-mediated enzymatic reactions, as well as via the interaction of nitric oxide (NO) with ROS (Grisham et al., 1999).
Although very little is known about the regulation of Ca2+ responses by RNS, most of the effects are attributed to posttranslational modifications that generate nitrosylation changes in cysteine residues of Ca2+ transporters, channels and pumps, altering their activity (Xu et al., 1998; Yoshida et al., 2006; Zhou et al., 2015). In addition, NO derivatives can induce irreversible oxidation (Adachi et al., 2004; Tong et al., 2008). These changes are functionally associated with the inhibition of Ca2+ pumps and activation of Ca2+ release and entry channels, thus increasing the overall cytoplasmic Ca2+ level. Experimental evidence in cancer cells suggests that RNS are involved in mediating anti-tumor effects through Ca2+-dependent cell death (Tokunaga et al., 2018); however, further studies are needed to elucidate the mechanisms of RNS-regulated Ca2+ dynamics.
Ca2+ Homeostasis and ROS Generation
In cancer cells, elevated ROS levels below the threshold causing cell damage stimulate tumorigenesis by activating signaling pathways that regulate cancer cell proliferation, angiogenesis and metabolic reprogramming. In contrast, excessive ROS leads to cell injury and death (Sullivan and Chandel, 2014). Thus, ROS levels must be kept under tight control by balancing ROS generation and scavenging.
Changes in Ca2+ levels may impact cancer progression by affecting protumorigenic ROS levels and inducing apoptosis. As a consequence of supporting mitochondrial metabolism, mitochondrial Ca2+ can impact ETC-dependent ROS production and redox signaling by directly stimulating mitochondrial ROS-generating enzymes, such as GPD-M and OGDH. Moreover, ER-mitochondrial Ca2+ transfer stimulates ROS mobilization from the mitochondria, leading to the formation of redox nanodomains at MAMs, which in turn modulate cytosolic Ca2+ oscillations (Booth et al., 2016).
Excessive oxidative stress by mitochondrial Ca2+ accumulation induces apoptosis (Wang et al., 2015), while moderate ROS levels following inactivation of MICU1 support tumor growth by sustaining protumoral signaling pathways (Marchi et al., 2019). Similarly, expression of the MCU regulator mitochondrial calcium uniporter regulator 1 (MCUR1) stimulates ROS-mediated cancer cell migration and survival, accelerating tumor growth (Ren et al., 2018; Jin et al., 2019; Liu et al., 2020). Targeting MCU-mediated ROS production reduces the invasive properties of cancer cells and suppresses tumor growth and metastasis in vivo (Tosatto et al., 2016; Ren et al., 2017). However, these effects are likely tumor-specific, as other studies have reported conflicting evidence. Depending on experimental conditions, no changes in ROS levels have been observed upon alteration of mitochondrial Ca2+ uptake (Hall et al., 2014). Moreover, Ca2+ can counterintuitively enhance ROS production when complex I is inhibited (Sousa et al., 2003).
In addition to MCU-mediated effects on ROS output, Ca2+ entry via transient receptor potential channels (TRPs) plays an important role in modulating mitochondrial ROS levels. Upon inhibition of TRP cation channel melastatin subfamily member 2 (TRPM2), decreased intracellular Ca2+ levels result in reduced expression of mitochondrial genes and mitochondrial dysfunction (Bao et al., 2016). High ROS levels in neuroblastoma cells expressing a dominant-negative form of TRPM2 are associated with increased susceptibility to chemotherapeutic drugs and reduced cell survival and tumor growth (Bao et al., 2016).
The binding of the glycolytic HK enzyme to VDAC has a protective antioxidant role. ADP generated by HK during the conversion of glucose to G6P is used in an ADP/ATP recycling process involving the direct interaction between VDAC and the adenine nucleotide translocator (ANT), keeping ADP at steady-state levels. This in turn sets the ΔΨm to lower levels and decreases O2– leakage (Figure 3B). When this cycling is affected, ROS production is increased and associated with oxidative stress (da-Silva et al., 2004). Thus, VDAC-HK association not only provides a metabolic advantage (see above) but also plays a major role in regulating apoptosis via redox signaling.
The regulation of the redox balance by VDAC also involves mitochondrial ROS efflux to the cytosol, as VDAC acts as a release channel for hydrophilic O2– (Han et al., 2003). Indeed, VDAC closure increases mitochondrial oxidative stress, sensitizing cells to Ca2+-induced mPTP opening and apoptosis (Tikunov et al., 2010). These findings have been corroborated by a recent study showing a positive correlation between VDAC levels and the expression of two subunits of ETC complex I, the major source of mitochondrial ROS (Mota-Martorell et al., 2020).
Finally, a yet unexplored possibility is that Ca2+ contributes to ROS production by regulating cellular antioxidant defenses. For example, increased intracellular Ca2+ concentrations may generate more ROS because of impaired levels of reduced glutathione (GSH), the most important antioxidant synthesized in cells. Interestingly, the existence of a Ca2+-sensitive GSH transporter in the ER has been recently postulated (Lizak et al., 2020), and its activity might be differentially regulated in cancer.
Conclusion
It has become increasingly evident that Ca2+ impacts multiple metabolic pathways within cells and that several Ca2+ channels are much more than simple ion transporters. An example is the activity of mitochondrial transporters VDAC and MCU, which have been found to be responsible for aberrant metabolism that supports cancer progression and therapy resistance (Azoulay-Zohar et al., 2004; Abu-Hamad et al., 2008; Chakraborty et al., 2017).
