
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 28 August 2020
Sec. Epigenomics and Epigenetics
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00842
 Gangqiang Guo1,2†
Gangqiang Guo1,2† Xinyu Shi2†
Xinyu Shi2† Huijing Wang3†Lele Ye2,4Xinya Tong2Kejing Yan2
Huijing Wang3†Lele Ye2,4Xinya Tong2Kejing Yan2 Ning Ding2
Ning Ding2 Chaosheng Chen5
Chaosheng Chen5 Huidi Zhang5*
Huidi Zhang5* Xiangyang Xue2*
Xiangyang Xue2*The emerging epitranscriptome plays an essential role in autoimmune disease. As a novel mRNA modification, N4-acetylcytidine (ac4C) could promote mRNA stability and translational efficiency. However, whether epigenetic mechanisms of RNA ac4C modification are involved in systemic lupus erythematosus (SLE) remains unclear. Herein, we detected eleven modifications in CD4+ T cells of SLE patients using mass spectrometry (LC-MS/MS). Furthermore, using samples from four CD4+ T cell pools, we identified lower modification of ac4C mRNA in SLE patients as compared to that in healthy controls (HCs). Meanwhile, significantly lower mRNA acetyltransferase NAT10 expression was detected in lupus CD4+ T cells by RT-qPCR. We then illustrated the transcriptome-wide ac4C profile in CD4+ T cells of SLE patients by ac4C-RIP-Seq and found ac4C distribution in mRNA transcripts to be highly conserved and enriched in mRNA coding sequence regions. Using bioinformatics analysis, the 3879 and 4073 ac4C hyper-acetylated and hypoacetylated peaks found in SLE samples, respectively, were found to be significantly involved in SLE-related function enrichments, including multiple metabolic and transcription-related processes, ROS-induced cellular signaling, apoptosis signaling, and NF-κB signaling. Moreover, we demonstrated the ac4C-modified regulatory network of gene biological functions in lupus CD4+ T cells. Notably, we determined that the 26 upregulated genes with hyperacetylation played essential roles in autoimmune diseases and disease-related processes. Additionally, the unique ac4C-related transcripts, including USP18, GPX1, and RGL1, regulate mRNA catabolic processes and translational initiation. Our study identified novel dysregulated ac4C mRNAs associated with critical immune and inflammatory responses, that have translational potential in lupus CD4+ T cells. Hence, our findings reveal transcriptional significance and potential therapeutic targets of mRNA ac4C modifications in SLE pathogenesis.
Systemic lupus erythematosus (SLE) is a heterogenous autoimmune disease with complicated clinical manifestations and high mortality, and is associated with development of autoantibodies and inflammatory responses resulting in damage to multiple organs and systems (Tsokos et al., 2016). Accumulating evidence indicates that the pathogenesis and etiology of this systemic disease are highly intricate, being influenced by several factors including genetic susceptibility, sex hormones, environmental risk factors, and immunological mechanisms (Rönnblom and Elkon, 2010; Liu and Davidson, 2012; Tsokos et al., 2016). Of these factors, imbalanced innate and adaptive immune responses, including immune cells (CD4+ T cells, B cells, and Th17 cells), cytokines (type I interferon and interleukin) and complement proteins contribute to inflammatory processes and tissue injury (Morel, 2017; Song et al., 2020). During this process, epigenetic abnormities can directly influence the autoimmune reaction by dysregulating immune cell function, particularly in CD4+ T lymphocytes (Wardowska, 2020).
Currently, our advanced understanding of the genetic mechanism underlying SLE indicated that epigenetic changes, such as disease-associated susceptibility gene regulation, non-coding RNA regulation, and post-transcriptional modifications (Meroni and Penatti, 2016; Mazzone et al., 2019), play important roles in SLE. For example, our previous study demonstrated that abnormally expressed microRNAs and non-coding circular RNAs (circRNAs) are involved in SLE pathogenesis and are potential diagnostic markers (Guo et al., 2018, 2019). Moreover, several studies have found that epigenetic changes, such as DNA and mRNA modifications are dysregulated and can alter gene expression in SLE CD4+ T cells (Zhang et al., 2013; Zhao et al., 2014, 2016). Recently, we also identified novel aberrant mRNA modification, 5-methylcytidine (m5C), linked to critical immune pathways in lupus CD4+ T cells (Guo et al., 2020).
Focusing on the epitranscriptome, mRNAs comprise a diverse array of posttranscriptional chemical modifications, which contribute to RNA structure and function alteration, as well as gene expression dysregulation (Peer et al., 2017; Zhao et al., 2017). Several regulatory complexities, including N6-methyladenosine (m6A), m5C, 5-hydroxymethylcytidine (hm5C), and N4-acetylcytidine (ac4C), have been identified as conserved in all domains of life (Delatte et al., 2016; Dominissini and Rechavi, 2017; Arango et al., 2018; Chen X. et al., 2019). Specifically, N-acetyltransferase 10 (NAT10) is reportedly the only known acetyltransferase for ac4C in human tRNAs and rRNAs (Chimnaronk et al., 2009; Ito et al., 2014). Moreover, a recent study identified ac4C as a novel mRNA modification catalyzed by NAT10, that promotes mRNA stability and translational efficiency (Arango et al., 2018). However, whether ac4C modification occurs in CD4+ T cells of SLE patients, and the specific correlation between mRNA ac4C modification and SLE pathogenesis is poorly understood.
In this study, using liquid chromatography-coupled tandem mass spectrometry (LC-MS/MS), we identified ac4C mRNA modification in lupus CD4+ T cells as a novel form of mRNA modification. Further, we utilized ac4C-RIP-Seq approach to measure global profile of ac4C modification in CD4+ T cells from SLE patients and healthy controls (HCs). Our results demonstrated prominent variation of ac4C patterns in SLE and showed that the modified mRNA was significantly enriched in disease-associated functional pathways. Taken together, we demonstrate that acetylation cytidine mRNA, a new class of the epitranscriptome in SLE, can potentially be a promising diagnostic marker and therapeutic target.
In this study, we recruited 39 patients with SLE from the First Affiliated Hospital of Wenzhou Medical University, between December 2019 and June 2020. All SLE patients fulfilled the diagnostic criteria of the American College of Rheumatology (ACR). In addition, clinical data were assessed at the time of blood collection. A total of 51 age- and sex-matched HCs without renal failure, heart failure, or arthralgia, and free from any inflammatory conditions, were recruited from the same hospital. For mRNA liquid chromatography-coupled triple quadrupole tandem mass spectrometry (LC-MS/MS) and RNA ac4C-RIP-Seq, 10 SLE patients and 18 HCs were divided into four groups as our previous report (Guo et al., 2020). For each SLE group, five SLE samples were mixed and sequenced; similarly, each control group comprised nine mixed and sequenced. To quantify NAT10 expression levels, samples from 20 SLE patients and 21 HCs were analyzed via RT-qPCR. Meanwhile, to validate genes with dys-acetylated sites, samples from 9 SLE patients and 12 HCs were utilized for ac4C-RIP-qPCR. This study was approved by the Medical Ethical Committees of the First Affiliated Hospital of Wenzhou Medical University (No. 2019121). All participants provided written informed consent.
Peripheral blood mononuclear cell (PBMCs) were isolated from HCs and SLE patients using human peripheral blood lymphocyte separation medium (Tianjin HaoYang Biological Manufacture, Tianjin, China) within 6 h of sample collection. CD4+ T cells were then purified following immunomagnetic separation using a human CD4+ T cell isolation kit (BD Biosciences, San Jose, CA, United States) according to the manufacturer’s instructions. For RNA purification, the isolated human CD4+ T cells were lysed in TRIzol reagent (Invitrogen Life Technologies, Grand Island, NY, United States). The extracted RNA was further digested by DNase I (Invitrogen, Waltham, MA, United States) to remove residual DNA, and subsequently separated from each sample using TRIzol reagent/RNeasy Mini kit (Qiagen, Hilden, Germany). Total extracted RNA was stored for use at −80°C.
Total RNA was quantified and qualified using NanoDrop ND-1000 (Thermo Fisher Scientific Inc., Waltham, MA, United States) and Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, United States). mRNA was isolated from total RNA using the NEBNext Poly(A) mRNA Magnetic Isolation Module (E7490; New England Biolabs, Inc., Ipswich, MA, United States) according to the manufacturer’s protocol. The detailed experimental methods and analyses monitoring peak information of utilized modified nucleosides were similar to those used in our recent study (Guo et al., 2020).
The NAT10 expression levels in CD4+ T cells of 41 individuals (21 HCs and 20 SLE patients) were measured by RT-qPCR using an Applied Biosystems QuantStudioTM 3 real-time PCR instrument (Thermo Fisher Scientific Inc.). All RT-qPCR experiments were performed using the QuantiNova SYBR Green PCR kit (Qiagen, Hilden, Germany). For each reaction, 1 μL of diluted cDNA was mixed with 10 μL of 2 × SYBR Green PCR Master Mix. A final volume of 20 μL was achieved by the addition of 1.4 μL forward and reverse primers (10 μmol). The conditions for PCR amplification were as follows: 95°C for 2 min, followed by 40 cycles each of 95°C for 5 s and 60°C for 10 s. All samples were tested in triplicates. The data were analyzed by the comparative threshold cycle (Ct) method. GAPDH was utilized as the control, and the relative quantification of NAT10 in CD4+ T cells was calculated using the following equation: amount of target = 2–ΔCt, where ΔCt = CtNAT10 − CtGAPDH. The gene-specific primers of NAT10 and GAPDH used for RT-qPCR were listed in Supplementary Table S1.
