 Wook Jin
Wook Jin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 09 July 2020
Sec. Molecular and Cellular Oncology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00613
This article is part of the Research Topic Repurposed Drugs Targeting Cancer Signaling Pathways: Dissecting New Mechanisms of Action Through in vitro and in vivo Analyses View all 25 articles
Ewing sarcoma (ES) is a rare, highly aggressive, bone, or soft tissue-associated tumor. Although this sarcoma often responds well to initial chemotherapy, 40% of the patients develop a lethal recurrence of the disease, with death recorded in 75–80% of patients with metastatic ES within 5 years, despite receiving high-dose chemotherapy. ES is genetically well-characterized, as indicated by the EWS-FLI1 fusion protein encoded as a result of chromosomal translocation in 80–90% of patients with ES, as well as in ES-related cancer cell lines. Recently, tyrosine kinases have been identified in the pathogenesis of ES. These tyrosine kinases, acting as oncoproteins, are associated with the clinical pathogenesis, metastasis, acquisition of self-renewal traits, and chemoresistance of ES, through the activation of various intracellular signaling pathways. This review describes the recent progress related to cellular and molecular functional roles of tyrosine kinases in the progression of ES.
The tyrosine kinase family, including both receptor tyrosine kinases (RTK) and non-receptor tyrosine kinases, triggers a cascade of downstream signaling pathways that control the complex biological process of cells, including proliferation, cellular organization, and differentiation. In normal cells, RTK activity is induced by the tightly regulated interaction between receptor-ligands, receptor-proteins, and reduced by negative regulation of protein tyrosine phosphatases (Zhang et al., 2009; Casaletto and McClatchey, 2012; Du and Lovly, 2018). For example, the tropomyosin-related kinase A (TrkA) receptor, which is activated via its ligand, nerve growth factor (NGF)-induced dimerization, contributes to the neuronal differentiation (Shen and Maruyama, 2011). Also, Platelet-derived growth factor (PDGF)-β-mediated activation of platelet-derived growth factor receptor (PDGFR) can activate intercellular pathways through complex formation between PDGFR and Src family tyrosine kinases (Kypta et al., 1990). Moreover, the density-enhanced phosphatase 1 (DEP1) blocks the internalization of vascular endothelial growth factor receptor (VEGFR) 2 by inducing dephosphorylation of VEGFR 2 (Lampugnani et al., 2003). However, dysregulation of RTKs (as identified in various human cancers) may result in their activation by ligand-dependent or independent autophosphorylation, gain-of-function mutations, and gene amplification-mediated overexpression (Zhang et al., 2009; Casaletto and McClatchey, 2012; Du and Lovly, 2018). RTK activation promotes cell proliferation, increases cell migration, motility, and disseminating ability, and confers drug resistance (Shibue et al., 2017; Jiao et al., 2018; Dongre and Weinberg, 2019). Hence, a variety of RTKs have been identified as therapeutic targets for anticancer drug development.
Notably, activation and overexpression of RTKs have been observed in Ewing sarcoma (ES), suggesting that tyrosine kinases may play key roles in the pathogenesis of ES. Clinical or preclinical inhibitors of tyrosine kinases have been developed and tested as molecular targeting therapy in ES (Gaspar et al., 2015; Grunewald et al., 2018). Herein, we explore recent advances in our understanding of the effects of tyrosine kinase activation in ES, particularly focusing on mechanisms by which tyrosine kinases are linked to the progression of ES. Determining molecular mechanisms through which tyrosine kinases influence the pathogenesis of ES may offer a novel strategy to treat ES, as well as to increase the efficacy of current therapeutics.
Although ES has a low incidence (accounting for less than 10% of human malignancies), it is the second most common round cell malignancy occurring in the primary soft tissues and bones of children, adolescents, and young adults, with 12% of pediatric malignancies (Grier, 1997). Additionally, ES has been detected in the ribs, spine, skull, and bones of hands, arms, legs, feet, and pelvis (Grunewald et al., 2018). Overall, 75% of ES patients with a localized tumor have demonstrated improved survival through a combination of multi-agent cytotoxic chemotherapy and local-control measures (surgical resection and radiotherapy) (Balamuth and Womer, 2010). Notably, the 5-year overall survival (OS) in ES patients is 87.5% at 10 years and 79.2% at 20 years, with patient mortality reported as 68.9% (Davenport et al., 2016). Approximately 20–25% of ES patients with local and distant treatment failure developed distant metastasis after completion of systemic and local treatment (Nesbit et al., 1990; Gaspar et al., 2015).
In another study, 30%–40% of ES patients presented local and distant recurrence, closely associated with poor prognosis. The 5-year survival rate of patients with metastases significantly decreases to less than 30%, and the most common recurrent sites include the bone (56.5%), lung (52.2%), brain (6.5%), and bone marrow (6.5%) (Rodriguez-Galindo et al., 2002; Riggi and Stamenkovic, 2007; Gaspar et al., 2015). Furthermore, chemotherapy and radiotherapy can induce secondary malignant neoplasms, and these secondary malignancies worsen the survival rates in ES patients (Longhi et al., 2012). In ES patients who underwent radiotherapy, local recurrence was observed in 30–40% of patients (Zucman et al., 1993; DuBois et al., 2015), after which the survival rate was significantly reduced to less than 20–25% (Ahrens et al., 1999; Ross et al., 2013).
A specific chromosomal translocation t(11;22) (q24;q12) occurs in ES (Delattre et al., 1992), and this chromosomal translocation results in the generation of a chimeric EWS-FLI1 fusion protein, created by the fusion of the amino-terminal portion of the FET family gene EWSR1 with the carboxyl-terminal DNA-binding domain of the ETS family gene, FLI1. Approximately 80–90% of patients with ES harbor the EWS-FLI1 fusion protein. This protein, as an aberrant transcription factor, is reportedly involved in the progression of ES (Delattre et al., 1992; Bonin et al., 1993; May et al., 1993). In the molecular diagnosis of ES, one key feature is the detection of translocation involving the EWSR1 gene on chromosome 22 band q12 by fluorescent in situ hybridization (FISH)-based detection or using the RT-PCR method (Grunewald et al., 2018). Additionally, the fusion of EWSR1 to the DNA-binding domain of ERG results in the EWS-ERG protein, which demonstrates functions similar to EWS-FLI1 (Sorensen et al., 1994), and EWS-ERG has been identified in 12.3% of ES patients (Delattre et al., 1994). EWS-ERG was detected in circulating tumor cells of ES patients with large tumors and has been correlated with reduced survival in these patients (Schleiermacher et al., 2003).
ES patients demonstrate a chromosomal abnormality as a prognostic indicator. In total, 77.6% (38/49), 26.5% (13/49), 26.5% (13/49), 26.5% (13/49), and 24.5% (12/49) of ES patients contained trisomy 8, 2, 5, 12, and 20, respectively. Specifically, trisomy 20 was closely associated with a worsened OS (Roberts et al., 2008). Moreover, ES induces the upregulation of the CD99 protein and caveolin 1 (CAV1), as diagnostic markers. CD 99 is a single-chain type-1 membrane glycoprotein, highly expressed in 90–97% of ES patients (Ambros et al., 1991; Riggi and Stamenkovic, 2007; Llombart-Bosch et al., 2009). CAV1, another diagnostic immunomarker, is highly expressed in 96% of ES patients, and its upregulation is significantly associated with CD99 expression. Additionally, CAV1 is detected in CD99-negative ES patients (Llombart-Bosch et al., 2009).
For the successful treatment of ES patients, most protocols of multi-agent cytotoxic chemotherapy involve vincristine/ifosfamide/doxorubicin/etoposide (VIDE) administration (Juergens et al., 2006). Furthermore, alternative multidrug chemotherapy protocols contain cyclophosphamide, topotecan, and etoposide. In standard-risk patients, the administration of vincristine/dactinomycin/ifosfamide/doxorubicin (VAIA) therapy presented no differences in the event-free survival (EFS) and OS hazard ratio when compared with VACA therapy (cyclophosphamide replacing ifosfamide). However, cyclophosphamide revealed a higher incidence of hematological toxicity. In high-risk patients who received chemotherapy including etoposide, the EFS and OS hazard ratio demonstrated a 17% and 15% reduction in the risk of an event or death relative to VAIA therapy, respectively (Paulussen et al., 2008). Moreover, the addition of vincristine/topotecan/cyclophosphamide (VTc) to the standard five-drug chemotherapy (vincristine/doxorubicin/cyclophosphamide/ifosfamide/etoposide [VDCIE]) presented better clinical benefits for ES patients. The addition of VTc to standard therapy demonstrated no toxicities, and in patients with ES, the 5-year OS and EFS were 88% and 79.5% when compared with standard 3-week cycles, respectively (Mascarenhas et al., 2016).
Insulin-like growth factor I receptor-1 mediated IGF-1R activation induces proliferation, epithelial-mesenchymal transition (EMT), metastasis, drug resistance, and tumor recurrence (Li et al., 2017). The promoter activity of IGF-1R is significantly activated by the binding of EWS-WT1 with the -331 to -40 region of the IGF-1R promoter in desmoplastic small round cell tumor (DSRCT), a malignant soft tissue sarcoma occurring in young children (Karnieli et al., 1996). This indicates that IGF-1R may promote the transcriptional expression of EWS fusion genes by inducing distinct cellular pathways involved in the pathogenesis of various types of cancer. In one study, IGF-1R was reportedly upregulated in all the tumor samples, including those from ES and synovial sarcoma patients, and inhibition of the IGF-1R signaling pathway resulted in a loss of the invasive ability of ES cells (Figure 1; Scotlandi et al., 1996; Xie et al., 1999; Asmane et al., 2012). Another report revealed that IGF-1R was upregulated in 93% of ES patients (Mora et al., 2012; Table 1). Additionally, IGF-1R activation is required for the EWS-1/FLI1-mediated transformation of ES cells (Toretsky et al., 1997). The sub-cellular localization of IGF-1R is associated with the poor survival observed in ES patients. Furthermore, nuclear localization of IGF-1R markedly increases prolonged progression-free survival (PFS) and OS in ES patients when compared with the cytoplasmic localization of IGF-1R (Asmane et al., 2012; Table 1).
