 Khanichi N. Charles1
Khanichi N. Charles1 Janis E. Shackelford1
Janis E. Shackelford1 Phyllis L. Faust2
Phyllis L. Faust2 Steven J. Fliesler3,4
Steven J. Fliesler3,4 Herbert Stangl5
Herbert Stangl5 Werner J. Kovacs6*
Werner J. Kovacs6*- 1Department of Biology, San Diego State University, San Diego, CA, United States
- 2Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, United States
- 3Departments of Ophthalmology and Biochemistry and Gradate Program in Neuroscience, University at Buffalo-The State University of New York (SUNY), Buffalo, NY, United States
- 4Research Service, Veterans Administration Western New York Healthcare System, Buffalo, NY, United States
- 5Department of Medical Chemistry, Center for Pathobiochemistry and Genetics, Medical University of Vienna, Vienna, Austria
- 6Institute of Molecular Health Sciences, ETH Zurich, Zurich, Switzerland
Cholesterol biosynthesis is a multi-step process involving several subcellular compartments, including peroxisomes. Cells adjust their sterol content by both transcriptional and post-transcriptional feedback regulation, for which sterol regulatory element-binding proteins (SREBPs) are essential; such homeostasis is dysregulated in peroxisome-deficient Pex2 knockout mice. Here, we compared the regulation of cholesterol biosynthesis in Chinese hamster ovary (CHO-K1) cells and in three isogenic peroxisome-deficient CHO cell lines harboring Pex2 gene mutations. Peroxisome deficiency activated expression of cholesterogenic genes, however, cholesterol levels were unchanged. 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) protein levels were increased in mutant cells, whereas HMGCR activity was significantly decreased, resulting in reduced cholesterol synthesis. U18666A, an inhibitor of lysosomal cholesterol export, induced cholesterol biosynthetic enzymes; yet, cholesterol synthesis was still reduced. Interestingly, peroxisome deficiency promoted ER-to-Golgi SREBP cleavage-activating protein (SCAP) trafficking even when cells were cholesterol-loaded. Restoration of functional peroxisomes normalized regulation of cholesterol synthesis and SCAP trafficking. These results highlight the importance of functional peroxisomes for maintaining cholesterol homeostasis and efficient cholesterol synthesis.
Introduction
Cholesterol is an essential lipid constituent of cellular membranes, in particular the plasma membrane, and an obligatory precursor for synthesis of steroid hormones, bile acids, and regulatory oxysterols, as well as a requisite ligand for activation of Hedgehog family proteins. Its synthesis involves more than 20 enzymes distributed over several subcellular compartments. Numerous studies assign the pre-squalene steps of cholesterol biosynthesis to peroxisomes (Figure 1A) [reviewed in Kovacs et al. (2002), Faust and Kovacs (2014)]. With the exception of HMGCR, the enzymes of the pre-squalene segment contain functional peroxisomal targeting signals (PTS) that mediate their import into the peroxisomal matrix (Kovacs et al., 2002; Faust and Kovacs, 2014). We showed that acetyl-CoA derived from peroxisomal β-oxidation of very long-chain fatty acids (i.e., [1,2,3,4-13C4]docosanoate) and dicarboxylic acids (i.e., [U-13C12]dodecanedioate) is channeled to cholesterol synthesis inside peroxisomes (Kovacs et al., 2007). The enzymes of the pre-squalene segment of the isoprenoid biosynthetic pathway are also localized to peroxisomes in plants [reviewed in Faust and Kovacs (2014), Henry et al. (2018)]. Furthermore, several enzymes of the isoprenoid biosynthetic pathway are localized within peroxisome-related microbodies called glycosomes in trypanosomatid parasites (Concepcion et al., 1998; Urbina et al., 2002; Colasante et al., 2006; Carrero-Lérida et al., 2009; Güther et al., 2014; Ferreira et al., 2016; Acosta et al., 2019).
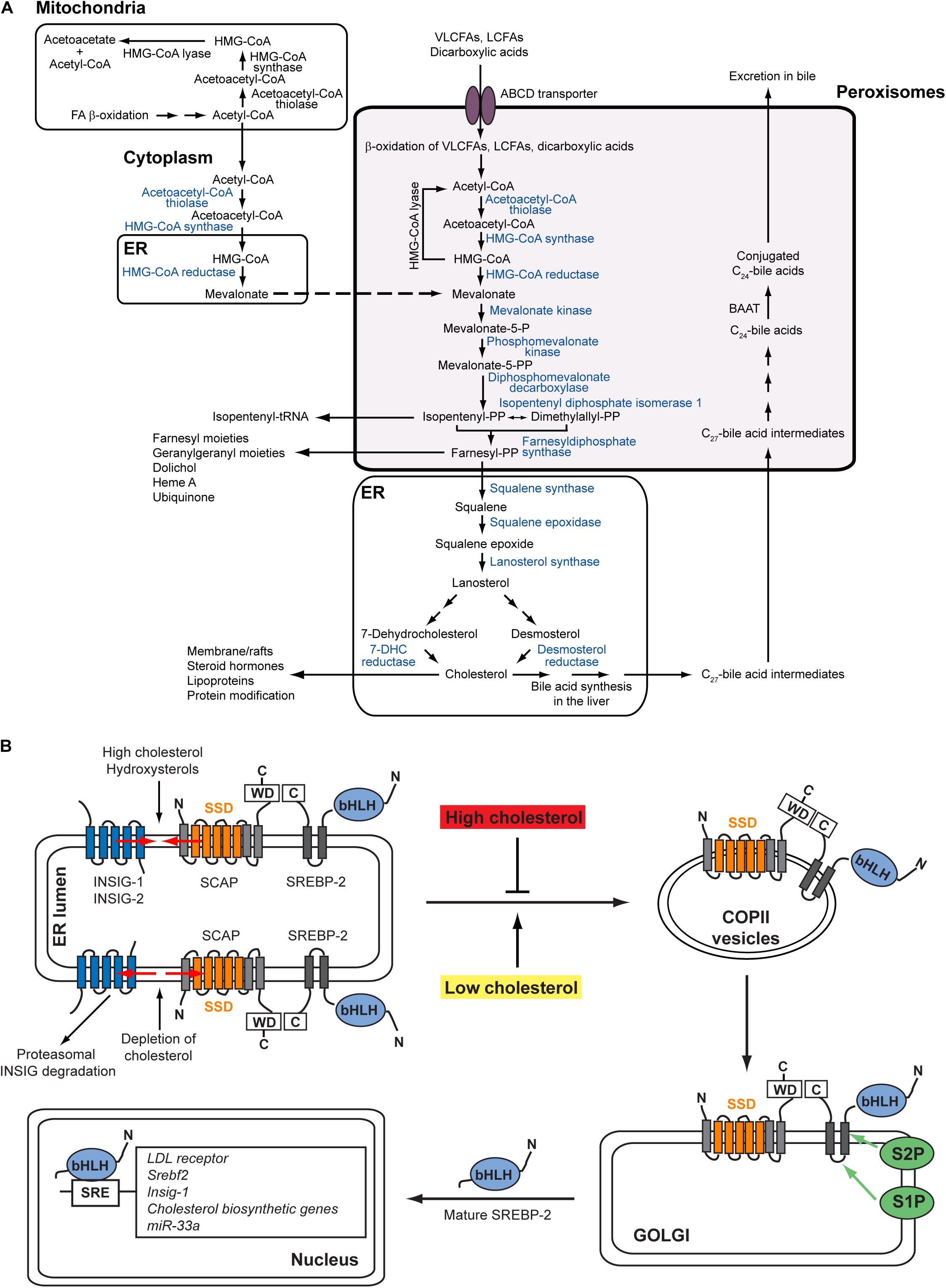
Figure 1. (A) Subcellular localization of the cholesterol biosynthetic pathway in mammalian cells. Blue fonts indicate SREBP-2-regulated cholesterol biosynthetic enzymes. With the exception of HMGCR, all enzymes for the conversion of acetyl-CoA to farnesyl diphosphate contain functional peroxisomal targeting signals. Acetyl-CoA derived from peroxisomal β-oxidation of very long-chain fatty acids and dicarboxylic acids is channeled to cholesterol synthesis inside peroxisomes (Weinhofer et al., 2006; Kovacs et al., 2007). Farnesyl diphosphate leaves the peroxisomes and is converted either to cholesterol in the ER or to nonsterol isoprenoids. The conversion of acetyl-CoA to HMG-CoA also occurs in the cytoplasm. The mitochondrial and peroxisomal acetoacetyl-CoA thiolase and the cytosolic and peroxisomal HMG-CoA synthase are encoded by single genes, respectively. In hepatocytes, the conversion of cholesterol to bile acids also partly takes place in peroxisomes. The primary C24-bile acids are formed from C27-bile acid intermediates by peroxisomal β-oxidation of the side chain, followed by conjugation of the C24-bile acids to glycine or taurine by the peroxisomal enzyme BAAT. Figure modified from Figure 1 in Faust and Kovacs (2014). (B) Regulation of the SREBP-2 pathway. The precursor SREBP-2 is embedded as inactive transcription factor in ER membranes. In the presence of cholesterol and hydroxysterols (e.g., 25-hydroxycholesterol, 27-hydroxycholesterol), the SCAP-SREBP-2 complex is retained in the ER by interaction with INSIG proteins. Whereas cholesterol binds to SCAP and induces a conformational change in SCAP through which it binds to INSIGs, hydroxysterols bind to INSIGs and cause INSIGs to bind to SCAP. In the absence of sterols, INSIGs dissociate from SCAP, whereupon the INSIGs are ubiquitinated and degraded in proteasomes. The SCAP-SREBP-2 complex is loaded into COPII-coated vesicles and transported to the Golgi. In the Golgi SREBPs are processed sequentially by Site-1 and Site-2 proteases to release the N-terminal transcription factor domain that then enters the nucleus and activates SRE (sterol regulatory element)-containing genes. Figure modified from Figure 2 in Faust and Kovacs (2014).
Cellular cholesterol homeostasis involves sensing sterol levels in the endoplasmic reticulum (ER) and thereby balancing its uptake, synthesis, efflux, and metabolic conversion to a wide variety of biologically significant products (Russell, 2003; Goldstein et al., 2006; Goldstein and Brown, 2015; Brown et al., 2018). For this cells have established an elaborate feedback system to adjust their sterol content, using both transcriptional and post-transcriptional feedback systems (Wangeline et al., 2017; Brown et al., 2018). The sterol regulatory element-binding protein (SREBP) family of transcription factors is central to this regulatory system (Goldstein et al., 2006). The SREBP family consists of SREBP-1a, SREBP-1c, and SREBP-2 in mammals. SREBP-1a and SREBP-1c are produced from a single gene by use of different promoters and alternative splicing. There is some functional overlap between the different isoforms, but SREBP-1c is mainly responsible for the expression of genes involved in fatty acid, triacylglycerol, and phospholipid biosynthesis, whereas SREBP-2 activates genes involved in the uptake and biosynthesis of cholesterol (Goldstein et al., 2006). SREBP-1a is a potent activator of all SREBP-responsive genes. Mammalian cells supply themselves with cholesterol through receptor-mediated endocytosis of low density lipoproteins (LDL), which carry cholesterol primarily in the form of fatty acid esters (Goldstein and Brown, 2015). After delivery to lysosomes, their cargo of cholesteryl esters is hydrolyzed, releasing free cholesterol, which is then exported from the lysosomes to other membrane-bound organelles and cellular membranes, notably the plasma membrane and the ER (Das et al., 2014; Iaea and Maxfield, 2015). In the ER cholesterol binds to SREBP cleavage-activating protein (SCAP), an ER protein that functions as a sterol sensor and regulates the cleavage and activation of membrane-bound SREBPs (Brown et al., 2018). In sterol-rich cells, SREBPs reside as transcriptionally inactive membrane-bound precursor proteins in the ER by interaction with SCAP and INSIG-1 (insulin-induced gene 1) proteins (Figure 1B). In sterol-depleted cells INSIG-1 dissociates from SCAP, which allows SCAP to escort SREBPs in COPII-coated vesicles from the ER to the Golgi apparatus (Figure 1B). In the Golgi SREBPs are processed sequentially by two proteases (Site-1 and Site-2 protease) to release the N-terminal transcription factor domain that then enters the nucleus and activates SRE (sterol regulatory element)-containing genes (Figure 1B; Horton et al., 2002). After dissociating from SCAP, INSIG-1 is rapidly ubiquitinated and degraded by the proteasome (Goldstein et al., 2006). Whereas cholesterol binds to SCAP and induces a conformational change in SCAP that causes it to bind to INSIG-1, hydroxycholesterols bind to INSIGs and cause INSIGs to bind to SCAP (Brown et al., 2018).
Using the peroxisome-deficient Pex2 knockout (Pex2–/–) mouse we have shown the importance of peroxisomes for the maintenance of normal cholesterol homeostasis (Kovacs et al., 2004, 2009, 2012). Total cholesterol levels were similar in the brain, kidney, spleen, heart, and lung of control (Pex2+/+ and Pex2+/–, hereafter referred to as Pex2+/) and Pex2–/– mice, however, hepatic cholesterol levels in 9-day-old (P9) Pex2–/– mice were decreased by 40%, relative to controls, while bile acid (BA) feeding normalized their hepatic cholesterol levels. HMG-CoA reductase (HMGCR), isopentenyl diphosphate isomerase 1 (IDI1), farnesyl diphosphate synthase (FDPS), and squalene synthase (FDFT1) activities and protein levels were significantly increased in the liver of P9-P10 Pex2–/– mice, while BA feeding attenuated these enzyme activities and protein levels. Interestingly, the increase in protein levels was significantly greater than the increase in the activities of these enzymes, and the “catalytic efficiency” of HMGCR and IDI1 was significantly decreased in newborn (P0) and untreated as well as BA-fed postnatal Pex2–/– mice (Kovacs et al., 2004, 2009, 2012). Notably, a different regulatory pattern of cholesterol enzyme activities was observed in kidneys from Pex2–/– vs. control mice: the activity of HMGCR was significantly decreased, whereas activities of other cholesterol biosynthetic enzymes (i.e., IDI1, FDPS, FDFT1) were increased (Kovacs et al., 2004, 2009, 2012). Using [3H]acetate as substrate, we showed that the rate of cholesterol synthesis in the liver, spleen, lung, and heart of P9 Pex2–/– mice was significantly increased compared to controls, which is probably due to the highly increased protein levels and activities of cholesterol biosynthetic enzymes in the tissues of Pex2–/– mice (Kovacs et al., 2004, 2009). Interestingly, the rate of cholesterol synthesis was significantly decreased in the brain and kidneys of P9 Pex2–/– mice (Kovacs et al., 2004, 2009). The hepatic expression of SREBP-2 target genes was significantly increased both in P0 and postnatal Pex2–/– mice. BA feeding reduced the expression of these genes significantly; yet, the mRNA levels were still increased (Kovacs et al., 2004, 2009, 2012). In addition, we showed that peroxisome deficiency activates hepatic ER stress pathways in Pex2–/– mice, especially the integrated stress response which is mediated by PKR-like endoplasmic reticulum kinase (PERK) and activating transcription factor-4 (ATF4) signaling; hence, we hypothesized that ER stress leads to dysregulation of the endogenous sterol response mechanism and SREBP-2 pathway induction (Kovacs et al., 2009, 2012; Faust and Kovacs, 2014).
Conflicting reports on the role of peroxisomes in cholesterol biosynthesis have been published from in vitro studies using fibroblasts from patients with peroxisomal biogenesis disorders as well as peroxisome-deficient rodent cells [reviewed in Kovacs et al. (2002)]. In summary, activities of peroxisomal cholesterol biosynthetic enzymes were either normal or decreased in fibroblasts of patients with Zellweger spectrum disorders. Three studies employing a total of 24 fibroblast cell lines from patients with disorders of the Zellweger spectrum demonstrated a significantly reduced rate of cholesterol biosynthesis compared to control cells, whereas two studies using a total of seven fibroblast cell lines found that cholesterol biosynthesis rates were similar or higher than those in control fibroblasts. HMGCR activity and the rates of both cholesterol and non-sterol (dolichol) biosynthesis were found to be significantly lower in a study using peroxisome-deficient Pex2–/– hamster cells (ZR-78.1C, ZR-82) (Aboushadi and Krisans, 1998), while in another study using ZR-82 cells, both HMGCR activity and the incorporation of [3H]acetate into sterols (cholesterol + lanosterol) were increased compared to wild-type CHO-K1 cells (van Heusden et al., 1992). However, in vitro studies have not investigated the regulation of cholesterol biosynthesis in peroxisome-deficient cells.
In this study, we investigated the transcriptional regulation of cholesterol biosynthesis in wild-type CHO-K1 cells, three isogenic peroxisome-deficient CHO cell lines (ZR-78.1C, ZR-82, ZR-87) with mutations in the Pex2 gene, and ZR cells with restored functional peroxisomes after complementation with wild-type Pex2 cDNA. We also determined the rate of cholesterol biosynthesis as well as activities and protein levels of cholesterol biosynthetic enzymes in these cell lines. Finally, we explored mechanisms that might lead to a dysregulation of the endogenous sterol response in peroxisome-deficient CHO cells.
Results
Peroxisome-Deficient CHO Cells
The Pex2 mutant and peroxisome-deficient CHO cell lines (ZR-78.1C, ZR-82, and ZR-87) were isolated from the CHO-K1 cell line used as control in this study (Zoeller and Raetz, 1986; Zoeller et al., 1989). PEX2 is anchored to the peroxisomal membrane by two membrane-spanning segments, with its N- and C-terminal regions exposed to the cytosol (Harano et al., 1999). The C-terminus of PEX2 contains a RING finger (C3HC4) motif. The point mutations of Pex2 in the ZR-78.1C and ZR-82 cell lines have been identified, whereas the mutation of Pex2 in the ZR-87 cell line is not known (Thieringer and Raetz, 1993). In ZR-78.1C nucleotide G at position 737 was mutated to A, resulting in the conversion of a cysteine residue into a tyrosine residue in the RING finger motif. The mutation in ZR-82 cells introduces a stop codon which leads to the translation of a truncated form of the PEX2 protein. This N-terminal fragment constitutes only one-fifth of the entire protein and lacks both membrane-spanning regions.
To assess if a complete or partial absence of peroxisomes exist in our cell lines, an immunofluorescence analysis was performed. The immunofluorescence pattern obtained for acyl-CoA oxidase 1 (ACOX1), a peroxisomal matrix protein involved in peroxisomal fatty acid β-oxidation, showed the characteristic punctuate peroxisomal distribution in CHO-K1 cells (Figure 2A) and a diffuse, cytoplasmic fluorescence in peroxisome-deficient CHO cells (Figure 2A), consistent with mislocalization of ACOX1 to the cytoplasm. A punctuate peroxisomal staining pattern for the peroxisomal membrane proteins PEX14 and ACBD5 was observed in CHO-K1 cells (Figures 2B,C). In peroxisome-deficient CHO cells, PEX14 and ACBD5 were present in less abundant cellular vesicles, consistent with peroxisome membrane ghosts (Figures 2B,C). These findings are consistent with the established function of PEX2 in the import of peroxisomal matrix proteins, but not peroxisomal membrane proteins.
