 Holly N. Wilkinson
Holly N. Wilkinson Matthew J. Hardman
Matthew J. Hardman- Centre for Atherothrombosis and Metabolic Disease, Hull York Medical School, University of Hull, Hull, United Kingdom
Cellular senescence is a fundamental stress response that restrains tumour formation. Yet, senescence cells are also present in non-cancerous states, accumulating exponentially with chronological age and contributing to age- and diabetes-related cellular dysfunction. The identification of hypersecretory and phagocytic behaviours in cells that were once believed to be non-functional has led to a recent explosion of senescence research. Here we discuss the profound, and often opposing, roles identified for short-lived vs. chronic tissue senescence. Transiently induced senescence is required for development, regeneration and acute wound repair, while chronic senescence is widely implicated in tissue pathology. We recently demonstrated that sustained senescence contributes to impaired diabetic healing via the CXCR2 receptor, which when blocked promotes repair. Further studies have highlighted the beneficial effects of targeting a range of senescence-linked processes to fight disease. Collectively, these findings hold promise for developing clinically viable strategies to tackle senescence in chronic wounds and other cutaneous pathologies.
Introduction
Senescence, a seminal discovery of Hayflick and Moorhead (1961), is a defined process that globally regulates cell fate. Cellular senescence is traditionally described as a terminal stress response, whereby cells are triggered to undergo stable and essentially irreversible cell cycle arrest following initiation by a diverse range of stress-inducing stimuli (Hernandez-Segura et al., 2018). Indeed, this process acts as an autonomous anti-tumour mechanism, halting incipient neoplastic transformation (Faget et al., 2019). Yet, senescent cells can be found in non-cancerous tissues, accumulating exponentially with increasing chronological age (Hudgins et al., 2018; McHugh and Gil, 2018). These non-proliferative cells retain metabolic capabilities, exhibiting a hypersecretory phenotype (Coppé et al., 2010). It has recently been shown that some senescent cells may even engulf their neighbouring cells, for a survival advantage (Tonnessen-Murray et al., 2019). These profound functional behaviours, identified in cells long thought to be non-functional, pose new questions around their tissue roles and consequences. This review will explore emerging roles for cellular senescence in normal and pathological wound repair, highlighting areas of potential therapeutic opportunity.
Senescence as an Anti-Proliferation Mechanism
It was originally thought that only mitotic cells, which may be highly proliferative, or spend large periods of time in quiescence, undergo senescence (Campisi and di Fagagna, 2007). This view has since been challenged, as features of senescence are observed in some differentiated cells (Jurk et al., 2012; von Zglinicki et al., 2020). The major age- and stress-related processes that induce cellular senescence include replicative exhaustion (Hayflick and Moorhead, 1961), mitogenic signals (Tchkonia et al., 2013), oxidative stress (Passos et al., 2010), DNA breaks (Di Micco et al., 2006), and epigenomic damage (Pazolli et al., 2012). These stressors subsequently initiate anti-tumourigenic networks, controlled by transcriptional regulators such as p53 (Vousden and Prives, 2009). p53 directly transactivates the cyclin dependent kinase (CDK) inhibitor, p21, to inhibit CDK2, CDK4, and CDK6-mediated retinoblastoma protein (pRb) phosphorylation (He et al., 2007). p16 similarly prevents pRb inactivation, but in a p53-independent manner (Chen et al., 2006). pRb naturally binds E2F/DP transcription factor complexes to block transcription of E2F target genes, thus failure to phosphorylate pRb halts cell cycle progression from the G1 to S phase (Narita et al., 2003).
It is important to note that, while simplified here, the role for p53 in cell survival is complex and somewhat contradictory, as p53 activation can actually suppress senescence, instead causing cell quiescence (Demidenko et al., 2010) or apoptosis (reviewed in Salminen et al., 2011). In this regard, a cell’s fate might be decided by the amount of damage sustained, and the expression of other senescence-linked factors. Molecular understanding of senescence is complicated further by the fact that the relative contribution of p21, p16, and other cell cycle regulators is thought to be context dependent (van Deursen, 2014).
Senescent Cell Characteristics
Morphologically, senescent cells exhibit flattened, elongated features, and may have multiple nuclei and enlarged vacuoles (Rhinn et al., 2019). Senescence-associated beta galactosidase is often used as an archetypal senescence biomarker (Dimri et al., 1995; Debacq-Chainiaux et al., 2009), yet its specificity has come under criticism (Krishna et al., 1999; Lee et al., 2006). For that reason, it is most often used in conjunction with other key biomarkers, such as p16 and p21, to confirm senescence (Baker et al., 2016; Matjusaitis et al., 2016; Biran et al., 2017).
Senescent cells may also possess regions of highly condensed chromatin (senescence-associated heterochromatic foci; Zhang et al., 2007) and DNA damage-induced chromatin alterations, including γH2AX and H3K9Me3 (Rodier and Campisi, 2011). Loss of histones, centrosome aberrations and the breakdown of the nuclear envelope (e.g., degradation of lamin B1) similarly occur in many senescent states to enable rearrangement of heterochromatin (Tigges et al., 2014; Wang et al., 2017). These chromatin modifications sequester E2F target genes to potentiate senescence (Shah et al., 2013). Moreover, senescence is reinforced by microRNA-mediated silencing of E2F target genes (Benhamed et al., 2012).
Experimental manipulation of epigenetic marks has demonstrably shown their importance in controlling the molecular induction of cellular senescence. H3K27me3, for example, represses p16 and p14 expression by silencing the INK4a-ARF locus (Kotake et al., 2007). Removal of H3K27me3, by JMJD3-induced demethylation (Agger et al., 2009; Sui et al., 2019) or pharmacological inhibition of the histone lysine methyltransferase, EZH2 (Ito et al., 2018), promotes p16 expression and senescence. Inhibition of EZH2 also leads to SASP production via enrichment of H3K27ac, and loss of H3K27me3, at SASP-related loci (Ito et al., 2018). Overexpression of another histone demethylase, UTX, can also silence H3K27me3 to promote cellular senescence (Perrigue et al., 2020).
Stressed cells are repressed at the transcriptional level to prevent the expansion of potentially harmful mutations. It is therefore understandable that regulators, such as p53, are not only responsible for initiating senescence, but also decide whether cells should instead enter temporary quiescence or undergo apoptosis (Salminen et al., 2011). Intriguingly, senescent cells may actually retain heightened resistance to apoptosis, first demonstrated in fibroblasts (Wang, 1995), possibly due to altered p53 signalling (Childs et al., 2014) and upregulation of pro-survival pathways (e.g., BCL-2 and ephrins, Zhu et al., 2015). Indeed, senescent keratinocytes are resistant to ultraviolet radiation-induced apoptosis (Chaturvedi et al., 2004) and senescent fibroblasts to thapsigargin-induced apoptosis (Ryu et al., 2007). Senescent endothelial cells, on the other hand, are more likely to undergo apoptosis than their non-senescent counterparts (Hampel et al., 2004). Clearly, this senescence trait is situational, and not ubiquitous to all cell types.
The hypersecretory phenotype of senescent cells is most often referred to as the senescence-associated secretory phenotype (SASP), an attribute closely linked to the positive or negative outcomes of tissue senescence that appears to be cell-type and context-dependent. Even though studies have characterised the SASP in multiple cell types, its detailed composition remains elusive. Broadly, the SASP comprises a collection of pro-inflammatory cytokines and chemokines, growth factors, proteases, lipids and extracellular matrix components (Freund et al., 2010; Elzi et al., 2012; Acosta et al., 2013; Lopes-Paciencia et al., 2019). It is thought to mainly be a feature of senescent cells that have undergone a DNA damage response, as a SASP is not apparent in cells that naturally senesce due to overexpression of p16 and p21 (Coppé et al., 2011). However, a DNA damage-independent SASP can occur in fibroblasts via p38MAPK phosphorylation, challenging previous preconceptions (Freund et al., 2011). Collectively, the secretome is the characteristic of senescent cells that confers most of their biological effects, significantly contributing to age-related functional decline (Rodier et al., 2009) and chronic disease (Zhu et al., 2014) in autocrine and paracrine manners.
The SASP is dynamically regulated by a number of factors that mostly converge on the NF-κB complex (Sun et al., 2018). Inflammatory cytokines, such as IL-1α, can form positive feedback loops with NF-κB and partner cascades to reinforce SASP release and senescence (Kuilman et al., 2008; Orjalo et al., 2009). In fact, multiple authors have demonstrated activation of NF-κB gene sets following senescence (Kuilman et al., 2010; Lujambio et al., 2013), while p53 and NF-κB are linked in coregulatory (in macrophages, Lowe et al., 2014) and antagonistic (HeLa cells, Huang et al., 2007) manners. The importance of NF-κB in senescence is highlighted by studies where NF-κB suppression allows oncogene-induced IMR-90 fibroblasts to bypass senescence (Chien et al., 2011) and reduces senescence in osteoarthritic cartilage (Wu et al., 2015).
Indeed, the SASP (e.g., TGFβ) can potentiate senescence in neighbouring cells (Acosta et al., 2013), but also promote senescent cell clearance by attracting immune cells (Kang et al., 2011; Tasdemir et al., 2016). The induction of senescence in, and clearance of, premalignant cells consequently reinforces tumour suppression. Paradoxically, the SASP can also drive pre-cancerous development in proximal tissues as many SASP proteins are potent mitogenic factors (e.g., VEGF, Coppé et al., 2006, 2010; Collado et al., 2007). The plasticity of the SASP across different microenvironments, cell types and stimuli (Campisi, 2013; Lupa et al., 2015; Maciel-Baron et al., 2016; Sun et al., 2018) further complicates our understanding of its role within tissues. However, it is clear that senescence, and the SASP, remain important regulators of normal physiology and pathology. Tissue consequences of senescent cells and their SASP are summarised in Figure 1.
