
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 22 September 2020
Sec. Molecular and Cellular Pathology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00854
This article is part of the Research Topic Ectopic Mineralization of Tissues: Mechanisms, Risk Factors, Diseases and Prevention View all 29 articles
 Carole-Anne Faraji-Bellée1†Axelle Cauliez1†Benjamin Salmon1,2†Olivier Fogel3†
Carole-Anne Faraji-Bellée1†Axelle Cauliez1†Benjamin Salmon1,2†Olivier Fogel3† Volha Zhukouskaya1,4
Volha Zhukouskaya1,4 Aurélie Benoit5Thorsten Schinke6Christian Roux3Agnès Linglart4,7,8Corinne Miceli-Richard3
Aurélie Benoit5Thorsten Schinke6Christian Roux3Agnès Linglart4,7,8Corinne Miceli-Richard3 Catherine Chaussain1,2,4Karine Briot3,4*
Catherine Chaussain1,2,4Karine Briot3,4* Claire Bardet1*
Claire Bardet1*X-linked hypophosphatemia (XLH) is characterized by rickets and osteomalacia, caused by inactivating mutations in the Phosphate-regulating endopeptidase homolog X-linked (PHEX) gene. With aging, adult patients develop paradoxical heterotopic calcifications of tendons and ligaments at their insertion sites (enthesophytes), and joint alterations. Understanding the progression of this structural damage that severely affects patients’ quality of life will help to improve the management of XLH. Here, we characterized the occurrence of enthesophytes and joint alterations through a 12 month in vivo micro-CT follow-up in the Hyp mouse, a murine model of XLH (n = 5 mice per group). Similar to adult patients with XLH, Hyp mice developed calcaneal enthesophytes, hip joint alterations, erosions of the sacroiliac joints and periarticular calcifications. These lesions were already present at month 3 and gradually worsened over time. In sharp contrast, no abnormalities were observed in control mice at early time points. Histological analyses confirmed the presence of bone erosions, calcifications and expansion of mineralizing enthesis fibrocartilage in Hyp mice and their absence in controls and suggested that new bone formation is driven by altered mechanical strain. Interestingly, despite a strong deformation of the curvature, none of the Hyp mice displayed enthesophyte at the spine. Peripheral enthesophytes and joint alterations develop at the early stages of the disease and gradually worsen overtime. Overall, our findings highlight the relevance of this preclinical model to test new therapies aiming to prevent bone and joint complications in XLH.
Mineralization defects and paradoxical mineralization of entheses are the hallmarks of X-linked hypophosphatemia (XLH) (Polisson et al., 1985; Reid et al., 1989), a rare skeletal disease caused by inactivating mutations in the phosphate-regulating endopeptidase homolog X-linked (PHEX) gene (Sabbagh et al., 2000; Gaucher et al., 2009). Mineralization defects manifest in XLH children as rickets with severe skeletal deformities and short stature (Wharton and Bishop, 2003; Carpenter et al., 2011; Glass et al., 2011; Fuente et al., 2017; Vakharia et al., 2018). In adulthood, the burden of disease is related to musculoskeletal symptoms due to osteomalacia (de Menezes Filho et al., 2006), osteoarthritis, and lower-limb muscle weakness, but also ossification of entheses (enthesophytes), this latter feature being a strong determinant of impairment in quality of life (QoL) in this population (Schott and Wills, 1976; Reid et al., 1989; Sullivan et al., 1992; Amora et al., 2006; Bergwitz and Juppner, 2009; Che et al., 2016; Skrinar et al., 2019). Enthesophytes form on the spine, peripheral joints and Achilles tendons, and are responsible for chronic pain, joint limitations, stiffness and disability (Figure 1; Che et al., 2016).
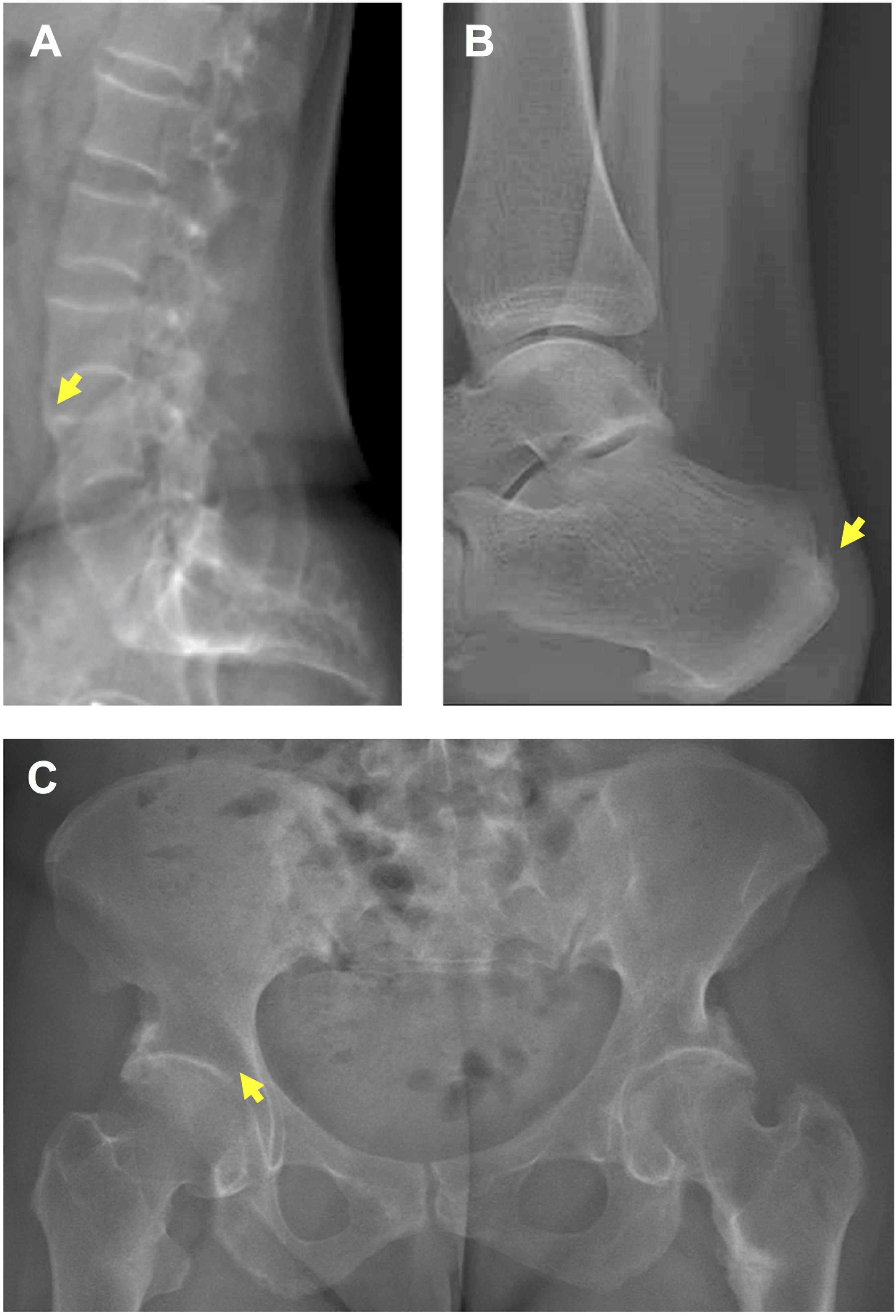
Figure 1. Enthesopathies and osteoarthritis in a 36 year-old female patient with XLH. (A) Spinal enthesophytes (yellow arrow). (B) Enthesophyte (yellow arrow) on the Achille’s tendon. (C) Early osteoarthritis (yellow arrow).
The current medical treatment, which is commonly prescribed from early childhood to the end of growth, seeks to address the deficiencies associated with this disease, i.e., oral phosphorus supplementation with multiple daily intakes to compensate for renal phosphate wasting and active vitamin D analogs (alfacalcidol or calcitriol) to counteract the 1,25-(OH)2-vitamin D deficiency (Glorieux et al., 1980; Friedman et al., 1993; Makitie et al., 2003; Liu et al., 2016). Nonetheless, despite this treatment during growth, musculoskeletal symptoms due to enthesopathies and osteoarthritis remain the main manifestations in the clinical progression of XLH (Carpenter, 2012; Linglart et al., 2014; Connor et al., 2015). It is unclear whether or not enthesophyte formation can be modified by using phosphate and vitamin D analogs. In an observational cross-sectional study in 52 adults with XLH, neither the presence nor the absence of enthesopathies was associated with conventional treatment of XLH, whatever the treatment duration (Connor et al., 2015). The pathogenesis underlying the development and progression of enthesopathies and osteoarthritis in patients with XLH has yet to be investigated.
The hypophosphatemic (Hyp) mouse, a murine model of XLH that harbors a spontaneous Phex mutation, recapitulates biochemical and skeletal features of XLH (Eicher et al., 1976; Strom et al., 1997). Although the expansion of mineralizing enthesis fibrocartilage of the calcaneus and osteoarthropathy of the tibial articular cartilage have been described (Liang et al., 2009; Liu et al., 2018), there are no data on spinal and other peripheral features in this model. Thus, in this study, we sought to describe the natural history of enthesopathies and joint alterations in XLH, using a 12 month (M) longitudinal follow-up of Hyp mice.