The hypothesis that cancer cells have dysfunctional mitochondrial metabolism and that the increase in glycolytic rate is a compensatory mechanism (Warburg’s theory) has been called into question by evidence that mitochondrial metabolism is essential for cancer biology. Increased mitochondrial Ca2+ uptake stimulates mitochondrial metabolism, which provides the building blocks and energy to sustain uncontrolled proliferation. Moreover, mitochondrial metabolism produces ROS, which activate signaling pathways supporting tumorigenesis, such as hypoxia and inflammation pathways (Tosatto et al., 2016; Liu et al., 2020).
An interesting observation is the mitochondrial Ca2+ addiction of cancer cells with defective oxidative metabolism. In these cells, Ca2+ transfer to mitochondria sustains survival by mediating the compensatory metabolic switch toward reductive carboxylation. Thus, specific anti-metabolic drugs may be effective as combination therapy with drugs that target mitochondrial Ca2+ transfer, regardless the OXPHOS status of cancer cells.
The relationship between glycolysis and Ca2+ is complex. In addition to the ATP-dependent regulation of Ca2+ pumps, the physical association of glycolytic intermediates with Ca2+ transporters modulates Ca2+ homeostasis, while changes in the subcellular localization of glycolytic enzymes influence cell fate by impacting Ca2+ homeostasis (Ciscato et al., 2020). Future investigations should focus on the cellular compartmentalization of metabolic intermediates as demonstrated for Ca2+. It is becoming highly feasible to analyze regional changes in metabolites and metabolic activities in a topographic dimension. The next step is to combine these imaging tools with those used to study Ca2+ compartmentalization.
Clearly, many gaps remain in terms of our understanding of Ca2+-metabolism communication. To this point, a key aspect that we should consider is experimental design. Early reports used Ca2+ concentrations exceeding normal physiological levels, providing evidence of regulatory mechanisms that may not be highly relevant to the normal physiological conditions that precede pathological transformation. Moreover, it is quite unlikely that findings obtained in (isolated) cells in culture would be valuable in organ systems as well.
One of the greatest challenges is restoring physiological Ca2+ levels or manipulating Ca2+ dynamics to selectively kill cancer cells. Unfortunately, the limited number of specific inhibitors has prevented the development of efficient anticancer strategies targeting aberrant Ca2+ signaling. An alternative strategy is to target the dysfunctional metabolic pathways that also regulate Ca2+ signaling, such as glycolysis. For example, targeting the glycolytic enzymes PFKFB3 and PKM2 which are highly expressed in cancer cells compared to normal cells, would provide a novel means of targeting Ca2+ pumps specifically in cancer cells. Given this possibility and the therapeutic potential of a synergistic approach to, for instance, reduce tumor acidification or induce Ca2+-dependent apoptosis in cancer cells during glucose deprivation, it is crucial to further dissect how Ca2+ levels and metabolism integrate to regulate tumorigenesis and cancer progression.
Finally, the possibility of targeting metabolic pathways and Ca2+ dynamics to manipulate the epigenome is a fascinating idea. New findings are emerging (Arif et al., 2017; Lombardi et al., 2019; Amsalem et al., 2020), and it will now be critical to understand the mechanisms by which concerted alterations in metabolism and Ca2+ signaling regulate epigenetic-mediated transcriptional control in cancer cells.
Author Contributions
ARC conceived the review and wrote, reviewed, and edited the text. CD provided a first draft of the manuscript and designed the figures. DG reviewed the text. All authors performed literature searching and contributed to discussion of the content.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
CD and ARC are supported by the European Regional Development Fund. DG was supported by the Institut Universitaire de France (IUF). ARC received funding from La Ligue Contre le Cancer (Pas de Calais) and Agence Nationale de la Recherche, Investissements d’avenir (ANR-16-IDEX-004-ULNE).
Acknowledgments
The authors thank M. Lebas for technical support, A. Fiorio Pla and L. Lemonnier for helpful discussions.
Abbreviations
6PG, 6-phosphogluconate; ADP, adenosine diphosphate; AMP, 5 ′ -adenosine monophosphate; AMPK, 5 ′ -AMP-activated protein kinase catalytic subunit alpha-2; ANT, adenine nucleotide translocator; ATP, adenosine triphosphate; Ca2+, calcium ions; CaMK-II, calcium/calmodulin-dependent protein kinase type II; DHAP, dihydroxyacetone phosphate; ER, endoplasmic reticulum; ETC, electron transport chain; F1,6-BP, fructose 1,6-bisphosphate; F6P, fructose 6-phosphate; FADH2, flavin adenine dinucleotide; G3P, glyceraldehyde 3-phosphate; G6P, glucose 6-phosphate; GPD-C, cytoplasmic glycerol-3-phosphate dehydrogenase; GPD-M, mitochondrial glycerol-3-phosphate dehydrogenase; GSH, reduced glutathione; H+, proton; H2O2, hydrogen peroxide; H6PD, hexose-6-phosphate dehydrogenase; HBP, hexosamine biosynthetic pathway; HK(s/1/2), hexokinase(s)-1/2; IDH, NAD-isocitrate dehydrogenase; IMM, inner mitochondrial membrane; IMS, intermembrane space; IP3, inositol 1,4,5-triphosphate; IP3R, inositol 1,4,5-triphosphate receptor; MAMs, mitochondria-associated endoplasmic reticulum membranes; MAS, malate/aspartate shuttle; MCU, mitochondrial calcium uniporter protein; MCUR1, mitochondrial calcium uniporter regulator 1; Mg2+, magnesium ions; MICU(1), mitochondrial calcium uptake protein (1); MPC, mitochondria pyruvate carrier; mPTP, mitochondrial permeability transition pore; NAD(H), nicotinamide adenine dinucleotide; NADP(H), nicotinamide adenine dinucleotide phosphate; NCLX, mitochondrial sodium/calcium exchanger protein; NFAT, nuclear factor of activated T-cells; NO, nitric oxide; NOS, nitric oxide synthase; NOX(s/1-5), NADPH oxidase(s) (1-5); O-GlcNAc, O-linked -beta-N-acetylglucosamine; O2, oxygen; O2–, oxygen superoxide anion; OGDH, mitochondrial 2-oxoglutarate dehydrogenase; OMM, outer mitochondrial membrane; OXPHOS, oxidative phosphorylation; PDH, pyruvate dehydrogenase; PDP, pyruvate dehydrogenase phosphatase; PEP, phosphoenolpyruvate; PFKFB3, phosphofructokinase fructose bisphosphatase-3; PKM2, pyruvate kinase M2; PMCAs, plasma membrane Ca2+-ATPases; PPP, pentose phosphate pathway; RNS, reactive nitrogen species; ROS, reactive oxygen species; RYR, ryanodine receptor; SERCAs, sarcoplasmic/endoplasmic reticulum calcium ATPases; TCA, tricarboxylic acid cycle; TRPA1, transient receptor potential cation channel subfamily A member 1; TRPM2, Transient receptor potential cation channel subfamily M member 2; TRPs, transient receptor potential channels; TRPV1, transient receptor potential cation channel subfamily V member 1; UDP-GlcNAc, uridine-diphosphate-N-acetylglucosamine; VDAC(1), voltage-dependent anion-selective channel protein (1); ΔΨ m, mitochondrial membrane potential.
