 Loic Auderset1Kimberley A. Pitman1
Loic Auderset1Kimberley A. Pitman1 Carlie L. Cullen1
Carlie L. Cullen1 Renee E. Pepper1
Renee E. Pepper1 Bruce V. Taylor1Lisa Foa2
Bruce V. Taylor1Lisa Foa2 Kaylene M. Young1*
Kaylene M. Young1*- 1Menzies Institute for Medical Research, University of Tasmania, Hobart, TAS, Australia
- 2School of Medicine, University of Tasmania, Hobart, TAS, Australia
Low-density lipoprotein receptor-related protein 1 (LRP1) is a large, endocytic cell surface receptor that is highly expressed by oligodendrocyte progenitor cells (OPCs) and LRP1 expression is rapidly downregulated as OPCs differentiate into oligodendrocytes (OLs). We report that the conditional deletion of Lrp1 from adult mouse OPCs (Pdgfrα-CreER :: Lrp1fl/fl) increases the number of newborn, mature myelinating OLs added to the corpus callosum and motor cortex. As these additional OLs extend a normal number of internodes that are of a normal length, Lrp1-deletion increases adult myelination. OPC proliferation is also elevated following Lrp1 deletion in vivo, however, this may be a secondary, homeostatic response to increased OPC differentiation, as our in vitro experiments show that LRP1 is a direct negative regulator of OPC differentiation, not proliferation. Deleting Lrp1 from adult OPCs also increases the number of newborn mature OLs added to the corpus callosum in response to cuprizone-induced demyelination. These data suggest that the selective blockade of LRP1 function on adult OPCs may enhance myelin repair in demyelinating diseases such as multiple sclerosis.
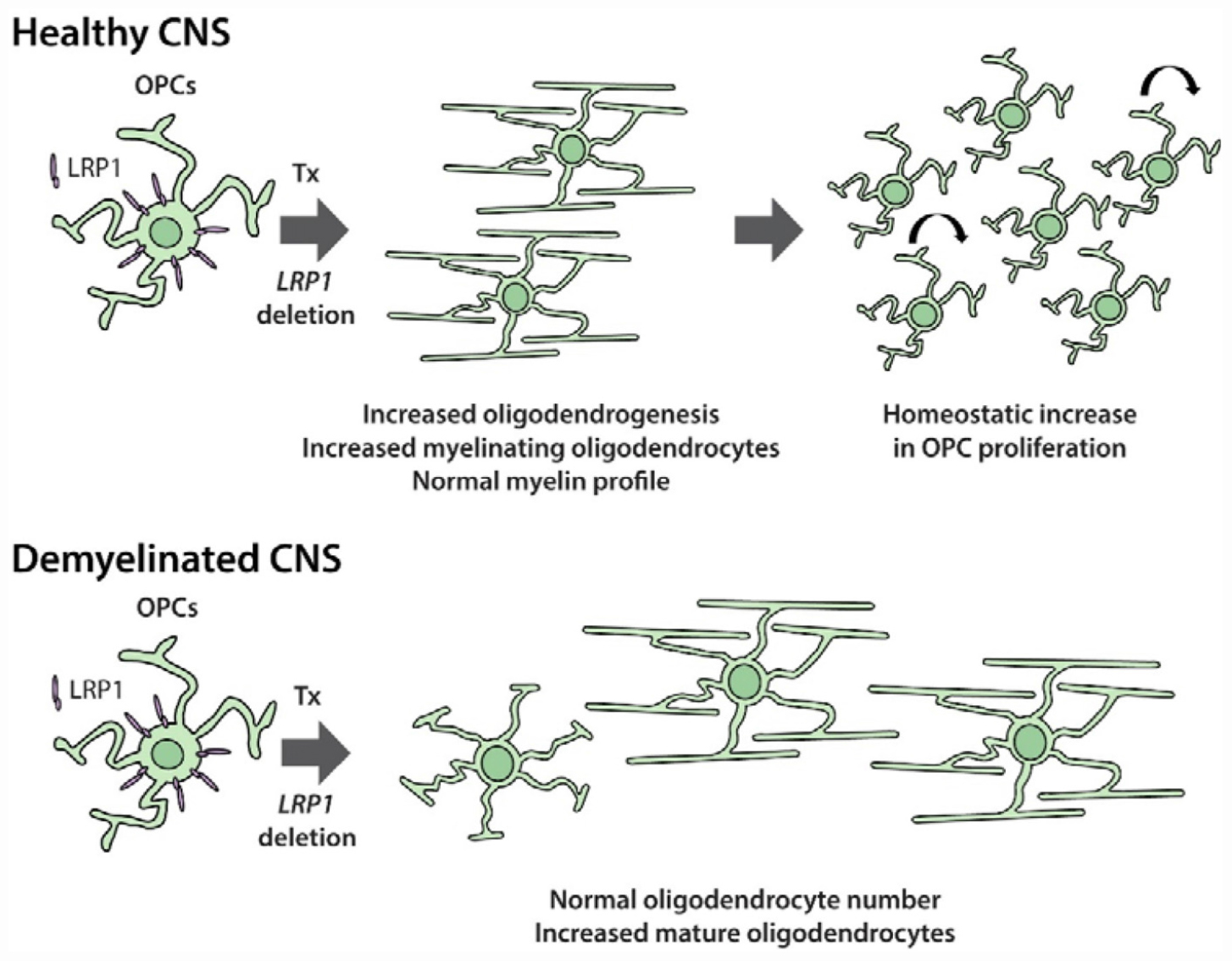
Graphical Abstract. LRP1 regulates adult oligodendrogenesis and remyelination.
Introduction
Oligodendrocytes (OLs) myelinate the central nervous system (CNS) to facilitate the saltatory conduction of action potentials and provide essential metabolic support to axons (reviewed by Pepper et al., 2018). The majority of OLs are produced from oligodendrocyte progenitor cells (OPCs) during development, however, adult OPCs also produce new OLs (Dimou et al., 2008; Rivers et al., 2008; Zhu et al., 2008; Kang et al., 2010; Hughes et al., 2013; Young et al., 2013; Hill et al., 2018) that add additional myelin to the mature CNS (Hill et al., 2018; Hughes et al., 2018). A number of signaling pathways have been identified that regulate developmental and adult OPC behavior and oligodendrogenesis, including Notch1 (Genoud et al., 2002; Givogri et al., 2002; Zhang et al., 2009), fibroblast growth factor 2 (Murtie et al., 2005; Zhou et al., 2006; Murcia-Belmonte et al., 2014), mammalian target of rapamycin (Zou et al., 2014; Jiang et al., 2016; Grier et al., 2017) and platelet-derived growth factor A (McKinnon, 2005; Rajasekharan, 2008; Chew et al., 2010) signaling. However, microarray (Cahoy et al., 2008) and RNA sequencing (Zhang et al., 2014; Hrvatin et al., 2018) experiments have uncovered a number of additional genes that are differentially expressed across OL development and have an unknown or partly characterized regulatory function in the OL lineage. One such gene is the low-density lipoprotein receptor related protein 1 (Lrp1).
LRP1, also known as CD91 or the α2 macroglobulin (α2M) receptor, is highly expressed by OPCs and is rapidly downregulated during OL differentiation (Auderset et al., 2016a). This large cell surface receptor, comprising a 515 kDa extracellular α-chain and an 85 kDa β-chain, could influence OPC behavior in a number of ways, as it interacts with a large variety of ligands, as well as extracellular and intracellular proteins, to facilitate signal transduction (Auderset et al., 2016b; Bres and Faissner, 2019). In other cell types, LRP1 acts as a receptor or co-receptor to initiate intracellular signal transduction, but also facilitates ligand endocytosis, transcytosis, or processing (Cam et al., 2005; Parkyn et al., 2008; Su et al., 2008; Yamamoto et al., 2014; Liu et al., 2017; Van Gool et al., 2019; Rauch et al., 2020), as well as receptor, channel and transporter trafficking (Parkyn et al., 2008; Maier et al., 2013; Nakajima et al., 2013; Liu et al., 2015; Boyé et al., 2017; Kadurin et al., 2017; Wujak et al., 2018) to influence blood brain barrier permeability (Polavarapu et al., 2007), lipid metabolism, glucose homeostasis, neuroinflammation (Brifault et al., 2017, 2019; Actis Dato and Chiabrando, 2018) and synaptic plasticity (Zhuo et al., 2000).
Lrp1 knockout mice are embryonic lethal, as the blastocysts fail to implant (Herz et al., 1992), but the conditional deletion of Lrp1 from cultured mouse neural stem and progenitor cells (NSPCs) impairs NSPC proliferation and reduces the number of OL lineage cells generated (Hennen et al., 2013; Safina et al., 2016). Furthermore, the conditional deletion of Lrp1 from Olig2+ cells (Olig2-Cre :: Lrp1fl/fl mice) impairs oligodendrogenesis in the developing mouse optic nerve, ultimately reducing the proportion of axons that are myelinated and myelin thickness by postnatal day (P)21 (Lin et al., 2017). As ∼20% of optic nerve axons are subsequently myelinated in control and Lrp1-deleted mice between P21 and P56, and the discrepancy in g-ratio lessens (Lin et al., 2017), this phenotype may represent a developmental delay in optic nerve myelination. This idea is supported by myelination being grossly normal in the brain and optic nerve of young adult mice following the developmental deletion of Lrp1 from Olig1+ cells (Fernandez-Castaneda et al., 2019).
As OPC physiology changes considerably between development and adulthood, and can also differ between CNS regions (Velez-Fort et al., 2010; Pitman et al., 2019; Spitzer et al., 2019), we examined the importance of LRP1 for adult OPC function. The conditional deletion of Lrp1 from adult OPCs (Pdgfrα-CreER :: Lrp1fl/fl) revealed that LRP1 is a negative regulator of oligodendrogenesis in the healthy adult mouse brain. Lrp1 deletion was associated with an increase in adult OPC proliferation and a significant increase in the number of newborn mature OLs added to the cortex and corpus callosum (see Graphical Abstract). Furthermore, Lrp1 deletion was associated with a larger number of newborn mature OLs being detected in the corpus callosum of cuprizone fed-mice and less of the corpus callosum showed overt demyelination.
Materials and Methods
Transgenic Mice and Their Housing
All animal experiments were approved by the University of Tasmania Animal Ethics (A0016151) and Institutional Biosafety Committees and were carried out in accordance with the Australian code of practice for the care and use of animals for scientific purposes. Pdgfrα-CreERT2 mice (Rivers et al., 2008) were a kind gift from Prof William D Richardson (University College London). Pdgfrα-CreERTM (Kang et al., 2010; RRID:IMSR_JAX:018280), Pdgfrα-H2BGFP [Pdgfrα-histGFP (Hamilton et al., 2003); RRID:IMSR_JAX:007669] and Lrp1fl/fl (Herz et al., 1992; RRID:IMSR_JAX:012604) mice were purchased from Jackson Laboratories. Cre-sensitive Rosa26-YFP (Srinivas et al., 2001; RRID: IMSR_JAX:006148) and Tau-mGFP (Hippenmeyer et al., 2005; RRID:IMSR_JAX021162) reporter mice were also purchased from Jackson laboratories. Mice were maintained on a C57BL/6 background and inter-crossed to generate male and female offspring for experimental use. All mice were weaned >P30 to ensure appropriate myelin development, were group housed with same-sex littermates in Optimice micro-isolator cages (Animal Care Systems, CO, United States) and were maintained on a 12-h light/dark cycle at 20°C, with uninhibited access to food and water.
Please note that two distinct Pdgfrα-CreER transgenic mouse lines were used in this study: the Pdgfrα-CreERTM transgenic mouse line (Kang et al., 2010) was used for the majority of experiments and the lower efficiency (LE) Pdgfrα-CreERT2 transgenic mouse line (Rivers et al., 2008), was used to perform the Tau-mGFP lineage tracing experiments, as we have previously demonstrated that the Pdgfrα-CreERTM transgenic mouse line cannot be used to induce OPC-specific recombination of the Tau-mGFP reporter, despite achieving the OPC-specific recombination of other transgenes (Pitman et al., 2019).
Genomic DNA Extraction and PCR Amplification
For genotyping, ear biopsies were digested overnight in DNA extraction buffer (100 mM Tris-HCl, 5 mM EDTA, 200 mM NaCl, 0.2% SDS and 120 ng of proteinase k) at 55°C. Cellular and histone proteins were precipitated by incubating samples with 6 M ammonium acetate (Sigma; A1542) on ice and the DNA was precipitated from the supernatant by incubating with isopropyl alcohol (Sigma; I9516). The DNA pellet was washed in 70% ethanol (Sigma; E7023), resuspended in sterile MilliQ water and used as template DNA for polymerase chain reaction (PCR). Each 25 μL reaction contained: 50–100 ng DNA; 0.5 μL of each primer (100 nmol/mL, GeneWorks); 12.5 μL of GoTaq green master mix (Promega) and MilliQ water. The following primers were used: Lrp1 5′ CATAC CCTCT CAAACC CCTT CCTG and Lrp1 3′ GCAAG CTCC CTGCTCA GACC TGGA; Rosa26 wildtype 5′ AAAGT CGCTC TGAGT TGTTAT, Rosa26 wildtype 3′ GGAGC GGGAG AAATG GATATG and Rosa26 mutant 5′ GCGAA GAGTT TGTCC TCAACC; Cre 5′ CAGGT CTCAG GAGCT ATGTC CAATT TACTG ACCGTA and Cre 3′ GGTGT TATAAG CAATCC CCAGAA, or GFP 5′ CCCTG AAGTTC ATCTG CACCAC and GFP 3′ TTCTC GTTGG GGTCT TTGCTC in a program of: 94°C for 4 min, and 34 cycles of 94°C for 30″, 60°C for 45″ (37 cycles for Rosa26-YFP genotyping) and 72°C for 60″, followed by 72°C for 10 min. Following gel electrophoresis [1% (w/v) agarose in TAE containing SYBR-safe (Thermo Fisher Scientific)], the DNA products were visualized using an Image Station 4000 M PRO gel system running Carestream software.
Tamoxifen Preparation and Administration
Tamoxifen (Sigma) was dissolved in corn oil (Sigma) to a concentration of 40 mg/ml by sonication in a water bath for 2 h at 21°C. Adult mice received tamoxifen (300 mg/kg) daily by oral gavage for 4 consecutive days.
Tissue Preparation and Immunohistochemistry
Mice were terminally anesthetized with an intraperitoneal (i.p) injection of sodium pentobarbital (30 mg/kg, Ilium) and were transcardially perfused with 4% (w/v) paraformaldehyde (PFA; Sigma) in phosphate buffered saline (PBS). Brains were cut into 2 mm-thick coronal slices using a 1 mm brain matrix (Kent Scientific) before being post-fixed in 4% (w/v) PFA in PBS at 21°C for 90 min. Tissue was cryoprotected in 20% sucrose (Sigma) in PBS and transferred to OCT (Thermo Fisher Scientific) before being snap frozen in liquid nitrogen and stored at −80°C.
Thirty μm coronal brain cryosections were collected and processed as floating sections (as per O’Rourke et al., 2016). Cryosections were exposed to primary antibodies diluted in blocking solution [10% (v/v) fetal calf serum (FCS, Serana) and 0.05% (v/v) triton ×100 in PBS] and incubated overnight at 4°C on an orbital shaker. Primary antibodies included: rabbit anti-LRP1 (1:500, Abcam ab92544; RRID:AB_2234877); goat anti-PDGFRα (1:100, R&D Systems AF1062; RRID:AB_2236879); rabbit anti-ASPA (1:200, Abcam ab97454; RRID:AB_10679051); rabbit anti-LRP2 (1:100, Abcam ab76969, RRID:AB_10673466); rat anti-GFP (1:2000, Nacalai Tesque 04404-26; RRID:AB_2314545); rat anti-MBP (1:100, Millipore MAB386; RRID:AB_94975), rabbit anti-OLIG2 (1:400, Abcam ab9610; RRID:AB_570666); guinea pig anti-IBA1 (1:250, Synaptic Systems 234004; RRID:AB_2493179), and mouse anti-NaBC1 (BCAS1; 1:200, Santa Cruz sc-136342; RRID:AB_10839529). After washing in PBS, cryosections were incubated with secondary antibodies conjugated to Fluors (Life Technologies Corporation), diluted in blocking solution, overnight at 4°C on an orbital shaker. Secondary antibodies included: donkey anti-rat 488 (1:500; RRID: AB_2535794); donkey anti-rabbit 488 (1:1000; RRID: AB_2535792); donkey anti-rabbit 568 (1:1000; RRID: AB_2534017); donkey anti-rabbit 647 (1:1000; RRID: AB_2536183); donkey anti-goat 647 (1:1000; RRID: AB_2535864), donkey anti-mouse 647 (1:1000; RRID AB_162542), or goat anti-guinea pig 488 (1:1000; RRID: AB_2534117). Cell nuclei were visualized by the inclusion of Hoechst 33342 (1:10,000, Invitrogen).
EdU Administration and Labeling
For the in vivo labeling of dividing cells, 5-Ethynly-2′-deoxyuridine (EdU; E10415, Thermo Fisher Scientific) was administered to mice via their drinking water at a concentration of 0.2 mg/ml for up to 21 consecutive days [as per Clarke et al. (2012)]. For in vitro labeling, cells were exposed to 2.5 μg/ml EdU in complete OPC medium (see below) for 10 h before the cells were fixed with 4% (w/v) PFA in PBS for 15 min at 21°C. The EdU developing cocktail was prepared according to the AlexaFluor-647 Click-IT EdU kit (Invitrogen) instructions, and brain slices were exposed to the developing reagent for 45 min at 21°C, while coverslips of cultured cells were exposed for 15 min. EdU developing was performed immediately after the secondary antibody was washed from tissue or cells during immunohistochemistry or immunocytochemistry.
Primary OPC Culture and in vitro Gene Deletion
The cortices of P1-10 mice were dissected into Earle’s Buffered Salt Solution (EBSS; Invitrogen, 14155-063), diced into pieces ∼1 mm3 and digested in 0.06 mg/ml trypsin (Sigma, T4799) in EBSS at 37°C for 10 min. The trypsin was inactivated by the addition of FCS, before the tissue was resuspended and triturated in EBSS containing 0.12 mg/ml DNAseI (Sigma, 5025). The cell preparation was filtered through a 40 μm sieve (Corning, 352340), centrifuged and resuspended in complete OPC medium [20 ng/ml human PDGF-AA (Peprotech), 10 ng/ml basic fibroblast growth factor (R&D Systems), 10 ng/ml human ciliary neurotrophic factor (Peprotech), 5 μg/ml N-acetyl cysteine (Sigma), 1 ng/ml neurotrophin-3 (Peprotech), 1 ng/ml biotin (Sigma), 10 μM forskolin (Sigma), 1× penicillin/streptomycin (Invitrogen), 2% B27 (Invitrogen), 50 μg/ml insulin (Sigma), 600 ng/ml progesterone (Sigma), 1 mg/ml transferrin (Sigma), 1 mg/ml BSA (Sigma), 400 ng/ml sodium selenite (Sigma) and 160 μg/ml putrescine (Sigma) in DMEM+Glutamax (Invitrogen)]. Cells were plated into 6 well plates coated with >300,000 MW Poly D Lysine (PDL; Sigma, P7405). After 7 DIV, the cells were dislodged by incubating in 1:5 TrypLE (Gibco) in EBSS for ∼10 min at 37°C, before the trypsin was inactivated by the addition of FCS, and cells were collected into EBSS. OPCs were then purified by immunopanning as previously described (Emery and Dugas, 2013). In brief, the cell suspension was transferred to a petri dish pre-coated with anti-PDGFRα (BD Pharmigen 558774; RRID:AB_397117) and the OPCs allowed to adhere for 45 min at 21°C. The non-adherent cells were then removed by rinsing with EBSS and the purified OPCs were stripped by treating with TrypLE diluted 1:5 with EBSS for 5 min at 37°C. The recovered cells were plated onto 13 mm glass coverslips in complete OPC medium.
