
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 06 August 2020
Sec. Molecular and Cellular Oncology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00760
This article is part of the Research Topic Exploring Metabolic Reprogramming in the Epithelial-Mesenchymal Transition in Cancer: New Perspectives in Therapy, Drug Discovery and Diagnostics View all 10 articles
 Xiaowei Lai1,2,3†
Xiaowei Lai1,2,3† Qian Li1,2,4†Fang Wu1,2,3
Qian Li1,2,4†Fang Wu1,2,3 Jiechun Lin1,2,4
Jiechun Lin1,2,4 Jiekai Chen1,2,3,4*
Jiekai Chen1,2,3,4* Hui Zheng1,2,3,4*
Hui Zheng1,2,3,4* Lin Guo1,2,3,4*
Lin Guo1,2,3,4*Epithelial-mesenchymal transition (EMT) and its critical roles during cancer progression have long been recognized and extensively reviewed. Recent studies on the generation of induced pluripotent stem cells (iPSCs) have established the connections among EMT, energy metabolism, DNA methylation, and histone modification. Since energy metabolism, DNA methylation, and histone modification are important for cancer development and there are common characteristics between cancer cells and stem cells, it is reasonable to identify mechanisms that have been established during both reprogramming and cancer progression. In the current review, we start from a brief review on EMT and related processes during cancer progression, and then switch to the EMT during somatic cell reprogramming. We summarize the connection between EMT and metabolic switch during reprogramming, and further review the involvements of DNA methylation and cell proliferation. The connections between EMT and mesenchymal-epithelial transition (MET) and cellular aspects including DNA methylation, histone modification and energy metabolism may provide potential new targets for cancer diagnosis and treatment.
Epithelial-mesenchymal transition (EMT) is defined as a biological process in which epithelial cells lose their characteristics and acquire mesenchymal features. During EMT, epithelial cells lose cell-cell junctions, apical-basal polarity, epithelial markers, and acquire cell motility, a spindle-cell shape, and mesenchymal markers. The concept of EMT was initially proposed as the epithelial-mesenchymal transformation by Elizabeth Hay in 1968 (Hay, 1968) as to describe the important cell changes in embryogenesis; it was later renamed EMT to distinguish it from neoplastic transformation (Nieto et al., 2016; Yang et al., 2020). EMT and its reverse process mesenchymal-epithelial transition (MET), display fundamental principles in diversified physiological and pathological progresses. During metazoan development, cells may sequentially undergo rounds of EMT and MET, as is seen in somite formation and heart development. EMT also occurs during wound healing in adults. Additional evidences related to development and wound healing has previously been reviewed extensively (Thiery et al., 2009; Lim and Thiery, 2012; Nieto et al., 2016). EMT also plays important roles in cancer progression and tissue fibrosis (Nieto et al., 2016; Pastushenko and Blanpain, 2019; Williams et al., 2019). Interestingly, during the processes of embryonic stem cells (ESCs) differentiation and induced pluripotent stem cells (iPSCs) formation, EMT, and MET are highly relevant to the loss and acquisition of pluripotency (Pei et al., 2019). EMT and MET are widely involved in various biological scenarios and display highly plastic and dynamic manners during cell fate transitions (Figure 1).
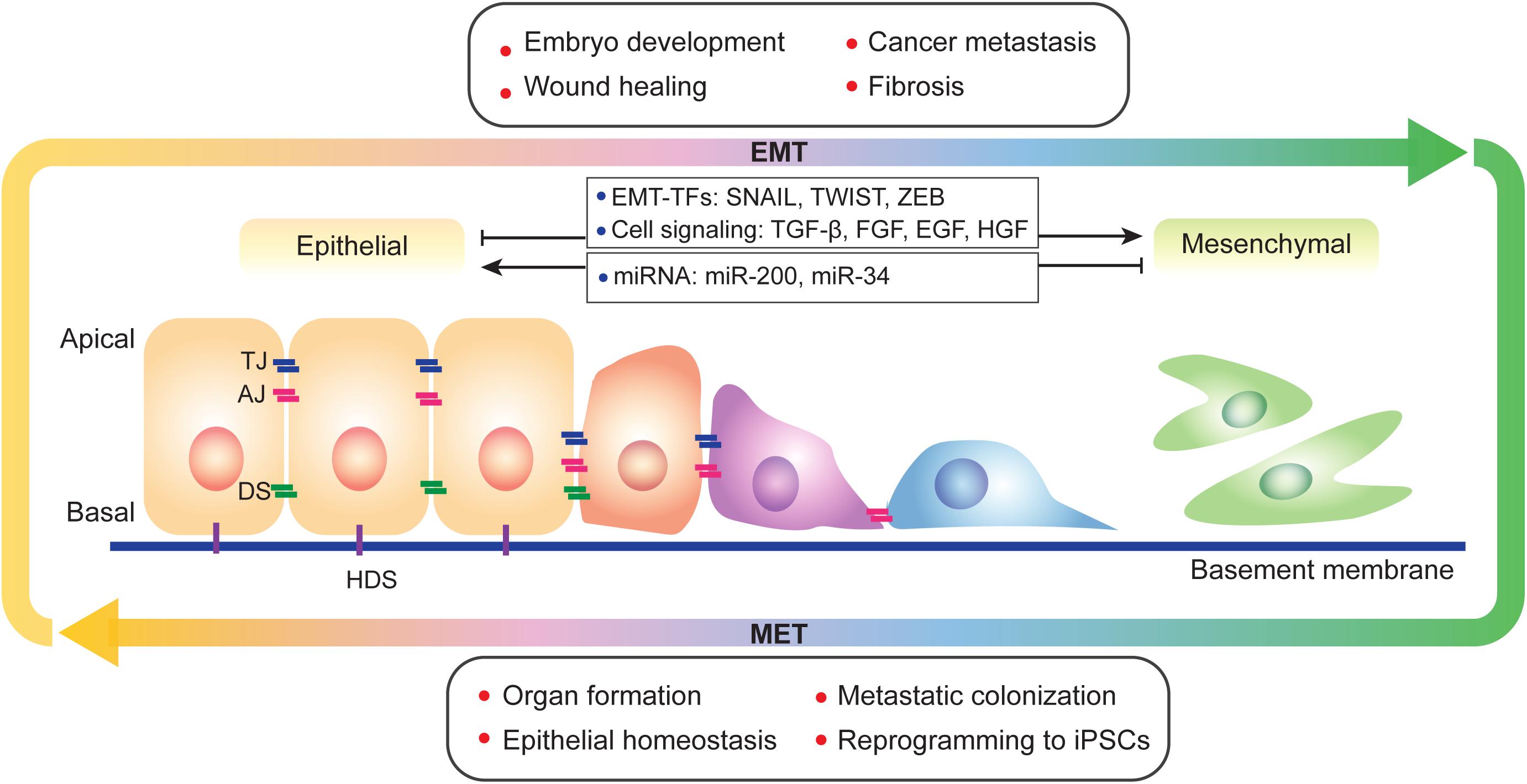
Figure 1. A brief view of EMT and MET. EMT and its reverse process MET occur during physiological and pathological progresses and display a spectral characteristic. During EMT, epithelial cells lost its apical-basal polarity and connections between cells and basement membrane. EMT is regulated by multiple factors, such as transcription factors, cell signaling and epigenetic modification. TJ, tight junction; AJ, adherens junction; DS, desmosome; and HDS, hemidesmosome.
Epithelial-mesenchymal transition is regulated at different levels by multiple factors, including cell signaling, transcriptional control, epigenetic modification, and post-translational modifications (Figure 1). Epithelial cells receive EMT-inducing signals from their niches. For example, cytokines such as transforming growth factor-β (TGF-β), fibroblast growth factor (FGF) family, epidermal growth factor (EGF), and hepatocyte growth factor (HGF) can induce or promote the EMT process (Zavadil and Bottinger, 2005; Thiery and Sleeman, 2006; Nieto et al., 2016; Williams et al., 2019). These EMT-inducing signals up-regulate specific transcription factors called EMT-TFs (e.g., SNAI, TWIST, and ZEB; Thiery and Sleeman, 2006; Nieto et al., 2016; Williams et al., 2019). EMT-TFs usually cooperate with miRNAs as well as epigenetic and/or post-translational regulators to control EMT (Ye and Weinberg, 2015; Nieto et al., 2016). For example, the miR-200 family, which includes miR-200a, miR-200b, miR-200c, miR-141, and miR-429 play important roles in repressing EMT by repressing the translation of ZEB1 and ZEB2 (Korpal et al., 2008; Park et al., 2008). Moreover, overexpression of miR-200 family members can upregulate E-cadherin expression (Park et al., 2008). miR-200 family expression is regulated by DNA methylation; hypermethylation of miR-200 loci lead to the silencing of miR-200 family expression, which promotes the EMT process and causes human tumors (Davalos et al., 2012). The miR-34 family is also known as to regulate EMT and suppress early phases of tumor metastasis. Ectopic expression of miR-34a prevents TGF-β-induced EMT, and miR-34a/b/c regulate SNAI1 expression by its 3’-UTR. miR-34a also suppresses SLUG and ZEB1. Conversely, SNAI1, and ZEB1 also regulate miR-34a/b/c expression by binding their promoters (Siemens et al., 2011). Additional details on EMT regulation by miRNA have been reviewed in multiple papers (Zaravinos, 2015; Suzuki, 2018; Asadzadeh et al., 2019).