The Hyp mouse model B6.Cg-Phex Hyp/J was obtained from the Jackson Laboratory (# 000528). Heterozygous breeding was carried out and tail snips were collected for genotyping. DNA was extracted from the snips using a DNeasy Blood and Tissue Kit (Qiagen, France) and the genotype was determined by PCR using primers for Phex (Sheen et al., 2012). Male wild-type (WT) and Hyp littermates were used for this study. Chemical methods were used for mice sacrifice according to the ethical protocol approved by the Animal Care Committee of French Veterinary Services (DPP Haut de Seine, France: agreement number C-9204901).
All mice were housed under conditions of controlled temperature (23 ± 2°C) and light (12:12 h light/dark cycle), with unlimited access to water and standard pelleted food (1.20% calcium and 0.83% phosphorus, rodent diet 3800PMS10, Provimi Kliba, Kaiseraugst, Switzerland).
Male WT and Hyp mice (n = 5 per group) were scanned at M3, 6, 9 and 12 using a high-resolution X-ray micro-computer tomography (micro-CT) system (Quantum FX Caliper, Life Sciences, Perkin Elmer, Waltham, MA) at the Life Imaging Platform (PIV), UR 2496, Montrouge, France. Standard acquisition settings were used (90 kV, 160 mA) and scans were performed with a field of view alternately focused on the right hind paw (180 s scan time, 20 μm3 voxel size) or hip (120 s, 50 μm3), or covering the full body (36 s, 236 μm3). Micro-CT data sets were analyzed using the built-in multiplanar reconstruction tool, OsiriX 5.8 (Pixmeo, Switzerland) in order to generate a series of images aligned over time for each anatomical region of each animal. Axial and coronal images of the sacroiliac joints and hip joints, sagittal images of the spine, and axial and sagittal images of the hind paw were reconstructed. The analysis was focused on these areas because they are the most common sites of structural involvement in adult patients with XLH.
Two rheumatologists specialized in the field of rare bone diseases and bone inflammatory diseases assessed hip joint alterations (defined as the presence of osteophytes on joint margins or narrowing of the joint space or altered shape of the bone ends), and enthesopathies (defined as new bone formation from the enthesis) on spine or paw, erosion of the sacroiliac joint and periarticular calcification. Both readers were blind to the status of the mouse (Hyp vs. WT) but were aware of the different analysis time points (M3, M6, M9, and M12). A semi-quantitative score was established, ranging from 0 (normal) to 3 (most severe feature assessed) for spinal enthesophytes and joint evaluation and from 0 (absent) to 2 (present) for peripheral calcifications and enthesophytes (see Table 1). When applicable, both sides were scored and a resulting mean was calculated. In the follow-up score, up to 1 point could be added in cases of a 50% increase in the size of the enthesophyte or calcification between two consecutive time points. A composite score (from 0 to 18) was built based on these parameters for each time point (see reading grid in Table 1). Observers were calibrated during a preliminary 3 h training session on micro-CT reading. In the event of disagreement between the two readers, a third also analyzed the image and a consensus was reached between the three readers.
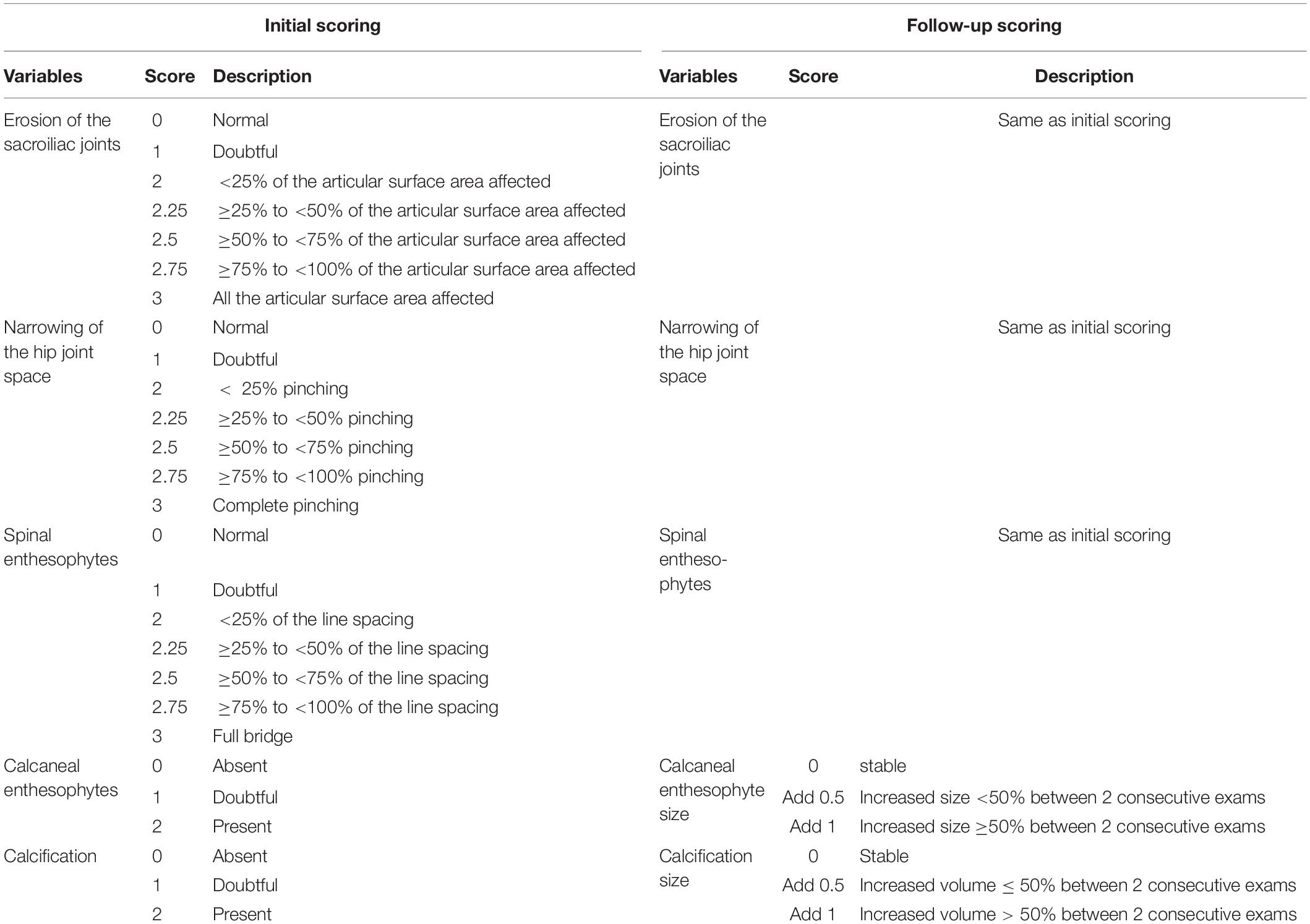
Table 1. Scoring grid of enthesopathies, erosions, ossifications, and narrowing of joint.
The angle of dorsolumbar kyphosis was defined for each mouse at M3, M6, M9, and M12. Using sagittal images of mice spines from full body CT scans, endplate orientations of thoracic and lumbar vertebrae were marked using ImageJ (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, MD, United States1, 1997–2016). The apical thoracic vertebrae of the rachis were identified and the kyphosis angle defined by the means of (1) the tangent of the cranial endplate of the 4th thoracic vertebra and (2) the tangent to the caudal endplate of the 4th lumbar vertebra, from the apical thoracic vertebra. A MATLAB script (MATLAB R2012b, The MathWorks Inc., Natick, MA, 2000) was used to measure these angles in all mice at each age.
Bones were fixed overnight at 4°C in 70% ethanol solution and dehydrated in a graded ethanol series. Undecalcified samples were embedded in methyl methacrylate (Merck, Rahway, NJ). Serial sections, 4 μm thick, were cut on a microtome (Polycut E microtome, Leica, Wetzlar, Germany). Series of consecutive sections representative of micro-CT images were stained respectively with toluidine blue (pH 3.8), von Kossa reagent (5% silver nitrate solution, Sigma-Aldrich, St Louis, MO), Masson’s trichrome staining (Sigma-Aldrich, St Louis, MO) and safranin O staining (Weigert’s iron hematoxylin, Sigma-Aldrich, St Louis, MO; Fast Green, Prolabo, Paris, France; and Safranin O solution, Sigma-Aldrich, St Louis, MO).
Sections embedded in methyl methacrylate were deplasticized in methyl glycol acetate and rehydrated in a graded ethanol series to pure distilled water. Sections were incubated in a moist chamber for 12 h at 4°C with goat anti-human osteopontin (OPN) antibody (R&D Systems, Minneapolis, MN, United States) diluted at 5 μg/mL in 10% goat serum/PBS-T, or goat anti-human sclerostin (SOST) primary antibody (R&D Systems, Minneapolis, MN, United States) diluted at 5 μg/mL in 10% donkey serum with 1% BSA/PBS-T. Sections were then incubated with peroxidase-conjugated anti-IgG diluted at 1/200 and Donkey anti Goat IgG secondary antibody Texas red (Thermo Fisher Scientific) diluted at 1/1000, respectively. Control incubations to assess non-specific staining involved the same procedures except that the primary antibody was replaced by non-immune serum.