For experiments where Lrp1 was deleted in vitro, OPCs were plated in complete OPC medium at a density of 20,000 cells per PDL-treated 13 mm coverslip and allowed to settle for 2 days. OPCs were then exposed to 1 μM TAT-Cre (Excellgen, EG-1001) in complete OPC medium at 37°C/5% CO2 for 90 min. The TAT-Cre-containing medium was then removed and replaced with fresh complete OPC medium and the cells returned to the incubator for 48 h. To induce differentiation, the complete OPC medium was removed and replaced with OPC differentiation medium [complete OPC medium lacking PDGF-AA and containing 4 μg/ml triiodothyronine (Sigma)] for 4 days before cells were fixed by exposure to 4% PFA (w/v) in PBS for 15 min at 21°C.
LRP1 ligands were reconstituted according to the manufacturer’s instructions. Human recombinant Apolipoprotein E4 (ApoE4; Sigma, A3234) was reconstituted to 0.7 mg/mL in sterile MQ water and was diluted to a concentration of 203 nM in the culture medium. Human recombinant tissue plasminogen activator (tPA; Abcam ab92637) was diluted to a final concentration of 20 nM in the culture medium. Human activated α-2 macroglobulin (∗α2M; Sigma, SRP6315) was reconstituted in MilliQ water and diluted to a final concentration of 60 mM in the culture medium.
Whole Cell Patch Clamp Electrophysiology
Acute coronal brain slices (300 μm) were generated from adult mice carrying the Pdgfrα-histGFP transgene using a VT1200s vibratome (Leica) as previously described (Pitman et al., 2019). Brain slices were transferred to a bath constantly perfused (2 ml/min) with ∼21°C artificial cerebral spinal fluid (ASCF) containing: 119 mM NaCl, 1.6 mM KCl, 1 mM NaH2PO4, 26.2 mM NaHCO3, 1.4 mM MgCl2, 2.4 mM CaCl2, and 11 mM glucose (300 ± 5 mOsm/kg), saturated with 95% O2/5% CO2, Whole cell patch clamp recordings of GFP+ cells in the motor cortex were collected using a HEKA Patch Clamp EPC800 amplifier and pCLAMP 10.5 software (Molecular devices; RRID: SCR_011323).
To record AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)/kainate receptor currents, recording electrodes (3–6 MΩ) were filled with an internal solution containing: 125 mM Cs-methanesulfonate, 5 mM TEA-Cl, 2 mM MgCl2, 8 mM HEPES, 9 mM EGTA, 10 mM phosphocreatine, 5 mM MgATP, and 1 mM Na2GTP, and set to a pH of 7.2 with CsOH and an osmolarity of 290 ± 5 mOsm/kg. Upon breakthrough, cells were held at −50 mV and a series of voltage steps (up to+30 mV) applied to determine the presence of a voltage-gated sodium channel current. GFP+ cells with a voltage-gated sodium current >100 pA were considered OPCs. All subsequent recordings were undertaken in ACSF containing 50 μm (2R)-amino-5-phosphonovaleric acid (APV; Sigma) and 1 μM tetrodotoxin (TTX; Sigma). Cells were held at −60 mV and currents elicited by applying 200 ms voltage steps from −80 to 20 mV (20 mV increments). After taking baseline recordings, currents were then elicited in ACSF containing 100 μM kainate. The mean steady state current (last 100 ms) of each voltage step was measured.
Voltage gated calcium channel (VGCC) current recordings were made using solutions previously described (Pitman et al., 2019). All other voltage gated currents (potassium and sodium) were blocked. To record L-type VGCC currents, OPCs were held at −50 mV and a series of 500 ms voltage steps (−60 to+30 mV) applied using a P/N subtraction protocol. The current-density relationship is presented as the average steady state current (the last 100 ms of the voltage steps) from ∼3 recordings per cell. To elicit currents through T-type VGCCs, OPCs were held at −50 mV and the cell was hyperpolarized to −120 mV for 200 ms before applying voltage steps from −70 to 30 mV (Fulton et al., 2010; Haberlandt et al., 2011). The maximum amplitude of the fast, transient inward current, revealed by the brief hyperpolarization, was measured from ∼3 recordings per cell.
Access resistance was measured before and after all recordings and an access resistance >20 MΩ resulted in exclusion of that recording. Due to the high membrane resistance of OPCs (>1 GΩ) during VGCC current recordings, recordings were made without series resistance compensation. However, series resistance compensation was applied for AMPA current recordings (60–80%). Measurements were made from each data file using Clampfit 10.5.
Cuprizone Administration and Black-Gold Myelin Staining
Mice were transferred onto a diet of crushed mouse food (Barrestock) containing 0.2% (w/w) cuprizone powder (C9012, Sigma), which was refreshed every 2 days for 5 weeks. Mice were perfusion fixed, their tissue processed as described above and 30 μm coronal brain floating cryosections collected into PBS. Cryosections were transferred onto glass microscope slides (Superfrost) and allowed to dry before being rehydrated in MilliQ water for 3 min and incubated with preheated 0.3% black-gold II stain (Millipore, AG105) at 60°C for 30 min. Slides were washed twice with MilliQ water before being incubated with preheated 1% (v/v) sodium thiosulphate solution at 60°C for 3 min, washed in MilliQ water (3 × 2 min), dehydrated using a series of graded alcohol steps, incubated in xylene (Sigma, 214736) for 3 min, and mounted with DPX mounting medium (Sigma, 06522).
Microscopy and Statistical Analyses
Fluorescent labeling was visualized using an UltraView Spinning Disk confocal microscope with Volocity software (Perkin Elmer, Waltham, United States). The motor cortex and corpus callosum were defined as regions of interest using anatomical markers identified in the Allen Mouse Brain Atlas, in brain sections collected between Bregma level 1.10 and −0.10 mm. Confocal images were collected using standard excitation and emission filters for DAPI, FITC (Alexa Fluor-488), TRITC (Alexa Fluor-568) and CY5 (Alexa Fluor-647). To quantify cell density, or the proportion of cells that proliferate or differentiate, a 10× or 20× air objective was used to collect images with 2 μm z-spacing that spanned the defined region of interest within a brain section. These images were stitched together using Volocity software to create a single image of that region for analysis. A minimum of 3 brain sections were imaged per mouse. To quantify oligodendrocyte morphology and measure myelin internodes a 40× (air) or 60× (water) objective was used to collect images with 1 μm z-spacing of individual mGFP+ OLs or single fields of view containing internodes within each region of interest. Black-gold myelin staining was imaged using a light microscope with a 2.5× objective and images were manually stitched together using Adobe Photoshop CS6 to recreate the region of interest. Cell counts were performed by manually evaluating the labeling of individual cells and area measurements were made by manually defining the region of interest within Photoshop CS6 (Adobe, San Jose, United States) or Image J (NIH, Bethesda, MD, United States). All measurements were made blind to the experimental group and treatment conditions.
Statistical comparisons were made using GraphPad Prism 8.0 (La Jolla, CA, United States; RRID: SCR_002798). Data were first assessed using the D’Agostino-Pearson normality test. Data that were normally distributed were analyzed by a parametric test [t-test, one-way analysis of variance (ANOVA) or two-way ANOVA for group comparisons with a Bonferroni post hoc test], and data that were not normally distributed were analyzed using a Mann–Whitney U test or Kolmogorov–Smirnov test. Lesion size (black-gold staining) data were analyzed using a t-test with a Welch’s correction to account for the uneven variance between groups. Data sets with n = 3 in any group were analyzed using parametric tests, as the non-parametric equivalents rely on ranking and are unreliable for small sample sizes (GraphPad Prism 8.0). To determine the rate at which OPCs become labeled with EdU over time, these data were analyzed by performing linear regression analyses. Details of the statistical comparisons are provided in each figure legend or in text when the data are not presented graphically. Statistical significance was defined as p < 0.05.
Results
Tamoxifen Administration Deletes Lrp1 From Essentially All OPCs in the Adult Mouse Brain
To determine the role that LRP1 plays in regulating adult myelination, Lrp1 was conditionally deleted from OPCs in young adult mice. Tamoxifen was administered to P50 control (Lrp1fl/fl) and Lrp1-deleted (Pdgfrα-CreERTM :: Lrp1fl/fl) mice and brain tissue examined 7 or 30 days later (at P50+7 and P50+30, respectively). Coronal brain cryosections from control (Figure 1A) and Lrp1-deleted mice (Figure 1B) were immunolabeled to detect LRP1 (red) and OPCs (PDGFRα, green). Consistent with our previous findings (Auderset et al., 2016a), essentially all OPCs in the corpus callosum of control mice expressed LRP1 (Figure 1C; P50+7: 99 ± 0.6%, mean ± SD for n = 4 mice; P50+30: 99.7 ± 0.3%, mean ± SD for n = 3 mice). However, in the corpus callosum of P50+7 Lrp1-deleted mice, only 2 ± 0.8% of PDGFRα+ OPCs expressed LRP1 (mean ± SD for n = 4 mice) and by P50+30, only 0.5 ± 0.5% of OPCs expressed LRP1 (mean ± SD for n = 3 mice; Figure 1C), confirming the successful deletion of Lrp1 from adult OPCs. Similarly, in the motor cortex of P50+7 control mice, 100 ± 0% of PDGFRα+ OPCs expressed LRP1 while only 0.4 ± 0.4% of PDGFRα+ OPCs expressed LRP1 in the motor cortex of Lrp1-deleted mice (mean ± SD for n = 4 mice per genotype). Other brain cell types that express LRP1, such as neurons and astrocytes, retained their expression of LRP1 (e.g., white arrows in Figure 1B). As the Cre-mediated recombination of the floxed Lrp1 gene deletes the extracellular coding region of Lrp1, recombination was also confirmed by performing a PCR analysis of genomic DNA from the brains of control (Lrp1fl/ +) and Lrp1-deleted (Pdgfrα-CreERTM :: Lrp1fl/fl) mice after tamoxifen treatment. Lrp1-deletion enabled the amplification of a recombination-specific DNA product from Lrp1-deleted brain DNA that was not amplified from control brain DNA (Figure 1D). These data confirm that tamoxifen administration to Pdgfrα-CreERTM :: Lrp1fl/fl transgenic mice efficiently and specifically deletes Lrp1 from adult OPCs.
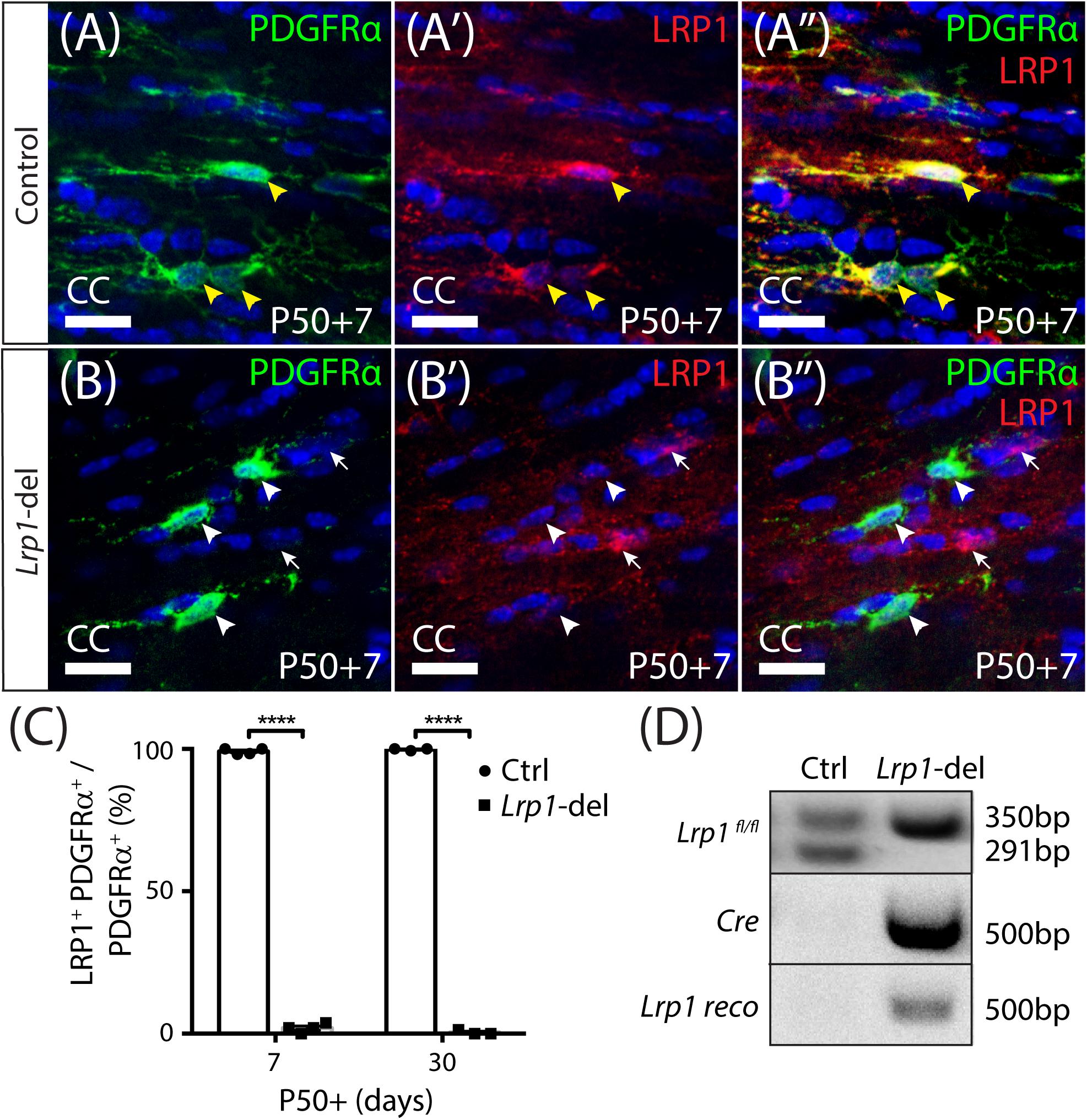
Figure 1. Lrp1 can be conditionally deleted from adult OPCs at high efficiency. Coronal brain sections from P57+7 and P57+30 control (Pdgfrα-CreERTM) and Lrp1-deleted (Pdgfrα-CreERTM :: Lrp1fl/fl) mice were immunolabeled to detect OPCs (PDGFRα, green), LRP1 (red), and cell nuclei (Hoechst 33342, blue). (A–A″) Compressed z-stack confocal image of LRP1+ OPCs (solid yellow arrow heads) in the corpus callosum (CC) of a P50+7 control mouse. (B–B″) Compressed z-stack confocal image of LRP1-neg OPCs (solid white arrow heads) in the CC of a P50+7 Lrp1-deleted mouse. White arrows indicate PDGFRα-neg cells that remain LRP1+ in the Lrp1-deleted mice. (C) The proportion (%) of PDGFRα+ OPCs that express LRP1 in P50+7 and P50+30 control and Lrp1-deleted mice [mean ± SD for n ≥ 3 mice per genotype per time-point; 2-way ANOVA: Genotype F (1,10) = 2.8, p < 0.0001; Time F (1,10) = 0.52, p = 0.5; Interaction F (1,10) = 3.44, p = 0.09]. Bonferroni multiple comparisons ****p ≤ 0.0001. (D) PCR amplification of genomic DNA extracted from the brain of a P50+7 control (Lrp1fl/+) and Lrp1-deleted (Pdgfrα-CreERTM :: Lrp1fl/fl) mouse indicates that recombination, producing the Lrp1 reco band, only occurs in Lrp1-deleted mice. Scale bars represent 17 μm.
Lrp1 Deletion Produces a Delayed Increase in Adult OPC Proliferation
Oligodendrocyte progenitor cells divide more frequently in white than gray matter regions of the adult mouse CNS (Psachoulia et al., 2009) and their homeostatic proliferation ensures that a stable pool of progenitors is maintained (Hughes et al., 2013). To determine whether LRP1 regulates the rate that adult OPCs re-enter the cell cycle to divide or the fraction of adult OPCs that proliferate, we delivered a thymidine analog, EdU, to P57+7 control and Lrp1-deleted mice via their drinking water for 2, 4, 6, or 20 days. Coronal brain cryosections from control (Figures 2A–D) and Lrp1-deleted (Figures 2E–H) mice were processed to detect PDGFRα+ OPCs (green) and EdU (red). When quantifying the proportion of OPCs that became EdU labeled over time, we found that 20 days of EdU-delivery resulted in EdU uptake by all OPCs in the corpus callosum of control and Lrp1-deleted mice (100 ± 0 and 100 ± 0%, respectively), indicating that the proportion of OPCs that can proliferate is not influenced by LRP1 signaling. Furthermore, the rate of EdU incorporation was equivalent for OPCs in the corpus callosum of control and Lrp1-deleted mice (Figure 2I), suggesting that LRP1 does not influence the rate at which OPCs enter or transition through the cell cycle to become EdU-labeled. OPCs in the motor cortex incorporated EdU at a slower rate than those in the corpus callosum (compare the slope of the regression lines in Figures 2I,J), but OPC proliferation in the motor cortex was also unaffected by Lrp1-deletion (Figure 2J).