Metabolic changes occur during development and cancer progression. For example, both ESCs and tumor cells prefer glycolysis, this similarity between stem cells and cancer cells indicate the intrinsic mechanism of stemness maintenance and metabolism. More recently, metabolic pathways including glycolysis, the TCA cycle, and amino acid and lipid metabolism have been reported as being involved in EMT, especially in tumor progression (Kang et al., 2019; Georgakopoulos-Soares et al., 2020; Sun and Yang, 2020). During tumor progression, cells prefer to obtain energy by increasing glucose conversion into lactate, even in oxygen-rich condition (Fischer and Bavister, 1993; Zhou et al., 2012; Setty et al., 2016). This specific energy metabolism pathway is called aerobic glycolysis. It was first observed in tumors and described by Otto Warburg in the 1920s, and thus, was named the Warburg effect (Warburg, 1956). This metabolic reprogramming depends upon an increase in glucose uptake and highly activated glycolysis. Glucose transporters GLUT1 and GLUT3 can be induced by hypoxia-inducible factor 1α (HIF-1α), which contributes to glucose uptake and promote EMT and tumor progression (Macheda et al., 2005). Enzymes participate in glycolysis, such as hexokinase 2 (HK2), phosphofructokinase (PFK), and pyruvate kinase M2 (PKM2) participate in glycolysis, play positive roles in glycolysis flux, and induce EMT (Luo et al., 2011; Patra et al., 2013; Kim et al., 2017). Tumor cells also show abnormal lipid metabolism, such as increased lipogenesis (Swinnen et al., 2006). Enzymes that participate in lipogenesis, such as acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN), and acyl-CoA synthetase long chain family member (ACSL), all show a close relationship with cancer and EMT (Georgakopoulos-Soares et al., 2020). For example, FASN, which synthesizes of palmitate from acetyl-CoA and malonyl-CoA, has been widely reported in various types of cancer. FASN reportedly promotes EMT through TGF-β signaling in lung cancer (Yang et al., 2016) and ErbB receptors in breast cancer (Zaravinos, 2015). Lipids are also important components of the plasma membrane. CTP-phosphocholine cytidylyltransferase (CTT), which is involved in phosphatidylcholine synthesis, is contributes to EMT in intestinal epithelial cells (Arsenault et al., 2013). Furthermore, amino acid metabolism is critical in EMT progression. Glutamine also plays important roles in energy supply. Glutaminase 1 (GLS1) and GLS2, which are involved in glutaminolysis can act as positive regulators of Snai1 (Choi and Park, 2018; Eiriksson et al., 2018). The important role of metabolism-related enzymes in cancer and EMT has resulted in many being selected as therapeutic targets. Additionally, metabolites from the pathways mentioned above can also regulate EMT through EMT-TFs and other epigenetic regulators, as we will discuss in this review.
This review discusses EMT progress and its regulators during cancer progression and iPSCs formation. We will also explore the relationship between metabolism and epigenetics in connection with EMT.
The important roles of EMT during tumorigenesis and metastasis have been demonstrated for decades. Most lethal human malignancies are derived from epithelial tissues, including the breast, colon, pancreas, and liver (Ye and Weinberg, 2015). Approximately about 90% of cancer-associated deaths are caused by metastatic disease rather than primary tumors (Lambert et al., 2017). The EMT program confers upon these epithelial cells properties critical to invasion and metastatic dissemination including notably increased motility, invasiveness, and the ability to degrade components of the extracellular matrix (ECM) components (Nieto et al., 2016). These complex metastatic cascades are orchestrated and coordinated by a series of master EMT-TFs that have been extensively explored (Craene and Berx, 2013; Lamouille et al., 2014). EMT-TFs facilitate the EMT process, but it is unclear whether EMT is indispensable for migration. Inhibiting EMT by overexpressing miR-200 did not affect lung metastasis when using an EMT lineage-tracing system in a spontaneous breast-to-lung metastasis model (Fischer et al., 2015). Furthermore, suppressing EMT by deleting Snai or Twist in a primary tumor did not decrease the invasion and metastasis of pancreatic carcinoma cells (Zheng et al., 2015). However, two subsequent have reports pointed out the shortcomings in these experiments (Aiello et al., 2017; Ye et al., 2017). These opposing opinions reflect the complexity of the EMT processes that must to be carefully investigated both in vivo and in vitro.
One of the difficulties in studying EMT arises because the transitions between epithelial and mesenchymal states are not binary. Instead, carcinoma cells often exhibit a spectrum of epithelial-mesenchymal characteristics (Jolly et al., 2017; Mathieu et al., 2017; Zhang and Weinberg, 2018). E-cadherin, occludins, and cytokeratins are the most commonly used markers for the epithelial traits, and N-cadherin and vimentin are the most commonly used for the mesenchymal state (Thiery et al., 2009). Recent studies have shown that some cancer cells, such as breast, pancreatic, renal, lung, and colorectal cancers, express both epithelial and mesenchymal markers (Bronsert et al., 2014; Sampson et al., 2014; Zhang et al., 2014; Schliekelman et al., 2015; Hiew et al., 2018). In fact, many cancer cells may not undergo a complete EMT process, but rather steadily acquire these intermediate, or hybrid epithelial/mesenchymal E/M phenotypes. The partial EMT or hybrid E/M phenotypes have multiple advantages compared to the complete EMT phenotype. It is suggested that these collectively migrating cells are the primary actors of metastasis (Jolly et al., 2015). To form distant metastases, cancer cells dissociate from the primary tumor, invade adjacent tissues, and intravasate into lymphatic and blood vessels to later colonize lymph nodes and distant organs (Nieto et al., 2016). Circulating tumor cells (CTCs) are defined as cancer cells that are released from a solid tumor and enter the peripheral blood; they are considered as biomarker of the metastatic process (Barcellos-Hoff et al., 2013). Most CTCs show hybrid E/M markers, suggesting an incomplete EMT. In addition, although EMT plays important roles in tumor progression, its reverse process, MET, also is significant in terms of dissemination. The last step of the invasion-metastasis cascade is termed colonization and largely depends on MET (Dongre and Weinberg, 2019).
The concept of cancer stem cells (CSCs) is based on the observation that not all cells in tumors are equal. A small number of tumor cells display two key features that distinguish them from the others: self-renewal and differentiation potential (Foster and Archer, 1979; Zhang and Wang, 2008). With these features, CSCs are hypothesized to play essential roles in tumor metastasis, heterogeneity, drug resistance, and recurrence. Since EMT and the metastatic cascade are inextricably linked, the relationship between EMT and CSCs has been investigated. A number of studies have now shown that EMT programs induce cancer cell stemness in many kinds of tissues (Dongre and Weinberg, 2019) and that EMT-TFs displayed the capacity to promote CSCs stemness in mouse and human models (Ye et al., 2015). Development of the cancer stem cell concept has been reviewed in several papers (Clevers, 2011; Batlle and Clevers, 2017). The isolation of CSCs from cancer cells and subsequent xenotransplantation assays provide evidence of the existence of CSCs. In the 1990s, CSCs were identified in acute myeloid leukemia (AML; Lapidot et al., 1994; Uckun et al., 1995; Bonnet and Dick, 1997). In 2003, CSCs were proved to be able to isolate from a solid tumor by a xenograft assay of small numbers of CD44+CD24–/low cells which isolated from human breast cancer (Al-Hajj et al., 2003). Interestingly, cancer cells are observed exhibiting a plasticity that demonstrates the ability to switch between CSCs and non-CSCs in different cancers (Thankamony et al., 2020). For example, human basal breast cancer cells can switch from non-CSCs to CSCs by regulating the ZEB1 promoter (Chaffer et al., 2013). Another example for CSC plasticity is from studies on colorectal cancer. LGR5 is a marker of colorectal CSCs; a recent colorectal cancer study showed that LGR5 negative cells can become CSCs and lead to metastasis (Fumagalli et al., 2020).
Epithelial-mesenchymal transition-TFs are often found accompanied by features of stemness. Slug (also known as Snai2) is proved to be a key inducer of stemness (Guo et al., 2012; Nassour et al., 2012). Other EMT-TFs, such as Twist, Snai1, and Six1 also induce stemness in various breast cancer models (McCoy et al., 2009; Morel et al., 2012; Ye et al., 2015). ZEB1 is required for stemness in pancreatic cancer; it inhibits members of the miR-200 family to maintain a stem-like phenotype (Wellner et al., 2009). EMT-related cell signaling factors, such as TGF-β, can be secreted in tumor stromal cells. This boosts metastatic potential and causes a poor-prognosis in colorectal cancer (Calon et al., 2015). Although the EMT process is associated with CSC characteristics, the relationship between EMT and stemness is still a controversial issue in tumorigenesis. Uncoupling EMT and stemness has been discussed in several studies and explained by the two events may occur in parallel (Nieto et al., 2016; Batlle and Clevers, 2017). EMT-TFs which critical in both EMT and CSCs are also provided evidence of uncoupling of the two events. For example, a study on Twist1 shows it is essential for initiate of skin tumorigenesis, however, Twist1 controlled tumor stemness independently of its EMT function (Beck et al., 2015). Recent studies have shown that cells with a hybrid E/M feature are much more likely to gain stemness (Bierie et al., 2017; Pastushenko et al., 2018). These studies may provide new insights for cancer therapy.
Metabolic reprogramming is one of the hallmarks of tumor progression. As discussed in the introduction, cancer cells change cell metabolism in diverse ways to gain energy and meet the requirements for proliferation. Glucose metabolism is considered the most studied metabolic change in CSCs. However, the glucose metabolism phenotype of CSCs, whether glycolysis or oxidative phosphorylation (OXPHOS), depends on both the tumor origin, and microenvironment (Thankamony et al., 2020). CSCs also show an interplay between glycolysis and OXPHOS (Yu et al., 2017). These metabolic characteristics increase the difficulty in providing effective cancer therapies; other metabolic pathways and metabolites must be investigated further.
Stemness or stem cells are functionally defined as cells with the ability to self-renew and differentiate. In addition to cancer stem cells, other kinds of stem cells are isolated during development and in adults. One well accepted classification is that of ESCs and adult stem cells (ASCs). ESCs are derived from the inner cell mass of blastocysts and can differentiate into three germ layers and form chimeras (Evans and Kaufman, 1981; Martin, 1981), while ASCs are isolated from adult tissues in special niches and are capable of differentiating into several cell types depending on their origins (Shyh-Chang et al., 2013; Clevers, 2015). Although ESCs, ASCs, and CSCs are all considered as stem cells, their stemness is displayed in different ways. For example, both ESCs and ASCs show their differentiation abilities by forming normal tissues or cells. However, CSCs displayed their differentiation potential by promoting tumor heterogeneity (Clarke et al., 2006). These stem cells differ in terms of their gene expression profiles, transcriptional regulatory networks, and epigenetic modifications; however, they show their stemness in their consecutive EMT-MET changes. During embryonic development, early mesoderm cells are formed by EMT and migrate to generate intermediate mesoderm, chorda-mesoderm, paraxial mesoderm, and lateral plate mesoderm. These mesoderm cells then undergo MET to give rise to the urogenital system, notochord, somite, and somatopleure (Nieto, 2013). In contrast, CSCs caused by EMT undergo MET upon reaching an appropriate niche to become a new tumor (Nieto, 2013). Intriguingly, this EMT-MET progress has also been reported in pluripotency setup (Liu et al., 2013). Mouse embryonic fibroblasts (MEFs) can be reprogrammed into iPSCs by introducing exogenous Oct4, Sox2, Klf4, and c-Myc. Furthermore, reprogramming efficiency can be improved by sequentially expressing these transcriptional factors. All these phenomena imply that epithelial or even mesenchymal cells can reach a more active mesenchymal state to promote their plasticity.