Briefly, mRNA enrichment from the total RNA was performed using the DynabeadsTM mRNA Purification kit (invitrogen) as per the manufacturer’s instructions and then subjected to immunoprecipitation (IP). RNA was randomly fragmented to ∼200 nt by RNA fragmentation reagents. The RNA fragments were incubated with the ac4C antibody (ab252215, Abcam, Cambridge, United Kingdom) by rotating at 4°C for 2 h. The mixture was then immunoprecipitated by incubation with Protein A/G beads (Thermo Fisher Scientific, MA, United States) at 4°C for an additional 2 h. Subsequently, the bound RNA were eluted from the beads and purified with Trizol reagent (Thermo Fisher) according to the manufacturer’s instructions. RNA libraries for IP and input samples were then constructed with NEBNext® Ultra II Directional RNA Library Prep kit (New England Biolabs, Inc., United States) according to the manufacturer’s instructions. Libraries were qualified using Agilent 2100 bioanalyzer and then subjected to 150-bp paired-end sequencing on an Illumina HiSeq 4000 sequencer (Illumina, Inc., San Diego, CA, United States), and were controlled by Q30. After 3′ adaptor-trimming and removing low quality reads using cutadapt software (v1.9.3), clean reads of all libraries were aligned to the reference genome (HG19) by Hisat2 software (v2.0.4) (Kim et al., 2015). Acetylated sites on RNAs (peaks) were identified by MACS software (Feng et al., 2012). Differentially acetylated peaks with a fold change cutoff > 2 and P < 0.0001 were identified by diffReps (Shen et al., 2013). Identified ac4C peaks were subjected to motif enrichment analysis by DREME (Bailey, 2011), and metagene ac4C distribution was characterized by R package MetaPlotR (Olarerin-George and Jaffrey, 2017). Moreover, NGS for mRNA profiling was also performed using input samples. Differentially expressed genes (DEGs) were defined with a fold change ≥ 1.5, and P < 0.05. The detailed analyses have been reported in our previous study (Guo et al., 2018).
To further explore the essential role of ac4C mRNA modification in SLE, DEGs were separated into four groups based on whether the modifications resulted in hyper- or hypoacetylation of ac4C, and based on the up- or down-regulation of gene expression (fold change > 1.5; P < 0.05). The signaling network in SLE was assessed with Thomson Reuters database1 (Schuierer et al., 2010). GO analysis was performed by Metascape2 and DAVID (version 6.83). GO terms with P < 0.05 were considered statistically significant. The PPI network analysis of DEGs from the four groups mentioned above for assessing generation, pathway enrichment, and visualization were performed as previously described (Han et al., 2020).
Twenty micrograms of total RNA was extracted from 9 SLE patients and 12 HCs and subjected to ac4C-RIP, as described above. The mRNA enriched with ac4C in each sample was determined via RT-qPCR, and quantified by normalizing to nine-fold input. The gene-specific qPCR primer sequences are presented in Supplementary Table S1.
Statistical analysis was performed using SPSS 22.0 software (IBM, Armonk, NY, United States). To assess the significant difference between two groups, a two-sided independent-samples t-test or two-sided Mann–Whitney U-test were performed as indicated. A two-sided P-value < 0.05 was regarded as a statistically significant difference.
We combined equal amounts of mRNA from the CD4+ T cells of five SLE patients or nine HCs, into one pool for each group, thus obtaining four separate CD4+ T cell pools as previously described (Zhao et al., 2014). We first utilized LC-MS/MS to generate mRNA modification for the four separate pools and identified mRNA modification in SLE patients and HCs. A total of 11 modifications, previously identified in mRNA, were detected in the two groups including ac4C, 2′-O-methylcytidine (Cm) and hm5C (Figures 1A,B). Compared with that in HCs, the inosine (I) level in CD4+ T cells of SLE was slightly elevated whereas that of other modifications were obviously decreased (Figure 1A). Based on a previous report (Arango et al., 2018), ac4C in eukaryotes functions in the regulation of gene expression by enhancing stability and translation of mRNA, potentially at the level of decoding efficiency. Therefore, we also evaluated the ac4C level in overall mRNA of patients with SLE. Compared to those of HCs, both the ac4C/C and ac4C/AUGC levels in poly (A) RNA of CD4+ T cells were significantly lower in SLE patients (P < 0.05; Figure 1C). Moreover, compared with that in HCs, the expression level of acetyltransferase NAT10 was markedly decreased in CD4+ T cells from SLE patients (P < 0.001; Figure 1D). These results suggested that NAT10 expression might be associated with the alternation of global ac4C levels in SLE.
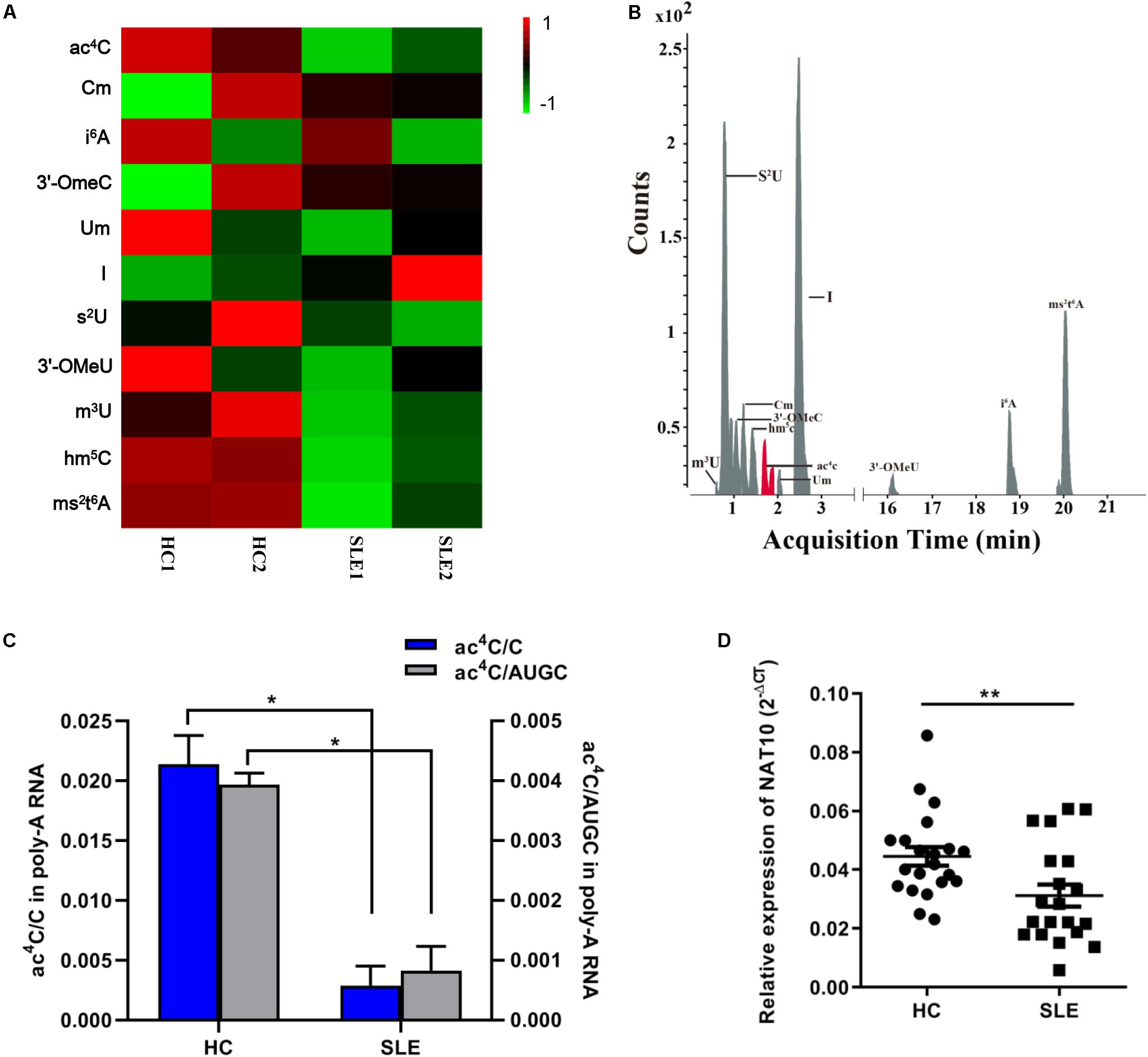
Figure 1. Overview of mRNA ac4C acetylation mapping between systemic lupus erythematosus (SLE) patients and healthy controls (HCs). (A) Heatmap of normalized abundance (modification/canonical nucleotide) of 11 mRNA modification forms detected through LC-MS/MS in CD4+ T cells from SLE patients and HCs. Green indicates a low z-score whereas red indicates a high z-score. (B). LC-MS/MS extracted ion chromatograms of modified mRNA in SLE patients and HCs. ac4C, N4-acetylcytidine; Cm, 2′-O-methylcytidine; i6A, N6-isopentenyladenosine; 3′-OmeC, 3′-O-methylcytidine; Um, 2′-O-methyluridine; I, inosine; s2U, 2-thiouridine; 3′-OMeA, 3′-O-methyladenosine; m3U, 3-methyluridine; hm5C, 5-hydroxymethylcytidine; ms2t6A, 2-methylthio-N6-threonylcarbamoyladenosine. (C) Comparation of calibrated ac4C/C and ac4C/AUGC levels in poly(A) RNA between HCs and SLE. Bars show the mean with SD of individual biological replicates (n = 2). *P < 0.05. (D) NAT10 mRNA expression level in CD4+ T cells from HCs (n = 21) and SLE patients (n = 20). Compared to HCs, NAT10 was downregulated in SLE patients. Data are presented as 2–ΔCt relative to GAPDH expression (mean with SEM; **P < 0.01).