GraphPad Prism (GraphPad Prism version 6.0 for Mac) was used for statistical analysis using 2-way ANOVA followed by multiple comparison tests. Differences were considered significant at p < 0.05. Data are expressed as mean ± SD.
A longitudinal follow-up was performed by micro-CT of the skeleton of Hyp and control mice. Hip joint alterations, enthesopathies on spine or hind paw, erosion of the sacroiliac joint and periarticular calcification were assessed following the semi-quantitative score detailed in the reading grid in Table 1. Enthesopathies, erosions and osteophytes were detected in all Hyp mice from M3 whereas no abnormalities were observed in control mice. The total composite score indicates, overall, a clear increase in structural severity over time in the Hyp mice (Figure 2A). On the other hand, the pattern of lesions differed between these mice (Figure 2B). All of them had a high sacroiliac joint score for bone erosion, i.e., a mean score of 2.5 and 2.85 out of 3 at M3 and M12, respectively. Four mice out of five had hip joint osteoarthritis and/or tarsal calcifications with a mean score of 0 and 1 at M3, and 1.8 and 2.1 out of 3 at M12, respectively. Structural damage of sacroiliac and hip joints was strictly symmetrical.
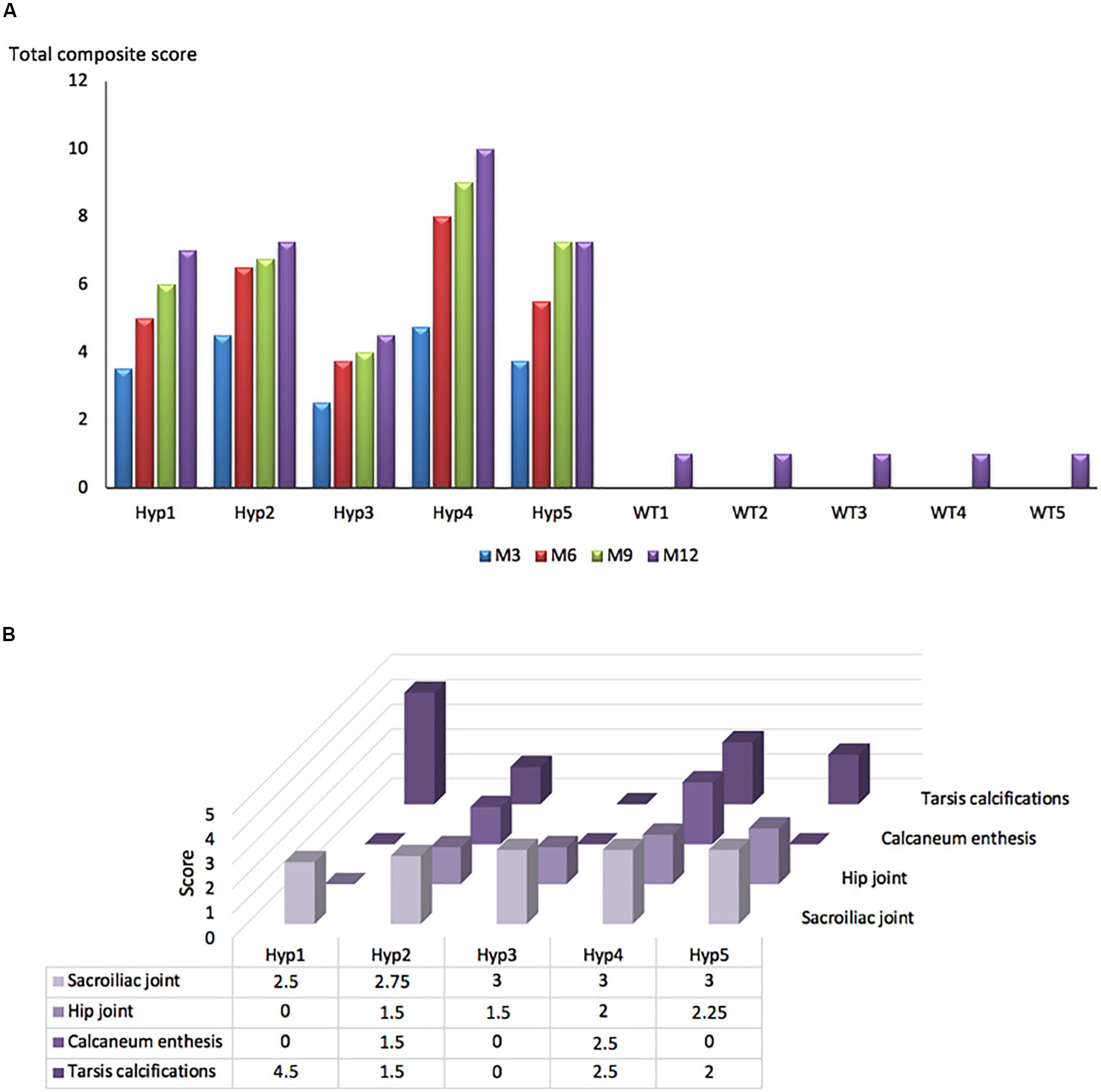
Figure 2. Longitudinal changes in the total composite score in Hyp and WT mice. (A) A total composite score was established following the scoring grid (Table 1) for each mouse at each time point (M3, 6, 9, 12). Enthesophytes, erosions and ossifications were observed in each Hyp mouse and no abnormalities were observed in controls. These lesions were already present at M3, the composite score increasing continuously over time. (B) Scores of enthesopathies, erosions, ossifications, and narrowing of joint established following the scoring grid (Table 1) for each Hyp mouse at M12. No spinal enthesophytes were observed. The pattern of lesions differed between Hyp mice.
Micro-CT follow-up revealed that calcaneal enthesophytes formed early and expanded over time in Hyp mice (Figure 3A). Two out of five already had visible calcaneal enthesophytes at M3. Histological sections showed mineralizing fibrochondrocytes expanding into both Achilles tendon and plantar fascia ligament insertions of calcaneal tuberosity in all Hyp mice at M12 (Figure 3B).
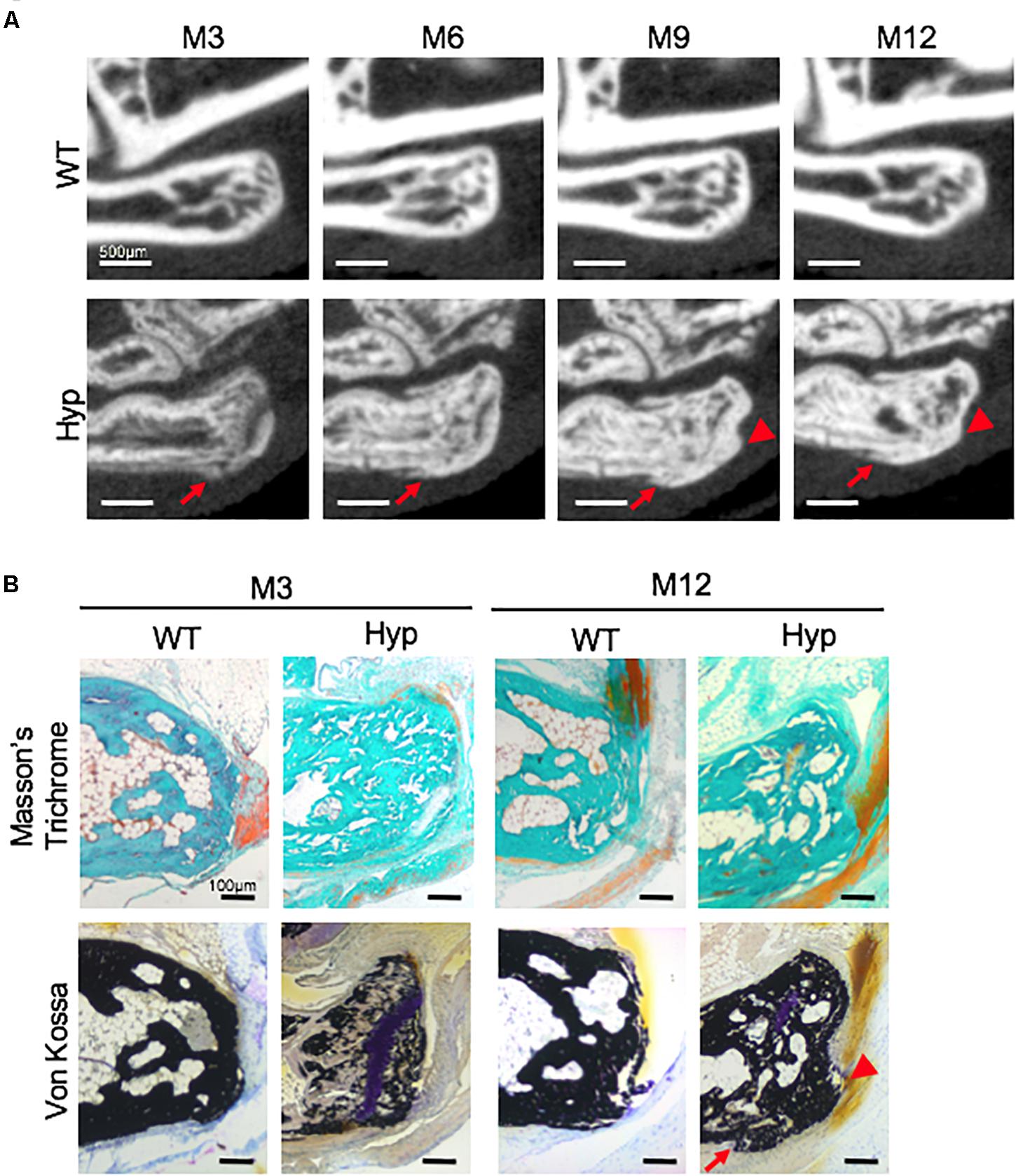
Figure 3. Calcaneal enthesophytes in Hyp mice. (A) Achilles (red arrowheads) and plantar fascia enthesophytes (red arrows) developed from M3 to M12 in Hyp mice (representative micro-CT sections from Hyp#4 mouse). In contrast, no enthesis ossification was found in WT littermates (scale bars, 500 μm). (B) Masson’s trichrome and Von Kossa staining of undecalcified sections of calcaneal area at M3 and M12 in WT and Hyp mice. Von Kossa staining confirmed the cellular expansion of mineralizing fibrochondrocytes into Achilles tendon (red arrowhead) and plantar fascia ligament (red arrow) insertions of the calcaneal tuberosity in Hyp mice (scale bars, 100 μm).