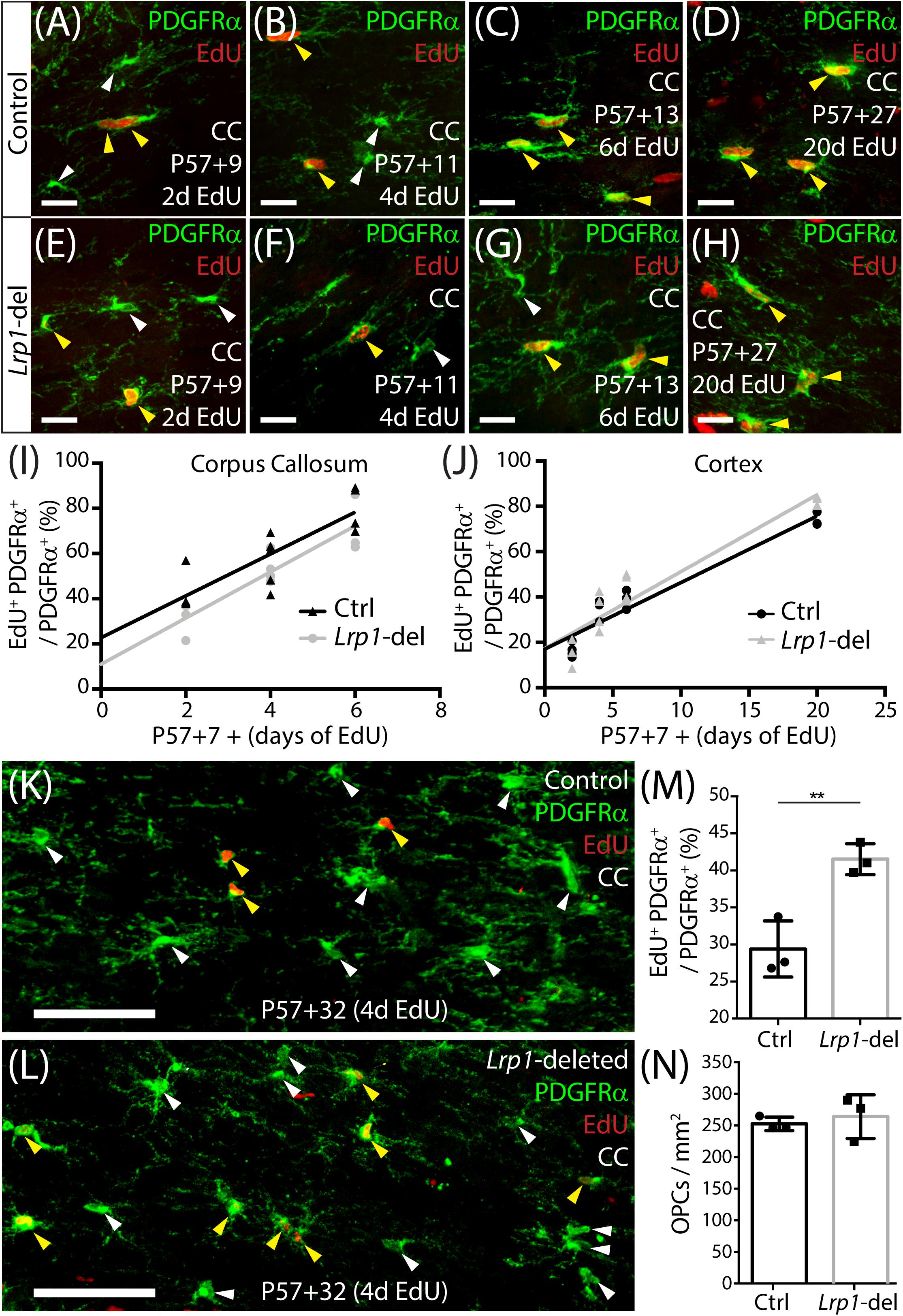
Figure 2. Adult OPC proliferation increases following Lrp1 deletion. (A–H) Compressed confocal z-stacks from the corpus callosum (CC) of control (Pdgfrα-CreERTM) and Lrp1-deleted (Pdgfrα-CreERTM :: Lrp1fl/fl) mice, immunolabeled to detect OPCs (PDGFRα, green) and EdU (red) after 2, 4, 6, or 20 days of EdU delivery. (I) Graph of the proportion (%) of OPCs that incorporate EdU, after 2, 4, or 6 days of delivery, in the CC of control (black) and Lrp1-deleted (gray) mice (n ≥ 3 mice per genotype per time-point). The rate of EdU uptake was unaffected by genotype (p = 0.7; linear regression for controls: m = 9.2 ± 1.8% per day and R2 = 0.7; linear regression for Lrp1-deleted: m = 10.2 ± 1.8% per day and R2 = 0.8). (J) Graph of the proportion (%) of OPCs that incorporate EdU, after 2, 4, 6, or 20 days of delivery, in the motor cortex of control (black) and Lrp1-deleted (gray) mice (n ≥ 3 mice per genotype per time-point). The rate of EdU uptake was unaffected by genotype (p = 0.3; linear regression for control: m = 2.9 ± 0.3 cells per day and R2 = 0.9; linear regression for Lrp1-deleted: m = 3.4 ± 0.3 cells per day and R2 = 0.9). (K,L) Compressed confocal z-stacks from the CC of P57+32 control and Lrp1-deleted mice that received EdU via the drinking water for 4 consecutive days (from P57+28), and were immunolabeled to detect OPCs (PDGFRα, green) and EdU (red). (M) The proportion (%) of OPCs that were EdU labeled in the CC of P57+32 control mice (black) and Lrp1-deleted mice (gray) that received 4 days of EdU labeling (mean ± SD, n = 3 mice per genotype; unpaired t-test, **p = 0.008). (N) Quantification of the density of OPCs in the CC of P57+32 control mice (black) and Lrp1-deleted mice (gray) (mean ± SD, n = 3 mice per genotype; unpaired t-test, p = 0.6). Solid white arrow heads indicate EdU-neg OPCs. Solid yellow arrowheads indicate EdU+ OPCs. Scale bars represent 17 μm (A–H) or 70 μm (K,L).
While the loss of LRP1 did not immediately influence OPC proliferation, LRP1 may regulate receptor and channel recycling at the cell membrane (Takayama et al., 2005; Liu et al., 2010; Pi et al., 2012; Maier et al., 2013; Nakajima et al., 2013), such that Lrp1-deletion may not immediately perturb OPC behavior. To explore this possibility, we delivered tamoxifen to young adult (P57) control and Lrp1-deleted mice and waited 28 days before administering EdU via the drinking water for 4 consecutive days. Coronal brain cryosections from P57+32 control (Figure 2K) and Lrp1-deleted (Figure 2L) mice were processed to detect PDGFRα+ OPCs (green) and EdU (red). The proportion of OPCs that incorporated EdU over the 4-day labeling period was significantly higher in the corpus callosum of Lrp1-deleted mice than controls (Figure 2M). This was also true in the motor cortex, where 15.0 ± 1.02% of control and 20.1 ± 2.53% of Lrp1-deleted OPCs had incorporated EdU (mean ± SD for n = 3 mice per genotype; unpaired t-test, p = 0.03). This increase in OPC proliferation was not accompanied by a change in the density of PDGFRα+ OPCs (Figure 2N), suggesting that the additional cells must either differentiate or die.
Lrp1 Deletion Increases the Number of New OLs Added to the Adult Mouse Brain
To determine whether LRP1 regulates OL production by adult OPCs, tamoxifen was given to P57 control (Pdgfrα-CreERTM :: Rosa26-YFP) and Lrp1-deleted (Pdgfrα-CreERTM :: Rosa26-YFP :: Lrp1fl/fl) mice to fluorescently label adult OPCs and the new OLs they produce. At P57+14, coronal brain cryosections were immunolabeled to detect YFP (green), PDGFRα (red), and OLIG2 (blue) and confirm the specificity of labeling (Supplementary Figure 1). Consistent with our previous findings in control mice (O’Rourke et al., 2016), all YFP+ cells in the corpus callosum of control and Lrp1-deleted mice were either PDGFRα+ OLIG2+ OPCs or PDGFRα-negative OLIG2+ newborn OLs (Supplementary Figure 1). In the motor cortex, the vast majority of YFP+ cells expressed OLIG2 (control: 96.2 ± 0.91%; Lrp1-deleted: 94.3 ± 1.02%, mean ± SD for n = 3 mice per genotype; Supplementary Figure 1). The small number of YFP+ OLIG2-negative cells identified in the cortex had the morphological characteristics of neurons, consistent with previous reports that the Pdgfrα promoter is active in a small subset of cortical neurons (Clarke et al., 2012). These cells were excluded from all subsequent analyses.
To determine whether LRP1 influences oligodendrogenesis, we quantified the proportion of YFP+ cells that were PDGFRα-negative OLIG2+ newborn OLs in the corpus callosum (Figures 3A–F) or motor cortex (Figures 3G–L) of P57+7, P57+14, P57+30 and P57+45 control and Lrp1-deleted mice. At P57+7 and P57+14 oligodendrogenesis was equivalent in the corpus callosum of control and Lrp1-deleted mice, however, by P57+30 a larger proportion of YFP+ cells had become newborn OLs in the corpus callosum of Lrp1-deleted mice, and this effect was sustained at P57+45 (Figure 3M). Similarly, for the first 2 weeks, OL production was equivalent for OPCs in the motor cortex of control and Lrp1-deleted mice, however, by P57+30, the proportion of YFP+ cells that were newborn OLs was higher in the motor cortex of Lrp1-deleted mice than controls (Figure 3N). This produced a corresponding increase in the density of new OLs detected in the corpus callosum (control: 107.2 ± 14.9 cells/mm2; Lrp1-deleted: 161.8 ± 27.4 cells/mm2; mean ± SD, n = 7 control and n = 4 Lrp1-deleted mice; t-test, p = 0.0005) and motor cortex (control: 42.55 ± 9.2 cells/mm2; Lrp1-del: 61.34 ± 7.0 cells/mm2; mean ± SD, n = 4 mice per genotype; t-test, p = 0.004) of P57+30 Lrp1-deleted mice compared to controls. By performing immunohistochemistry to detect YFP (green) and the mature OL marker, aspartoacylase (ASPA, red) (Figures 4A–L), we confirmed that Lrp1-deletion increased the number of newborn mature OLs added to the adult mouse corpus callosum and motor cortex (Figure 4M).
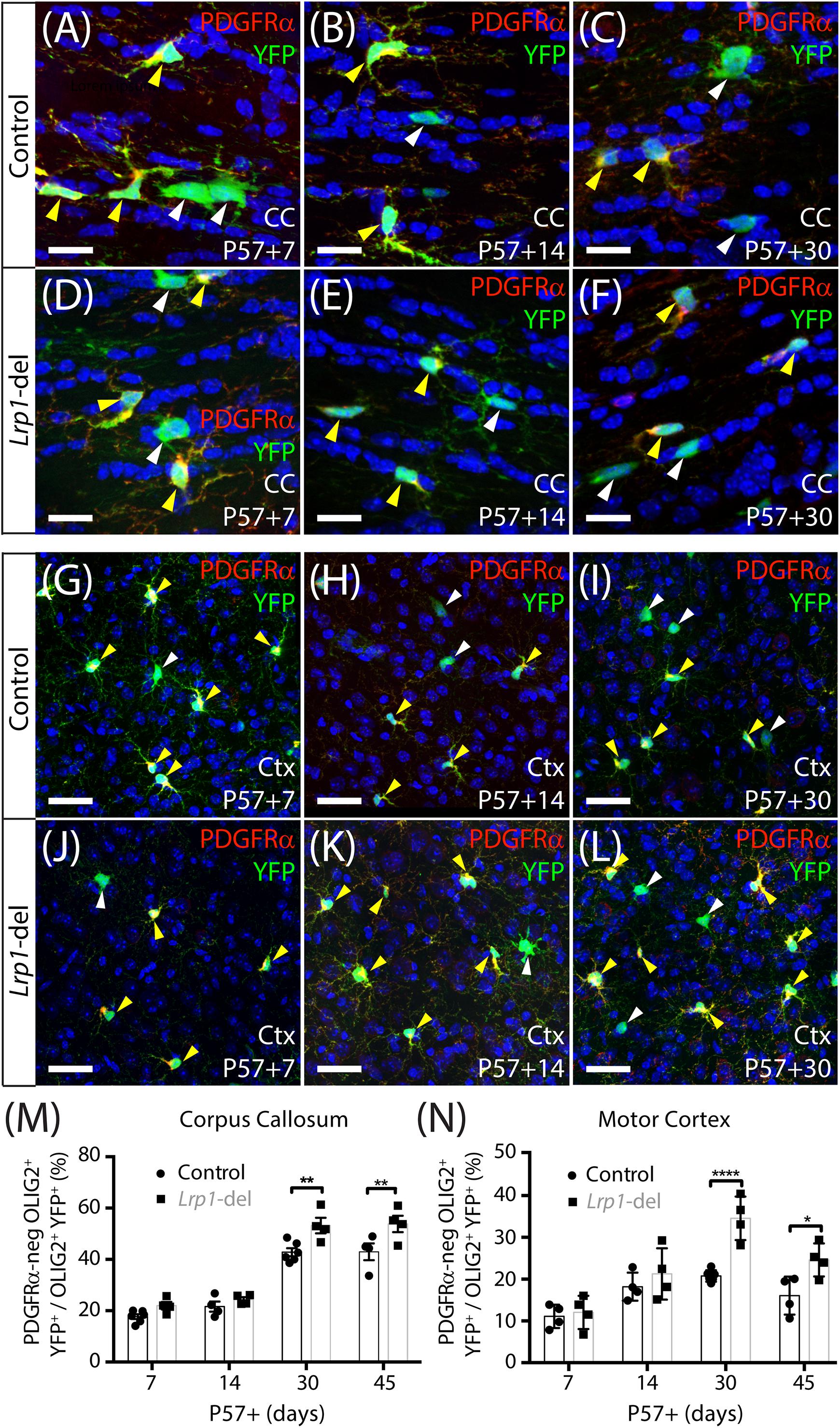
Figure 3. Lrp1 deletion increases the number of new OLs added to the adult mouse corpus callosum and motor cortex. (A–L) Confocal images of the corpus callosum (CC; A–F) and motor cortex (Ctx; G–L) of P57+7, P57+14, and P57+30 control (Pdgfrα-CreERTM :: Rosa26-YFP) and Lrp1-deleted (Pdgfrα-CreERTM :: Rosa26-YFP :: Lrp1fl/fl) mice immunolabeled to detect PDGFRα (red), YFP (green), and the nuclear marker Hoechst 33342 (blue). Solid yellow arrowheads indicate YFP+ PDGFRα+ OPCs. Solid white arrowheads indicate YFP+ PDGFRα-neg newborn OLs. (M) Graph of the proportion (%) of YFP+ cells that are YFP+ PDGFRα-neg OLIG2+ newborn OLs in the CC of control and Lrp1-deleted mice [mean ± SD for n ≥ 4 mice per genotype per time-point; 2-way ANOVA: Genotype F (1, 28) = 22.3, p < 0.0001; Time F (3, 28) = 109.7, p < 0.0001; Interaction F (3, 28) = 1.9, p = 0.15]. (N) Graph of the proportion (%) of YFP+ cells that are YFP+ PDGFRα-neg OLIG2+ newborn OLs in the motor cortex of control and Lrp1-deleted mice [mean ± SD for n ≥ 4 mice per genotype per time-point; 2-way ANOVA: Genotype F (1, 26) = 22.5, p < 0.0001; Time F (3, 26) = 23.4, p < 0.0001; Interaction F (3, 26) = 4.56, p = 0.011]. Bonferroni multiple comparisons: *p < 0.05, **p < 0.01, ****p < 0.0001. Scale bars represent 17 μm (A–F) and 34 μm (G–L).
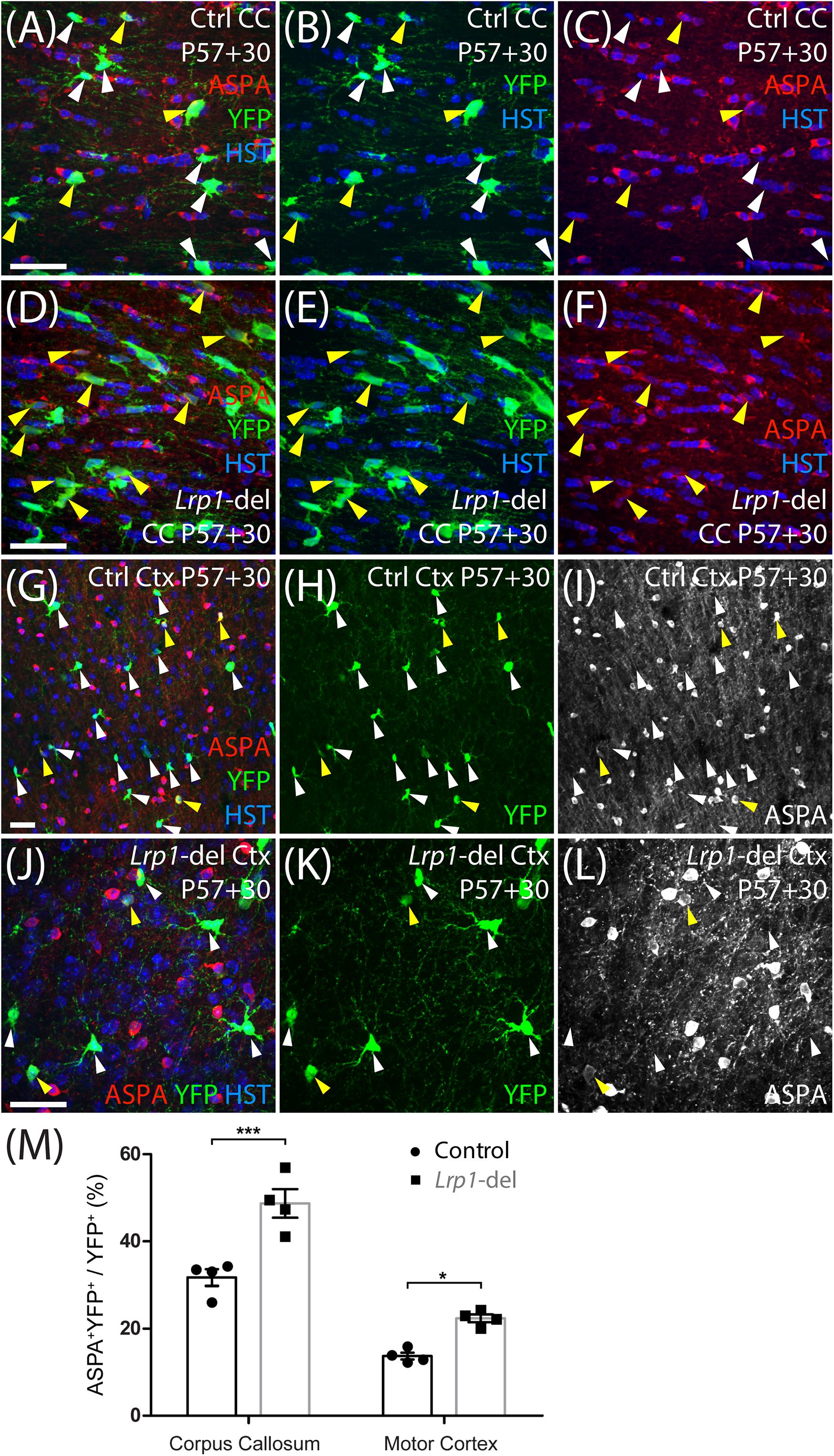
Figure 4. Lrp1 deletion increases the number of newborn ASPA+ OLs in the adult mouse corpus callosum and motor cortex. (A–L) Confocal images from the corpus callosum (CC; A–F) or motor cortex (Ctx; G–L) of P57+30 control (Pdgfrα-CreERTM:: Rosa26-YFP) and Lrp1-deleted (Pdgfrα-CreERTM:: Rosa26-YFP:: Lrp1fl/fl) mice that were immunolabeled to detect ASPA (red), YFP (green), and the nuclear marker Hoechst 33342 (HST, blue). Solid yellow arrowheads indicate YFP+ ASPA+ mature OLs. Solid white arrowheads indicate YFP+ ASPA-neg cells. (M) Quantification of the proportion (%) of YFP+ cells that are YFP+ ASPA+ newborn mature OLs in the CC and Ctx of P57+30 control and Lrp1-deleted mice [mean ± SD for n = 4 mice per genotype; 2-way ANOVA: Genotype F (1, 12) = 125.3, p < 0.0001; Brain region F (1, 12) = 42.1, p < 0.0001; Interaction F (3, 28) = 4.5, p = 0.056]. Bonferroni multiple comparisons: *p < 0.05, ***p < 0.001. Scale bars represent 30 μm (A–J).
Myelinating OLs Produced by Lrp1-Deleted OPCs Elaborate a Normal Number of Internodes
As OPCs differentiate, they rapidly downregulate their expression of PDGFRα, the NG2 proteoglycan and voltage-gated sodium channels (NaV) (Richardson et al., 1988; Pringle et al., 1989; De Biase et al., 2010; Kukley et al., 2010; Clarke et al., 2012). They become highly ramified pre-myelinating OLs that either die or continue to mature into myelinating OLs and elaborate myelin internodes (Trapp et al., 1997; Psachoulia et al., 2009; Hughes et al., 2013; Young et al., 2013; Tripathi et al., 2017; Pitman et al., 2019). To grossly examine the myelin profile of adult-born OLs in control and Lrp1-deleted mice, we fluorescently labeled a subset of OPCs in the adult mouse brain with a membrane-targeted form of green fluorescent protein (GFP). We have previously shown that tamoxifen delivery to adult Pdgfrα-CreERTM :: Tau-GFP mice does not result in the specific fluorescent labeling of OPCs and their progeny (Pitman et al., 2019). Therefore, for this experiment, we instead delivered tamoxifen to adult LE-control (Pdgfrα-CreERT2 :: Tau-GFP) and LE-Lrp1-deleted (Pdgfrα-CreERT2 :: Tau-GFP :: Lrp1fl/fl) mice. The Pdgfrα-CreERT2 transgenic mouse (Rivers et al., 2008) has a lower recombination efficiency (LE) than the Pdgfrα-CreERTM transgenic mouse (Kang et al., 2010), so we first evaluated the efficiency of Lrp1 deletion using this mouse model. Coronal brain cryosections from P57+30 LE-control and LE-Lrp1-deleted mice were immunolabeled to detect PDGFRα and LRP1 (Figures 5A,B). While 100 ± 0% of PDGFRα+ OPCs expressed LRP1 in the motor cortex of LE-control mice, only 35 ± 9% of PDGFRα+ OPCs expressed LRP1 in the motor cortex of LE-Lrp1-deleted mice (mean ± SD for n = 3 mice per genotype). The recombination efficiency was similar in the corpus callosum with 100 ± 0% of OPCs expressing LRP1 in LE-control mice and only 37 ± 7% in LE-Lrp1-deleted mice (Figure 5C).