Direct reprogramming of somatic cells into iPSCs by defined factors is known as reprogramming (Takahashi and Yamanaka, 2006). A noticeable change that occurs during reprogramming is the transformation of MEF cells from a mesenchymal morphology into tightly packed colonies (Shu and Pei, 2014). This transition is defined as MET and is a critical step in acquiring pluripotency (Li et al., 2010; Samavarchi-Tehrani et al., 2010). Exogenous transcription factors Oct4, Sox2, and c-Myc are orchestrated to suppress Snai1, Tgfb, and Tgfbr to retain the non-mesenchymal characteristics of MEFs; Klf4 induces epithelial properties by directly up-regulating E-cadherin (Li et al., 2010). Activating BMP signaling or repressing TGF-β signaling is beneficial for iPSC generation by promoting MET progress (Li et al., 2010; Samavarchi-Tehrani et al., 2010). Not long after the discovery of the essential role of MET during reprogramming, another publication revealed that a sequential EMT–MET mechanism at the beginning of reprogramming can enhancer reprogramming efficiency (Liu et al., 2013). Briefly, the sequential introduction of Oct4 and Klf4 first, followed by c-Myc, then Sox2 (OK+M+S), can induce Snai2 up-regulation and inhibit E-cadherin expression in the early stage of reprogramming. Further analysis revealed that this temporary EMT can generate iPSCs with a high efficiency of about 600% of basal level (Liu et al., 2013). Coincidentally, transient exogenous C/EBPα expression followed by the activation of Yamanaka factors (OSKM) in mouse primary B cells is capable of initiating EMT, which induces a 100-fold increase in reprogramming efficiency (Stefano et al., 2014). Hence, although MET is an essential event for reprogramming, having a more mesenchymal state at the early stage may provide a more facilitated state for cell fate transition.
The generation of iPSCs is a multi-step process. In addition to MET, another important change is the metabolic switch that occurs during reprogramming. Unlike somatic cells that use OXPHOS to gain energy, pluripotent stem cells, including ESCs and iPSCs, and prefer glycolysis (Zhang et al., 2012). Using small molecules to activate glycolysis and inhibit OXPHOS or upregulate glycolytic gene expression can promote the metabolic switch from oxidative phosphorylation to glycolysis (OGS) and accelerate reprogramming (Folmes et al., 2011; Zhang et al., 2012; Prigione et al., 2014; Cao et al., 2015). Since both OKSM-induced EMT-MET processes and facilitated OGS accelerate reprogramming, it remains an open question whether EMT and OGS cooperate to regulate iPSCs.
A recent study explored the relationship between early EMT and OGT (Sun et al., 2020). This study used 5C medium, a chemically-defined medium (He et al., 2015), to promote reprogramming by inducing a sequential EMT-MET process. Further analysis revealed that 5C medium increases glycolysis, inhibits OXPHOS, and accelerates the OGS process compared to a serum-containing medium. Early EMT and OGS may help cells overcome epigenetic barriers during reprogramming by upregulating five epigenetic factors, Bmi1, Ctcf, Ezh2, Kdm2b, and Wdr5, which have been proven to facilitate reprogramming in previous studies (Ang et al., 2011; Moon et al., 2011; Wang et al., 2011, 2017; Onder et al., 2012; Figure 2). The barriers that trapped cells at the pre-iPSC stage during reprogramming in serum containing medium (Chen et al., 2013b) may be overcome by the five epigenetic factors present in 5C medium. However, because the positive feedback loop between early EMT and facilitated OGS is too strong, further reprogramming is inhibited at the late stage by the induction of EMT and glycolysis to a level that is too high for further reprogramming and by preventing necessary MET (Sun et al., 2020). These findings are consistent other works showing that EMT induced at the late stage of reprogramming significantly impairs the generation of pluripotency (Liu et al., 2013). Furthermore, HIF2α, which is upregulated in 5C medium, represses reprogramming at later stages (Mathieu et al., 2014).
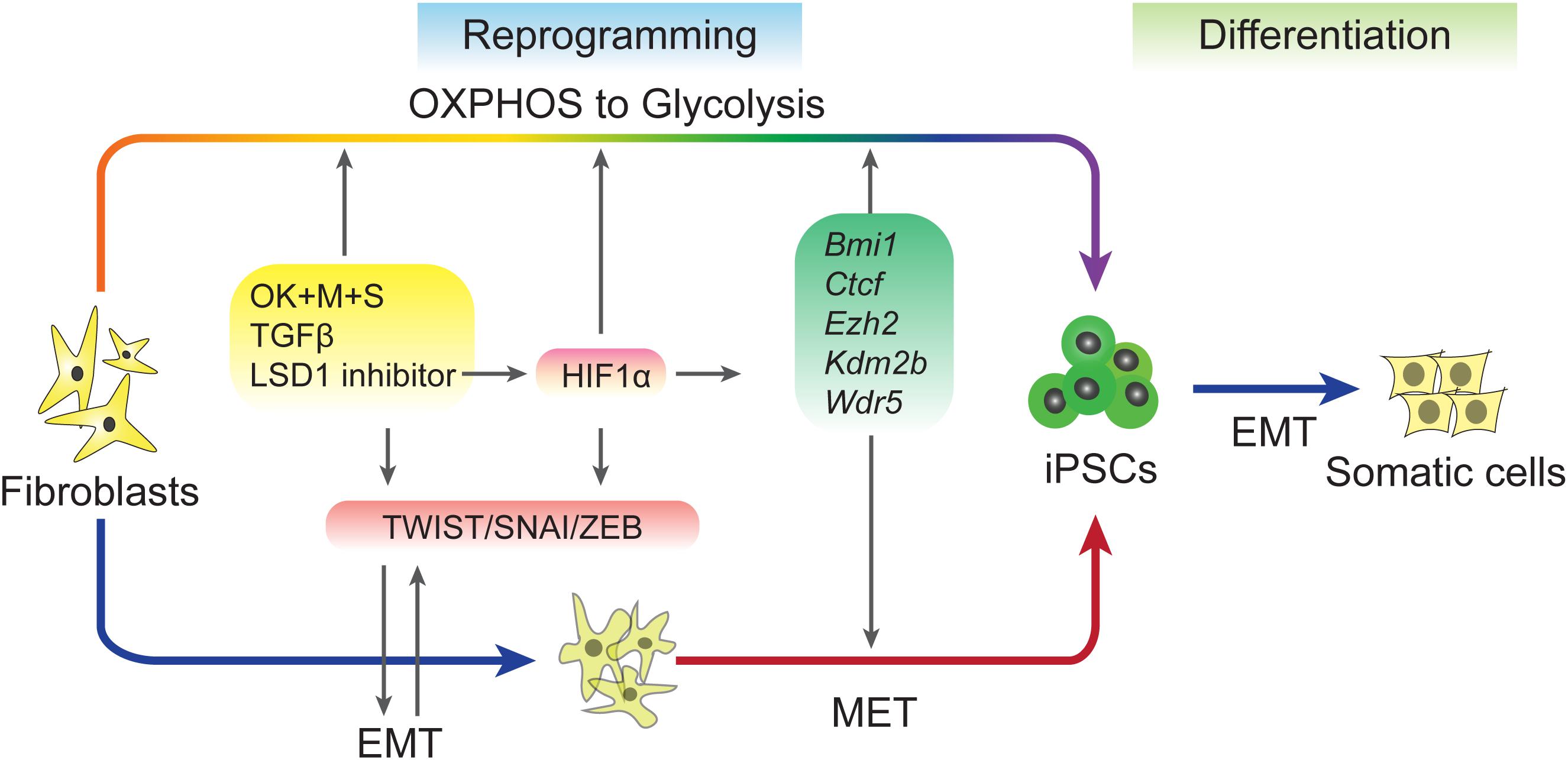
Figure 2. Regulation of EMT–MET during reprogramming. Induced pluripotent stem cells (iPSCs) can be accelerated by sequential EMT–MET process. Early EMT can be induced by sequential expression of Yamanaka factors, adding TGF-β or inhibiting of LSD1. These treatments up-regulation of EMT-TFs and also proved metabolic switch from OXPHOS to Glycolysis (OGS). EMT and OGS coordinate to facilitate the reprogramming through an epigenetic regulation way.
In addition to glycolysis, other metabolic pathways and metabolites are involved in EMT during reprogramming. A recent report showed that phospholipid remodeling is important for acquiring pluripotency by facilitating MET (Wu et al., 2019). This study demonstrated that the CDP-ethanolamine (CDP-Etn) pathway mechanically promotes reprogramming at the early stage through the CDP-Etn-Pebp1 axis to inhibit mesenchymal genes. Since metabolic regulation plays critical roles in EMT regulation and tumor progression, additional metabolic and EMT-MET connections in reprogramming need to be studied.
As discussed above, while OGS upregulates five epigenetic factors in an HIF1α-dependent manner, early EMT promotes the expression of EMT-TFs that are enriched by both the five epigenetic factors and glycolytic genes (Sun et al., 2020). These reveal that the mesenchymal state may act as a link between metabolic and epigenetic regulation. Since metabolites generated during multiple metabolic processes are utilized in enzymatic reactions leading to epigenetic modifications and transcriptional regulation as previously reviewed (Ryall et al., 2015), we will focus on the crosstalk between EMT-MET, metabolism and epigenetic regulation during somatic cell reprogramming.
A vital role for metabolism in regulating cell fate has been derived from studies documenting rapid and dynamic changes in substrate utilization. Metabolites, including α-ketoglutarate (αKG), acetyl-coA, S-adenosyl methionine (SAM), and flavin adenine dinucleotide (FAD), regulate many of the important cell fate conversions by epigenetic catalytic reactions (Ryall et al., 2015; Wu et al., 2016; Figure 3). For example, glycolysis-produced acetyl-coA have been reportedly controls the early differentiation of ESCs by regulation histone acetylation (Moussaieff et al., 2015).