To observe the global profile of transcriptome-wide ac4C modification, we further performed ac4C-RIP-Seq on the mRNA samples extracted from CD4+ T cell pools of SLE patients and HCs, which were the same samples as those used for the LC-MS/MS analyses. The number of overlaps and differences in ac4C-modified transcripts among the four separate biological pools are displayed in a Venn diagram (Figure 2A). A total of 3485 and 4352 acetylated genes transcripts were detected in HC replicates and SLE patients, respectively. Among all ac4C-represented transcripts within the two groups, only 1778 ac4C-modified mRNAs were identified. Compared with HCs, the SLE group lost 3269 representative acetylated gene transcripts and gained 3384 new ac4C-modified mRNAs. When compared with that in HCs, the number of ac4C-modified mRNAs were increased in CD4+ T cells from SLE patients (Figure 2B).
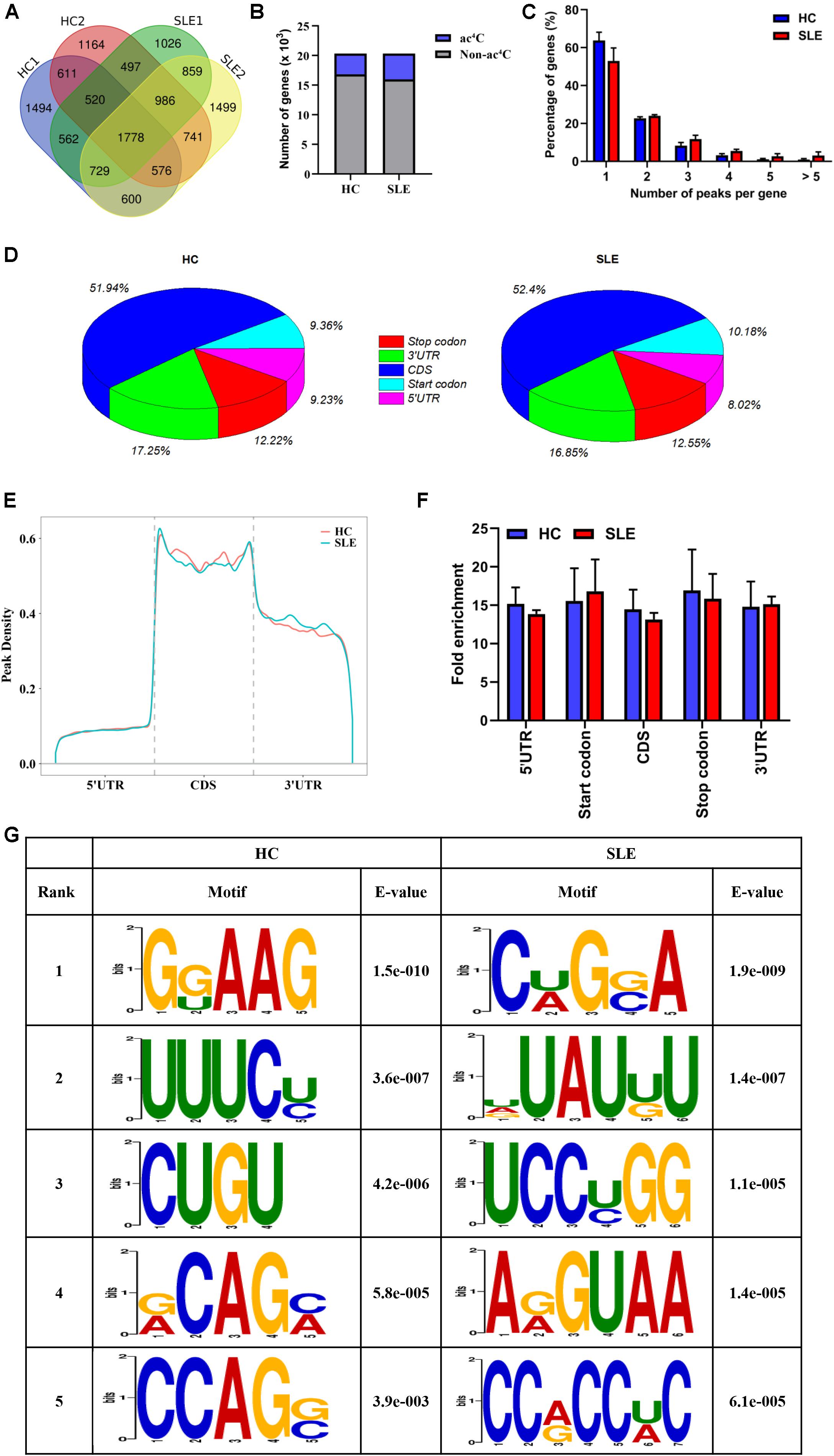
Figure 2. Transcriptome-wide ac4C-RIPSeq and distribution of ac4C peaks. (A) Venn diagram of ac4C acetylated genes in HCs and SLE patients. The numbers of HC-unique, SLE-unique, and common ac4C modified genes are shown. (B) Bar charts indicating the number of genes with no (gray) or at least one significant (blue) ac4C peak at SLE group or HC group. (C) Proportion of genes harboring different numbers of ac4C peaks in the two groups. The majority of transcripts harbor only one or two ac4C peaks per gene in HC and SLE groups. (D) Pie charts show ac4C peaks distribution in different gene context in HCs and SLE patients. The averages of percentages of mRNA ac4C peaks within 5′UTR, StartC, CDS, StopC, and 3′UTR regions in transcriptomes are shown. (E) Distribution of ac4C peaks along mRNA transcripts. The moving averages of percentages of mRNA ac4C peaks are shown. (F) Statistics of fold enrichment of ac4C peaks in five segments in the two groups. Error bars represent the mean with SD. (G) Sequence logo showing the top five differential mode motifs enriched across changed ac4C peaks identified from HCs and SLE patients.
Next, we further analyzed the distribution of ac4C peaks per gene between the two groups. Intriguingly, in the SLE and HC groups, the majority of ac4C-modified transcripts were observed to have one or two ac4C peak(s) per gene (77.06 and 86.53%, respectively), while a very small number of transcripts contained three or more ac4C peaks (Figure 2C), which is similar to the trend of m6A characteristics previously reported in abnormal mouse livers (Luo et al., 2019). Furthermore, the proportion of ac4C-modified transcripts harboring only one ac4C peak was lower in CD4+ T cells of SLE groups (63.87% vs. 53.03%) than in HCs; whereas, the percentage of transcripts harboring two or more ac4C peaks was higher. By analyzing the distribution profile of ac4C peaks within mRNAs of SLE and HC samples, ac4C peaks were divided into five segments, according to their location in RNA transcripts (Figure 2D). Notably, the ac4C peaks were primarily enriched in coding sequences (CDS), stop codons (StopC), and 3′-untranslated regions (3′UTR) in both SLE and HC groups (Figure 2E), which differs from previous observations in HeLa cells (Arango et al., 2018). Compared with those of HCs, the fold enrichment of ac4C peaks showed no distinct difference in the five regions of lupus CD4+ T cells (Figure 2F). Furthermore, we investigated ac4C-modified peaks in the consensus sequences shared between the SLE and HC groups, using HOMER software. The ac4C peaks were characterized by the GRAAG motif in HCs, and CRGRA motif in SLE patients, of which, the top five ac4C peak motifs in SLE patients comprised CCRCCR (Figure 2G), which is similar to the results previously observed in HeLa cells (Arango et al., 2018).