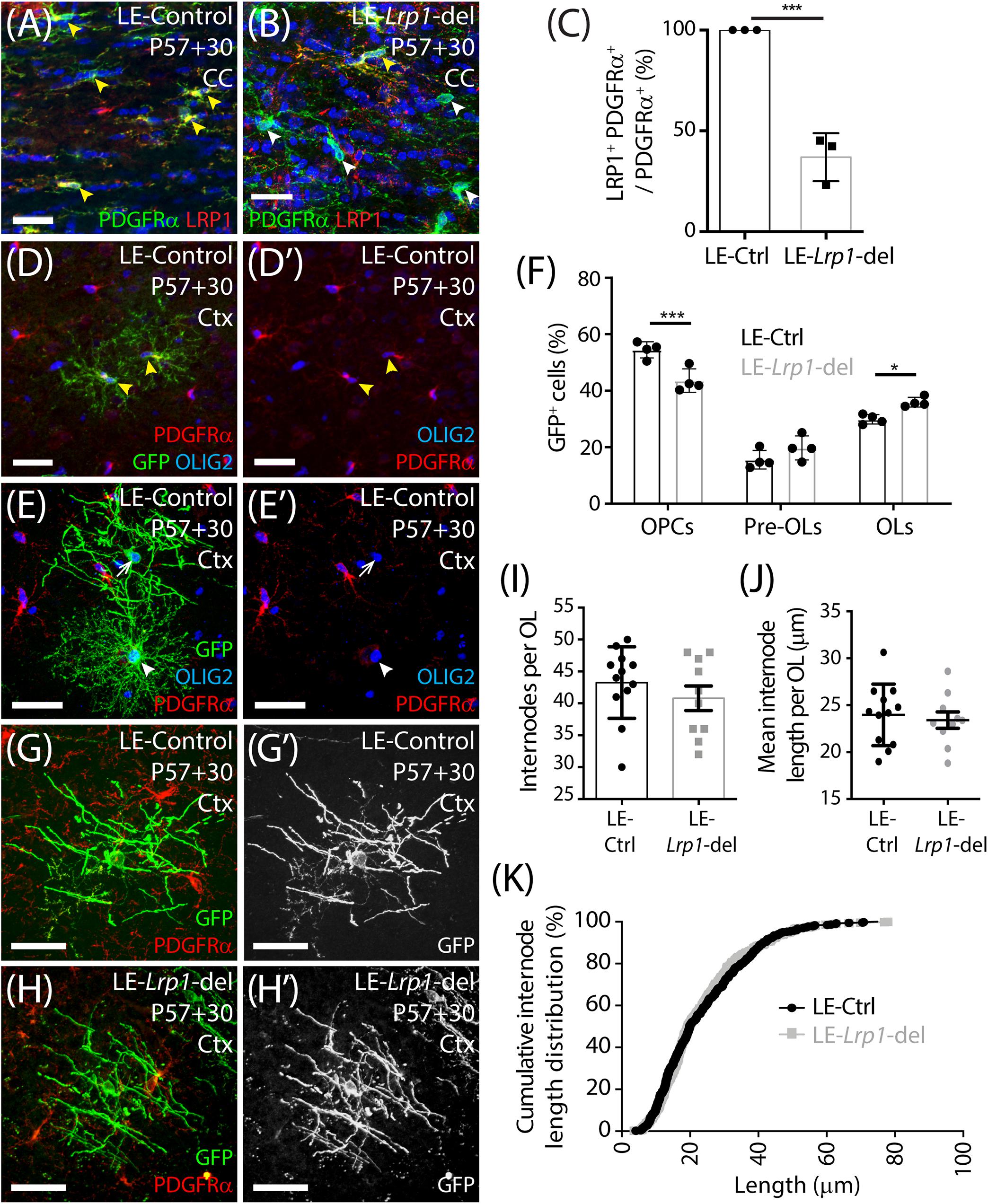
Figure 5. Lrp1 deletion increases the number of mature, myelinating OLs added to the motor cortex of adult mice. (A,B) Compressed confocal z-stack from the corpus callosum (CC) of P57+30 LE-control (Pdgfrα-CreERT2) and LE-Lrp1-deleted (Pdgfrα-CreERT2 :: Lrp1fl/fl) mice that were immunolabeled to detect OPCs (PDGFRα, green), LRP1 (red), and Hoechst 33342 (blue). Solid yellow arrowheads indicate OPCs that express LRP1. Solid white arrowheads indicate OPCs that do not express LRP1. (C) The proportion (%) of PDGFRα+ OPCs in the CC of LE-control and the LE-Lrp1-deleted mice that express LRP1 (mean ± SD for n = 3 mice per group; unpaired t-test, ***p < 0.001). (D–E′) Compressed confocal z-stack from the motor cortex (Ctx) of a P57+30 control (LE-Pdgfrα-CreERT2 :: Tau-mGFP) mouse immunolabeled to detect PDGFRα (red), GFP (green), and OLIG2 (blue). Solid yellow arrowheads indicate GFP+ PDGFRα+ OLIG2+ OPCs. Solid white arrowhead indicates a GFP+ PDGFRα-neg OLIG2+ newborn pre-myelinating OL. The white arrow indicates a GFP+ PDGFRα-neg OLIG2+ newborn myelinating OL. (F) Quantification of the proportion (%) of GFP+ cells that are PDGFRα+ OLIG2+ OPCs, PDGFRα-neg OLIG2+ premyelinating OLs (pre-OLs) and PDGFRα-neg OLIG2+ myelinating OLs (OLs) [mean ± SD for n = 4 mice per genotype; 2-way ANOVA: Maturation stage F (2,18) = 195.1, p < 0.0001; Genotype F (1, 18) = 0.032, p = 0.85; Interaction F (2, 18) = 17.1, p < 0.0001]. Bonferroni multiple comparisons: *p = 0.046 and ***p = 0.0004. (G–H′) Compressed confocal z-stack showing GFP+ PDGFRα-neg myelinating OLs in the motor cortex of P57+30 LE-control and LE-Lrp1-deleted mice. (I) The number of internodes elaborated by individual GFP+ myelinating OLs in the motor cortex of LE-control and LE-Lrp1-deleted mice (mean ± SEM for n ≥ 10 OLs from n = 3 mice per genotype; Mann–Whitney Test, p = 0.38). (J) The average length of internodes elaborated by individual GFP+ myelinating OLs in LE-control and LE-Lrp1-deleted mice (mean ± SEM for n ≥ 10 OLs from n = 3 mice per genotype; unpaired t-test, p = 0.67). (K) Cumulative length distribution plot for GFP+ internodes measured in the motor cortex of P57+30 LE-control and LE-Lrp1-deleted mice (n = 519 LE-control GFP+ internodes and n = 408 LE-Lrp1-deleted GFP+ internodes measured from n = 3 mice per genotype; K-S test, D = 0.053, p = 0.5). Scale bars represent 34 μm (A,B) or 17 μm (G,H).
Only ∼65% of OPCs lacked LRP1 in the LE-Lrp1-deleted mice, but this was sufficient to increase the number of newborn OLs detected in the brain. When P57+30 LE-control and LE-Lrp1-deleted mouse brain cryosections were immunolabeled to detect GFP (green), PDGFRα (red) and OLIG2 (blue) (Figures 5D,E), we found that the proportion of GFP+ cells that became PDGFRα-negative OLIG2+ newborn OLs was significantly elevated in the motor cortex of LE-Lrp1-deleted mice (56.3 ± 2.06%) compared to control mice (49.2 ± 1.51%, mean ± SD for n = 4 mice per genotype; unpaired t-test p = 0.03). Using the membrane targeted GFP to further subdivide the newborn OLs into premyelinating and myelinating OLs, we determined that Lrp1 deletion significantly increased the proportion of GFP+ cells that had become myelinating OLs (Figure 5F).
Despite the difference in overall cell number, the morphology of the myelinating OLs added to the brain of control and Lrp1-deleted mice was equivalent (Figures 5G,H). Our detailed morphological analysis of individual GFP+ myelinating OLs in the motor cortex of LE-control and LE-Lrp1-deleted mice revealed that neither the average number of internodes elaborated by GFP+ myelinating OLs (Figure 5I) or the mean length of internodes elaborated by GFP+ myelinating OLs (Figure 5J) was changed by Lrp1 deletion. Additionally, LRP1 expression did not influence the length distribution for internodes elaborated by newborn myelinating OLs in the motor cortex (Figure 5K). These data indicate that LRP1 negatively regulates the number of myelinating OLs added to the healthy adult mouse brain but does not influence their gross myelination profile.
Lrp1 Deletion Does Not Influence NaV, AMPA Receptor, L- or T-Type VGCC, PDGFRα or LRP2 Expression by OPCs
LRP1 has the potential to influence a number of signaling pathways that directly or indirectly regulate oligodendrogenesis. The conditional deletion of Lrp1 from neurons in vitro and in vivo increases AMPA receptor turnover and reduces expression of the GluA1 subunit of the AMPA receptor (Gan et al., 2014). Adult OPCs express AMPA receptors (Gallo et al., 1996; Gudz, 2006; Zonouzi et al., 2011) that enhance the survival of premyelinating oligodendrocytes during development (Kougioumtzidou et al., 2017) and glutamatergic signaling regulates OPC proliferation, differentiation (Gallo et al., 1996; Fannon et al., 2015) and migration (Gudz, 2006). To determine whether LRP1 regulates AMPA receptor signaling in OPCs we obtained whole cell patch clamp recordings from GFP-labeled OPCs in the motor cortex of P57+30 control (Lrp1fl/fl :: Pdgfrα-histGFP) and Lrp1-deleted (Pdgfrα-CreERTM :: Lrp1fl/fl :: Pdgfrα-histGFP) mice (Figure 6). OPCs elicit a large inward voltage-gated sodium current (INa) in response to a series of voltage-steps (Figure 6A). We found that INa amplitude (Figure 6B) and OPC capacitance (approximation of cell size; Figure 6C) were not affected by LRP1 expression. AMPA receptors were subsequently activated by the bath application of 100 μm kainate, which evoked a large depolarizing current in control and Lrp1-deleted OPCs (Figures 6D,E). The amplitude of the evoked current was equivalent for control and Lrp1-deleted OPCs across all voltages examined (Figure 6E), suggesting that Lrp1 deletion has no effect on the composition or cell-surface expression of AMPA/kainate receptors.
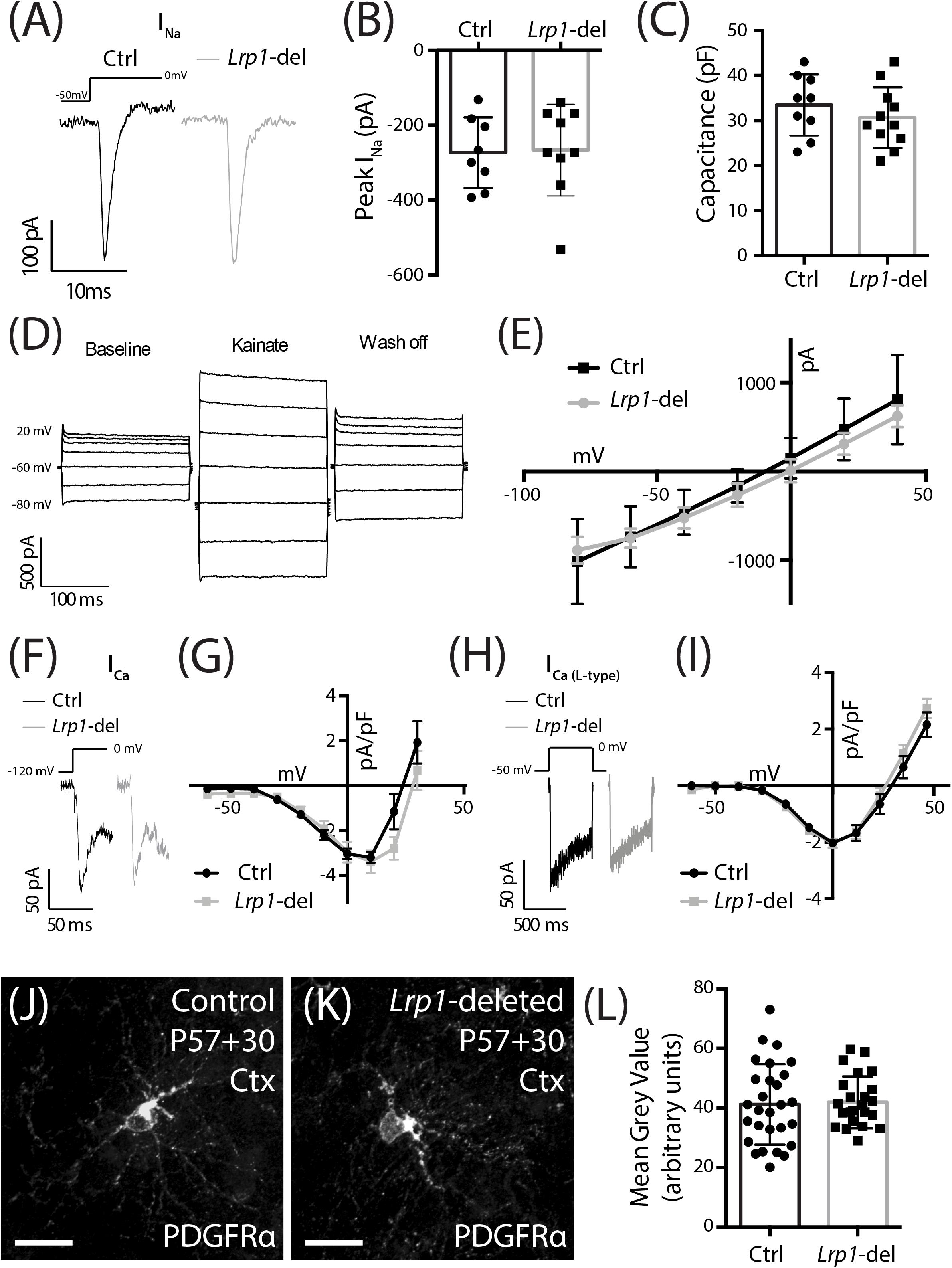
Figure 6. LRP1 does not alter functional NaV, VGCC, or AMPA/kainate receptor expression, or total PDGFRα expression in OPCs. (A) Representative traces of voltage-gated sodium channels currents (INa) evoked in GFP+ OPCs in the motor cortex of P57+30 control (Pdgfrα-histGFP :: Lrp1fl/fl) and Lrp1-deleted (Pdgfrα-CreERTM :: Pdgfrα-histGFP :: Lrp1fl/fl) mice. (B) Quantification of peak inward INa (n ≥ 8 GFP+ OPCs analyzed from n = 3 mice per genotype; unpaired t-test, p = 0.8). (C) Quantification of cell capacitance (n ≥ 9 GFP+ OPCs analyzed from n = 3 mice per genotype; unpaired t-test, p = 0.9). (D) Representative trace from a control GFP+ OPC responding to the bath application of 100 μM kainate. (E) The current density-voltage relationship of AMPA/kainate receptors in control (n = 3 GFP+ OPCs) and Lrp1-deleted (n = 3 GFP+ OPCs) cells [mean ± SEM; 2-way repeated measures ANOVA: Genotype F (1, 28) = 0.91, p = 0.3; Voltage F (6, 28) = 31.3, p < 0.0001; Interaction F (6, 28) = 0.25, p = 0.9]. (F) Representative traces show the fast inactivating leak subtracted ICa evoked in GFP+ OPCs in response to a depolarizing step. (G) The current density-voltage relationship for the leak subtracted ICa (peak amplitude) recorded from control cells (dark circles, n = 11 GFP+ OPCs across n = 3 mice) and Lrp1-deleted cells (gray squares, n = 10 GFP+ OPCs across n = 3 mice) [mean ± SEM; 2-way repeated measures ANOVA: Genotype F (1, 190) = 2.85, p = 0.09; Voltage F (9, 190) = 23.5, p < 0.0001; Interaction F (9, 190) = 1.14, p = 0.3]. (H) Representative traces show the leak subtracted ICa L-type evoked in GFP+ OPCs in response to a depolarizing step. (I) The current density-voltage relationship for leak subtracted ICa L-type (mean sustained current) recorded from control cells (dark circles, n = 7 GFP+ OPCs across n = 3 mice) and Lrp1-deleted cells (gray squares, n = 11 GFP+ OPCs across n = 3 mice) [mean ± SEM; 2-way repeated measures ANOVA: Genotype F (1, 176) = 1.03, p = 0.3; Voltage F (10, 176) = 66.8, p < 0.0001; Interaction F (10, 176) = 0.62, p = 0.8]. (J,K) Compressed z-stack confocal image of PDGFRα+ OPCs in the motor cortex (Ctx) of P57+30 control and Lrp1-deleted mice. (L) The mean gray value of PDGFRα staining for individual OPCs measured in the motor cortex of P57+30 control and Lrp1-deleted mice (mean ± SD; n ≥ 24 OPCs measured across n = 3 mice per genotype; unpaired t-test, p = 0.8). Scale bars represent 17 μm.
LRP1 regulates the cell surface expression and distribution of N-type voltage gated calcium channels (VGCC) by interacting with the α2δ subunit (Kadurin et al., 2017). In adult OPCs, the closely related L-type VGCCs reduce OPC proliferation in the motor cortex and corpus callosum (Pitman et al., 2019) and influence OL maturation in vitro (Cheli et al., 2015). The other major VGCCs expressed by OPCs are T-type VGCCs (Williamson et al., 1997; Fulton et al., 2010) which are activated at lower (hyperpolarized) voltages than L-type channels and inactivate quickly (transient). To determine whether the cell surface expression of VGCCs is altered following Lrp1 deletion, we performed whole cell patch clamp electrophysiology and measured the current density (pA/pF) in OPCs from control and Lrp1-deleted mice (Figures 6F–I). We found that the VGCC current density was equivalent for OPCs in the motor cortex of control and Lrp1-deleted mice (Figures 6F,G). The current density was also equivalent between OPCs from control and Lrp1-deleted mice when measured currents were elicited selectively through L-type VGCCs (Figures 6H,I), indicating that LRP1 does not influence L- or T-type VGCC expression in adult OPCs.
Tissue plasminogen activator (tPA) is an LRP1 ligand (Bu et al., 1992; Orth et al., 1992) and its addition to astrocytic cultures increases PDGF-CC cleavage and activation (Su et al., 2008). While PDGF-CC is a ligand of PDGFRα, a key receptor regulating OPC proliferation, survival and migration (Noble et al., 1988; Richardson et al., 1988; Pringle et al., 1989), increased mitogenic stimulation would not account for LRP1 reducing adult OPC proliferation. In other cell types, LRP1 has instead been shown to influence the cell surface expression of PDGFRβ (Takayama et al., 2005; Muratoglu et al., 2010), a receptor that is closely related to PDGFRα. When performing immunohistochemistry using an antibody against the intracellular domain of PDGFRα, it is not possible to specifically quantify the cell surface expression of PDGFRα in OPCs with and without LRP1, however, we were able to quantify PDGFRα expression (mean gray value; Figures 6J–L), and determined that LRP1 did not influence total PDGFRα expression.