Hyp mice developed a significantly larger kyphosis angle from M9 compared to that in WT mice (Figure 4). Although no spine ossifications were observed, we highlighted hypertrophy of the vertebral body on histological staining of Hyp mouse vertebrae (Supplementary Figure 1).
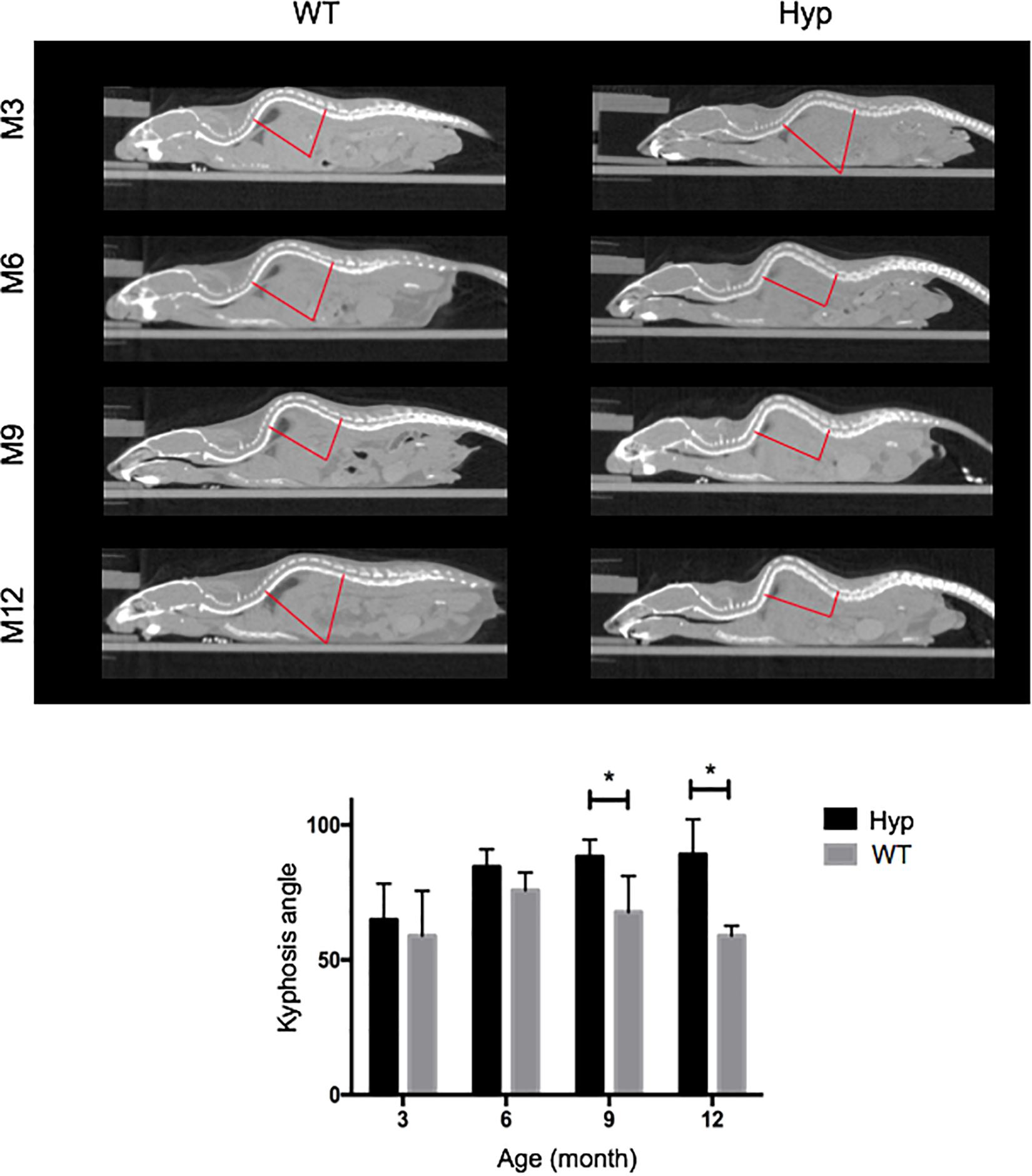
Figure 4. Angle of dorsolumbar kyphosis of mice at M3, 6, 9, and 12. The kyphosis angle was significantly larger from M9 in Hyp mice than WT mice (*p < 0.05) (representative micro-CT sections from Hyp#2 mouse).
Ossification delay at the femoral head was observed at M3 and M6 (Figure 5A) and confirmed by histological analyses at M3 (Figure 5B). Safranin O staining showed an abnormal thick layer of hypertrophic chondrocytes at M3 in Hyp mice, whereas complete ossification of femoral heads was observed in WT mice as confirmed by Von Kossa staining. Micro-CT analyses and Von Kossa staining showed bony outgrowths at the margin of the hip joint in Hyp mice at M12 (Figure 5).
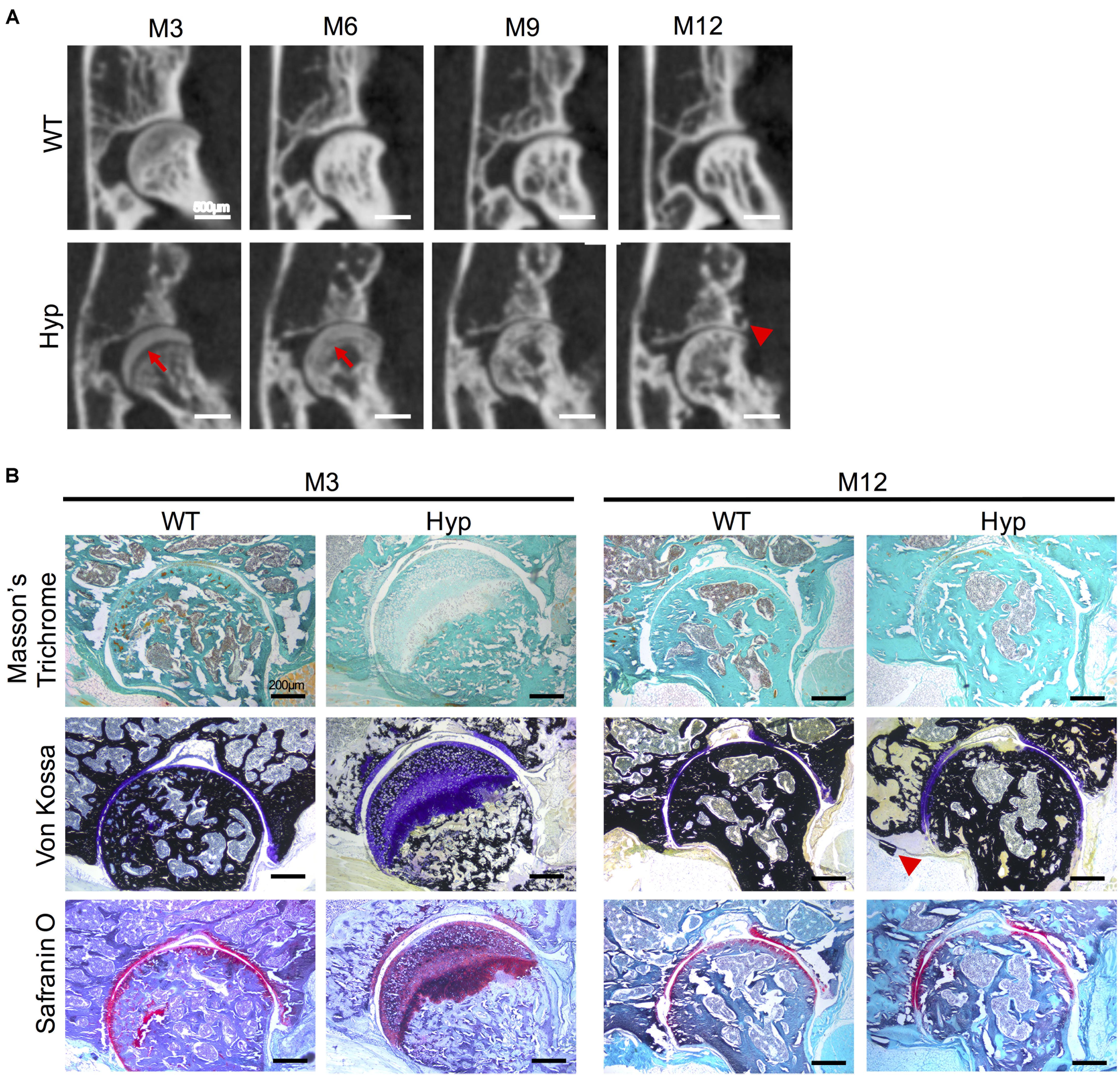
Figure 5. Features of coxofemoral joint of Hyp and WT mice. (A) Delayed ossification was observed in Hyp mice on micro-CT (red arrows). New bone formation was noticeable at M12 in Hyp mice (red arrowhead) (representative micro-CT sections from Hyp#2 mouse) (scale bars, 500 μm). (B) Histological staining confirmed micro-CT observations. High thickness of the non-mineralized cartilage at M3 on Von Kossa staining was evident in Hyp mice which is confirmed by safranin O staining. At M12, the new bone formation was also observable on Von Kossa staining in Hyp mice (red arrowhead) (scale bars, 200 μm).