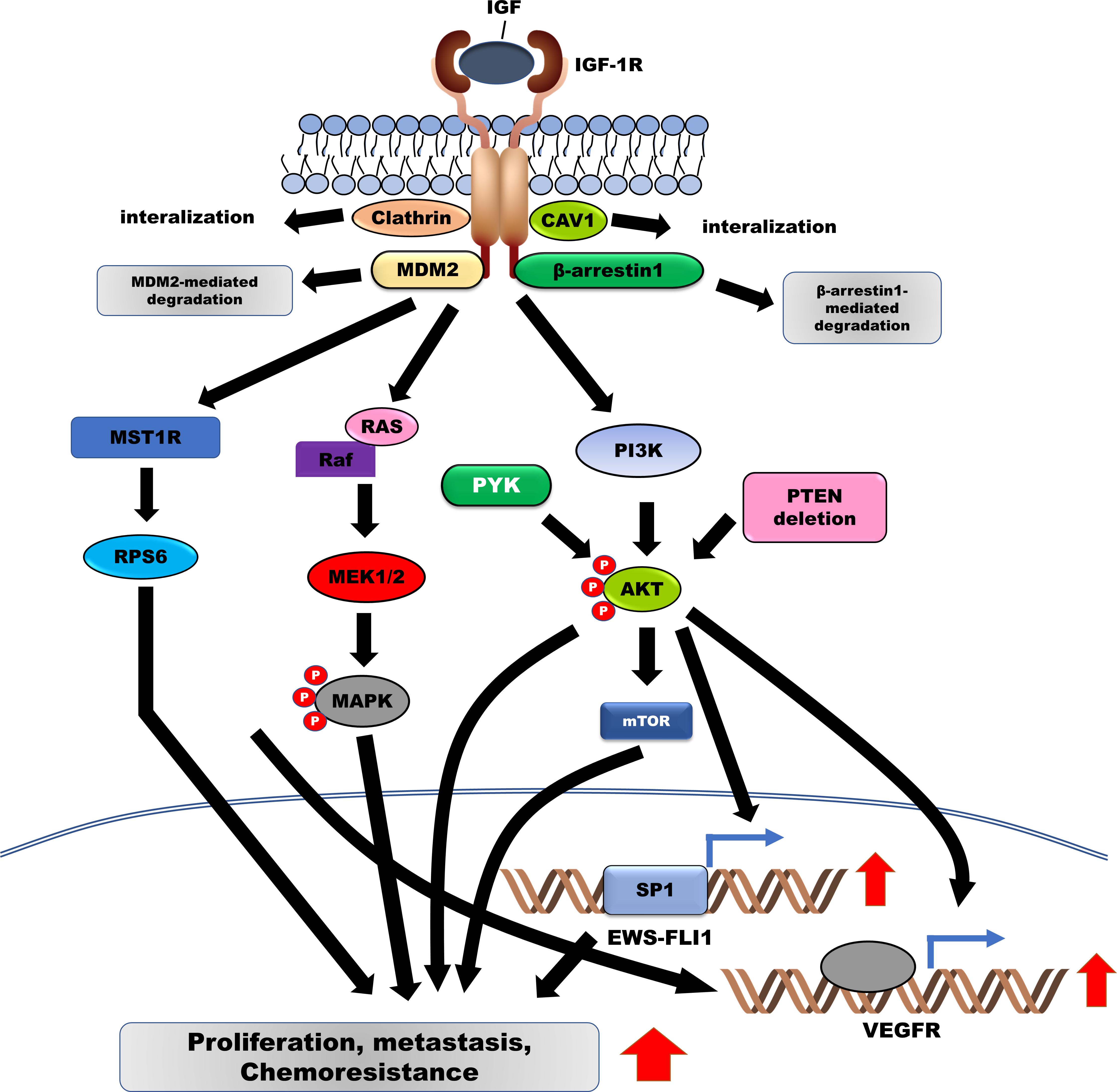
Figure 1. Induced activation of IGF-1R facilitates the survival, metastasis, and chemoresistance in Ewing sarcoma (ES) by activating downstream signaling pathways. IGF/IGF-1R enhances the pathogenesis of ES by upregulating EWS-FLI-1 and VEGFR via the activation of the RAS/MAPK/ERK and PI3K/AKT signaling pathways. Additionally, IGF-1R is degraded by MDM2 and β-arrestin-1. IGF-1R, Insulin-like growth factor 1 receptor; VEGFR, vascular endothelial growth factor; MDM2, murine double minute-2.
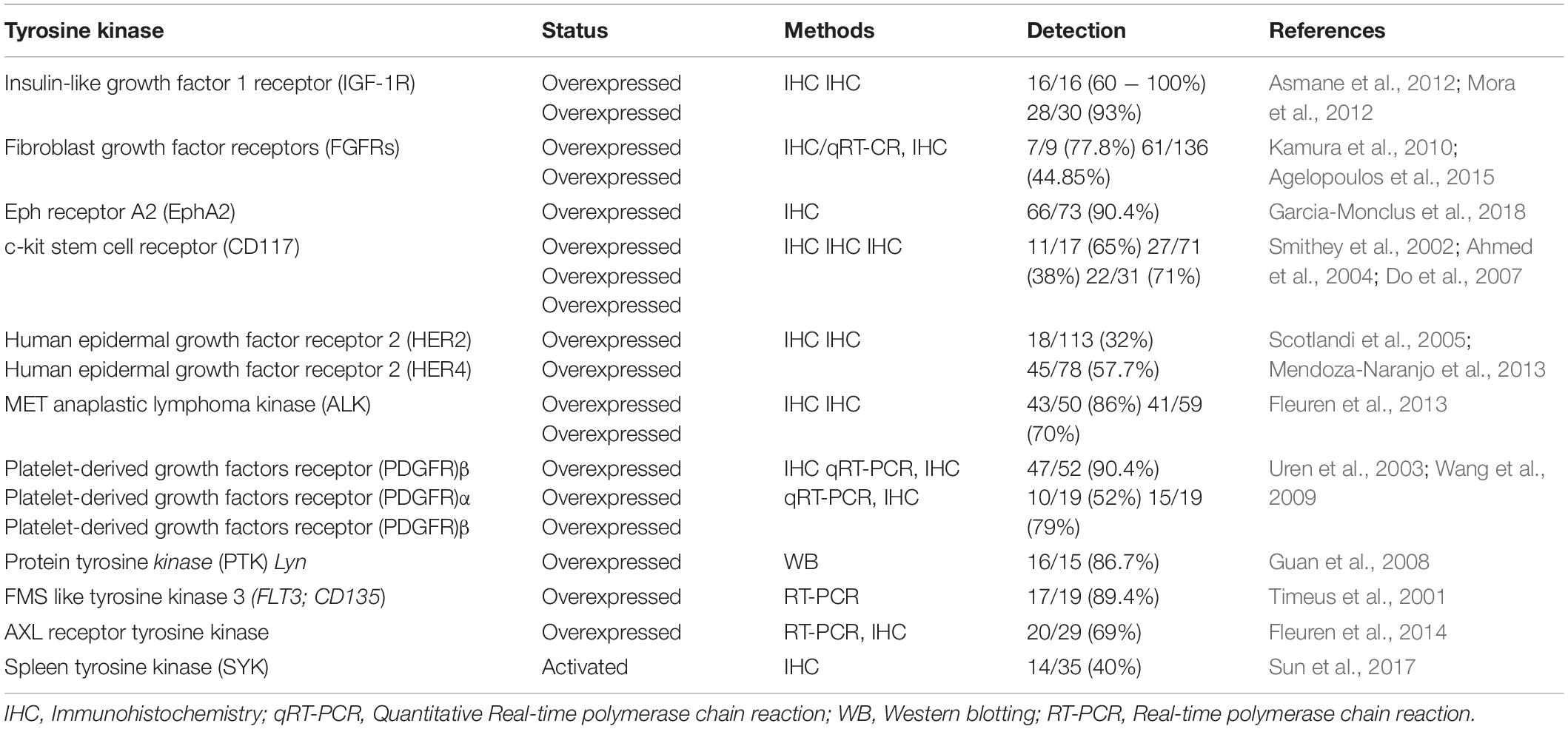
Table 1. Involvement of tyrosine kinases in Ewing sarcoma.
Importantly, activation of IGF-1R signaling prevents the apoptosis of ES cells induced by the cytotoxic activities of anticancer drugs, such as doxorubicin, by activating phosphoinositide-3-OH kinase/AKT pathway (Toretsky et al., 1999) and enhancing the transformation of mouse fibroblasts through transcriptional induction of EWS-FLI1 via the PI3K/AKT-mediated upregulation of specificity protein 1 (SP1) transcription factor (Toretsky et al., 1997; Giorgi et al., 2015). Introduction of antisense IGF-1R decreased cell proliferation and tumor growth of ES cells in vivo and in vitro (Scotlandi et al., 2002b). Both in vivo and in vitro, the expression of the IGF-I receptor dominant-negative mutant in ES cells markedly reduced the growth rate, tumor formation, metastases, and chemoresistance against doxorubicin (Scotlandi et al., 2002a). Activation of IGF-1R by IGF-1 activates AKT and extracellular-signal-regulated kinases (ERK) 1/2 in ES cells. However, inhibition of RAS/MEK/MAPK activation failed to rescue the proliferation of ES cells induced by IGF-1 treatment, indicating that compensatory activation of the RAS/MEK/MAPK signaling pathway may inhibit only IGF-1R (Benini et al., 2004). Moreover, investigation of 56 ES patients receiving neoadjuvant chemotherapy for prognostic and therapeutic purposes demonstrated positive correlations between the levels of IGF-1R and phospho-ERK (ρ = 0.286, p = 0.031, respectively), IGF-1R and phospho-AKT (ρ = 0.269, p = 0.045, respectively), phospho-AKT and phospho-ERK (ρ = 0.460, p = 0.000, respectively), and phospho-ERK and phospho-mTOR (mammalian target of rapamycin) (ρ = 0.273, p = 0.038, respectively) (van de Luijtgaarden et al., 2013). Furthermore, activation of IGF-1R and mTOR and expression of EWS-FLI1 were strongly correlated with high BMI-1 levels, which is overexpressed in 80% of ES patients and induces tumor growth in a xenograft model (van Doorninck et al., 2009).