References
Abu-Hamad, S., Zaid, H., Israelson, A., Nahon, E., and Shoshan-Barmatz, V. (2008). Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J. Biol. Chem. 283, 13482–13490. doi: 10.1074/jbc.m708216200
Adachi, T., Weisbrod, R. M., Pimentel, D. R., Ying, J., Sharov, V. S., Schoneich, C., et al. (2004). S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 10, 1200–1207. doi: 10.1038/nm1119
Amaya, M. J., and Nathanson, M. H. (2013). Calcium signaling in the liver. Compr. Physiol. 3, 515–539.
Amsalem, Z., Arif, T., Shteinfer-Kuzmine, A., Chalifa-Caspi, V., and Shoshan-Barmatz, V. (2020). The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link. Cancers 12:1031. doi: 10.3390/cancers12041031
Arif, T., Krelin, Y., Nakdimon, I., Benharroch, D., Paul, A., Dadon-Klein, D., et al. (2017)., VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro. Oncol. 19, 951–964. doi: 10.1093/neuonc/now297
Azoulay-Zohar, H., Israelson, A., Abu-Hamad, S., and Shoshan-Barmatz, V. (2004). In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 377, 347–355. doi: 10.1042/bj20031465
Bao, L., Chen, S. J., Conrad, K., Keefer, K., Abraham, T., Lee, J. P., et al. (2016)., Depletion of the Human Ion Channel TRPM2 in Neuroblastoma Demonstrates Its Key Role in Cell Survival through Modulation of Mitochondrial Reactive Oxygen Species and Bioenergetics. J. Biol. Chem. 291, 24449–24464. doi: 10.1074/jbc.m116.747147
Bathori, G., Csordas, G., Garcia-Perez, C., Davies, E., and Hajnoczky, G. (2006). Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem. 281, 17347–17358. doi: 10.1074/jbc.m600906200
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21. doi: 10.1038/35036035
Bidasee, K. R., Zhang, Y., Shao, C. H., Wang, M., Patel, K. P., Dincer, U. D., et al. (2004). Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 53, 463–473. doi: 10.2337/diabetes.53.2.463
Booth, D. M., Enyedi, B., Geiszt, M., Varnai, P., and Hajnoczky, G. (2016). Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 63, 240–248. doi: 10.1016/j.molcel.2016.05.040
Boyman, L., Karbowski, M., and Lederer, W. J. (2020). Regulation of Mitochondrial ATP Production: Ca(2+) Signaling and Quality Control. Trends Mol. Med. 26, 21–39. doi: 10.1016/j.molmed.2019.10.007
Brini, M., Cali, T., Ottolini, D., and Carafoli, E. (2013). The plasma membrane calcium pump in health and disease. FEBS J. 280, 5385–5397. doi: 10.1111/febs.12193
Brzozowski, J. S., and Skelding, K. A. (2019). The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention. Pharmaceuticals 12:8 doi: 10.3390/ph12010008
Camello-Almaraz, C., Gomez-Pinilla, P. J., Pozo, M. J., and Camello, P. J. (2006). Mitochondrial reactive oxygen species and Ca2+ signaling. Am. J. Physiol. Cell Physiol. 291, C1082–C1088.
Cardenas, C., Lovy, A., Silva-Pavez, E., Urra, F., Mizzoni, C., Ahumada-Castro, U., et al. (2020). Cancer cells with defective oxidative phosphorylation require endoplasmic reticulum-to-mitochondria Ca(2+) transfer for survival. Sci. Signal 13:eaay1212. doi: 10.1126/scisignal.aay1212
Cardenas, C., Miller, R. A., Smith, I., Bui, T., Molgo, J., Muller, M., et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. doi: 10.1016/j.cell.2010.06.007
Cardenas, C., Muller, M., McNeal, A., Lovy, A., Jana, F., Bustos, G., et al. (2016). Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep. 14, 2313–2324. doi: 10.1016/j.celrep.2016.02.030
Cardenas, C., Pinton, P., and Bultynck, G. (2018). Editorial: Inter-Organelle Calcium Communication in Cancer. Front. Oncol. 8:14. doi: 10.3389/fonc.2018.00014
Caride, A. J., Penheiter, A. R., Filoteo, A. G., Bajzer, Z., Enyedi, A., and Penniston, J. T. (2001). The plasma membrane calcium pump displays memory of past calcium spikes. Differences between isoforms 2b and 4b. J. Biol. Chem. 276, 39797–39804. doi: 10.1074/jbc.m104380200
Chakraborty, P. K., Mustafi, S. B., Xiong, X., Dwivedi, S. K. D., Nesin, V., Saha, S., et al. (2017). MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 8:14634.