Micro-CT follow-up showed a progressive erosion of the sacroiliac joint with irregular articular surfaces reflecting progressive structural damage in Hyp mice compared to controls (Figure 6). These types of damage were present in all the Hyp mice as soon as M3 and worsened up to M12. Detailed images obtained from this in vivo imaging approach provided a valuable tool to study sacroiliac joint alterations (Figure 6A). Both Masson’s trichrome and Von Kossa staining showed severe osteomalacia at Hyp sacroiliac bone and revealed that Hyp bone is mainly composed of osteoid matrix at M3 and M12. Safranin O staining showed joint surface damage in Hyp mice, characterized by an altered staining of articular cartilage appearing discontinuous along joint surface from M3 and M12 samples (Figure 6B).
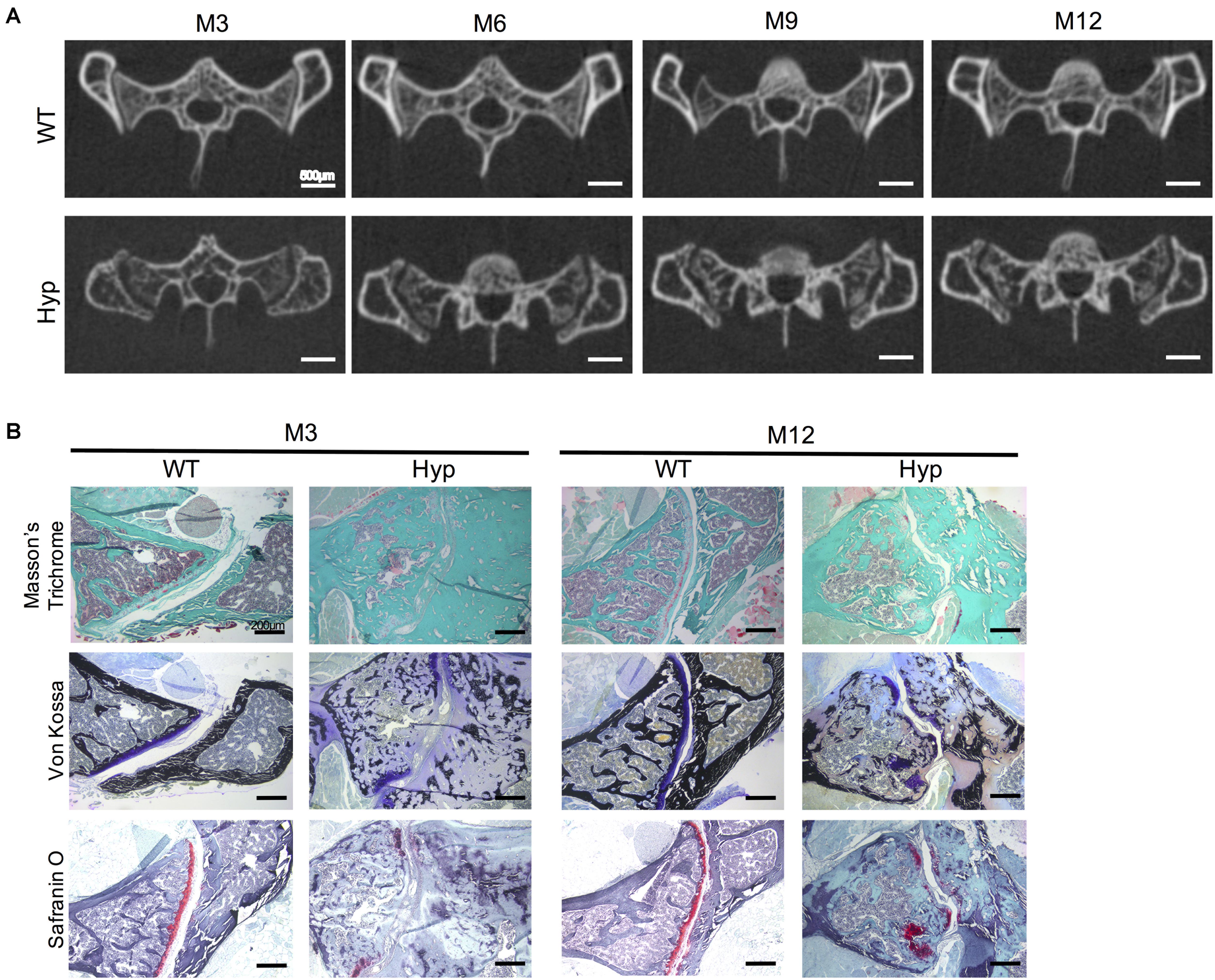
Figure 6. Features of sacroiliac joint of Hyp and WT mice. (A) Micro-CT analysis showed bone erosion of the sacroiliac joint in Hyp mice which appears at early stages and progressed up to M12 (representative micro-CT sections from Hyp#2 mouse) (scale bars, 500 μm). (B) Masson’s trichrome staining displayed that sacroiliac bone of Hyp mice presented a large amount of collagen matrix compared to WT mice. Von Kossa staining showed a weak mineralization (in dark) of the Hyp collagen matrix revealing an enlarged osteoid characteristic of features of osteomalacia bone. Safranin O staining revealed high wear of cartilage at M3 in Hyp mice (scale bars, 200 μm).
Micro-CT follow up revealed abundant new bone formation on the anterior-inferior side of the tibia in Hyp mice, already detectable at M3 and worsening over time up to M12 (Supplementary Figure 2A). This bone formation was confirmed by Masson’s trichrome and Von Kossa staining revealing mineralized collagenous matrix at M3 and M12 (Supplementary Figure 2B).
Periarticular calcifications were also observed in various regions such as the tarsus in the hind paw (Supplementary Figure 3). In contrast, no ectopic bone formation was observed in WT mice even at the end of follow-up.
In order to investigate the mechanisms underlying abnormal bone formation in XLH, proteins involved in the mineralization process, namely, osteopontin (OPN) and sclerostin, were analyzed in the calcaneus and the sacroiliac joints in control and Hyp mice (Moester et al., 2010; Boukpessi et al., 2017). Faint expression of OPN was observed in the Hyp calcaneal enthesis, likely associated with the ossification of the enthesis, whereas in WT mice the calcaneum displayed strong OPN labeling (Figure 7A). In the sacroiliac joint, OPN immunostaining appeared stronger than that in WT mice and was associated with the mineralized matrix rather than the enlarged osteoid matrix (Figure 7B). In Hyp mice, a very faint expression of sclerostin was observed in osteocytes and fibrochondrocytes in the calcaneum and in osteocytes in the cortical bone of the sacroiliac joint, whereas a strong staining of both cells was seen in WT mice (Figure 8).