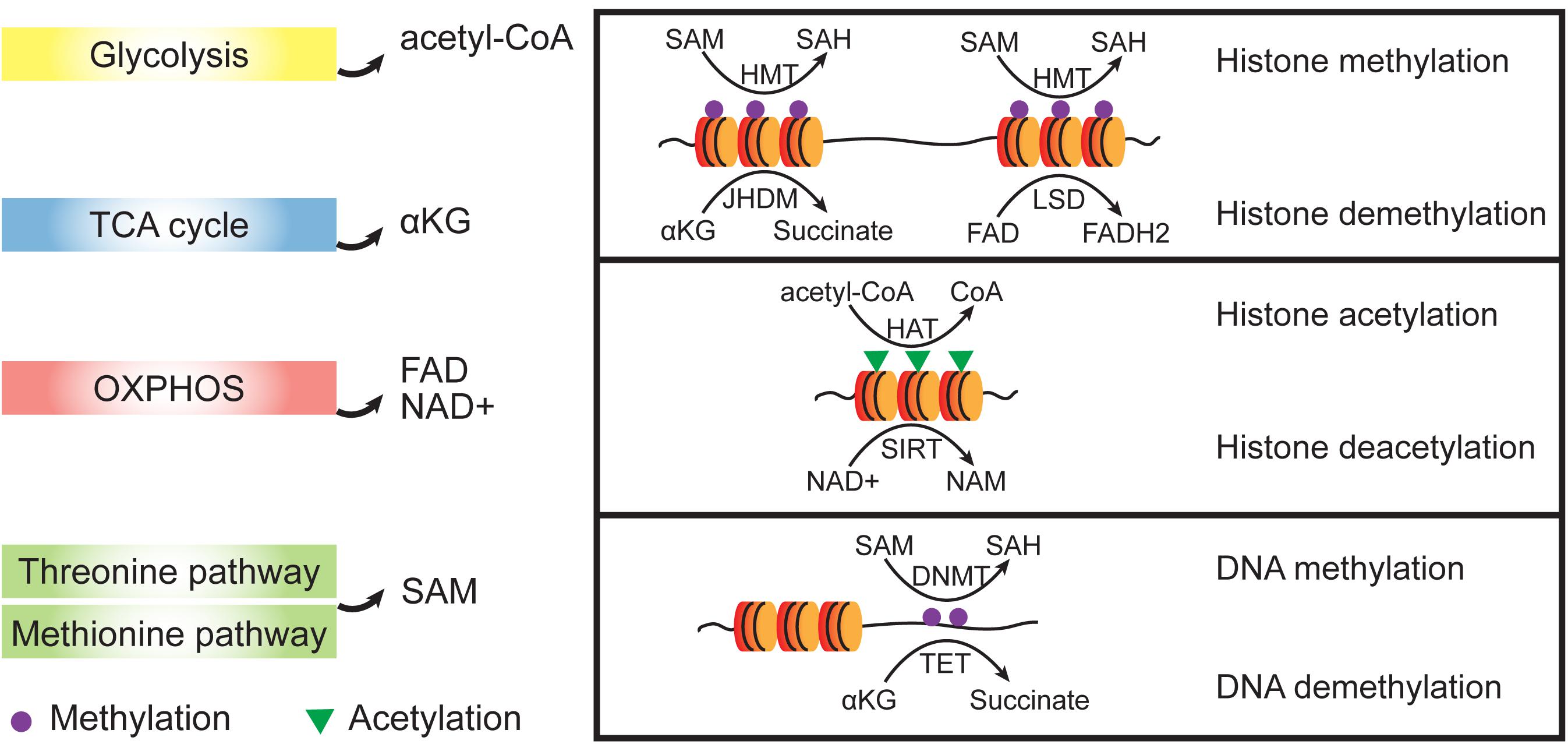
Figure 3. Crosstalk between metabolic and epigenetic regulation. Metabolites from different metabolic pathways participate epigenetic regulation as cofactors. Dioxygenases such as HMTs and DNMTs use SAM as methyl group donor and the reverse demethylation reactions use αKG or FAD to receive the methyl groups. Acetylation of histones is achieved by HATs which transfer acetyl groups from acetyl-CoA. To remove acetyl group from histone proteins, NAD+ is used as a cofactor to support the SIRT activities which belongs to histone deacetylases (HDACs). SAM, S-adenosyl methionine; SAH, S-adenosyl homocysteine; NAD+, oxidized nicotinamide adenine dinucleotide; NAM, nicotinamide; FAD, flavin adenine dinucleotide; αKG, α-ketoglutarate; HMT, histone methyltransferase; HAT, histone acetyltransferase; and SIRT, Sirtuin.
S-adenosyl methionine, which functions as a methyl donor for both DNA and histone methylation, is important for the maintaining of pluripotency in both mouse and human ESCs (Ng et al., 2013; Shiraki et al., 2014). The cellular αKG/succinate ratio contributes to the ability of ESCs to suppress differentiation (Carey et al., 2015). αKG/succinate ratios are involved in the regulation of a large family of αKG-dependent dioxygenases, such as Jumonji C (JmjC)-domain-containing histone demethylases (Carey et al., 2015; Figure 4). JHDM1A and JHDM1B (also known as KDM2A and KDM2B, respectively) belong to the Jumonji family of proteins and have been shown to demethylate H3K36me2/3 (He et al., 2008). Early studies showed that JHDM1A/1B inhibit Ink4a/Arf and activate miR-302/367 to promote reprogramming (Wang et al., 2011). A recent investigation showed that JHDM1B works as a component of the PRC1.1 complex and cooperates with BMP signaling to upregulate reprogramming efficiency by promoting MET (Zhou et al., 2017). Consistently, histone demethylases JMJD1 and JMJD2 (also known as KDM3A and KDM4A, respectively) function as the on/off switch for pre-iPSC fate by regulating H3K9 methylation status at core pluripotency loci (Chen et al., 2013b). In addition, lysine-specific demethylase 1 (LSD1, also known as KDM1A) functions through an FAD-dependent oxidative reaction to specifically catalyze the demethylation of H3K4me1/2 or H3K9me1/2 (Shi et al., 2004; Amente et al., 2013). Accordingly, inhibiting LSD1 by shRNA or a small molecular inhibitor can promote reprogramming at the early stage by increasing both OGS and exogenous transcriptional factors expression (Sun et al., 2016).
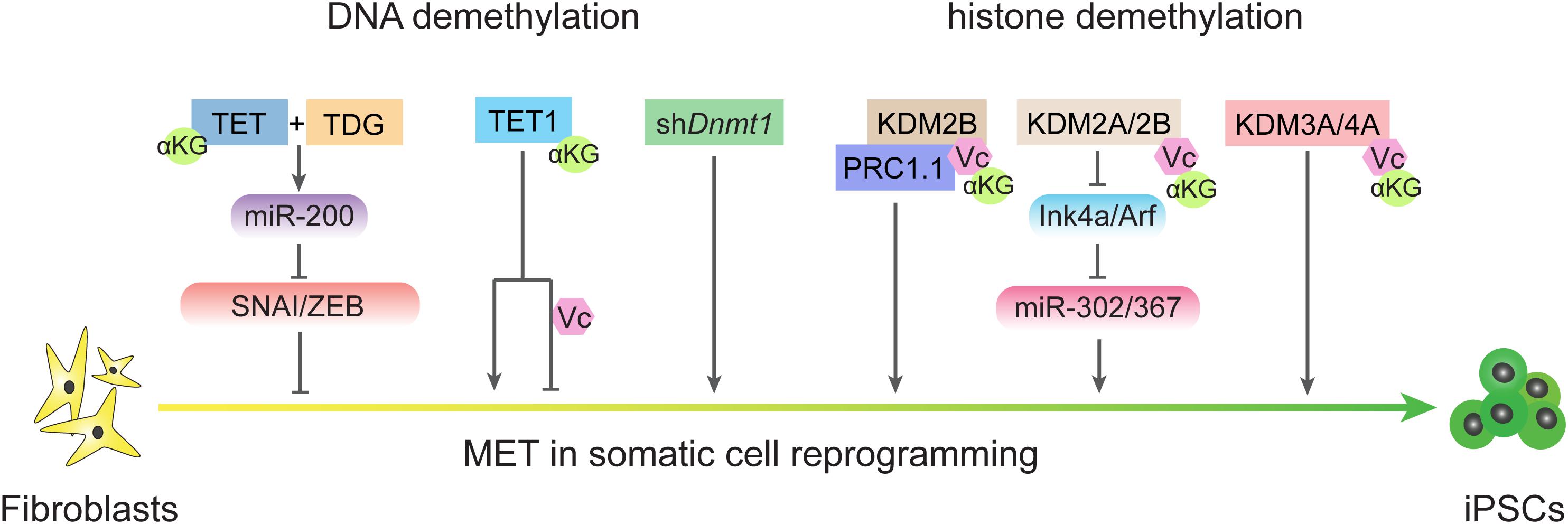
Figure 4. DNA demethylation and histone demethylation regulate MET process during reprogramming. TET and TDG are essential for early MET through demethylating miR-200 loci and TET1 plays different roles in reprogramming in condition of the present or absent of vitamin C. Passive DNA demethylation through knockdown the expression of Dnmt1 also promote MET. Histone demethylases also regulate MET process in different ways.
Histones can also be acetylated and deacetylated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. HATs mediate the acetyl group transfer from acetyl-CoA, a metabolite from glycolysis. A higher level of acetyl-CoA is found in mouse ESCs compared to that of differentiated cells (Wang et al., 2020) and reportedly controls the early differentiation of ESCs by regulating histone acetylation (Moussaieff et al., 2015). In addition to histone acetylation, non-histone acetylation is also related to pluripotency. A recent study showed that the inhibition of CBP/EP300, which is closely related HATs, can repress the expression of mesenchymal transcription factor PRRX1 and promote reprogramming (Ebrahimi et al., 2019). HDACs are grouped into four classes. Class I (HDACs 1–3 and 8), class II (HDACs 4–7, 9, and 10), and class IV (HDAC 11) are zinc-dependent metalloproteins; class III (SIRT1-7) are NAD+-dependent proteins (Emmett and Lazar, 2019). Class I HDAC1-HDAC2 can form multiprotein complexes named SIN3A, NuRD, and CoREST, respectively. These HDAC containing complexes play different roles in establishing and maintaining pluripotency in a context-dependent manner. For example, the SIN3A/HDAC2 complex cooperates with NANOG to promote reprogramming (Saunders et al., 2017). However, HDAC2 reportedly acts as a barrier to reprogramming in other studies (Anokye-Danso et al., 2011; Wei et al., 2015). Interestingly, we noticed that miR-302/367 can promote reprogramming by inhibiting HDAC2 (Anokye-Danso et al., 2011), but in a contemporaneous study by Liao et al., miR-302/367 accelerated MET by inhibiting TGFBR2 and promoting E-cadherin expression (Liao et al., 2011). These studies indicate that the relationship between HDACs and EMT, and indeed, deacetylation by HDACs, can occur without histones. Snai1 interaction with HDAC1 and HDAC2 is dependent upon its SNAG domain and can mediate E-cadherin repression (Peinado et al., 2004). Sirtuins (SIRT1–7) belong to class III HDACs and depend on NAD+ to exhibit their enzyme activity. SIRT1 enhances reprogramming via SOX2 hypoacetylation (Mu et al., 2015), as well as through the miR34a-SIRT1-p53 axis (Lee et al., 2012). STRT7 reportedly reverse metastatic phenotypes in tumors (Malik et al., 2015) and may affect MET during reprogramming (Hsu et al., 2018).