Chu, H., Puchulu-Campanella, E., Galan, J. A., Tao, W. A., Low, P. S., and Hoffman, J. F. (2012). Identification of cytoskeletal elements enclosing the ATP pools that fuel human red blood cell membrane cation pumps. Proc. Natl. Acad. Sci. U S A 109, 12794–12799. doi: 10.1073/pnas.1209014109
Ciscato, F., Filadi, R., Masgras, I., Pizzi, M., Marin, O., Damiano, N., et al. (2020). Hexokinase 2 displacement from mitochondria-associated membranes prompts Ca(2+) -dependent death of cancer cells. EMBO Rep. 21:e49117.
Cole, J. T., Kean, W. S., Pollard, H. B., Verma, A., and Watson, W. D. (2012). Glucose-6-phosphate reduces calcium accumulation in rat brain endoplasmic reticulum. Front. Mol. Neurosci. 5:51. doi: 10.3389/fnmol.2012.00051
Contreras, L., Drago, I., Zampese, E., and Pozzan, T. (2010). Mitochondria: the calcium connection. Biochim. Biophys. Acta 1797, 607–618. doi: 10.1016/j.bbabio.2010.05.005
Das, A. M., and Harris, D. A. (1990). Control of mitochondrial ATP synthase in heart cells: inactive to active transitions caused by beating or positive inotropic agents. Cardiovasc. Res. 24, 411–417. doi: 10.1093/cvr/24.5.411
da-Silva, W. S., Gomez-Puyou, A., de Gomez-Puyou, M. T., Moreno-Sanchez, R., De Felice, F. G., de Meis, L., et al. (2004). Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 279, 39846–39855. doi: 10.1074/jbc.m403835200
Dayton, T. L., Jacks, T., and Vander Heiden, M. G. (2016). PKM2, cancer metabolism, and the road ahead. EMBO Rep. 17, 1721–1730.
De Pinto, V., Reina, S., Gupta, A., Messina, A., and Mahalakshmi, R. (2016). Role of cysteines in mammalian VDAC isoforms’ function. Biochim. Biophys. Acta 1857, 1219–1227. doi: 10.1016/j.bbabio.2016.02.020
Denton, R. M. (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316. doi: 10.1016/j.bbabio.2009.01.005
Dong, Z., Shanmughapriya, S., Tomar, D., Siddiqui, N., Lynch, S., Nemani, N., et al. (2017). Mitochondrial Ca(2+) Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 65, 1014–1028.e7.
Duchen, M. R. (2000). Mitochondria and calcium: from cell signalling to cell death. J. Physiol. 529(Pt 1), 57–68. doi: 10.1111/j.1469-7793.2000.00057.x
Erickson, J. R., Joiner, M. L., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Erickson, J. R., Pereira, L., Wang, L., Han, G., Ferguson, A., Dao, K., et al. (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376. doi: 10.1038/nature12537
Fagian, M. M., da Silva, L. P., and Vercesi, A. E. (1986). Inhibition of oxidative phosphorylation by Ca2+ or Sr2+: a competition with Mg2+ for the formation of adenine nucleotide complexes. Biochim. Biophys. Acta 852, 262–268. doi: 10.1016/0005-2728(86)90231-8
Fink, B. D., Bai, F., Yu, L., and Sivitz, W. I. (2017). Regulation of ATP production: dependence on calcium concentration and respiratory state. Am. J. Physiol. Cell Physiol. 313, C146–C153.
Finkel, T., Menazza, S., Holmstrom, K. M., Parks, R. J., Liu, J., Sun, J., et al. (2015). The ins and outs of mitochondrial calcium. Circ. Res. 116, 1810–1819.
Gaspers, L. D., Pierobon, N., and Thomas, A. P. (2019). Intercellular calcium waves integrate hormonal control of glucose output in the intact liver. J. Physiol. 597, 2867–2885. doi: 10.1113/jp277650
Gellerich, F. N., Gizatullina, Z., Trumbekaite, S., Korzeniewski, B., Gaynutdinov, T., Seppet, E., et al. (2012). Cytosolic Ca2+ regulates the energization of isolated brain mitochondria by formation of pyruvate through the malate-aspartate shuttle. Biochem. J. 443, 747–755. doi: 10.1042/bj20110765
Gerencser, A. A., Mookerjee, S. A., Jastroch, M., and Brand, M. D. (2017). Positive Feedback Amplifies the Response of Mitochondrial Membrane Potential to Glucose Concentration in Clonal Pancreatic Beta Cells. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1054–1065. doi: 10.1016/j.bbadis.2016.10.015
Glancy, B., Willis, W. T., Chess, D. J., and Balaban, R. S. (2013). Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 52, 2793–2809. doi: 10.1021/bi3015983
Glass-Marmor, L., Penso, J., and Beitner, R. (1999). Ca2+-induced changes in energy metabolism and viability of melanoma cells. Br. J. Cancer 81, 219–224. doi: 10.1038/sj.bjc.6690680
Grisham, M. B., Jourd’Heuil, D., Wink, D. A., and Nitric oxide, I. (1999). Physiological chemistry of nitric oxide and its metabolites:implications in inflammation. Am. J. Physiol. 276, G315–G321.