To explore the potential biological function of ac4C modification in SLE, the abundance of the ac4C-modified peaks between SLE and HC samples was compared. Among all ac4C peaks detected in both the groups, a total of 7952 significantly dysregulated acetylated peaks were selected for sequential study (fold change > 2; P < 0.0001). A total of 3879 ac4C hyperacetylated, and 4073 ac4C hypoacetylated peaks were detected in SLE patients and compared with those in HCs (Figure 3A). The distribution of altered ac4C peaks in the two groups revealed that the differentially ac4C-modified peaks were transcribed from all chromosomes, particularly chr1, chr2, and chr3 (Figure 3B). We further performed Pathway analysis in dys-acetylated peaks (hypo- or hyper-) from CD4+ T cells of SLE patients using GO, KEGG pathway, and Thomson Reuters database analysis. GO process results showed that both the dys-acetylated peaks were significantly enriched in the regulation of metabolic processes, including cellular metabolic processes, primary metabolic processes, and nitrogen compound metabolic processes (Figure 3C). In the Pathway map analysis, the hyperacetylated peaks were markedly associated with the modulation of TGF-β receptor signaling, oxidative stress ROS-induced cellular signaling, and signal transduction pathways involving NF-κB activation (Figure 3D). Moreover, the hypoacetylated peaks involved multiple biological pathways, including immune response B cell antigen receptor (BCR) pathway, signal transduction NF-κB activation pathway, and antigen presentation by MHC II (Figure 3D). Furthermore, the top scored network analyses of hyperacetylated peaks were found to be involved in translational processes including nucleic acid-templated transcription (18.6%), DNA-templated transcription (18.6%), and transcription by RNA polymerase II (16.3%) (Figure 3E and Supplementary Table S2). The top scored networks of hypoacetylated peaks were enriched in positive regulation of multi-organism processes (70.8%), positive regulation of responses to external stimulus (72.9%), positive regulation of NF-κB transcription factor activity (52.1%), and I-κB kinase/NF-κB signaling (50.0%) (Figure 3E and Supplementary Table S2), which agrees with the pathway map analysis. Notably, we selected the top scored networks for the hypoacetylated peaks (including ATP2C1, IKBKB, NFKB1, MAPK3, and TLR2) that were enriched in positive regulation of NF-κB transcription factor activity and I-κB kinase/NF κB signaling, and quantified the acetylation levels of these peaks by ac4C-RIP-qPCR. All of these peaks in CD4+ T cells of SLE patients were found to be hypoacetylated when compared to those in HCs. Particularly, IKBKB, NFKB1, and MAPK3 in CD4+ T cells of SLE patients were significantly hypoacetylated, which was consistent with the results of the ac4C-RIP profiles (Supplementary Figure S1). Taken together, it indicated that these differentially acetylated peaks are significantly enriched in crucial disease-related functional pathways.
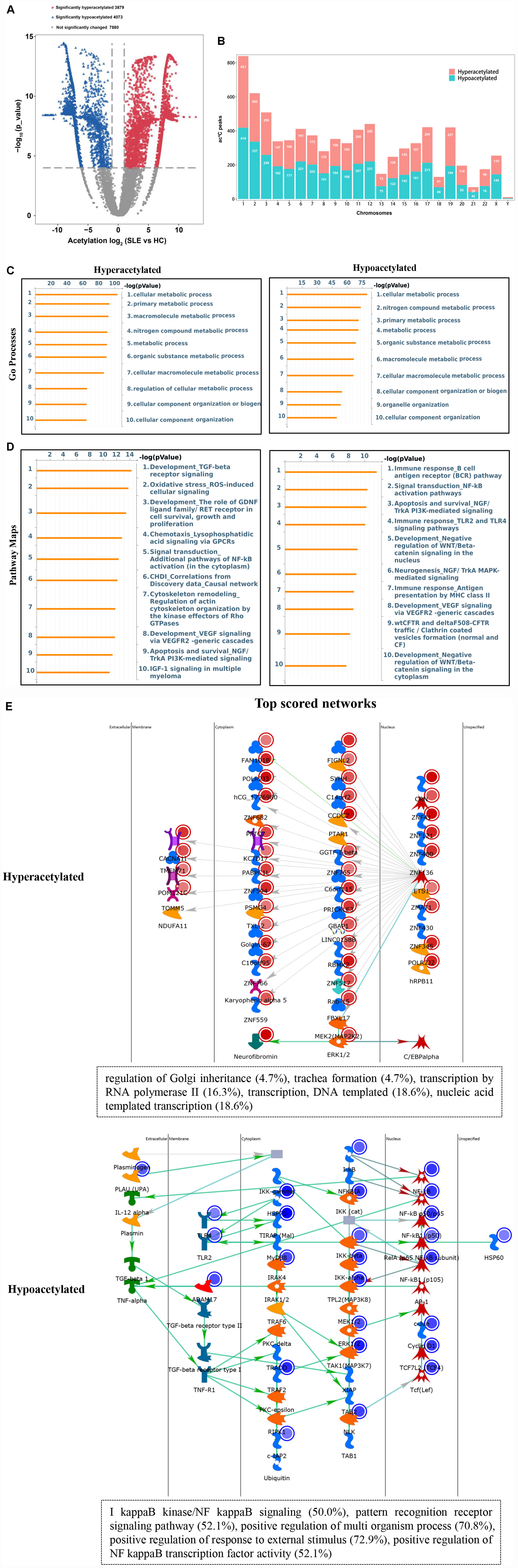
Figure 3. Pathway analysis of differentially acetylated genes. (A) Volcano plots display the differential ac4C peaks with statistical significance (fold change > 2; P < 0.0001). Blue points indicate significantly hypoacetylated transcripts and red points indicate significantly hyperacetylated transcripts. (B) The distributions of altered ac4C peaks in chromosomes of SLE patients. (C) Gene Ontology (GO) process analysis of differentially acetylated genes. Left panel represents hyperacetylated transcripts and right panel represents hypoacetylated transcripts. (D) Pathway Maps analysis of differentially acetylated genes. Left panel represents hyperacetylated transcripts and right panel represents hypoacetylated transcripts. (E) Top scored networks analysis of differentially acetylated genes. Top panel represents hyperacetylated transcripts and bottom panel represents hypoacetylated transcripts.
To obtain the disease-related regulatory pathway of altered genes in lupus CD4+ T cells, we further investigated the functional enrichment of DEGs using Pathway maps, and Process Networks analyses. By analyzing the abnormally expressed mRNA profiles of the CD4+ T cells from SLE and HC groups, a total of 674 upregulated mRNAs, and 292 downregulated mRNAs were identified (fold change > 1.5; P < 0.05; Supplementary Figure S2A). Similar to previous reports on peripheral blood mononuclear cells (PBMCs) of SLE patients (Guo et al., 2018), pathway analysis showed that DEGs were associated with immune-related pathways (NETosis in SLE, antigen presentation by MHC class I and class II, IL-5 signaling via JAK/STAT, and IFN-alpha/beta signaling via MAPKs) and inflammatory processes (neutrophil activation, phagocytosis, chemotaxis, complement system) (Supplementary Figures S2B,C). Consistent with differentially acetylated peaks, DEGs were also found to be relevant to several functional pathways including signal transduction, antigen presentation by MHC II, and positive regulation of NF-kB transcription factor activity.
We then investigated whether ac4C-modified transcripts were associated with gene expression levels. To this end, we combined the RNA-Seq data of altered genes with the RNA ac4C-RIP-Seq data for the ac4C hyperacetylation or hypoacetylation, and found that approximately 75% (249/329) of the genes with ac4C hyperacetylation or hypoacetylation exhibited upregulated expression levels. Next, we defined all abnormally expressed/modified genes into four sets: upregulated genes with ac4C hypoacetylation (hypo-up set, n = 109), upregulated genes with ac4C hyperacetylation (hyper-up set, n = 140), downregulated genes with ac4C hypoacetylation (hypo-down set, n = 54), and downregulated genes with ac4C hyperacetylation (hyper-down set, n = 26) (Supplementary Figure S3). However, the data did not show a linear relationship between acetylation and expression (R = 0.139). To further investigate the biological and molecular function of ac4C-modified DEGs in SLE patients, comprehensive functional enrichment analysis was carried out. The PPI network and GO analyses of processes associated with the four gene sets apparently linked multiple functional processes and pathways (Figure 4 and Supplementary Tables S3, S4). We found that the genes of the hyper-down set were associated with macrophage chemotaxis, negative regulation of epithelial cell migration, and amino acid catabolic process; whereas, genes in the hypo-down set were associated with calcium signaling pathway and regulation of small GTPase mediated signal transduction. Genes in the hypo-up set were associated with multiple metabolic activities, including regulated exocytosis, small molecule catabolic processes, and secondary metabolite biosynthetic processes. Meanwhile, the genes included in the hyper-up set were significantly associated with macrophage activity and cytokine-mediated signaling pathway, thereby contributing to the recruitment and activation of inflammatory cells and immune responses. These results suggested that DEGs with ac4C modification affect key functional processes and pathways that related to immunology.