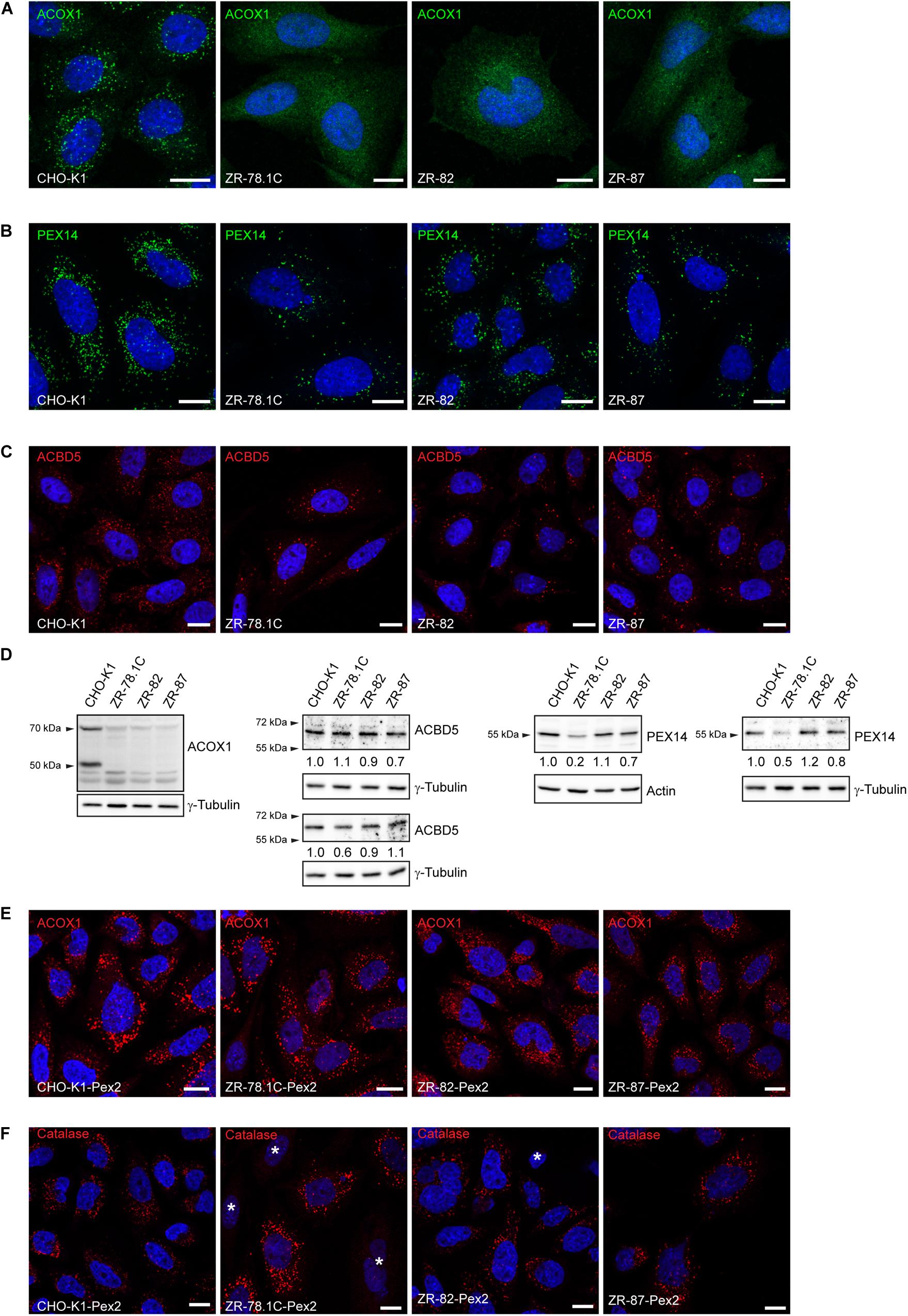
Figure 2. Peroxisome-deficient CHO cells contain peroxisome membraneghosts. (A) Peroxisomes were detected with an antibody against the peroxisomal matrix protein ACOX1. The nuclei were stained with DAPI (blue). Note the cytoplasmic localization of Acox1 in the peroxisome-deficient ZR-78.1C, ZR-82, and ZR-87 cells. (B) Peroxisomes were detected with an antibody against the peroxisomal membrane protein PEX14. Note the presence of peroxisome membrane ghosts in peroxisome-deficient CHO cells. The number of peroxisome membrane ghosts in peroxisome-deficient CHO cells is significantly lower than the number of peroxisomes in CHO-K1 cells. (C) Peroxisomes were visualized with an antibody against the peroxisomal tail-anchored protein ACBD5. Note that ACBD5 localizes to peroxisome membrane ghosts in peroxisome-deficient CHO cells. (D) Immunoblots of total cell lysates with antibodies against peroxisomal matrix and membrane proteins. Numbers at the bottom of the blots indicate the fold change in protein levels in peroxisome-deficient cells relative to that in CHO-K1 cells, which were arbitrarily defined as 1. (E,F) Functional peroxisomes are restored in peroxisome-deficient CHO mutants (ZR-78.1C, ZR-82, ZR-87) upon complementation with rat Pex2 cDNA. Cells were immunostained with antibodies against the peroxisomal matrix proteins ACOX1 (E) and catalase (F). The nuclei were stained with DAPI (blue). An asterisk indicates non-transfected cells without import-competent peroxisomes. The scale bars represent 10 μm.
Acyl-CoA oxidase 1 is synthesized as a precursor protein of 72 kDa and undergoes proteolytic cleavage after import into peroxisomes, resulting in subunits of 52 and 20.5 kDa (Miyazawa et al., 1987). In peroxisome-deficient CHO cells only a slight band of the uncleaved precursor protein was seen (Figure 2D), consistent with lack of peroxisomal protein import and rapid degradation outside of peroxisomes (Schram et al., 1986; Zoeller et al., 1989). Interestingly, the protein levels of ACBD5 were not significantly altered in peroxisome-deficient cells (Figure 2D), whereas PEX14 levels were decreased in ZR-78.1C and ZR-87 cells (Figure 2D).
In order to restore functional peroxisomes, we transfected the peroxisome-deficient CHO cells with wild-type rat Pex2 cDNA and stably selected transfected cells with G418. Transfection with Pex2 restored peroxisomal matrix protein import, since a normal punctate fluorescence pattern was observed with ACOX1 and catalase staining (Figures 2E,F; cf. also Supplementary Figures S1A,B). The peroxisomes of the Pex2-transfected mutant cell lines were morphologically indistinguishable from peroxisomes found in the wild-type CHO-K1 and Pex2-transfected CHO-K1 cells. Hereafter we refer to these transfected cells as ZR-78.1C-Pex2, ZR-82-Pex2, and ZR-87-Pex2.
Transcriptional Activity of Cholesterogenic Promoter-Reporters Is Increased in Peroxisome-Deficient CHO Cells
In order to determine whether sterol-regulated transcription of cholesterogenic genes is affected in peroxisome-deficient CHO cells, luciferase reporter constructs containing the sterol response element (SRE) of SREBP-2 (Srebf2), HMG-CoA reductase (Hmgcr), farnesyl diphosphate synthase (Fdps), or squalene synthase (Fdft1) were transiently transfected into wild-type CHO-KI and peroxisome-deficient ZR-82 and ZR-87 cells. For this cells were incubated in medium supplemented with either 10% FCS or 5% lipoprotein-deficient serum (LPDS), the latter a condition known to upregulate the SREBP-2 pathway. Expression of the Srebf2 and Fdps luciferase reporter genes was increased significantly in all cell lines when incubated in medium with LPDS (Figure 3A), indicating the responsiveness of SREBP to low cholesterol levels. However, Fdps luciferase activity was higher in peroxisome-deficient cells compared with CHO-K1 cells. It is interesting to note that the expression of the Hmgcr, Fdps, and Fdft1 luciferase reporter constructs in peroxisome-deficient cells was significantly increased compared with CHO-K1 even when the cells were incubated in medium containing 10% FCS and therefore sufficient amount of extracellular cholesterol was available (Figure 3A). The expression of the Hmgcr luciferase reporter constructs was only slightly enhanced further in peroxisome-deficient cells incubated in medium with LPDS compared to FCS-cultured cells, whereas the expression was not increased at all in LPDS-cultured CHO-K1 cells. It has been shown that LDL in FCS provides enough cholesterol to suppress SREBP-2 cleavage in wild-type CHO and HEK-293 cells (Hannah et al., 2001; Lee and Ye, 2004). Whereas the expression of the Fdft1 luciferase reporter gene was increased in FCS-cultured peroxisome-deficient cells compared with CHO-K1, culturing all cell lines in medium with 5% LPDS did not increase the expression of the Fdft1 luciferase reporter (Figure 3A).
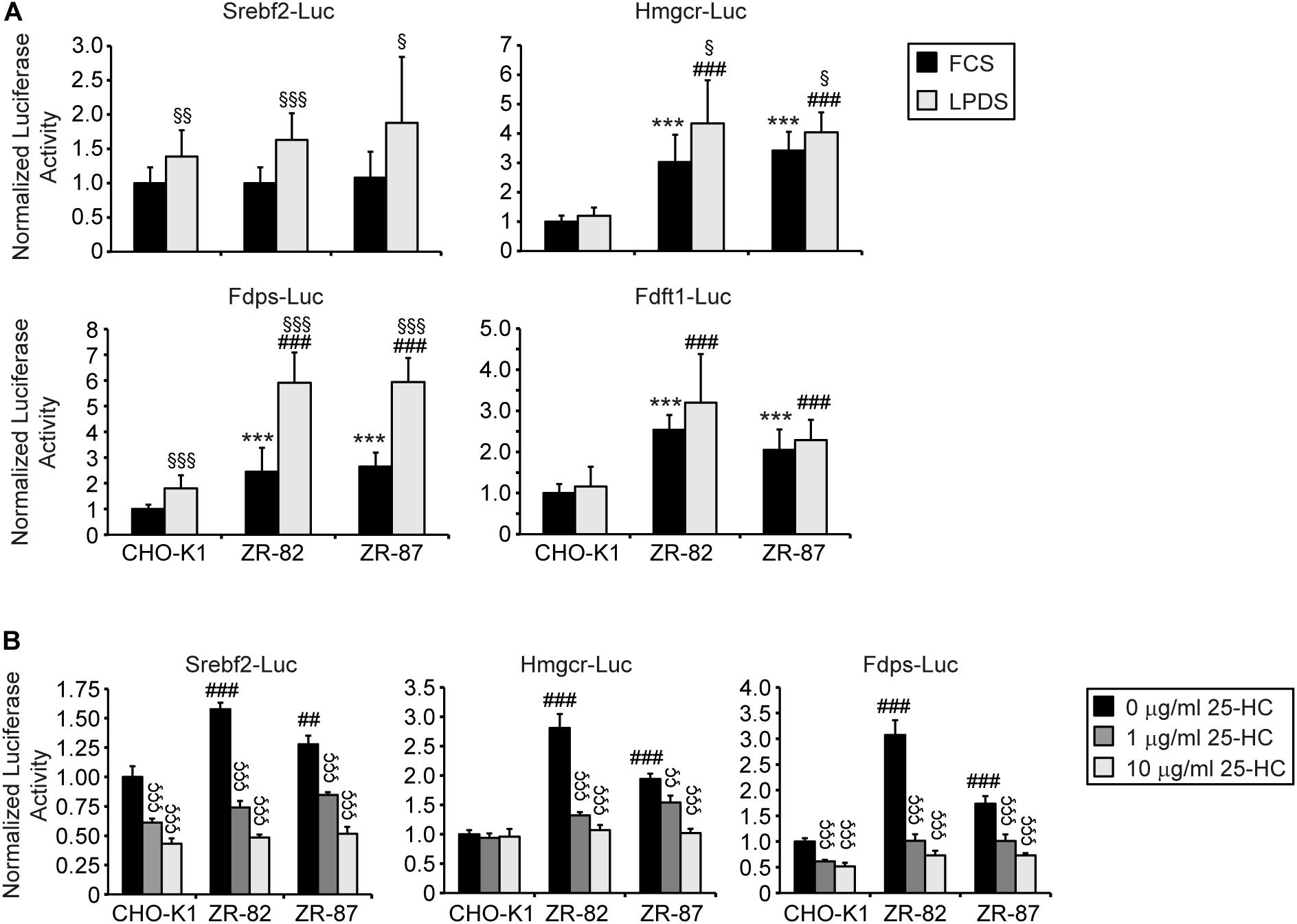
Figure 3. Promoter activities of Srebf2 and the cholesterol biosynthetic genes Hmgcr, Fdps, and Fdft1 in CHO-K1 and peroxisome-deficient CHO cells. (A) The indicated plasmids were transfected into cells as described in Materials and Methods. 24 hours after transfection, the medium was switched to medium containing either 10% FCS or 5% LPDS and incubated for 24 h, after which the cells were harvested and assayed for dual luciferase activities as described in section “Materials and Methods.” (B) The indicated plasmids were transfected into cells and 24 hours after transfection the medium was switched to medium supplemented with 5% LPDS and 0, 1, or 10 μg/ml 25-hydroxycholesterol (25-HC). After 24 h incubation, cells were harvested and assayed for dual luciferase activities. Data are presented as firefly luciferase activity normalized to Renilla luciferase activity and each value represents the mean ± SD of three independent transfection experiments done in quadruplicate. Statistical analysis was performed using Student’s t-test or Student’s t-test with Welch’s correction or one-way ANOVA followed by Dunnett’s multiple comparisons test. *** p < 0.001 vs. FCS-cultured CHO-K1. ##p < 0.01; ###p < 0.001 vs. LPDS-cultured CHO-K1. § p < 0.05; §§ p < 0.01; §§§ p < 0.001 vs. corresponding FCS-cultured cell line. ÇÇ p < 0.01; ÇÇÇ p < 0.001 vs. corresponding cell line incubated in medium containing 5% LPDS without 25-HC.
We then tested whether cholesterogenic promoter activity in peroxisome-deficient cells can be suppressed by incubation with 5% LPDS supplemented with 1 and 10 μg/ml 25-hydroxycholesterol (25-HC) (Figure 3B). Unlike LDL-borne cholesterol, 25-HC added to the culture medium in solvent enters the cell and reaches the ER without first traversing lysosomes (Abi-Mosleh et al., 2009). 25-HC suppressed Srebf2, Hmgcr, and Fdps promoter activity both in CHO-K1 and peroxisome-deficient ZR-82 and ZR-87 cells (Figure 3B) indicating the responsiveness of SREBP to hydroxycholesterols. Taken together, these results show that peroxisome deficiency activates expression of cholesterogenic promoter-reporter genes, even though total cholesterol levels are unchanged when compared to wild-type CHO-K1 cells (Table 1).
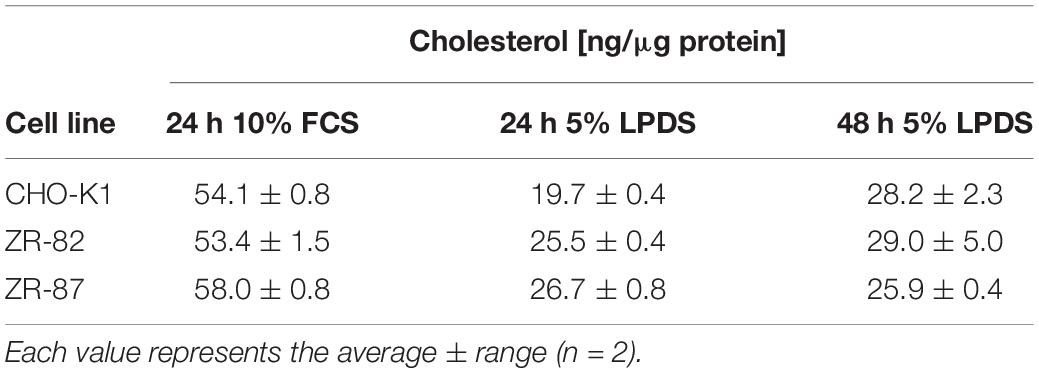
Table 1. Cholesterol levels in CHO-K1 and peroxisome-deficient CHO cells.
Expression of Cholesterol Biosynthetic Genes Is Increased in Peroxisome-Deficient CHO Cells
Next, we determined the mRNA expression of genes involved in cholesterol biosynthesis and their regulation, in cholesterol efflux, and in fatty acid synthesis in CHO-K1 and peroxisome-deficient CHO cells cultured in medium containing 10% FCS or 5% LPDS for 24 h. Again, the expression of Srebf2, Insig1, cholesterol biosynthetic genes (Hmgcs, Hmgcr, Idi1, Fdft1, Sqle, Lss, Dhcr24), Ldlr, and Fasn was significantly increased in all cell lines when incubated in medium containing LPDS (Figure 4A). However, the expression of these genes was much more strongly induced in peroxisome-deficient cells, especially in ZR-82 cells, than in CHO-K1 cells. Importantly, the expression of cholesterol biosynthetic genes and its regulators Srebf2 and Insig1 also was significantly increased in peroxisome-deficient cells compared with CHO-K1 when the cells were incubated in medium containing 10% FCS (Figure 4A; cf. also Supplementary Figure S2). These data corroborate the promoter studies shown in Figure 3. Next, exit of cholesterol from lysosomes requires the membrane-bound Niemann-Pick C1 (NPC1) protein (Kwon et al., 2009); however, we did not detect any changes in Npc1 expression at least in peroxisome-deficient cells (Figure 4A). Hence, although it is well-known that all cholesterol biosynthetic genes are regulated by SREBP-2 (Horton et al., 2002), the magnitude of response in their expression varied significantly among the genes tested, indicating that their levels do not depend on the expression level of SREBP-2 alone.