Halestrap, A. P. (1989). The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro and its role in the control of mitochondrial metabolism. Biochim. Biophys. Acta 973, 355–382. doi: 10.1016/s0005-2728(89)80378-0
Hall, D. D., Wu, Y., Domann, F. E., Spitz, D. R., and Anderson, M. E. (2014). Mitochondrial calcium uniporter activity is dispensable for MDA-MB-231 breast carcinoma cell survival. PLoS One 9:e96866. doi: 10.1371/journal.pone.0096866
Han, D., Antunes, F., Canali, R., Rettori, D., and Cadenas, E. (2003). Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 278, 5557–5563. doi: 10.1074/jbc.m210269200
Hardin, C. D., Raeymaekers, L., and Paul, R. J. (1992). Comparison of endogenous and exogenous sources of ATP in fueling Ca2+ uptake in smooth muscle plasma membrane vesicles. J. Gen. Physiol. 99, 21–40. doi: 10.1085/jgp.99.1.21
Hardin, C. D., Zhang, C., Kranias, E. G., Steenaart, N. A., Raeymaekers, L., and Paul, R. J. (1993). Regulation of glycolytically fueled Ca2+ uptake in smooth muscle plasmalemmal vesicles by phosphorylation. Am. J. Physiol. 265, H1326–H1333.
Ho, P. C., Bihuniak, J. D., Macintyre, A. N., Staron, M., Liu, X., Amezquita, R., et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228. doi: 10.1016/j.cell.2015.08.012
Hook, S. S., and Means, A. R. (2001). Ca(2+)/CaM-dependent kinases: from activation to function. Annu. Rev. Pharmacol. Toxicol. 41, 471–505.
Hubbard, M. J., and McHugh, N. J. (1996). Mitochondrial ATP synthase F1-beta-subunit is a calcium-binding protein. FEBS Lett. 391, 323–329. doi: 10.1016/0014-5793(96)00767-3
James, A. D., Chan, A., Erice, O., Siriwardena, A. K., and Bruce, J. I. (2013). Glycolytic ATP fuels the plasma membrane calcium pump critical for pancreatic cancer cell survival. J. Biol. Chem. 288, 36007–36019. doi: 10.1074/jbc.m113.502948
James, A. D., Patel, W., Butt, Z., Adiamah, M., Dakhel, A., Latif, C., et al. (2015). The Plasma Membrane Calcium Pump in Pancreatic Cancer Cells Exhibiting the Warburg Effect Relies on Glycolytic ATP. J. Biol. Chem. 290, 24760–24771. doi: 10.1074/jbc.m115.668707
James, A. D., Richardson, I. W., Oh, P., Sritangos, T., Attard, L., Barrett et al (2020). Cutting off the fuel supply to calcium pumps in pancreatic cancer cells: role of pyruvate kinase-M2 (PKM2). Br. J. Cancer 122, 266–278. doi: 10.1038/s41416-019-0675-3
Jana, F., Bustos, G., Rivas, J., Cruz, P., Urra, F., Basualto-Alarcon, C., et al. (2019). Complex I and II are required for normal mitochondrial Ca(2+) homeostasis. Mitochondrion 49, 73–82. doi: 10.1016/j.mito.2019.07.004
Jin, M., Wang, J., Ji, X., Cao, H., Zhu, J., Chen, Y., et al. (2019). MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 38:136.
Joseph, S. K., Young, M. P., Alzayady, K., Yule, D. I., Ali, M., Booth, D. M., et al. (2018). Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J. Biol. Chem. 293, 17464–17476. doi: 10.1074/jbc.ra118.005624
Jouaville, L. S., Pinton, P., Bastianutto, C., Rutter, G. A., and Rizzuto, R. (1999). Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. U S A 96, 13807–13812. doi: 10.1073/pnas.96.24.13807
Jozwiak, P., Forma, E., Brys, M., and Krzeslak, A. (2014). O-GlcNAcylation and Metabolic Reprograming in Cancer. Front. Endocrinol. 5:145. doi: 10.3389/fendo.2014.00145
Korzeniewski, B. (2017). Regulation of oxidative phosphorylation through each-step activation (ESA): Evidences from computer modeling. Prog. Biophys. Mol. Biol. 125, 1–23.
Kostic, M., Katoshevski, T., and Sekler, I. (2018). Allosteric Regulation of NCLX by Mitochondrial Membrane Potential Links the Metabolic State and Ca(2+) Signaling in Mitochondria. Cell Rep. 25, 3465–3475.e4.
Lai, J. C., DiLorenzo, J. C., and Sheu, K. F. (1988). Pyruvate dehydrogenase complex is inhibited in calcium-loaded cerebrocortical mitochondria. Neurochem. Res. 13, 1043–1048. doi: 10.1007/bf00973148
Lavery, G. G., Walker, E. A., Turan, N., Rogoff, D., Ryder, J. W., Shelton, J. M., et al. (2008). Deletion of hexose-6-phosphate dehydrogenase activates the unfolded protein response pathway and induces skeletal myopathy. J. Biol. Chem. 283, 8453–8461. doi: 10.1074/jbc.m710067200
Lemeshko, V. (2015). The Warburg Effect as a VDAC−Hexokinase−Mediated Electrical Suppression of Mitochondrial Energy Metabolism. FASEB J. 29, (Suppl. 1) 725.27.
Liu, Y., Jin, M., Wang, Y., Zhu, J., Tan, R., Zhao, J., et al. (2020). MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Signal Transduct. Target Ther. 5:59.
Lizak, B., Birk, J., Zana, M., Kosztyi, G., Kratschmar, D. V., Odermatt, A., et al. (2020). Ca(2+) mobilization-dependent reduction of the endoplasmic reticulum lumen is due to influx of cytosolic glutathione. BMC Biol. 18:19. doi: 10.1186/s12915-020-0749-y
Lombardi, A. A., Gibb, A. A., Arif, E., Kolmetzky, D. W., Tomar, D., Luongo, T. S., et al. (2019). Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat. Commun. 10:4509.