The low-density lipoprotein receptor related protein 2 (LRP2) is a large cell surface receptor that is closely related to LRP1, with a number of common ligands (Spuch et al., 2012). LRP2 can increase the proliferation of neural precursor cells in the subependymal zone (Gajera et al., 2010) and the proliferation and survival of skin cancer cells (Andersen et al., 2015), however, it is unclear whether cells of the OL lineage express LRP2 (Cahoy et al., 2008; Zhang et al., 2014; Hrvatin et al., 2018), or whether Lrp1 deletion could alter LRP2 expression. We examined this possibility by performing immunohistochemistry on coronal brain cryosections from P57+30 control and Lrp1-deleted mice to detect LRP2 and PDGFRα or ASPA (Supplementary Figure 2). We determined that LRP2 is not expressed by OPCs or OLs in mice of either genotype, despite the robust expression of LRP2 by Iba1+ microglia (Supplementary Figure 2). These data indicate that compensation from LRP2 or a change in LRP2 expression by OPCs is not responsible for the elevated OPC proliferation and differentiation observed in Lrp1-deleted mice.
Lrp1 Deletion Does Not Alter EdU Incorporation by OPCs in vitro
Our data suggest that in the healthy adult mice, Lrp1 deletion either increases OPC proliferation which then results in an increased number of newborn OLs, or increases OPC differentiation which subsequently triggers a homeostatic increase in OPC proliferation to maintain the OPC population. Previous studies have shown that Lrp1 deletion enhances the proliferation of retinal endothelial cells (Mao et al., 2016) and reduces the proliferation of cells within embryonic mouse neurospheres (Safina et al., 2016), while the activation of LRP1 by tPA enhances the proliferation of interstitial fibroblasts (Lin et al., 2010). To determine whether LRP1 directly suppresses OPC proliferation, we generated primary OPC cultures from the cortex of P0–P5 control (Pdgfrα-hGFP) or Lrp1-deleted (Pdgfrα-hGFP: Lrp1fl/fl) mice. To delete Lrp1 in vitro, we used a cell-permeable form of Cre-recombinase, TAT-Cre, which is a fusion peptide of Cre-recombinase and the HIV cell-penetrating TAT peptide. After 7 days in vitro (DIV), OPCs were incubated with 1 μM TAT-Cre for 90 min and LRP1 expression was determined at 9 DIV by performing immunocytochemistry to detect PDGFRα (red), GFP (green), and LRP1 (blue) (Figures 7A,B). Following Tat-Cre treatment all OPCs cultured from control mice expressed LRP1, however, only ∼21% of PDGFRα+ OPCs cultured from Lrp1-deleted mice retained LRP1 expression (Figure 7C). At the same time-point, additional control and Lrp1-deleted OPC cultures were exposed to EdU to label all cells that entered the S-phase of the cell cycle over a 10-h period. By performing immunocytochemistry to detect GFP (green), LRP1 (red) and EdU (Figures 7D,E), we found that LRP1 expression did not influence OPC proliferation in vitro, as the fraction of OPCs that were EdU+ was equivalent in control and Lrp1-deleted cultures (Figure 7F).
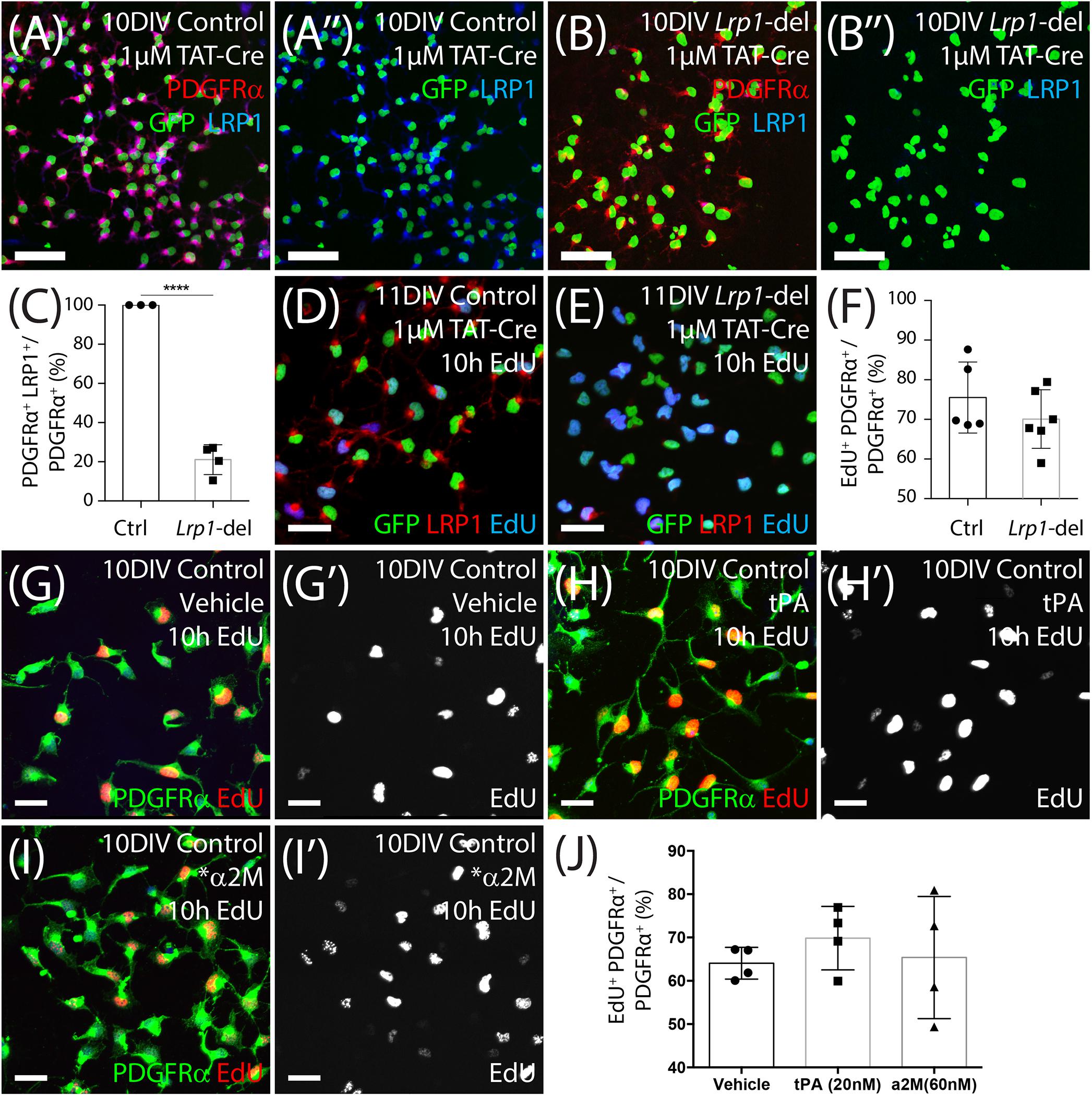
Figure 7. LRP1 does not affect OPC proliferation in vitro. (A–B″) Tat-Cre-treated OPCs cultured from the cortex of early postnatal control (Pdgfrα-histGFP) and Lrp1-deleted (Pdgfrα-histGFP :: Lrp1fl/fl) mice were immunolabeled to detect PDGFRα (red), LRP1 (blue), and GFP (green). (C) Quantification of the proportion (%) of control and Lrp1-deleted OPCs that express LRP1 48 h after TAT-Cre treatment (mean ± SEM, n ≥ 3 independent cultures per genotype; unpaired t-test, ****p < 0.0001). (D,E) Tat-Cre-treated OPCs from control and Lrp1-deleted mice exposed to EdU for 10 h and immunolabeled to detect GFP (green), LRP1 (red), and EdU (blue). (F) Quantification of the proportion (%) of control and Lrp1-deleted OPCs that become EdU-labeled over a 10-h period (mean ± SEM, n ≥ 5 independent cultures per genotype; unpaired t-test, p = 0.3). (G–I′) Compressed confocal z-stack showing OPCs cultured from control mice that were exposed to EdU and either vehicle (MilliQ water; G,G′), 20 nM tPa (H,H′) or 60 nM *α2M (I,I′) for 10 h, before being processed to detect EdU (red) and PDGFRα (green). (J) Quantification of the proportion (%) of control OPCs that incorporated EdU when treated with vehicle, tPa or *α2M for 10 h (mean ± SEM, n ≥ 4 independent cultures; 1-way ANOVA: Treatment F (2, 9) = 0.42, p = 0.66). Scale bars represent 17 μm. DIV = days in vitro; tPA = tissue plasminogen activator; *α2M = activated α-2 macroglobulin.
To determine whether LRP1 activation by ligands could directly influence OPC proliferation, we added vehicle (MilliQ water) or the LRP1 ligands tPA (20 nM) or activated α-2 macroglobulin (∗α2M; 60 mM) to OPC primary cultures for 10 h, along with EdU (Figures 7G–J). By performing immunocytochemistry to detect PDGFRα (green) and EdU (red), we determined that the proportion of OPCs that became EdU-labeled did not change with the addition of tPA or ∗α2M (Figure 7J), indicating that LRP1 activation by these ligands is unable to acutely modify OPC proliferation in vitro.
Lrp1 Deletion Increases the Proportion of OPCs That Differentiate in vitro
In vitro, OPCs can be triggered to differentiate by withdrawing the mitogen PDGF-AA and providing triiodothyronine (T3) in the culture medium. To determine whether Lrp1 deletion can enhance OPC differentiation, TAT-Cre-treated control and Lrp1-deleted OPCs were transferred into differentiation medium for 4 days before they were immuno-labeled to detect PDGFRα+ OPCs (red) and MBP+ OLs (green) (Figures 8A,B). We determined that the proportion of cells that were PDGFRα+ OPCs was reduced in the Lrp1-deleted cultures, while the proportion of cells that were MBP+ OLs was significantly increased compared with control cultures (Figure 8C).
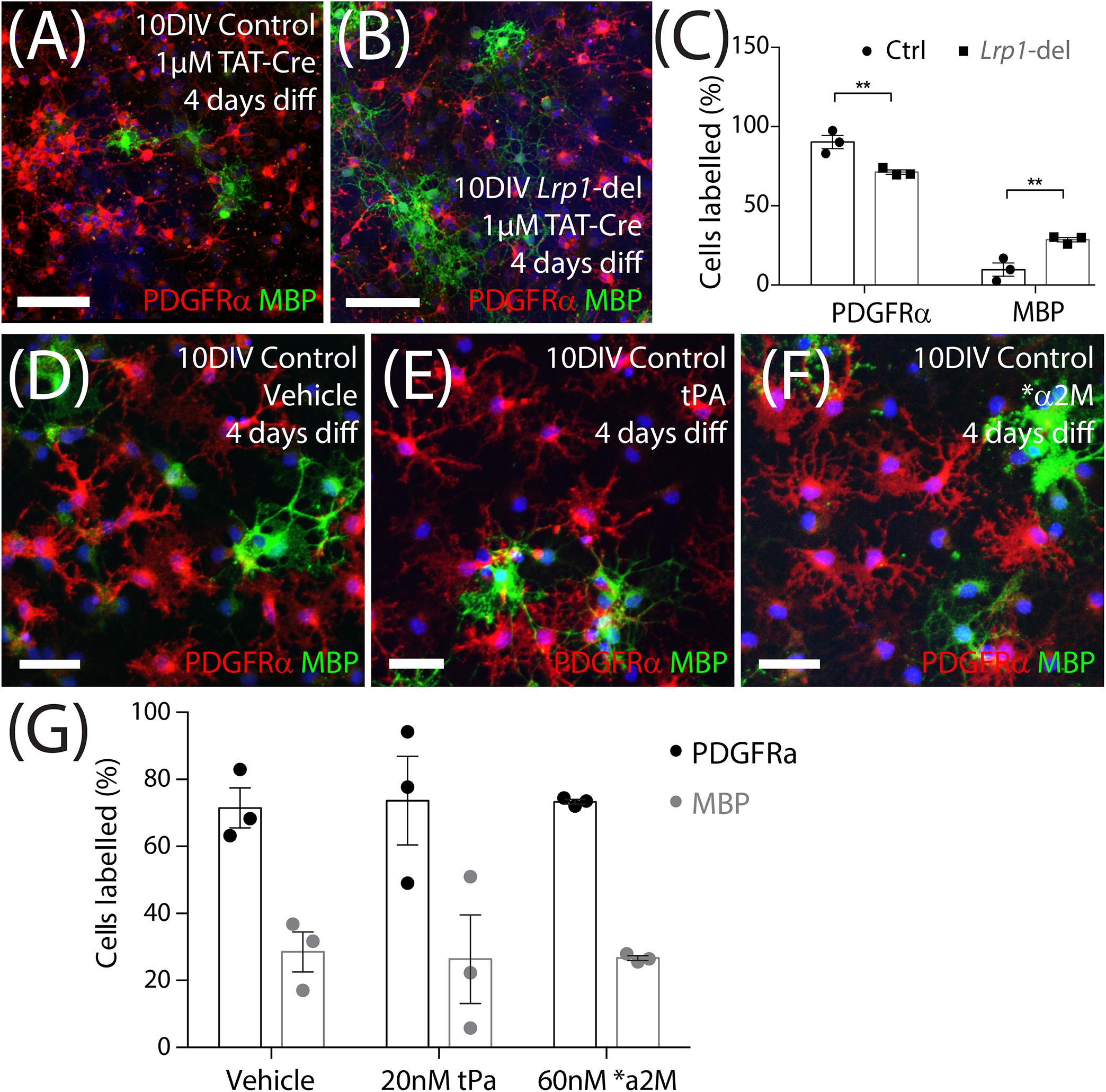
Figure 8. Lrp1 deletion increases OPC differentiation in vitro. (A,B) Control and Lrp1-deleted OPCs were exposed to Tat-Cre, cultured for a further 48-h, and then transferred to differentiation medium for 4 days. Compressed confocal z-stack showing differentiated control and Lrp1-deleted (Lrp1fl/fl) OPC cultures immunolabeled to detect OPCs (PDGFRα, red), OLs (MBP, green), and all cell nuclei (Hoechst 33342, blue). (C) Quantification of the proportion (%) of cells that were PDGFRα+ OPCs or MBP+ OLs in control and Lrp1-deleted cultures [mean ± SEM for n = 3 independent cultures per genotype; 2-way ANOVA: Cell type F (1, 8) = 212, p < 0.0001; Genotype F (1, 8) = 0.001, p = 0.97; Interaction F (1, 8) = 19.9, p = 0.02]. Bonferroni multiple comparisons: **p = 0.005. (D–F) Compressed confocal z-stack shows OPCs from control mice that were transferred into differentiation medium and exposed to vehicle (MilliQ water; D), tPa (E), or *α2M (F) for 4 days before being immunolabeled to detect OPCs (PDGFRα, red), OLs (MBP, green), and all cell nuclei (Hoechst 33342, blue). (G) Quantification of the proportion (%) of OPCs that became PDGFRα+ OPCs or MBP+ OLs after 4 days in differentiation medium with vehicle, tPa or *α2M [mean ± SEM, n = 3 independent cultures per treatment; 2-way ANOVA: Cell type F (1, 12) = 44.6, p < 0.0001; Treatment F (2, 10) = 0, p = 1; Interaction F (2, 12) = 0.03, p = 0.97]. tPa = tissue plasminogen activator; *α2M = activated α2 macroglobulin. Scale bars represent 34 μm (A,B) or 17 μm (D–F).
To determine whether the ligand activation of LRP1 was sufficient to suppress OPC differentiation, OPC primary cultures were transferred into differentiation medium containing vehicle (MilliQ water), tPA (20 nM) or ∗α2M (60 nM) for 4 days. By performing immunocytochemistry to detect PDGFRα+ OPCs and MBP+ OLs (Figures 8D–F) we found that the activation of LRP1 by tPA or ∗α2M had no impact on the proportion of cells that differentiated over time (Figure 8G). As ApoE4 produces an LRP1-dependent increase in Erk and Akt phosphorylation to increase the number of O4+ OLs produced by differentiating embryonic mouse neurospheres (Safina et al., 2016), we also added vehicle (MilliQ water) or ApoE4 (203 nM) to a distinct set of proliferating and differentiating OPC cultures. We report that ApoE4 had no effect on the proportion (%) of OPCs that incorporated EdU (vehicle: 56.2 ± 11.5%; ApoE4: 57.2 ± 7.3%, mean ± SEM for n = 4 independent cultures; paired t-test t = 0.2, p = 0.8). Furthermore, the proportion (%) of cells that were MBP+ OLs (vehicle, 29.1 ± 4.3% and ApoE4, 35.9 ± 3.4%) after 4 days of differentiation, was equivalent in vehicle and ApoE4-treated cultures [mean ± SEM for n = 4 independent cultures; paired t-test t = 1.0, p = 0.4]. These data suggest that the LRP1 ligands tPA, α2M and ApoE4, do not regulate OPC differentiation in vitro.
Lrp1 Deletion Increases the Number of Newborn Mature OLs Added to the Corpus Callosum During Cuprizone Feeding
As Lrp1 deletion enhances OL addition to the healthy adult mouse brain, we next aimed to determine whether the deletion of Lrp1 from adult OPCs could similarly increase OL generation in response to OL loss and demyelination. Control (Pdgfrα-CreERTM :: Rosa26-YFP) and Lrp1-deleted (Pdgfrα-CreERTM :: Rosa26-YFP :: Lrp1fl/fl) mice received tamoxifen by oral gavage at P57 and at P57+7 were transferred onto a diet containing 0.2% (w/w) cuprizone for 5 weeks to induce significant OL loss, demyelination of the corpus callosum and oligodendrogenesis. After 5 weeks of cuprizone feeding, control and Lrp1-deleted mice were perfusion fixed and coronal brain cryosections processed to detect myelin by black-gold staining (Figure 9). We identified and measured areas of overt demyelination in the corpus callosum of control and Lrp1-deleted mice (Figures 9A–D) and found that this represented a smaller proportion of the corpus callosum in Lrp1-deleted mice compared with controls (Figure 9E).