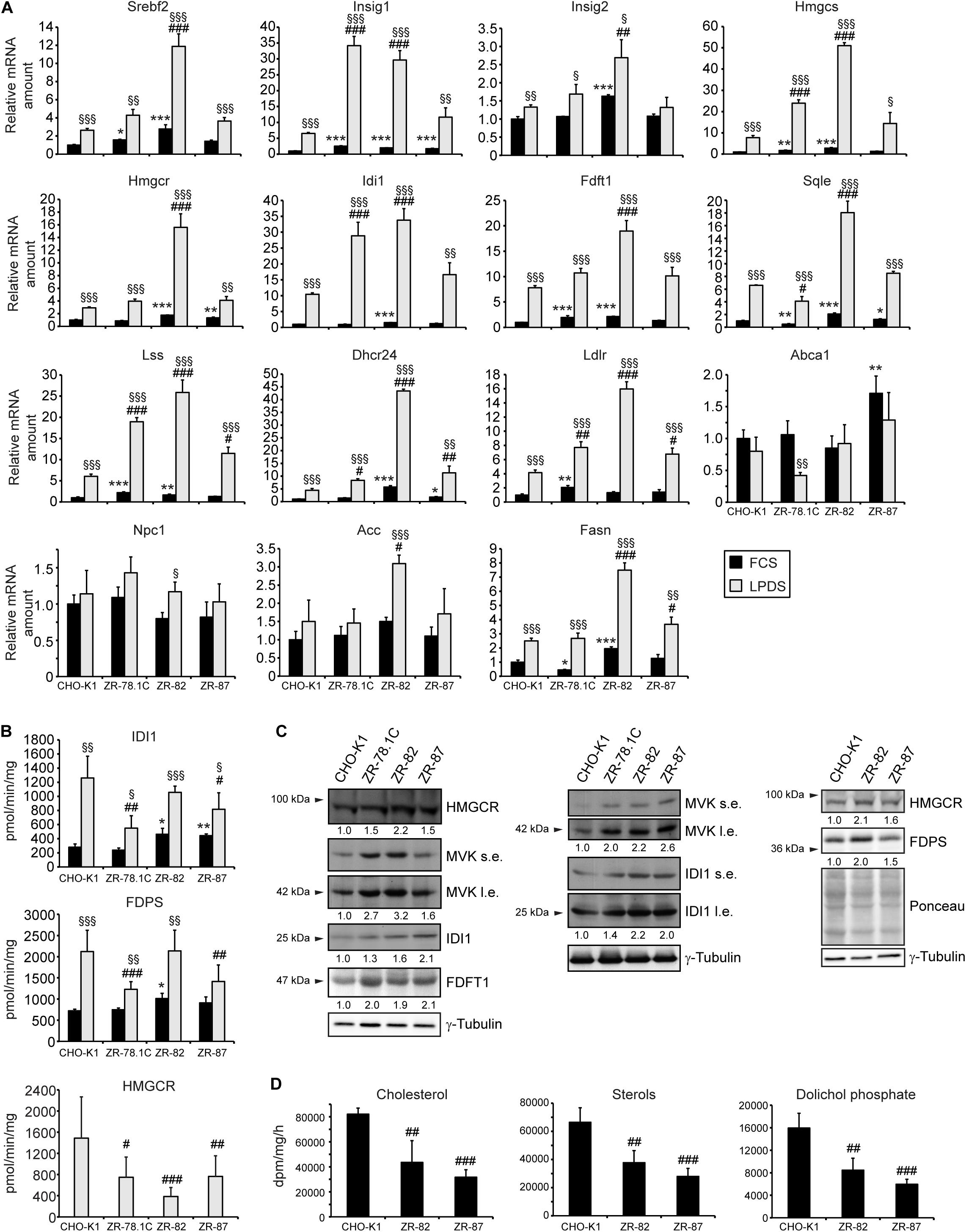
Figure 4. (A) Expression of genes involved in cholesterol biosynthesis and its regulation, cholesterol efflux, and fatty acid synthesis in CHO-K1 and peroxisome-deficient CHO cells cultured in medium containing 10% FCS or 5% LPDS for 24 h. Each value represents the amount of mRNA relative to that in FCS-cultured CHO-K1, which was arbitrarily defined as 1. Data are mean ± SD (n = 3). (B) Activities of the cholesterol biosynthetic enzymes HMGCR, IDI1, and FDPS. Each value represents the average of 10-17 experiments, performed in duplicate. Data are mean ± SD. (C) Immunoblot analysis of cholesterol biosynthetic enzymes HMGCR, MVK, IDI1, FDPS, and squalene synthase (FDFT1) in cells after incubation in medium containing 5% LPDS for 24 h. Numbers at the bottom of the blots indicate the fold change in protein levels in peroxisome-deficient cells relative to that in CHO-K1 cells, which were arbitrarily defined as 1. l.e., long exposure; s.e., short exposure. (D) Rate of biosynthesis of cholesterol, total sterols, and dolichols in CHO-K1 and peroxisome-deficient ZR-82 and ZR-87 cells as measured by the incorporation of [14C]acetate. Data are mean ± SD of three experiments, performed in duplicate. Statistical analysis was performed using Student’s t-test or Student’s t-test with Welch’s correction or one-way ANOVA followed by Dunnett’s multiple comparisons test. *, p < 0.05; **, p < 0.01; ***, p < 0.001 vs. FCS-cultured CHO-K1. #p < 0.05; ##p < 0.01; ###p < 0.001 vs. LPDS-cultured CHO-K1. § p < 0.05; §§ p < 0.01; §§§ p < 0.001 vs. corresponding FCS-cultured cell line.
Activities of Cholesterol Biosynthetic Enzymes and Protein Levels Are Altered in Peroxisome-Deficient CHO Cells
To examine the effect of peroxisome deficiency on the activities of cholesterol biosynthetic enzymes, we measured the activities of HMGCR, IDI1, and FDPS in CHO-K1 and peroxisome-deficient cells (Figure 4B). HMGCR, which catalyzes the rate-limiting step of sterol biosynthesis, is localized mainly in the ER, whereas IDI1 and FDPS are localized predominantly in peroxisomes (Kovacs et al., 2002; Faust and Kovacs, 2014). HMGCR activity was decreased 50% in ZR-78.1C and ZR-87 cells and 75% in ZR-82 cells, corroborating previously published data (Aboushadi and Krisans, 1998). The activity of IDI1 and FDPS was similar or increased in FCS-cultured peroxisome-deficient cells compared with CHO-K1 cells. IDI1 and FDPS activities were significantly increased in all cell lines when incubated in medium containing LPDS, however, the activities were somewhat lower in peroxisome-deficient cells compared with CHO-K1 cells.
Western blot analysis of proteins involved in cholesterol biosynthesis was performed to determine whether the measured activities are a reflection of the protein levels. The protein levels of HMGCR, mevalonate kinase (MVK), IDI1, FDPS, and FDFT1 were significantly increased in peroxisome-deficient cells compared with CHO-K1 (Figure 4C). Thus, decreased HMGCR and IDI1 activities in the peroxisomal-deficient cells are not a reflection of their protein levels, however, gene expression data gave reason to expect even higher protein levels of cholesterol biosynthetic genes in peroxisome-deficient cells.
Rates of Cholesterol and Dolichol Biosynthesis Are Decreased in Peroxisome-Deficient Cells
To evaluate the effects of the decrease in HMGCR, IDI1, and FDPS activities in peroxisome-deficient cells, we measured the rate of sterol (cholesterol) and non-sterol (dolichols) biosynthesis in CHO-K1, ZR-82, and ZR-87 cells after incubating the cells with [14C]acetate. The rates of cholesterol and dolichol biosynthesis were significantly reduced in the peroxisomal-deficient cells as compared to the CHO-K1 cells (Figure 4D and Supplementary Table S1). These data corroborate a previously published study showing that cholesterol and dolichol biosynthesis rates were decreased in ZR-78.1C and ZR-82 cells when incubated with either [3H]acetate or [3H]mevalonate (Aboushadi and Krisans, 1998). However, despite decreased biosynthesis rates, total cholesterol levels were similar in CHO-K1 and peroxisome-deficient cells cultured in medium containing either 10% FCS or 5% LPDS for 24 and 48 h (Table 1).
U18666A Treatment Activates the SREBP-2 Pathway in CHO-K1 and Peroxisome-Deficient CHO Cells
A recent study suggested that peroxisomes play a critical role in the transport of LDL-derived cholesterol from the lysosome to the plasma membrane and other intracellular compartments via a lysosome-peroxisome membrane contact site (Chu et al., 2015). Knockdown of ABCD1, PEX1, and PEX26 in HeLa cells using shRNAs led to accumulation of cholesterol in lysosomes to a similar extent as in cells from Niemann-Pick disease type C (NPC) patients. Therefore, we aimed to determine the effect of U18666A on the SREBP-2 pathway in CHO-K1 and peroxisome-deficient cells. The amphiphilic compound U18666A has been shown to bind to Niemann-Pick C1 (NPC1) and inhibit cholesterol export from lysosomes (Liscum and Faust, 1989; Liscum, 1990; Lu et al., 2016), leading to SREBP-2 activation (Colgan et al., 2007). If peroxisome deficiency leads to an accumulation of cholesterol in lysosomes (Chu et al., 2015), U18666A should activate the SREBP-2 pathway only in CHO-K1 cells, but should not lead to further activation in ZR-78.1C, ZR-82, and ZR-87 cells. The expression of SREBP-2 target genes was determined in cells that were grown in cholesterol-containing medium (10% FCS) supplemented with 10 μM U18666A for 24 h. U18666A significantly increased the mRNA levels of Srebf2, Insig1, and cholesterol biosynthetic genes both in CHO-K1 and peroxisome-deficient cells compared with vehicle-treated cells (Figure 5A). The increase was similar in CHO-K1, ZR-78.1C, and ZR-87 cells, whereas ZR-82 cells again showed a much stronger increase, even though with a larger standard deviation. U18666A treatment significantly decreased the expression of the cholesterol efflux transporter Abca1 (ATP-binding cassette transporter A1) in all cell lines (Figure 5A). The addition of 10 μg/ml 25-HC significantly suppressed the activation of SREBP-2 target genes in U18666A-treated cells (Figure 5A), indicating that the sensitivity towards 25-HC is functional in peroxisome-deficient cells.
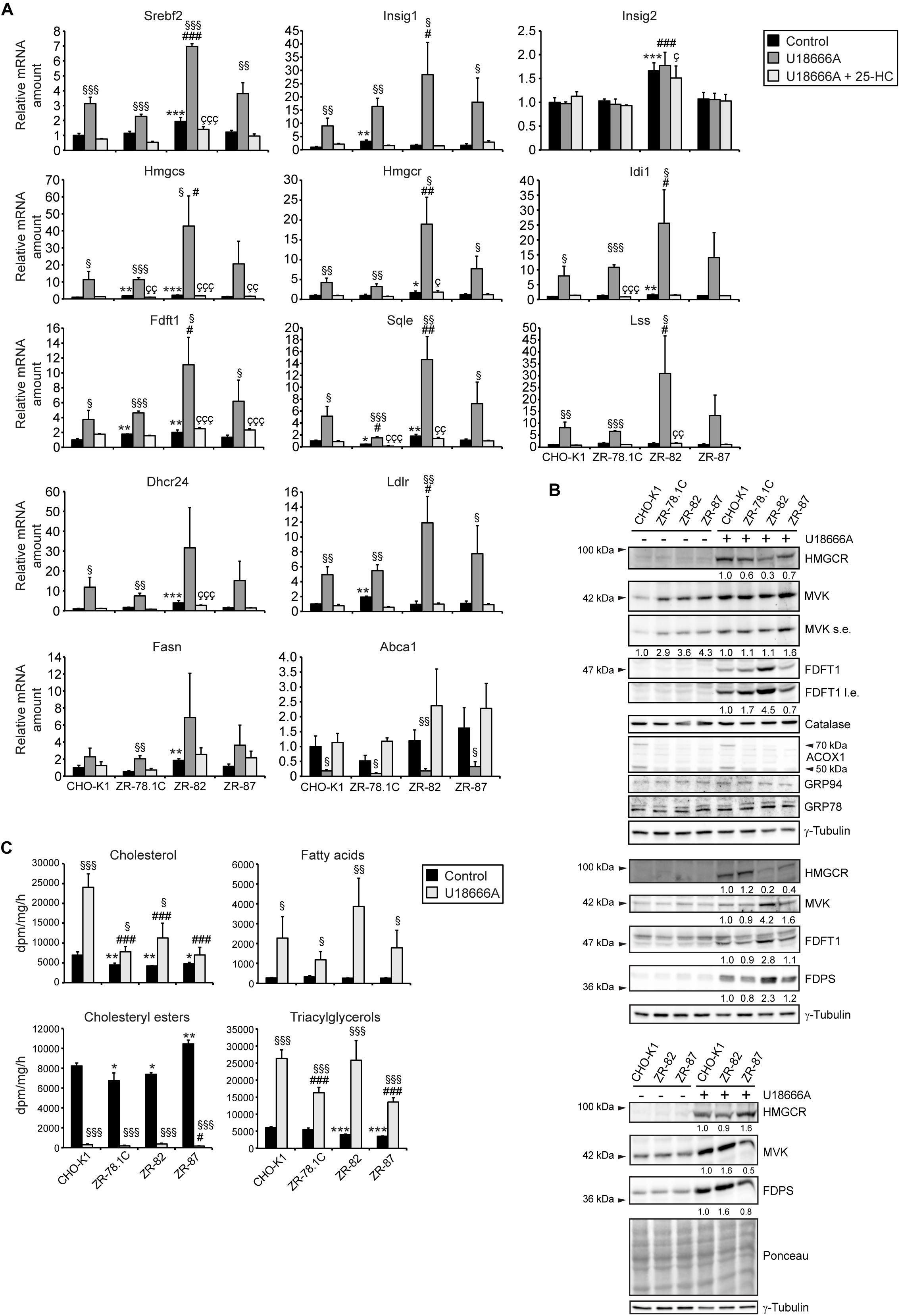
Figure 5. (A) Expression of genes involved in cholesterol biosynthesis and its regulation, cholesterol efflux, and fatty acid synthesis in CHO-K1 and peroxisome-deficient CHO cells cultured in medium containing 10% FCS and 0.1% EtOH as solvent control or 10 μM U18666A or 10 μM U18666A and 10 μg/ml 25-hydroxycholesterol (25-HC) for 24 h. Each value represents the amount of mRNA relative to that in control CHO-K1, which was arbitrarily defined as 1. Data are mean ± SD (n = 3). (B) Immunoblots of total cell lysates with antibodies against cholesterol biosynthetic enzymes (HMGCR, MVK, FDPS, and FDFT1), peroxisomal (catalase, ACOX1) and ER (GRP78, GRP94) proteins after treatment with 10 μM U18666A for 24 h. Shown are immunoblots from three experiments. (C) Rate of biosynthesis of cholesterol, cholesteryl esters, fatty acids, and triacylglycerols in vehicle- and U18666A-treated CHO-K1 and peroxisome-deficient ZR-78.1C, ZR-82, and ZR-87 cells as measured by the incorporation of [14C]acetate. Lipids were separated by thin-layer chromatography after [14C]acetate labeling. Data are mean ± SD (n = 3–6). Statistical analysis was performed using Student’s t-test or Student’s t-test with Welch’s correction or one-way ANOVA followed by Dunnett’s multiple comparisons test. *p < 0.05; **p < 0.01; ***p < 0.001 vs. vehicle (EtOH)-treated CHO-K1. #p < 0.05; ##p < 0.01; ###p < 0.001 vs. U18666A-treated CHO-K1. Çp < 0.05; ÇÇ p < 0.01; ÇÇÇ p < 0.001 vs. U18666A- and 25-HC-treated CHO-K1. § p < 0.05; §§ p < 0.01; §§§ p < 0.001 vs. corresponding vehicle-treated cell line.
U18666A treatment increased the protein levels of the cholesterol biosynthetic enzymes MVK, FDPS, and FDFT1 in all cell lines compared with vehicle-treated cells (Figure 5B). Interestingly, despite highly elevated mRNA levels, HMGCR protein levels in U18666A-treated ZR-82 cells were lower compared with CHO-K1, ZR-78.1C, and ZR-87 cells, whereas the FDPS and FDFT1 protein levels were higher in ZR-82 cells. The protein levels of catalase and the ER proteins GRP78 and GRP94 were similar in CHO-K1 and peroxisome-deficient cells.
To assess levels of de novo lipogenesis upon U18666A treatment we measured total de novo lipid production by quantifying incorporation of radioactively labeled [14C]acetate into lipids. Cholesterol synthesis was significantly decreased in vehicle-treated peroxisome-deficient cells compared with CHO-K1 cells (Figure 5C). U18666A treatment significantly increased the rate of cholesterol biosynthesis in all cell lines, however, cholesterol biosynthesis was significantly lower in peroxisome-deficient cells compared with CHO-K1 cells (Figure 5C). U18666A treatment significantly decreased as expected the rate of cholesteryl ester synthesis in all cell lines, whereas the rates of fatty acid and triacylglycerol synthesis were increased (Figure 5C). Interestingly, the rate of cholesteryl ester synthesis was similar in all cell lines cultivated in FCS, suggesting that they have a similar supply of cholesterol in the ER. In summary, U18666A induced mRNA and protein levels of cholesterol biosynthetic enzymes in peroxisome-deficient cells, however, again the rates of cholesterol synthesis were significantly lower than in CHO-K1 cells.
Expression of SREBP-2 Target Genes in Extrahepatic Tissues of SW/129 Pex2–/– Mice
We reported previously that total cholesterol levels were similar in the lung, kidney, heart, and spleen of P10 SW/129 Pex2–/– mice when compared with control mice, whereas the rate of cholesterol synthesis was significantly increased in the lung, heart, and spleen and decreased in the kidney (Kovacs et al., 2004, 2009). As total cholesterol levels were similar in P10 SW/129 Pex2–/– and control mice, we examined the expression of Srebf2, Insig1, Insig2, and cholesterol biosynthetic genes (Hmgcr, Pmvk, Mvd, Idi1, Fdps, Fdft1, Sqle, Sc4mol, Lss) in these tissues as well as skeletal muscle. Indeed, the mRNA levels of all these genes were significantly increased in Pex2–/– mice (Figures 6A–E). In summary, there is a very similar mRNA up-regulation of Srebf2 and its target genes in extrahepatic tissues compared with livers of P0 and postnatal Pex2–/– mice (Kovacs et al., 2004, 2009, 2012). Taken together, these data corroborate our findings in CHO cell lines, which indicate that functional peroxisomes are necessary for efficient cholesterol biosynthesis.
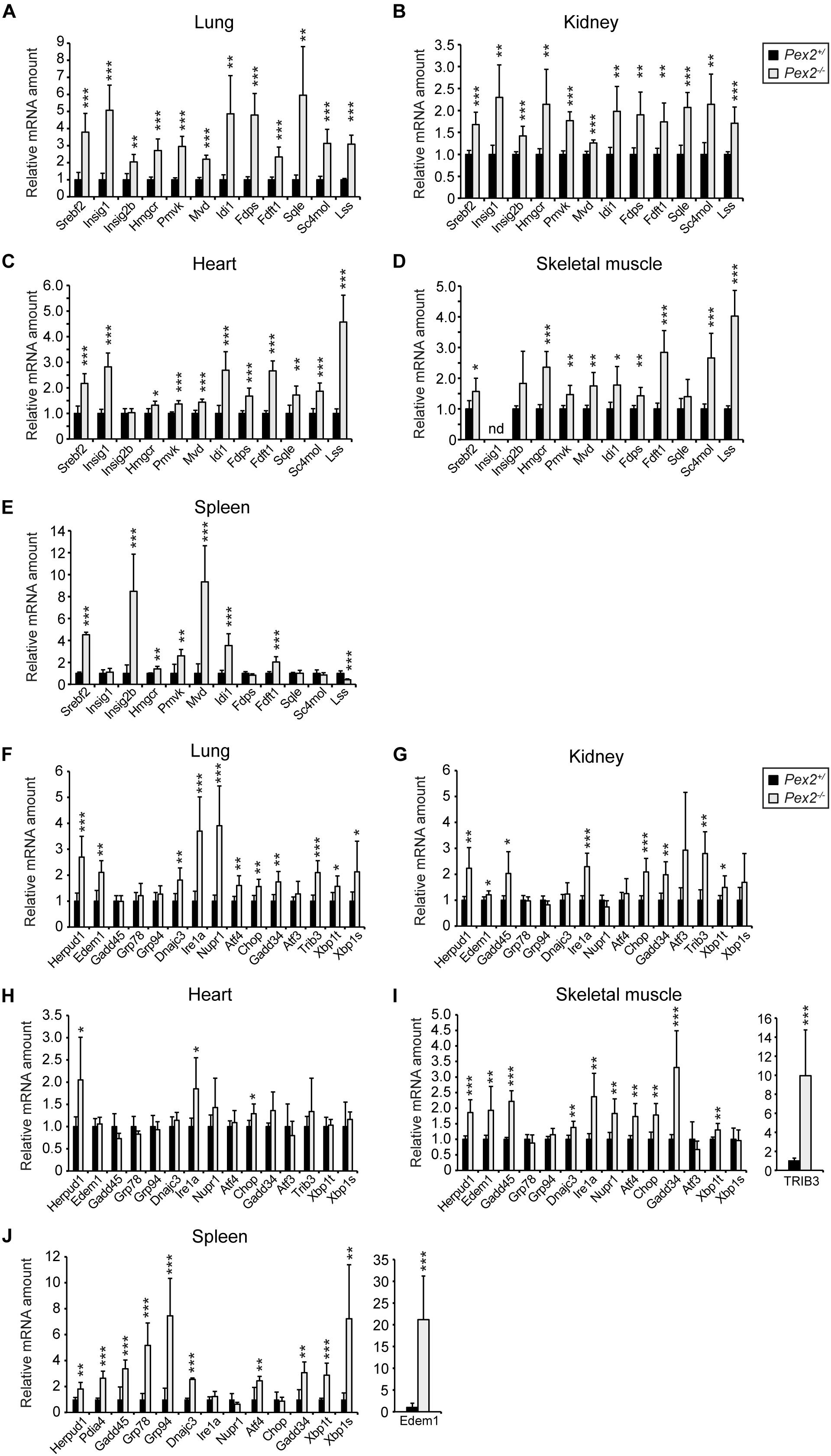
Figure 6. Expression of SREBP-2 regulated genes in lung (A), kidney (B), heart (C), skeletal muscle (D), and spleen (E) from P10 SW/129 control and Pex2–/– mice. (F–J) Expression of UPR target genes in lung (F), kidney (G), skeletal muscle (H), heart (I), and spleen (J) from P10 SW/129 control and Pex2–/– mice. Each value represents the amount of mRNA relative to that in control mice, which was arbitrarily defined as 1. Data are mean ± SD (n = 6 for control and Pex2–/– mice). Statistical analysis was performed using Student’s t-test or Student’s t-test with Welch’s correction. *p < 0.05; **p < 0.01; ***p < 0.001 vs. control mice. nd, not detected.