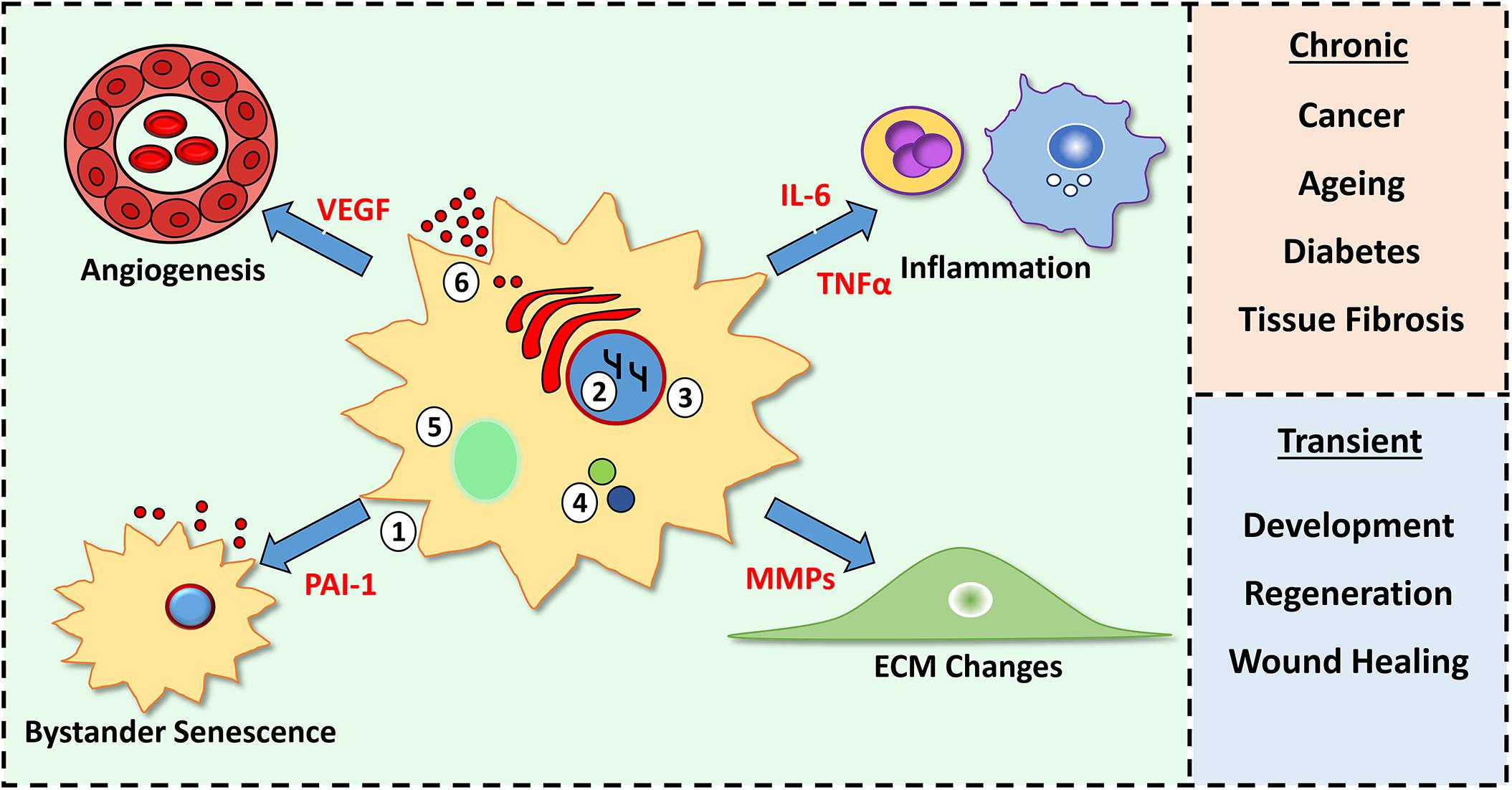
Figure 1. Characteristics of senescent cells and their tissue consequences. Senescent cells feature morphological changes (1), chromatin modifications (2), loss of nuclear envelope lamin B1 (3), increased p16 and p21 (4), senescence-associated beta galactosidase activity (5) and production of a senescence-associated secretory phenotype (SASP, 6). SASP components (red) alter cellular processes (e.g., inflammation and angiogenesis) within the microenvironment. A chronic SASP causes negative outcomes, while a short-lived, transient SASP is beneficial.
Roles for Senescence During Development and Regeneration
Many crucial biological processes require cells to undergo cell cycle arrest and differentiation to terminal states. For example, developmental lineage-specification requires cells to differentiate in a temporospatial manner in order to properly form tissues (Da Silva-Álvarez et al., 2019). In the skin, basal keratinocytes first proliferate, and then differentiate, transiting through the epidermis to replenish the non-viable stratum corneum (Fuchs and Byrne, 1994). In fact, to allow effective keratinocyte differentiation, p21 is activated initially (Missero et al., 1996), but then suppressed (Di Cunto et al., 1998). It is therefore unsurprising that tumour suppressor genes also aid development and regeneration through control of quiescence, terminal differentiation, apoptosis and senescence (e.g., Di Giovanni et al., 2006; Watkins et al., 2013).
Three main roles have been put forward for the presence of senescent cells during embryogenesis: (1) to promote the regression of transient structures; (2) to balance cell populations and/or; (3) to act as a signalling hub to regulate tissue morphogenesis (Da Silva-Álvarez et al., 2019). In embryonic development, temporal induction of senescence is required for tissue patterning in the developing limb bud. Here, p21 induction leads to SASP factor expression (e.g., FGF), stimulating cell proliferation and tissue formation. Resulting senescent (and apoptotic) cells are then effectively cleared by macrophages, prior to tissue remodelling. Indeed, genetic knockdown of p21 to attenuate senescence leads to mild patterning defects in murine limbs (Storer et al., 2013). In a corroborating study, p21 was shown to contribute to senescence-linked development in a p53-independent manner in human and murine embryos (Muñoz-Espín et al., 2013). In this case, however, loss of p21 was compensated for by increased apoptosis. Thus, p21-regulated senescence and apoptosis can perform synergistic roles during organismal development.
Akin to development, lower organisms and anamniotes are able to regenerate their tissues to full form and function, either as juveniles or throughout their lives (Brockes and Kumar, 2005). In fact, it has recently been shown that senescence may play an important role in these regenerative processes. In salamanders, senescence induction occurs at the intermediate stages of limb regeneration and then diminishes due to effective clearance by macrophages (Yun et al., 2015). Senescence is similarly invoked during pectoral fin regeneration in zebrafish, and impaired when senescence is blocked with the senolytic compound, ABT-263 (Da Silva-Álvarez et al., 2020). Given the importance of senescence in regulating tissue formation throughout development and regeneration, it is logical to ask whether senescence could play a role in the reparative responses of higher vertebrates.
Senescence in Normal Tissue Repair
Tissue repair is necessary for all life. While it seldom leads to full regeneration, the process prevents exsanguination and infection, and aids structural and functional restoration required for survival. Tissue repair is rapid and highly dynamic, comprising multiple cell types and overlapping processes that broadly include haemastasis, inflammation, cell proliferation and dermal remodelling (Wilkinson and Hardman, 2017). During haemastasis, an insoluble blood clot is formed and endothelial cells from damaged vasculature enter the wound, depositing a temporary fibrin scaffold and releasing factors to attract both circulating immune cells and resident skin cells (Velnar et al., 2009). Inflammatory cells are rapidly recruited to the site of damage, first dominated by neutrophils and pro-inflammatory macrophages to remove bacteria and necrotic tissue (Young and McNaught, 2011). Later stage healing is characterised by a switch to anti-inflammatory macrophages, which phagocytose any remaining pro-inflammatory cells, supporting fibroplasia and wound resolution (Korns et al., 2011). To allow effective repair, keratinocytes undergo partial epithelial-to-mesenchymal transition and begin migrating to close the wound gap, a process known as re-epithelialisation (Shaw and Martin, 2016). Formation of new vasculature (angiogenesis) is essential to provide sustenance during the highly proliferative stage of healing (Baum and Arpey, 2005). Finally, the immature matrix laid down during early healing is replaced by stronger scaffold proteins, such as mature collagen fibres produced and remodelled by fibroblasts (Li et al., 2007). Each stage of wound repair involves extensive cellular communication, orchestrated by cytokines, chemokines, growth factors and components of the extracellular milieu. The plasticity of the response, and the cellular behaviours that occur, are homologous to those observed in cancer (e.g., immune cell infiltration, invasion and epithelial-to-mesenchymal transition, Schäfer and Werner, 2008). It is therefore not unreasonable to suggest that senescence, and associated mechanisms, could significantly contribute to wound healing.
Indeed, pertinent roles for senescence in tissue injury have been emerging, largely focusing on the beneficial, transient initiation of senescence during repair. Here, induction of senescence following liver damage (Krizhanovsky et al., 2008) and cutaneous injury (Jun and Lau, 2010) was shown to prevent excessive fibrosis that would otherwise cause tissue dysfunction. Krizhanovsky et al. (2008) confirmed that reduced fibrosis was the result of senescence-linked fibrolytic enzyme production, and immune-regulated clearance of injury-expanded cell populations that would otherwise contribute to excessive matrix deposition. Likewise, senescence decreased fibrosis in a model of cardiac injury, where genetic ablation of p53 and p16 accelerated fibrosis (Meyer et al., 2016). Ectopic expression of Ccn1, which increased cardiac senescence, also limited fibrosis in this model. Interestingly, Jun and Lau (2010), the first authors to observe transient senescence during skin repair, revealed that Ccn1 causes fibroblast senescence via an oxidative-stress dependent mechanism. Upregulation of Ccn1 was vitally important to prevent excessive fibrosis. More recently, the same authors demonstrated that topical application of another Ccn family member, Ccn2, similarly actuates senescence and reduces fibrosis in cutaneous murine wounds (Jun and Lau, 2017).
By contrast, when Demaria et al. (2014) ablated p16- and p21-expressing cells in mice they observed impaired extracellular matrix deposition and a decreased rate of wound closure. Intriguingly, by day 15 post-injury, these senescent-deficient wounds were excessively fibrotic. Similar to previous research (Jun and Lau, 2010), transient senescence appeared limited to fibroblast-like cells, which produced a PDGFA-enriched SASP to stimulate appropriate skin repair (Demaria et al., 2014). Studies continue to explore the importance of transient senescence during acute wound healing, with Hiebert et al. (2018) recently reporting that overexpression of nrf2 promotes fibroblast senescence, which is accompanied by accelerated wound re-epithelialisation and extracellular matrix deposition. Although at present limited to murine models, these key investigations provide clear evidence that temporal induction of senescence is necessary for effective skin repair. Yet, many questions remain unanswered. For instance, does transient wound-induced senescence arise through intrinsic cell factors or environmental influences? And how are these senescent cells so effectively cleared once they are no longer required?
Senescence in Aged and Diabetic Wound Healing
The above studies provide substantial insight into the importance of senescence for the healing of experimental wounds. What they do not address is the potential differential influences of acute vs. chronic senescence to tissue repair, nor how senescence could be involved in pathological healing. These are important considerations for the clinical setting, where effective healing can mean the difference between life or death (Han and Ceilley, 2017). Chronic, non-healing wounds are a huge socioeconomic burden, reducing quality of life and costing billions each year to treat (Guest et al., 2015). Considered a “silent epidemic” (Lindholm and Searle, 2016), chronic wounds display diverse aetiology, with incomplete molecular and cellular understanding (Frykberg and Banks, 2015). Inadequate current treatments mean it is fundamentally important to further understand why chronic wounds fail to heal, and ultimately develop more effective therapies.
It has long been appreciated that chronic wound pathology is almost entirely restricted to those who are elderly and/or diabetic. This is fascinating, as the biological processes of ageing and diabetes are themselves notably linked to senescence (Wilkinson and Hardman, 2020). Senescence is both a characteristic feature of Baker et al. (2013), Xu et al. (2015), and Helman et al. (2016) and contributor to Baker et al. (2008, 2011) widespread tissue ageing. Epigenetic modifications are one feature of ageing that is linked to senescence. Genomic instability and DNA methylation changes correlate with chronological ageing in mice (Stubbs et al., 2017) and humans (Horvath, 2013). Interestingly, the repressive mark, H3K27me3, showed altered DNA coverage on aged vs. young stem cells (Liu et al., 2013; Sun et al., 2014), which may contribute to their reduced renewal capacity.
Another attribute of normal metabolic ageing that is experimentally linked to senescence is cumulative oxidative damage. For example, human diploid fibroblasts (Duan et al., 2005) and endothelial cells (Ruan et al., 2014) undergo senescence in the presence of heightened reactive oxygen species (ROS), while replicative lifespan can be extended in cell culture by lowering oxygen tension (Parrinello et al., 2003). More notably, exposure to ultraviolet radiation simulates photoageing by increasing ROS production in skin (Herrling et al., 2006), while ROS upregulates p16 in skin cells (Jenkins et al., 2011). Skin ageing is also characterised by cell accumulation of p16 (Waaijer et al., 2012) and senescence-associated beta galactosidase (Dimri et al., 1995; Ressler et al., 2006). This association is causally corroborated by Xu et al. (2018), who demonstrated that transplantation of senescent cells to young mice accelerated ageing, while Baker et al. (2011) revealed that eradication of p16-positive cells alleviated features of premature ageing in a murine progeroid model.