Clinical and preclinical IGF-1R inhibitors are being utilized as monotherapy or in combination with anti-neoplastic inhibitors to inhibit ES (Table 2). Internalization and ubiquitination of IGF-1R are required for enhancing the sensitivity of the IGF-1R inhibitor. Both clathrin- and caveolin-1 (CAV-1)–dependent endocytosis-mediated internalization of IGF-1R are localized in the cytoplasm via complex formation between IGF-1R and both clathrin and CAV-1. Inhibition of clathrin- and CAV-1-dependent IGF-1R internalization following treatment with endocytosis inhibitors such as chlorpromazine (CPMZ) and methyl-β-cyclodextrin (MCD) rendered ES cells more sensitive to NVP-AEW541, an IGF-1R inhibitor (Martins et al., 2011). PU-71, heat shock protein (HSP) 90 inhibitor, is currently under evaluation for patients with solid metastatic tumors, lymphoma, and myeloma (ClinicalTrials.gov identifier: NCT01393509, NCT03166085, and NCT01269593). PU-71 treatment markedly suppressed the survival and metastatic ability of ES cells through inhibition of IGF-1R, AKT, and pERK activation, as well as downregulation of RAF-1, c-MYC, c-KIT, IGF1R, hTERT, and EWS-FLI1 expression. Furthermore, the combination of PU-71 and bortezomib, as a proteasome inhibitor, significantly inhibited tumor growth and metastasis in ES xenograft models (Ambati et al., 2014).

Table 2. Efficacy of Insulin-like growth factor 1 receptor (IGF-1R) inhibitors in combination with other inhibitors in treating Ewing sarcoma.
In patients with ES, IGF-1R inhibitors in combination with mTOR inhibitors could enhance the clinical benefits of therapy. The deletion of phosphatase and tensin homolog on chromosome 10 (PTEN) was identified in 25% of the ES patients, consistent with the PTEN deletion in ES cells, which eventually resulted in the reduced sensitivity to IGF-1R inhibitors, including NVP-AEW541 and OSI-906, and activation of the AKT pathway in the ES cells. Interestingly, the loss of PTEN expression in ES cells potentiated the autophagic response by increasing responsiveness to temsirolimus, a mammalian target of rapamycin (mTOR) inhibitor (Patel et al., 2014; Niemeyer et al., 2015).
In ES cells, constitutive activation of IGF-1R and its downstream PI3K/AKT/mTOR pathway results in the resistance to bromodomain and extra-terminal domain (BET) inhibitors. However, combining IGF-1R inhibitors with BET inhibitors, or mTOR inhibitors, presents a synergistic effect on the inhibition of survival and tumor growth of ES cells both in vivo and in vitro (Loganathan et al., 2019). Additionally, IGF-1R inhibitor-resistance in ES cells was closely correlated with the activation of IRS1, PI3K, signal transducer and activator of transcription (STAT)-3, Src, and p38 MAPK. Both IGF-1R inhibitor and mTOR inhibitor treatment significantly reduced the tumor growth in ES xenografts through inhibition of IGF-1R/PI3K/AKT/mTOR, and other signaling pathways, including MEK1/2, JAK/STAT3, TGF-β, and G-protein coupled receptors (Lamhamedi-Cherradi et al., 2016). Treatment with BMS-536924, an IGF-1R inhibitor, induced cell death, and cell cycle arrest in ES cells by suppressing activation of IGF-1R and RPS6. However, activation of MST1R, which is highly expressed in 50% of ES patients, blocked the BMS-536924-mediated cell death of ES cells by activating RPS6, a distal IGF1R effector. Treatment with rapamycin, a mTOR1 inhibitor, markedly reduced RPS6 activation in BMS-536924-resistant ES cells, inducing cell death in BMS-536924-resistant ES cells (Potratz et al., 2010). A combination of IMC-A12 (an IGF-1R) and rapamycin presented an enhanced therapeutic effect, demonstrating a prolonged response when compared with IMC-A12 treatment alone (Kolb et al., 2012). Moreover, patients who acquired resistance to a combination of IMC-A12 and temsirolimus (an mTOR inhibitor) (ClinicalTrials.gov identifier: NCT00678769) revealed activation of mTOR, STAT3, and AKT pathways after receiving this combination treatment (Subbiah et al., 2011).
Treatment with TAE226, a dual inhibitor of IGF-1R and focal adhesion kinase (FAK), significantly suppressed proliferation and tumor growth of ES cells in vivo and in vitro by inducing apoptosis by inhibiting IGF-1R, FAK, and AKT activation when compared with PF-562,271, a dual inhibitor of FAK and proline-rich tyrosine kinase 2 (Pyk2). TAE226 inhibited the activation of the brain-derived neurotrophic factor (BDNF), epidermal growth factor receptor (EGFR), and transforming growth factor (TGF) -β signaling pathways, regulated by IGF-1R and FAK (Moritake et al., 2019). Loss of CDK4 as an ES-selective dependency gene markedly reduced the growth of ES cells, but IGF-1R overexpression promoted the acquisition of resistance against the cyclin-dependent kinase (CDK) 4/6 inhibitor. Combination treatment with an IGF-1R inhibitor and CDK4/6 inhibitor demonstrated synergistic activity by inhibiting tumor growth and survival of ES cells in vivo and in vitro through the suppression of PKC/AKT/mTOR and activation of RB (Guenther et al., 2019). Furthermore, AZD3463, as a dual inhibitor of anaplastic lymphoma kinase (ALK) and IGF-1R, in combination with vorinostat, a histone deacetylase inhibitor, and temozolomide, an alkylating agent, induce cell death in ES cells by significantly inhibiting the activation of STAT3/AKT activation (Sampson et al., 2015).
N-linked glycosylation of IGF-1R is essential for its translocation to the cell surface. Treatment of IGF-1R with glycosylation inhibitors, including tunicamycin and the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, lovastatin, reduced the activation and expression of IGF-1R, subsequently reducing the survival of ES cells (Girnita et al., 2000b). Treatment with RITA (reactivating p53 and inducing tumor apoptosis), a furanic compound, significantly suppressed cell growth and metastatic potential in ES cells via RITA-induced downregulation of IGF-1R as a transcriptional target of p53, as well as the degradation of IGF-1R by RITA-mediated upregulation of murine double minute-2 (MDM2) (Di Conza et al., 2012).
Treatment with teprotumumab (R1507), an anti-IGF-1R antibody, completely inhibited the growth of ES cells by suppressing activation of the IGF-1R/IRS-1/AKT axis (Huang et al., 2011). Following this treatment, the complete response rate was 10% of the objective response rate (ORR), and the median OS was 7.6 months, indicating that teprotumumab treatment is well-tolerated. Teprotumumab has an extremely safe toxicity profile in patients with recurrent or refractory ES (ClinicalTrials.gov identifier: NCT00642941) (Pappo et al., 2011); however, it was reportedly inactive in patients presenting other types of sarcoma (Pappo et al., 2014). In January 2020, the FDA-approved teprotumumab as the first drug for the treatment of patients with thyroid eye disease (U.S.F.A.D. Administration, 2020a).
Figitumumab, a monoclonal anti-IGF-1R antibody, significantly reduces cell proliferation of ES cells by suppressing signaling pathways downstream to IGF-1R, including MAPK/ERK and PI3K/AKT pathways (Zheng et al., 2012). Additionally, figitumumab treatment increased β-arrestin1 expression, which inhibited the survival of ES cells by decreasing IGF-1R expression via β-arrestin1-mediated ubiquitination, following the formation of the IGF-1R/β-arrestin1 complex (Zheng et al., 2012). Figitumumab was evaluated in ES patients with metastases in phase 1/2 clinical trials. Reportedly, the complete response rate (disease stabilization) and partial response rate were 23% and 14.2% of the ORR, respectively. Additionally, in a phase 2 trial, figitumumab showed prolonged OS in ES patients (ClinicalTrials.gov identifier: NCT00560235) (Juergens et al., 2011).
Furthermore, human monoclonal antibodies have been developed and evaluated for the treatment of solid tumors, including ES. As cixutumumab (an IGF-1R inhibitor) monotherapy demonstrated limited single-agent activity in a phase 2 trial (ClinicalTrials.gov identifier: NCT00831844) in ES patients, a combination of cixutumumab and temsirolimus was used, revealing that 31% of patients achieved stable disease (SD) for more than 5 months and 29% of patients demonstrated more than 20% tumor regression (Naing et al., 2012). A phase 2 clinical trial of ganitumab (ClinicalTrials.gov identifier: NCT00563680), a monoclonal anti-IGF-1R antibody, revealed its well-tolerated and safe profile. In the trial, patients with a 49% ORR demonstrated SD with a good response, while those with 6% ORR exhibited partial response (Tap et al., 2012). However, IGF-1R-directed monotherapies, including figitumumab, cixutumumab, teprotumumab, ganitumumab, or cixutumumab, in combination with temsirolimus, presented only modest efficacy in ES patients (van Maldegem et al., 2016).
Currently, ganitumab monotherapy and a combination of palbociclib and ganitumab are under evaluation in phase 3 clinical trial as add-on therapy in patients with metastatic ES (ClinicalTrials.gov identifier: NCT02306161 and NCT04129151). Although anti-IGF-1R antibodies have been tested in clinical trials, these antibodies remain unapproved by the FDA.
Trabectedin (ET-743), an FDA-approved drug for the treatment of unresectable or metastatic liposarcoma or leiomyosarcoma (ClinicalTrials.gov identifier: ET743-SAR-3007 and NCT01343277) (Gordon et al., 2016; Barone et al., 2017), demonstrates antitumor activity against ES cells by inhibiting the binding of EWS-FLI1 to the promoter of its target genes, TGFβR2 and CD99. Additionally, trabectedin in combination with OSI-906 (linsitinib), an IGF-1R inhibitor, markedly increases cytotoxic effects in ES cells by synergistically interacting with the anti-IGF-1R antibody or trabectedin (Amaral et al., 2015). Notably, ES patients with a genetic alteration owing to the CDKN2A/B loss and a FUS-ERG fusion-derived orthotopic xenograft (PDOX) model exhibited a similar histomorphological appearance. A combination of OSI-906 and Palbociclib (a CDK4/6 inhibitor) markedly decreased tumor growth in a PDOX model by suppressing the CDK4/6 and IGF-1R pathways (Murakami et al., 2016).