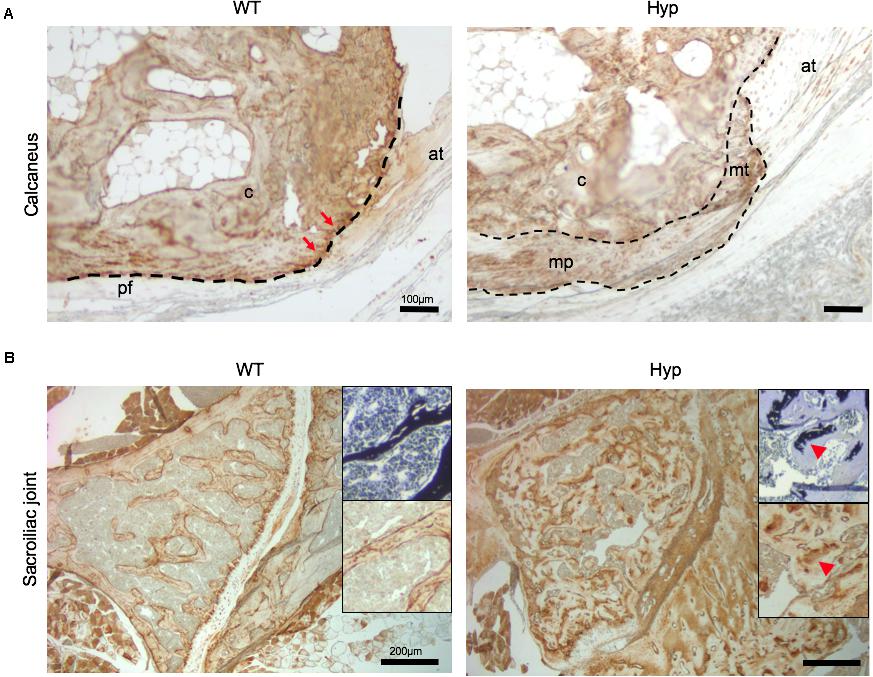
Figure 7. OPN expression in osteoarticular lesions in WT and Hyp mice. (A) WT calcaneal enthesis showed a strong OPN expression at tidemark (red arrows) between uncalcified and calcified fibrocartilage compared to Hyp mice at M12. Cellular expansion of mineralizing fibrochondrocytes of both Achilles tendon and plantar fascia ligament insertions into the calcaneal tuberosity showed faint OPN expression in Hyp mice. pf, plantar fascia ligament; at, Achilles tendon; mt, mineralized fibrochondrocytes of Achilles tendon; mp, mineralized fibrochondrocytes of plantar fascia ligament; c, calcaneal tuberosity; t, tendon (scale bars, 100 μm). (B) OPN immunostaining in Hyp mice showed a strong expression in sacroiliac joint compared to that in WT mice at M3. The staining was associated with the mineralized matrix rather than the enlarged osteoid matrix (red arrowheads) (scale bars, 200 μm).
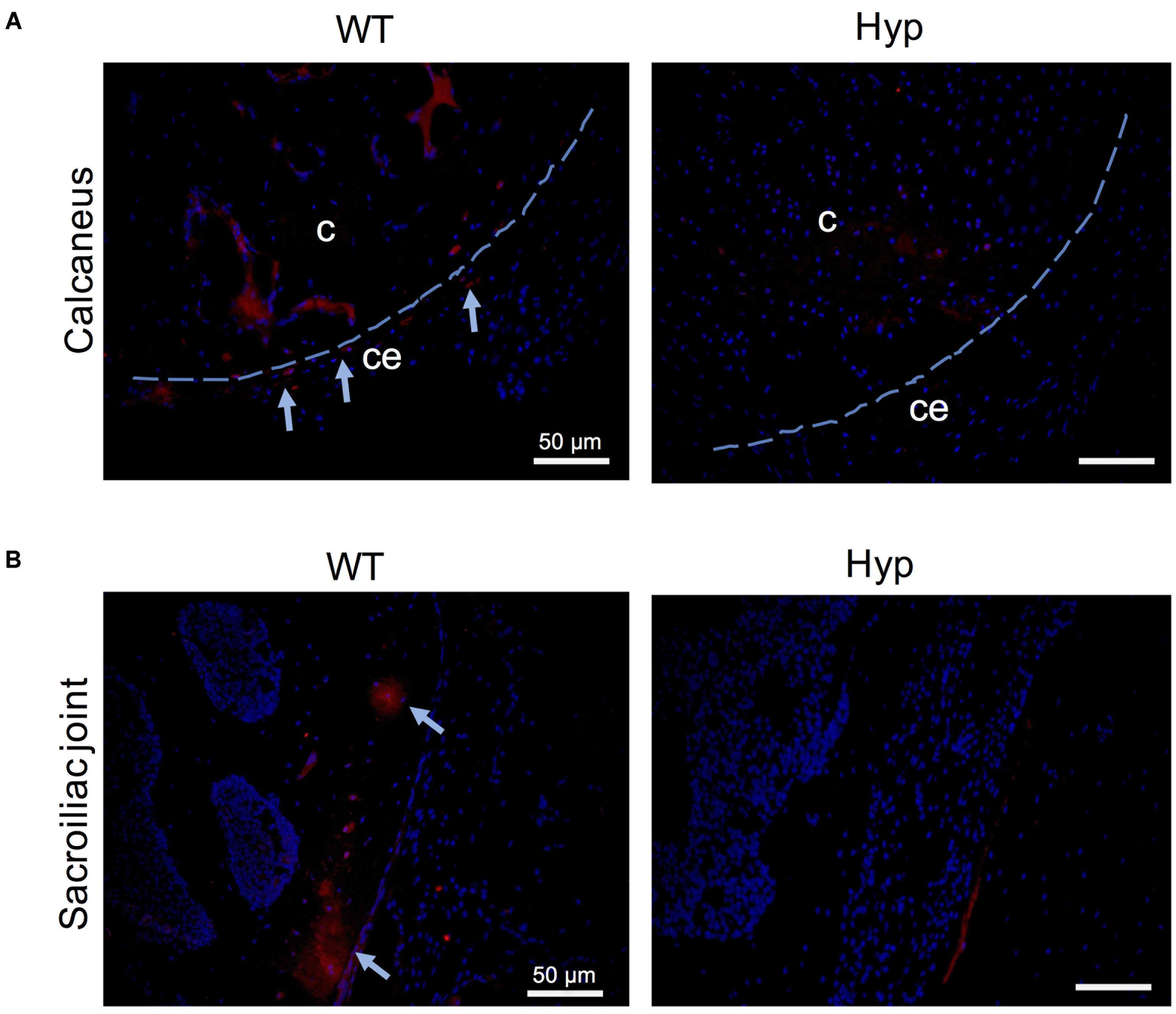
Figure 8. Sclerostin expression in osteoarticular lesions in Hyp mice at M3. In WT mice, sclerostin immunolabeled osteocytes and fibrochondrocytes were observed in the calcaneum (A) and in the cortical bone of the sacroiliac joint (B) (both in the bony part and in the transitional zone between bone and enthesis, indicated with blue arrows). In contrast, very weak expression of sclerostin was seen in these cells in the Hyp calcaneal enthesis and sacroiliac joint. c, calcaneus; ce, calcaneal enthesis (scale bars, 50 μm).
In the present study, we report that Hyp mice develop enthesopathies, joint ossifications and bone erosions relatively early in life, unlike WT mice. In addition, a progressive worsening of these manifestations was observed over time. These skeletal complications related to early osteoarthritis and enthesopathies phenocopy skeletal features of adult patients with XLH (Figure 1) (Skrinar et al., 2019). Despite their variable severity, these manifestations strongly impair the QoL of adults with XLH similar to the impairment seen in patients with chronic diseases (Che et al., 2016), introducing a novel parameter for the monitoring of therapeutic interventions in XLH. Employing intraindividual follow-up with high-resolution micro-CT and histology on undecalcified bone samples, we documented the natural history of manifestations such as osteoarthritis and enthesopathies in Hyp mice. Detailed images obtained by in vivo imaging provide a valuable tool for studying structural damage of the sacroiliac joints.
Early features of osteoarthritis are found in up to 85.4% of young adult patients with XLH (Che et al., 2016). In our study, degenerative lesions of the sacroiliac joint were common and developed early in Hyp mice. Histological sections revealed that this area was strongly affected in Hyp mice, including osteomalacia with accumulation of osteoid matrix and insufficient mineralized tissue. The joints between the sacrum and the ilium are joined by several ligaments and consist of a lower fibrocartilaginous part and an upper ligamentous part. As such, this region is prone to mechanical stress. In the underlying bone under normal conditions, osteocytes are entrapped within the mineralized bone matrix showing a specific morphology and molecular signature. These features allow them to serve as a bone response mechanism for mechanical stress in their microenvironment. Sclerostin, a primarily osteocyte product, is widely considered a key mechanotransduction molecule, whose expression is suppressed by mechanical loading (Moester et al., 2010). Interestingly, we detected a weak expression of sclerostin in Hyp mouse sacroiliac joints. This finding could be associated with (1) altered transduction of the mechanical loading due to osteomalacia, or (2) an immature osteocyte phenotype due to the inhibition of the extracellular matrix mineralization. Similar mechanisms have been described when mineralization of osteoid is inhibited by administration of bisphosphonate etidronate (Irie et al., 2008). It is worth noting that a recent study suggests a key role of sclerostin in FGF23 regulation, phosphate metabolism and XLH pathobiology (Carpenter and Ross, 2020).