Madreiter-Sokolowski, C. T., Gottschalk, B., Parichatikanond, W., Eroglu, E., Klec, C., Waldeck-Weiermair, M., et al. (2016). Resveratrol Specifically Kills Cancer Cells by a Devastating Increase in the Ca2+ Coupling Between the Greatly Tethered Endoplasmic Reticulum and Mitochondria. Cell Physiol. Biochem. 39, 1404–1420. doi: 10.1159/000447844
Mallilankaraman, K., Cardenas, C., Doonan, P. J., Chandramoorthy, H. C., Irrinki, K. M., Golenar, T., et al. (2012). MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 14, 1336–1343. doi: 10.1038/ncb2622
Malyala, S., Zhang, Y., Strubbe, J. O., and Bazil, J. N. (2019). Calcium phosphate precipitation inhibits mitochondrial energy metabolism. PLoS Comput. Biol. 15:e1006719. doi: 10.1371/journal.pcbi.1006719
Mankad, P., James, A., Siriwardena, A. K., Elliott, A. C., and Bruce, J. I. (2012). Insulin protects pancreatic acinar cells from cytosolic calcium overload and inhibition of plasma membrane calcium pump. J. Biol. Chem. 287, 1823–1836. doi: 10.1074/jbc.m111.326272
Marchi, S., Corricelli, M., Branchini, A., Vitto, V. A. M., Missiroli, S., Morciano, G., et al. (2019). Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca(2+) levels and tumor growth. EMBO J. 38:e99435.
Marcu, R., Wiczer, B. M., Neeley, C. K., and Hawkins, B. J. (2014). Mitochondrial matrix Ca(2)(+) accumulation regulates cytosolic NAD(+)/NADH metabolism, protein acetylation, and sirtuin expression. Mol. Cell Biol. 34, 2890–2902. doi: 10.1128/mcb.00068-14
Markwardt, M. L., Seckinger, K. M., and Rizzo, M. A. (2016). Regulation of Glucokinase by Intracellular Calcium Levels in Pancreatic beta Cells. J. Biol. Chem. 291, 3000–3009. doi: 10.1074/jbc.m115.692160
Marreiros, B. C., Calisto, F., Castro, P. J., Duarte, A. M., Sena, F. V., Silva, A. F., et al. (2016). Exploring membrane respiratory chains. Biochim. Biophys. Acta 1857, 1039–1067. doi: 10.1016/j.bbabio.2016.03.028
Moreno-Felici, J., Hyrossova, P., Arago, M., Rodriguez-Arevalo, S., Garcia-Roves, P. M., Escolano, C., et al. (2019). Phosphoenolpyruvate from Glycolysis and PEPCK Regulate Cancer Cell Fate by Altering Cytosolic Ca(2). Cells 9:18 doi: 10.3390/cells9010018
Mota-Martorell, N., Jove, M., Pradas, I., Sanchez, I., Gomez, J., Naudi, A., et al. (2020). Low abundance of NDUFV2 and NDUFS4 subunits of the hydrophilic complex I domain and VDAC1 predicts mammalian longevity. Redox Biol. 34:101539. doi: 10.1016/j.redox.2020.101539
Mukherjea, D., Jajoo, S., Whitworth, C., Bunch, J. R., Turner, J. G., Rybak, L. P., et al. (2008). Short interfering RNA against transient receptor potential vanilloid 1 attenuates cisplatin-induced hearing loss in the rat. J. Neurosci. 28, 13056–13065. doi: 10.1523/jneurosci.1307-08.2008
Nemani, N., Dong, Z., Daw, C. C., Madaris, T. R., Ramachandran, K., Enslow, B. T., et al. (2020). Mitochondrial pyruvate and fatty acid flux modulate MICU1-dependent control of MCU activity. Sci. Signal 13:eaaz6206. doi: 10.1126/scisignal.aaz6206
Nosek, M. T., Dransfield, D. T., and Aprille, J. R. (1990). Calcium stimulates ATP-Mg/Pi carrier activity in rat liver mitochondria. J. Biol. Chem. 265, 8444–8450.
Ozcan, L., Wong, C. C., Li, G., Xu, T., Pajvani, U., Park, S. K., et al. (2012). Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 15, 739–751. doi: 10.1016/j.cmet.2012.03.002
Palmieri, L., Pardo, B., Lasorsa, F. M., del Arco, A., Kobayashi, K., Iijima, M., et al. (2001). Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 20, 5060–5069. doi: 10.1093/emboj/20.18.5060
Pan, X., Liu, J., Nguyen, T., Liu, C., Sun, J., Teng, Y., et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell. Biol. 15, 1464–1472. doi: 10.1038/ncb2868
Pandya, J. D., Nukala, V. N., and Sullivan, P. G. (2013). Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Front. Neuroenergetics 5:10. doi: 10.3389/fnene.2013.00010
Park, K. C., Kim, S. W., Jeon, J. Y., Jo, A. R., Choi, H. J., Kim, J., et al. (2018). Survival of Cancer Stem-Like Cells Under Metabolic Stress via CaMK2alpha-mediated Upregulation of Sarco/Endoplasmic Reticulum Calcium ATPase Expression. Clin. Cancer Res. 24, 1677–1690. doi: 10.1158/1078-0432.ccr-17-2219
Park, K. S., Wiederkehr, A., Kirkpatrick, C., Mattenberger, Y., Martinou, J. C., Marchetti, P., et al. (2008). Selective actions of mitochondrial fission/fusion genes on metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 283, 33347–33356. doi: 10.1074/jbc.m806251200
Paul, R. J., Hardin, C. D., Raeymaekers, L., Wuytack, F., and Casteels, R. (1989). Preferential support of Ca2+ uptake in smooth muscle plasma membrane vesicles by an endogenous glycolytic cascade. FASEB J. 3, 2298–2301. doi: 10.1096/fasebj.3.11.2528493
Peters, A. A., Milevskiy, M. J., Lee, W. C., Curry, M. C., Smart, C. E., Saunus, J. M., et al. (2016). The calcium pump plasma membrane Ca(2+)-ATPase 2 (PMCA2) regulates breast cancer cell proliferation and sensitivity to doxorubicin. Sci. Rep. 6:25505.