In a phase − clinical trial for picropodophyllin (AXL1717), a novel IGF-1R inhibitor, performed in patients with relapsed malignant astrocytomas, 44% of patients demonstrated prolonged SD for 12 months (ClinicalTrials.gov identifier: NCT01721577) (Aiken et al., 2017). Picropodophyllin inhibits cell survival in ES cells by suppressing IGF-1R-mediated AKT activation (Wu et al., 2015).
Metformin (1,1-dimethyl biguanide hydrochloride) is an FDA-approved drug indicated for the treatment of type 2 diabetes, reducing glucose concentration by inhibiting mitochondrial respiration and mitochondrial glycerophosphate dehydrogenase (Foretz et al., 2019). Recently, metformin was assessed in combination with chemotherapeutic drugs and it reportedly inhibited the mTOR pathway via suppression of IGF-1R activation, by decreasing the levels of insulin-like growth factor (IGF) (Engelman and Cantley, 2010). Metformin, in combination with 5-fluorouracil and oxaliplatin (FuOX), induced cell death in chemoresistant colon cancer cells by suppressing cancer stem cells (CSC) characteristics, STAT3, and NF-κB activation (Nangia-Makker et al., 2014) and suppressing STAT3 and NF-κB activation (Lin et al., 2013; Feng et al., 2014; Yue et al., 2015; Esparza-Lopez et al., 2019). ES demonstrates high metabolic and glycolytic activity, and a combination of 2-deoxy-D-glucose (2DG) with metformin induces cell death in ES via metformin-induced inhibition of mitochondrial respiration and 2DG-induced inhibition of aerobic glycolysis (Dasgupta et al., 2017). Furthermore, a combination of metformin and imatinib reduced the proliferation of ES cells by inducing cell cycle arrest and inhibiting tumor growth and metastasis in mice (Nan et al., 2020).
Natural killer (NK) cells and chimeric antigen receptor (CAR)-based immunotherapy for adoptive cellular immunotherapy have been applied for the effective treatment of ES, inducing the activation and expansion of NK cells by inhibiting IGF-1R expression, without affecting the immunophenotypes of NK cells and their degranulation response to ES cells, and subsequently maintaining potent antitumor effects against ES cells (Jamitzky et al., 2015). As NK cell therapy has reported safety and efficacy, as well as good tolerance, in leukemia patients, clinical trials of allogeneic NK cell infusions in patients with metastatic ES and high-risk rhabdomyosarcoma (RMS) are currently ongoing (ClinicalTrials.gov identifier: NCT02409576). IGF-1R and tyrosine kinase-like orphan receptor 1 (ROR1) are highly expressed in ES cells, and the application of adoptive T-cell therapy using IGF-1R-CAR-T and ROR1-CAT-T cells derived from sarcoma patients significantly increases ES cell death and reduces tumor growth of sarcoma xenografts (Huang et al., 2015).
Activation of FGFRs by their ligand, FGF, promotes tumor progression through the activation of the PI3K/AKT, RAS-MAPK, and JAK/STAT signaling pathways (Figure 2) (Babina and Turner, 2017). FGFR1 is highly expressed in ES cells, and moderate-to-high levels of FGFR1 activation were detected in 77.8% of ES patients. FGF treatment induces metastasis in ES cells by activating the PI3K/Rac1 pathway (Table 1; Kamura et al., 2010). Another report revealed that FGFR1 was upregulated in 54.3% of ES patients (Table 1; Agelopoulos et al., 2015), inducing marked EWS-FLI1 upregulation (Girnita et al., 2000a) compared with other ligands, such as insulin-like growth factor-1 (IGF-1), platelet-derived growth factor (PDGF)-BB, hepatocyte growth factor (HGF), epidermal growth factor (EGF), and TGF-β1. However, suppression of FGFR activation, by the addition of the FGF-neutralizing antibody, leads to a marked reduction in EWS-FLI-1 expression (Figure 1; Girnita et al., 2000a). Expression of SPRY1, a negative feedback inhibitor of the FGFR-activated RAS/MAPK/ERK signaling pathway, was markedly reduced by EWS-FLI1; however, its upregulated expression suppressed cell proliferation and aggressiveness of ES cells by downregulating EWS-FLI1 through the inhibition of RAS/MAPK/ERK activation (Figure 1; Cidre-Aranaz et al., 2017).
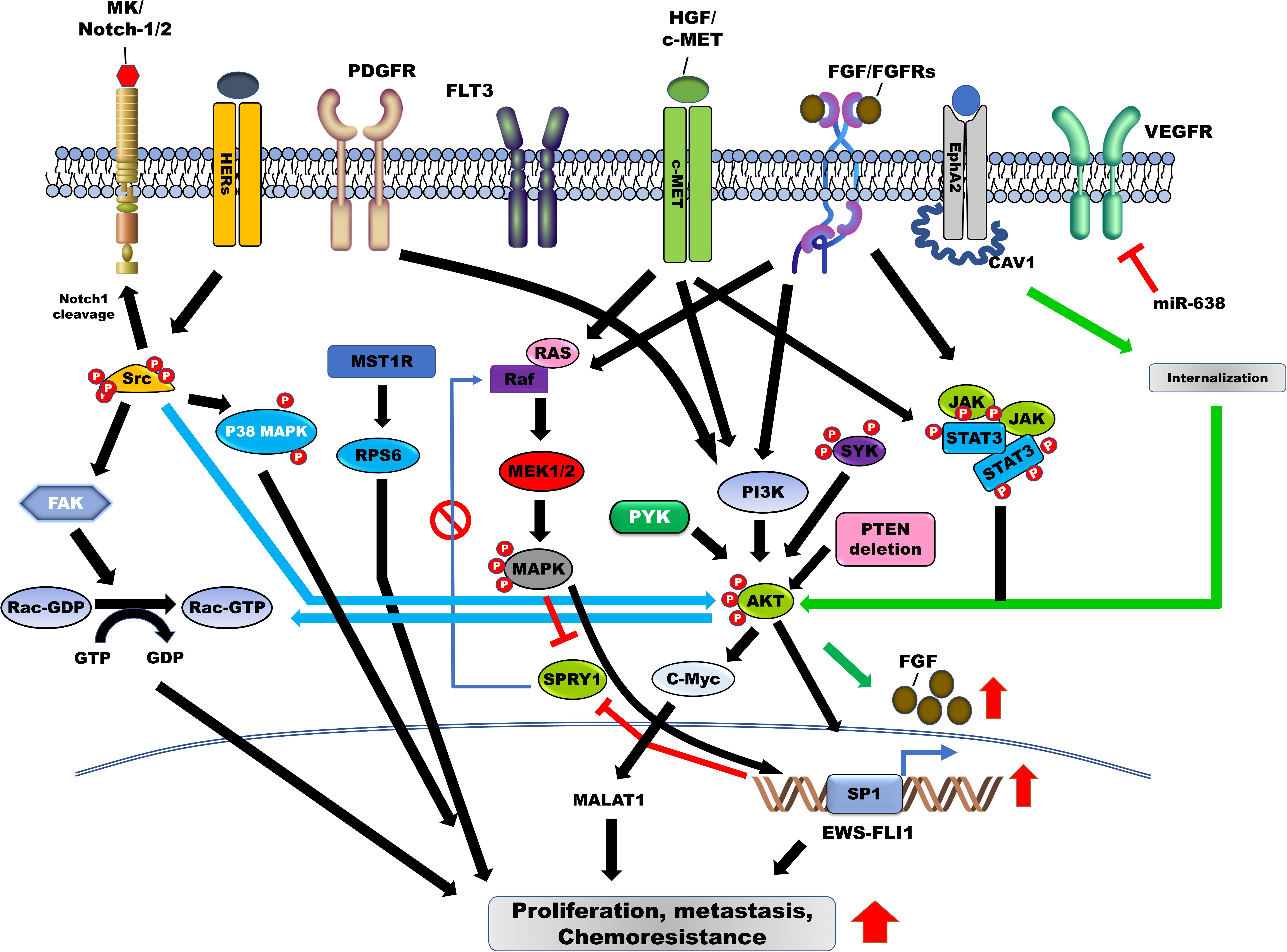
Figure 2. Contribution of tyrosine kinases in the induction of survival, metastasis, and chemoresistance in Ewing sarcoma (ES) through the activation of downstream signaling pathways. Tyrosine kinase receptors and non-receptor tyrosine kinase, including HGF/MET, EphA2, c-kit, HER2/3/4, ALK, PDGFR/PDGF, JAK/STAT, SFK, PYK22, FLT3, AXL, and SYK are involved in the pathogenesis of ES by blocking feedback inhibition of RAS/MAPK/ERK by SPRY1 and upregulation of EWS-FLI-1 through the activation of RAS/MAPK/ERK, PI3K/AKT, and JAK/STAT3 signaling pathways. Additionally, Src activation leads to cell proliferation and metastasis of ES cells via induction of Rac1 activity through the activation of p38 MAPK, FAK, and PI3K/AKT signaling pathways. HGF, hepatocyte growth factor; EphA2, erythropoietin-producing hepatoma receptor A2; STAT3, signal transducer and activator of transcription-3; FAK, focal adhesion kinase; MAPK, mitogen-activated protein kinase; JAK, Janus kinase.