The link between diabetes and senescence is less well-established, but is an area of intense current research. As previously mentioned, senescent cells cause widespread disruption to normal tissue architecture by virtue of their SASP (Coppé et al., 2010). Major SASP constituents influence senescence by targeting immunological pathways, such as NF-κB (Salminen et al., 2011). This leads to matrix proteolysis and increased inflammation, primary features of aged and diabetic wounds (Makrantonaki et al., 2017; Wilkinson et al., 2019c). Indeed, growing evidence suggests that a heightened intrinsic immune response, or “sterile” inflammation, contributes to age- and diabetes-related pathology (reviewed in Prattichizzo et al., 2016). Characteristic features of diabetes that drive immune cell accumulation, and therefore potentiate senescence, include obesity and hyperglycaemia (Yokoi et al., 2006; Minamino et al., 2009; Maeda et al., 2015; Schafer et al., 2016). These processes most likely promote senescence via increasing advanced glycation end-products and causing widespread oxidative damage (Coughlan et al., 2011; Fang et al., 2016).
Turning specifically to the skin, it is clear that in diabetic and aged tissue, accumulation of senescent cells extends to both uninjured skin and wounds (Ressler et al., 2006; Waaijer et al., 2012; Wilkinson et al., 2019a). Previous authors have demonstrated that chronic venous leg ulcers harbour senescent fibroblasts (Mendez et al., 1998; Vande Berg et al., 1998; Agren et al., 1999; Wall et al., 2008). The presence of senescent fibroblasts in chronic wounds may even exacerbate pathology, where it was shown that ulcers containing over 15% senescent cells were hard to heal (Stanley and Osler, 2001). We recently reported a novel mechanistic link between senescence and healing in diabetic wounds (Wilkinson et al., 2019a). Here, intrinsically senescent macrophages were observed to promote impaired wound healing in a non-aged, murine model of diabetic pathological repair.
Indeed, many SASP factors attract monocytes and macrophages (e.g., MCP-1; Kamei et al., 2006; Coppé et al., 2008; Prattichizzo et al., 2018), often with a pro-inflammatory phenotype (Mosser and Edwards, 2008; Lujambio et al., 2013). Excessive immune cell recruitment and inappropriate retention is a hallmark of chronic wound pathology. This may be even be exacerbated by other local factors, such as iron, which induces a pro-inflammatory phenotype in macrophages and leads to fibroblast senescence in chronic venous leg ulcers (Sindrilaru et al., 2011). Thus, macrophages are likely a nexus for uncontrolled local inflammation in both diabetic pathogenesis and senescence, ultimately delivering poor wound healing. Moreover, the impaired function of macrophages (and other immune cell types) in aged (Swift et al., 2001) and diabetic (Wilkinson et al., 2019b) wounds likely contributes to prolonged, rather than transient, senescence due to ineffective clearance mechanisms.
Senescence in the wound environment is probably not limited to fibroblasts and macrophages, as other wound cells, including keratinocytes (Smirnov et al., 2016) and endothelial cells (Ruan et al., 2014), are capable of undergoing senescence in response to environment cues. Senescent keratinocytes are certainly observed in aged skin (Velarde et al., 2012) and are suggested to influence the reduced regenerative capacity of aged epidermis (Zouboulis et al., 2008). Chronic wounds also harbour pathogenic microorganisms (Kalan et al., 2019) that may contribute to senescence by stimulating ROS production in keratinocytes and exacerbating inflammation (Grange et al., 2009). Indeed, this may occur via specific bacterial virulence factors, as pyocyanin from Pseudomonas aeruginosa can induce senescence in fibroblasts (Muller et al., 2009).
It is clear that the chronic ulcer milieu, which is rich in pro-inflammatory factors, indirectly causes senescence via exacerbating inflammation. However, as wound fluid from venous leg ulcers directly induces senescence in neonatal fibroblasts (Mendez et al., 1999), it is likely that the local microenvironment also stimulates cellular senescence. Intrinsically senescent wounds cells, such as fibroblasts, are similarly capable of potentiating senescence across neighbouring cell types in a paracrine manner, via their SASP (Acosta et al., 2013; Wilkinson et al., 2019a). Moreover, in local environments where the SASP is insufficient to directly induce cellular senescence, it may still promote pathological cellular phenotypes, such as epidermal hyperproliferation (Albanesi et al., 2018) and excessive dermal proteolysis (via MMPs; Caley et al., 2015).
To add a further level of complexity, evidence for the disparities between transient and chronic senescence is beginning to emerge, with clear implications for wound healing. For instance, stemness and reprogramming in keratinocytes is promoted by a transient SASP, yet inhibited when the SASP becomes chronic (Ritschka et al., 2017). Transient senescence also encourages matrix deposition following tissue injury (Demaria et al., 2014), but prevents excessive fibrosis (Jun and Lau, 2010), while chronic senescence is linked fibrotic disease (Yanai et al., 2015). Taken together, published and emerging studies are certainly challenging the dogma that senescence is primarily limited to age-related dysfunction and cancer. Indeed, evolving understanding of the concept of transient vs. chronic senescence is likely to deliver important new insight into the processes that occur during acute and pathological repair. Current understanding of senescence contribution to normal and pathological wound healing is summarised in Figure 2.
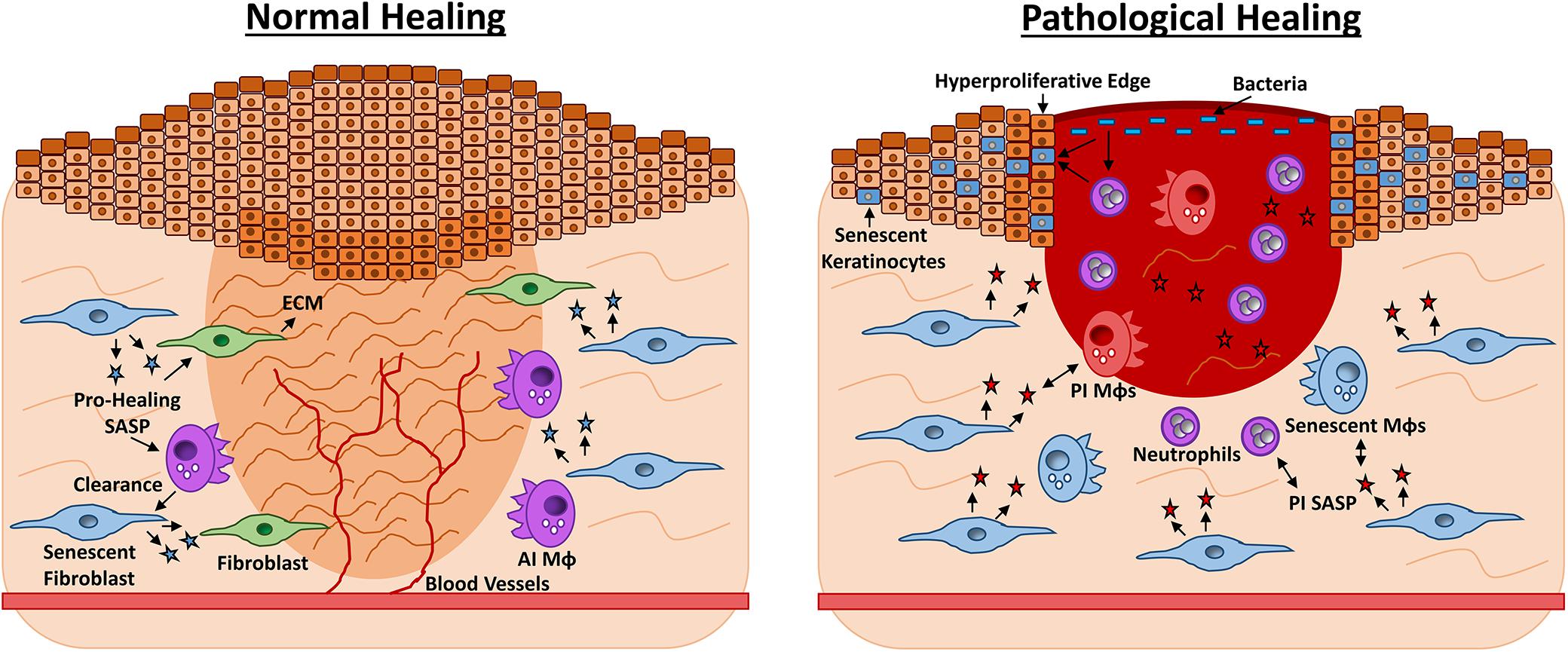
Figure 2. Roles for senescence in acute vs. chronic wound repair. Late-stage wound healing is characterised by extracellular matrix (ECM) deposition, full wound closure and blood vessel perfusion (left). Senescent fibroblasts appear during late-stage healing and contribute to ECM deposition by producing a pro-healing senescence-associated secretory phenotype (SASP, blue stars) containing PDGFA. Senescent fibroblasts prevent excessive fibrosis via CCN1 and CCN2. The senescent cells are then effectively cleared by anti-inflammatory macrophages (AI MΦs) and the skin is restored. Chronic healing wounds feature wound edge hyperproliferation and excessive inflammation (right). Here senescent fibroblasts and MΦs (blue) exacerbate inflammation via a pro-inflammatory (PI) SASP of cytokines and proteases. The SASP may also exert a paracrine effect, causing senescence in other wound cell types. High bacterial load stimulates further inflammation and oxidative stress, which can cause hyperproliferation or senescence in keratinocytes. Senescent cells are not effectively cleared by the dysfunctional chronic wound immune cells, thus tissue damage persists and the wound fails to heal.
Cellular Senescence as a Therapeutic Target in Pathological Wounds
The widespread causative biological effects of cellular senescence in tissue ageing pathology make the therapeutic modulation of senescence an attractive target for a plethora of age-related diseases. Genetic studies positively support this idea, with inducible knockdown of p16 alleviating hallmark features of ageing in progeroid murine models (Baker et al., 2011; Sato et al., 2015). In fact, the well-documented effects of caloric restriction, which both extends mammalian lifespan (Sohal and Weindruch, 1996) and delays the onset of age-related disease (Weindruch and Walford, 1982; Colman et al., 2014), may be a physical manifestation of tissue senescence modulation. Caloric restriction has been shown to reduce cardiac senescence (Shinmura et al., 2011), and senescence in hepatocytes and intestinal crypt cells in vivo (Wang et al., 2010). At the epigenetic level, caloric restriction protects against age-related changes in DNA methylation (Hahn et al., 2017). Caloric restriction also decreases senescence partly by upregulating the epigenetically linked sirtuin pathway, promoting anti-apoptosis and anti-inflammatory mechanisms (Bonda et al., 2011). Subsequent effects include slowing metabolic processes that contribute to cellular ageing (e.g., oxidative stress, Yang et al., 2016), increasing antioxidant production (Meydani et al., 2011), and increasing autophagy to remove damaged and unimportant intracellular components (reviewed in Cuervo, 2008). Moreover, sirtuins may play important roles in preventing age-related decline in skin repair, as SIRT1 deficiency exacerbates healing pathology in diabetic wounds (Thandavarayan et al., 2015).