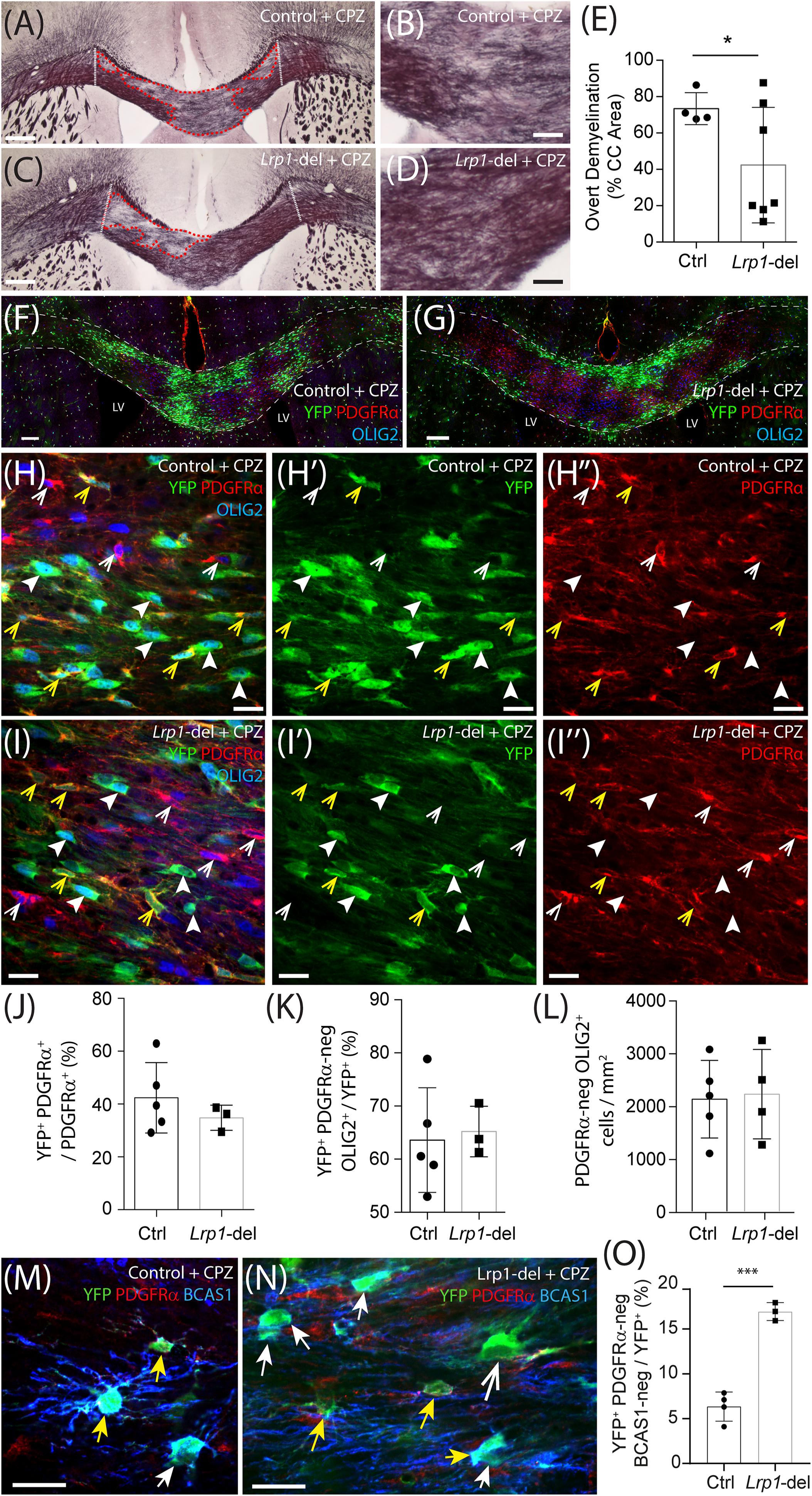
Figure 9. Lrp1 deletion increases the number of mature OLs added to the mouse corpus callosum during cuprizone feeding. (A–D) Young adult control (Pdgfrα-CreERTM :: Rosa26-YFP) and Lrp1-deleted (Pdgfrα-CreERTM :: Rosa26-YFP :: Lrp1fl/fl) mice were fed a diet containing 0.2% cuprizone for 5 weeks. At the end of 5 weeks, coronal brain sections were collected and stained with black-gold to visualize myelin in the corpus callosum. Dotted white lines define the lateral limits of the region of the corpus callosum analyzed. Red dashed lines denote areas of overt demyelination. (E) The proportion (% area) of the corpus callosum with faint or absent black-gold staining (overt demyelination) in cuprizone-fed control and Lrp1-deleted mice (mean ± SD, n ≥ 4 mice per genotype; unpaired with Welch’s correction, *p = 0.04). (F–I″) Compressed confocal z-stack shows the corpus callosum in cuprizone-fed control and Lrp1-deleted mice, immunolabeled to detect YFP (green), PDGFRα (red), and OLIG2 (blue). White dashed lines indicate the dorsal and ventral boundaries of the corpus callosum; yellow arrowheads indicate YFP+ PDGFRα+ parenchymal OPCs; solid white arrowheads indicate YFP+ PDGFRα-negative newborn OLs; white arrowheads indicate YFP-negative PDGFRα+ stem cell-derived OPCs. (J) Quantification of the proportion (%) of OPCs in the corpus callosum of cuprizone-fed control and Lrp1-deleted mice that are YFP+, PDGFRα+ parenchymal OPCs (mean ± SD, n ≥ 3 mice per genotype; unpaired t-test, p = 0.4). (K) Quantification of the proportion of YFP+ cells in the corpus callosum of cuprizone-fed control and Lrp1-deleted mice that are PDGFRα-negative OLIG2+ newborn OLs (mean ± SD, n ≥ 3 mice per genotype; unpaired t-test, p = 0.9). (L) Quantification of the density of PDGFRα-negative OLIG2+ OLs in the corpus callosum of cuprizone-fed control and Lrp1-deleted mice (mean ± SD, n ≥ 4 mice per genotype; unpaired t-test, p = 0.9). (M,N) Single z-plane confocal images showing the corpus callosum of cuprizone-fed control and Lrp1-deleted mice stained to detect YFP (green), PDGFRα (red), and BCAS1 (blue). Solid yellow arrows indicate parenchymal OPCs (YFP+ PDGFRα+ ± BCAS1). Solid white arrows indicate newborn premyelinating OLs derived from parenchymal OPCs (YFP+ PDGFRα-neg BCAS1+). Large white arrow indicates newborn mature OL derived from parenchymal OPCs (YFP+ PDGFRα-neg BCAS1-neg). (O) Quantification of the proportion of YFP+ cells in the corpus callosum of cuprizone-fed control and Lrp1-deleted mice that are YFP+ PDGFRα-neg BCAS1-neg newborn mature OLs (mean ± SD for n ≥ 3 mice per genotype; unpaired t-test, t = 9.8, ***p = 0.0002). Scale bars represent 150 μm (A,C), 30 μm (B,D), 100 μm (F,G), 17 μm (H,I), or 20 μm (M,N). LV, lateral ventricle.
When EdU was delivered via the drinking water from week 2 to week 5 of cuprizone feeding, it was incorporated into the vast majority of OLIG2+ cells in the corpus callosum of control and Lrp1-deleted mice (Supplementary Figure 3). Despite being newborn cells, many of the PDGFRα+ OPCs (red) and PDGFRα-negative OLIG2+ OLs (blue) within the corpus callosum of control and Lrp1-deleted mice did not co-label with YFP (green) (Figures 9F,G), indicating that these cells were not derived from the YFP+ parenchymal OPC population. Following cuprizone-induced demyelination, both parenchymal OPCs (YFP-labeled) and neural stem cell-derived OPCs (YFP-negative) contribute to OL replacement and remyelination (Xing et al., 2014). After cuprizone feeding the proportion of OPCs that were YFP+ parenchymal OPCs was equivalent in the corpus callosum of control and Lrp1-deleted mice (Figure 9J). As total OPC density was also unaffected by genotype (838 ± 165 OPCs/mm2 in control and 740 ± 134 OPCs/mm2 in Lrp1-deleted corpus callosum; mean ± SD for n = 5 control and n = 3 Lrp1-deleted mice; unpaired t-test, p = 0.67), the expression of LRP1 by parenchymal OPCs does not appear to influence OPC production by neural stem cells.
Following demyelination, YFP+ parenchymal OPCs present in the corpus callosum of Lrp1-deleted mice lacked LRP1, however, the YFP-negative neural stem cell-derived OPCs had intact LRP1 expression (Supplementary Figure 3). Surprisingly, YFP+ parenchymal OPCs no longer generated more OLs in Lrp1-deleted mice compared to controls, as 60 ± 15% of YFP+ cells were PDGFRα-negative OLIG2+ newborn OLs in the corpus callosum of control mice and 65 ± 5% of YFP+ cells were PDGFRα-negative OLIG2+ newborn OLs in the corpus callosum of Lrp1-deleted mice (Figure 9K; mean ± SD for n = 5 control and n = 3 Lrp1-deleted mice). As total OL density was also equivalent in the corpus callosum of control and Lrp1-deleted mice (Figure 9L), a change in oligodendrogenesis did not appear to account for Lrp1-deleted mice having less overt demyelination. However, by performing immunohistochemistry to detect YFP, the OPC marker PDGFRα, and Breast Carcinoma Amplified Sequence 1 (BCAS1), a protein expressed by some OPCs and all pre-myelinating OLs (Fard et al., 2017; Ferreira et al., 2020), we found that the fraction of YFP+ cells that were mature OLs (YFP+ PDGFRα-neg BCAS1-neg) was increased in the corpus callosum of Lrp1-deleted mice compared to controls (Figures 9M–O). These data suggest that LRP1 expression by adult OPCs impairs newborn OL maturation in the cuprizone-injured CNS.
Discussion
Within the OL lineage, LRP1 is highly expressed by OPCs and is rapidly down-regulated upon differentiation (Cahoy et al., 2008; Zhang et al., 2014; Auderset et al., 2016a; Fernandez-Castaneda et al., 2019), suggesting that LRP1 regulates the function or behavior of the progenitor cells. As LRP1 can signal in a number of different ways (Lillis et al., 2008; Auderset et al., 2016b; Bres and Faissner, 2019) and has been shown to influence cellular behaviors relevant to OPCs, such as proliferation, differentiation (Boucher et al., 2007; Mao et al., 2016; Safina et al., 2016) and migration (Mantuano et al., 2010, 2015; Barcelona et al., 2013; Ferrer et al., 2016; Sayre and Kokovay, 2019), we took a conditional gene deletion approach to determine whether Lrp1 influenced the behavior of adult mouse OPCs. We report that Lrp1-deletion produces a delayed increase in adult OPC proliferation and increases the number of OPCs that differentiate into mature, myelinating OLs in the motor cortex and corpus callosum. Lrp1 deletion does not, however, alter the number or length of internodes produced by the newborn, myelinating OLs. Furthermore, during cuprizone-induced demyelination, Lrp1 deletion does not influence the total number of newborn OLs added to the corpus callosum, but a larger proportion of the newborn cells are mature OLs.
Does LRP1 Have a Different Effect on Developmental and Adult OPCs in the Healthy CNS?
New OLs are added to the adult mouse CNS throughout life (Dimou et al., 2008; Rivers et al., 2008; Kang et al., 2010; Zhu et al., 2011), and when we followed the fate of adult OPCs after Lrp1 deletion, we observed a significant increase in the number of new OLs added to the corpus callosum and motor cortex at P57+30 and P57+45 (Figure 3). In the healthy adult mouse brain, there is a significant population of pre-myelinating OLs (Xiao et al., 2016; Fard et al., 2017) that are constantly turned over, as ∼78% of newly generated pre-myelinating OLs survive for less than 2 days (Hughes et al., 2018). However, Lrp1 deletion did not simply increase the pool of premyelinating OLs, as deleting Lrp1 from adult OPCs ultimately increased the number of newborn mature ASPA+ OLs added to the brain (Figure 4). Furthermore, by using LE-Pdgfrα-CreERT2 :: Tau-mGFP transgenic mice to visualize the full morphology of the newly generated OLs, we were able to confirm that Lrp1 deletion effectively increased the number of newborn OLs that had myelinating morphology (Figure 5). The gross myelination profile of individual myelinating OLs appeared unaffected by LRP1 expression, as OLs in the cortex of Lrp1-deleted mice supported the same number of internodes of an equivalent length as those elaborated by newborn myelinating OLs in the cortex of control mice (Figure 5).
These data suggest that LRP1 expression by adult OPCs has the capacity to influence the number of OPCs that differentiate and survive as newborn, myelinating OLs in the healthy mouse brain, without significantly impacting their gross myelination profile. This was unexpected, as it suggests that LRP1 plays a different role in adult OPCs compared with developmental OPCs. In the developing mouse optic nerve, deleting Lrp1 from cells of the OL lineage (Olig2-Cre :: Lrp1fl/fl) reduces the number of OLs produced and results in hypomyelination by P21 (Lin et al., 2017). As LRP1 is expressed by radial glia and neural stem cells (Hennen et al., 2013; Safina et al., 2016; Auderset et al., 2016a), and the Olig2 gene promoter can be active in some neural stem cells and transiently active in some astrocytes (Marshall et al., 2005; Cai et al., 2007), it is possible that the Olig2-Cre transgenic mouse results in the unintended deletion of Lrp1 from some neural stem cells. This would be expected to reduce OL generation, as cultured embryonic NSPCs lacking Lrp1 produce fewer cells of the OL lineage (Hennen et al., 2013; Safina et al., 2016). However, a series of in vitro experiments suggest that the constitutive loss of Lrp1 from OPCs in development, results in longer-lasting detrimental changes within the lineage. After 2 days of differentiation, primary brain OPCs derived from early postnatal Olig1-Cre and Olig1-Cre :: Lrp1fl/fl mice, have an equivalent level of Myrf, Mbp, and CNPase mRNA expression (Fernandez-Castaneda et al., 2019), Similarly, after 3 days in differentiation medium, OPCs from control and conditionally-deleted (Olig2-Cre :: Lrp1fl/fl) mice contain an equivalent number of NG2+ OPCs or CNPase+ OLs (Lin et al., 2017). By 5 days of differentiation, however, cultures from the conditionally-deleted mice contain significantly fewer PLP+ or MBP+ OLs and these cells had impaired myelin biogenesis (Lin et al., 2017). Sterol-regulatory element binding protein-2 expression was elevated and peroxisomal biogenesis factor-2 expression reduced in cultures generated from Olig2-Cre :: Lrp1fl/fl mice, but the combined delivery of cholesterol and pioglitazone rescued this phenotype (Lin et al., 2017). Therefore, it is likely that LRP1 plays an important role in cholesterol homeostasis and peroxisome biogenesis in developmental OPCs (Lin et al., 2017). As Lrp1 deletion did not disrupt the gross myelinating morphology of newborn OLs added to the adult mouse CNS, this function of LRP1 does not appear to be retained by adult OPCs. However, this could only be truly ruled out by performing an ultrastructural analysis of the newly elaborated myelin.
Why Does Lrp1 Deletion Have a Delayed Effect on OPC Proliferation in the Healthy Adult Mouse CNS?
At any one time, the majority of OPCs in the healthy adult mouse CNS are in the G0 phase of the cell cycle (Psachoulia et al., 2009). In young adulthood, all OPCs in the corpus callosum re-enter the cell cycle and divide at least once in a 10-day period, but a similar level of turnover takes ∼38 days for OPCs in the cortex (Young et al., 2013). In this study, we found that Lrp1-deletion increased the rate at which OPCs re-entered the cell cycle and became EdU-labeled, but the onset of this phenotype was not coincident with the loss of LRP1 (Figures 1, 2). More specifically, when EdU labeling commenced at P57+7, the proportion of OPCs that became EdU labeled over time was the same for control and Lrp1-deleted mice (Figure 2). However, when EdU delivery was delayed and instead commenced at P57+28, the proportion of OPCs that incorporated EdU was elevated in the corpus callosum or motor cortex of Lrp1-deleted mice compared with controls. It is feasible that LRP1 directly suppresses adult OPC proliferation. LRP1 is known to modulate the proliferation of other cell types (Boucher et al., 2007; Basford et al., 2009; Mao et al., 2016; Safina et al., 2016; Yang et al., 2018; Zucker et al., 2019), suppressing the hypoxia-induced proliferation of mouse and human retinal endothelial cells by regulating the activity of poly (ADP-ribose) polymerase-1 (PARP-1; Mao et al., 2016), and suppressing the proliferation of cultured mouse vascular smooth muscle cells by reducing PDGFRβ activity (Boucher et al., 2003; Basford et al., 2009). However, the inability of Lrp1 deletion to acutely influence OPC proliferation in vivo or directly influence OPC proliferation in vitro (Figure 7; Lin et al., 2017), may suggest that LRP1 affects OPC proliferation indirectly.
OPC proliferation is intimately linked to OPC differentiation in vivo (Hughes et al., 2013). As the number of new OLs detected in the adult mouse brain increases following the conditional deletion of Lrp1 (Figures 3–5), it is possible that Lrp1-deletion directly enhances OPC differentiation and, in doing so, produces a homeostatic increase in OPC proliferation (see Figure 10). Further supporting this idea, the deletion of Lrp1 from OPCs in vitro, directly increased the number of MBP+ OLs generated (Figure 8).
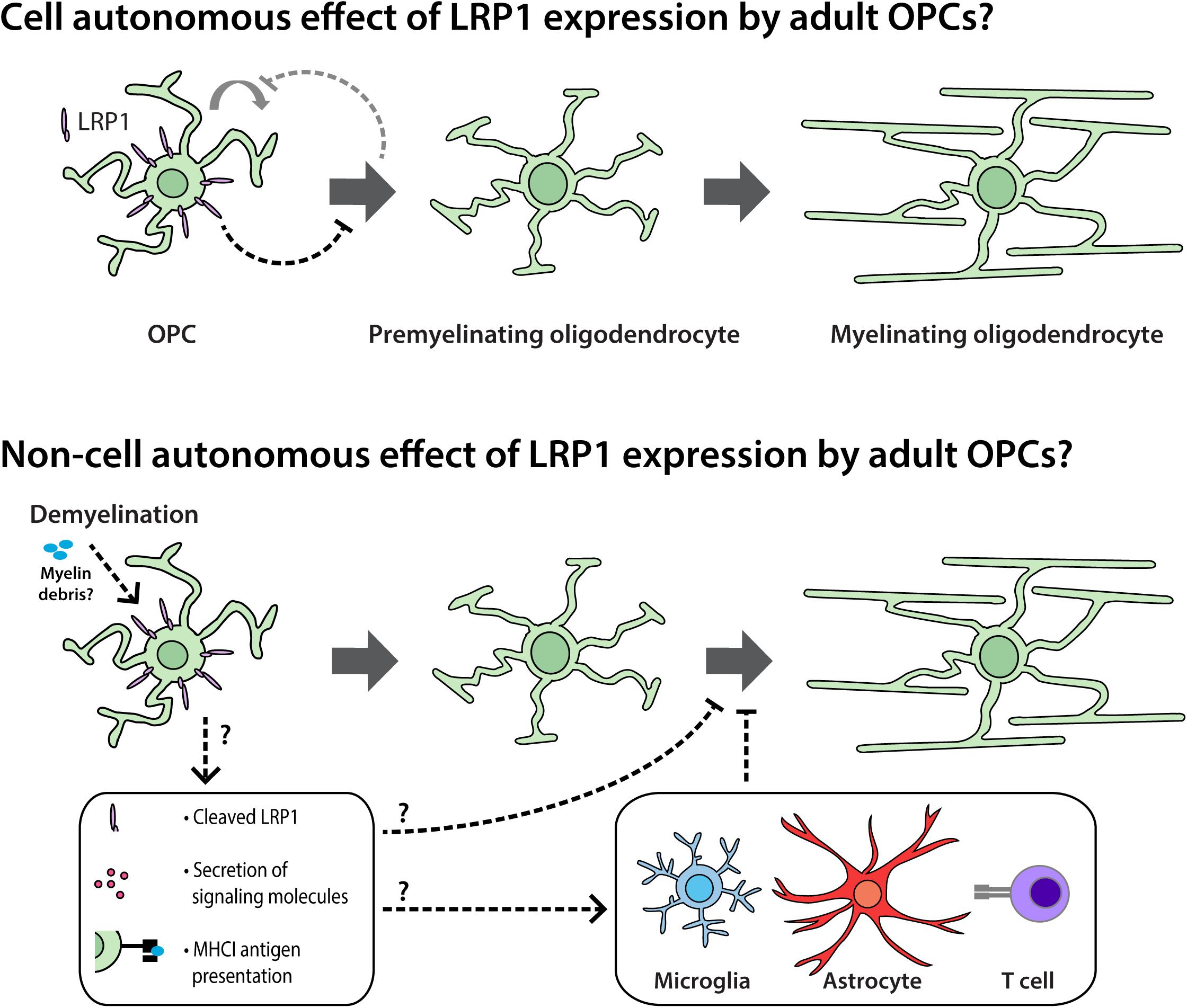
Figure 10. LRP1 signaling in adult OPCs may have cell autonomous and non-cell autonomous effects on myelination and remyelination. Cell autonomous: in vivo, LRP1 signaling by adult mouse OPCs reduces OPC proliferation and the number of newborn OLs added to the brain. As LRP1 suppresses OPC differentiation in vitro, we propose that LRP1 can directly suppress OPC differentiation. This cell autonomous effect would also be expected to reduce OPC proliferation, as these processes are closely and homeostatically linked in vivo. Non-cell autonomous: during cuprizone-induced demyelination, LRP1 expression by OPCs hinders OL maturation. This may be the result of a cell autonomous function of LRP1 in adult OPCs that has lasting effects during differentiation, but may also be the result of a less direct signaling mechanism. For example, the release of soluble LRP1 from the surface of OPCs; the LRP1-directed secretion of proteins from OPCs; or an LRP1-directed increase in OPC-mediated antigen presentation could influence the maturation of pre-myelinating OLs or the behavior of other cell types such as microglia, astrocytes or lymphocytes.