DNA methylation is another area of crosstalk between metabolism and epigenetics that also plays critical roles in regulating EMT-MET during reprogramming (Figure 2). In mammals, methyl groups supplied by SAM are added at the 5-carbon of cytosines by the catalysis of DNA methyltransferases (DNMTs; Wu and Zhang, 2014). DNA methylation can be removed by both TETs-mediated active DNA demethylation and cell cycle-dependent passive DNA demethylation (Wu and Zhang, 2014, 2017). Since reprogramming occurs along with DNA demethylation in pluripotency-related loci, the mechanisms of DNA demethylation during iPSCs formation has been invested. TET proteins belong to αKG-dependent dioxygenases and require Fe2+ and vitamin C (Vc) as assistant (Ngo et al., 2019). Both TET1 and TET2 promote reprogramming (Doege et al., 2012; Costa et al., 2013; Gao et al., 2013), and under certain conditions, Tet1 even could replace Oct4 (Gao et al., 2013). Furthermore, active demethylation promoted by TETs and TDG is essential to reactive the miR-200 family that enables MET by inhibiting SNAI/ZEB in the reprogramming process (Hu et al., 2014). Repressing DNA methylation by shDnmt1 or promoting passive demethylation by shp53-induced proliferation acceleration also promotes MET and facilitates iPSCs generation (He et al., 2017b, 2019). Further analysis has revealed that both shDnmt1-induced passive DNA demethylation and TET1-induced active DNA demethylation prefer hemi-methylated CpG sites that are enriched at the loci of core pluripotency genes and epithelial markers (He et al., 2019). The effects of DNA methylation on EMT-TFs were also studied in several types of cancer (Lee and Kong, 2016). Thus, metabolites from diverse metabolic pathways contribute to cell fate conversions by modulating epigenetic properties and show a high correlation with EMT-MET.
Intriguingly, further study of TET1 found that the physiological concentration of vitamin C increases TET1 activity, which in turn impairs the MET process and inhibits reprogramming (Chen et al., 2013a). In the absence of vitamin C, TET1 induces DNA demethylation on the loci of core pluripotency genes and epithelial markers rather than mesenchymal markers, which results in MET and accelerated reprogramming. The DNA demethylation activity of TET1 is enhanced in the presence of vitamin C. Because DNA methylation on the loci of core pluripotency genes and epithelial markers is already at a low level, the expression of core pluripotency genes and epithelial markers is not further affected. However, increased TET1 activity can induce DNA demethylation on the loci of mesenchymal markers, which in turn suppresses the expression of core pluripotency genes and epithelial markers. In addition to their DNA demethylation function, TET proteins also have important functions that are independent of catalytic activity. For example, TET1 could be recruited by Polycomb repressive complex 2 (PRC2) at H3K27me3 positive regions in ESCs (Neri et al., 2013). TET1 also interacts with SIN3A/HDAC complex (Vella et al., 2013) which is reported to promote pluripotency (Saunders et al., 2017). TET proteins including TET1, TET2, and TET3, are reported associated with the O-GlcNAc transferase (OGT) (Deplus et al., 2013; Vella et al., 2013; Chen et al., 2013c) in ESCs. OGT or O-GlcNAcylation could further recruit SET/COMPASS (Deplus et al., 2013) or modified transcription factors by O-GlcNAc, such as Pou5f1 (Constable et al., 2017), that may promote somatic cell reprogramming. Recent studies show that O-GlcNAcylation effects EMT through multiple pathways and promote cancer metastasis (Lucena et al., 2016; Harosh-Davidovich and Khalaila, 2018; Jiang et al., 2019; Feng et al., 2020), these may set up the relationship between TET and EMT. Tsai et al. reports TET1 could interacts with HIF-1α and HIF-2α to regulate hypoxia-induced EMT (Tsai et al., 2014).
Therefore, regulation of DNA methylation can affect both EMT and its reverse process, MET.
Although the field of EMT research has developed rapidly, many critical questions remain unanswered. For example, the driving force initiating EMT and the manner by which dose metabolism influences EMT, as well as the function of the mesenchymal state during the EMT-MET process are still unknown. Since transcription factors are important for cell identity, EMT-TFs are considered critical for losing epithelial characteristics and gaining mesenchymal phenotypes (Nieto, 2013). Hence, the regulation of EMT-TFs cooperating with cell signaling in vitro and transcriptional and epigenetic controls in vivo need to be carefully considered when studying EMT.
The concept of EMT comes from studies on early embryogenesis and is now widely investigated in cancer and stem cell biology. The transitions between mesenchymal and epithelial states demonstrate the plasticity of the cells. The sequential transition of EMT-MET not only occurs during tumor metastasis and somatic cell reprogramming as discussed above, but is also observed in cell differentiation and transdifferentiation. For example, a sequential EMT-MET drives the differentiation of human ESCs toward hepatocytes (Li et al., 2017). Activin A-induced formation of definitive endoderm (DE) accompanies a synchronous EMT mediated by autocrine TGF-β signaling and activates SNAI1 to initiate EMT followed by MET (Li et al., 2017). In addition, a temporary EMT-MET can increase the conversion efficiency of mouse astrocytes into induced dopamine neurons (Cervo et al., 2017) and human gastric epithelial cells into multipotent endodermal progenitors (Wang et al., 2016). Interestingly, in the early study of 5C medium, sequential EMT-MET was also observed during the transition of MEFs to neuron-like cells (He et al., 2017a). All of this evidence suggests that the mesenchymal state may be a necessary state for cell fate transition.
A recent study revealed the cooperation of EMT-MET and OGS during reprogramming as discussed above (Sun et al., 2020). However, coordinated regulation between EMT-MET and OGS should be investigated in other types of cell fate transition for common regulatory mechanisms. Metabolic shifts can control EMT during tumor metastasis, and metabolites from different metabolic pathways are involved in epigenetic regulation (Ryall et al., 2015; Kang et al., 2019). Both cancer cells and ESCs prefer to obtain energy through glycolysis, implying an intrinsic connection between the two kinds of cells. Furthermore, lessons can be learned from somatic cell reprogramming. For example, AML can be caused by the mutation of isocitrate dehydrogenase (IDH) IDH1 and IDH2 or TET2 (Figueroa et al., 2010; Quivoron et al., 2011). TET2 is a dioxygenase and its activity depends upon αKG, Fe2+, and Vc (Blaschke et al., 2013; Yin et al., 2013; Chen et al., 2013a). IDH1 and IDH2 convert isocitrate to αKG, which supports TET2 activity. A mutation in IDH1 or IDH2 causes the accumulation of 2-hydroxyglutarate (2-HG) and inhibits TET2 enzyme activity, resulting increased DNA methylation that drives AML progression (Lu et al., 2012). Previous studies on TETs in ESCs and reprogramming have showed that Vc can activate TETs as a cofactor, and a high dose of Vc has rescued TET2 deletion in a mouse model of leukemia (Agathocleous et al., 2017; Cimmino et al., 2017).
Recent studies on CSCs focusing on their plasticity and resistance to therapy have been discussed in several other reviews (Batlle and Clevers, 2017; Najafi et al., 2019; Thankamony et al., 2020). Firstly, enzymes involved in metabolic reprogramming during tumor progression can be used as biomarkers for the therapeutic targeting of cancers (Sun and Yang, 2020). In addition to IDH, FASN in lipid synthesis and GLS1 in glutamine metabolism have been reported as targets for breast cancer and colorectal cancer, respectively, Sun and Yang (2020). Secondly, regulators involved in EMT can be used biomarkers and for therapeutic targeting (Williams et al., 2019). For example, both the transcriptional and epigenetic regulation of EMT-TFs could be treatment targets (Sun and Yang, 2020). Thirdly, since CSCs contribute to tumor resistance and recurrence, special attention must be paid to CSCs. Therefore, this review has discussed EMT in the context of both cancer and reprogramming, and explores the relationship between EMT, metabolism, and epigenetic regulation. These mechanisms could provide new scenarios for regenerative medicine and cancer therapy.