Poburko, D., and Demaurex, N. (2012). Regulation of the mitochondrial proton gradient by cytosolic Ca(2)(+) signals. Pflugers Arch. 464, 19–26. doi: 10.1007/s00424-012-1106-y
Prevarskaya, N., Skryma, R., and Shuba, Y. (2018). Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 98, 559–621. doi: 10.1152/physrev.00044.2016
Qin, F., Siwik, D. A., Lancel, S., Zhang, J., Kuster, G. M., Luptak, I., et al. (2013). Hydrogen peroxide-mediated SERCA cysteine 674 oxidation contributes to impaired cardiac myocyte relaxation in senescent mouse heart. J. Am. Heart Assoc. 2:e000184.
Rasola, A., and Bernardi, P. (2015). Reprint of “The mitochondrial permeability transition pore and its adaptive responses in tumor cells”. Cell Calcium. 58, 18–26. doi: 10.1016/j.ceca.2015.03.004
Ren, T., Wang, J., Zhang, H., Yuan, P., Zhu, J., Wu, Y., et al. (2018). MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxid. Redox. Signal 28, 1120–1136. doi: 10.1089/ars.2017.6990
Ren, T., Zhang, H., Wang, J., Zhu, J., Jin, M., Wu, Y., et al. (2017). mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 36, 5897–5909. doi: 10.1038/onc.2017.167
Rengifo, J., Gibson, C. J., Winkler, E., Collin, T., and Ehrlich, B. E. (2007). Regulation of the inositol 1,4,5-trisphosphate receptor type I by O-GlcNAc glycosylation. J. Neurosci. 27, 13813–13821. doi: 10.1523/jneurosci.2069-07.2007
Richardson, D. A., Sritangos, P., James, A. D., Sultan, A., and Bruce, J. I. E. (2020). Metabolic regulation of calcium pumps in pancreatic cancer: role of phosphofructokinase-fructose-bisphosphatase-3 (PFKFB3). Cancer. Metab. 8:2.
Robb-Gaspers, L. D., Burnett, P., Rutter, G. A., Denton, R. M., Rizzuto, R., and Thomas, A. P. (1998). Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 17, 4987–5000. doi: 10.1093/emboj/17.17.4987
Rossi, A., Pizzo, P., and Filadi, R. (2019). Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 1866, 1068–1078. doi: 10.1016/j.bbamcr.2018.10.016
Rutter, G. A., and Rizzuto, R. (2000). Regulation of mitochondrial metabolism by ER Ca2+ release: an intimate connection. Trends Biochem. Sci. 25, 215–221. doi: 10.1016/s0968-0004(00)01585-1
Samad, A., James, A., Wong, J., Mankad, P., Whitehouse, J., Patel, W., et al. (2014). Insulin protects pancreatic acinar cells from palmitoleic acid-induced cellular injury. J. Biol. Chem. 289, 23582–23595. doi: 10.1074/jbc.m114.589440
Santo-Domingo, J., and Demaurex, N. (2010). Calcium uptake mechanisms of mitochondria. Biochim. Biophys. Acta 1797, 907–912. doi: 10.1016/j.bbabio.2010.01.005
Schieber, M., and Chandel, N. S. (2014). ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462.
Sharov, V. S., Dremina, E. S., Galeva, N. A., Williams, T. D., and Schoneich, C. (2006). Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem. J. 394, 605–615. doi: 10.1042/bj20051214
Shoshan-Barmatz, V., Zakar, M., Rosenthal, K., and Abu-Hamad, S. (2009). Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim. Biophys. Acta 1787, 421–430. doi: 10.1016/j.bbabio.2008.11.009
Song, J. H., Jung, S. Y., Hong, S. B., Kim, M. J., and Suh, C. K. (2003). Effect of high glucose on basal intracellular calcium regulation in rat mesangial cell. Am. J. Nephrol. 23, 343–352. doi: 10.1159/000072916
Song, X., Liu, B. C., Lu, X. Y., Yang, L. L., Zhai, Y. J., Eaton, A. F., et al. (2014). Lovastatin inhibits human B lymphoma cell proliferation by reducing intracellular ROS and TRPC6 expression. Biochim. Biophys. Acta 1843, 894–901. doi: 10.1016/j.bbamcr.2014.02.002
Sousa, S. C., Maciel, E. N., Vercesi, A. E., and Castilho, R. F. (2003). Ca2+-induced oxidative stress in brain mitochondria treated with the respiratory chain inhibitor rotenone. FEBS Lett. 543, 179–183. doi: 10.1016/s0014-5793(03)00421-6
Sritangos, P., Pena Alarcon, E., James, A. D., Sultan, A., Richardson, D. A., and Bruce, J. I. E. (2020). Plasma Membrane Ca(2+) ATPase Isoform 4 (PMCA4) Has an Important Role in Numerous Hallmarks of Pancreatic Cancer. Cancers 12:218. doi: 10.3390/cancers12010218
Sullivan, L. B., and Chandel, N. S. (2014). Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2:17.