Although caloric restriction (without malnutrition) provides a multitude of health benefits, it retains poor feasibility as a clinical intervention, requiring high compliance and patient discipline. Many lifestyle choices, such as obesity, are actually strongly associated with social status (Drewnowski and Specter, 2004). Similarly, those suffering from uncontrolled type II diabetes and severe chronic wounds are often from socially deprived backgrounds (Anderson et al., 2018), a difficult population in which to manage compliance. For all of these reasons, a considerably more attractive proposition is the use of senescence-targeted drugs, otherwise known as senolytics. These drugs affect unique features of senescent cells, such as resistance to apoptosis (Salminen et al., 2011). Senescent cells upregulate prosurvival pathways, particularly BCL-2 (Ovadya and Krizhanovsky, 2018). This opens up drug repurposing opportunities around the numerous BCL-2 inhibitors that were developed for the treatment of cancer (Roberts et al., 2016; Montero and Letai, 2018). Results have been promising. Targeting BCL-2 in vivo induces apoptosis and thus eliminates senescent cells in the lung following irradiation (Yosef et al., 2016) and throughout the body following irradiation or natural ageing (Chang et al., 2016). Chang et al. (2016) further established that senescent human and murine fibroblasts, and human renal epithelial cells, are more susceptible to BCL-2 inhibitor (ABT-263) than non-senescent cells, proposing potent and specific effects. Unfortunately, traditional BCL-2 inhibitors possess activity against other BCL class proteins, such as BCL-XL, raising questions around off-target effects in the clinic, including thrombocytopenia and neutropenia. As a result, more specific BCL-2 inhibitors with lower toxicity are being tested (King et al., 2017). It has even been suggested that low-dose, combinatorial use of senolytics may be an effective and less harmful alternative (Ovadya and Krizhanovsky, 2018).
Other senolytics that have demonstrated experimental efficacy include the tyrosine kinase inhibitor, Dasatinib, used to treat leukaemia (Keskin et al., 2016), and the flavonoid p53 activator, Quercetin (Khan et al., 2016). Combinatorial treatment with Dasatinib and Quercetin extends lifespan, alleviates frailty (Xu et al., 2018), and improves vasomotor function (Roos et al., 2016) in aged mice. Dasatinib and Quercetin have also shown promise in a phase I trial in diabetic kidney disease patients, where reduced senescent cells and circulating SASP factors were observed following administration (Hickson et al., 2019). Alternative flavonoids are now being tested for their potential senolytic effects, such as Fisetin, which is able to eliminate senescent cells and, crucially, restore tissue function in aged mice (Yousefzadeh et al., 2018).
The importance of transient senescence for effective healing should not be underestimated. As noted previously, temporary induction of senescence aids rapid tissue reformation (Demaria et al., 2014; Hiebert et al., 2018). During a normal damage response, these senescent cells are effectively cleared by natural killer cells (Krizhanovsky et al., 2008) and macrophages (Yun et al., 2015). Nevertheless, in chronic situations, senescent cells persist, likely due to elevated immunosenescence and resulting impaired immunological functions (Hall et al., 2016). It follows that treatments to boost immune system function, for instance by aiding senescent cell recognition, could be beneficial in the context of transient senescence and tissue repair. Generally, senescent cells express stimulatory ligands that bind to NK2GD receptors on natural killer cells, thus initiating a killing response (Sagiv et al., 2016). However, senescent fibroblasts in aged skin have recently been shown to express HLA-E, which bypasses recognition and clearance by natural killer and T cells (Pereira et al., 2019). Here, approaches developed in the cancer field may also be useful, for example engineering T cells to express receptors that target specific cellular (tumour) proteins (reviewed in June et al., 2018). Studies to identify and validate new senescent cell receptors will be essential to the development and clinical application of such immune-regulated approaches.
Indeed, the emergence of global profiling methodologies, such as single-cell RNA sequencing, could provide the basis to understanding senescence-linked changes in ageing and pathology by identifying unique cell-based transcriptomic signatures within tissues. Kimmel et al. (2019) used this approach to compare cell frequency, heterogeneity and age-related transcriptomic changes between aged and young murine tissues. Similarly, Angelidis et al. (2019) combined transcriptomics and proteomics to not only identify the epigenetic and transcriptional consequences of ageing in the lung, but also determine their functional implications. Future harnessing of these technologies could therefore facilitate the identification and targeting of key senescence-linked receptors and biomarkers in a tissue and pathology-specific manner.
Alternative strategies to diminish or limit senescence and alleviate pathology instead target the SASP or specific senescence-linked receptors directly (summarised in Figure 3). Certainly, the SASP is transcriptionally regulated by NF-κB and others (Salminen et al., 2011), and contributes heavily to tissue deterioration, both driving widespread destruction and reinforcing senescence (Rodier et al., 2009; Acosta et al., 2013). SASP inhibitors affect key transcriptional mediators, blocking signalling and preventing SASP production (Moiseeva et al., 2013). Interestingly, Metformin, a widely used anti-diabetic drug, is an effective SASP inhibitor (reviewed in Rena et al., 2017) able to directly accelerate healing in diabetic mice (Han et al., 2017). Rapamycin, another SASP inhibitor, was the first drug revealed to extend lifespan in mice (Harrison et al., 2009), and enhance the replicative lifespan of human keratinocytes (Horvath et al., 2019) and skin fibroblasts in vitro (Sodagam et al., 2017). Although these studies suggest potential skin-related benefits of SASP inhibitors, removal of the SASP could be deleterious, impairing the healing response and preventing senescent cell clearance (von Kobbe, 2019). Consequently, it may be more advantageous to target particular SASP components known to impact tissue function, either with antibodies (e.g., IL-1α, Orjalo et al., 2009), or specific inhibitors (e.g., against CXCR2, Wilkinson et al., 2019a).
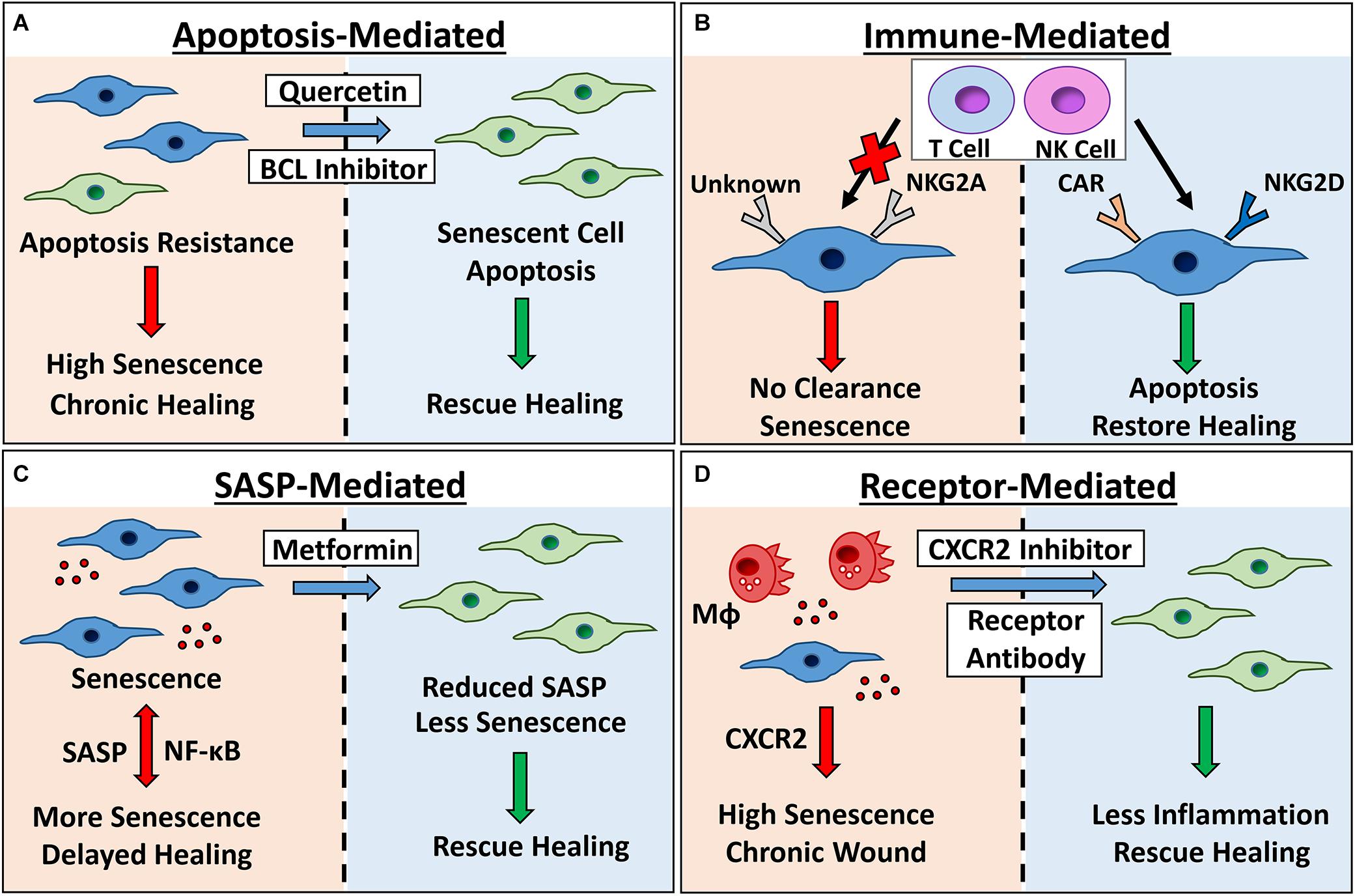
Figure 3. Therapeutic targeting of senescence for chronic healing wounds. Senescent cells accumulate in chronic healing wounds, contributing to inflammation and poor healing. Senescence can be targeted by: (A) inhibiting pro-survival pathways with BCL inhibitors and broad spectrum drugs (e.g., quercetin) to cause apoptosis; (B) engineering chimeric antigen receptor (CAR) T cells to target senescent cell receptors, or modulating expression of natural killer (NK) cell receptors NKG2A and NKG2D to increase clearance; (C) using Metformin or other SASP inhibitors to reduce NF-κB-mediated inflammation and bystander senescence and; (D) inhibiting receptors known to potentiate wound senescence (e.g., CXCR2). Red arrows/left panels = negative outcomes. Green arrows/right panels = positive outcomes. MΦ= macrophage. Senescent cells = blue.
We remain a long way from implementing senescence-targeted treatments for pathological wound healing, yet it is reassuring to see that current senolytic drugs display efficacy across a wide range of tissues and pathologies. In a number of studies, systemic senolytic treatments have been shown to have clear effects in peripheral target tissues across a range of treatment regimens. For example, a single dose of BCL inhibitor (Yosef et al., 2016), and dosing over consecutive days (Chang et al., 2016), was able to reverse irradiation-induced senescence in different tissues. In the work by Xu et al. (2018), aged mice showed improved physical performance following biweekly oral treatments of Dasatinib and Quercetin for 4 months, yet reduced SASP was observed in human ex vivo cultured adipose tissue within 48 h of treatment. Moreover, a single 3 day oral treatment of Dasatinib and Quercetin was able to reduce senescence in the adipose tissue of diabetic patients in a phase I trial (Hickson et al., 2019). These studies therefore suggest that senolytic treatments not only have rapid effects in target peripheral tissues, but can overcome established tissue senescence.