LG, XL, and QL wrote this manuscript. HZ, JC, FW, and JL helped to improve this manuscript. JC, HZ, and LG approved the final version of this submission. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key R&D Program of China (Nos. 2016YFA0100400 and 2019YFA0110200); National Natural Science Foundation of China (Nos. 31900699 and 81702445); the Guangzhou Key Area Research and Development Project (202007030003), the Key Research Program of Frontier Sciences of Chinese Academy of Sciences, No. QYZDB-SSW-SMC031; the International Partnership Program of Chinese Academy of Sciences, No. 154144KYSB20190034; the Key Research & Development Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory, No. 2018GZR110104008, and the Science and Technology Planning Project of Guangdong Province, No. 2017B030314056; and the Youth Innovation Promotion Association from Chinese Academy of Sciences (to LG).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Agathocleous, M., Meacham, C., Burgess, R., Piskounova, E., Zhao, Z., Crane, G., et al. (2017). Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481. doi: 10.1038/nature23876
Aiello, N., Brabletz, T., Kang, Y., Nieto, M., Weinberg, R., and Stanger, B. (2017). Upholding a role for EMT in pancreatic cancer metastasis. Nature 547, E7–E8. doi: 10.1038/nature22963
Al-Hajj, M., Wicha, M., Benito-Hernandez, A., Morrison, S., and Clarke, M. (2003). Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 3983–3988. doi: 10.1073/pnas.0530291100
Amente, S., Lania, L., and Majello, B. (2013). The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim. Biophys. Acta 1829, 981–986. doi: 10.1016/j.bbagrm.2013.05.002
Ang, Y.-S., Tsai, S.-Y., Lee, D.-F., Monk, J., Su, J., Ratnakumar, K., et al. (2011). Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell 145, 183–197. doi: 10.1016/j.cell.2011.03.003
Anokye-Danso, F., Trivedi, C. M., Juhr, D., Gupta, M., Cui, Z., Tian, Y., et al. (2011). Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 8, 376–388. doi: 10.1016/j.stem.2011.03.001
Arsenault, D. J., Yoo, B. H., Rosen, K. V., and Ridgway, N. D. (2013). ras-Induced up-regulation of CTP:phosphocholine cytidylyltransferase alpha contributes to malignant transformation of intestinal epithelial cells. J. Biol. Chem. 288, 633–643. doi: 10.1074/jbc.M112.347682
Asadzadeh, Z., Mansoori, B., Mohammadi, A., Aghajani, M., Haji-Asgarzadeh, K., Safarzadeh, E., et al. (2019). microRNAs in cancer stem cells: biology, pathways, and therapeutic opportunities. J. Cell. Physiol. 234, 10002–10017. doi: 10.1002/jcp.27885
Barcellos-Hoff, M., Lyden, D., and Wang, T. (2013). The evolution of the cancer niche during multistage carcinogenesis. Nat. Rev. Cancer 13, 511–518. doi: 10.1038/nrc3536
Batlle, E., and Clevers, H. (2017). Cancer stem cells revisited. Nat. Med. 23, 1124–1134. doi: 10.1038/nm.4409
Beck, B., Lapouge, G., Rorive, S., Drogat, B., Desaedelaere, K., Delafaille, S., et al. (2015). Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 16, 67–79. doi: 10.1016/j.stem.2014.12.002
Bierie, B., Pierce, S. E., Kroeger, C., Stover, D. G., Pattabiraman, D. R., Thiru, P., et al. (2017). Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. U.S.A. 114, E2337–E2346. doi: 10.1073/pnas.1618298114
Blaschke, K., Ebata, K., Karimi, M., Zepeda-Martínez, J., Goyal, P., Mahapatra, S., et al. (2013). Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500, 222–226. doi: 10.1038/nature12362
Bonnet, D., and Dick, J. E. (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737. doi: 10.1038/nm0797-730
Bronsert, P., Enderle-Ammour, K., Bader, M., Timme, S., Kuehs, M., Csanadi, A., et al. (2014). Cancer cell invasion and EMT marker expression: a three-dimensional study of the human cancer-host interface. J. Pathol. 234, 410–422. doi: 10.1002/path.4416
Calon, A., Lonardo, E., Berenguer-Llergo, A., Espinet, E., Hernando-Momblona, X., Iglesias, M., et al. (2015). Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 47, 320–329. doi: 10.1038/ng.3225
Cao, Y., Guo, W., Tian, S., He, X., Wang, X., Liu, X., et al. (2015). miR-290/371-Mbd2-Myc circuit regulates glycolytic metabolism to promote pluripotency. EMBO J. 34, 609–623. doi: 10.15252/embj.201490441
Carey, B. W., Finley, L. W. S., Cross, J. R., Allis, C. D., and Thompson, C. B. (2015). Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. doi: 10.1038/nature13981
Cervo, P. R. D. V., Romanov, R. A., Spigolon, G., Masini, D., Martin-Montanez, E., Toledo, E. M., et al. (2017). Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 35, 444–452. doi: 10.1038/nbt.3835
Chaffer, C. L., Marjanovic, N. D., Lee, T., Bell, G., Kleer, C. G., Reinhardt, F., et al. (2013). Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154, 61–74. doi: 10.1016/j.cell.2013.06.005
Chen, J., Guo, L., Zhang, L., Wu, H., Yang, J., Liu, H., et al. (2013a). Vitamin C modulates TET1 function during somatic cell reprogramming. Nat. Genet. 45, 1504–1509. doi: 10.1038/ng.2807
Chen, J., Liu, H., Liu, J., Qi, J., Wei, B., Yang, J., et al. (2013b). H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 45, 34–42. doi: 10.1038/ng.2491
Chen, Q., Chen, Y., Bian, C., Fujiki, R., and Yu, X. (2013c). TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564. doi: 10.1038/nature11742
Choi, Y. K., and Park, K. G. (2018). Targeting glutamine metabolism for cancer treatment. Biomol. Ther. 26, 19–28. doi: 10.4062/biomolther.2017.178
Cimmino, L., Dolgalev, I., Wang, Y., Yoshimi, A., Martin, G., Wang, J., et al. (2017). Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095.e20. doi: 10.1016/j.cell.2017.07.032
Clarke, M., Dick, J., Dirks, P., Eaves, C., Jamieson, C., Jones, D., et al. (2006). Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 66, 9339–9344. doi: 10.1158/0008-5472.CAN-06-3126
Clevers, H. (2011). The cancer stem cell: premises, promises and challenges. Nat. Med. 17, 313–319. doi: 10.1038/nm.2304
Clevers, H. (2015). STEM CELLS. What is an adult stem cell? Science 350, 1319–1320. doi: 10.1126/science.aad7016
Constable, S., Lim, J., Vaidyanathan, K., and Wells, L. (2017). O-GlcNAc transferase regulates transcriptional activity of human Oct4. Glycobiology 27, 927–937. doi: 10.1093/glycob/cwx055
Costa, Y., Ding, J., Theunissen, T., Faiola, F., Hore, T., Shliaha, P., et al. (2013). NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 495, 370–374. doi: 10.1038/nature11925
Craene, B. D., and Berx, G. (2013). Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 13, 97–110. doi: 10.1038/nrc3447
Davalos, V., Moutinho, C., Villanueva, A., Boque, R., Silva, P., Carneiro, F., et al. (2012). Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 31, 2062–2074. doi: 10.1038/onc.2011.383
Deplus, R., Delatte, B., Schwinn, M., Defrance, M., Mendez, J., Murphy, N., et al. (2013). TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 32, 645–655. doi: 10.1038/emboj.2012.357
Doege, C., Inoue, K., Yamashita, T., Rhee, D., Travis, S., Fujita, R., et al. (2012). Early-stage epigenetic modification during somatic cell reprogramming by Parp1 and Tet2. Nature 488, 652–655. doi: 10.1038/nature11333
Dongre, A., and Weinberg, R. (2019). New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84. doi: 10.1038/s41580-018-0080-4
Ebrahimi, A., Sevinc, K., Gurhan Sevinc, G., Cribbs, A. P., Philpott, M., Uyulur, F., et al. (2019). Bromodomain inhibition of the coactivators CBP/EP300 facilitate cellular reprogramming. Nat. Chem. Biol. 15, 519–528. doi: 10.1038/s41589-019-0264-z
Eiriksson, F. F., Rolfsson, O., Ogmundsdottir, H. M., Haraldsson, G. G., Thorsteinsdottir, M., and Halldorsson, S. (2018). Altered plasmalogen content and fatty acid saturation following epithelial to mesenchymal transition in breast epithelial cell lines. Int. J. Biochem. Cell Biol. 103, 99–104. doi: 10.1016/j.biocel.2018.08.003
Emmett, M. J., and Lazar, M. A. (2019). Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell Biol. 20, 102–115. doi: 10.1038/s41580-018-0076-0
Evans, M. J., and Kaufman, M. H. (1981). Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156. doi: 10.1038/292154a0
Feng, D., Sheng-Dong, L., Tong, W., and Zhen-Xian, D. (2020). O-GlcNAcylation of RAF1 increases its stabilization and induces the renal fibrosis. Biochim. Biophys. Acta 1866:165556. doi: 10.1016/j.bbadis.2019.165556
Figueroa, M., Abdel-Wahab, O., Lu, C., Ward, P., Patel, J., Shih, A., et al. (2010). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567. doi: 10.1016/j.ccr.2010.11.015
Fischer, B., and Bavister, B. D. (1993). Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J. Reprod. Fertil. 99, 673–679. doi: 10.1530/jrf.0.0990673
Fischer, K., Durrans, A., Lee, S., Sheng, J., Li, F., Wong, S., et al. (2015). Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476. doi: 10.1038/nature15748
Folmes, C., Nelson, T., Martinez-Fernandez, A., Arrell, D., Lindor, J., Dzeja, P., et al. (2011). Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 14, 264–271. doi: 10.1016/j.cmet.2011.06.011
Foster, J., and Archer, S. (1979). Birth order and intelligence: an immunological interpretation. Percept. Mot. Skills 48, 79–93. doi: 10.2466/pms.1979.48.1.79
Fumagalli, A., Oost, K. C., Kester, L., Morgner, J., Bornes, L., Bruens, L., et al. (2020). Plasticity of Lgr5-negative cancer cells drives metastasis in colorectal cancer. Cell Stem Cell 26, 569–578.e7. doi: 10.1016/j.stem.2020.02.008
Gao, Y., Chen, J., Li, K., Wu, T., Huang, B., Liu, W., et al. (2013). Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell 12, 453–469. doi: 10.1016/j.stem.2013.02.005
Georgakopoulos-Soares, I., Chartoumpekis, D. V., Kyriazopoulou, V., and Zaravinos, A. (2020). EMT factors and metabolic pathways in cancer. Front. Oncol. 10:499. doi: 10.3389/fonc.2020.00499
Guo, W., Keckesova, Z., Donaher, J., Shibue, T., Tischler, V., Reinhardt, F., et al. (2012). Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028. doi: 10.1016/j.cell.2012.02.008
Harosh-Davidovich, S., and Khalaila, I. (2018). O-GlcNAcylation affects beta-catenin and E-cadherin expression, cell motility and tumorigenicity of colorectal cancer. Exp. Cell Res. 364, 42–49. doi: 10.1016/j.yexcr.2018.01.024
Hay, E. (1968). “Organization and fine structure of epithelium and mesenchyme in the developing chick embryos,” in Proceedings of the 18th Hahnemann Symposium Epithelial-MesenchymalInteractions, eds R. Fleischmajer and R. E. Billingham (Baltimore, Maryland: Williams & Wilkins), 31–55.