OPN is an acidic, phosphorylated, calcium-binding, extracellular matrix protein from the small integrin-binding ligand, N-linked glycoprotein (SIBLING) family (Fisher and Fedarko, 2003; Qin et al., 2004), and has been proposed as a key player in XLH pathobiology. OPN mineralization-inhibiting functions are regulated through its degradation by the endopeptidase PHEX for which it is a substrate (Addison et al., 2010; Barros et al., 2013). Indeed, OPN and its mineralization-inhibiting peptides have been shown to accumulate in XLH/Hyp bone matrix, including in periosteocytic lesions (Barros et al., 2013; Boukpessi et al., 2016), and in interglobular dentin (Boukpessi et al., 2010; Salmon et al., 2014), contributing locally to the matrix mineralization defect independent of systemic Pi−regulating factors (Hoac et al., 2020). In the present study, the investigation of the sacroiliac joint showed that, in addition to this abnormal accumulation of OPN in hypomineralized bone regions, this protein was also strongly expressed within the mineralized matrix (Salmon et al., 2014; Boukpessi et al., 2016). Taken together, this might suggest a differential regulation of OPN depending on the mechanical constraints the tissue studied is under.
Enthesophytes are diagnosed in up to 85% of adults with XLH on X-ray and are usually associated with a poor QoL (Che et al., 2016). Here, we performed for the first time a follow-up of enthesopathies by micro-CT in Hyp mice over 12 months. Our study allowed the identification of severe calcaneal enthesopathies in 2/5 mice, consistent with the considerable variability observed in clinical manifestations in humans (Martos Moreno et al., 2016). Histological sections showed mineralizing fibrochondrocytes of the calcaneum in Hyp mice (Liang et al., 2009; Liu et al., 2018). Interestingly, sclerostin, which is a potent inhibitor of bone formation down-regulated by mechanical loading, was weakly expressed in Hyp calcaneal enthesis. Moreover, OPN localization also appeared to be altered in Hyp calcaneal enthesis compared to that in WT mice. Our results support the view that enthesis in XLH patients present an abnormal balance of mineralization factors. Enthesis lesions might develop because of additional stimulation such as biomechanical stress (Jacques et al., 2014).
In contrast, we did not observe axial enthesophytes at the spine in Hyp mice, although there was spinal curvature deformation. In humans, however, spinal enthesophytes are a major concern in relation to adult manifestations and QoL, being present in ∼64% of adults with XLH (Che et al., 2016). This specific feature of the Hyp spine might be explained by the quadruped locomotion, which strongly modifies spinal constraints compared to those in humans, suggesting that axial enthesophyte formation is probably associated with impaired mechanical loading in humans.
Our study widens the prospects of XLH treatments. Specifically, the conventional medical treatment (phosphorus supplementation and active vitamin D analogs) is commonly prescribed from early childhood to the end of growth and seeks to counteract consequences of FGF23 excess (Linglart et al., 2014). Although biological tests to monitor XLH are performed to avoid complications such as hypercalciuria or hyperparathyroidism, to date, there is no consensus on the indications and duration of treatment in adults (Carpenter et al., 2011). This murine model of XLH appears to be a suitable preclinical model for assessing the long-term effect of therapies on adult skeletal manifestations and developing effective preventive strategies.
A recent therapeutic strategy was developed to neutralize FGF23 action using monoclonal anti-FGF23 antibodies in children and adults with XLH (Carpenter et al., 2014; Imel et al., 2019). In Hyp mice, repeated s.c. administration of FGF23 antibody rescues the bone and mineral phenotype, including circulating phosphate levels. The effect of anti-FGF23 antibodies on severe XLH manifestations that strongly impact patient QoL remains to be explored. In particular, the Hyp mouse model will make it possible to study the local effect of the new drugs on extracellular matrix mineralization defects due to PHEX deficiency (Coyac et al., 2018). In this context, the study of FGF23 involvement in mineralization defect at the tissue level through the Klotho/FGF-R pathway will be all the more important.
In conclusion, we report early and progressive axial deformation and peripheral enthesophytes in Hyp mice. This appears to be a relevant model (i) to investigate pathobiological mechanisms underlying development and severity of enthesopathies and joint structural damage, and (ii) for preclinical studies seeking to test the impact of conventional and new therapies on the development of such manifestations. Further, these findings suggest a new paradigm in which mechanical constraints contribute to the development of enthesophytes and bone structural alterations in XLH. This opens avenues for a new field of mechanotransduction-associated pathways in XLH and improves knowledge of the natural history of this disease that ranks alongside causal understanding in importance for prevention and control.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
The animal study was reviewed and approved by the Animal Care Committee of French Veterinary Services (DPP Haut de Seine, France: agreement number C-9204901).
BS, CM-R, KB, CC, and CB contributed to the design of the experiments. CB, AC, BS, OF, VZ, AB, TS, CR, AL, CM-R, CC, KB, and CB performed and analyzed experiments. KB and CB wrote the manuscript with contributions from all authors. KB and CB supervised the project. All authors reviewed and approved the final version of the manuscript.
This study was supported by the Agence Nationale de la Recherche (Grant HYPOSKEL 18-CE14-0018-01), Fondation pour la Recherche Médicale (Grant DGE20111123012), and Axelle Cauliez was supported by the Société Française d’Endocrinologie et de Diabétologie Pédiatrique (SFEDP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MM declared a past collaboration with several of the authors CC, CB, and AL to the handling Editor.
We gratefully acknowledge Lotfi Slimani and Jérémy Sadoine (UR 2496 and Plateforme d’Imagerie du Vivant (PIV), Université de Paris, France for their help producing Micro-CT data, Annie Llorens, Brigitte Baroukh, Sandy Ribes for their kind assistance for histological and immunofluorescence techniques, and the Société Française de Biologie des Tissus Minéralisés (SFBTM).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.00854/full#supplementary-material
Addison, W. N., Masica, D. L., Gray, J. J., and Mckee, M. D. (2010). Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J. Bone Miner. Res. 25, 695–705.
Amora, D. N., Sousa, T. M., Martins, A. M., Barbosa, P. S., Magalhaes, M. R., Toyama, M. H., et al. (2006). Effects of Crotalus durissus collilineatus venom in the isolated rat kidney. Toxicon 47, 260–264. doi: 10.1016/j.toxicon.2005.10.007
Barros, N. M., Hoac, B., Neves, R. L., Addison, W. N., Assis, D. M., Murshed, M., et al. (2013). Proteolytic processing of osteopontin by PHEX and accumulation of osteopontin fragments in Hyp mouse bone, the murine model of X-linked hypophosphatemia. J. Bone Miner. Res. 28, 688–699. doi: 10.1002/jbmr.1766
Bergwitz, C., and Juppner, H. (2009). Disorders of phosphate homeostasis and tissue mineralisation. Endocr. Dev. 16, 133–156. doi: 10.1159/000223693
Boukpessi, T., Gaucher, C., Leger, T., Salmon, B., Le Faouder, J., Willig, C., et al. (2010). Abnormal presence of the matrix extracellular phosphoglycoprotein-derived acidic serine- and aspartate-rich motif peptide in human hypophosphatemic dentin. Am. J. Pathol. 177, 803–812. doi: 10.2353/ajpath.2010.091231
Boukpessi, T., Hoac, B., Coyac, B. R., Leger, T., Garcia, C., Wicart, P., et al. (2016). Osteopontin and the dento-osseous pathobiology of X-linked hypophosphatemia. Bone 95, 151–161.
Boukpessi, T., Hoac, B., Coyac, B. R., Leger, T., Garcia, C., Wicart, P., et al. (2017). Osteopontin and the dento-osseous pathobiology of X-linked hypophosphatemia. Bone 95, 151–161. doi: 10.1016/j.bone.2016.11.019
Carpenter, K. A., and Ross, R. D. (2020). Sclerostin antibody treatment increases bone mass and normalizes circulating phosphate levels in growing hyp mice. J. Bone Miner. Res. 35, 596–607. doi: 10.1002/jbmr.3923
Carpenter, T. O. (2012). The expanding family of hypophosphatemic syndromes. J. Bone Miner. Metab. 30, 1–9. doi: 10.1007/s00774-011-0340-2
Carpenter, T. O., Imel, E. A., Holm, I. A., Jan De Beur, S. M., and Insogna, K. L. (2011). A clinician’s guide to X-linked hypophosphatemia. J. Bone Miner. Res. 26, 1381–1388.
Carpenter, T. O., Imel, E. A., Ruppe, M. D., Weber, T. J., Klausner, M. A., Wooddell, M. M., et al. (2014). Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J. Clin. Invest. 124, 1587–1597. doi: 10.1172/jci72829
Che, H., Roux, C., Etcheto, A., Rothenbuhler, A., Kamenicky, P., Linglart, A., et al. (2016). Impaired quality of life in adults with X-linked hypophosphatemia and skeletal symptoms. Eur. J. Endocrinol. 174, 325–333. doi: 10.1530/eje-15-0661
Connor, J., Olear, E. A., Insogna, K. L., Katz, L., Baker, S., Kaur, R., et al. (2015). Conventional therapy in adults With X-linked hypophosphatemia: effects on enthesopathy and dental disease. J. Clin. Endocrinol. Metab. 100, 3625–3632. doi: 10.1210/jc.2015-2199
Coyac, B. R., Hoac, B., Chafey, P., Falgayrac, G., Slimani, L., Rowe, P. S., et al. (2018). Defective mineralization in X-linked hypophosphatemia dental pulp cell cultures. J. Dent. Res. 97, 184–191. doi: 10.1177/0022034517728497
de Menezes Filho, H., De Castro, L. C., and Damiani, D. (2006). Hypophosphatemic rickets and osteomalacia. Arq. Bras. Endocrinol. Metabol. 50, 802–813.