ER Stress Does Not Activate the SREBP-2 Pathway in CHO-K1 Cells and Peroxisome-Deficient CHO Cells
We showed that peroxisome deficiency activates hepatic ER stress pathways, especially the integrated stress response mediated by PERK and ATF4 signaling, in livers of newborn and postnatal Pex2–/– mice (Kovacs et al., 2009, 2012). Studies have shown that ER stress induces SREBP-2 activation independently of intracellular cholesterol concentration in various cell lines (Werstuck et al., 2001; Lee and Ye, 2004; Colgan et al., 2007; Lhoták et al., 2012). The analysis of the mRNA expression of several unfolded protein response (UPR) target genes in extrahepatic tissues of P10 Pex2–/– mice showed that ER stress is also present in the lung, kidney, skeletal muscle, heart, and spleen (Figures 6F–J). To determine whether ER stress is also present in peroxisome-deficient CHO cells, we examined the mRNA expression of several UPR target genes in cells cultured in medium containing 10% FCS or 5% LPDS for 24 h (Figure 7A). The expression of most UPR target genes was, with the exception of Chop (C/EBP homologous protein), somewhat similar in CHO-K1 and peroxisome-deficient cells cultured in FCS- or LPDS-containing medium. Chop was significantly increased in all peroxisome-deficient cells compared with CHO-K1 when grown in medium containing 10% FCS. Culturing cell in 5% LPDS increased the expression of Xbp1t (total) and Xbp1u (unspliced) in all cell lines, however, the expression levels of the active transcription factor Xbp1s (spliced) were very low (CT > 38) and could not be analyzed. Interestingly, Xbp1t and Xbp1u mRNA levels were increased in peroxisome-deficient CHO cells, but the expression of Xbp1s was again too low to be evaluated.
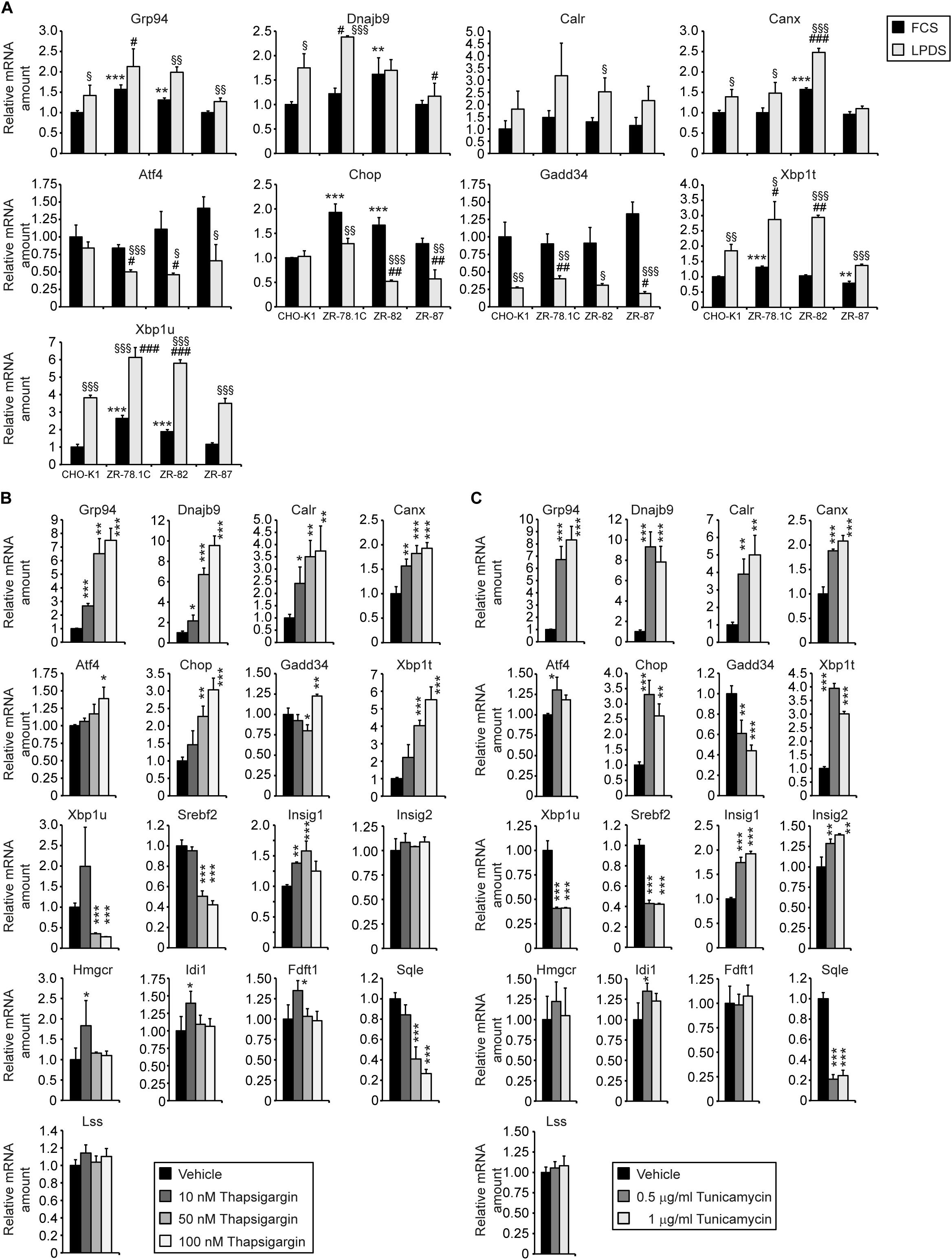
Figure 7. ER stress does not induce the SREBP-2 pathway in CHO-K1 cells. (A) Expression of UPR target genes in CHO-K1 and peroxisome-deficient CHO cells cultured in medium containing 10% FCS or 5% LPDS for 24 h. Each value represents the amount of mRNA relative to that in FCS-cultured CHO-K1, which was arbitrarily defined as 1. Data are mean ± SD (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001 vs. FCS-cultured CHO-K1. #p < 0.05; ##p < 0.01; ###p < 0.001 vs. LPDS-cultured CHO-K1. § p < 0.05; §§ p < 0.01; §§§ p < 0.001 vs. corresponding FCS-treated cell line. (B,C) CHO-K1 cells were cultured in medium containing 10% FCS and DMSO as solvent control or various concentrations of thapsigargin (B) and tunicamycin (C) for 18 h. Each value represents the amount of mRNA relative to that in control CHO-K1, which was arbitrarily defined as 1. Data are mean ± SD (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001 vs. solvent (DMSO)-treated CHO-K1. Statistical analysis was performed using Student’s t-test or Student’s t-test with Welch’s correction or one-way ANOVA followed by Dunnett’s multiple comparisons test.
Next, we investigated whether induction of ER stress activates the SREBP-2 pathway in CHO-K1 cells. CHO-K1 cells were treated with various concentrations of the ER stress inducers thapsigargin (Tg), an inhibitor of the ER Ca2+-dependent ATPase, or tunicamycin (Tm), an inhibitor of protein N-glycosylation (the latter resulting in misfolding of proteins that normally would be glycosylated). Quantitative real-time PCR analysis revealed that Tg and Tm induced a significant dose-dependent increase in mRNA levels of various ER stress markers (Grp94, Dnajb9, Calr, Canx, Atf4, Chop, Gadd34, Xbp1t), however, the expression of SREBP-2 target genes was in general not induced in Tg- or Tm-treated CHO-K1 cells (Figures 7B,C). The mRNA levels of Srebf2 and Sqle were decreased in Tg- and Tm-treated cells, and only Insig1 expression was increased.
In summary, the increased expression of cholesterol biosynthetic genes in peroxisome-deficient CHO cells cannot be ascribed to ER stress, and activation of ER stress in CHO-K1 cells does not lead to an activation of the SREBP-2 pathway.
mTORC1 and MAPK Pathways Do Not Activate the SREBP-2 Pathway in Peroxisome-Deficient CHO Cell
Next, we investigated whether activated mTORC1 (mechanistic target of rapamycin complex 1) triggers activation of the SREBP-2 pathway in peroxisome-deficient cells. mTORC1 has been shown to activate SREBP-2 in mammalian cells through several mechanisms, including, at least in some cell types, through p70 ribosomal S6 kinase (p70 S6K) (Peterson et al., 2011; Wang et al., 2011; Eid et al., 2017). The only kinase known to be activated directly by mTORC1 is p70 S6K, which subsequently activates ribosomal S6 protein (S6) (Laplante and Sabatini, 2012). We assessed mTORC1 activity by analyzing the phosphorylation of S6 and p70 S6K in lysates from CHO-K1 and peroxisome-deficient cells treated with U18666A or cultured in the presence of FCS or LPDS for 24 h (Figures 8A–C). Phosphorylation of S6 on Ser235/236 was decreased in peroxisome-deficient cells compared with CHO-K1 in all three culture conditions. Phosphorylation of p70 S6K on Thr421/Ser424 was decreased in ZR-82 cells, while it tended to be decreased in ZR-78.1C and increased in ZR-87 cells. These data suggest that mTORC1 activity is inherently lower in peroxisome-deficient cell lines. Importantly, in accordance with our data, a recent study suggested that acetyl-CoA derived from peroxisomal β-oxidation promotes Raptor acetylation and mTORC1 activation in hepatocytes, and hepatic Acox1 deficiency resulted in inhibition of mTORC1 (He et al., 2020).
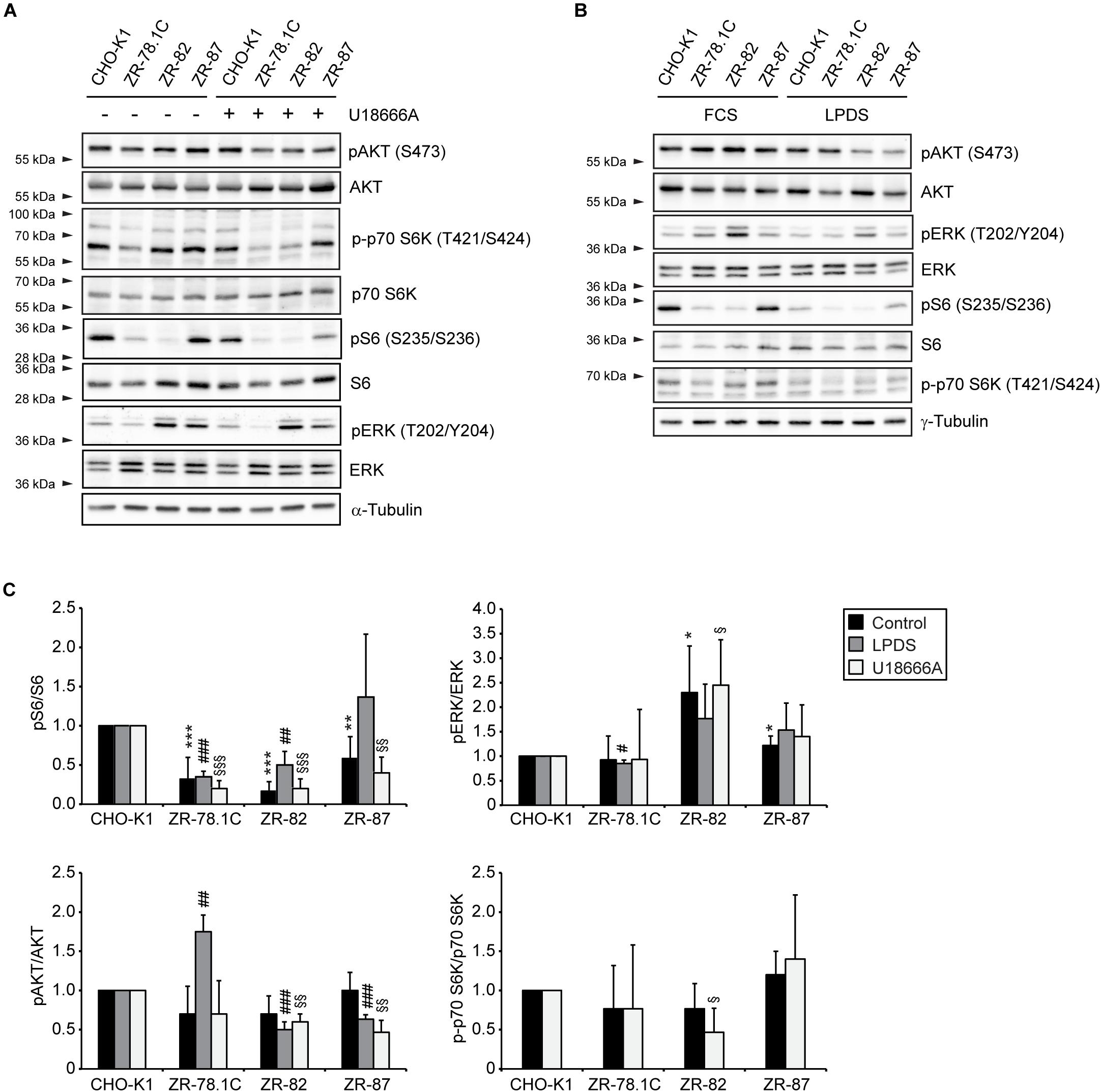
Figure 8. mTORC1, AKT, and MAPK signaling in peroxisome-deficient CHO cells. (A,B) Immunoblot analysis of the mTORC1, AKT, and MAPK pathways of total cell lysates after treatment with 10 μM U18666A (A) or incubation in medium supplemented with 5% LPDS (B) for 24 h. Western blots from a representative of 2–3 independent experiments are shown in panels A,B. (C) For quantification, pAKT, pS6, pERK, and p-p70 S6K levels were normalized to the corresponding total AKT, S6, ERK, and p70 S6K levels, respectively. Protein ratios are expressed relative to that in CHO-K1 cells, which were arbitrarily defined as 1 for each treatment. Data are mean ± SD (n = 2–3). Statistical analysis was performed using Student’s t-test or Student’s t-test with Welch’s correction. ***p < 0.001 vs. FCS-cultured CHO-K1. #p < 0.05; ##p < 0.01; ###p < 0.001 vs. LPDS-cultured CHO-K1. § p < 0.05; §§ p < 0.01; §§§ p < 0.001 vs. U18666A-treated CHO-K1.
The phosphatidylinositol 3-kinase (PI3K)/AKT pathway is involved in SREBP-2 transport to the Golgi and thereby contributes to the control of SREBP-2 activation (Du et al., 2006). To compare AKT phosphorylation in CHO-K1 and peroxisome-deficient cells, immunoblot analyses were performed on lysates from cells treated with U18666A or cultured in FCS and LPDS for 24 h (Figures 8A–C). AKT phosphorylation on Ser473 was similar in FCS-cultured cells and slightly decreased in LPDS- or U18666A-treated ZR-82 and ZR-87 cells compared with CHO-K1 cells. Phosphorylation of AKT was only increased in LPDS-cultured ZR-78.1C cells.
Major signaling pathways that couple transcription factors to extracellular stimuli include the mitogen-activated protein (MAP) kinase cascades through extracellular signal-regulated kinase (ERK1/2). It has been shown that the mature form of SREBP-2 is a substrate of ERK-MAPK in vitro, affecting its trans-activity (Kotzka et al., 2000, 2004). In order to examine a possible link between the MAPK pathway and increased SREBP-2 activity and expression of cholesterol biosynthetic genes, we determined whether the phosphorylation of p44/42 ERK was affected in peroxisome-deficient CHO cells. We measured the amounts of total and phosphorylated ERK proteins in cells treated with U18666A or cultured in the presence of FCS or LPDS for 24 h (Figures 8A–C). Total ERK levels were similar in CHO-K1 and peroxisome-deficient cells; however, phosphorylated ERK levels were equally increased in ZR-82 cells under all three culture conditions.
We conclude that the activities of the PI3K/AKT and mTORC1 pathways are not, in general, increased in peroxisome-deficient cells, and also are not the cause of the observed increased expression of cholesterol biosynthetic genes. That said, however, phosphorylated ERK could contribute to higher activity of SREBP-2 in ZR-82 cells.
Increased Transfer of GFP-SCAP From the ER to the Golgi in Peroxisome-Deficient CHO Cells
SCAP, the sterol sensor, plays an essential role in SREBP-2 activation by mediating its ER-to-Golgi transport. To test if peroxisome deficiency enhances ER-to-Golgi trafficking of SCAP/SREBP, we analyzed SCAP trafficking in CHO-K1 and peroxisome-deficient CHO cells using a GFP-SCAP fusion protein, which allows determination of the subcellular distribution of GFP-SCAP by confocal microscopy. Cells stably expressing GFP-SCAP were cultured in medium either containing 10% FCS or 5% LPDS or, alternatively, 10% FCS and 10 μM U18666A (Figures 9A–D). GFP-SCAP showed a reticular ER pattern in CHO-K1 cells grown in cholesterol-containing medium. Incubating cells in medium with 5% LPDS for 24 h or treatment with U18666A for 12 h led to increased juxtanuclear staining and co-immunolocalization with giantin (also known as GOLGB1), a Golgi marker (Linstedt and Hauri, 1993), in addition to the reticular ER pattern (Figure 9A). Peroxisome-deficient ZR-78.1C and ZR-87 cells showed already an increased juxtanuclear staining when cultured in medium containing 10% FCS; incubating cells in medium containing 5% LPDS or treatment with U18666A led to a strong colocalization of GFP-SCAP with giantin (Figures 9B,D). In ZR-82 cells GFP-SCAP localized independently from the culture conditions only to the Golgi (Figure 9C). In general, the data show that deficiency of functional peroxisomes promotes GFP-SCAP trafficking to the Golgi.