Szibor, M., Gizatullina, Z., Gainutdinov, T., Endres, T., Debska-Vielhaber, G., Kunz, M., et al. (2020). Cytosolic, but not matrix, calcium is essential for adjustment of mitochondrial pyruvate supply. J. Biol. Chem. 295, 4383–4397. doi: 10.1074/jbc.ra119.011902
Takahashi, N., Chen, H. Y., Harris, I. S., Stover, D. G., Selfors, L. M., Bronson, R. T., et al. (2018). Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 33, 985–1003e7.
Territo, P. R., Mootha, V. K., French, S. A., and Balaban, R. S. (2000). Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am. J. Physiol. Cell Physiol. 278, C423–C435.
Tikunov, A., Johnson, C. B., Pediaditakis, P., Markevich, N., Macdonald, J. M., Lemasters, J. J., et al. (2010). Closure of VDAC causes oxidative stress and accelerates the Ca(2+)-induced mitochondrial permeability transition in rat liver mitochondria. Arch. Biochem. Biophys. 495, 174–181. doi: 10.1016/j.abb.2010.01.008
Tokunaga, T., Ando, T., Suzuki-Karasaki, M., Ito, T., Onoe-Takahashi, A., Ochiai, T., et al. (2018). Plasma-stimulated medium kills TRAIL-resistant human malignant cells by promoting caspase-independent cell death via membrane potential and calcium dynamics modulation. Int. J. Oncol. 52, 697–708.
Tong, X., Ying, J., Pimentel, D. R., Trucillo, M., Adachi, T., and Cohen, R. A. (2008). High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J. Mol. Cell Cardiol. 44, 361–369. doi: 10.1016/j.yjmcc.2007.10.022
Tosatto, A., Sommaggio, R., Kummerow, C., Bentham, R. B., Blacker, T. S., Berecz, T., et al. (2016). The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol. Med. 8, 569–585. doi: 10.15252/emmm.201606255
Tsachaki, M., Mladenovic, N., Stambergova, H., Birk, J., and Odermatt, A. (2018). Hexose-6-phosphate dehydrogenase controls cancer cell proliferation and migration through pleiotropic effects on the unfolded-protein response, calcium homeostasis, and redox balance. FASEB J. 32, 2690–2705. doi: 10.1096/fj.201700870rr
Ushio-Fukai, M. (2006). Localizing NADPH oxidase-derived ROS. Sci. STKE 2006:re8. doi: 10.1126/stke.3492006re8
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. doi: 10.1126/science.1160809
Waller, A. P., Kalyanasundaram, A., Hayes, S., Periasamy, M., and Lacombe, V. A. (2015). Sarcoplasmic reticulum Ca2+ ATPase pump is a major regulator of glucose transport in the healthy and diabetic heart. Biochim. Biophys. Acta 1852, 873–881. doi: 10.1016/j.bbadis.2015.01.009
Wang, W., Xie, Q., Zhou, X., Yao, J., Zhu, X., Huang, P., et al. (2015). Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 358, 47–58. doi: 10.1016/j.canlet.2014.12.025
Wang, Y., Li, G., Goode, J., Paz, J. C., Ouyang, K., Screaton, R., et al. (2012). Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 485, 128–132. doi: 10.1038/nature10988
Wayman, G. A., Tokumitsu, H., Davare, M. A., and Soderling, T. R. (2011). Analysis of CaM-kinase signaling in cells. Cell Calcium. 50, 1–8. doi: 10.1016/j.ceca.2011.02.007
Wescott, A. P., Kao, J. P. Y., Lederer, W. J., and Boyman, L. (2019). Voltage-energized Calcium-sensitive ATP Production by Mitochondria. Nat. Metab. 1, 975–984. doi: 10.1038/s42255-019-0126-8
Wright, D. C., Hucker, K. A., Holloszy, J. O., and Han, D. H. (2004). Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes 53, 330–335. doi: 10.2337/diabetes.53.2.330
Xu, L., Eu, J. P., Meissner, G., and Stamler, J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234–237. doi: 10.1126/science.279.5348.234
Yoshida, T., Inoue, R., Morii, T., Takahashi, N., Yamamoto, S., Hara, Y., et al. (2006). Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2, 596–607. doi: 10.1038/nchembio821
Zaidi, A., Barron, L., Sharov, V. S., Schoneich, C., Michaelis, E. K., and Michaelis, M. L. (2003). Oxidative inactivation of purified plasma membrane Ca2+-ATPase by hydrogen peroxide and protection by calmodulin. Biochemistry 42, 12001–12010. doi: 10.1021/bi034565u
Zhao, R. Z., Jiang, S., Zhang, L., and Yu, Z. B. (2019). Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 44, 3–15.
Zhao, S., Xu, W., Jiang, W., Yu, W., Lin, Y., Zhang, T., et al. (2010). Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004.
Zhou, M. H., Bavencoffe, A., and Pan, H. L. (2015). Molecular Basis of Regulating High Voltage-Activated Calcium Channels by S-Nitrosylation. J. Biol. Chem. 290, 30616–30623. doi: 10.1074/jbc.m115.685206
Keywords: calcium, signaling, metabolism, interplay, cancer
Citation: Dejos C, Gkika D and Cantelmo AR (2020) The Two-Way Relationship Between Calcium and Metabolism in Cancer. Front. Cell Dev. Biol. 8:573747. doi: 10.3389/fcell.2020.573747
Received: 17 June 2020; Accepted: 12 October 2020;
Published: 13 November 2020.
Edited by:
José Lozano, University of Málaga, SpainReviewed by:
Rafael Franco, University of Barcelona, SpainFrank Thévenod, Witten/Herdecke University, Germany
Copyright © 2020 Dejos, Gkika and Cantelmo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Rita Cantelmo, YW5uYS1yaXRhLmNhbnRlbG1vQHVuaXYtbGlsbGUuZnI=