He, J., Kallin, E. M., Tsukada, Y. I., and Zhang, Y. (2008). The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15Ink4b(Ink4b). Nat. Struct. Mol. Biol. 15, 1169–1175. doi: 10.1038/nsmb.1499
He, S., Chen, J., Zhang, Y., Zhang, M., Yang, X., Li, Y., et al. (2017a). Sequential EMT-MET induces neuronal conversion through Sox2. Cell Discov. 3:17017. doi: 10.1038/celldisc.2017.17
He, S., Guo, Y., Zhang, Y., Li, Y., Feng, C., Li, X., et al. (2015). Reprogramming somatic cells to cells with neuronal characteristics by defined medium both in vitro and in vivo. Cell Regen. 4:12. doi: 10.1186/s13619-015-0027-6
He, S., Sun, H., Lin, L., Zhang, Y., Chen, J., Liang, L., et al. (2017b). Passive DNA demethylation preferentially up-regulates pluripotency-related genes and facilitates the generation of induced pluripotent stem cells. J. Biol. Chem. 292, 18542–18555. doi: 10.1074/jbc.M117.810457
He, S., Wang, F., Zhang, Y., Chen, J., Liang, L., Li, Y., et al. (2019). Hemi-methylated CpG sites connect Dnmt1-knockdown-induced and Tet1-induced DNA demethylation during somatic cell reprogramming. Cell Discov. 5:11. doi: 10.1038/s41421-018-0074-6
Hiew, M., Cheng, H., Huang, C., Chong, K., Cheong, S., Choo, K., et al. (2018). Incomplete cellular reprogramming of colorectal cancer cells elicits an epithelial/mesenchymal hybrid phenotype. J. Biomed. Sci. 25:57. doi: 10.1186/s12929-018-0461-1
Hsu, Y. C., Wu, Y. T., Tsai, C. L., and Wei, Y. H. (2018). Current understanding and future perspectives of the roles of sirtuins in the reprogramming and differentiation of pluripotent stem cells. Exp. Biol. Med. 243, 563–575. doi: 10.1177/1535370218759636
Hu, X., Zhang, L., Mao, S.-Q., Li, Z., Chen, J., Zhang, R.-R., et al. (2014). Tet and TDG Mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem Cell 14, 512–522. doi: 10.1016/j.stem.2014.01.001
Jiang, M., Xu, B., Li, X., Shang, Y., Chu, Y., Wang, W., et al. (2019). O-GlcNAcylation promotes colorectal cancer metastasis via the miR-101-O-GlcNAc/EZH2 regulatory feedback circuit. Oncogene 38, 301–316. doi: 10.1038/s41388-018-0435-5
Jolly, M., Boareto, M., Huang, B., Jia, D., Lu, M., Ben-Jacob, E., et al. (2015). Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 5:155. doi: 10.3389/fonc.2015.00155
Jolly, M. K., Ware, K. E., Gilja, S., Somarelli, J. A., and Levine, H. (2017). EMT and MET: necessary or permissive for metastasis? Mol. Oncol. 11, 755–769. doi: 10.1002/1878-0261.12083
Kang, H., Kim, H., Lee, S., Youn, H., and Youn, B. (2019). Role of metabolic reprogramming in epithelial-mesenchymal transition (EMT). Int. J. Mol. Sci. 20, 2042. doi: 10.3390/ijms20082042
Kim, N. H., Cha, Y. H., Lee, J., Lee, S. H., Yang, J. H., Yun, J. S., et al. (2017). Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat. Commun. 8:14374. doi: 10.1038/ncomms14374
Korpal, M., Lee, E. S., Hu, G., and Kang, Y. (2008). The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 283, 14910–14914. doi: 10.1074/jbc.C800074200
Lambert, A. W., Pattabiraman, D. R., and Weinberg, R. A. (2017). Emerging Biological Principles of Metastasis. Cell 168, 670–691. doi: 10.1016/j.cell.2016.11.037
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. doi: 10.1038/nrm3758
Lapidot, T., Sirard, C., Vormoor, J., Murdoch, B., Hoang, T., Caceres-Cortes, J., et al. (1994). A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648. doi: 10.1038/367645a0
Lee, J. Y., and Kong, G. (2016). Roles and epigenetic regulation of epithelial-mesenchymal transition and its transcription factors in cancer initiation and progression. Cell. Mol. Life Sci. 73, 4643–4660. doi: 10.1007/s00018-016-2313-z
Lee, Y. L., Peng, Q., Fong, S. W., Chen, A. C., Lee, K. F., Ng, E. H., et al. (2012). Sirtuin 1 facilitates generation of induced pluripotent stem cells from mouse embryonic fibroblasts through the miR-34a and p53 pathways. PLoS One 7:e45633. doi: 10.1371/journal.pone.0045633
Li, Q., Hutchins, A. P., Chen, Y., Li, S., Shan, Y., Liao, B., et al. (2017). A sequential EMT-MET mechanism drives the differentiation of human embryonic stem cells towards hepatocytes. Nat. Commun. 3:15166. doi: 10.1038/ncomms15166
Li, R., Liang, J., Ni, S., Zhou, T., Qing, X., Li, H., et al. (2010). A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63. doi: 10.1016/j.stem.2010.04.014
Liao, B., Bao, X., Liu, L., Feng, S., Zovoilis, A., Liu, W., et al. (2011). MicroRNA cluster 302-367 enhances somatic cell reprogramming by accelerating a mesenchymal-to-epithelial transition. J. Biol. Chem. 286, 17359–17364. doi: 10.1074/jbc.C111.235960
Lim, J., and Thiery, J. P. (2012). Epithelial-mesenchymal transitions: insights from development. Development 139, 3471–3486. doi: 10.1242/dev.071209
Liu, X., Sun, H., Qi, J., Wang, L., He, S., Liu, J., et al. (2013). Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat. Cell Biol. 15, 829–838. doi: 10.1038/ncb2765
Lu, C., Ward, P., Kapoor, G., Rohle, D., Turcan, S., Abdel-Wahab, O., et al. (2012). IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. doi: 10.1038/nature10860
Lucena, M., Carvalho-Cruz, P., Donadio, J., Oliveira, I., de, Q. R., Marinho-Carvalho, M., et al. (2016). Epithelial mesenchymal transition induces aberrant glycosylation through hexosamine biosynthetic pathway activation. J. Biol. Chem. 291, 12917–12929. doi: 10.1074/jbc.M116.729236
Luo, W., Hu, H., Chang, R., Zhong, J., Knabel, M., O’Meally, R., et al. (2011). Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145, 732–744. doi: 10.1016/j.cell.2011.03.054
Macheda, M. L., Rogers, S., and Best, J. D. (2005). Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 202, 654–662. doi: 10.1002/jcp.20166
Malik, S., Villanova, L., Tanaka, S., Aonuma, M., Roy, N., Berber, E., et al. (2015). SIRT7 inactivation reverses metastatic phenotypes in epithelial and mesenchymal tumors. Sci. Rep. 5:9841. doi: 10.1038/srep09841
Martin, G. R. (1981). Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. U.S.A. 78, 7634–7638. doi: 10.1073/pnas.78.12.7634
Mathieu, J., Zhou, W., Xing, Y., Sperber, H., Ferreccio, A., Agoston, Z., et al. (2014). Hypoxia-inducible factors have distinct and stage-specific roles during reprogramming of human cells to pluripotency. Cell Stem Cell 14, 592–605. doi: 10.1016/j.stem.2014.02.012
Mathieu, J., Zhou, W., Xing, Y., Sperber, H., Ferreccio, A., Agoston, Z., et al. (2017). EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 11, 755–769.
McCoy, E., Iwanaga, R., Jedlicka, P., Abbey, N., Chodosh, L., Heichman, K., et al. (2009). Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J. Clin. Invest. 119, 2663–2677. doi: 10.1172/JCI37691
Moon, J. H., Heo, J. S., Kim, J. S., Jun, E. K., Lee, J. H., Kim, A., et al. (2011). Reprogramming fibroblasts into induced pluripotent stem cells with Bmi1. Cell Res. 21, 1305–1315. doi: 10.1038/cr.2011.107
Morel, A., Hinkal, G., Thomas, C., Fauvet, F., Courtois-Cox, S., Wierinckx, A., et al. (2012). EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genet. 8:e1002723. doi: 10.1371/journal.pgen.1002723
Moussaieff, A., Rouleau, M., Kitsberg, D., Cohen, M., Levy, G., Barasch, D., et al. (2015). Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 21, 392–402. doi: 10.1016/j.cmet.2015.02.002
Mu, W. L., Wang, Y. J., Xu, P., Hao, D. L., Liu, X. Z., Wang, T. T., et al. (2015). Sox2 Deacetylation by Sirt1 Is Involved in Mouse Somatic Reprogramming. Stem Cells 33, 2135–2147. doi: 10.1002/stem.2012
Najafi, M., Farhood, B., and Mortezaee, K. (2019). Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell. Physiol. 234, 8381–8395. doi: 10.1002/jcp.27740
Nassour, M., Idoux-Gillet, Y., Selmi, A., Come, C., Faraldo, M., Deugnier, M., et al. (2012). Slug controls stem/progenitor cell growth dynamics during mammary gland morphogenesis. PLoS One 7:e53498. doi: 10.1371/journal.pone.0053498
Neri, F., Incarnato, D., Krepelova, A., Rapelli, S., Pagnani, A., Zecchina, R., et al. (2013). Genome-wide analysis identifies a functional association of Tet1 and Polycomb repressive complex 2 in mouse embryonic stem cells. Genome Biol. 14:R91. doi: 10.1186/gb-2013-14-8-r91
Ng, S.-C., Locasale, J. W., Lyssiotis, C. A., Zheng, Y., Teo, R. Y., Ratanasirintrawoot, S., et al. (2013). Influence of threonine metabolism on S-Adenosylmethionine and histone methylation. Science 339, 222–226. doi: 10.1126/science.1226603
Ngo, B., Van, R. J., Cantley, L., and Yun, J. (2019). Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 19, 271–282. doi: 10.1038/s41568-019-0135-7
Nieto, M. A. (2013). Epithelial plasticity: a common theme in embryonic and cancer cells. Science 342:1234850. doi: 10.1126/science.1234850
Nieto, M. A., Huang, R. Y., Jackson, R. A., and Thiery, J. P. (2016). Emt: 2016. Cell 166, 21–45. doi: 10.1016/j.cell.2016.06.028
Onder, T. T., Kara, N., Cherry, A., Sinha, A. U., Zhu, N., Bernt, K. M., et al. (2012). Chromatin-modifying enzymes as modulators of reprogramming. Nature 483, 598–602. doi: 10.1038/nature10953
Park, S. M., Gaur, A. B., Lengyel, E., and Peter, M. E. (2008). The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 22, 894–907. doi: 10.1101/gad.1640608
Pastushenko, I., and Blanpain, C. (2019). EMT transition states during tumor progression and metastasis. Trends Cell Biol. 29, 212–226. doi: 10.1016/j.tcb.2018.12.001
Pastushenko, I., Brisebarre, A., Sifrim, A., Fioramonti, M., Revenco, T., Boumahdi, S., et al. (2018). Identification of the tumour transition states occurring during EMT. Nature 556, 463–468. doi: 10.1038/s41586-018-0040-3
Patra, K. C., Wang, Q., Bhaskar, P. T., Miller, L., Wang, Z., Wheaton, W., et al. (2013). Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24, 213–228. doi: 10.1016/j.ccr.2013.06.014
Pei, D., Shu, X., Gassama-Diagne, A., and Thiery, J. P. (2019). Mesenchymal-epithelial transition in development and reprogramming. Nat. Cell Biol. 21, 44–53. doi: 10.1038/s41556-018-0195-z
Peinado, H., Ballestar, E., Esteller, M., and Cano, A. (2004). Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell Biol. 24, 306–319. doi: 10.1128/mcb.24.1.306-319.2004
Prigione, A., Rohwer, N., Hoffmann, S., Mlody, B., Drews, K., Bukowiecki, R., et al. (2014). HIF1alpha modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells 32, 364–376. doi: 10.1002/stem.1552