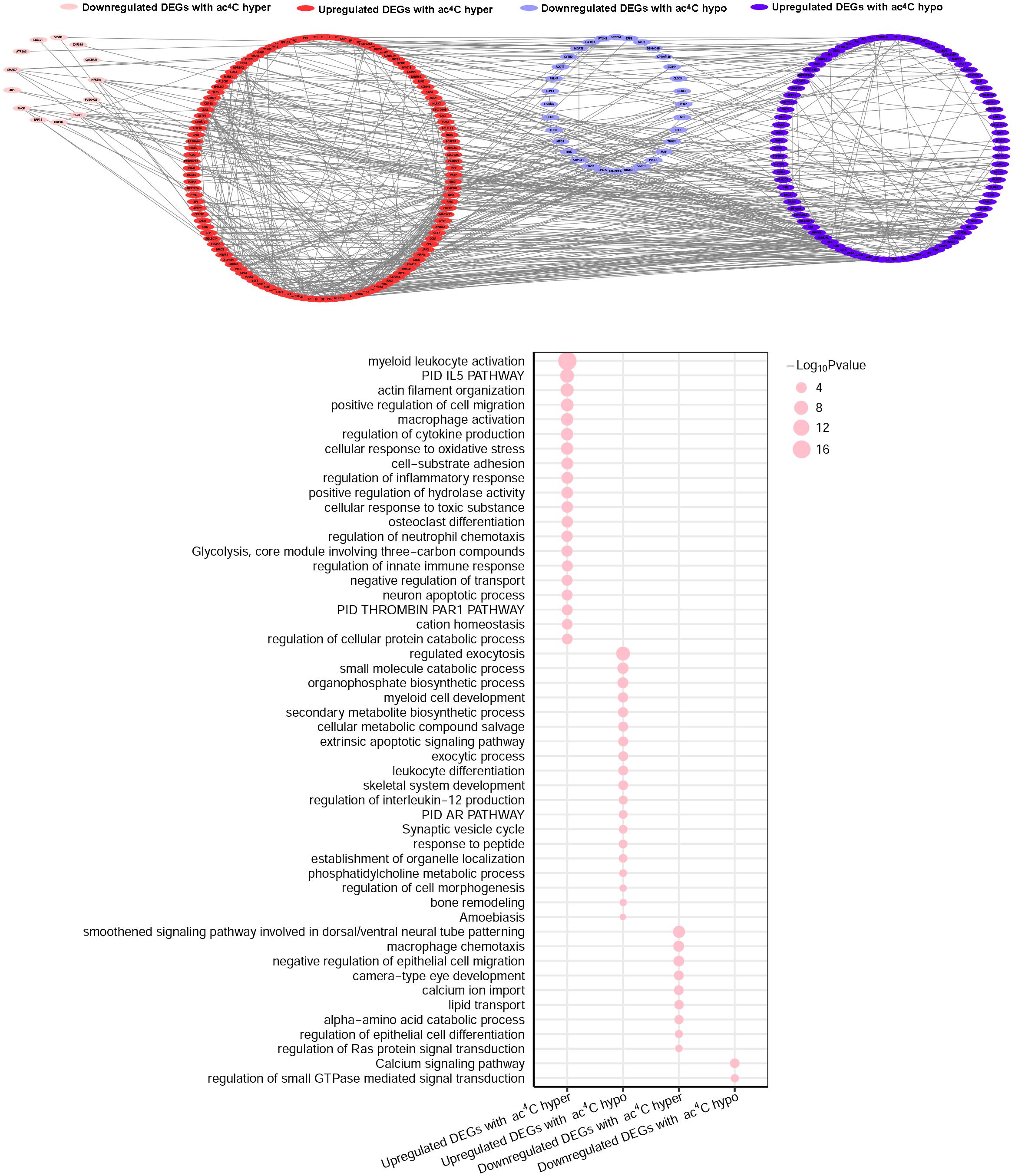
Figure 4. Gene Ontology enrichment map for ac4C-modified DEGs. Groups of upregulated DEGs with ac4C hyperacetylation (red), downregulated DEGs with ac4C hyper (pink), upregulated DEGs with ac4C hypo (dark blue), and downregulated DEGs with ac4C hypo (light blue) are marked in the network (P < 0.01; for details, see Supplementary Tables S3, S4) (top panel). Bubble diagram of GO biological process categories enriched for DEGs with ac4C hyper- or hypo-acetylation. The pink circle represents the –log10 P-value (down panel).
Intriguingly, hyper-up DEGs were found to participate in diverse important SLE-related biological processes, including inflammatory and immune responses, as well as regulation of cytokines and cellular response to oxidative stress. Moreover, the pathway enrichment results for the four DEGs sets identified a special cluster of DEGs, which were highly correlated with autoimmune diseases in the hyper-up set, consisting of 26 specific genes (Figure 5A and Supplementary Table S5). Therefore, we further selected these specific 26 upregulated DEGs with ac4C hyperacetylation to pursue the potential effects by GO analyses in CD4+ T cells of SLE patients. As shown in Figure 5B, the selected genes are notably abundant in vital functions including biological processes (innate immune response, inflammatory response, and cytokine-mediated signaling pathway), cellular components (plasma membrane and extracellular exosome), and molecular functions (drug binding and receptor activity). Particularly in biological process analyses, the results are consistent with those of the functional enrichment results of DEGs in SLE, which indicated that the 26 ac4C modified genes are highly relevant to the pathogenesis of SLE. Moreover, disease marker analysis of these 26 genes clearly showed that a series of more important genes (14/26) are directly related to SLE, including CD180, MDM2, KLHDC7B, USP18, TLR8, DDX60, CD163, PXK, JAK2, GSTP1, CISH, GPX1, RGL1, and CCR1 (Figure 5C). In addition, the top scored network analyses indicated that three of the 26 selected genes, namely USP18, GPX1, and RGL1, were primarily enriched in transcription-related processes (translational initiation, and nuclear-transcribed mRNA catabolic process) (Figure 5D and Supplementary Table S6). These observations indicate that the important role of these three specific mRNAs with ac4C modification in lupus CD4+ T cells might be highly relevant to SLE by regulating mRNA catabolic processes and translational initiation. In summary, we assume that mRNA ac4C modification may function as a key factor in the pathogenesis of SLE.
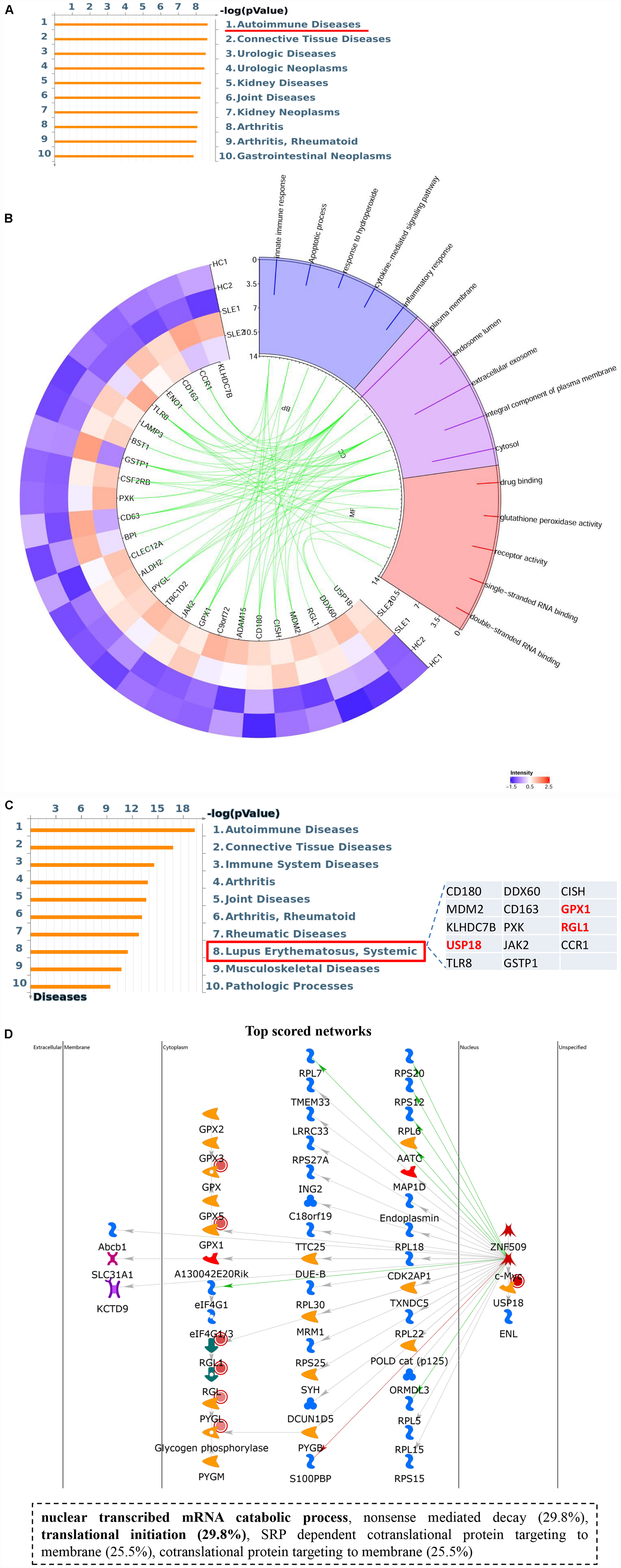
Figure 5. Upregulated gene set with ac4C hyperacetylation associated with SLE. (A) Pathway enriched analyses of upregulated DEGs with ac4C hyper. Twenty-six genes of upregulated DEGs with ac4C hyper-associated with autoimmune disease. (B) GO analysis of 26 upregulated DEGs with ac4C hyper. (C) Disease marker analysis of 26 upregulated DEGs with ac4C hyper. Fourteen of 26 genes of upregulated DEGs with ac4C hype associated with SLE. (D) The top scored networks analysis of 26 upregulated DEGs with ac4C hyper. GPX1, RGL1, and USP18 were involved in the top scored networks.