Experimental studies do show beneficial effects of modulating senescence in the skin. For example, elimination of senescent cells from the epidermis restored proliferative capacity in hair follicle stem cells (Yosef et al., 2016), known to participate in wound healing (Joost et al., 2018). Further, blockade of the potential senescence receptor, CXCR2 (Acosta et al., 2008), directly accelerated healing in human ex vivo skin wounds and diabetic murine wounds in vivo (Wilkinson et al., 2019a). Here, a CXCR2 antagonist was administered to wounds topically (ex vivo) and subcutaneously (in vivo), suggesting direct delivery to the wound site as a viable administration route. Indeed, elevated CXCR2 has previously been observed in diabetic wounds (Wetzler et al., 2000), and more recently in T cells from human diabetic patients (Lau et al., 2019). We note with interest that pharmacological inhibition of CXCR1/2 additionally prevents inflammation-mediated damage to pancreatic islets, thus prohibiting streptozocin-induced diabetes in mice (Citro et al., 2015). Therefore, CXCR2 appears a common factor in both the ontology and local pathology of diabetes. Senolytics should certainly be considered for the treatment of human chronic wounds characterised by high levels of senescence (Stanley and Osler, 2001). However, given that knockdown of CXCR2 (Devalaraja et al., 2000) and ablation of senescent cells (Demaria et al., 2014) actually delays acute wound healing, future senescence-targeted therapies should be reserved for the treatment of chronic conditions.
Conclusion
Despite seemingly contradictory roles in many cancers, the detrimental contribution of cumulative senescence to ageing and age-related disease is now well-established. By contrast, the short-lived, transient senescence observed to benefit tissue development, regeneration and repair, remains significantly less well-characterised. In wound repair, a paradigm is emerging where local transient senescence predominately constrains fibrosis, while chronic senescence drives diabetic wound pathology. Indeed, experimentally blocking the senescence-linked receptor, CXCR2, in vivo reverses pathology and accelerates diabetic healing. These observations now pave the way to explore the beneficial effects of senescence-targeted therapies for the treatment of chronic wounds.
Author Contributions
HW wrote the manuscript and prepared the figures. MH provided critical appraisal. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Medical Research Council (United Kingdom) Ph.D. studentship (MR/M016307/1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acosta, J. C., Banito, A., Wuestefeld, T., Georgilis, A., Janich, P., Morton, J. P., et al. (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990. doi: 10.1038/ncb2784
Acosta, J. C., O’Loghlen, A., Banito, A., Guijarro, M. V., Augert, A., Raguz, S., et al. (2008). Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018. doi: 10.1016/j.cell.2008.03.038
Agger, K., Cloos, P. A., Rudkjær, L., Williams, K., Andersen, G., Christensen, J., et al. (2009). The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A–ARF locus in response to oncogene-and stress-induced senescence. Genes Dev. 23, 1171–1176. doi: 10.1101/gad.510809
Agren, M. S., Steenfos, H. H., Dabelsteen, S., Hansen, J. B., and Dabelsteen, E. (1999). Proliferation and mitogenic response to PDGF-BB of fibroblasts isolated from chronic venous leg ulcers is ulcer-age dependent. J. Invest. Dermatol. 112, 463–469. doi: 10.1046/j.1523-1747.1999.00549.x
Albanesi, C., Madonna, S., Gisondi, P., and Girolomoni, G. (2018). The interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front. Immunol. 9:1549. doi: 10.3389/fimmu.2018.01549
Anderson, S. G., Shoo, H., Saluja, S., Anderson, C. D., Khan, A., Livingston, M., et al. (2018). Social deprivation modifies the association between incident foot ulceration and mortality in type 1 and type 2 diabetes: a longitudinal study of a primary-care cohort. Diabetologia 61, 959–967. doi: 10.1007/s00125-017-4522-x
Angelidis, I., Simon, L. M., Fernandez, I. E., Strunz, M., Mayr, C. H., Greiffo, F. R., et al. (2019). An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 10:963. doi: 10.1038/s41467-019-08831-9
Baker, D. J., Childs, B. G., Durik, M., Wijers, M. E., Sieben, C. J., Zhong, J., et al. (2016). Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189. doi: 10.1038/nature16932
Baker, D. J., Perez-Terzic, C., Jin, F., Pitel, K. S., Niederländer, N. J., Jeganathan, K., et al. (2008). Opposing roles for p16 Ink4a and p19 Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 10, 825–836. doi: 10.1038/ncb1744
Baker, D. J., Weaver, R. L., and van Deursen, J. M. (2013). p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep. 3, 1164–1174. doi: 10.1016/j.celrep.2013.03.028
Baker, D. J., Wijshake, T., Tchkonia, T., LeBrasseur, N. K., Childs, B. G., Van De Sluis, B., et al. (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 47, 232–236. doi: 10.1038/nature10600
Baum, C. L., and Arpey, C. J. (2005). Normal cutaneous wound healing: clinical correlation with cellular and molecular events. Dermatol. Surg. 31, 674–686. doi: 10.1111/j.1524-4725.2005.31612
Benhamed, M., Herbig, U., Ye, T., Dejean, A., and Bischof, O. (2012). Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat. Cell Biol. 14, 266–275. doi: 10.1038/ncb2443
Biran, A., Zada, L., Abou Karam, P., Vadai, E., Roitman, L., Ovadya, Y., et al. (2017). Quantitative identification of senescent cells in aging and disease. Aging Cell 16, 661–671. doi: 10.1111/acel.12592
Bonda, D. J., Lee, H. G., Camins, A., Pallàs, M., Casadesus, G., Smith, M. A., et al. (2011). The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations. Lancet Neurol. 10, 275–279. doi: 10.1016/S1474-4422(11)70013-8
Brockes, J. P., and Kumar, A. (2005). Appendage regeneration in adult vertebrates and implications for regenerative medicine. Science 310, 1919–1923. doi: 10.1126/science.1115200
Caley, M. P., Martins, V. L., and O’Toole, E. A. (2015). Metalloproteinases and wound healing. Adv. Wound Care 4, 225–234. doi: 10.1089/wound.2014.0581
Campisi, J. (2013). Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705. doi: 10.1146/annurev-physiol-030212-183653
Campisi, J., and di Fagagna, F. D. A. (2007). Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740. doi: 10.1038/nrm2233
Chang, J., Wang, Y., Shao, L., Laberge, R. M., Demaria, M., Campisi, J., et al. (2016). Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83. doi: 10.1038/nm.4010
Chaturvedi, V., Qin, J. Z., Stennett, L., Choubey, D., and Nickoloff, B. (2004). Resistance to UV-induced apoptosis in human keratinocytes during accelerated senescence is associated with functional inactivation of p53. J. Cell. Physiol. 198, 100–109. doi: 10.1002/jcp.10392
Chen, J., Huang, X., Halicka, D., Brodsky, S., Avram, A., Eskander, J., et al. (2006). Contribution of p16 INK4a and p21 CIP1 pathways to induction of premature senescence of human endothelial cells: permissive role of p53. Am. J. Physiol. Heart Circ. Physiol. 290, H1575–H1586. doi: 10.1152/ajpheart.00364.2005
Chien, Y., Scuoppo, C., Wang, X., Fang, X., Balgley, B., Bolden, J. E., et al. (2011). Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 25, 2125–2136. doi: 10.1101/gad.17276711
Childs, B. G., Baker, D. J., Kirkland, J. L., Campisi, J., and Van Deursen, J. M. (2014). Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 15, 1139–1153. doi: 10.15252/embr.201439245
Citro, A., Cantarelli, E., and Piemonti, L. (2015). The CXCR1/2 pathway: involvement in diabetes pathophysiology and potential target for T1D interventions. Curr. Diab. Rep. 15:68. doi: 10.1007/s11892-015-0638-x
Collado, M., Blasco, M. A., and Serrano, M. (2007). Cellular senescence in cancer and aging. Cell 130, 223–233. doi: 10.1016/j.cell.2007.07.003
Colman, R. J., Beasley, T. M., Kemnitz, J. W., Johnson, S. C., Weindruch, R., and Anderson, R. M. (2014). Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 5:3557. doi: 10.1038/ncomms4557
Coppé, J.-P., Desprez, P.-Y., Krtolica, A., and Campisi, J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118.
Coppé, J.-P., Kauser, K., Campisi, J., and Beauséjour, C. M. (2006). Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 281, 29568–29574. doi: 10.1074/jbc.M603307200
Coppé, J.-P., Patil, C. K., Rodier, F., Sun, Y., Muñoz, D. P., Goldstein, J., et al. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6:2853–2868. doi: 10.1371/journal.pbio.0060301
Coppé, J.-P., Rodier, F., Patil, C. K., Freund, A., Desprez, P.-Y., and Campisi, J. (2011). Tumor suppressor and aging biomarker p16INK4a induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 286, 36396–36403. doi: 10.1074/jbc.M111.257071
Coughlan, M. T., Yap, F. Y., Tong, D. C., Andrikopoulos, S., Gasser, A., Thallas-Bonke, V., et al. (2011). Advanced glycation end products are direct modulators of β-cell function. Diabetes Metab. Res. Rev. 60, 2523–2532. doi: 10.2337/db10-1033
Cuervo, A. M. (2008). Autophagy and aging: keeping that old broom working. Trends Genet. 24, 604–612. doi: 10.1016/j.tig.2008.10.002
Da Silva-Álvarez, S., Guerra-Varela, J., Sobrido-Cameán, D., Quelle, A., Barreiro-Iglesias, A., Sánchez, L., et al. (2020). Cell senescence contributes to tissue regeneration in zebrafish. Aging Cell 19:e13052. doi: 10.1111/acel.13052
Da Silva-Álvarez, S., Picallos-Rabina, P., Antelo-Iglesias, L., Triana-Martínez, F., Barreiro-Iglesias, A., Sánchez, L., et al. (2019). The development of cell senescence. Exp. Gerontol. 128:110742. doi: 10.1016/j.exger.2019.110742
Debacq-Chainiaux, F., Erusalimsky, J. D., Campisi, J., and Toussaint, O. (2009). Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 4, 1798–1806. doi: 10.1038/nprot.2009.191
Demaria, M., Ohtani, N., Youssef, S. A., Rodier, F., Toussaint, W., Mitchell, J. R., et al. (2014). An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733. doi: 10.1016/j.devcel.2014.11.012
Demidenko, Z. N., Korotchkina, L. G., Gudkov, A. V., and Blagosklonny, M. V. (2010). Paradoxical suppression of cellular senescence by p53. Proc. Natl. Acad. Sci. U.S.A. 107, 9660–9664. doi: 10.1073/pnas.1002298107
Devalaraja, R. M., Nanney, L. B., Qian, Q., Du, J., Yu, Y., Devalaraja, M. N., et al. (2000). Delayed wound healing in CXCR2 knockout mice. J. Invest. Dermatol. 115, 234–244. doi: 10.1046/j.1523-1747.2000.00034.x
Di Cunto, F., Topley, G., Calautti, E., Hsiao, J., Ong, L., Seth, P. K., et al. (1998). Inhibitory function of p21Cip1/WAF1 in differentiation of primary mouse keratinocytes independent of cell cycle control. Science 280, 1069–1072. doi: 10.1126/science.280.5366.1069
Di Giovanni, S., Knights, C. D., Rao, M., Yakovlev, A., Beers, J., Catania, J., et al. (2006). The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 25, 4084–4096. doi: 10.1038/sj.emboj.7601292
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C., et al. (2006). Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642. doi: 10.1038/nature05327
Dimri, G. P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367. doi: 10.1073/pnas.92.20.9363
Drewnowski, A., and Specter, S. E. (2004). Poverty and obesity: the role of energy density and energy costs. Am. J. Clin. Nutr. 79, 6–16. doi: 10.1093/ajcn/79.1.6
Duan, J., Duan, J., Zhang, Z., and Tong, T. (2005). Irreversible cellular senescence induced by prolonged exposure to H2O2 involves DNA-damage-and-repair genes and telomere shortening. Int. J. Biochem. Cell Biol. 37, 1407–1420. doi: 10.1016/j.biocel.2005.01.010
Elzi, D. J., Lai, Y., Song, M., Hakala, K., Weintraub, S. T., and Shiio, Y. (2012). Plasminogen activator inhibitor 1-insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc. Natl. Acad. Sci. U.S.A. 109, 12052–12057. doi: 10.1073/pnas.1120437109
Faget, D. V., Ren, Q., and Stewart, S. A. (2019). Unmasking senescence: context-dependent effects of SASP in cancer. Nat. Rev. Cancer 19, 439–453. doi: 10.1038/s41568-019-0156-2
Fang, M., Wang, J., Li, S., and Guo, Y. (2016). Advanced glycation end-products accelerate the cardiac aging process through the receptor for advanced glycation end-products/transforming growth factor-β-Smad signaling pathway in cardiac fibroblasts. Geriatr. Gerontol. Int. 16, 522–527. doi: 10.1111/ggi.12499
Freund, A., Orjalo, A. V., Desprez, P.-Y., and Campisi, J. (2010). Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246. doi: 10.1016/j.molmed.2010.03.003
Freund, A., Patil, C. K., and Campisi, J. (2011). p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548. doi: 10.1038/emboj.2011.69
Frykberg, R. G., and Banks, J. (2015). Challenges in the treatment of chronic wounds. Adv. Wound Care 4, 560–582. doi: 10.1089/wound.2015.0635
Fuchs, E., and Byrne, C. (1994). The epidermis: rising to the surface. Curr. Opin. Genet. Dev. 4, 725–736. doi: 10.1016/0959-437x(94)90140-x
Grange, P. A., Chéreau, C., Raingeaud, J., Nicco, C., Weill, B., Dupin, N., et al. (2009). Production of superoxide anions by keratinocytes initiates P. acnes-induced inflammation of the skin. PLoS Pathog. 5:7. doi: 10.1371/journal.ppat.1000527
Guest, J. F., Ayoub, N., McIlwraith, T., Uchegbu, I., Gerrish, A., Weidlich, D., et al. (2015). Health economic burden that wounds impose on the National Health Service in the UK. BMJ Open 5:e009283.