Quivoron, C., Couronne, L., Della Valle, V., Lopez, C. K., Plo, I., Wagner-Ballon, O., et al. (2011). TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38. doi: 10.1016/j.ccr.2011.08.006
Ryall, J. G., Cliff, T., Dalton, S., and Sartorelli, V. (2015). Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell 17, 651–662. doi: 10.1016/j.stem.2015.11.012
Samavarchi-Tehrani, P., Golipour, A., David, L., Sung, H. K., Beyer, T. A., Datti, A., et al. (2010). Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7, 64–77. doi: 10.1016/j.stem.2010.04.015
Sampson, V., David, J., Puig, I., Patil, P., Herreros, A., Thomas, G., et al. (2014). Wilms’ tumor protein induces an epithelial-mesenchymal hybrid differentiation state in clear cell renal cell carcinoma. PLoS One 9:e102041. doi: 10.1371/journal.pone.0102041
Saunders, A., Huang, X., Fidalgo, M., Reimer, M. H. Jr., Faiola, F., Ding, J., et al. (2017). The SIN3A/HDAC corepressor complex functionally cooperates with NANOG to promote pluripotency. Cell Rep 18, 1713–1726. doi: 10.1016/j.celrep.2017.01.055
Schliekelman, M., Taguchi, A., Zhu, J., Dai, X., Rodriguez, J., Celiktas, M., et al. (2015). Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res. 75, 1789–1800. doi: 10.1158/0008-5472.CAN-14-2535
Setty, M., Tadmor, M. D., Reich-Zeliger, S., Angel, O., Salame, T. M., Kathail, P., et al. (2016). Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol. 34, 637–645. doi: 10.1038/nbt.3569
Shi, Y. J., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear arnine oxidase homolog LSD1. Cell 119, 941–953. doi: 10.1016/j.cell.2004.12.012
Shiraki, N., Shiraki, Y., Tsuyama, T., Obata, F., Miura, M., Nagae, G., et al. (2014). Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 19, 780–794. doi: 10.1016/j.cmet.2014.03.017
Shu, X., and Pei, D. (2014). The function and regulation of mesenchymal-to-epithelial transition in somatic cell reprogramming. Curr. Opin. Genet. Dev. 28, 32–37. doi: 10.1016/j.gde.2014.08.005
Shyh-Chang, N., Daley, G., and Cantley, L. (2013). Stem cell metabolism in tissue development and aging. Development 140, 2535–2547. doi: 10.1242/dev.091777
Siemens, H., Jackstadt, R., Hunten, S., Kaller, M., Menssen, A., Gotz, U., et al. (2011). miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 10, 4256–4271. doi: 10.4161/cc.10.24.18552
Stefano, B. D., Sardina, J., Oevelen, C. V., Collombet, S., Kallin, E., Vicent, G., et al. (2014). C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature 506, 235–239. doi: 10.1038/nature12885
Sun, H., Liang, L., Li, Y., Feng, C., Li, L., Zhang, Y., et al. (2016). Lysine-specific histone demethylase 1 inhibition promotes reprogramming by facilitating the expression of exogenous transcriptional factors and metabolic switch. Sci. Rep. 6:30903. doi: 10.1038/srep30903
Sun, H., Yang, X., Liang, L., Zhang, M., Li, Y., Chen, J., et al. (2020). Metabolic switch and epithelial-mesenchymal transition cooperate to regulate pluripotency. EMBO J. 39:e102961. doi: 10.15252/embj.2019102961
Sun, N. Y., and Yang, M. H. (2020). Metabolic reprogramming and epithelial-mesenchymal plasticity: opportunities and challenges for cancer therapy. Front. Oncol. 10:792. doi: 10.3389/fonc.2020.00792
Suzuki, H. I. (2018). MicroRNA control of TGF-beta signaling. Int. J. Mol. Sci. 19:1901. doi: 10.3390/ijms19071901
Swinnen, J. V., Brusselmans, K., and Verhoeven, G. (2006). Increased lipogenesis in cancer cells: new players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care 9, 358–365. doi: 10.1097/01.mco.0000232894.28674.30
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Thankamony, A. P., Saxena, K., Murali, R., Jolly, M. K., and Nair, R. (2020). Cancer stem cell plasticity - a deadly deal. Front. Mol. Biosci. 7:79. doi: 10.3389/fmolb.2020.00079
Thiery, J. P., Acloque, H., Huang, R. Y., and Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890. doi: 10.1016/j.cell.2009.11.007
Thiery, J. P., and Sleeman, J. P. (2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142. doi: 10.1038/nrm1835
Tsai, Y., Chen, H., Chen, S., Cheng, W., Wang, H., Shen, Z., et al. (2014). TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 15:513. doi: 10.1186/s13059-014-0513-0
Uckun, F. M., Sather, H., Reaman, G., Shuster, J., Land, V., Trigg, M., et al. (1995). Leukemic cell growth in SCID mice as a predictor of relapse in high-risk B-lineage acute lymphoblastic leukemia. Blood 85, 873–878.
Vella, P., Scelfo, A., Jammula, S., Chiacchiera, F., Williams, K., Cuomo, A., et al. (2013). Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell 49, 645–656. doi: 10.1016/j.molcel.2012.12.019
Wang, F., Han, J., Wang, L., Jing, Y., Zhu, Z., Hui, D., et al. (2017). CCCTC-binding factor transcriptionally targets Wdr5 to mediate somatic cell reprogramming. Stem Cells Dev. 26, 743–750. doi: 10.1089/scd.2016.0309
Wang, T., Chen, K., Zeng, X., Yang, J., Wu, Y., Shi, X., et al. (2011). The Histone Demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell 9, 575–587. doi: 10.1016/j.stem.2011.10.005
Wang, Y., Dong, C., and Zhou, B. P. (2020). Metabolic reprogram associated with epithelial-mesenchymal transition in tumor progression and metastasis. Genes Dis. 7, 172–184. doi: 10.1016/j.gendis.2019.09.012
Wang, Y., Qin, J., Wang, S., Zhang, W., Duan, J., Zhang, J., et al. (2016). Conversion of human gastric epithelial cells to multipotent endodermal progenitors using defined small molecules. Cell Stem Cell 19, 449–461. doi: 10.1016/j.stem.2016.06.006
Warburg, O. (1956). On the origin of cancer cells. Science 123, 309–314. doi: 10.1126/science.123.3191.309
Wei, T., Chen, W., Wang, X., Zhang, M., Chen, J., Zhu, S., et al. (2015). An HDAC2-TET1 switch at distinct chromatin regions significantly promotes the maturation of pre-iPS to iPS cells. Nucleic Acids Res. 43, 5409–5422. doi: 10.1093/nar/gkv430
Wellner, U., Schubert, J., Burk, U., Schmalhofer, O., Zhu, F., Sonntag, A., et al. (2009). The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 11, 1487–1495. doi: 10.1038/ncb1998
Williams, E. D., Gao, D., Redfern, A., and Thompson, E. W. (2019). Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 19, 716–732. doi: 10.1038/s41568-019-0213-x
Wu, H., and Zhang, Y. (2014). Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156, 45–68. doi: 10.1016/j.cell.2013.12.019
Wu, J., Ocampo, A., and Belmonte, J. (2016). Cellular Metabolism and Induced Pluripotency. Cell 166, 1371–1385. doi: 10.1016/j.cell.2016.08.008
Wu, X., and Zhang, Y. (2017). TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 18, 517–534. doi: 10.1038/nrg.2017.33
Wu, Y., Chen, K., Xing, G., Li, L., Ma, B., Hu, Z., et al. (2019). Phospholipid remodeling is critical for stem cell pluripotency by facilitating mesenchymal-to-epithelial transition. Sci. Adv. 5:eaax7525. doi: 10.1126/sciadv.aax7525
Yang, J., Antin, P., Berx, G., Blanpain, C., Brabletz, T., Bronner, M., et al. (2020). Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 21, 341–352. doi: 10.1038/s41580-020-0237-9
Yang, L., Zhang, F., Wang, X., Tsai, Y., Chuang, K. H., Keng, P. C., et al. (2016). A FASN-TGF-beta1-FASN regulatory loop contributes to high EMT/metastatic potential of cisplatin-resistant non-small cell lung cancer. Oncotarget 7, 55543–55554. doi: 10.18632/oncotarget.10837
Ye, X., Brabletz, T., Kang, Y., Longmore, G., Nieto, M., Stanger, B., et al. (2017). Upholding a role for EMT in breast cancer metastasis. Nature 547, E1–E3. doi: 10.1038/nature22816
Ye, X., Tam, W. L., Shibue, T., Kaygusuz, Y., Reinhardt, F., Ng Eaton, E., et al. (2015). Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 525, 256–260. doi: 10.1038/nature14897
Ye, X., and Weinberg, R. A. (2015). Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 25, 675–686. doi: 10.1016/j.tcb.2015.07.012
Yin, R., Mao, S.-Q., Zhao, B., Chong, Z., Yang, Y., Zhao, C., et al. (2013). Ascorbic acid enhances Tet-Mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 135, 10396–10403. doi: 10.1021/ja4028346
Yu, L., Lu, M., Jia, D., Ma, J., Ben-Jacob, E., Levine, H., et al. (2017). Modeling the genetic regulation of cancer metabolism: interplay between glycolysis and oxidative phosphorylation. Cancer Res. 77, 1564–1574. doi: 10.1158/0008-5472.CAN-16-2074
Zaravinos, A. (2015). The regulatory role of MicroRNAs in EMT and cancer. J. Oncol. 2015:865816. doi: 10.1155/2015/865816
Zavadil, J., and Bottinger, E. P. (2005). TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24, 5764–5774. doi: 10.1038/sj.onc.1208927
Zhang, H., and Wang, Z. (2008). Mechanisms that mediate stem cell self-renewal and differentiation. J. Cell. Biochem. 103, 709–718. doi: 10.1002/jcb.21460
Zhang, J., Nuebel, E., Daley, G., Koehler, C., and Teitell, M. (2012). Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 11, 589–595. doi: 10.1016/j.stem.2012.10.005
Zhang, J., Tian, X., Zhang, H., Teng, Y., Li, R., Bai, F., et al. (2014). TGF-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal. 7:ra91. doi: 10.1126/scisignal.2005304
Zhang, Y., and Weinberg, R. A. (2018). Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front. Med. 12, 361–373. doi: 10.1007/s11684-018-0656-6
Zheng, X., Carstens, J., Kim, J., Scheible, M., Kaye, J., Sugimoto, H., et al. (2015). Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530. doi: 10.1038/nature16064
Zhou, W., Choi, M., Margineantu, D., Margaretha, L., Hesson, J., Cavanaugh, C., et al. (2012). HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 31, 2103–2116. doi: 10.1038/emboj.2012.71
Keywords: EMT, cancer, reprogramming, energy metabolism, glycolysis, OXPHOS
Citation: Lai X, Li Q, Wu F, Lin J, Chen J, Zheng H and Guo L (2020) Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons From Somatic Cell Reprogramming. Front. Cell Dev. Biol. 8:760. doi: 10.3389/fcell.2020.00760
Received: 27 May 2020; Accepted: 20 July 2020;
Published: 06 August 2020.
Edited by:
Marco Fiorillo, University of Salford, United KingdomReviewed by:
Anup Kumar Singh, Beckman Research Institute, City of Hope, United StatesCopyright © 2020 Lai, Li, Wu, Lin, Chen, Zheng and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiekai Chen, Y2hlbl9qaWVrYWlAZ2liaC5hYy5jbg==; Hui Zheng, emhlbmdfaHVpQGdpYmguYWMuY24=; Lin Guo, Z3VvX2xpbjA5QGdpYmguYWMuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.