As an emerging epigenetic field, epitranscriptome presents a powerful insight that diverse reversible posttranscriptional chemical modifications of mRNA are important regulators in human disease. Recently, great effort has been devoted to establishing the significance of the epitranscriptome in the regulation of eukaryotic gene expression, particularly in cancer and autoimmune diseases (O’Connell et al., 2015; Chen X. Y. et al., 2019). As recently reported, acetylation of cytidine has been identified as a mode of mRNA modification catalyzed by acetyltransferase NAT10, which enhances mRNA stability and elevates translational efficiency (Arango et al., 2018). However, the specific role of ac4C modifications in human disease is largely unknown. Therefore, we sought to explore the relationship between ac4C modification and SLE by performing epitranscriptome ac4C-modified profiles of lupus CD4+ T cells. As the proportion of CD4+ T cells in peripheral blood is small and epigenetics can vary from cell to cell, we pooled RNA of CD4+ T cells from 5/9 subjects, equally, into a single pool, to average the extremes and obtain a variety of profiles as described in previous study (Zhao et al., 2014; Guo et al., 2020).
Here, to the best of our knowledge, for the first time, we illustrated the new mRNA modification types in a global epitranscriptomic map of CD4+ T cells from SLE patients versus HCs, uncovering multiple modifications in CD4+ T cells of SLE patients that are distinctly different from those in CD4+ T cells of HCs. A significantly lower total ac4C level, as well as a decreased NAT10 expression level, was observed in the SLE group compared to HCs. NAT10 has been identified as the sole known human enzyme in eukaryotic cells to have both acetyltransferase and RNA binding activities, and which plays a crucial part in ribosome biogenesis (Sleiman and Dragon, 2019). Therefore, we speculated that the total ac4C level in SLE was primarily determined by the expression level of NAT10. However, we found a larger number of ac4C-modified transcripts in SLE than in HCs, by analyzing ac4C-RIP-Seq. The overall ac4C modification level of one sample is primarily determined by the numbers of ac4C modified transcripts in that sample, as well as the level of ac4C modification of each transcript. Considering the lower level of ac4C, and greater number of ac4C-modified transcripts in SLE patients, an increased number of ac4C-modified transcripts in one pool does not necessarily indicate an overall increased level of modified ac4C.
ac4C peaks are globally enriched within the CDS and 3′UTR regions of mRNAs in both HCs and SLE patients, which indicates that ac4C is a conserved internal transcriptional modification. However, ac4C sites in HeLa cells were observed proximal to translation start sites (5′UTRs and CDS region) in a previous study (Arango et al., 2018), which differs slightly from our observations. Similarly, differences in m6A peak distribution in different cells are evident in the following example. m6A peaks in osteosarcoma stem cells were mainly enriched in CDS, stop codon, and the beginning of 3′UTRs, whereas in clear cell renal cell carcinoma, they were enriched in the vicinity of transcription start site (TSS), CDS, and the stop codon (Wang et al., 2019; Chen et al., 2020). This was similar to what was observed in m5C sites in SLE patients, which were enriched in CDS and 3′UTR regions (Guo et al., 2020); however, the enrichment was primarily in the vicinity of the binding regions within 3′UTRs and regions immediately downstream of translation initiation in HeLa cells (Yang et al., 2017). Overall, different mRNA modifications are enriched in CDS region but have slightly biased distribution in 3′UTR, 5′UTR, and TSS regions, suggesting that the difference may be influenced by disease types, sample types, or sample sizes. Therefore, the localization of ac4C modifications in SLE may be a disease-related characteristic. Indeed, the top motifs identified for ac4C peaks, “CRGRA” and “CCRCCRC,” in lupus CD4+ T cells provide potential targets that differ from those in HCs. These specific motifs in SLE patients imply the potential presence of an as-yet unknown writer and reader, which needs to be validated in the future. However, ac4C peaks in HeLa cells present four obligate cytidines separated by two non-obligate nucleotides (CXX) (Arango et al., 2018), which in part is distinct from the patterns of ac4C modification in SLE. These differing results may be due to the different samples from different diseases, as well as the limitation of small sample size and heterogeneous sample pools in this study.
A total of 7952 differently ac4C-acetylated peaks were detected in SLE group compared to the HC group, including 3879 hyperacetylated and 4073 hypoacetylated peaks. Additionally, several differentially acetylated peaks were validated by ac4C-RIP-qPCR, which demonstrate the accuracy of our ac4C-RIP profiles. In contrast with the lower NAT10 expression in SLE patients, hyperacetylated peaks were observed in SLE patients. Similar to the methyltransferases of mRNA m5C modification in SLE, downregulated methyltransferase expressions along with aberrant mRNA m5C modification (hyper- and hypo-) were also found in SLE patients (Guo et al., 2020). Similar results have been obtained in bladder cancer, zebrafish, and PM2.5-induced pulmonary fibrosis mouse models (Chen X. Y. et al., 2019; Yang et al., 2019; Han et al., 2020). However, although an overall lower level of modified ac4C was observed in SLE patients, ac4C modified transcripts were not necessarily lower in any one sample. Moreover since it has been reported that several mRNA m6A methyltransferases, including METTL3/14, WTAP, and RBM15/15B, participate in installation of m6A modification in mRNA (He et al., 2019), we postulated that, in addition to NAT10, other “writer(s)” exist that regulate ac4C acetylation in SLE, accounting for the generation of more ac4C-peaks or hyperacetylated mRNA. Such differentially expressed ac4C-modified profiles might result from abnormal expression of multiple acetyltransferases. Bioinformatics analysis showed that these dys-acetylated peaks are involved in the regulation of cellular metabolism and several important immune and inflammatory pathways in lupus CD4+ T cells. The alterations in metabolism of CD4+ T involves regulation of normal immune responses (Choi et al., 2016); therefore, the metabolic processes associated with abnormally acetylated peaks in CD4+ T cells provide new insight into the molecular mechanisms of SLE. Among these inflammatory pathways, NF-κB signaling is involved in the transcription of numerous inflammatory genes and has been shown to be a molecular target in lupus T cells (Wong et al., 1999). Extensive research demonstrated fundamental roles for TGF-β receptor signaling in T cells contributing to autoimmunity (Morianos et al., 2019). Moreover, the top scored networks analysis showed that the hyperacetylated peaks participate in several central transcription processes, which has been verified in HeLa cells (Arango et al., 2018). These results imply that both hyperacetylated and hypoacetylated peaks are highly associated with lupus pathogenesis.
Previous reports have described a strong relationship between mRNA modification and gene expression (Dai et al., 2018; Chen X. Y. et al., 2019; Yang et al., 2019; Han et al., 2020), and hence, we sought to characterize the functional role of the ac4C modification in mRNA of lupus CD4+ T cells. Furthermore, as reported previously (Chen X. Y. et al., 2019), increased global mRNA m5C content in tumor samples relative to normal controls was identified by via LC-MC/MC analyses. The association of m5C modifications with gene expression was investigated and four different set of genes were found to be present. Chen X. Y. et al. (2019) further identified several oncogenes, which displayed both m5C hypermethylation and mRNA upregulation, and showed a significant correlation with bladder cancer. This phenomena has also been described in PM2.5-induced mouse pulmonary fibrosis (Han et al., 2020). Except for the ac4C modification, we could not delineate the integrated impact of other regulatory factors on the transcriptome profile in SLE, such as m5C and m6A modification (Chen et al., 2020; Guo et al., 2020). Moreover, as reported in our previous study (Guo et al., 2018, 2019), we found abnormal non-coding RNAs, such as miRNA and circRNAs, which serve as pivotal regulators in SLE, indicating that comprehensive factors contribute to the development of SLE. Intriguingly, large changes in RNA abundance were likely mediated by ac4C hyperacetylation or hypoacetylation in lupus CD4+ T cells. Taken together, ac4C modification may regulate expression of the modified upregulated mRNAs related to the pathogenesis of SLE.