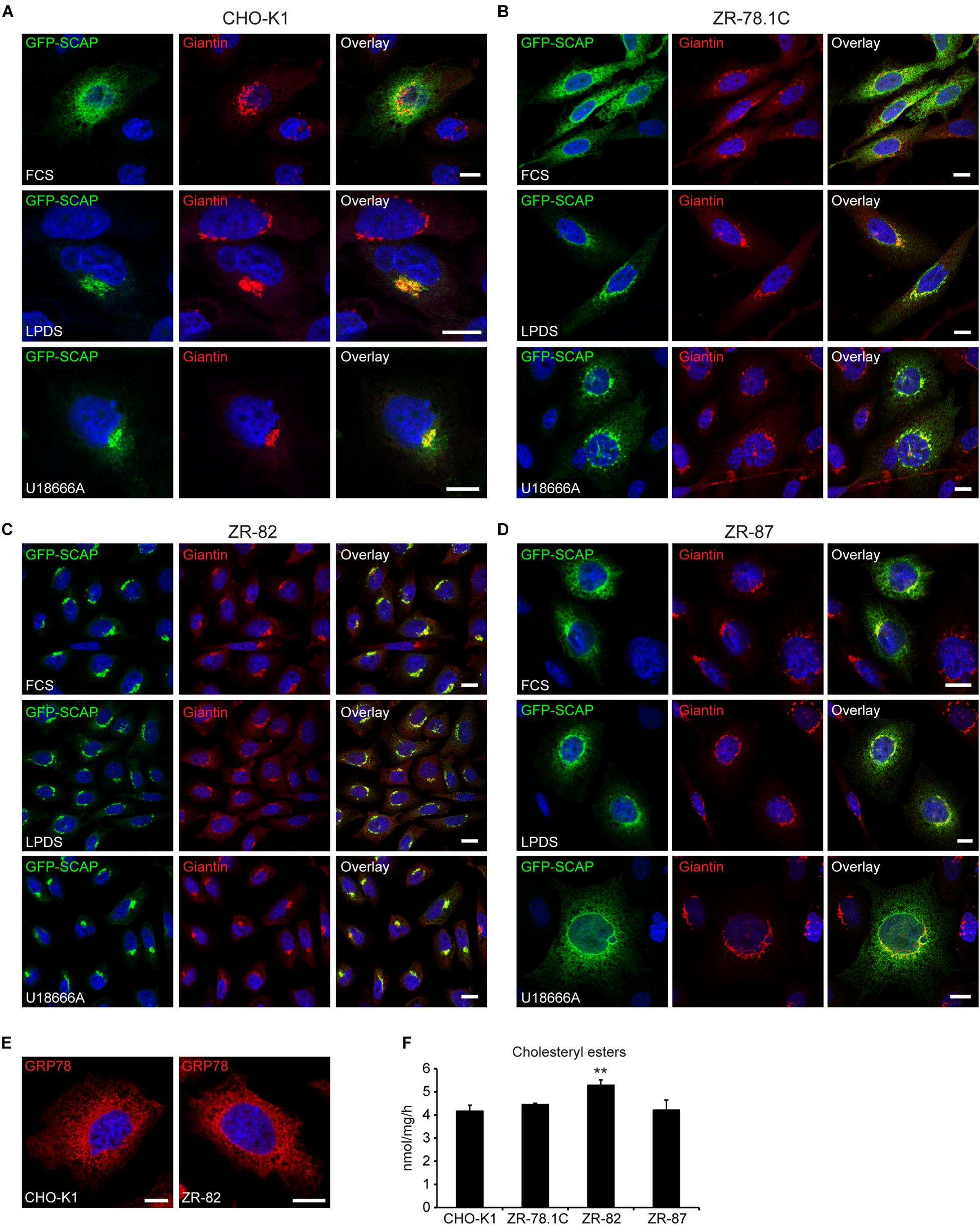
Figure 9. Peroxisome deficiency promotes ER-to-Golgi trafficking of GFP-SCAP. Confocal microscopy images show GFP-SCAP trafficking related to the Golgi marker giantin in CHO-K1 (A), ZR-78.1C (B), ZR-82 (C), and ZR-87 (D) cells. Cells were either cultured in 10% FCS or 5% LPDS for 24 h or treated with 10 μM U18666A for 12 h. GFP-SCAP was imaged using a chicken polyclonal anti-GFP antibody followed by Alexa488 secondary antibody; Golgi was visualized by rabbit anti-giantin followed by Alexa594 secondary antibody. The nuclei were stained with DAPI (blue). Representative fields are shown for each condition. The scale bars represent 10 μm. (E) The endoplasmic reticulum was detected with an antibody against GRP78. The scale bars represent 10 μm. (F) Cholesterol esterification in CHO-K1 and peroxisome-deficient cells. Cells cultured in 10% FCS were incubated for 4 h with [14C]palmitate (∼8000 dpm/nmol). Cholesterol ester levels were analyzed by TLC. Data are mean ± SD (n = 3). Statistical analysis was performed using one-way ANOVA followed by Dunnett’s multiple comparisons test. **p < 0.01 vs. FCS-cultured CHO-K1.
To exclude the possibility that the altered GFP-SCAP localization in ZR-82 cells cultured in medium containing 10% FCS was caused by general changes in ER morphology, we stained the ER with an antibody against the glucose-regulated protein 78 (GRP78). However, the morphology of the ER was comparable in CHO-K1 and ZR-82 cells (Figure 9E).
Radhakrishnan et al. (2008) determined the concentration of cholesterol in the ER that is sufficient to retain the SCAP-SREBP-2 complex in the ER and suppress SREBP-2 processing. They showed that SCAP transported SREBP-2 from the ER to the Golgi when ER cholesterol was ≤5 mol% of ER lipids, and ER-to-Golgi transport of SCAP was inhibited when the cholesterol content of the ER rose above the sharp 5% threshold. An open question is whether the cholesterol level in the ER is reduced in peroxisome-deficient cells, which could explain the increased ER-to-Golgi trafficking of SCAP and SREBP-2 activation in peroxisome-deficient cells even when total cholesterol levels are comparable to control cells. Therefore, we determined the rate of cholesterol esterification as a surrogate marker for ER cholesterol levels (Figure 9F). The enzyme acyl-coenzyme A:cholesterol acyltransferase (SOAT) is located in the ER and synthesizes cholesteryl esters, using cholesterol and fatty acyl-CoA as its substrates (Chang et al., 2006). Two Soat genes have been identified in mammals (Soat1 and Soat2), and SOAT1 is the major isoenzyme in CHO cells. Assuming that SOAT is located in the ER and that its activity is controlled by cholesterol availability, SOAT activity in intact cells has been used to monitor cholesterol arriving at the ER in cholesterol trafficking studies (Chang et al., 2006). We found that the esterification of cholesterol was similar in CHO-K1 cells and peroxisome-deficient cell lines (Figure 9F), suggesting that ER cholesterol levels are normal in peroxisome-deficient cells.
To exclude the possibility that the altered GFP-SCAP localization in ZR-82 cells cultured in medium containing 10% FCS was caused by general changes in ER morphology, we examined the localization of activating transcription factor 6 (ATF6) in CHO-K1 and ZR-82 cells. ATF6 resides as a transcriptionally inactive membrane-bound precursor protein in the ER by its interaction with GRP78. In response to ER stress and dissociation of GRP78, ATF6 is trafficked from the ER to the Golgi where it is sequentially cleaved by the proteases S1P and S2P to produce an active transcription factor, a process similar to that involving SREBP-2 (Ye et al., 2000; Chen et al., 2002). However, SCAP is not required for the ER-to-Golgi translocation of ATF6, but a similar molecular escort may bind ATF6 (Ye et al., 2000; Chen et al., 2002). We transfected CHO-K1 and ZR-82 cells with a GFP-ATF6 construct where the S1P site was mutated [GFP-ATF6(S1P-)] (Chen et al., 2002). The S1P cleavage blockade due to the S1P site mutation causes a longer retention of GFP-ATF6 in the Golgi after ER stress-induced ER-to-Golgi transfer (Chen et al., 2002). The cells were analyzed by fluorescence microscopy and the staining of GFP-ATF6(S1P-) in the perinuclear and cytoplasmic regions of FCS-cultured CHO-K1 and ZR-82 cells was consistent with ER localization (Figure 10A). Treatment with thapsigargin (Figure 10B) and tunicamycin (Figure 10C) for 12 h caused the movement of GFP-ATF6(S1P-) to perinuclear locations consistent with a Golgi marker staining pattern. GFP-ATF6(S1P-) colocalized with the Golgi marker giantin in both cell lines in response to ER stress induction (Figures 10B,C), but GFP-ATF6(S1P-) was still detectable in the ER in both cell lines. These data show that the altered SCAP localization in peroxisome-deficient cells is specific for SCAP, while other proteins normally subject to ER-to-Golgi transport were not affected, indicating that defective ER-to-Golgi transport is not a generalized phenomenon.
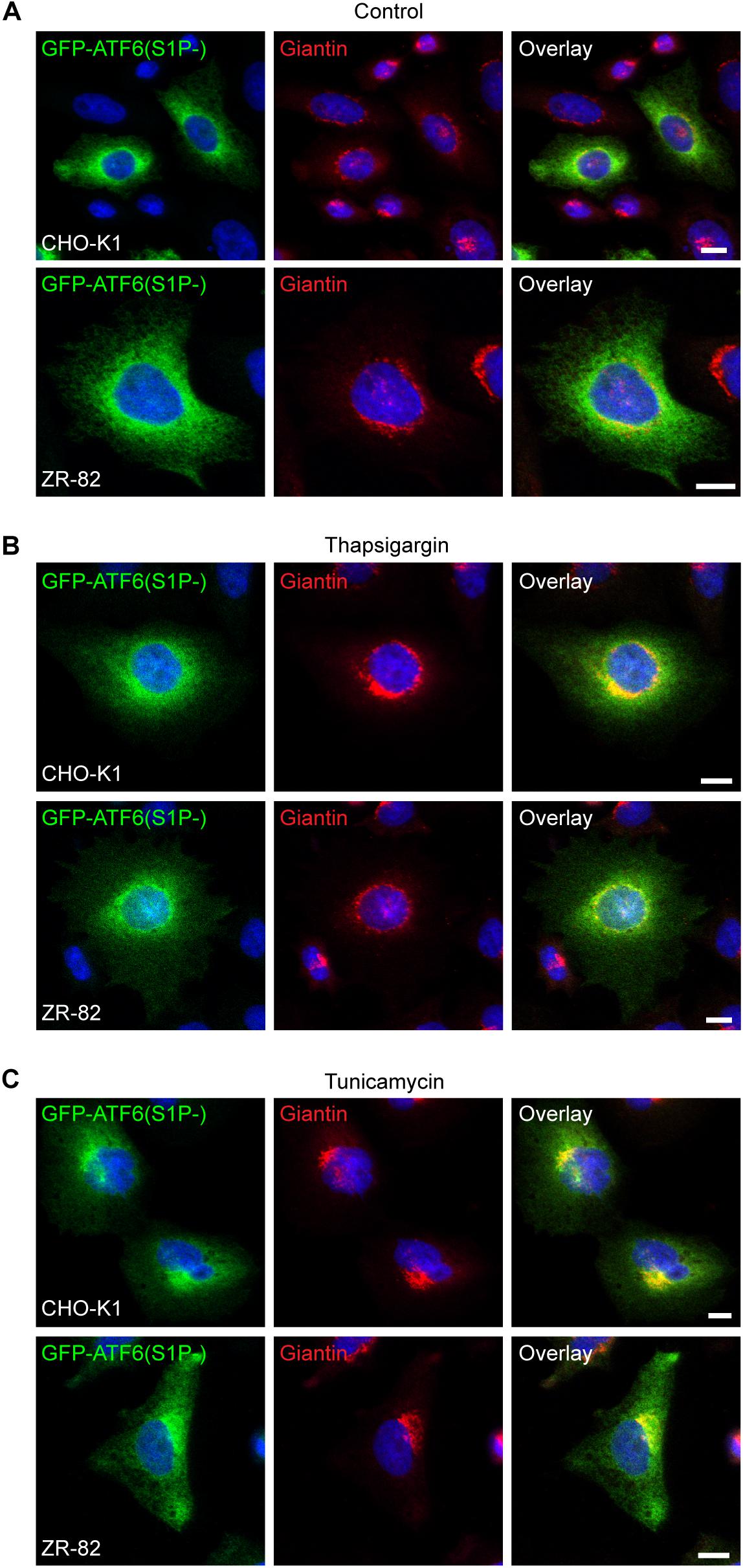
Figure 10. Peroxisome deficiency does not enhance ER-to-Golgi trafficking of GFP-ATF6(S1P-). Confocal microscopy images show GFP-ATF6(S1P-) trafficking related to the Golgi marker giantin in CHO-K1 and ZR-82 cells. Cells were either cultured in 10% FCS (Control) (A) or treated with 100 nM thapsigargin (B) or 0.5 μg/ml tunicamycin (C) for 12 h. GFP-ATF6 was visualized using a chicken polyclonal anti-GFP antibody; the Golgi was visualized by rabbit anti-giantin; DAPI was used for nuclear staining. Representative fields are shown for each condition. The scale bars represent 10 μm.
Normally, SCAP is retrieved from the Golgi and moves back to the ER by COPI-mediated Golgi-to-ER retrograde trafficking after processing of SREBP under sterol-deficient conditions (Takashima et al., 2015). To provide further evidence that SCAP in peroxisome-deficient cells increasingly moves to the Golgi, we used brefeldin A, which causes the relocation of Golgi proteins into the ER (Lippincott-Schwartz et al., 1989; Klausner et al., 1992). GFP-SCAP-transfected ZR-82 cells cultured in medium containing 10% FCS were treated with brefeldin A for 30, 60, and 90 min. GFP-SCAP showed a Golgi-like staining pattern and co-localization with giantin in ZR-82 cells cultured in medium containing 10% FCS (Figure 11A). Even by 30 min, treatment with brefeldin A caused the pattern to disperse and return to a more ER-like pattern (Figure 11A). However, as expected, the staining pattern of the ER marker GRP78 was not affected by brefeldin A treatment (Figure 11B).
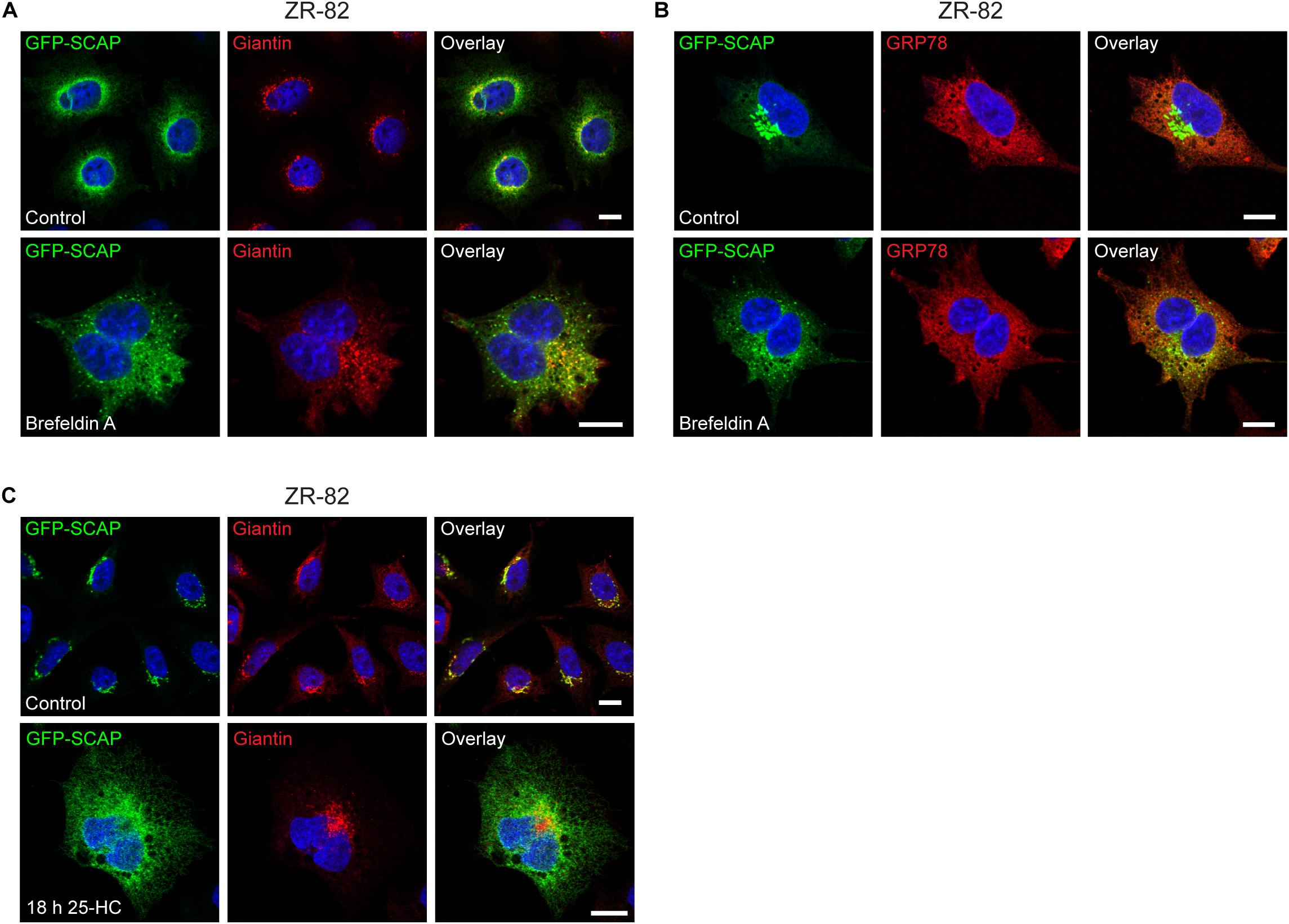
Figure 11. Brefeldin A and 25-HC sensitivity of GFP-SCAP localization in ZR-82 cells. (A,B) ZR-82 cells transfected with GFP-SCAP were treated with brefeldin A (5 μg/ml) for 30 min. (A) Cells were immunostained for GFP-SCAP using a chicken polyclonal anti-GFP antibody and giantin. (B) Cells were immunostained for GFP-SCAP and GRP78. (C) ZR-82 cells transfected with GFP-SCAP were treated with 10 μg/ml 25-HC for 18 h and immunostained for GFP-SCAP and giantin. DAPI was used for nuclear staining. Representative fields are shown for each condition. The scale bars represent 10 μm.
Next, we examined if treatment with 25-HC affects the GFP-SCAP localization in ZR-82 cells. We have shown that 25-HC suppressed both cholesterogenic promoter activity in LPDS-treated (Figure 3B) and activation of SREBP-2 target genes in U18666A-treated (Figure 5A) CHO-K1 and peroxisome-deficient cells. GFP-SCAP-transfected ZR-82 cells cultured in medium containing 10% FCS were treated with 10 μg/ml 25-HC for 90 min, 12, 15, and 18 h. GFP-SCAP showed a Golgi-like staining pattern and co-localization with giantin in vehicle-treated ZR-82 cells (Figure 11C) and cells treated with 25-HC for 90 min (data not shown). Treatment with 25-HC for 12 and 15 h caused the pattern to disperse and return to a more ER-like pattern (data not shown), and GFP-SCAP showed a reticular ER pattern after an 18 h treatment with 25-HC (Figure 11C).
Restoring Functional Peroxisomes Normalized the Transcriptional Regulation of the Cholesterol Biosynthetic Pathway, the Rate of Cholesterol Synthesis, and SCAP Trafficking
To examine whether restoring functional peroxisomes normalizes the transcriptional regulation of the cholesterol biosynthetic pathway, the expression of SREBP-2 target genes was determined in Pex2-transfected cells cultured in medium containing 10% FCS, 5% LPDS, or 10% FCS plus 10 μM U18666A for 24 h (Figure 12A). Incubation in medium containing 5% LPDS or 10 μM U18666A significantly increased the mRNA levels of SREBP-2 target genes in all cell lines compared with FCS-cultured cells. However, the expression levels in CHO-K1-Pex2 cells and Pex2-complemented ZR cells with restored functional peroxisomes were similar under all conditions. Next, the protein levels of HMGCR, MVK, and FDPS were determined in CHO-K1-Pex2 and Pex2-complemented ZR cell lines cultured in medium containing 10% FCS, 5% LPDS, or 10 μM U18666A for 24 h (Figure 12B). The cultivation of the cells in medium containing 5% LPDS and the treatment with U18666A increased the protein levels of HMGCR, MVK and FDPS in all cell lines as expected. MVK and FDPS protein levels tended to be lower in ZR-78.1C-Pex2 cells, whereas HMGCR protein levels tended to be lower in LPDS-cultured and U18666A-treated ZR-82-Pex2 cells (Figure 12B). Interestingly, the protein levels of PEX14 were increased in Pex2-transfected ZR-82 cells. In summary, compared to peroxisome-deficient cells, no significant increase in protein levels of cholesterol biosynthesis enzymes compared to CHO-K1 cells was observed in Pex2-transfected ZR cells after restoring peroxisome functionality.