Eicher, E. M., Southard, J. L., Scriver, C. R., and Glorieux, F. H. (1976). Hypophosphatemia: mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets. Proc. Natl. Acad. Sci. U.S.A. 73, 4667–4671. doi: 10.1073/pnas.73.12.4667
Fisher, L. W., and Fedarko, N. S. (2003). Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect. Tissue Res. 44(Suppl. 1), 33–40. doi: 10.1080/713713644
Friedman, N. E., Lobaugh, B., and Drezner, M. K. (1993). Effects of calcitriol and phosphorus therapy on the growth of patients with X-linked hypophosphatemia. J. Clin. Endocrinol. Metab. 76, 839–844. doi: 10.1210/jc.76.4.839
Fuente, R., Gil-Pena, H., Claramunt-Taberner, D., Hernandez, O., Fernandez-Iglesias, A., Alonso-Duran, L., et al. (2017). X-linked hypophosphatemia and growth. Rev. Endocr. Metab. Disord. 18, 107–115.
Gaucher, C., Walrant-Debray, O., Nguyen, T. M., Esterle, L., Garabedian, M., and Jehan, F. (2009). PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum. Genet. 125, 401–411. doi: 10.1007/s00439-009-0631-z
Glass, L. R., Dagi, T. F., and Dagi, L. R. (2011). Papilledema in the setting of x-linked hypophosphatemic rickets with craniosynostosis. Case Rep. Ophthalmol. 2, 376–381. doi: 10.1159/000334941
Glorieux, F. H., Marie, P. J., Pettifor, J. M., and Delvin, E. E. (1980). Bone response to phosphate salts, ergocalciferol, and calcitriol in hypophosphatemic vitamin D-resistant rickets. N. Engl. J. Med. 303, 1023–1031. doi: 10.1056/nejm198010303031802
Hoac, B., Ostergaard, M., Wittig, N. K., Boukpessi, T., Buss, D. J., Chaussain, C., et al. (2020). Genetic ablation of osteopontin in osteomalacic Hyp mice partially rescues the deficient mineralization without correcting hypophosphatemia. J. Bone Miner. Res. [Epub ahead of print].
Imel, E. A., Glorieux, F. H., Whyte, M. P., Munns, C. F., Ward, L. M., Nilsson, O., et al. (2019). Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet 393, 2416–2427.
Irie, K., Ejiri, S., Sakakura, Y., Shibui, T., and Yajima, T. (2008). Matrix mineralization as a trigger for osteocyte maturation. J. Histochem. Cytochem. 56, 561–567. doi: 10.1369/jhc.2008.950527
Jacques, P., Lambrecht, S., Verheugen, E., Pauwels, E., Kollias, G., Armaka, M., et al. (2014). Proof of concept: enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann. Rheum Dis. 73, 437–445. doi: 10.1136/annrheumdis-2013-203643
Liang, G., Katz, L. D., Insogna, K. L., Carpenter, T. O., and Macica, C. M. (2009). Survey of the enthesopathy of X-linked hypophosphatemia and its characterization in Hyp mice. Calcif. Tissue Int. 85, 235–246. doi: 10.1007/s00223-009-9270-6
Linglart, A., Biosse-Duplan, M., Briot, K., Chaussain, C., Esterle, L., Guillaume-Czitrom, S., et al. (2014). Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr. Connect. 3, R13–R30.
Liu, E. S., Martins, J. S., Raimann, A., Chae, B. T., Brooks, D. J., Jorgetti, V., et al. (2016). 1,25-dihydroxyvitamin D alone improves skeletal growth, microarchitecture, and strength in a murine model of XLH, despite enhanced FGF23 expression. J. Bone Miner. Res. 31, 929–939. doi: 10.1002/jbmr.2783
Liu, E. S., Martins, J. S., Zhang, W., and Demay, M. B. (2018). Molecular analysis of enthesopathy in a mouse model of hypophosphatemic rickets. Development 145:dev163519. doi: 10.1242/dev.163519
Makitie, O., Doria, A., Kooh, S. W., Cole, W. G., Daneman, A., and Sochett, E. (2003). Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J. Clin. Endocrinol. Metab. 88, 3591–3597. doi: 10.1210/jc.2003-030036
Martos Moreno, G. A., Aparicio, C., De Lucas, C., Gil Pena, H., and Argente, J. (2016). [X-linked hypophosphatemic rickets due to mutations in PHEX: clinical and evolutionary variability]. An. Pediatr. 85, 41–43. doi: 10.1016/j.anpede.2016.04.002
Moester, M. J., Papapoulos, S. E., Lowik, C. W., and Van Bezooijen, R. L. (2010). Sclerostin: current knowledge and future perspectives. Calcif. Tissue Int. 87, 99–107. doi: 10.1007/s00223-010-9372-1
Polisson, R. P., Martinez, S., Khoury, M., Harrell, R. M., Lyles, K. W., Friedman, N., et al. (1985). Calcification of entheses associated with X-linked hypophosphatemic osteomalacia. N. Engl. J. Med. 313, 1–6. doi: 10.1056/nejm198507043130101
Qin, C., Baba, O., and Butler, W. T. (2004). Post-translational modifications of sibling proteins and their roles in osteogenesis and dentinogenesis. Crit. Rev. Oral Biol. Med. 15, 126–136. doi: 10.1177/154411130401500302
Reid, I. R., Hardy, D. C., Murphy, W. A., Teitelbaum, S. L., Bergfeld, M. A., and Whyte, M. P. (1989). X-linked hypophosphatemia: a clinical, biochemical, and histopathologic assessment of morbidity in adults. Medicine 68, 336–352. doi: 10.1097/00005792-198911000-00002
Sabbagh, Y., Jones, A. O., and Tenenhouse, H. S. (2000). PHEXdb, a locus-specific database for mutations causing X-linked hypophosphatemia. Hum. Mutat. 16, 1–6. doi: 10.1002/1098-1004(200007)16:1<1::aid-humu1>3.0.co;2-j
Salmon, B., Bardet, C., Coyac, B. R., Baroukh, B., Naji, J., Rowe, P. S., et al. (2014). Abnormal osteopontin and matrix extracellular phosphoglycoprotein localization, and odontoblast differentiation, in X-linked hypophosphatemic teeth. Connect. Tissue Res. 55(Suppl. 1), 79–82. doi: 10.3109/03008207.2014.923864
Schott, G. D., and Wills, M. R. (1976). Muscle weakness in osteomalacia. Lancet 1, 626–629. doi: 10.1016/s0140-6736(76)90428-1
Sheen, C. R., Pilarowski, G. O., Wang, W., and Millan, J. L. (2012). Molecular characterisation of the Hyp deletion and an improved assay for its detection. Bone 50, 592–595. doi: 10.1016/j.bone.2011.11.018
Skrinar, A., Dvorak-Ewell, M., Evins, A., Macica, C., Linglart, A., Imel, E. A., et al. (2019). The lifelong impact of X-linked hypophosphatemia: results from a burden of disease survey. J. Endocr. Soc. 3, 1321–1334. doi: 10.1210/js.2018-00365
Strom, T. M., Francis, F., Lorenz, B., Boddrich, A., Econs, M. J., Lehrach, H., et al. (1997). Pex gene deletions in Gy and Hyp mice provide mouse models for X-linked hypophosphatemia. Hum. Mol. Genet. 6, 165–171. doi: 10.1093/hmg/6.2.165
Sullivan, W., Carpenter, T., Glorieux, F., Travers, R., and Insogna, K. (1992). A prospective trial of phosphate and 1,25-dihydroxyvitamin D3 therapy in symptomatic adults with X-linked hypophosphatemic rickets. J. Clin. Endocrinol. Metab. 75, 879–885. doi: 10.1210/jc.75.3.879
Vakharia, J. D., Matlock, K., Taylor, H. O., Backeljauw, P. F., and Topor, L. S. (2018). Craniosynostosis as the presenting feature of X-linked hypophosphatemic rickets. Pediatrics 141, S515–S519.
Keywords: Hyp mouse, natural history, enthesophyte, joint alteration, XLH
Citation: Faraji-Bellée C-A, Cauliez A, Salmon B, Fogel O, Zhukouskaya V, Benoit A, Schinke T, Roux C, Linglart A, Miceli-Richard C, Chaussain C, Briot K and Bardet C (2020) Development of Enthesopathies and Joint Structural Damage in a Murine Model of X-Linked Hypophosphatemia. Front. Cell Dev. Biol. 8:854. doi: 10.3389/fcell.2020.00854
Received: 05 June 2020; Accepted: 10 August 2020;
Published: 22 September 2020.
Edited by:
Svetlana Komarova, McGill University, CanadaReviewed by:
Toshimi Michigami, Osaka Women’s and Children’s Hospital, JapanCopyright © 2020 Faraji-Bellée, Cauliez, Salmon, Fogel, Zhukouskaya, Benoit, Schinke, Roux, Linglart, Miceli-Richard, Chaussain, Briot and Bardet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claire Bardet, Y2xhaXJlLmJhcmRldEB1LXBhcmlzLmZy; Karine Briot, a2FyaW5lLmJyaW90QGFwaHAuZnI=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.