Hahn, O., Grönke, S., Stubbs, T. M., Ficz, G., Hendrich, O., Krueger, F., et al. (2017). Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 18:56. doi: 10.1186/s13059-017-1187-1
Hall, B. M., Balan, V., Gleiberman, A. S., Strom, E., Krasnov, P., Virtuoso, L. P., et al. (2016). Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 8, 1294–1315. doi: 10.18632/aging.100991
Hampel, B., Malisan, F., Niederegger, H., Testi, R., and Jansen-Dürr, P. (2004). Differential regulation of apoptotic cell death in senescent human cells. Exp. Gerontol. 39, 1713–1721. doi: 10.1016/j.exger.2004.05.010
Han, G., and Ceilley, R. (2017). Chronic wound healing: a review of current management and treatments. Adv. Ther. 34, 599–610. doi: 10.1007/s12325-017-0478-y
Han, X., Tao, Y., Deng, Y., Yu, J., Sun, Y., and Jiang, G. (2017). Metformin accelerates wound healing in type 2 diabetic db/db mice. Mol. Med. Rep. 16, 8691–8698. doi: 10.3892/mmr.2017.7707
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. doi: 10.1038/nature08221
Hayflick, L., and Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621. doi: 10.1016/0014-4827(61)90192-6
He, L., He, X., Lim, L. P., De Stanchina, E., Xuan, Z., Liang, Y., et al. (2007). A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134. doi: 10.1038/nature05939
Helman, A., Klochendler, A., Azazmeh, N., Gabai, Y., Horwitz, E., Anzi, S., et al. (2016). p16 Ink4a-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 22, 412–420. doi: 10.1038/nm.4054
Hernandez-Segura, A., Nehme, J., and Demaria, M. (2018). Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453. doi: 10.1016/j.tcb.2018.02.001
Herrling, T., Jung, K., and Fuchs, J. (2006). Measurements of UV-generated free radicals/reactive oxygen species (ROS) in skin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 63, 840–845. doi: 10.1016/j.saa.2005.10.013
Hickson, L. J., Langhi Prata, L. G. P., Bobart, S. A., Evans, T. K., Giorgadze, N., Hashmi, S. K., et al. (2019). Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456. doi: 10.1016/j.ebiom.2019.08.069
Hiebert, P., Wietecha, M. S., Cangkrama, M., Haertel, E., Mavrogonatou, E., Stumpe, M., et al. (2018). Nrf2-mediated fibroblast reprogramming drives cellular senescence by targeting the matrisome. Dev. Cell 46, 145–161. doi: 10.1016/j.devcel.2018.06.012
Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biol. 14:3156. doi: 10.1186/gb-2013-14-10-r115
Horvath, S., Lu, A. T., Cohen, H., and Raj, K. (2019). Rapamycin retards epigenetic ageing of keratinocytes independently of its effects on replicative senescence, proliferation and differentiation. Aging 11, 3238–3249. doi: 10.18632/aging.101976
Huang, W.-C., Ju, T.-K., Hung, M.-C., and Chen, C.-C. (2007). Phosphorylation of CBP by IKKα promotes cell growth by switching the binding preference of CBP from p53 to NF-κB. Mol. Cell 26, 75–87. doi: 10.1016/j.molcel.2007.02.019
Hudgins, A. D., Tazearslan, C., Tare, A., Zhu, Y., Huffman, D., and Suh, Y. (2018). Age-and tissue-specific expression of senescence biomarkers in mice. Front. Genet. 9:59. doi: 10.3389/fgene.2018.00059
Ito, T., Teo, Y. V., Evans, S. A., Neretti, N., and Sedivy, J. M. (2018). Regulation of cellular senescence by polycomb chromatin modifiers through distinct DNA damage-and histone methylation-dependent pathways. Cell Rep. 22, 3480–3492. doi: 10.1016/j.celrep.2018.03.002
Jenkins, N. C., Liu, T., Cassidy, P., Leachman, S. A., Boucher, K. M., Goodson, A. G., et al. (2011). The p16 INK4A tumor suppressor regulates cellular oxidative stress. Oncogene 30, 265–274. doi: 10.1038/onc.2010.419
Joost, S., Jacob, T., Sun, X., Annusver, K., La Manno, G., Sur, I., et al. (2018). Single-Cell Transcriptomics of traced epidermal and hair follicle stem cells reveals rapid adaptations during wound healing. Cell Rep. 25, 585–597. doi: 10.1016/j.celrep.2018.09.059
Jun, J.-I., and Lau, L. F. (2010). The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12, 676–685. doi: 10.1038/ncb2070
Jun, J. I., and Lau, L. F. (2017). CCN2 induces cellular senescence in fibroblasts. J. Cell Commun. Signal. 11, 15–23. doi: 10.1007/s12079-016-0359-1
June, C. H., O’Connor, R. S., Kawalekar, O. U., Ghassemi, S., and Milone, M. C. (2018). CAR T cell immunotherapy for human cancer. Science 359, 1361–1365. doi: 10.1126/science.aar6711
Jurk, D., Wang, C., Miwa, S., Maddick, M., Korolchuk, V., Tsolou, A., et al. (2012). Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004. doi: 10.1111/j.1474-9726.2012.00870.x
Kalan, L. R., Meisel, J. S., Loesche, M. A., Horwinski, J., Soaita, I., Chen, X., et al. (2019). Strain-and species-level variation in the microbiome of diabetic wounds is associated with clinical outcomes and therapeutic efficacy. Cell Host Microbe 25, 641–655. doi: 10.1016/j.chom.2019.03.006
Kamei, N., Tobe, K., Suzuki, R., Ohsugi, M., Watanabe, T., Kubota, N., et al. (2006). Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 281, 26602–26614. doi: 10.1074/jbc.M601284200
Kang, T. W., Yevsa, T., Woller, N., Hoenicke, L., Wuestefeld, T., Dauch, D., et al. (2011). Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551. doi: 10.1038/nature10599
Keskin, D., Sadri, S., and Eskazan, A. E. (2016). Dasatinib for the treatment of chronic myeloid leukemia: patient selection and special considerations. Drug Des. Dev. Ther. 10, 3355–3361. doi: 10.2147/DDDT.S85050
Khan, F., Niaz, K., Maqbool, F., Ismail Hassan, F., Abdollahi, M., Nagulapalli Venkata, K. C., et al. (2016). Molecular targets underlying the anticancer effects of quercetin: an update. Nutrients 8:529. doi: 10.3390/nu8090529
Kimmel, J. C., Penland, L., Rubinstein, N. D., Hendrickson, D. G., Kelley, D. R., and Rosenthal, A. Z. (2019). Murine single-cell RNA-seq reveals cell-identity-and tissue-specific trajectories of aging. Genome Res. 29, 2088–2103. doi: 10.1101/gr.253880.119
King, A. C., Peterson, T. J., Horvat, T. Z., Rodriguez, M., and Tang, L. A. (2017). Venetoclax: a first-in-class oral BCL-2 inhibitor for the management of lymphoid malignancies. Ann. Pharmacother. 51, 410–416. doi: 10.1177/1060028016685803
Korns, D., Frasch, S. C., Fernandez-Boyanapalli, R., Henson, P. M., and Bratton, D. L. (2011). Modulation of macrophage efferocytosis in inflammation. Front. Immunol. 2:57. doi: 10.3389/fimmu.2011.00057
Kotake, Y., Cao, R., Viatour, P., Sage, J., Zhang, Y., and Xiong, Y. (2007). pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4a tumor suppressor gene. Genes Dev. 21, 49–54. doi: 10.1101/gad.1499407
Krishna, D. R., Sperker, B., Fritz, P., and Klotz, U. (1999). Does pH 6 β-galactosidase activity indicate cell senescence? Mech. Ageing Dev. 109, 113–123. doi: 10.1016/s0047-6374(99)00031-7
Krizhanovsky, V., Yon, M., Dickins, R. A., Hearn, S., Simon, J., Miething, C., et al. (2008). Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667. doi: 10.1016/j.cell.2008.06.049
Kuilman, T., Michaloglou, C., Mooi, W. J., and Peeper, D. S. (2010). The essence of senescence. Genes Dev. 24, 2463–2479. doi: 10.1101/gad.1971610
Kuilman, T., Michaloglou, C., Vredeveld, L. C., Douma, S., van Doorn, R., Desmet, C. J., et al. (2008). Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031. doi: 10.1016/j.cell.2008.03.039
Lau, E. Y. M., Carroll, E. C., Callender, L. A., Hood, G. A., Berryman, V., Pattrick, M., et al. (2019). Type 2 diabetes is associated with the accumulation of senescent T cells. Clin. Exp. Immunol. 197, 205–213. doi: 10.1111/cei.13344
Lee, B. Y., Han, J. A., Im, J. S., Morrone, A., Johung, K., Goodwin, E. C., et al. (2006). Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 5, 187–195. doi: 10.1111/j.1474-9726.2006.00199.x
Li, J., Chen, J., and Kirsner, R. (2007). Pathophysiology of acute wound healing. Clin. Dermatol. 25, 9–18. doi: 10.1016/j.clindermatol.2006.09.007
Lindholm, C., and Searle, R. (2016). Wound management for the 21st century: combining effectiveness and efficiency. Int. Wound J. 13, 5–15. doi: 10.1111/iwj.12623
Liu, L., Cheung, T. H., Charville, G. W., Hurgo, B. M. C., Leavitt, T., Shih, J., et al. (2013). Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 4, 189–204. doi: 10.1016/j.celrep.2013.05.043
Lopes-Paciencia, S., Saint-Germain, E., Rowell, M.-C., Ruiz, A. F., Kalegari, P., and Ferbeyre, G. (2019). The senescence-associated secretory phenotype and its regulation. Cytokine 117, 15–22.