Figure 12. Restoring functional peroxisomes normalized the transcriptional regulation of the cholesterol biosynthetic pathway, the rate of cholesterol synthesis, and SCAP trafficking. (A) Expression of genes involved in cholesterol biosynthesis and its regulation in Pex2-transfected CHO-K1 and Pex2-complemented peroxisome-deficient CHO cells (ZR-78.1C-Pex2, ZR-82-Pex2, ZR-87-Pex2) cultured in medium containing 10% FCS, 5% LPDS and 10 μM U18666A for 24 h. Each value represents the amount of mRNA relative to that in FCS-cultured CHO-K1-Pex2, which was arbitrarily defined as 1. Data are mean ± SD (n = 6). (B) Immunoblots of total cell lysates with antibodies against cholesterol biosynthetic enzymes after culturing cells in medium containing 10% FCS, 5% LPDS and 10 μM U18666A for 24 h. (C) Rate of biosynthesis of cholesterol, cholesteryl esters, fatty acids, and triacylglycerols in vehicle- and U18666A-treated CHO-K1-Pex2 and Pex2-complemented peroxisome-deficient CHO cells (ZR-78.1C-Pex2, ZR-82-Pex2, ZR-87-Pex2) as measured by the incorporation of [14C]acetate. Lipids were separated by thin-layer chromatography after [14C]acetate labeling. Data are mean ± SD (n = 9). Statistical analysis was performed using one-way ANOVA followed by Dunnett’s multiple comparisons test. *p < 0.05; **p < 0.01; ***p < 0.001 vs. FCS-cultured CHO-K1-Pex2. #p < 0.05; ##p < 0.01; ###p < 0.001 vs. U18666A-cultured CHO-K1-Pex2. § p < 0.05; §§ p < 0.01; §§§ p < 0.001 vs. LPDS-cultured CHO-K1-Pex2.
To assess whether restoring functional peroxisomes in ZR cells also normalizes de novo cholesterol biosynthesis, we measured de novo lipogenesis by quantifying incorporation of [14C]acetate into lipids in CHO-K1-Pex2, ZR-78.1C-Pex2, ZR-82-Pex2, and ZR-87-Pex2 cells cultured in cholesterol-containing medium with 10 μM U18666A. De novo cholesterol synthesis was similar in CHO-K1-Pex2 and ZR-82-Pex2 cells and significantly increased in ZR-87-Pex2 cells, whereas cholesterol synthesis was still significantly decreased in ZR-78.1C-Pex2 cells (Figure 12C). However, mutant ZR-78.1C, in contrast to ZR-82 and ZR-87 cells, have secondary mutations, are ouabain-resistant, and hypoxanthine phosphoribosyltransferase (HGPRT)-negative in hybridization studies (Ralph Zoeller, personal communication); hence, these alterations might prevent the normalization of cholesterol synthesis in ZR-78.1C-Pex2 cells, even though the dysregulation of the SREBP-2 pathway is corrected.
Finally, we examined if the enhanced localization of GFP-SCAP to the Golgi in peroxisome-deficient cells could be prevented by restoring functional peroxisomes. Since GFP-SCAP localized independently of culture conditions exclusively to the Golgi complex in ZR-82 cells, ZR-82-Pex2 cells were transiently transfected with GFP-SCAP. GFP-SCAP showed a reticular ER pattern both in CHO-K1-Pex2 and ZR-82-Pex2 cells grown in cholesterol-containing medium (Figure 13). Importantly, none of the transfected ZR-82-Pex2 cells showed localization of GFP-SCAP to the Golgi complex.
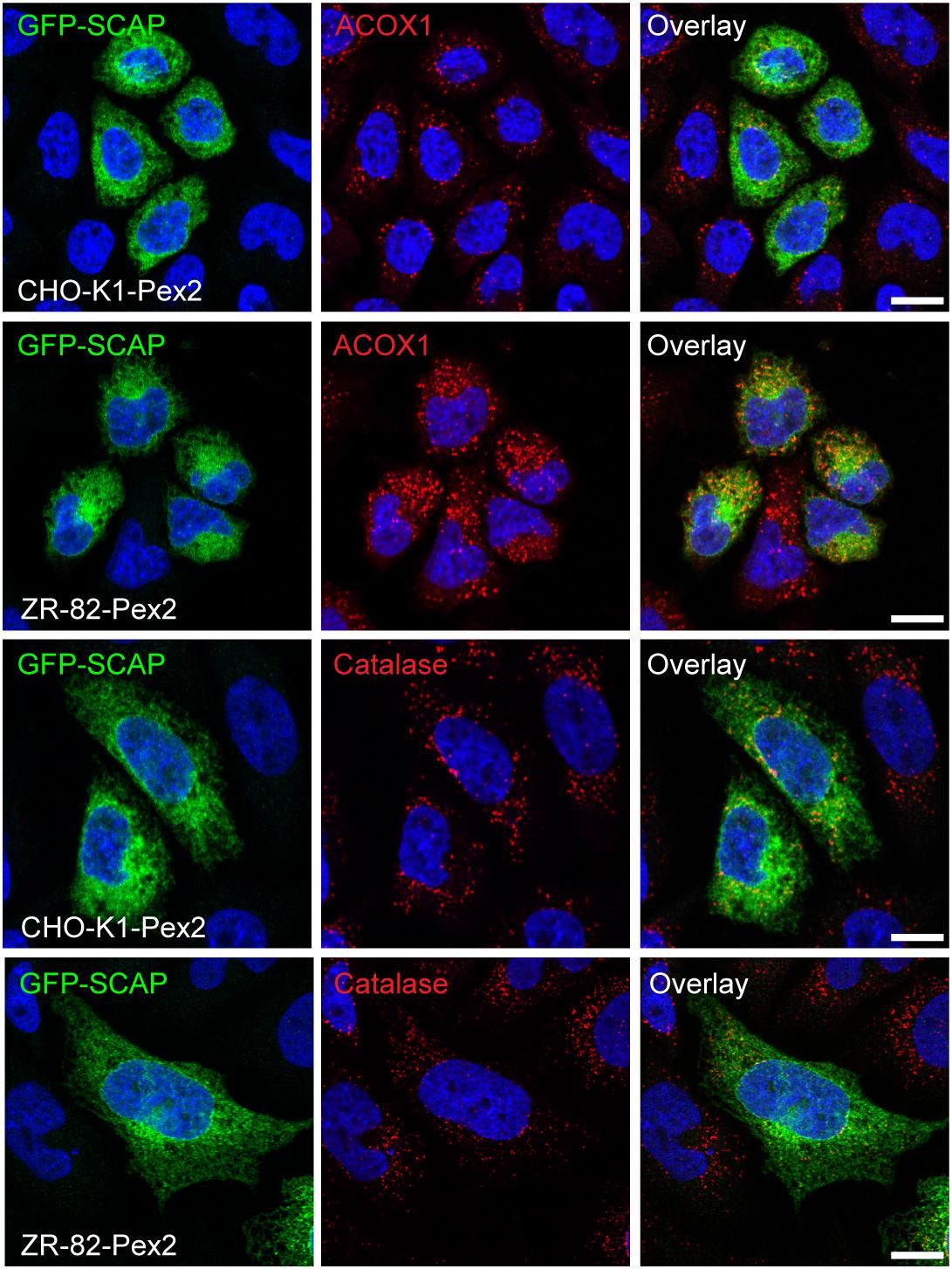
Figure 13. Restoring functional peroxisomes in ZR-82 cells prevents enhanced ER-to-Golgi trafficking of GFP-SCAP. CHO-K1-Pex2 and ZR-82-Pex2 cells were transiently transfected with GFP-SCAP and cultured in 10% FCS. GFP-SCAP was imaged using a chicken polyclonal anti-GFP antibody followed by Alexa488 secondary antibody; peroxisomes were visualized by rabbit anti-ACOX1 and rabbit anti-catalase followed by Alexa594 secondary antibody; DAPI was used for nuclear staining. The scale bars represent 10 μm.
Taken together, restoring functional peroxisomes in these cells resulted in a coordinated transcriptional and post-transcriptional regulation of the cholesterol biosynthetic pathway, demonstrating the importance of peroxisome functionality for this pathway.
Discussion
Herein, we have provided evidence that functional peroxisomes are necessary for efficient cholesterol synthesis and that peroxisome deficiency dysregulates the SREBP-2 pathway. Cells maintain cholesterol homeostasis by multiple feedback controls that act through transcriptional and post-transcriptional mechanisms, especially transcriptionally through SREBPs (Goldstein et al., 2006) and sterol-accelerated ubiquitination and degradation of HMGCR (DeBose-Boyd, 2008). Cholesterol synthesis and uptake are tightly regulated by SREBP-2 (Horton et al., 2003), a membrane-bound transcription factor that undergoes sterol-regulated export from the ER to the Golgi apparatus for proteolytic processing. The transcriptionally inactive SREBP-2 precursor protein is retained in the ER membrane through the interaction with SCAP and INSIG proteins. In sterol-depleted cells, such as would occur under conditions of starvation or where the extracellular supply of cholesterol is diminished substantially, SCAP undergoes a conformational change and it escorts SREBP-2 from the ER to the Golgi in COPII vesicles, where it is processed to its mature form. The ER contains relatively low levels of sterols compared to the whole cell or plasma membrane, making it a highly effective environment for the high-affinity cholesterol sensor SCAP to reside (Brown et al., 2018). Radhakrishnan et al. (2008) showed that SCAP transports SREBP-2 to the Golgi when the ER cholesterol content was less than 5 mol% of the total ER lipids. SCAP transport was inhibited when cholesterol content of ER membranes rose above this minimum threshold. The SREBP-2 pathway is activated in peroxisome-deficient CHO cells even when grown in cholesterol-replete medium. Accordingly, stronger activation of the SREBP-2 pathway is seen when peroxisome-deficient CHO cells are grown in cholesterol-deficient medium or if cholesterol is sequestered in the lysosomes, e.g., upon U18666A treatment. However, peroxisome-deficient cells are still sensitive towards 25-HC, indicating that the sterol-dependent mechanism for activation of the SREBP-2 pathway is still intact.
What is the cause of the increased transfer of SCAP from the ER to the Golgi and the dysregulation of the SREBP-2 pathway in peroxisome-deficient cells? Dysregulation of SREBP activation has been associated with ER stress and activation of the unfolded protein response (Colgan et al., 2011). In contrast to observations in the liver and extrahepatic tissues of peroxisome-deficient Pex2–/– mice (Kovacs et al., 2009, 2012; Figures 6F–J), we did not observe an induction of ER stress markers in peroxisome-deficient CHO cells; this might be due to adaptation of the stable cell lines during the course of propagation. Another explanation for why no ER stress and UPR activation was observed in peroxisome-deficient CHO cells, in contrast to, e.g., postnatal Pex2–/– mouse tissues or biopsies obtained from patients with peroxisomal disorders (Kovacs et al., 2009, 2012; Launay et al., 2017), might be the lack of accumulation of ER stress-inducing metabolites. Indeed, it has been shown that ER stress and UPR are induced in human primary skin fibroblast cell lines from X-linked adrenoleukodystrophy patients with ABCD1 mutations and impaired peroxisomal β-oxidation only after incubation with very long-chain fatty acids (van de Beek et al., 2017). All three parallel branches of the UPR− governed by the ER-stress sensors IRE1, PERK, and ATF6, respectively, are activated upon pharmacologically induced ER stress (Lin et al., 2007; Zhang et al., 2015). However, also acute ER stress, which was chemically induced in the in vitro system used in the present study, did not activate SREBP-2 in CHO-K1 cells.
Mechanistic target of rapamycin complex 1 has emerged as a regulator of lipid biosynthesis (Laplante and Sabatini, 2012). Porstmann et al. (2005) showed that activation of Akt, an upstream activator of mTOR, induced the expression of SREBP-1 and its target genes, but the effect on SREBP-2 was less pronounced. The mTOR inhibitor rapamycin has been shown to have opposing effects on SREBP-2 processing and the expression of HMGCR in different cell types (Gueguen et al., 2007; Sharpe and Brown, 2008; Ma et al., 2010). It has been shown that SREBP-2 processing and cholesterol biosynthetic gene expression are coordinately regulated by the eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) and p70 S6K, and that the relative contribution of each mTORC1 effector varies between cell types (Wang et al., 2011). It has been reported that an increase in mTORC1 activity is accompanied by a decrease in ER cholesterol and by SREBP-2 activation (Eid et al., 2017). mTORC1, through its regulation of autophagy and endosomal membrane trafficking, activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells (Eid et al., 2017). On the other hand, cholesterol has been shown to drive mTORC1 recruitment and activation at the lysosomal surface through a mechanism that is, at least in part, distinct from growth factor signaling (Castellano et al., 2017). Our data suggest that mTORC1 does not play a role in SREBP-2 activation or in the increased expression of cholesterol biosynthetic genes in peroxisome-deficient CHO cells, since the mTORC1 activity is inherently lower in peroxisome-deficient cell lines (i.e., decreased phosphorylation of S6 and p70 S6K). However, we cannot exclude the possibility that another target of mTORC1 could be involved in the activation of SREBP-2 in CHO cells instead of p70 S6K. Importantly, in accordance with our data, a recent study suggested that acetyl-CoA derived from peroxisomal β-oxidation promotes Raptor acetylation and mTORC1 activation in hepatocytes, and hepatic Acox1 deficiency resulted in inhibition of mTORC1 (He et al., 2020).
Despite the sensitivity of the SREBP-2 pathway to ER cholesterol levels, the rate of cholesterol synthesis is lower in peroxisome-deficient cells compared with CHO-K1 cells, illustrating the fact that functional peroxisomes and probably the compartmentalization of cholesterol biosynthetic enzymes in peroxisomes are necessary to ensure optimal biosynthesis of cholesterol. Consequently, plasma cholesterol levels are reduced both in patients with peroxisome biogenesis disorders and in 10-day-old Pex2–/– mice (Kovacs et al., 2002, 2004). Indeed, restoring functional peroxisomes by complementing peroxisome-deficient CHO cells with rat Pex2 cDNA enabled efficient cholesterol synthesis, at levels comparable to those of control (CHO-K1) cells.
Interestingly, loss of intact peroxisomes leads to enhanced ER-to-Golgi trafficking of GFP-SCAP. SCAP trafficking was normalized after restoration of functional peroxisomes, which goes also hand in hand with a normalization of the expression of SREBP-2 target genes and the rate of cholesterol synthesis. Thus, intact peroxisomes are a prerequisite for efficient cholesterol synthesis and their loss leads to defective cholesterol homeostasis.
Peroxisome-deficient ZR-78.1C, ZR-82, and ZR-87 cells have mutations in Pex2, a gene encoding an integral peroxisomal membrane protein, resulting in nonfunctional PEX2 (Zoeller et al., 1989; Thieringer and Raetz, 1993). PEX2 is involved in the translocation of peroxisomal matrix proteins following the docking of PTS receptors to the peroxisome membrane. These mutant cells exhibit typical peroxisomal membrane “ghosts”− abnormal membrane-delimited compartments that contain integral peroxisomal membrane proteins, but lack peroxisomal matrix proteins (Santos et al., 1988, 2000). In such cases (e.g., Zellwegers Syndrome, and other peroxisomal deficiency diseases), peroxisomal matrix proteins such as catalase and ACOX1 are not appropriately compartmentalized in peroxisomes; instead, they are diffusely distributed throughout the cytoplasm, consistent with the inability of these mutant cells to import peroxisomal matrix proteins (Figure 2). The number of peroxisomal ghosts in the peroxisome-deficient cells is lower compared with the number of functional peroxisomes in CHO-K1 cells, however, the number of ghosts also varies between the peroxisome-deficient cells Figures 2B,C). Recently, tethers between the ER and peroxisomes have been identified in mammalian cells, involving the peroxisomal tail-anchored proteins ACBD4 and ACBD5 and the ER-resident vesicle-associated membrane protein-associated proteins (VAPs) (Costello et al., 2017a,b; Hua et al., 2017). In immunostaining experiments in peroxisome-deficient cells, ACBD5 and PEX14 were present in cellular vesicles, suggesting that membrane contact sites between peroxisomal membrane ghosts and the ER might exist in peroxisome-deficient CHO cells (Figures 2B, C). Hua et al. (2017) showed that tethering of peroxisomes to the ER via ACBD5-VAP-mediated contact sites is necessary for the maintenance of cellular cholesterol and plasmalogen levels: depletion of these tethers resulted in a decrease of total cholesterol levels. While total cholesterol levels in peroxisome-deficient CHO cells were normal, it is tempting to speculate that a transfer of farnesyl diphosphate to the ER in order to be used as substrate of FDFT1 might occur via ACBD5-VAP-mediated contact sites.
The cholesterol transfer from endocytic organelles to the ER likely involves several parallel pathways (Pfisterer et al., 2016). Recently, it has been suggested that LDL-derived cholesterol traffics from lysosomes via peroxisomes to the ER and that membrane contact sites facilitate this cholesterol transfer (Chu et al., 2015; Hu et al., 2018; Xiao et al., 2019). Tethering between the lysosomes and peroxisomes is facilitated by peroxisomal phosphatidylinositol-4,5-biphosphate (PI(4,5)P2) and synaptotagmin 7 on lysosomes, and disruption of the lysosome-peroxisome membrane contacts caused cholesterol accumulation in lysosomes (Chu et al., 2015). Since ZR-78.1C, ZR-82, and ZR-87 cells lack functional peroxisomes, but contain abundant peroxisomal membrane ghosts, lysosome-peroxisome membrane contact sites might be able to form in such cells. This raises a fundamental biological question: if cholesterol traffics from the lysosome to peroxisomes for further distribution in the cell, are peroxisomal matrix proteins necessary (i.e., must the cholesterol go into the matrix) or does the transfer occur only via the peroxisomal membrane? Here, we demonstrated that total cholesterol levels were similar in CHO-K1 and peroxisome-deficient CHO cells. Interestingly, we and others have reported that cholesterol levels are either unaffected or decreased in peroxisome biogenesis-defective human fibroblasts, mouse tissues or serum (Kovacs et al., 2002; Faust and Kovacs, 2014). These findings are at odds with those of Chu et al. (2015); the reasons for this disparity are not yet clear. Cholesterol is distributed among multiple cellular pools, and any newly appearing cholesterol (arising either by de novo synthesis or by extracellular uptake) is rapidly incorporated into many types of cellular membranes by both vesicular and non-vesicular means (Lange and Steck, 2016). It might be that peroxisomal dysfunction results in subcellular cholesterol redistribution without a net increase in cholesterol content. Furthermore, we cannot exclude the possibility that peroxisome deficiency alters the equilibrium among different cellular cholesterol pools. In particular, cholesterol represents only about 5% of the total lipid mass of ER membranes, making them exquisitely sensitive to changes in the levels of cholesterol in cells (Radhakrishnan et al., 2008). However, peroxisome-deficient CHO cells are sensitive to inhibition of SREBP-2 activation by 25-HC, which enters cells and reaches the ER without traversing the lysosomes (and by a mechanism that does not involve membrane vesicles). This suggest that the regulation of SREBP-2 processing via hydroxysterols is still responsive in peroxisome-deficient cells. However, peroxisome deficiency leads to relocalization of SCAP to the Golgi, thereby altering the SCAP-Insig-SREBP-2 cholesterol sensing complex. In conclusion, functional peroxisomes are essential for efficient cholesterol sensing and synthesis.