Furthermore, the functional enrichment results of RNA-Seq combined with our previous data (Guo et al., 2018), suggest that a few dysregulated mRNAs are significantly enriched in the immune and inflammatory responses in either lupus PBMCs or CD4+ T cells, whereas certain mRNAs, which participate in NF-κB transcription factor activity, and antigen presentation by MHC II were found only in CD4+ T cells, parallel to the biological process of differently ac4C-acetylated peaks. The PPI network and GO analysis of all DEGs with ac4C modification indicate that they were involved in multiple essential processes. Therefore, we speculate that mRNA ac4C modification may be associated with pathogenic factors regulating SLE. Particularly, the GO analyses showed that only 26 specific hyper-up genes were associated with the pathogenesis of autoimmune diseases by regulating immune and inflammatory responses, and cytokine-mediated signaling pathways, which support the importance of ac4C modification in SLE. Moreover, we found that 14 specific genes out of 26 selected mRNAs are related to SLE, including USP18, GPX1, and RGL1. In light of the top scored network, 3 (USP18, GPX1, and RGL1) of the 26 selected mRNAs are intricately connected with the translational process. Therefore, we suspected that the three upregulated mRNAs with ac4C hyperacetylation in lupus CD4+ T cells were highly associated with SLE by regulating mRNA catabolic processes and translational initiation. Among the three common genes, GPX1 has been reported in lupus endothelial dysfunction (Huang et al., 2020). RGL1 has been identified as a novel target in inflammation-associated pathways of lymphocyte networks (Kirkby et al., 2014). In addition, USP18 DNA are differentially methylated in interferon-regulated genes in lupus T cells, directly suggesting a mechanism for type I interferon hyper-responsiveness in SLE (Coit et al., 2013). Furthermore, the relationship between USP18 DNA hypomethylated and USP18 RNA ac4C hyperacetylated in SLE should be explored in the future. It has been reported that engineered gain of USP18 decreases cancer growth by destabilizing growth-regulatory proteins (Mustachio et al., 2018), which suggests that USP18 may alter transcribed mRNA catabolic process and translational initiation to facilitate the pathogenesis of SLE. In contrast, a previous study has discovered that the engineered loss of USP18 increases ISGylation (François-Newton et al., 2011); therefore, upregulated USP18 might reduce ISGylation to inactivate the interferon response, implying the function of ac4C modification in SLE which should be identified in further studies. However, the development of SLE depends on integrated modulation of various genes rather than a single gene. Hence, USP18, GPX1, and RGL1 as they relate to SLE, may be potential ac4C-modified targets to explore the pathogenesis of SLE, which provides promising perspectives for additional study of ac4C-related pathogenic mechanisms. Although lower NAT10 expression directly resulted in lower levels of overall ac4C and hypoacetylated mRNAs in CD4+ T cells of SLE, we considered that other acetyltransferase(s) or integrated elements may also contribute to the hyperacetylated peaks. Based on the significance of GO analyses and a role of ac4C in the regulation of mRNA translation, it is necessary to further explore the mechanism of the 26 hyper-up mRNAs in SLE patients.
Taken together, these observations suggest that ac4C modification may participate in mRNA metabolism processing in SLE, leading to the activation of immune cells and inflammatory responses. In particular, mRNA ac4C modification might positively affect mRNA stabilization by upregulating certain SLE-related genes. Therefore, we consider that mRNA ac4C modification may act as an effective factor regulating the pathogenesis of SLE. However, certain limitations were noted in this study, including the highly heterogeneous sample pool, relative small sample size and lack of mRNA ac4C modification with SLE disease severity. Subsequent research using a larger sample size is needed to further validate the global ac4C alterations, and to explore the association of mRNA ac4C modification with the clinical outcome (disease severity) in SLE. As the most abundant, prevalent, and functionally relevant internal modification of RNA in eukaryotic cells, the regulatory function of m6A or m5C should also be jointly analyzed with ac4C modification in SLE. Additionally, association analyses between ac4C acetylation and related components (including length and GC content) would be essential for further understanding the biological function of ac4C acetylation. Above all, the impact of the specific ac4C-modified mRNAs on SLE pathogenesis should be investigated in future studies.
In summary, a transcriptome-wide ac4C modified panel of CD4+ T cells from HCs and SLE patients was illustrated to the best of our knowledge, for the first time, uncovering gene expression and SLE-related pathological pathways regulated by aberrated ac4C RNA modification. Our results provide potential proof of an association between mRNA ac4C modification and the SLE pathogenic mechanism, which should prove useful in prospective research to elucidate the mechanisms underlying ac4C modification in SLE. Moreover, the targeted genes of ac4C modifications are likely to serve as promising therapeutic targets for SLE.
The datasets generated for this study can be found in NCBI SRA accession PRJNA649884.
This study was approved by the Medical Ethical Committees of the First Affiliated Hospital of Wenzhou Medical University (No. 2019121). All participants in this research provided written informed consent.
GG analyzed and interpreted the data. GG and HW drafted the manuscript. XS performed the experiments and statistical analysis. LY, XT, KY, ND, and CC acquired the data and provided material support. HZ and XX contributed to the conception and design of the study, analyzed and interpreted the data, supervised the study, provided the project funding, revised the manuscript, and finally approved the version of the manuscript for publication. All authors read and approved the final manuscript.
This study was supported by Zhejiang Provincial Natural Science Foundation of China (grant number LY20H100003), Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (grant number 2019KY453), Zhejiang Science and Technology Bureau (grant number 2012C33126), and Wenzhou Municipal Science and Technology Bureau (grant number Y20180158).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the patients and healthy volunteers who participated in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.00842/full#supplementary-material
FIGURE S1 | Gene-specifc ac4C qPCR validation of ac4C level changes of five representative hypo-acetylated genes in SLE patients and HCs. *P < 0.05; **P < 0.01.
FIGURE S2 | Functional enrichment analysis of differentially expressed genes in CD4+ T cells from HCs and SLE patients. (A) Volcano plots display the differentially expressed genes with statistical significance (fold change > 1.5; P < 0.05). Red points indicate significantly upregulated genes and blue points indicate significantly downregulated genes. (B) Pathway maps and Process Networks analysis of DEGs.
FIGURE S3 | Four quadrant graph shows the relationship between mRNA ac4C acetylated levels and its mRNA expressed levels. Yellow indicates upregulated DEGs with ac4C hypoacetylation (Hypo-Up), red indicates upregulated DEGs with ac4C hyperacetylation (Hyper-Up), green indicates downregulated DEGs with ac4C hypoacetylation (Hypo-Down), and blue indicates downregulated DEGs with ac4C hyperacetylation (Hyper-Down).
TABLE S1 | Primers used for RT-qPCR and ac4C-RIP-qPCR.
TABLE S2 | Most relevant networks.
TABLE S3 | PPI of DEGs with ac4C Hyper or Hypo.
TABLE S4 | Biological process and pathway categories of DEGs with ac4C hyper or hypo.
TABLE S5 | Twenty-six specific genes correlated with autoimmune diseases.
TABLE S6 | Most relevant networks for 26 autoimmune diseases related genes analyses.