How Does LRP1 Expression by Adult OPCs Affect OL Production in Response to an Injury?
As Lrp1 deletion increased the number of new OLs detected in the healthy adult mouse brain, we predicted that Lrp1 deletion would also enhance the number of new OLs produced in response to OL loss. However, OPCs can behave differently in the healthy versus injured CNS. It was previously reported that the delivery of tamoxifen to adult CAG-CreERTM :: Lrp1fl/fl mice, which reduced LRP1 expression by ∼50%, reduced the remyelination of a lysolecithin-induced callosal lesion (Hennen et al., 2013). This could have been the result of LRP1 loss from OPCs, microglia, astrocytes or neurons (May et al., 2004; Casse et al., 2012; Chuang et al., 2016; Yang et al., 2016; Liu et al., 2017; He et al., 2020), however, focal lesion repair was also impaired when Lrp1 was conditionally deleted from adult OPCs (Pdgfrα-CreERTM :: Lrp1fl/fl mice) (Hennen et al., 2013). While the level and specificity of Lrp1 deletion was not reported in this study, these data suggest that LRP1 signaling in adult OPCs could improve myelin repair (Hennen et al., 2013).
We report that LRP1 experts an altogether different effect on adult OPCs following cuprizone-mediated OL loss and demyelination. By performing cre-lox lineage tracing to follow the fate of adult OPCs, we were able to quantify the number of new OLs added to the corpus callosum of control and Lrp1-deleted mice during cuprizone feeding. While the number of OPC-derived OLs was equivalent in control and Lrp1-deleted mice, a larger proportion of the new OLs had matured in the corpus callosum of Lrp1-deleted mice. Furthermore, the area of the corpus callosum showing overt demyelination was reduced in Lrp1-deleted mice compared with controls. Consistent with our findings, adult Olig1-Cre and Olig1-Cre: Lrp1fl/fl mice, subjected to 5 weeks of cuprizone feeding also had an equivalent number of OLs in the corpus callosum, however, within 3.5 days of cuprizone withdrawal, the corpus callosum of Olig1-Cre: Lrp1fl/fl mice contained more OLs than controls and had greater MBP coverage (Fernandez-Castaneda et al., 2019). Cuprizone-induced demyelination is a robust stimulus for OPC differentiation (Xing et al., 2014; Baxi et al., 2017) and could readily mask the lesser effect of Lrp1 deletion on new OL addition. These data also suggest that LRP1 expression by OPCs impair newborn OL maturation, which could reflect a cell intrinsic function of LRP1 within the OL lineage or could reflect an alternative role for LRP1 allowing OPCs to regulate the maturation of adjacent maturing OLs by a less direct, perhaps inflammatory mechanism (see Figure 10).
Neuroinflammation impairs OL generation (Miron et al., 2011) and OPCs can modulate neuroinflammation, releasing cytokines in response to interleukin 17 receptor signaling (Wang et al., 2017). OPCs also express genes associated with antigen processing and presentation (Falcao et al., 2018; Kirby et al., 2019). While this was not investigated here, previous research has shown that LRP1 can bind and phagocytose myelin debris (Gaultier et al., 2009; Fernandez-Castaneda et al., 2013; Stiles et al., 2013) and that LRP1 expression by OPCs can influence the inflammatory nature of the remyelinating environment (Fernandez-Castaneda et al., 2019). RNA profiling of the remyelinating corpus callosum of Olig1-Cre and Olig1-Cre: Lrp1fl/fl mice, 3.5 days after cuprizone withdrawal, revealed that inflammatory gene expression was lower in Olig1-Cre :: Lrp1fl/fl mice (Fernandez-Castaneda et al., 2019). As LRP1 can be cleaved, it is possible that OPCs release soluble, cleaved LRP1 to enhance the inflammatory response of nearby microglia (Brifault et al., 2017, 2019). However, LRP1 may also facilitate antigen presentation by OPCs, as the deletion of Lrp1 from OPCs reduces their expression of MHC class I antigen presenting genes in the corpus callosum of cuprizone-demyelinated mice and reduces the ability of OPCs to cross-present antigens to lymphocytes in vitro (Fernandez-Castaneda et al., 2019). Therefore, further research is required to fully elucidate the cell autonomous effect of LRP1 expression in OPCs and the capacity for LRP1 signaling on OPCs to influence the behavior of other nearby CNS cell types.
Data Availability Statement
All datasets presented in this study are included in the article/Supplementary Material.
Ethics Statement
The animal study was reviewed and approved by The University of Tasmania Animal Ethics Committee.
Author Contributions
LA, KY, LF, and BT developed the project and wrote the manuscript. LA, CC, RP, and KP carried out the experiments. KY and LF obtained the funding. LA, CC, KP, and KY performed the statistical analyses and generated the figures. KY, LF, and BT provided supervision. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by grants from the National Health and Medical Research Council of Australia (NHMRC; 1077792 and 1139041), MS Research Australia (11-014, 16-105, and 17-007), and the Australian Research Council (DP180101494). LA was supported by an Australian Postgraduate Award. KP was supported by a fellowship from the NHMRC (1139180). CC was supported by a fellowship from MS Research Australia and the Penn Foundation (15-054). RP was supported by a scholarship from the Menzies Institute for Medical Research. KY and BT were supported by a paired fellowship from MS Research Australia and the Macquarie Group Foundation (17-0223).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our colleagues at the University of Tasmania for providing animal services support or providing constructive feedback and suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.564351/full#supplementary-material
References
Actis Dato, V., and Chiabrando, G. A. (2018). The role of low-density lipoprotein receptor-related Protein 1 in lipid metabolism, glucose homeostasis and inflammation. Int. J. Mol. Sci. 19:1780. doi: 10.3390/ijms19061780
Andersen, R. K., Hammer, K., Hager, H., Christensen, J. N., Ludvigsen, M., Honoré, B., et al. (2015). Melanoma tumors frequently acquire LRP2/megalin expression, which modulates melanoma cell proliferation and survival rates. Pigment Cell Melanoma Res. 28, 267–280. doi: 10.1111/pcmr.12352
Auderset, L., Cullen, C. L., and Young, K. M. (2016a). Low density lipoprotein-receptor related protein 1 is differentially expressed by neuronal and glial populations in the developing and mature mouse central nervous system. PLoS One 11:e155878. doi: 10.1371/journal.pone.0155878
Auderset, L., Landowski, L. M., Foa, L., and Young, K. M. (2016b). Low density lipoprotein receptor related proteins as regulators of neural stem and progenitor cell function. Stem Cells Int. 2016:2108495.
Barcelona, P. F., Jaldin-Fincati, J. R., Sanchez, M. C., and Chiabrando, G. A. (2013). Activated 2-macroglobulin induces Muller glial cell migration by regulating MT1-MMP activity through LRP1. FASEB J. 27, 3181–3197. doi: 10.1096/fj.12-221598
Basford, J. E., Moore, Z. W. Q., Zhou, L., Herz, J., and Hui, D. Y. (2009). Smooth muscle LDL receptor-related protein-1 inactivation reduces vascular reactivity and promotes injury-induced neointima formation. Arterioscler. Thromb. Vasc. Biol. 29, 1772–1778. doi: 10.1161/atvbaha.109.194357
Baxi, E. G., DeBruin, J., Jin, J., Strasburger, H. J., Smith, M. D., Orthmann-Murphy, J. L., et al. (2017). Lineage tracing reveals dynamic changes in oligodendrocyte precursor cells following cuprizone-induced demyelination. Glia 65, 2087–2098. doi: 10.1002/glia.23229
Boucher, P., Gotthardt, M., Li, W. P., Anderson, R., and Herz, J. L. R. P. (2003). Role in vascular wall integrity and protection from atherosclerosis. Science 300, 329–332. doi: 10.1126/science.1082095
Boucher, P., Li, W.-P., Matz, R. L., Takayama, Y., Auwerx, J., Anderson, R. G. W., et al. (2007). LRP1 functions as an atheroprotective integrator of TGFβ and PDGF signals in the vascular wall: implications for marfan syndrome. PLoS One 2:e448. doi: 10.1371/journal.pone.0000448
Boyé, K., Pujol, N., Alves, D. I., Chen, Y.-P., Daubon, T., Lee, Y.-Z., et al. (2017). The role of CXCR3/LRP1 cross-talk in the invasion of primary brain tumors. Nat. Commun. 8:1571.
Bres, E. E., and Faissner, A. (2019). Low density receptor-related protein 1 interactions with the extracellular matrix: more than meets the eye. Front. Cell Dev. Biol. 7:1916. doi: 10.3389/fcell.2019.00031
Brifault, C., Gilder, A. S., Laudati, E., Banki, M., and Gonias, S. L. (2017). Shedding of membrane-associated LDL receptor-related protein-1 from microglia amplifies and sustains neuroinflammation. J. Biol. Chem. 292, 18699–18712. doi: 10.1074/jbc.m117.798413
Brifault, C., Kwon, H., Campana, W. M., and Gonias, S. L. (2019). LRP1 deficiency in microglia blocks neuro-inflammation in the spinal dorsal horn and neuropathic pain processing. Glia 67, 1210–1224. doi: 10.1002/glia.23599
Bu, G., Morton, P. A., and Schwartz, A. L. (1992). Identification and partial characterization by chemical cross-linking of a binding protein for tissue-type plasminogen activator (t-PA) on rat hepatoma cells. A plasminogen activator inhibitor type 1-independent t-PA receptor. J. Biol. Chem. 267, 15595–15602.
Cahoy, J. D., Emery, B., Kaushal, A., Foo, L. C., Zamanian, J. L., Christopherson, K. S., et al. (2008). A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278. doi: 10.1523/jneurosci.4178-07.2008
Cai, J., Chen, Y., Cai, W.-H., Hurlock, E. C., Wu, H., Kernie, S. G., et al. (2007). A crucial role for Olig2 in white matter astrocyte development. Development 134, 1887–1899. doi: 10.1242/dev.02847
Cam, J. A., Zerbinatti, C. V., Li, Y., and Bu, G. (2005). Rapid endocytosis of the low density lipoprotein receptor-related protein modulates cell surface distribution and processing of the -amyloid precursor protein. J. Biol. Chem. 280, 15464–15470. doi: 10.1074/jbc.m500613200
Casse, F., Bardou, I., Danglot, L., Briens, A., Montagne, A., Parcq, J., et al. (2012). Glutamate controls tPA recycling by astrocytes, which in turn influences glutamatergic signals. J. Neurosci. 32, 5186–5199. doi: 10.1523/jneurosci.5296-11.2012
Cheli, V. T., Santiago González, D. A., Spreuer, V., and Paez, P. M. (2015). Voltage-gated Ca2+ entry promotes oligodendrocyte progenitor cell maturation and myelination in vitro. Exp. Neurol. 265, 69–83. doi: 10.1016/j.expneurol.2014.12.012
Chew, L.-J., Coley, W., Cheng, Y., and Gallo, V. (2010). Mechanisms of regulation of oligodendrocyte development by p38 mitogen-activated protein kinase. J. Neurosci. 30, 11011–11027. doi: 10.1523/jneurosci.2546-10.2010
Chuang, T.-Y., Guo, Y., Seki, S. M., Rosen, A. M., Johanson, D. M., Mandell, J. W., et al. (2016). LRP1 expression in microglia is protective during CNS autoimmunity. Acta Neuropathol. Commun. 4:68.
Clarke, L. E., Young, K. M., Hamilton, N. B., Li, H., Richardson, W. D., and Attwell, D. (2012). Properties and fate of oligodendrocyte progenitor cells in the corpus callosum, motor cortex, and piriform cortex of the mouse. J. Neurosci. 32, 8173–8185. doi: 10.1523/jneurosci.0928-12.2012
De Biase, L. M., Nishiyama, A., and Bergles, D. E. (2010). Excitability and synaptic communication within the oligodendrocyte lineage. J. Neurosci. 30, 3600–3611. doi: 10.1523/jneurosci.6000-09.2010
Dimou, L., Simon, C., Kirchhoff, F., Takebayashi, H., and Gotz, M. (2008). Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J. Neurosci. 28, 10434–10442. doi: 10.1523/jneurosci.2831-08.2008
Emery, B., and Dugas, J. C. (2013). Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb. Protoc. 2013, 854–868.
Falcao, A. M., van Bruggen, D., Marques, S., Meijer, M., Jäkel, S., Agirre, E., et al. (2018). Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 24, 1837–1844. doi: 10.1038/s41591-018-0236-y
Fannon, J., Tarmier, W., and Fulton, D. (2015). Neuronal activity and AMPA-type glutamate receptor activation regulates the morphological development of oligodendrocyte precursor cells. Glia 63, 1021–1035. doi: 10.1002/glia.22799
Fard, M. K., van der Meer, F., Sánchez, P., Cantuti-Castelvetri, L., Mandad, S., Jäkel, S., et al. (2017). BCAS1 expression defines a population of early myelinating oligodendrocytes in multiple sclerosis lesions. Sci. Transl. Med. 9:eaam7816. doi: 10.1126/scitranslmed.aam7816
Fernandez-Castaneda, A., Arandjelovic, S., Stiles, T. L., Schlobach, R. K., Mowen, K. A., Gonias, S. L., et al. (2013). Identification of the low density lipoprotein (LDL) receptor-related Protein-1 interactome in central nervous system myelin suggests a role in the clearance of necrotic cell debris. J. Biol. Chem. 288, 4538–4548. doi: 10.1074/jbc.m112.384693
Fernandez-Castaneda, A., Chappell, M. S., Rosen, D. A., Seki, S. M., Beiter, R. M., Johanson, D. M., et al. (2019). The active contribution of OPCs to neuroinflammation is mediated by LRP1. Acta Neuropathol. 139, 365–382. doi: 10.1007/s00401-019-02073-1
Ferreira, S., Pitman, K. A., Summers, B. S., Wang, S., Young, K. M., and Cullen, C. L. (2020). Oligodendrogenesis increases in hippocampal grey and white matter prior to locomotor or memory impairment in an adult mouse model of tauopathy. Eur. J. Neurosci. doi: 10.1111/ejn.14726 [Epub ahead of print].
Ferrer, D. G., Actis Dato, V., Jaldín Fincati, J. R., Lorenc, V. E., Sánchez, M. C., and Chiabrando, G. A. (2016). Activated α2 -macroglobulin induces mesenchymal cellular migration of Raw264.7 cells through low-density lipoprotein receptor-related Protein 1. J. Cell Biochem. 118, 1810–1818. doi: 10.1002/jcb.25857
Fulton, D., Paez, P. M., Fisher, R., Handley, V., Colwell, C. S., and Campagnoni, A. T. (2010). Regulation of L-type Ca++ currents and process morphology in white matter oligodendrocyte precursor cells by golli-myelin proteins. Glia 58, 1292–1303. doi: 10.1002/glia.21008
Gajera, C. R., Emich, H., Lioubinski, O., Christ, A., Beckervordersandforth-Bonk, R., Yoshikawa, K., et al. (2010). LRP2 in ependymal cells regulates BMP signaling in the adult neurogenic niche. J. Cell Sci. 123(Pt 11), 1922–1930. doi: 10.1242/jcs.065912
Gallo, V., Zhou, J. M., McBain, C. J., Wright, P., Knutson, P. L., and Armstrong, R. C. (1996). Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J. Neurosci. 2659–2670. doi: 10.1523/jneurosci.16-08-02659.1996
Gan, M., Jiang, P., McLean, P., Kanekiyo, T., and Bu, G. (2014). Low-density lipoprotein receptor-related Protein 1 (LRP1) regulates the stability and function of GluA1 α-Amino-3-Hydroxy-5-Methyl-4-Isoxazole propionic acid (AMPA) receptor in neurons. PLoS One 9:e113237. doi: 10.1371/journal.pone.0113237
Gaultier, A., Wu, X., Le Moan, N., Takimoto, S., Mukandala, G., Akassoglou, K., et al. (2009). Low-density lipoprotein receptor-related protein 1 is an essential receptor for myelin phagocytosis. J. Cell Sci. 122(Pt 8), 1155–1162. doi: 10.1242/jcs.040717
Genoud, S., Lappe-Siefke, C., Goebbels, S., Radtke, F., Aguet, M., Scherer, S. S., et al. (2002). Notch1 control of oligodendrocyte differentiation in the spinal cord. J. Cell Biol. 158, 709–718. doi: 10.1083/jcb.200202002
Givogri, M. I., Costa, R. M., Schonmann, V., Silva, A. J., Campagnoni, A. T., and Bongarzone, E. R. (2002). Central nervous system myelination in mice with deficient expression of notch1 receptor. J. Neurosci. Res. 67, 309–320. doi: 10.1002/jnr.10128
Grier, M. D., West, K. L., Kelm, N. D., Fu, C., Does, M. D., Parker, B., et al. (2017). Loss of mTORC2 signaling in oligodendrocyte precursor cells delays myelination. PLoS One 12:e0188417. doi: 10.1371/journal.pone.0188417
Gudz, T. I. (2006). Glutamate stimulates oligodendrocyte progenitor migration mediated via an v integrin/myelin proteolipid protein complex. J. Neurosci. 26, 2458–2466. doi: 10.1523/jneurosci.4054-05.2006
Haberlandt, C., Derouiche, A., Wyczynski, A., Haseleu, J., Pohle, J., Karram, K., et al. (2011). Gray matter NG2 cells display multiple Ca2+-signaling pathways and highly motile processes. PLoS One 6:e17575. doi: 10.1371/journal.pone.0017575
Hamilton, T. G., Klinghoffer, R. A., Corrin, P. D., and Soriano, P. (2003). Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol. Cell. Biol. 23, 4013–4025. doi: 10.1128/mcb.23.11.4013-4025.2003
He, Y., Ruganzu, J. B., Zheng, Q., Wu, X., Jin, H., Peng, X., et al. (2020). Silencing of LRP1 exacerbates inflammatory response Via TLR4/NF-κB/MAPKs signaling pathways in APP/PS1 transgenic mice. Mol. Neurobiol. 57, 3727–3743. doi: 10.1007/s12035-020-01982-7
Hennen, E., Safina, D., Haussmann, U., Worsdorfer, P., Edenhofer, F., Poetsch, A., et al. (2013). A LewisX glycoprotein screen identifies the low density lipoprotein receptor-related Protein 1 (LRP1) as a modulator of oligodendrogenesis in mice. J. Biol. Chem. 288, 16538–16545. doi: 10.1074/jbc.m112.419812
Herz, J., Clouthier, D. E., and Hammer, R. E. (1992). LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 71, 411–421. doi: 10.1016/0092-8674(92)90511-a
Hill, R. A., Li, A. M., and Grutzendler, J. (2018). Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci. 21, 683–695. doi: 10.1038/s41593-018-0120-6
Hippenmeyer, S., Vrieseling, E., Sigrist, M., Portmann, T., Laengle, C., Ladle, D. R., et al. (2005). A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 3:e159. doi: 10.1371/journal.pbio.0030159
Hrvatin, S., Hochbaum, D. R., Nagy, M. A., Cicconet, M., Robertson, K., Cheadle, L., et al. (2018). Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat. Neurosci. 21, 120–129. doi: 10.1038/s41593-017-0029-5
Hughes, E. G., Kang, S. H., Fukaya, M., and Bergles, D. E. (2013). Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 16, 668–676. doi: 10.1038/nn.3390
Hughes, E. G., Orthmann-Murphy, J. L., Langseth, A. J., and Bergles, D. E. (2018). Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci. 21, 696–706. doi: 10.1038/s41593-018-0121-5
Jiang, M., Liu, L., He, X., Wang, H., Lin, W., Wang, H., et al. (2016). Regulation of PERK-eIF2α signalling by tuberous sclerosis complex-1 controls homoeostasis and survival of myelinating oligodendrocytes. Nat. Commun. 7:12185.