The current experiments were performed not only in medium containing LPDS, but also in the presence of 10% serum (FCS), which provides an abundant source of cholesterol and fatty acids and usually suppresses the activation of all three SREBPs (Hannah et al., 2001). Despite the presence of lipid-replete medium, peroxisome deficiency led to activation of SREBP-specific reporter genes and increased expression of SREBP target genes. However, as mentioned above, it is not known whether cholesterol obligatorily enters the peroxisomal matrix in order to be distributed to other intracellular compartments or if the redistribution only occurs via the peroxisomal membrane. Blocking the exit of cholesterol from the lysosomes by treatment with U18666A resulted in activation of the SREBP-2 pathway, both in CHO-K1 and peroxisome-deficient cells, however, the increase of SREBP-2 target genes was more pronounced in peroxisome-deficient CHO cells, whereas cholesterol synthesis was lower in these cell lines.
Taken together, our findings demonstrate that disruption of peroxisomal function directly leads to a disarray in cholesterol metabolism and regulation, despite normal cholesterol content and sensitivity to external stimuli via 25-HC. Thus, intact (functional) peroxisomes are of utmost importance for a balance in the cholesterol homeostatic pathways.
Materials and Methods
Materials
All chemicals were reagent grade and were purchased from Sigma/Aldrich (St. Louis, MO) unless otherwise indicated. Fetal bovine lipoprotein-deficient serum (LPDS) was obtained from Intracel (Frederick, MD, United States) or Sigma (S5394). U18666A (3-β-[2-(diethylamino)ethoxy]androst-5-en-17-one) (BML-S200; Enzo Life Sciences, Switzerland) was dissolved in ethanol and stored as a 10 mM stock solution at −20°C. 25-hydroxycholesterol (25-HC) (H1015; Sigma/Aldrich) was dissolved in ethanol as a 10 mg/ml stock solution and stored at −20°C. Tunicamycin (T7765; Sigma/Aldrich) was dissolved in DMSO as a 4 mg/ml stock solution and stored at −20°C. Thapsigargin (BML-PE180; Enzo Life Sciences) was dissolved in DMSO as a 1 mM stock solution and stored at −20°C. Brefeldin A (B6542; Sigma/Aldrich) was dissolved in DMSO as 10 mg/ml stock solution and stored at −20°C.
Cell Culture
The CHO cell line, CHO-K1, and the peroxisome-deficient mutant CHO lines ZR-78.1C, ZR-82, and ZR-87 were obtained from Dr. Raphael A. Zoeller (Boston University School of Medicine, Boston, MA, United States) (Zoeller and Raetz, 1986). The mutant CHO cell lines were isolated from the CHO-K1 cell line used as control in this study. Cells were maintained in monolayer culture at 37 °C in 5% CO2 in Ham’s F-12 medium (Cat. no. 21765-029; Life Technologies, United Kingdom) containing 100 units/ml penicillin and 100 μg/ml streptomycin sulfate supplemented with 10% (v/v) fetal calf serum (FCS).
Animals
Homozygous Pex2–/– mice were obtained by breeding Pex2 heterozygotes (Pex2+/–) on a hybrid Swiss Webster-129 (SW/129) background (Kovacs et al., 2004, 2009). Mice had access to food and water ad libitum and were exposed to a 12:12-hour light-dark cycle. For the purposes of this study, control mice consisted of Pex2+/+ and Pex2+/– genotypes (hereafter referred to as Pex2+/), as their biochemical characteristics were comparable to one another (Kovacs et al., 2004, 2009). All protocols for animal use and experiments were reviewed and approved by the Institutional Animal Care and Use Committee of San Diego State University.
Transient Transfections of Promoter-Luciferase Reporters
The Firefly luciferase gene reporter plasmids for Hmgcr (pREDluc, hereafter called HMGCR-Luc) (Vallett et al., 1996) and Srebf2 (-4316/SREBP-2.Luc, hereafter called Srebf2-Luc) (Shin and Osborne, 2003), Fdps (pGL2-FPPS, hereafter called Fdps-Luc) (Ericsson et al., 1996), and Fdft1 (pHSS1kb-Luc, hereafter called Fdft1-Luc) (Guan et al., 1995) promoters were kind gifts from Tim Osborne (University of California, Irvine, CA, United States), Peter Edwards (University of California, Los Angeles, CA, United States), and Ishaiahu Shechter (Uniformed Services University of the Health Sciences, Bethesda, MD, United States), respectively.
Transient transfections were performed in 12-well plates using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, United States) according to the manufacturer’s instructions. Cells were seeded at a density of 5 × 104 cells/well. Cells were transfected with 500 ng of the luciferase reporter plasmids. Transfection efficiencies were normalized by co-transfecting in the ratio of 1:10 with a control plasmid encoding Renilla luciferase driven by the HSV-thymidine kinase promoter (pRL-TK, Cat. no. E2241, Promega, Madison, WI, United States). After transfection cells were cultured in medium with either 10% FCS or 5% LPDS for 24 h. After incubation cells were washed twice with PBS and harvested in 1× Passive Lysis Buffer (Promega). Firefly and Renilla luciferase activities in the cell lysates were measured with the Dual-Luciferase® reporter assay system (Promega) according to the manufacturer’s protocol. Photon production was detected as relative light units by using an Analytical Luminescence Laboratory MonolightTM 2010 Luminometer. The values presented are the means of at least three separate transfections done in quadruplicate. The amount of Firefly luciferase activity in relative light units was normalized to the amount of Renilla luciferase activity from the same test tube.
Western Blot Analysis
Cells were washed 3x in phosphate buffered saline (PBS) and lysed in RIPA buffer (20 mM Tris, pH 7.5; 150 mM NaCl; 1 mM EDTA; 1 mM EGTA; 1% NP-40; 1% sodium deoxycholate) containing protease and phosphatase inhibitors (cOmplete and PhosSTOP, respectively, Roche Diagnostics, Mannheim, Germany). Lysates were incubated on ice for 30 min and centrifuged at 20,000 × g for 20 min at 4 °C. Protein concentration was determined by the BCA method (Pierce, Rockford, IL, United States). Equal amounts of protein were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred to Amersham Protran Supported 0.2 μM nitrocellulose (#10600015; GE Healthcare, Glattbrugg, Switzerland). After blocking for 1 h in TBST (Tris-buffered saline with 0.05% Tween 20) containing 1% bovine serum albumin (BSA), membranes were probed with the indicated primary antibodies (see section “Results” above) overnight at 4°C. The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Goat anti-rabbit (#401393) and goat anti-mouse (#401253) from Sigma-Aldrich; goat anti-rat (#629520) and rabbit anti-goat (#611620) from Invitrogen) and were visualized using enhanced chemiluminescence. Membranes were exposed either to Super RX autoradiographic films (Fuji, Düsseldorf, Germany) or the Fusion Solo S imaging system. Antibodies are listed in Supplementary Table S2. Blots were semi-quantitatively analyzed by densitometry using ImageJ 1.52 v (National Institutes of Health).
Immunoblot analysis of HMG-CoA reductase was performed as described by Kovacs et al. (2004). Briefly, trichloroacetic acid-precipitated proteins were first resuspended in 25 μl Tris–HCl (125 mM, pH 6.8), 1% SDS, 0.1 M NaOH, followed by 125 μl of sample buffer containing 7 M urea, 8% SDS, and 1.1 M 2β-mercaptoethanol, and separated on 7.5% acrylamide gels.
Enzyme Assays
3-hydroxy-3-methylglutaryl (HMG)-CoA reductase (HMGCR; EC 1.1.1.34), farnesyldiphosphate synthase (FDPS; EC 2.5.1.1), and isopentenyldiphosphate isomerase (IDI1; EC 5.3.3.2) activities were assayed as described previously (Kovacs et al., 2004).
Quantitative Real-Time RT-PCR
Total RNA was prepared from cells and frozen tissues with RNeasy Mini Kit (QIAGEN, Hilden, Germany) and treated with DNase I. Quantitative RT-PCR (qRT-PCR) assays were performed as described previously (Kovacs et al., 2012). First-strand cDNA was synthesized with random hexamer primers using Ready-To-Go You-Prime First-Strand Beads (GE Healthcare, Glattbrugg, Switzerland) and the High-Capacity RNA-to-cDNA Kit (No. 4368813; Applied Biosystems). Real-time RT-PCR was performed on a Roche LightCycler LC480 instrument in duplicates using 10 ng cDNA, 7.5 pmol forward and reverse primers, and the 2x KAPA SYBR FAST qPCR Mastermix (No. KK4601; KAPA Biosystems). Thermal cycling was carried out with a 5 min denaturation step at 95°C, followed by 45 three-step cycles: 10 s at 95°C, 10 s at 60°C, and 10 s at 72°C. Melt curve analysis was carried out to confirm the specific amplification of a target gene and absence of primer dimers. Primer sequences are listed in Supplementary Table S3. Relative mRNA amount was calculated using the comparative threshold cycle (CT) method (Livak and Schmittgen, 2001). Gapdh and actin were used as the invariant control for CHO cells, and 18S rRNA and cyclophilin (Ppia) were used as the invariant control for mouse tissues.
Cells Stably Expressing GFP-SCAP, GFP-ATF6(S1P-) and Pex2
pGFP-SCAP plasmid, encoding GFP fused to wild-type hamster SCAP under the control of the CMV promoter, was a gift from Dr. Peter Espenshade (Johns Hopkins University School of Medicine, Baltimore, MD, United States) (Nohturfft et al., 2000). pEGFP-ATF6-(S1P-) was a gift from Ron Prywes (Addgene plasmid # 32956) (Chen et al., 2002). CHO-K1, ZR-78.1C, ZR-82, and ZR-87 cells stably expressing GFP-SCAP were generated by transfection with pGFP-SCAP using polyethylenimine (PEI), followed by selection with G418. After selection with G418 GFP-SCAP-positive cells were sorted on a BD FACSAriaTM IIIu BL1 sorter (BD Biosciences). The plasmid (PYN3218) containing full-length rat Pex2 cDNA was a gift from Masaki Ito (Saga Medical School, Saga, Japan).
Determination of Rate of Biosynthesis of Lipids
Cells were incubated with 5 μCi [1-14C]acetate (58 mCi/mmol; PerkinElmer, Boston, MA, United States) for 4 h. After incubation, the cells were rinsed three times with PBS, and lipids were extracted with hexane:isopropanol (3:2). Manipulative losses of lipids were accounted for by addition of a known amount of [3H]cholesterol as internal standard. After lipid extraction cells were lysed in 0.1 M NaOH for protein determination using the BCA method. Acetate incorporation into specific lipids was analyzed after separation of lipids by thin-layer chromatography (TLC). Therefore, organic phases were evaporated to dryness under a nitrogen stream. Lipids were resuspended in chloroform and spotted together with appropriate lipid standards on silica gel 60 plates (Merck, Darmstadt, Germany). For separation of neutral lipids, plates were developed in heptanes:diethylether:acetic acid (90:30:1) as solvent. Lipid samples and standards were visualized by iodine vapor. The lipid fractions were scraped from the plate and their radioactive content was determined by liquid scintillation counting. Values were normalized for sample protein content.
After incubation with 5 μCi [1-14C]acetate incorporation into cholesterol was also analyzed by reverse-phase radio-HPLC as previously described (Kovacs et al., 2004). Briefly, following saponification and petroleum ether extraction, the non-saponifiable lipids were analyzed by reverse-phase radio-HPLC, and the specific activities of the radiolabeled products (e.g., sterols, squalene) were determined.
Cholesterol Esterification
[1-14C]palmitic acid (56.1 mCi/mmol, NEC075H050UC) was purchased from PerkinElmer. A 2.5 mM stock solution of [1-14C]palmitic acid was prepared by complexing labeled and unlabeled palmitate to fatty acid-free albumin as described previously (Knobloch et al., 2017). The reaction mixture was prepared by diluting the palmitate/BSA stock with cell culture medium and contained 0.4 μCi [1-14C]palmitic acid and 100 μM sodium palmitate in 1 ml medium. Cells were seeded in 6-well plates in medium containing 10% FCS (v/v). The reaction was initiated by replacing the culture medium with 1 ml of the reaction mixture. The cells were incubated for 4 h at 37°C. After incubation, lipids were extracted with hexane:isopropanol (3:2) and cholesterol ester levels were analyzed by TLC as described above for the determination of the rate of lipid biosynthesis.
Immunofluorescence Staining
Immunofluorescence staining was performed as described previously (Kovacs et al., 2007). As secondary antibodies, Alexa Fluor 488 donkey anti-chicken IgY (#703-545-155; Jackson ImmunoResearch), Alexa Fluor 488 goat anti-rabbit IgG (A11070; Invitrogen), Alexa Fluor 594 goat anti-rabbit IgG (A11072; Invitrogen), and Alexa Fluor 594 donkey anti-goat IgG (A11058; Invitrogen) were used in a dilution of 1:400. Images were taken with a Leica SP8-AOBS confocal laser scanning microscope (Leica Microsystems GmbH, Wetzlar, Germany) using a 63× oil immersion objective and the Leica LAS Lite Software. Acquirement settings were optimized for each experiment, and identical settings were used for all images throughout one experiment. Fluorescent dyes were imaged sequentially in frame interlace mode to eliminate crosstalk between the channels. Image processing was performed off-line using Fiji software (Schindelin et al., 2012).
Statistical Analyses
Data are expressed as mean ± SD. When two groups where compared, statistical significance was evaluated by an unpaired, two-tailed Student’s t-test or an unpaired, two-tailed Student’s t-test with Welch’s correction when variances where significantly different. For multiple group analysis one-way ANOVA followed by Dunnett’s multiple comparisons test was performed. Data were assumed to be normally distributed. Statistical analyses were performed using GraphPad Prism version 8.2.0.
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of San Diego State University.
Author Contributions
HS and WK: conception and design. KC, JS, PF, SF, HS, and WK: acquisition, analysis, and interpretation of data. PF, SF, and WK: resources. WK: writing – original draft and study supervision. PF, SF, HS, and WK: writing – review and editing. All authors approved the submitted manuscript.
Funding
We dedicate this manuscript to the memory of our friend, colleague, and mentor Prof. Skaidrite K. Krisans (San Diego State University). This work was supported by the Swiss National Science Foundation (SNSF) Grants Nos. 31003A_132982 and 31003A_166245 to WK. KC was supported by a fellowship from SDSU-MARC funded by the National Institutes of General Medical Sciences/National Institutes of Health under Grant No. T34GM008303. Parts of this study have been supported by the NIH grants DK58238 and DK58040, and the R01 minority supplement R01DK058238-03S1. Imaging was performed on instruments of ScopeM, the Scientific Center for Optical and Electron Microscopy of ETH Zurich. SF was supported, in part, by a Research Career Scientist Award from the United States Department of Veterans Affairs, BLR&D Service. The opinions expressed herein do not represent those of the Department of Veterans Affairs or the United States Government.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.560266/full#supplementary-material
Abbreviations
25-HC, 25-hydroxycholesterol; Abca1, ATP-binding cassette transporter A1; ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; BAAT, bile acid-coenzyme A:amino acid N-acyltransferase; bHLH, basic helix-loop-helix; Calr, calreticulin; Canx, calnexin; CHO, Chinese hamster ovary; Chop, C/EBP-homologous protein; COPII, coat protein complex II; DHCR24, 24-dehydrocholesterol reductase; Dnajb9, DnaJ heat shock protein family (Hsp40) member B9; ER, endoplasmic reticulum; FASN, fatty acid synthase; FDFT1, squalene synthase; FDPS, farnesyl diphosphate synthase; Gadd34, growth arrest and DNA damage-inducible 34; GRP78, glucose-regulated protein 78; GRP94, glucose-regulated protein 94; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; HMGCS, 3-hydroxy-3-methylglutaryl-CoA synthase; IDI1, isopentenyl diphosphate isomerase 1; INSIG, insulin-induced gene; LDLR, low density lipoprotein receptor; LPDS, lipoprotein-deficient serum; LSS, lanosterol synthase; MVD, mevalonate pyrophosphate decarboxylase; MVK, mevalonate kinase; NPC1, Niemann-Pick C1; PERK, PKR-like endoplasmic reticulum kinase; Pex, peroxin; PMVK, phosphomevalonate kinase; S1P, Site-1 protease; S2P, Site-2 protease; SC4MOL, methylsterol monoxygenase 1; SCAP, SREBP cleavage-activating protein; SQLE, squalene epoxidase; SRE, sterol regulatory element; SREBF2, sterol regulatory element-binding protein 2; SREBP, sterol regulatory element-binding protein; SSD, sterol-sensing domain; WD, tryptophan-aspartate repeat; Xbp1, X-box -binding protein 1.
References
Abi-Mosleh, L., Infante, R. E., Radhakrishnan, A., Goldstein, J. L., and Brown, M. S. (2009). Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. U.S.A. 106, 19316–19321. doi: 10.1073/pnas.0910916106
Aboushadi, N., and Krisans, S. K. (1998). Analysis of isoprenoid biosynthesis in peroxisomal-deficient Pex2 CHO cell lines. J. Lipid Res. 39, 1781–1791.