An FGFR1 mutant (FGFR1 N546K), which occurs in the tyrosine kinase domain of FGFR1 as a result of an activating mutation, has been identified in pineal tumor, peritoneal sarcoma, pilocytic astrocytoma, gastrointestinal stromal tumors (GISTs), pheochromocytomas, liposarcoma, breast sarcoma, malignant peripheral nerve sheath tumor, neuroblastoma, paraganglioma, and glioblastoma (Lew et al., 2009; Jones et al., 2013; Helsten et al., 2016; Toledo et al., 2016; Nannini et al., 2017). This mutant has also been identified in 0.02% (1/50) of ES patients, and 31.7% (23/50) of ES patients demonstrated a gain of copy number of FGFR1. FGFR1 N546K mutation promotes the constitutive activation of FRFR1 (Lew et al., 2009; Katoh, 2019), and the introduction of FGFR1 N546K in NIH3T3 cells significantly increased the proliferation and the aggressiveness of cells relative to wild-type FGFR1 (Agelopoulos et al., 2015). Furthermore, FGFR4 (FGFR4 G388A) was identified in ES and breast cancer patients, but its functional role remains unclear. Further, FGFR3 (K650E), a product of an activating mutation, was identified in glioma, non-small-cell lung carcinoma (NSCLC), ES, and liposarcoma (LPS), and correlated with poor survival in patients with LPS (Helsten et al., 2016; Li et al., 2016; Lee et al., 2018).
Table 3 shows preclinical or clinical trials using FGFR inhibitors, with or without anti-neoplastic inhibitors as targeted therapeutics against ES. Treatment with FGFR inhibitors (PD173074 [PD-74], PD166866 [PD-66], SU5402 [SU54], and NVP-BGJ398 [BG-98]) reduced the proliferation of ES cells; PD173074 in particular suppressed the proliferation of ES cells and tumor formation in mice (Cidre-Aranaz et al., 2017). Additionally, treatment with ponatinib, a known FGFR inhibitor targeting FGFR1, elicited a good response by suppressing glucose uptake in ES tumors (Agelopoulos et al., 2015).
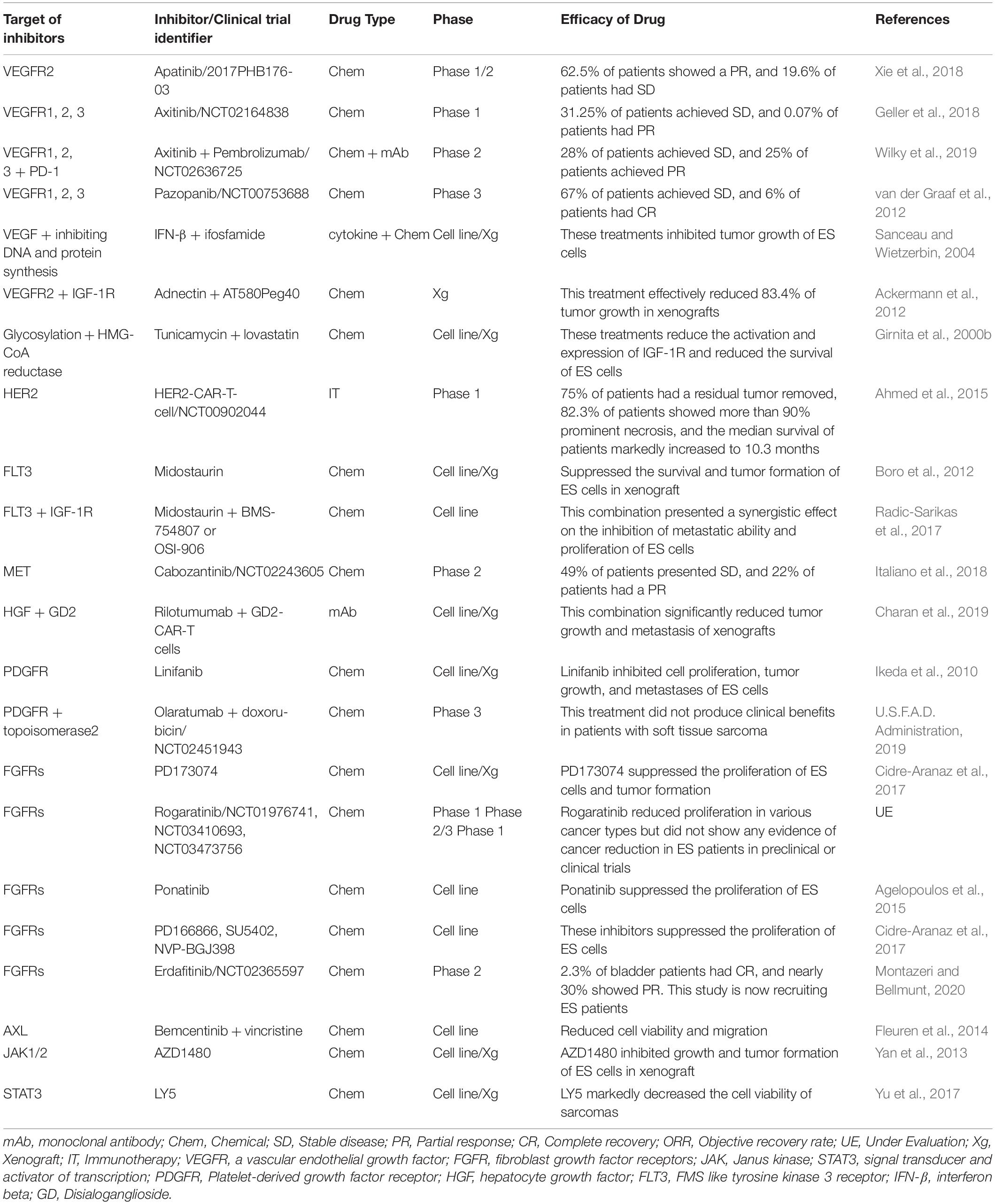
Table 3. Efficacy of tyrosine kinase targeting inhibitors in combination with other inhibitors in treating ES.
In preclinical or clinical trials, rogaratinib, an ATP-competitive inhibitor of FGFR1-4, failed to demonstrate any antitumor activity in ES conducted both in vitro or in vivo. However, rogaratinib potently inhibited the kinase of FGFR1-4 in various cancers (Collin et al., 2018; Grunewald et al., 2019). These results support the inhibitory activity of rogaratinib, which is currently undergoing phase 2/3 clinical trials (ClinicalTrials.gov identifier: NCT01976741, NCT03410693, NCT03473756).
In April 2019, a clinical trial investigated FDA-approved erdafitinib, the first targeted therapy for the treatment of patients with metastatic bladder cancer with FGFR2 or FGFR3 mutations or gene fusions. Treatment with erdafitinib (ClinicalTrials.gov identifier: NCT02365597) demonstrated an ORR of 32.2%, with 2.3% of these patients eliciting a complete response and nearly 30% showing partial response (Montazeri and Bellmunt, 2020). Erdafitinib, as well as rogaratinib, exhibited antitumor activity against ES in vitro or in vivo in non-clinical studies. Clinical trials for the same are currently ongoing in patients with ES (NCT03210714).
Human epidermal growth factor receptors (HERs; ERBBs) have been identified as inducers of ES (Figure 2). HER2 (ERBB2) is overexpressed in various human cancers and is associated with poor patient survival (Moasser, 2007). Additionally, HER2 is upregulated in 16% of ES patients and 83% of ES cell lines, and this upregulation is associated with a reduced survival rate in patients. Moreover, in 78% of ES patients, HER2 expression was associated with P-glycoprotein expression (Table 1; Scotlandi et al., 2005). Furthermore, HER2 expression is associated with the tumorigenicity of ES and is overexpressed in ES cells, conferring resistance against topoisomerase II-targeting anticancer drugs, including etoposide and adriamycin. However, treatment with adenovirus early region 1A (E1A) or adenovirus-E1A therapy markedly decreased tumor formation in mice and enhanced sensitivity to etoposide and adriamycin by downregulating HER2 and upregulating topoisomerase IIα (Zhou et al., 2001; Zhou et al., 2002).
Human epidermal growth factor receptor 3 plays a crucial role in the progression of cancer as an inducer of chemoresistance, through heterodimerization with other HER receptors or through ligand binding (Baselga and Swain, 2009). In ES patients, analysis of RTK expression profiles revealed that HER3 (ERBB3) is widely expressed; however, EGFR, its heterodimer-partner, demonstrated low-level expression in ES patients. Reportedly, the expression of HER3 is associated with reduced survival in ES patients (Potratz et al., 2016).
The expression of HER4, a member of the EGFR family, is associated with ES progression. HER4 is strongly expressed in metastatic ES cells, 57.7% of primary ES patients, and 78.9% of metastatic ES patients, relative to primary ES patients. Treatment with lapatinib and a pan-ErbB inhibitor reduced the proliferation and metastatic potential of ES cells (Table 1; Mendoza-Naranjo et al., 2012; Mendoza-Naranjo et al., 2013). Moreover, HER4 enhanced the invasiveness of metastatic ES cells and increased their metastases in vivo by increasing Rac1 GTPase activity by activating the PI3K/AKT and FAK pathways (Mendoza-Naranjo et al., 2012, 2013). Expression of microRNA (MiR)-193b suppressed anchorage-independent growth in ES cells by downregulating HER4 as its direct target (Moore et al., 2017).
HER2 chimeric antigen receptors and lapatinib (as a HER2 and HER4 inhibitor) have been evaluated in the treatment of solid tumors, including ES. HER2-specific chimeric antigen receptor (HER2-CAR) T-cells from healthy donors markedly reduced the proliferation of HER2-positive medulloblastoma cells, and HER2-CAR-T-cells from HER2-positive patients killed primary autologous HER2-positive medulloblastoma cells in vivo and in vitro by the inducing interferon (IFN)-γ production (Ahmed et al., 2007; Thanindratarn et al., 2020). Immunotherapy with antigen-specific T-cells (HER2-CAR-T-cells) was developed and tested in patients with metastatic or recurrent sarcoma in phase I/II clinical trial (ClinicalTrials.gov identifier: NCT00902044). Reportedly, 23.5% of patients receiving HER2-CAR-T-cells demonstrated SD for 3−14 months, and 75% of the patients who had residual tumors removed reported superior outcomes. Additionally, 82.3% of patients showed more than 90% necrosis of tumor cells, and the median survival of patients increased to 10.3 months (Table 3; Ahmed et al., 2015). Recently, FDA-approved neratinib, a new HER2 inhibitor for the treatment of patients with metastatic HER2-positive breast cancer (ClinicalTrials.gov identifier: NCT01808573) (U.S.F.A.D. Administration, 2020b), decreased the risk of disease progression or death by 24% when compared with lapatinib or capecitabine, indicating that this inhibitor might provide a promising therapeutic option to improve host cytotoxicity and advance the clinical treatment of sarcoma. BDTX-189, osimertinib, and DZD9008, as novel ERBB inhibitors, are now under evaluation in phase 1/2 clinical trial in patients with solid tumors and NSCLC with mutations or alternations in HER2 or EGFR, respectively (ClinicalTrials.gov identifier: NCT04209465, NCT03586453, NCT03974022). However, these inhibitors have not been evaluated in ES.