Lowe, J. M., Menendez, D., and Fessler, M. B. (2014). A new inflammatory role for p53 in human macrophages. Cell Cycle 13, 2983–2984. doi: 10.4161/15384101.2014.959852
Lujambio, A., Akkari, L., Simon, J., Grace, D., Tschaharganeh, D. F., Bolden, J. E., et al. (2013). Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460. doi: 10.1016/j.cell.2013.03.020
Lupa, D. M. W., Kalfalah, F., Safferling, K., Boukamp, P., Poschmann, G., Volpi, E., et al. (2015). Characterization of skin aging–associated secreted proteins (SAASP) produced by dermal fibroblasts isolated from intrinsically aged human skin. J. Invest. Dermatol. 135, 1954–1968. doi: 10.1038/jid.2015.120
Maciel-Baron, L., Morales-Rosales, S., Aquino-Cruz, A., Triana-Martínez, F., Galván-Arzate, S., Luna-López, A., et al. (2016). Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age 38:26. doi: 10.1007/s11357-016-9886-1
Maeda, M., Hayashi, T., Mizuno, N., Hattori, Y., and Kuzuya, M. (2015). Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: role of superoxide production by NADPH oxidase. PLoS One 10:e0123169. doi: 10.1371/journal.pone.0123169
Makrantonaki, E., Wlaschek, M., and Scharffetter-Kochanek, K. (2017). Pathogenesis of wound healing disorders in the elderly. J. Dtsch. Dermatol. Ges. 15, 255–275. doi: 10.1111/ddg.13199
Matjusaitis, M., Chin, G., Sarnoski, E. A., and Stolzing, A. (2016). Biomarkers to identify and isolate senescent cells. Ageing Res. Rev. 29, 1–12. doi: 10.1016/j.arr.2016.05.003
McHugh, D., and Gil, J. (2018). Senescence and aging: causes, consequences, and therapeutic avenues. J. Cell Biol. 217, 65–77. doi: 10.1083/jcb.201708092
Mendez, M. V., Raffetto, J. D., Phillips, T., Menzoian, J. O., and Park, H. Y. (1999). The proliferative capacity of neonatal skin fibroblasts is reduced after exposure to venous ulcer wound fluid: a potential mechanism for senescence in venous ulcers. J. Vasc. Surg. 30, 734–743. doi: 10.1016/S0741-5214(99)70113-8
Mendez, M. V., Stanley, A., Park, H.-Y., Shon, K., Phillips, T., and Menzoian, J. O. (1998). Fibroblasts cultured from venous ulcers display cellular characteristics of senescence. J. Vasc. Surg. 28, 876–883. doi: 10.1016/s0741-5214(98)70064-3
Meydani, M., Das, S., Band, M., Epstein, S., and Roberts, S. (2011). The effect of caloric restriction and glycemic load on measures of oxidative stress and antioxidants in humans: results from the CALERIE Trial of Human Caloric Restriction. J. Nutr. Health Aging. 15, 456–460. doi: 10.1007/s12603-011-0002-z
Meyer, K., Hodwin, B., Ramanujam, D., Engelhardt, S., and Sarikas, A. (2016). Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J. Am. Coll. Cardiol. 67, 2018–2028. doi: 10.1016/j.jacc.2016.02.047
Minamino, T., Orimo, M., Shimizu, I., Kunieda, T., Yokoyama, M., Ito, T., et al. (2009). A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 15, 1082–1087. doi: 10.1038/nm.2014
Missero, C., Di Cunto, F., Kiyokawa, H., Koff, A., and Dotto, G. P. (1996). The absence of p21Cip1/WAF1 alters keratinocyte growth and differentiation and promotes ras-tumor progression. Genes Dev. 10, 3065–3075. doi: 10.1101/gad.10.23.3065
Moiseeva, O., Deschênes-Simard, X., St-Germain, E., Igelmann, S., Huot, G., Cadar, A. E., et al. (2013). Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 12, 489–498. doi: 10.1111/acel.12075
Montero, J., and Letai, A. (2018). Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 25, 56–64. doi: 10.1038/cdd.2017.183
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. doi: 10.1038/nri2448
Muller, M., Li, Z., and Maitz, P. K. (2009). Pseudomonas pyocyanin inhibits wound repair by inducing premature cellular senescence: role for p38 mitogen-activated protein kinase. Burns 35, 500–508. doi: 10.1016/j.burns.2008.11.010
Muñoz-Espín, D., Cañamero, M., Maraver, A., Gómez-López, G., Contreras, J., Murillo-Cuesta, S., et al. (2013). Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118. doi: 10.1016/j.cell.2013.10.019
Narita, M., Nuñez, S., Heard, E., Narita, M., Lin, A. W., Hearn, S. A., et al. (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716. doi: 10.1016/s0092-8674(03)00401-x
Orjalo, A. V., Bhaumik, D., Gengler, B. K., Scott, G. K., and Campisi, J. (2009). Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. U.S.A. 106, 17031–17036. doi: 10.1073/pnas.0905299106
Ovadya, Y., and Krizhanovsky, V. (2018). Strategies targeting cellular senescence. J. Clin. Invest. 128, 1247–1254. doi: 10.1172/JCI95149
Parrinello, S., Samper, E., Krtolica, A., Goldstein, J., Melov, S., and Campisi, J. (2003). Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 5, 741–747. doi: 10.1038/ncb1024
Passos, J. F., Nelson, G., Wang, C., Richter, T., Simillion, C., Proctor, C. J., et al. (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6:347. doi: 10.1038/msb.2010.5
Pazolli, E., Alspach, E., Milczarek, A., Prior, J., Piwnica-Worms, D., and Stewart, S. A. (2012). Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 72, 2251–2261.
Pereira, B. I., Devine, O. P., Vukmanovic-Stejic, M., Chambers, E. S., Subramanian, P., Patel, N., et al. (2019). Senescent cells evade immune clearance via HLA-E-mediated NK and CD8. Nat. Commun. 10:2387. doi: 10.1038/s41467-019-10335-5
Perrigue, P. M., Rakoczy, M., Pawlicka, K. P., Belter, A., Giel-Pietraszuk, M., Naskrêt-Barciszewska, M., et al. (2020). Cancer stem cell-inducing media activates senescence reprogramming in fibroblasts. Cancers 12:1745. doi: 10.3390/cancers12071745
Prattichizzo, F., De Nigris, V., La Sala, L., Procopio, A. D., Olivieri, F., and Ceriello, A. (2016). “Inflammaging” as a druggable target: a senescence-associated secretory phenotype-centered view of type 2 diabetes. Oxid. Med. Cell. Longev. 2016:1810327. doi: 10.1155/2016/1810327
Prattichizzo, F., De Nigris, V., Mancuso, E., Spiga, R., Giuliani, A., Matacchione, G., et al. (2018). Short-term sustained hyperglycaemia fosters an archetypal senescence-associated secretory phenotype in endothelial cells and macrophages. Redox Biol. 15, 170–181. doi: 10.1016/j.redox.2017.12.001
Rena, G., Hardie, D. G., and Pearson, E. R. (2017). The mechanisms of action of metformin. Diabetologia 60, 1577–1585. doi: 10.1007/s00125-017-4342-z
Ressler, S., Bartkova, J., Niederegger, H., Bartek, J., Scharffetter-Kochanek, K., Jansen-Dürr, P., et al. (2006). p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 5, 379–389. doi: 10.1111/j.1474-9726.2006.00231.x
Rhinn, M., Ritschka, B., and Keyes, W. M. (2019). Cellular senescence in development, regeneration and disease. Development 146:dev151837. doi: 10.1242/dev.151837
Ritschka, B., Storer, M., Mas, A., Heinzmann, F., Ortells, M. C., Morton, J. P., et al. (2017). The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 31, 172–183. doi: 10.1101/gad.290635.116
Roberts, A. W., Davids, M. S., Pagel, J. M., Kahl, B. S., Puvvada, S. D., Gerecitano, J. F., et al. (2016). Targeting BCL2 with Venetoclax in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 374, 311–322. doi: 10.1056/NEJMoa1513257
Rodier, F., and Campisi, J. (2011). Four faces of cellular senescence. J. Cell Biol. 192, 547–556. doi: 10.1083/jcb.201009094
Rodier, F., Coppé, J.-P., Patil, C. K., Hoeijmakers, W. A., Muñoz, D. P., Raza, S. R., et al. (2009). Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979. doi: 10.1038/ncb1909
Roos, C. M., Zhang, B., Palmer, A. K., Ogrodnik, M. B., Pirtskhalava, T., Thalji, N. M., et al. (2016). Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15, 973–977. doi: 10.1111/acel.12458
Ruan, Y., Wu, S., Zhang, L., Chen, G., and Lai, W. (2014). Retarding the senescence of human vascular endothelial cells induced by hydrogen peroxide: effects of 17beta-estradiol (E2) mediated mitochondria protection. Biogerontology 15, 367–375. doi: 10.1007/s10522-014-9507-2
Ryu, S., Oh, Y., and Park, S. (2007). Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 14, 1020–1028. doi: 10.1038/sj.cdd.4402091
Sagiv, A., Burton, D. G., Moshayev, Z., Vadai, E., Wensveen, F., Ben-Dor, S., et al. (2016). NKG2D ligands mediate immunosurveillance of senescent cells. Aging 8, 328–344. doi: 10.18632/aging.100897
Salminen, A., Hyttinen, J. M., and Kaarniranta, K. (2011). AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J. Mol. Med. 89, 667–676. doi: 10.1007/s00109-011-0748-0
Sato, S., Kawamata, Y., Takahashi, A., Imai, Y., Hanyu, A., Okuma, A., et al. (2015). Ablation of the p16(INK4a) tumour suppressor reverses ageing phenotypes of klotho mice. Nat. Commun. 6:7035. doi: 10.1038/ncomms8035
Schäfer, M., and Werner, S. (2008). Cancer as an overhealing wound: an old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 9, 628–638. doi: 10.1038/nrm2455
Schafer, M. J., White, T. A., Evans, G., Tonne, J. M., Verzosa, G. C., Stout, M. B., et al. (2016). Exercise prevents diet-induced cellular senescence in adipose tissue. Diabetes Metab. Res. Rev. 65, 1606–1615. doi: 10.2337/db15-0291
Shah, P. P., Donahue, G., Otte, G. L., Capell, B. C., Nelson, D. M., Cao, K., et al. (2013). Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 27, 1787–1799. doi: 10.1101/gad.223834.113
Shaw, T. J., and Martin, P. (2016). Wound repair: a showcase for cell plasticity and migration. Curr. Opin. Cell Biol. 42, 29–37. doi: 10.1016/j.ceb.2016.04.001
Shinmura, K., Tamaki, K., Sano, M., Murata, M., Yamakawa, H., Ishida, H., et al. (2011). Impact of long-term caloric restriction on cardiac senescence: caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. J. Mol. Cell. Cardiol. 50, 117–127. doi: 10.1016/j.yjmcc.2010.10.018
Sindrilaru, A., Peters, T., Wieschalka, S., Baican, C., Baican, A., Peter, H., et al. (2011). An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Invest. 121, 985–997. doi: 10.1172/JCI44490
Smirnov, A., Panatta, E., Lena, A., Castiglia, D., Di Daniele, N., Melino, G., et al. (2016). FOXM1 regulates proliferation, senescence and oxidative stress in keratinocytes and cancer cells. Aging 8, 1384–1396. doi: 10.18632/aging.100988
Sodagam, L., Lewinska, A., Wnuk, M., and Rattan, S. I. S. (2017). Chronic exposure to rapamycin and episodic serum starvation modulate ageing of human fibroblasts in vitro. Biogerontology 18, 841–854. doi: 10.1007/s10522-017-9730-8
Sohal, R. S., and Weindruch, R. (1996). Oxidative stress, caloric restriction, and aging. Science 273, 59–63. doi: 10.1126/science.273.5271.59
Stanley, A., and Osler, T. (2001). Senescence and the healing rates of venous ulcers. J. Vasc. Surg. 33, 1206–1211. doi: 10.1067/mva.2001.115379
Storer, M., Mas, A., Robert-Moreno, A., Pecoraro, M., Ortells, M. C., Di Giacomo, V., et al. (2013). Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130. doi: 10.1016/j.cell.2013.10.041
Stubbs, T. M., Bonder, M. J., Stark, A. K., Krueger, F., von Meyenn, F., Stegle, O., et al. (2017). Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 18:68.