Arango, D., Sturgill, D., Alhusaini, N., Dillman, A. A., Sweet, T. J., Hanson, G., et al. (2018). Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 175, 1872.e24–1886.e24. doi: 10.1016/j.cell.2018.10.030
Bailey, T. L. (2011). DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27, 1653–1659. doi: 10.1093/bioinformatics/btr261
Chen, X., Li, A., Sun, B. F., Yang, Y., Han, Y. N., Yuan, X., et al. (2019). 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 21, 978–990. doi: 10.1038/s41556-019-0361-y
Chen, X. Y., Zhang, J., and Zhu, J. S. (2019). The role of m(6)A RNA methylation in human cancer. Mol. Cancer 18:103. doi: 10.1186/s12943-019-1033-z
Chen, Y., Zhou, C., Sun, Y., He, X., and Xue, D. (2020). m(6)A RNA modification modulates gene expression and cancer-related pathways in clear cell renal cell carcinoma. Epigenomics 12, 87–99. doi: 10.2217/epi-2019-0182
Chimnaronk, S., Suzuki, T., Manita, T., Ikeuchi, Y., Yao, M., Suzuki, T., et al. (2009). RNA helicase module in an acetyltransferase that modifies a specific tRNA anticodon. EMBO J. 28, 1362–1373. doi: 10.1038/emboj.2009.69
Choi, S. C., Titov, A. A., Sivakumar, R., Li, W., and Morel, L. (2016). Immune cell metabolism in systemic lupus erythematosus. Curr. Rheumatol. Rep. 18:66. doi: 10.1007/s11926-016-0615-7
Coit, P., Jeffries, M., Altorok, N., Dozmorov, M. G., Koelsch, K. A., Wren, J. D., et al. (2013). Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J. Autoimmun. 43, 78–84. doi: 10.1016/j.jaut.2013.04.003
Dai, D., Wang, H., Zhu, L., Jin, H., and Wang, X. (2018). N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 9:124. doi: 10.1038/s41419-017-0129-x
Delatte, B., Wang, F., Ngoc, L. V., Collignon, E., Bonvin, E., Deplus, R., et al. (2016). RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 351, 282–285. doi: 10.1126/science.aac5253
Dominissini, D., and Rechavi, G. (2017). 5-methylcytosine mediates nuclear export of mRNA. Cell Res. 27, 717–719. doi: 10.1038/cr.2017.73
Feng, J., Liu, T., Qin, B., Zhang, Y., and Liu, X. S. (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 7, 1728–1740. doi: 10.1038/nprot.2012.101
François-Newton, V., Magno de Freitas Almeida, G., Payelle-Brogard, B., Monneron, D., Pichard-Garcia, L., Piehler, J., et al. (2011). USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLoS One 6:e22200. doi: 10.1371/journal.pone.0022200
Guo, G., Wang, H., Shi, X., Ye, L., Wu, K., Lin, K., et al. (2018). NovelmiRNA-25 inhibits AMPD2 in peripheral blood mononuclear cells of patients with systemic lupus erythematosus and represents a promising novel biomarker. J. Transl. Med. 16:370. doi: 10.1186/s12967-018-1739-5
Guo, G., Wang, H., Shi, X., Ye, L., Yan, K., Chen, Z., et al. (2020). Disease activity-associated alteration of mRNA m5C Methylation in CD4+ T cells of systemic lupus erythematosus. Front. Cell Dev. Biol. 8:430. doi: 10.3389/fcell.2020.00430
Guo, G., Wang, H., Ye, L., Shi, X., Yan, K., Lin, K., et al. (2019). Hsa_circ_0000479 as a novel diagnostic biomarker of systemic lupus erythematosus. Front. Immunol. 10:2281. doi: 10.3389/fimmu.2019.02281
Han, X., Liu, H., Zhang, Z., Yang, W., Wu, C., Liu, X., et al. (2020). Epitranscriptomic 5-Methylcytosine Profile in PM2.5-induced mouse pulmonary fibrosis. Genomics Proteomics Bioinformatics 18, 41–51. doi: 10.1016/j.gpb.2019.11.005
He, L., Li, H., Wu, A., Peng, Y., Shu, G., and Yin, G. (2019). Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 18:176. doi: 10.1186/s12943-019-1109-9
Huang, H., Liu, X., Chen, D., Lu, Y., Li, J., Du, F., et al. (2020). Melatonin prevents endothelial dysfunction in SLE by activating the nuclear receptor retinoic acid-related orphan receptor-α. Int. Immunopharmacol. 83:106365. doi: 10.1016/j.intimp.2020.106365
Ito, S., Horikawa, S., Suzuki, T., Kawauchi, H., Tanaka, Y., Suzuki, T., et al. (2014). Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J. Biol. Chem. 289, 35724–35730. doi: 10.1074/jbc.C114.602698
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kirkby, N. S., Lundberg, M. H., Wright, W. R., Warner, T. D., Paul-Clark, M. J., and Mitchell, J. A. (2014). COX-2 protects against atherosclerosis independently of local vascular prostacyclin: identification of COX-2 associated pathways implicate Rgl1 and lymphocyte networks. PLoS One 9:e98165. doi: 10.1371/journal.pone.0098165
Liu, Z., and Davidson, A. (2012). Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat. Med. 18, 871–882. doi: 10.1038/nm.2752
Luo, Z., Zhang, Z., Tai, L., Zhang, L., Sun, Z., and Zhou, L. (2019). Comprehensive analysis of differences of N(6)-methyladenosine RNA methylomes between high-fat-fed and normal mouse livers. Epigenomics 11, 1267–1282. doi: 10.2217/epi-2019-0009
Mazzone, R., Zwergel, C., Artico, M., Taurone, S., Ralli, M., Greco, A., et al. (2019). The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenetics 11:34. doi: 10.1186/s13148-019-0632-2
Meroni, P. L., and Penatti, A. E. (2016). Epigenetics and systemic lupus erythematosus: unmet needs. Clin. Rev. Allergy Immunol. 50, 367–376. doi: 10.1007/s12016-015-8497-4
Morel, L. (2017). Immunometabolism in systemic lupus erythematosus. Nat. Rev. Rheumatol. 13, 280–290. doi: 10.1038/nrrheum.2017.43
Morianos, I., Papadopoulou, G., Semitekolou, M., and Xanthou, G. (2019). Activin-A in the regulation of immunity in health and disease. J. Autoimmun. 104:102314. doi: 10.1016/j.jaut.2019.102314
Mustachio, L. M., Lu, Y., Kawakami, M., Roszik, J., Freemantle, S. J., Liu, X., et al. (2018). Evidence for the ISG15-specific deubiquitinase USP18 as an antineoplastic target. Cancer Res. 78, 587–592. doi: 10.1158/0008-5472.CAN-17-1752
O’Connell, M. A., Mannion, N. M., and Keegan, L. P. (2015). The epitranscriptome and innate immunity. PLoS Genet. 11:e1005687. doi: 10.1371/journal.pgen.1005687
Olarerin-George, A. O., and Jaffrey, S. R. (2017). MetaPlotR: a Perl/R pipeline for plotting metagenes of nucleotide modifications and other transcriptomic sites. Bioinformatics 33, 1563–1564. doi: 10.1093/bioinformatics/btx002
Peer, E., Rechavi, G., and Dominissini, D. (2017). Epitranscriptomics: regulation of mRNA metabolism through modifications. Curr. Opin. Chem. Biol. 41, 93–98. doi: 10.1016/j.cbpa.2017.10.008
Rönnblom, L., and Elkon, K. B. (2010). Cytokines as therapeutic targets in SLE. Nat. Rev. Rheumatol. 6, 339–347. doi: 10.1038/nrrheum.2010.64
Schuierer, S., Tranchevent, L. C., Dengler, U., and Moreau, Y. (2010). Large-scale benchmark of endeavour using metacore maps. Bioinformatics 26, 1922–1923. doi: 10.1093/bioinformatics/btq307
Shen, L., Shao, N. Y., Liu, X., Maze, I., Feng, J., and Nestler, E. J. (2013). diffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLoS One 8:e65598. doi: 10.1371/journal.pone.0065598
Sleiman, S., and Dragon, F. (2019). Recent advances on the structure and function of RNA acetyltransferase Kre33/NAT10. Cells 8:1035. doi: 10.3390/cells8091035
Song, K., Liu, L., Zhang, X., and Chen, X. (2020). An update on genetic susceptibility in lupus nephritis. Clin. Immunol. 210:108389. doi: 10.1016/j.clim.2020.108389
Tsokos, G. C., Lo, M. S., Costa Reis, P., and Sullivan, K. E. (2016). New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 12, 716–730. doi: 10.1038/nrrheum.2016.186
Wang, Y., Zeng, L., Liang, C., Zan, R., Ji, W., Zhang, Z., et al. (2019). Integrated analysis of transcriptome-wide m(6)A methylome of osteosarcoma stem cells enriched by chemotherapy. Epigenomics 11, 1693–1715. doi: 10.2217/epi-2019-0262
Wardowska, A. (2020). The epigenetic face of lupus: focus on antigen-presenting cells. Int. Immunopharmacol. 81:106262. doi: 10.1016/j.intimp.2020.106262
Wong, H. K., Kammer, G. M., Dennis, G., and Tsokos, G. C. (1999). Abnormal NF-kappa B activity in T lymphocytes from patients with systemic lupus erythematosus is associated with decreased p65-RelA protein expression. J. Immunol. 163, 1682–1689.
Yang, X., Yang, Y., Sun, B. F., Chen, Y. S., Xu, J. W., Lai, W. Y., et al. (2017). 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 27, 606–625. doi: 10.1038/cr.2017.55
Yang, Y., Wang, L., Han, X., Yang, W. L., Zhang, M., Ma, H. L., et al. (2019). RNA 5-Methylcytosine facilitates the Maternal-to-Zygotic transition by preventing maternal mRNA decay. Mol. Cell 75, 1188.e11–1202.e11. doi: 10.1016/j.molcel.2019.06.033
Zhang, Y., Zhao, M., Sawalha, A. H., Richardson, B., and Lu, Q. (2013). Impaired DNA methylation and its mechanisms in CD4(+)T cells of systemic lupus erythematosus. J. Autoimmun. 41, 92–99. doi: 10.1016/j.jaut.2013.01.005
Zhao, B. S., Roundtree, I. A., and He, C. (2017). Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42. doi: 10.1038/nrm.2016.132
Zhao, M., Liu, S., Luo, S., Wu, H., Tang, M., Cheng, W., et al. (2014). DNA methylation and mRNA and microRNA expression of SLE CD4+ T cells correlate with disease phenotype. J. Autoimmun. 54, 127–136. doi: 10.1016/j.jaut.2014.07.002
Keywords: epitranscriptome, systemic lupus erythematosus, N4-acetylcytidine (ac4C), CD4+ T cells, NAT10
Citation: Guo G, Shi X, Wang H, Ye L, Tong X, Yan K, Ding N, Chen C, Zhang H and Xue X (2020) Epitranscriptomic N4-Acetylcytidine Profiling in CD4+ T Cells of Systemic Lupus Erythematosus. Front. Cell Dev. Biol. 8:842. doi: 10.3389/fcell.2020.00842
Received: 28 May 2020; Accepted: 05 August 2020;
Published: 28 August 2020.
Edited by:
Hehuang Xie, Virginia Tech, United StatesReviewed by:
George Bertsias, University of Crete, GreeceCopyright © 2020 Guo, Shi, Wang, Ye, Tong, Yan, Ding, Chen, Zhang and Xue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huidi Zhang, aGRfemhhbmdAMTYzLmNvbQ==; Xiangyang Xue, d3p4eHkwMDFAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.