Activation of MET by HGF, and that of ALK by PTN or MK as its ligands, renders cancer more aggressive by activating multiple downstream pathways, including the PI3/AKT and ERK pathway (Hallberg and Palmer, 2013; Duplaquet et al., 2018). MET and ALK expression are involved in the pathogenesis of ES and synovial sarcoma (Dirks et al., 2002; Fleuren et al., 2013). MET and ALK are strongly expressed in the cytoplasm of 86% ES patients, as well as in the membranes of 70% ES patients. Membranous MET expression and high ALK expression have been associated with poor survival in ES patients (Table 1; Fleuren et al., 2013). Using phosphoproteomic profiling, MET and ALK have been identified as novel targets of synovial sarcoma and are highly expressed in 58% and 14% of ES patients, respectively (Fleuren et al., 2017). In MET, missense mutations were identified in various cancer types, including ES. Patients with ES (11.1%) exhibited MET mutations such as T1010I and N375S; however, these mutations failed to demonstrate functional roles in ES (Jiang et al., 2014). Additionally, these mutations have been observed in other cancers. In breast cancer, the MET T1010I mutation increased the metastatic potential of mammary epithelial cells by induction of AKT, MAPK, and STAT3 activation. Moreover, this mutant induced tumor formation in xenografts (Figure 1; Liu et al., 2015). Furthermore, the MET N375S mutation modulates sensitivity and confers resistance to MET inhibitors, eventually suppressing MET inhibitor-mediated apoptosis in lung cancer cells (Krishnaswamy et al., 2009).
Table 3 shows preclinical and clinical trials using MET/HGF inhibitors with or without anti-neoplastic inhibitors as targeted therapeutics against ES. The FDA has approved crizotinib and cabozantinib (MET inhibitors) for patients with metastatic NSCLC, advanced renal cell carcinoma (RCC), and hepatocellular carcinoma (HCC), along with sorafenib or anti-angiogenic therapy. Crizotinib and cabozantinib have been tested in ES patients, and results indicated that they significantly reduced the viability of ES cells (Fleuren et al., 2013). Furthermore, 26% of ES patients treated with cabozantinib in a phase 2 trial demonstrated an objective response, 49% of patients showed SD, with 22% demonstrating a partial response for 6 months, thus indicating that cabozantinib possesses antitumor effects in ES patients (ClinicalTrials.gov identifier: NCT02243605) (Italiano et al., 2018).
Disialoganglioside (GD2) was upregulated in 20% of ES patients; a combination of GD2-CAR-T cells and all-trans retinoic acid (ATRA) significantly reduced tumor formation and prolonged survival in mice by suppressing the population of monocytic and granulocytic myeloid-derived suppressor cells (MDSCs) (Long et al., 2016). In ES cells, HGF treatment significantly increased the tumor mass and metastasis. Moreover, a combination of GD2-CAR-T cells and rilotumumab, an anti-HGF antibody (AMG102), increased mice survival by suppressing tumor growth and metastasis (Charan et al., 2019).
PDGFR and its ligand (PDGF) are known therapeutic targets promoting proliferation and progression of cancer. Additionally, they are associated with the pathogenesis of ES, with PDGFRβ highly upregulated in ES cells (90.4%) and ES patients (79%) (Table 1; Uren et al., 2003). Activation of PDGFR by its ligand leads to the induction of motility in ES cells, by activation and induction of downstream targets, including PI3K/AKT and PLCγ (Figure 2; Uren et al., 2003). Additional reports revealed that the α- and β-subunits of PDGFR were strongly expressed in tissues of ES patients. High levels of PDGFRα were observed in 100% of ES patients, with high levels of PDGFRβ observed in 79% of these patients. The knockdown of PDGFRβ in ES cells markedly reduced pulmonary metastases and reduced the size of primary chest wall ES tumors in an ES orthotopic xenograft model (Table 1; Wang et al., 2009). Bone marrow (BM) cells migrate to Ewing tumors and differentiate into the vascular smooth muscle/pericyte-like cells and endothelial cells expressing α-smooth muscle actin, desmin, and PDGFRβ, as well as a vascular endothelial growth factor (VEGFR)-2, and are subsequently associated with tumor vessel endothelium by PDFGRβ to support tumor vasculature (Reddy et al., 2008).
Table 3 shows PDGFR inhibitors in combination with or without other inhibitors for the treatment of ES. Functional crosstalk between PDGF-BB/PDGFR and stromal-derived growth factor 1α (SDF-1α)/C-X-C motif chemokine receptor 4 (CXCR4) requires enhanced cancer tumorigenesis and metastasis. Autocrine activation of PDGFRα induced tumorigenesis and metastasis in advanced skin squamous cell carcinomas (SCCs) via upregulation and secretion of SDF-1α, and then SDF-1/CXCR4 induced PDGFR-induced tumor cell invasion and metastasis (Bernat-Peguera et al., 2019). Furthermore, PDGFRβ inhibitors inhibited SDF-1α-induced chemotaxis in glioblastoma (Sciaccaluga et al., 2013), and PDGF-BB induced CXCR4 and CXCR7 expression in pericytes (Xiang et al., 2019).
Plerixafor (AMD3100), a CXCR4 antagonist, inhibits the BM-mediated tumor vasculature in ES by reducing PDGFRβ expression through the disruption of the SDF-1α/CXCR4 axis (Hamdan et al., 2014). CXCR4 was markedly upregulated in 64% of therapy-naïve ES patients and 47% of metastatic ES patients. SDF-1α (CXCL12), a ligand of CXCR4, was significantly increased in 95% of therapy-naïve and metastatic ES patients, and high CXCR4 expression was correlated with reduced survival in ES patients. ES cell proliferation was markedly increased following treatment with SDF-1α; however, the addition of plerixafor suppressed the proliferation of ES cells (Berghuis et al., 2012).
The efficacy of PDGFR inhibitors against ES has been evaluated. Linifanib (ABT-869) is a multitargeting tyrosine kinase inhibitor, which targets tyrosine kinases such as VEGFR, FLT3, and PDGFR. Linifanib is clinically active in patients with HCC, acute myeloid leukemia (AML), renal cell cancer, and NSCLC (Lowery et al., 2018), inhibiting cell proliferation, tumor growth, and metastases in ES cells in xenografts by suppressing PDGFRβ and c-kit activation (Ikeda et al., 2010). PDGFRβ signaling-mediated pericyte recruitment facilitated the formation of the vascular-like structure by ES cells. However, treatment with imatinib- or a PDGFR-blocking antibody inhibited pericyte recruitment by suppressing PDGFRβ signaling, subsequently reducing tumor growth in ES cells in vivo (Thijssen et al., 2018).
Olaratumab, a PDGFRα inhibitor, delayed tumor formation in pediatric bone and soft tissue tumors in mice (Lowery et al., 2018); however, a combination of olaratumab and doxorubicin (ClinicalTrials.gov identifier: NCT02451943) failed to demonstrate clinical benefits in patients with soft tissue sarcoma (U.S.F.A.D. Administration, 2019). Instead, FDA-approved avapritinib (Ayvakit), another PDGFRα inhibitor, was investigated in clinical trials to treat patients with gastrointestinal stromal tumors (GISTs) presenting a PDGFRA D842V mutation, demonstrating a high response rate in 85% of patients. To date, no such clinical trials or preclinical examinations have been conducted in ES patients.
FMS like tyrosine kinase 3 (FLT3; CD135) is associated with poor clinical outcome in AML as it promotes cell proliferation and survival through the activation of the PI3K, RAS, and STAT3 signaling pathways (Stirewalt and Radich, 2003), and is reportedly mutated in approximately 30% of AML patients (Figure 2; Daver et al., 2019). These mutations lead to ligand-independent activation of FLT3 and its downstream pathway (Stirewalt and Radich, 2003). The expression of FLT3 and its ligand was markedly upregulated in 89.4% of ES patients (Table 1; Timeus et al., 2001).
Table 3 shows the FLT3 inhibitors investigated for the treatment of ES. Midostaurin (PKC412), an FLT3 inhibitor, first approved by the FDA for treatment of AML patients with FLT3 mutations, suppresses the survival of ES cells and tumor growth in mice (Boro et al., 2012). A combination of midostaurin and IGF-1R/INSR inhibitors (BMS-754807 and OSI-906), identified through specific drug-drug interactions by a parallel combinatorial drug screen, has demonstrated a synergistic effect by inhibiting the metastatic ability and proliferation of cells through the suppression of PKC/AKT/mTOR and MAPK signaling pathways (Radic-Sarikas et al., 2017). Recently, gilteritinib (XOSPATA), a novel FLT3 inhibitor, was approved by the FDA for relapsed or refractory AML patients with FLT mutation (ClinicalTrials.gov identifier: NCT0241939). In AML patients receiving gilteritinib, the OS (9.3 vs. 5.6 months) and EFS (2.8 vs. 0.7 months) rates were significantly increased relative to the salvage chemotherapy group. Moreover, the number of patients with complete remission, with full or partial hematologic recovery, was 2.2 fold higher when compared with the salvage chemotherapy group (Perl et al., 2019). Treatment with gilteritinib may contribute to the better survival of ES patients, although its effects have not been investigated in ES patients.
Treatment with IFN-β induces activation of p38 mitogen-activated protein kinase (MAPK) by increasing serine phosphorylation of STAT1, inducing apoptosis in ES cells by activating caspase-7 through the induction of interferon response factor (IRF)-1, thereby inducing cell cycle arrest in cooperation with p53 through transactivation of p21waf1/CIP1 (Sanceau et al., 2000).