Sui, A., Xu, Y., Pan, B., Guo, T., Wu, J., Shen, Y., et al. (2019). Histone demethylase KDM6B regulates 1, 25-dihydroxyvitamin D3-induced senescence in glioma cells. J. Cell. Physiol. 234, 17990–17998. doi: 10.1002/jcp.28431
Sun, D., Luo, M., Jeong, M., Rodriguez, B., Xia, Z., Hannah, R., et al. (2014). Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 14, 673–688. doi: 10.1016/j.stem.2014.03.002
Sun, Y., Coppé, J.-P., and Lam, E. W.-F. (2018). Cellular senescence: the sought or the unwanted? Trends Mol. Med. 24, 871–885. doi: 10.1016/j.molmed.2018.08.002
Swift, M. E., Burns, A. L., Gray, K. L., and DiPietro, L. A. (2001). Age-related alterations in the inflammatory response to dermal injury. J. Invest. Dermatol. 117, 1027–1035. doi: 10.1046/j.0022-202x.2001.01539.x
Tasdemir, N., Banito, A., Roe, J. S., Alonso-Curbelo, D., Camiolo, M., Tschaharganeh, D. F., et al. (2016). BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov. 6, 612–629. doi: 10.1158/2159-8290.CD-16-0217
Tchkonia, T., Zhu, Y., Van Deursen, J., Campisi, J., and Kirkland, J. L. (2013). Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972. doi: 10.1172/JCI64098
Thandavarayan, R. A., Garikipati, V. N., Joladarashi, D., Suresh Babu, S., Jeyabal, P., Verma, S. K., et al. (2015). Sirtuin-6 deficiency exacerbates diabetes-induced impairment of wound healing. Exp. Dermatol. 24, 773–778. doi: 10.1111/exd.12762
Tigges, J., Krutmann, J., Fritsche, E., Haendeler, J., Schaal, H., Fischer, J. W., et al. (2014). The hallmarks of fibroblast ageing. Mech. Ageing Dev. 138, 26–44. doi: 10.1016/j.mad.2014.03.004
Tonnessen-Murray, C. A., Frey, W. D., Rao, S. G., Shahbandi, A., Ungerleider, N. A., Olayiwola, J. O., et al. (2019). Chemotherapy-induced senescent cancer cells engulf other cells to enhance their survival. J. Cell Biol. 218, 3827–3844. doi: 10.1083/jcb.201904051
van Deursen, J. M. (2014). The role of senescent cells in ageing. Nature 509, 439–446. doi: 10.1038/nature13193
Vande Berg, J. S., Rudolph, R., Hollan, C., and Haywood-Reid, P. L. (1998). Fibroblast senescence in pressure ulcers. Wound Repair Regen. 6, 38–49. doi: 10.1046/j.1524-475x.1998.60107.x
Velarde, M. C., Flynn, J. M., Day, N. U., Melov, S., and Campisi, J. (2012). Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 4, 3–12. doi: 10.18632/aging.100423
Velnar, T., Bailey, T., and Smrkolj, V. (2009). The wound healing process: an overview of the cellular and molecular mechanisms. J. Int. Med. Res. 37, 1528–1542. doi: 10.1177/147323000903700531
von Kobbe, C. (2019). Targeting senescent cells: approaches, opportunities, challenges. Aging 11, 12844–12861. doi: 10.18632/aging.102557
von Zglinicki, T., Wan, T., and Miwa, S. (2020). Senescence in post-mitotic cells: A driver of ageing? Antioxid. Redox Signal. doi: 10.1089/ars.2020.8048 [Epub ahead of print].
Vousden, K. H., and Prives, C. (2009). Blinded by the light: the growing complexity of p53. Cell 137, 413–431. doi: 10.1016/j.cell.2009.04.037
Waaijer, M. E., Parish, W. E., Strongitharm, B. H., van Heemst, D., Slagboom, P. E., de Craen, A. J., et al. (2012). The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 11, 722–725. doi: 10.1111/j.1474-9726.2012.00837.x
Wall, I. B., Moseley, R., Baird, D. M., Kipling, D., Giles, P., Laffafian, I., et al. (2008). Fibroblast dysfunction is a key factor in the non-healing of chronic venous leg ulcers. J. Invest. Dermatol. 128, 2526–2540. doi: 10.1038/jid.2008.114
Wang, A. S., Ong, P. F., Chojnowski, A., Clavel, C., and Dreesen, O. (2017). Loss of lamin B1 is a biomarker to quantify cellular senescence in photoaged skin. Sci. Rep. 7:15678. doi: 10.1038/s41598-017-15901-9
Wang, C., Maddick, M., Miwa, S., Jurk, D., Czapiewski, R., Saretzki, G., et al. (2010). Adult-onset, short-term dietary restriction reduces cell senescence in mice. Aging 2, 555–566. doi: 10.18632/aging.100196
Wang, E. (1995). Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 55, 2284–2292.
Watkins, T. A., Wang, B., Huntwork-Rodriguez, S., Yang, J., Jiang, Z., Eastham-Anderson, J., et al. (2013). DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. U.S.A. 110, 4039–4044. doi: 10.1073/pnas.1211074110
Weindruch, R., and Walford, R. L. (1982). Dietary restriction in mice beginning at 1 year of age: effect on life-span and spontaneous cancer incidence. Science 215, 1415–1418. doi: 10.1126/science.7063854
Wetzler, C., Kämpfer, H., Stallmeyer, B., Pfeilschifter, J., and Frank, S. (2000). Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: prolonged persistence of neutrophils and macrophages during the late phase of repair. J. Invest. Dermatol. 115, 245–253. doi: 10.1046/j.1523-1747.2000.00029.x
Wilkinson, H. N., Clowes, C., Banyard, K. L., Matteuci, P., Mace, K. A., and Hardman, M. J. (2019a). Elevated local senescence in diabetic wound healing is linked to pathological repair via CXCR2. J. Invest. Dermatol. 139, 1171–1181. doi: 10.1016/j.jid.2019.01.005
Wilkinson, H. N., and Hardman, M. J. (2017). The role of estrogen in cutaneous ageing and repair. Maturitas 103, 60–64. doi: 10.1016/j.maturitas.2017.06.026
Wilkinson, H. N., and Hardman, M. J. (2020). Wound senescence: a functional link between diabetes and ageing? Exp. Dermatol. doi: 10.1111/exd.14082 [Epub ahead of print].
Wilkinson, H. N., Roberts, E. R., Stafford, A. R., Banyard, K. L., Matteucci, P., Mace, K. A., et al. (2019b). Tissue iron promotes wound repair via M2 macrophage polarization and the chemokine (CC motif) ligands 17 and 22. Am. J. Pathol. 189, 2196–2208. doi: 10.1016/j.ajpath.2019.07.015
Wilkinson, H. N., Upson, S. E., Banyard, K. L., Knight, R., Mace, K. A., and Hardman, M. J. (2019c). Reduced iron in diabetic wounds: an oxidative stress-dependent role for STEAP3 in extracellular matrix deposition and remodeling. J. Invest. Dermatol. 139, 2368–2377. doi: 10.1016/j.jid.2019.05.014
Wu, Y., Chen, L., Wang, Y., Li, W., Lin, Y., Yu, D., et al. (2015). Overexpression of Sirtuin 6 suppresses cellular senescence and NF-κB mediated inflammatory responses in osteoarthritis development. Sci. Rep. 5:17602. doi: 10.1038/srep17602
Xu, M., Pirtskhalava, T., Farr, J. N., Weigand, B. M., Palmer, A. K., Weivoda, M. M., et al. (2018). Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256. doi: 10.1038/s41591-018-0092-9
Xu, M., Tchkonia, T., Ding, H., Ogrodnik, M., Lubbers, E. R., Pirtskhalava, T., et al. (2015). JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. U.S.A. 112, E6301–E6310. doi: 10.1073/pnas.1515386112
Yanai, H., Shteinberg, A., Porat, Z., Budovsky, A., Braiman, A., Ziesche, R., et al. (2015). Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging 7, 664–672. doi: 10.18632/aging.100807
Yang, L., Licastro, D., Cava, E., Veronese, N., Spelta, F., Rizza, W., et al. (2016). Long-term calorie restriction enhances cellular quality-control processes in human skeletal muscle. Cell Rep. 14, 422–428. doi: 10.1016/j.celrep.2015.12.042
Yokoi, T., Fukuo, K., Yasuda, O., Hotta, M., Miyazaki, J., Takemura, Y., et al. (2006). Apoptosis signal-regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes Metab. Res. Rev. 55, 1660–1665. doi: 10.2337/db05-1607
Yosef, R., Pilpel, N., Tokarsky-Amiel, R., Biran, A., Ovadya, Y., Cohen, S., et al. (2016). Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 7:11190. doi: 10.1038/ncomms11190
Young, A., and McNaught, C.-E. (2011). The physiology of wound healing. Surgery 29, 475–479. doi: 10.1016/j.mpsur.2011.06.011
Yousefzadeh, M. J., Zhu, Y., McGowan, S. J., Angelini, L., Fuhrmann-Stroissnigg, H., Xu, M., et al. (2018). Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28. doi: 10.1016/j.ebiom.2018.09.015
Yun, M. H., Davaapil, H., and Brockes, J. P. (2015). Recurrent turnover of senescent cells during regeneration of a complex structure. eLife 4:e05505. doi: 10.7554/eLife.05505
Zhang, R., Chen, W., and Adams, P. D. (2007). Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell Biol. 27, 2343–2358. doi: 10.1128/MCB.02019-06
Zhu, Y., Armstrong, J. L., Tchkonia, T., and Kirkland, J. L. (2014). Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr. Opin. Clin. Nutr. Metab. Care 17, 324–328. doi: 10.1097/MCO.0000000000000065
Zhu, Y. I., Tchkonia, T., Pirtskhalava, T., Gower, A. C., Ding, H., Giorgadze, N., et al. (2015). The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658. doi: 10.1111/acel.12344
Keywords: senescence, ageing, diabetes, wound healing, senolytics
Citation: Wilkinson HN and Hardman MJ (2020) Senescence in Wound Repair: Emerging Strategies to Target Chronic Healing Wounds. Front. Cell Dev. Biol. 8:773. doi: 10.3389/fcell.2020.00773
Received: 21 May 2020; Accepted: 22 July 2020;
Published: 11 August 2020.
Edited by:
Richard George Faragher, University of Brighton, United KingdomReviewed by:
Oliver Bischof, Institut Pasteur, FranceSuk-Won Jin, Gwangju Institute of Science and Technology, South Korea
Copyright © 2020 Wilkinson and Hardman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew J. Hardman, bS5oYXJkbWFuQGh1bGwuYWMudWs=