Acosta, H., Burchmore, R., Naula, C., Gualdrón-López, M., Quintero-Troconis, E., Cáceres, A. J., et al. (2019). Proteomic analysis of glycosomes from Trypanosoma cruzi epimastigotes. Mol. Biochem. Parasitol. 229, 62–74. doi: 10.1016/j.molbiopara.2019.02.008
Brown, M. S., Radhakrishnan, A., and Goldstein, J. L. (2018). Retrospective on cholesterol homeostasis: the central role of scap. Annu. Rev. Biochem. 87, 783–807. doi: 10.1146/annurev-biochem-062917-011852
Carrero-Lérida, J., Pérez-Moreno, G., Castillo-Acosta, V. M., Ruiz-Pérez, L. M., and González-Pacanowska, D. (2009). Intracellular location of the early steps of the isoprenoid biosynthetic pathway in the trypanosomatids Leishmania major and Trypanosoma brucei. Int. J. Parasitol. 39, 307–314. doi: 10.1016/j.ijpara.2008.08.012
Castellano, B. M., Thelen, A. M., Moldavski, O., Feltes, M., van der Welle, R. E. N., Mydock-McGrane, L., et al. (2017). Lysosomal cholesterol activates mTORC1 via an SLC38A9–Niemann-Pick C1 signaling complex. Science 355, 1306–1311. doi: 10.1126/science.aag1417
Chang, T.-Y., Chang, C. C. Y., Ohgami, N., and Yamauchi, Y. (2006). Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 22, 129–157. doi: 10.1146/annurev.cellbio.22.010305.104656
Chen, X., Shen, J., and Prywes, R. (2002). The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol.l Chem. 277, 13045–13052. doi: 10.1074/jbc.M110636200
Chu, B.-B., Liao, Y.-C., Qi, W., Xie, C., Du, X., Wang, J., et al. (2015). Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161, 291–306. doi: 10.1016/j.cell.2015.02.019
Colasante, C., Ellis, M., Ruppert, T., and Voncken, F. (2006). Comparative proteomics of glycosomes from bloodstream form and procyclic culture form Trypanosoma brucei brucei. Proteomics 6, 3275–3293. doi: 10.1002/pmic.200500668
Colgan, S. M., Al-Hashimi, A. A., and Austin, R. C. (2011). Endoplasmic reticulum stress and lipid dysregulation. Exp. Rev. Mol. Med. 13, e4. doi: 10.1017/S1462399410001742
Colgan, S. M., Tang, D., Werstuck, G. H., and Austin, R. C. (2007). Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int. J. Biochem. Cell Biol. 39, 1843–1851. doi: 10.1016/j.biocel.2007.05.002
Concepcion, J. L., Gonzalez-Pacanowska, D., and Urbina, J. A. (1998). 3-Hydroxy-3-methyl-glutaryl-CoA reductase in trypanosoma (Schizotrypanum) cruzi:Subcellular localization and kinetic properties. Arch. Biochem. Biophys. 352, 114–120. doi: 10.1006/abbi.1998.0577
Costello, J. L., Castro, I. G., Hacker, C., Schrader, T. A., Metz, J., Zeuschner, D., et al. (2017a). ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol. 216, 331–342. doi: 10.1083/jcb.201607055
Costello, J. L., Castro, I. G., Schrader, T. A., Islinger, M., and Schrader, M. (2017b). Peroxisomal ACBD4 interacts with VAPB and promotes ER-peroxisome associations. Cell Cycle 16, 1039-1045. doi: 10.1080/15384101.2017.1314422
Das, A., Brown, M. S., Anderson, D. D., Goldstein, J. L., and Radhakrishnan, A. (2014). Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. eLife 3, e02882. doi: 10.7554/eLife.02882
Du, X., Kristiana, I., Wong, J., and Brown, A. J. (2006). Involvement of Akt in ER-to-Golgi transport of SCAP/SREBP: a link between a key cell proliferative pathway and membrane synthesis. Mol. Biol. Cell 17, 2735–2745. doi: 10.1091/mbc.e05-11-1094
DeBose-Boyd, R. A. (2008). Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG-CoA reductase. Cell Res. 18, 609–621. doi: 10.1038/cr.2008.61
Eid, W., Dauner, K., Courtney, K. C., Gagnon, A., Parks, R. J., Sorisky, A., et al. (2017). mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 114, 7999–8004. doi: 10.1073/pnas.1705304114
Ericsson, J., Jackson, S. M., Lee, B. C., and Edwards, P. A. (1996). Sterol regulatory element binding protein binds to a cis element in the promoter of the farnesyl diphosphate synthase gene. PNAS 93, 945–950. doi: 10.1073/pnas.93.2.945
Faust, P. L., and Kovacs, W. J. (2014). Cholesterol biosynthesis and ER stress in peroxisome deficiency. Biochimie 98, 75–85. doi: 10.1016/j.biochi.2013.10.019
Ferreira, ÉR., Horjales, E., Bonfim-Melo, A., Cortez, C., da Silva, C. V., De Groote, M., et al. (2016). Unique behavior of Trypanosoma cruzi mevalonate kinase: a conserved glycosomal enzyme involved in host cell invasion and signaling. Sci. Rep. 6:24610. doi: 10.1038/srep24610
Goldstein, J. L., and Brown, M. S. (2015). A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161, 161–172. doi: 10.1016/j.cell.2015.01.036
Goldstein, J. L., DeBose-Boyd, R. A., and Brown, M. S. (2006). Protein Sensors for Membrane Sterols. Cell 124, 35–46. doi: 10.1016/j.cell.2005.12.022
Guan, G., Jiang, G., Koch, R. L., and Shechter, I. (1995). Molecular cloning and functional analysis of the promoter of the human squalene synthase gene. J. Biol. Chem. 270, 21958–21965. doi: 10.1074/jbc.270.37.21958
Gueguen, Y., Ferrari, L., Souidi, M., Batt, A.-M., Lutton, C., Siest, G., et al. (2007). Compared effect of immunosuppressive drugs cyclosporine a and rapamycin on cholesterol homeostasis key enzymes CYP27A1 and HMG-CoA Reductase. Basic Clin. Pharmacol. Toxicol. 100, 392–397. doi: 10.1111/j.1742-7843.2007.00066.x
Güther, M. L. S., Urbaniak, M. D., Tavendale, A., Prescott, A., and Ferguson, M. A. J. (2014). High-Confidence Glycosome Proteome for Procyclic Form Trypanosoma brucei by Epitope-Tag Organelle Enrichment and SILAC Proteomics. Journal of Proteome Research 13, 2796–2806. doi: 10.1021/pr401209w
Hannah, V. C., Ou, J., Luong, A., Goldstein, J. L., and Brown, M. S. (2001). Unsaturated fatty acids down-regulate SREBP isoforms 1a and 1c by two mechanisms in HEK-293 cells. J. Biol. Chem. 276, 4365–4372. doi: 10.1074/jbc.M007273200
Harano, T., Shimizu, N., Otera, H., and Fujiki, Y. (1999). transmembrane topology of the peroxin, Pex2p, an essential component for the peroxisome assembly1. J. Biochem. 125, 1168–1174. doi: 10.1093/oxfordjournals.jbchem.a022400
He, A., Chen, X., Tan, M., Chen, Y., Lu, D., Zhang, X., et al. (2020). Acetyl-CoA derived from hepatic peroxisomal β-oxidation inhibits autophagy and promotes steatosis via mTORC1 activation. Mol. Cell 79, 30.e–42.e. doi: 10.1016/j.molcel.2020.05.007
Henry, L. K., Thomas, S. T., Widhalm, J. R., Lynch, J. H., Davis, T. C., Kessler, S. A., et al. (2018). Contribution of isopentenyl phosphate to plant terpenoid metabolism. Nat. Plants 4, 721–729. doi: 10.1038/s41477-018-0220-z
Horton, J. D., Goldstein, J. L., and Brown, M. S. (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131. doi: 10.1172/jci0215593
Horton, J. D., Shah, N. A., Warrington, J. A., Anderson, N. N., Park, S. W., Brown, M. S., et al. (2003). Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. U.S.A. 100, 12027–12032. doi: 10.1073/pnas.1534923100
Hu, A., Zhao, X.-T., Tu, H., Xiao, T., Fu, T., Wang, Y., et al. (2018). PIP4K2A regulates intracellular cholesterol transport through modulating PI(4,5)P2 homeostasis. J. Lipid Res. 59, 507–514. doi: 10.1194/jlr.M082149
Hua, R., Cheng, D., Coyaud, É, Freeman, S., Di Pietro, E., Wang, Y., et al. (2017). VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 216, 367–377. doi: 10.1083/jcb.201608128
Iaea, D. B., and Maxfield, F. R. (2015). Cholesterol trafficking and distribution. Essays Biochem. 57, 43–55. doi: 10.1042/bse0570043
Klausner, R. D., Donaldson, J. G., and Lippincott-Schwartz, J. (1992). Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116, 1071–1080. doi: 10.1083/jcb.116.5.1071
Knobloch, M., Pilz, G.-A., Ghesquière, B., Kovacs, W. J., Wegleiter, T., Moore, D. L., et al. (2017). A fatty acid oxidation-dependent metabolic shift regulates adult neural stem cell activity. Cell Rep. 20, 2144–2155. doi: 10.1016/j.celrep.2017.08.029
Kotzka, J., Lehr, S., Roth, G., Avci, H., Knebel, B., and Muller-Wieland, D. (2004). Insulin-activated Erk-mitogen-activated protein kinases phosphorylate sterol regulatory element-binding Protein-2 at serine residues 432 and 455 in Vivo. J. Biol. Chem. 279, 22404–22411. doi: 10.1074/jbc.M401198200
Kotzka, J., Müller-Wieland, D., Roth, G., Kremer, L., Munck, M., Schürmann, S., et al. (2000). Sterol regulatory element binding proteins (SREBP)-1a and SREBP-2 are linked to the MAP-kinase cascade. J. Lipid Res. 41, 99–108.
Kovacs, W., Tape, K., Shackelford, J., Duan, X., Kasumov, T., Kelleher, J., et al. (2007). Localization of the pre-squalene segment of the isoprenoid biosynthetic pathway in mammalian peroxisomes. Histochem. Cell Biol. 127, 273–290. doi: 10.1007/s00418-006-0254-6
Kovacs, W. J., Charles, K. N., Walter, K. M., Shackelford, J. E., Wikander, T. M., Richards, M. J., et al. (2012). Peroxisome deficiency-induced ER stress and SREBP-2 pathway activation in the liver of newborn PEX2 knock-out mice. Biochim. Biophys. Acta 1821, 895–907. doi: 10.1016/j.bbalip.2012.02.011
Kovacs, W. J., Olivier, L. M., and Krisans, S. K. (2002). Central role of peroxisomes in isoprenoid biosynthesis. Prog. Lipid Res. 41, 369–391. doi: 10.1016/s0163-7827(02)00002-4
Kovacs, W. J., Shackelford, J. E., Tape, K. N., Richards, M. J., Faust, P. L., Fliesler, S. J., et al. (2004). Disturbed cholesterol homeostasis in a peroxisome-deficient Pex2 knockout mouse model. Mol. Cell Biol. 24, 1—-13.
Kovacs, W. J., Tape, K. N., Shackelford, J. E., Wikander, T. M., Richards, M. J., Fliesler, S. J., et al. (2009). Peroxisome deficiency causes a complex phenotype because of hepatic srebp/insig dysregulation associated with endoplasmic reticulum stress. J. Biol. Chem. 284, 7232–7245. doi: 10.1074/jbc.M809064200
Kwon, H. J., Abi-Mosleh, L., Wang, M. L., Deisenhofer, J., Goldstein, J. L., Brown, M. S., et al. (2009). Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137, 1213–1224. doi: 10.1016/j.cell.2009.03.049
Lange, Y., and Steck, T. L. (2016). Active membrane cholesterol as a physiological effector. Chem. Phys. Lipids 199, 74–93. doi: 10.1016/j.chemphyslip.2016.02.003
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Launay, N., Ruiz, M., Grau, L., Ortega, F. J., Ilieva, E. V., Martínez, J. J., et al. (2017). Tauroursodeoxycholic bile acid arrests axonal degeneration by inhibiting the unfolded protein response in X-linked adrenoleukodystrophy. Acta Neuropathol. 133, 283–301. doi: 10.1007/s00401-016-1655-9
Lee, J. N., and Ye, J. (2004). Proteolytic activation of sterol regulatory element-binding protein induced by cellular stress through depletion of Insig-1. J. Biol. Chem. 279, 45257–45265. doi: 10.1074/jbc.m408235200
Lhoták, Š, Sood, S., Brimble, E., Carlisle, R. E., Colgan, S. M., Mazzetti, A., et al. (2012). ER stress contributes to renal proximal tubule injury by increasing SREBP-2-mediated lipid accumulation and apoptotic cell death. Am. J. Physiol. Renal Physiol. 303, F266–F278. doi: 10.1152/ajprenal.00482.2011
Lin, J. H., Li, H., Yasumura, D., Cohen, H. R., Zhang, C., Panning, B., et al. (2007). IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949. doi: 10.1126/science.1146361
Linstedt, A. D., and Hauri, H. P. (1993). Giantin, a novel conserved Golgi membrane protein containing a cytoplasmic domain of at least 350 kDa. Mol. Biol. Cell 4, 679–693. doi: 10.1091/mbc.4.7.679
Lippincott-Schwartz, J., Yuan, L. C., Bonifacino, J. S., and Klausner, R. D. (1989). Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56, 801–813. doi: 10.1016/0092-8674(89)90685-5
Liscum, L. (1990). Pharmacological inhibition of the intracellular transport of low-density lipoprotein-derived cholesterol in Chinese hamster ovary cells. Biochim. Biophys. Acta 1045, 40–48. doi: 10.1016/0005-2760(90)90201-8
Liscum, L., and Faust, J. R. (1989). The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J. Biol. Chem. 264, 11796–11806.
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, F., Liang, Q., Abi-Mosleh, L., Das, A., De Brabander, J. K., Goldstein, J. L., et al. (2016). Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. eLife 4:e12177. doi: 10.7554/eLife.12177
Ma, K. L., Varghese, Z., Ku, Y., Powis, S. H., Chen, Y., Moorhead, J. F., et al. (2010). Sirolimus inhibits endogenous cholesterol synthesis induced by inflammatory stress in human vascular smooth muscle cells. Am. J. Physiol. Heart Circul. Physiol. 298, H1646–H1651. doi: 10.1152/ajpheart.00492.2009
Miyazawa, S., Hayashi, H., Hijikata, M., Ishii, N., Furuta, S., Kagamiyama, H., et al. (1987). Complete nucleotide sequence of cDNA and predicted amino acid sequence of rat acyl-CoA oxidase. J. Biol. Chem. 262, 8131–8137.
Nohturfft, A., Yabe, D., Goldstein, J. L., Brown, M. S., and Espenshade, P. J. (2000). Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell 102, 315–323. doi: 10.1016/s0092-8674(00)00037-4
Peterson, T. R., Sengupta, S. S., Harris, T. E., Carmack, A. E., Kang, S. A., Balderas, E., et al. (2011). mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420. doi: 10.1016/j.cell.2011.06.034
Pfisterer, S. G., Peränen, J., and Ikonen, E. (2016). LDL–cholesterol transport to the endoplasmic reticulum: current concepts. Curr. Opin. Lipidol. 27, 282–287. doi: 10.1097/mol.0000000000000292
Porstmann, T., Griffiths, B., Chung, Y.-L., Delpuech, O., Griffiths, J. R., Downward, J., et al. (2005). PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 24, 6465–6481. doi: 10.1038/sj.onc.1208802
Radhakrishnan, A., Goldstein, J. L., McDonald, J. G., and Brown, M. S. (2008). Switch-like control of SREBP-2 transport triggered by small changes in er cholesterol: a delicate balance. Cell Metab. 8, 512–521. doi: 10.1016/j.cmet.2008.10.008
Russell, D. W. (2003). The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 72, 137–174. doi: 10.1146/annurev.biochem.72.121801.161712
Santos, M., Imanaka, T., Shio, H., Small, G., and Lazarow, P. (1988). Peroxisomal membrane ghosts in Zellweger syndrome–aberrant organelle assembly. Science 239, 1536–1538. doi: 10.1126/science.3281254
Santos, M. J., Henderson, S. C., Moser, A. B., Moser, H. W., and Lazarow, P. B. (2000). Peroxisomal ghosts are intracellular structures distinct from lysosomal compartments in Zellweger syndrome: a confocal laser scanning microscopy study. Biol. Cell 92, 85–94. doi: 10.1016/s0248-4900(00)89016-4
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Schram, A. W., Strijland, A., Hashimoto, T., Wanders, R. J., Schutgens, R. B., van den Bosch, H., et al. (1986). Biosynthesis and maturation of peroxisomal beta-oxidation enzymes in fibroblasts in relation to the Zellweger syndrome and infantile Refsum disease. Proc. Natl. Acad. Sci. U.S.A. 83, 6156–6158. doi: 10.1073/pnas.83.16.6156
Sharpe, L. J., and Brown, A. J. (2008). Rapamycin down-regulates LDL-receptor expression independently of SREBP-2. Biochem. Biophys. Res. Commun. 373, 670–674. doi: 10.1016/j.bbrc.2008.06.108
Shin, D.-J., and Osborne, T. F. (2003). Thyroid hormone regulation and cholesterol metabolism are connected through sterol regulatory element-binding protein-2 (SREBP-2). J. Biol. Chem. 278, 34114–34118.
Takashima, K., Saitoh, A., Funabashi, T., Hirose, S., Yagi, C., Nozaki, S., et al. (2015). COPI-mediated retrieval of SCAP is crucial for regulating lipogenesis under basal and sterol-deficient conditions. J. Cell Sci. 128, 2805–2815. doi: 10.1242/jcs.164137
Thieringer, R., and Raetz, C. (1993). Peroxisome-deficient Chinese hamster ovary cells with point mutations in peroxisome assembly factor-1. J. Biol. Chem. 268, 12631–12636.
Urbina, J. A., Concepcion, J. L., Rangel, S., Visbal, G., and Lira, R. (2002). Squalene synthase as a chemotherapeutic target in Trypanosoma cruzi and Leishmania mexicana. Mol. Biochem. Parasitol. 125, 35–45. doi: 10.1016/S0166-6851(02)00206-2
Vallett, S. M., Sanchez, H. B., Rosenfeld, J. M., and Osborne, T. F. (1996). A direct role for sterol regulatory element binding protein in activation of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase Gene. J. Biol. Chem. 271, 12247–12253. doi: 10.1074/jbc.271.21.12247
van de Beek, M.-C., Ofman, R., Dijkstra, I., Wijburg, F., Engelen, M., Wanders, R., et al. (2017). Lipid-induced endoplasmic reticulum stress in X-linked adrenoleukodystrophy. Biochim. Biophys. Acta1863, 2255–2265. doi: 10.1016/j.bbadis.2017.06.003
van Heusden, G. P. H., van Beckhoven, J. C. M., Thieringer, R., Raetz, C. R. H., and Wirtz, K. W. A. (1992). Increased cholesterol synthesis in Chinese hamster ovary cells deficient in peroxisomes. Biochim. Biophys. Acta 1126, 81–87. doi: 10.1016/0005-2760(92)90220-P
Wang, B. T., Ducker, G. S., Barczak, A. J., Barbeau, R., Erle, D. J., and Shokat, K. M. (2011). The mammalian target of rapamycin regulates cholesterol biosynthetic gene expression and exhibits a rapamycin-resistant transcriptional profile. Proc. Natl. Acad. Sci. U.S.A. 108, 15201–15206. doi: 10.1073/pnas.1103746108
Wangeline, M. A., Vashistha, N., and Hampton, R. Y. (2017). Proteostatic tactics in the strategy of sterol regulation. Annu. Rev. Cell Dev. Biol. 33, 467–489. doi: 10.1146/annurev-cellbio-111315-125036
Weinhofer, I., Kunze, M., Stangl, H., Porter, F. D., and Berger, J. (2006). Peroxisomal cholesterol biosynthesis and Smith-Lemli-Opitz syndrome. Biochem. Biophys. Res. Commun. 345, 205–209. doi: 10.1016/j.bbrc.2006.04.078
Werstuck, G. H., Lentz, S. R., Dayal, S., Hossain, G. S., Sood, S. K., Shi, Y. Y., et al. (2001). Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Invest. 107, 1263–1273. doi: 10.1172/jci11596
Xiao, J., Luo, J., Hu, A., Xiao, T., Li, M., Kong, Z., et al. (2019). Cholesterol transport through the peroxisome-ER membrane contacts tethered by PI(4,5)P2 and extended synaptotagmins. Sci. China Life Sci. 62, 1117–1135. doi: 10.1007/s11427-019-9569-9
Ye, J., Rawson, R. B., Komuro, R., Chen, X., Davé, U. P., Prywes, R., et al. (2000). ER Stress Induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364. doi: 10.1016/S1097-2765(00)00133-7
Zhang, S. X., Ma, J. H., Bhatta, M., Fliesler, S. J., and Wang, J. J. (2015). The unfolded protein response in retinal vascular diseases: implications and therapeutic potential beyond protein folding. Prog. Ret. Eye Res. 45, 111–131. doi: 10.1016/j.preteyeres.2014.12.001
Zoeller, R. A., Allen, L. A., Santos, M. J., Lazarow, P. B., Hashimoto, T., Tartakoff, A. M., et al. (1989). Chinese hamster ovary cell mutants defective in peroxisome biogenesis. Comparison to Zellweger syndrome. J. Biol. Chem. 264, 21872–21878.
Keywords: cholesterol synthesis, CHO cells, ER-to-Golgi transport, peroxisomes, PEX2, SCAP, SREBP-2, Zellweger syndrome
Citation: Charles KN, Shackelford JE, Faust PL, Fliesler SJ, Stangl H and Kovacs WJ (2020) Functional Peroxisomes Are Essential for Efficient Cholesterol Sensing and Synthesis. Front. Cell Dev. Biol. 8:560266. doi: 10.3389/fcell.2020.560266
Received: 08 May 2020; Accepted: 22 September 2020;
Published: 06 November 2020.
Edited by:
Maya Schuldiner, Weizmann Institute of Science, IsraelReviewed by:
Joe Costello, University of Exeter, United KingdomMarkus Islinger, Heidelberg University, Germany
Copyright © 2020 Charles, Shackelford, Faust, Fliesler, Stangl and Kovacs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Werner J. Kovacs, d2VybmVyLmtvdmFjc0BiaW9sLmV0aHouY2g=