As the FDA has approved IFN-based therapies for multiple sclerosis (English and Aloi, 2015) in the U.S., treatment with IFN-β (Betaseron, Extavia), which has direct and indirect effects on tumor inhibition, will soon be utilized as a new therapeutic strategy for cancer (Borden, 2019). IFN-β demonstrates potential anti-tumorigenic effects on tumor cells, as well as immune-infiltrating cells (Musella et al., 2017), reducing tumor cell growth in various type of cancers through by suppressing PI3K/AKT activation, reducing anti-apoptotic proteins, including FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (c-FLIP) and caspase-8, and activating the TNF-related-apoptosis-inducing ligand (TRAIL)-dependent pathway (Chawla-Sarkar et al., 2001; Dedoni et al., 2010; Apelbaum et al., 2013). Moreover, IFN-β repressed the acquisition of CSC characteristics (Doherty and Jackson, 2018); treatment with IFN-β markedly suppressed tumor growth in ES cells (Table 3). Furthermore, IFN-β treatment combined with ifosfamide demonstrated a strong antitumor effect by potently inhibiting tumor growth in ES cells in a xenograft model (Sanceau et al., 2002), by drastically reducing the expression of angiogenesis mediators, including VEGF, matrix metalloproteinase (MMP)-9, and urokinase plasminogen activator receptor (uPAR) (Sanceau and Wietzerbin, 2004).
Vascular Endothelial Cell Growth Factor A and its ligand VEGF are involved in the pathogenesis of ES. VEGF isoform (VEGF165) expression significantly increased cell proliferation and tumor growth in ES cells by stimulating the migration of BM to the tumor, by increasing tumor vessel density (Lee et al., 2006). Furthermore, miR-638, which inhibits cell proliferation by targeting Sox-2, suppressed the expression of VEGFA in ES cells through direct targeting (Figure 2; Ma et al., 2014; Zhou et al., 2018). Moreover, the secretion and expression of VEGFA in ES cells were induced by IGF-mediated activation of PI3K/AKT and MAPK signaling pathways (Figure 1; Strammiello et al., 2003).
Additionally, VEGFR inhibitors were evaluated as vascular targeting agents for the treatment of ES (Table 3). Apatinib (rivoceranib), a novel VEGFR2 inhibitor, showed stabilized disease symptoms in 19.6% sarcoma patients and a partial response in 62.5% patients. The ORR with apatinib was 70% for ES, 40.9% for osteosarcoma, 100% for chondrosarcoma, and 71.4% for soft tissue sarcoma (Xie et al., 2018), indicating that combination therapy with IGF-1R may be therapeutically more efficient for cancer patients.
Following the administration of axitinib, another VEGFR 1–3 inhibitor, 31.25% of recurrent or refractory solid tumor patients, including ES, achieved SD, and 0.07% of patients showed partial response (ClinicalTrials.gov identifier: NCT02164838) (Geller et al., 2018). Additionally, axitinib in combination with pembrolizumab, an anti-programmed cell death protein 1 (PD-1) monoclonal antibody for advanced sarcoma patients, demonstrated that 28% of patients achieved SD and 25% showed partial response (ClinicalTrials.gov, number NCT02636725) (Wilky et al., 2019).
Pazopanib is an inhibitor of multiple protein tyrosine kinases, including VEGFR 1-3, c-kit, and PDGFR. In phase 3 clinical trial (ClinicalTrials.gov, number: NCT00753688), treatment with pazopanib, a synthetic indazolpyrimidine, revealed a 2.87-fold higher PFS in sarcoma patients when compared with the placebo group. Pazopanib treatment resulted in SD in 67% of patients, demonstrating a partial response in 6% of patients (van der Graaf et al., 2012). In metastatic ES patients, the administration of pazopanib produced significant responsiveness with complete tumor regression and prolonged survival for more than 26 months (Mori et al., 2018). Moreover, pazopanib maintenance therapy in ES patients, following high-dose chemotherapy with topotecan, significantly prolonged PFS, with near-complete remission for 1 year (Tamura et al., 2019).
Adnectin, a VEGFR2 inhibitor, in combination with AT580Peg40, an IGF-1R inhibitor, reduced tumor growth by 83.4% in mice by normalizing the microvascular architecture via inhibition of autocrine VEGF-secretion and downregulation of IGF-1R. Moreover, AT580Peg40 and adnectin monotherapies yielded 37.18% and 73.39% tumor growth inhibition, respectively (Ackermann et al., 2012). These monotherapies or a combination therapy presented manageable toxicities; therefore, such therapies can be promising new treatment options.
AXL receptor tyrosine kinase, together with its ligand, growth-arrest-specific protein 6 (Gas6), is a new therapeutic target overexpressed in various cancers and significantly associated with tumor proliferation, metastasis, EMT, and acquisition of CSC characteristics (Gay et al., 2017; Zhu et al., 2019). Moreover, AXL expression is associated with the pathogenesis of ES. Medium and high levels of AXL were detected in 69% of ES patients (Table 1). High AXL expression worsened the survival rate in ES patients. Bemcentinib (BGB324), an AXL inhibitor, in combination with vincristine (a mitotic inhibitor), showed a synergistic therapeutic effect, reducing cell viability and migration of high AXL- and Gas6-expressing ES cells (Table 3; Fleuren et al., 2014).
The activation of the erythropoietin-producing hepatoma (EPH) receptor A2 (EphA2) is involved in FGF production. EphA2 is highly expressed in ES cells and ES patients. CAV1, an activator of EphA2, increased EphA2 activation through proper localization of EphA2, via the formation of the CAV1/EphA2 complex. CAV1-mediated EphA2 activation suppressed cell death and impaired vascularization in ES xenograft models by increasing the migration of endothelial cells and inducing the expression and secretion of FGF through AKT activation (Figure 2; Sainz-Jaspeado et al., 2013). Phosphorylation at S897 (p-EphA2S897) of EphA2 correlated with the aggressiveness of ES. High p-EphA2S897 levels significantly increased the proliferation and migration of ES cells when compared with low phosphorylation levels; however, its mutant markedly suppressed the metastatic potential of ES cells. EphA2 expression was observed in 90.4% of ES patients, and this phosphorylation was associated with a lower survival rate in ES patients. Additionally, EphA2 demonstrated a strong correlation with cellular movement and survival-related signaling, such as the EGFR signaling pathway (Table 1; Garcia-Monclus et al., 2018).
c-kit (CD117), a tyrosine kinase receptor, plays a crucial role in the development of melanogenesis, hematopoiesis, and gonadogenesis (Nocka et al., 1989), and is highly expressed in soft tissue sarcoma cells of neuroectodermic origin, such as ES and peripheral neuroectodermal tumors (PNET). Activation of c-kit by its ligand stem cell factor (SCF) subsequently induces cell proliferation (Ricotti et al., 1998). Previously, two separate reports revealed that c-kit was significantly upregulated in 65% of ES patients and 38% of ES patients, respectively (Table 1; Ahmed et al., 2004; Do et al., 2007). Another report documented that 71% of ES and 100% of synovial sarcoma patients were positive for c-kit; additionally, 29 and 64% of ES and synovial sarcoma patients showed strong and diffuse staining for c-kit, respectively (Table 1; Smithey et al., 2002). However, treatment with soluble SCF, presenting biological activities distinct from membrane-bound SCF, markedly decreased cell migration, metastasis to the lungs and extrapulmonary sites, and death due to metastasis in mice (Landuzzi et al., 2000).
Imatinib inhibits tumor growth of ES cells in vivo and in vitro by suppressing activation of SCF-mediated c-kit (Merchant et al., 2002); however, a clinical trial evaluating imatinib failed to demonstrate clinical benefits in patients with ES (ClinicalTrials.gov Identifier: NCT00154388) (Chugh et al., 2009). In another study, 14.3% of ES patients exhibited partial response without progression (ClinicalTrials.gov Identifier: NCT00062205) (Chao et al., 2010). Regorafenib treatment, as a multikinase inhibitor, including c-Kit, VEGFR1-3, BRAF, FGFR1, significantly increased the EFS in 100% (9/9) of sarcoma models, including osteosarcoma, rhabdomyosarcoma, and ES (Harrison et al., 2019), and is currently under evaluation in phase 2 clinical trial for patients with sarcoma subtypes, including ES (ClinicalTrials.gov identifier: NCT02048371).
The JAK/STAT pathway plays a major role in the progression of various types of cancers (Jones and Jenkins, 2018), including ES. STAT3 activation was identified in 51% of ES patients and primary ES cells. However, STAT3 activation failed to correlate with EWS-FLI1 (Lai et al., 2006). Another report demonstrated that STAT3 was activated in 58% of ES patients and ES cells (Behjati et al., 2012). The activation of STAT3 was markedly increased by a W775 stop germline mutation of protein tyrosine phosphatase delta, a tumor suppressor, identified in 37.5% of metastatic ES patients (Veeriah et al., 2009; Jiang et al., 2013; Jiang et al., 2014). Treatment with epigallocatechin gallate, major catechin present in green tea, inhibited proliferation, survival, and invasiveness of ES cells by suppressing its downstream pathways, including PI3/AKT, JAK2/STAT3, and by RAS/ERK signaling through the inhibition IGF-1R activation (Kang et al., 2010).
The secretion of cytokines enhances the progression of ES. Oncostatin M (OSM), a cytokine of the interleukin-6 (IL-6) family, as well as its receptors, induced cell proliferation in ES cells by activation of STAT3 and Myc (David et al., 2012). Furthermore, the high levels of IL-6, secreted by ES-stroma crosstalk in ES tumor microenvironment, activated the JAK/STAT3 signaling and inducing resistance to the IGF-1R blockade; however, the combination of IGF-1R and STAT3 inhibitors significantly increased sensitivity against the resistance to IGF-1R (Lee et al., 2015; Santoro et al., 2017).