Kadurin, I., Rothwell, S. W., Lana, B., Nieto-Rostro, M., and Dolphin, A. C. (2017). LRP1 influences trafficking of N-type calcium channels via interaction with the auxiliary α2δ-1 subunit. Sci. Rep. 7:43802.
Kang, S. H., Fukaya, M., Yang, J. K., Rothstein, J. D., and Bergles, D. E. (2010). NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681. doi: 10.1016/j.neuron.2010.09.009
Kirby, L., Jin, J., Cardona, J. G., Smith, M. D., Martin, K. A., Wang, J., et al. (2019). Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 10:3887.
Kougioumtzidou, E., Shimizu, T., Hamilton, N. B., Tohyama, K., Sprengel, R., Monyer, H., et al. (2017). Signalling through AMPA receptors on oligodendrocyte precursors promotes myelination by enhancing oligodendrocyte survival. eLife 6:31.
Kukley, M., Nishiyama, A., and Dietrich, D. (2010). The fate of synaptic input to NG2 glial cells: neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J. Neurosci. 30, 8320–8331. doi: 10.1523/jneurosci.0854-10.2010
Lillis, A. P., Van Duyn, L. B., Murphy-Ullrich, J. E., and Strickland, D. K. (2008). LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918. doi: 10.1152/physrev.00033.2007
Lin, J.-P., Mironova, Y. A., Shrager, P., and Giger, R. J. (2017). LRP1 regulates peroxisome biogenesis and cholesterol homeostasis in oligodendrocytes and is required for proper CNS myelin development and repair. eLife 6:e30498.
Lin, L., Bu, G., Mars, W. M., Reeves, W. B., Tanaka, S., and Hu, K. (2010). tPA activates LDL receptor-related protein 1-mediated mitogenic signaling involving the p90RSK and GSK3beta pathway. Am. J. Pathol. 177, 1687–1696. doi: 10.2353/ajpath.2010.100213
Liu, C. C., Hu, J., Tsai, C. W., Yue, M., Melrose, H. L., Kanekiyo, T., et al. (2015). Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J. Neurosci. 35, 5851–5859. doi: 10.1523/jneurosci.5180-14.2015
Liu, C.-C., Hu, J., Zhao, N., Wang, J., Na, W., Cirrito, J. R., et al. (2017). Astrocytic LRP1 mediates brain Aβ clearance and impacts amyloid deposition. J. Neurosci. 37, 4023–4031. doi: 10.1523/jneurosci.3442-16.2017
Liu, Q., Trotter, J., Zhang, J., Peters, M. M., Cheng, H., Bao, J., et al. (2010). Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J. Neurosci. 30, 17068–17078. doi: 10.1523/jneurosci.4067-10.2010
Maier, W., Bednorz, M., Meister, S., Roebroek, A., Weggen, S., Schmitt, U., et al. (2013). LRP1 is critical for the surface distribution and internalization of the NR2B NMDA receptor subtype. Mol. Neurodegener. 8:25. doi: 10.1186/1750-1326-8-25
Mantuano, E., Jo, M., Gonias, S. L., and Campana, W. M. (2010). Low density lipoprotein receptor-related protein (LRP1) regulates Rac1 and RhoA reciprocally to control Schwann cell adhesion and migration. J. Biol. Chem. 285, 14259–14266. doi: 10.1074/jbc.m109.085126
Mantuano, E., Lam, M. S., Shibayama, M., Campana, W. M., and Gonias, S. L. (2015). The NMDA receptor functions independently and as an LRP1 co-receptor to promote Schwann cell survival and migration. J. Cell Sci. 128, 3478–3488. doi: 10.1242/jcs.173765
Mao, H., Lockyer, P., Townley-Tilson, W. H. D., Xie, L., and Pi, X. (2016). LRP1 regulates retinal angiogenesis by inhibiting PARP-1 activity and endothelial cell proliferation. Arterioscler. Thromb. Vasc. Biol. 36, 350–360. doi: 10.1161/atvbaha.115.306713
Marshall, C. A. G., Novitch, B. G., and Goldman, J. E. (2005). Olig2 directs astrocyte and oligodendrocyte formation in postnatal subventricular zone cells. J. Neurosci. 25, 7289–7298. doi: 10.1523/jneurosci.1924-05.2005
May, P., Rohlmann, A., Bock, H. H., Zurhove, K., Marth, J. D., Schomburg, E. D., et al. (2004). Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol. Cell. Biol. 24, 8872–8883. doi: 10.1128/mcb.24.20.8872-8883.2004
McKinnon, R. D. (2005). PDGF-receptor signal strength controls an RTK rheostat that integrates phosphoinositol 3′-kinase and phospholipase C pathways during oligodendrocyte maturation. J. Neurosci. 25, 3499–3508. doi: 10.1523/jneurosci.5049-04.2005
Miron, V. E., Kuhlmann, T., and Antel, J. P. (2011). Cells of the oligodendroglial lineage, myelination, and remyelination. BBA Mol. Basis Dis. 1812, 184–193. doi: 10.1016/j.bbadis.2010.09.010
Muratoglu, S. C., Mikhailenko, I., Newton, C., Migliorini, M., and Strickland, D. K. (2010). Low density lipoprotein receptor-related protein 1 (LRP1) forms a signaling complex with platelet-derived growth factor receptor-beta in endosomes and regulates activation of the MAPK pathway. J. Biol. Chem. 285, 14308–14317. doi: 10.1074/jbc.m109.046672
Murcia-Belmonte, V., Medina-Rodríguez, E. M., Bribián, A., de Castro, F., and Esteban, P. F. (2014). ERK1/2 signaling is essential for the chemoattraction exerted by human FGF2 and human anosmin-1 on newborn rat and mouse OPCs via FGFR1. Glia 62, 374–386. doi: 10.1002/glia.22609
Murtie, J. C., Zhou, Y. X., Le, T. Q., and Armstrong, R. C. (2005). In vivo analysis of oligodendrocyte lineage development in postnatal FGF2 null nice. Glia 49, 542–554. doi: 10.1002/glia.20142
Nakajima, C., Kulik, A., Frotscher, M., Herz, J., Schäfer, M., Bock, H. H., et al. (2013). Low density lipoprotein receptor-related protein 1 (LRP1) modulates N-Methyl-d-aspartate (n.d.) receptor-dependent intracellular signaling and NMDA-induced regulation of postsynaptic protein complexes. J. Biol. Chem. 288, 21909–21923. doi: 10.1074/jbc.m112.444364
Noble, M., Murray, K., Stroobant, P., Waterfield, M. D., and Riddle, P. (1988). Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte/type-2 astrocyte progenitor cell. Nature 333, 560–562. doi: 10.1038/333560a0
O’Rourke, M., Cullen, C. L., Auderset, L., Pitman, K. A., Achatz, D., Gasperini, R., et al. (2016). Evaluating tissue-specific recombination in a Pdgfrα-CreERT2 transgenic mouse line. PLoS One 11:e0162858. doi: 10.1371/journal.pone.0162858
Orth, K., Madison, E. L., Gething, M. J., Sambrook, J. F., and Herz, J. (1992). Complexes of tissue-type plasminogen activator and its serpin inhibitor plasminogen-activator inhibitor type 1 are internalized by means of the low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor. Proc. Natl. Acad. Sci. U.S.A. 89, 7422–7426. doi: 10.1073/pnas.89.16.7422
Parkyn, C. J., Vermeulen, E. G. M., Mootoosamy, R. C., Sunyach, C., Jacobsen, C., Oxvig, C., et al. (2008). LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J. Cell Sci. 121, 773–783. doi: 10.1242/jcs.021816
Pepper, R. E., Pitman, K. A., Cullen, C. L., and Young, K. M. (2018). How do cells of the oligodendrocyte lineage affect neuronal circuits to influence motor function, memory and mood? Front. Cell Neurosci. 12:399. doi: 10.3389/fncel.2018.00399
Pi, X., Schmitt, C. E., Xie, L., Portbury, A. L., Wu, Y., Lockyer, P., et al. (2012). LRP1-dependent endocytic mechanism governs the signaling output of the bmp system in endothelial cells and in angiogenesis. Circ. Res. 111, 564–574. doi: 10.1161/circresaha.112.274597
Pitman, K. A., Ricci, R., Gasperini, R., Beasley, S., Pavez, M., Charlesworth, J., et al. (2019). The voltage-gated calcium channel CaV1.2 promotes adult oligodendrocyte progenitor cell survival in the mouse corpus callosum but not motor cortex. Glia 22, 54.
Polavarapu, R., Gongora, M. C., Yi, H., Ranganthan, S., Lawrence, D. A., Strickland, D., et al. (2007). Tissue-type plasminogen activator-mediated shedding of astrocytic low-density lipoprotein receptor-related protein increases the permeability of the neurovascular unit. Blood 109, 3270–3278. doi: 10.1182/blood-2006-08-043125
Pringle, N., Collarini, E. J., Mosley, M. J., Heldin, C. H., Westermark, B., and Richardson, W. D. (1989). PDGF A chain homodimers drive proliferation of bipotential (O-2A) glial progenitor cells in the developing rat optic nerve. EMBO J. 8, 1049–1056. doi: 10.1002/j.1460-2075.1989.tb03472.x
Psachoulia, K., Jamen, F., Young, K. M., and Richardson, W. D. (2009). Cell cycle dynamics of NG2 cells in the postnatal and ageing brain. Neuron Glia Biol. 5, 57–67.
Rajasekharan, S. (2008). Intracellular signaling mechanisms directing oligodendrocyte precursor cell migration. J. Neurosci. 28, 13365–13367. doi: 10.1523/jneurosci.4931-08.2008
Rauch, J. N., Luna, G., Guzman, E., Audouard, M., Challis, C., Sibih, Y. E., et al. (2020). LRP1 is a master regulator of tau uptake and spread. Nature 580, 381–385. doi: 10.1038/s41586-020-2156-5
Richardson, W. D., Pringle, N., Mosley, M. J., Westermark, B., and Dubois-Dalcg, M. (1988). A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell 53, 309–319. doi: 10.1016/0092-8674(88)90392-3
Rivers, L. E., Young, K. M., Rizzi, M., Jamen, F., Psachoulia, K., Wade, A., et al. (2008). PDGFRα/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat. Neurosci. 11, 1392–1401. doi: 10.1038/nn.2220
Safina, D., Schlitt, F., Romeo, R., Pflanzner, T., Pietrzik, C. U., Narayanaswami, V., et al. (2016). Low−density lipoprotein receptor−related protein 1 is a novel modulator of radial glia stem cell proliferation, survival, and differentiation. Glia 64, 1363–1380. doi: 10.1002/glia.23009
Sayre, N., and Kokovay, E. (2019). Loss of lipoprotein receptor LRP1 ablates neural stem cell migration to ischemic lesions. FASEB J. 33:lb613.
Spitzer, S. O., Sitnikov, S., Kamen, Y., Evans, K. A., Kronenberg-Versteeg, D., Dietmann, S., et al. (2019). Oligodendrocyte progenitor cells become regionally diverse and heterogeneous with age. Neuron 101:459. doi: 10.1016/j.neuron.2018.12.020
Spuch, C., Ortolano, S., and Navarro, C. (2012). LRP-1 and LRP-2 receptors function in the membrane neuron. Trafficking mechanisms and proteolytic processing in Alzheimer’s disease. Front Physiol. 6:3. doi: 10.3389/fphys.2012.00269
Srinivas, S., Watanabe, T., Lin, C. S., William, C. M., Tanabe, Y., Jessell, T. M., et al. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1:4. doi: 10.1186/1471-213X-1-4
Stiles, T. L., Dickendesher, T. L., Gaultier, A., Fernandez-Castaneda, A., Mantuano, E., Giger, R. J., et al. (2013). LDL receptor-related protein-1 is a sialic-acid-independent receptor for myelin-associated glycoprotein that functions in neurite outgrowth inhibition by MAG and CNS myelin. J. Cell Sci. 126(Pt 1), 209–220. doi: 10.1242/jcs.113191
Su, E. J., Fredriksson, L., Geyer, M., Folestad, E., Cale, J., Andrae, J., et al. (2008). Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat. Med. 14, 731–737. doi: 10.1038/nm1787
Takayama, Y., May, P., Anderson, R. G. W., and Herz, J. (2005). Low density lipoprotein receptor-related protein 1 (LRP1) controls endocytosis and c-CBL-mediated ubiquitination of the platelet-derived growth factor receptor beta (PDGFR beta). J. Biol. Chem. 280, 18504–18510. doi: 10.1074/jbc.m410265200
Trapp, B. D., Nishiyama, A., Cheng, D., and Macklin, W. (1997). Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J. Cell Biol. 137, 459–468. doi: 10.1083/jcb.137.2.459
Tripathi, R. B., Jackiewicz, M., McKenzie, I. A., Kougioumtzidou, E., Grist, M., and Richardson, W. D. (2017). Remarkable Stability of myelinating oligodendrocytes in mice. Cell Rep. 21, 316–323. doi: 10.1016/j.celrep.2017.09.050
Van Gool, B., Storck, S. E., Reekmans, S. M., Lechat, B., Gordts, P. L. S. M., Pradier, L., et al. (2019). LRP1 has a predominant role in production over clearance of Aβ in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 56, 7234–7245. doi: 10.1007/s12035-019-1594-2
Velez-Fort, M., Maldonado, P. P., Butt, A. M., Audinat, E., and Angulo, M. C. (2010). Postnatal switch from synaptic to extrasynaptic transmission between interneurons and NG2 cells. J. Neurosci. 30, 6921–6929. doi: 10.1523/jneurosci.0238-10.2010
Wang, C., Zhang, C.-J., Martin, B. N., Bulek, K., Kang, Z., Zhao, J., et al. (2017). IL-17 induced NOTCH1 activation in oligodendrocyte progenitor cells enhances proliferation and inflammatory gene expression. Nat. Commun. 8, 15508–15516.
Williamson, A. V., Compston, D. A., and Randall, A. D. (1997). Analysis of the ion channel complement of the rat oligodendrocyte progenitor in a commonly studied in vitro preparation. Eur. J. Neurosci. 9, 706–720. doi: 10.1111/j.1460-9568.1997.tb01419.x
Wujak, L., Böttcher, R. T., Pak, O., Frey, H., Agha, El E., Chen, Y., et al. (2018). Low density lipoprotein receptor-related protein 1 couples β1 integrin activation to degradation. Cell Mol. Life Sci. 75, 1671–1685. doi: 10.1007/s00018-017-2707-6
Xiao, L., Ohayon, D., McKenzie, I. A., Sinclair-Wilson, A., Wright, J. L., Fudge, A. D., et al. (2016). Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat. Neurosci. 19, 1210–1217. doi: 10.1038/nn.4351
Xing, Y. L., Röth, P. T., Stratton, J. A. S., Chuang, B. H. A., Danne, J., Ellis, S. L., et al. (2014). Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J. Neurosci. 34, 14128–14146. doi: 10.1523/jneurosci.3491-13.2014
Yamamoto, K., Owen, K., Parker, A. E., Scilabra, S. D., Dudhia, J., Strickland, D. K., et al. (2014). Low density lipoprotein receptor-related protein 1 (LRP1)-mediated endocytic clearance of a disintegrin and metalloproteinase with thrombospondin motifs-4 (ADAMTS-4): functional differences of non-catalytic domains of ADAMTS-4 and ADAMTS-5 in LRP1 binding. J. Biol. Chem. 289, 6462–6474. doi: 10.1074/jbc.m113.545376
Yang, L., Liu, C.-C., Zheng, H., Kanekiyo, T., Atagi, Y., Jia, L., et al. (2016). LRP1 modulates the microglial immune response via regulation of JNK and NF-kappa B signaling pathways. J. Neuroinflamm. 13:304.
Yang, T.-H., St John, L. S., Garber, H. R., Kerros, C., Ruisaard, K. E., Clise-Dwyer, K., et al. (2018). Membrane-associated proteinase 3 on granulocytes and acute myeloid leukemia inhibits T cell proliferation. J. Immunol. 201, 1389–1399. doi: 10.4049/jimmunol.1800324
Young, K. M., Psachoulia, K., Tripathi, R. B., Dunn, S.-J., Cossell, L., Attwell, D., et al. (2013). Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77, 873–885. doi: 10.1016/j.neuron.2013.01.006
Zhang, Y., Argaw, A. T., Gurfein, B. T., Zameer, A., Snyder, B. J., Ge, C., et al. (2009). Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc. Natl. Acad. Sci. U.S.A. 106, 19162–19167. doi: 10.1073/pnas.0902834106
Zhang, Y., Chen, K., Sloan, S. A., Bennett, M. L., Scholze, A. R., O’Keeffe, S., et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. doi: 10.1523/jneurosci.1860-14.2014
Zhou, Y.-X., Flint, N. C., Murtie, J. C., Le, T. Q., and Armstrong, R. C. (2006). Retroviral lineage analysis of fibroblast growth factor receptor signaling in FGF2 inhibition of oligodendrocyte progenitor differentiation. Glia 54, 578–590. doi: 10.1002/glia.20410
Zhu, X., Bergles, D. E., and Nishiyama, A. (2008). NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development 135, 145–157. doi: 10.1242/dev.004895
Zhu, X., Hill, R. A., Dietrich, D., Komitova, M., Suzuki, R., and Nishiyama, A. (2011). Age-dependent fate and lineage restriction of single NG2 cells. Development 138, 745–753. doi: 10.1242/dev.047951
Zhuo, M., Holtzman, D. M., Li, Y., Osaka, H., DeMaro, J., Jacquin, M., et al. (2000). Role of tissue plasminogen activator receptor LRP in hippocampal long-term potentiation. J. Neurosci. 20, 542–549. doi: 10.1523/jneurosci.20-02-00542.2000
Zonouzi, M., Renzi, M., Farrant, M., and Cull-Candy, S. G. (2011). Bidirectional plasticity of calcium-permeable AMPA receptors in oligodendrocyte lineage cells. Nat. Neurosci. 14, 1430–1438. doi: 10.1038/nn.2942
Zou, Y., Jiang, W., Wang, J., Li, Z., Zhang, J., Bu, J., et al. (2014). Oligodendrocyte precursor cell-intrinsic effect of Rheb1 controls differentiation and mediates mTORC1-dependent myelination in brain. J. Neurosci. 34, 15764–15778. doi: 10.1523/jneurosci.2267-14.2014
Keywords: LRP1, oligodendrocyte, myelin, proliferation, NG2 glia, remyelination, cuprizone, alpha-2-macroglobulin
Citation: Auderset L, Pitman KA, Cullen CL, Pepper RE, Taylor BV, Foa L and Young KM (2020) Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) Is a Negative Regulator of Oligodendrocyte Progenitor Cell Differentiation in the Adult Mouse Brain. Front. Cell Dev. Biol. 8:564351. doi: 10.3389/fcell.2020.564351
Received: 21 May 2020; Accepted: 14 September 2020;
Published: 13 November 2020.
Edited by:
Tim Czopka, University of Edinburgh, United KingdomReviewed by:
Nathalie Sol-Foulon, INSERM U1127 Institut du Cerveau et de la Moelle épinière (ICM), FranceAkiko Nishiyama, University of Connecticut, United States
Copyright © 2020 Auderset, Pitman, Cullen, Pepper, Taylor, Foa and Young. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaylene M. Young, a2F5bGVuZS55b3VuZ0B1dGFzLmVkdS5hdQ==