Janus Kinase/STAT inhibitors have been investigated as monotherapy for the treatment of ES (Table 3). ViscumTT treatment, a whole mistletoe extract, significantly reduced tumor growth in ES cells in vivo through the JAK/STAT3 pathway and its downstream targets, including BIRC5 and Myc (Twardziok et al., 2016; Kleinsimon et al., 2018). AZD1480, a JAK1/2 inhibitor, exhibited antitumor activity by inhibiting the growth of ES cells and tumor formation in xenograft models via the suppression of the IL-6/JAK2/STAT3 signaling pathway. Furthermore, AZD1480 treatment significantly increased the survival of ES tumor-bearing xenograft mice (Yan et al., 2013). Another small molecule inhibitor, LY5, which inhibits homodimerization by blocking the phosphotyrosine site of the STAT3 SH2 domain, inhibited STAT3 activation by decreasing the cell viability of sarcomas, including osteosarcoma, ES, and RMS. LY5 treatment showed excellent oral bioavailability in both mice and dogs, with 69.5% bioavailability observed following intraperitoneal administration and 78.6% with oral administration. Although treatment with LY5 inhibits the activation of STAT3 in metastatic pulmonary lesions of sarcoma xenografts, it failed to suppress lung metastasis in vivo (Yu et al., 2017).
In cancer pathogenesis, SFK is essential for growth and metastasis (Kim et al., 2009) and its roles have been well established in cancer, with several studies reporting the functional role of SFK in the pathogenesis of ES. Although Src is highly expressed and activated in ES cells and ES patients, its expression demonstrated no significant correlation with histologic subtypes. Dasatinib, an Src inhibitor, induced apoptosis and reduced the invasiveness of ES cells by suppressing the downstream signaling pathways, such as FAK and p130CAS, via inhibition of Src activity (Figure 1; Shor et al., 2007). Using ES cells, phosphoproteomics has shown that SFK is an abundant phosphorylated tyrosine kinase, and dasatinib treatment markedly reduced cell viability in ES cells by inhibiting the activation of PDGFRα and Src (Bai et al., 2012). Furthermore, peptide-based tyrosine kinase array revealed that Src is highly expressed and phosphorylated in CIC-DUX4 sarcoma (CDS) patient-derived xenografts, which is a high-grade Ewing-like small round cell sarcoma (Oyama et al., 2017). Lyn, a member of SFK, was upregulated and activated in 86.7% of ES patients and ES cells (Table 1). However, the downregulation of SFK and treatment with AP23994, an SFK inhibitor, suppressed tumor growth, bone tumor lysis, and metastases in ES cells and the xenograft model (Guan et al., 2008). Microenvironmental stress, including growth constraints, serum deprivation, and hypoxia, induced the invasiveness of ES cells through the formation of invadopodia by Src activation, not through the induction of EMT markers and cell proliferation (Bailey et al., 2016).
Secretion of the extracellular matrix protein, tenascin C, upon microenvironmental stress, induced cell-cell interaction, and growth of ES cells by promoting invadopodia formation by Src activation. In contrast, dasatinib treatment suppressed the environmental stress-mediated invasiveness of ES cells by blocking invadopodia formation associated with Src activation and tenascin (Bailey et al., 2016; Hawkins et al., 2019). SI221, a new pyrazolo[3,4-d]pyrimidine SFK inhibitor, reduced cell proliferation and migration of ES cells by inhibiting NOTCH1 cleavage and activating P38 MAPK (Indovina et al., 2017). Additionally, dasatinib has been evaluated in phase 2 clinical trial in sarcoma patients (ClinicalTrials.gov Identifier: NCT00464620), revealing that the 6 month PFS rate was 48%, and more than 50% of patients with alveolar soft part sarcoma (ASPS) and epithelioid sarcoma showed disease-free progression for more than 6 months (Schuetze et al., 2017). Furthermore, dasatinib, in combination with other conventional agents (ifosfamide, carboplatin, and etoposide), ids currently under investigation in phases 1 and 2 trials for patients with solid tumors, including sarcoma (ClinicalTrials.gov Identifier: NCT00788125).
Focal adhesion kinase has been identified as a therapeutic target for cancer, inducing tumor growth, metastasis, acquisition of CSC traits, and drug resistance (Sulzmaier et al., 2014; Yoon et al., 2015). FAK is significantly upregulated in ES cells (Moritake et al., 2003) and highly activated in ES patients and ES cells. Loss of FAK and treatment with a FAK inhibitor (PF-562271) suppressed tumor growth and the invasion ability of ES cells in vivo and in vitro by suppressing Crkl-associated substrate (CAS) activity and the AKT/mTOR pathway (Crompton et al., 2013). EWS-FLI1-mediated upregulation of ezrin enhanced the viability and invasion of ES cells via FAK activation (Steinestel et al., 2020). Furthermore, the upregulation of ERBB4 in metastatic ES patients was closely associated with poor survival in ES patients, and ERBB4-mediated activation of the PI3K/AKT/FAK/RAC1 pathway promoted the metastatic capacity of ES cells (Mendoza-Naranjo et al., 2013).
Moreover, rapamycin treatment or loss of TORC1 and TORC2, as mTOR complexes, markedly reduced IGF-1-mediated activation of FAK in ES cells (Liu et al., 2008). Furthermore, aurora kinase B (AURKB) inhibitors have been identified as a synergistic drug in combination with FAK inhibitors by utilizing high-throughput drug combination screening, with AURKB highly expressed in ES patients and ES cells. AURKB inhibitor significantly suppressed cell viability, and the combination of the FAK and AURKB inhibitors demonstrated synergistic activity by inhibiting activation of the mTOR pathway (Wang et al., 2019).
Clinical activity of GSK2256098, a FAK inhibitor, has been evaluated in cancer patients. GSK2256098 monotherapy was well-tolerated in patients with relapsed glioblastoma, and SD was observed in 27% (3/11) of patients (ClinicalTrials.gov: NCT01138033) (Brown et al., 2018). In patients with mesothelioma or solid tumors, combination treatment with GSK2256098 and trametinib, a MEK inhibitor, demonstrated that 38% (13/34) of patients presented SD and 50% (3/6) patients demonstrated more than a 70% decrease in phosphorylated FAK/FAK (ClinicalTrials.gov Identifier: NCT01938443) (Mak et al., 2019). Another FAK inhibitor, defatinib (VS-6063), was well-tolerated to patients with solid tumors, with 33% (3/9) patients presenting SD (Shimizu et al., 2016). The combination of defatinib with RO5126766 (RAF/MEK inhibitor) or pembrolizumab (PD-1 blocking antibody) is currently under evaluation for patients with solid tumors (ClinicalTrials.gov Identifier: NCT03875820, NCT02546531, NCT02758587), but remains to be evaluated in ES patients.
Activation of ERK 1 and 2 by EWS/FLI-1 induces the transformation of cells. Moreover, a dominant-negative RAS and EWS/FLI-1 mutant, defective in DNA-binding and transcriptional activation, significantly decreased activation of ERK1/2 and transformation of cells (Silvany et al., 2000).
The C-terminal portion of proline-rich tyrosine kinase (PYK2), involved in the pathogenesis of many cancers, interacts with the N-terminal region (1-233 amino acid) of EWS and eventually regulates G-protein-coupled receptor signaling (Felsch et al., 1999). Additionally, suppression of PYK2 activity inhibited the AKT/mTOR pathway (Crompton et al., 2013).
Previous studies have demonstrated that activation of spleen tyrosine kinase (SYK) increases the risk of chemotherapy failure or relapse of B cell acute lymphoblastic leukemia (B-ALL) (Perova et al., 2014), and its inhibitors, PRT318 and P505-15, inhibited the survival and migration of chronic lymphocytic leukemia (CLL) by blocking the activation of chemokine receptors (CXCR4, CXCR5) and SYK (Hoellenriegel et al., 2012). SYK was identified as a clinical target through a high-throughput approach using a siRNA library and a small-molecule library, and was reportedly activated in 40% of ES patients (Table 1). SYK enhanced cell survival and tumor growth in xenografts. Additionally, MALAT1, a non-coding RNA, transcriptionally activated through SYK/c-MYC pathway, enhanced the malignant traits of ES cells; however, its inhibitors suppressed cell viability and tumor formation in ES cells in vivo and in vitro via the inhibition of the AKT pathway (Figure 1; Sun et al., 2017).
Recent studies have demonstrated that activation of several tyrosine kinases, identified as potential therapeutic targets, enhances tumor growth and metastasis, developing chemoresistance in ES cells through the activation of various intracellular downstream signaling pathways. Moreover, drugs targeting specific tyrosine kinases have been developed and approved. Although clinical observations and preclinical trials have reported that monotherapy, as well as combination therapy with such inhibitors, can efficiently combat ES, further investigations with novel multifaceted therapeutic strategies are required. IGF-1R directed therapies have demonstrated only modest efficacy in ES patients; monotherapy and combination therapy with tyrosine kinase inhibitors remain under evaluation. Additionally, the inhibition of kinase activity in ES reduced the sensitivity to the tyrosine kinase inhibitor by developing resistance. Cancer cells acquire drug resistance via mutations that abrogate inhibitor binding. These mutations upregulate oncogenes and activate alternative kinase pathways via compensatory cross-signaling through the tyrosine kinase-induced activation of target networks. The subsequent development of therapeutic resistance to cancer increases the evasion of tyrosine kinase inhibitors (Zhang et al., 2009). Additional mutations on resistant mutants, to inhibit the developed drug resistance, can increase their sensitivity to cancer cells and prolong patient survival (Ferguson and Gray, 2018). Several strategies have been proposed to overcome drug-resistant mutations by developing inhibitors such as nilotinib and dasatinib, which possess increased affinity to the ATP binding site and can recognize active conformations of tyrosine kinase. These two compounds have high cellular activity and inhibit all known BCR-ABL1 mutants (Weisberg et al., 2005; Karaman et al., 2008; Zhang et al., 2009; Ferguson and Gray, 2018).
Adoptive cell therapy and a combination of tyrosine kinase targeting agents, along with other conventional agents, may provide novel options to cancer patients to overcome drug resistance and optimize their efficacy and cytotoxicity. The FDA has approved axicabtagene ciloleucel and tisagenlecleucel as CD19 targeting CAR-T cell immunotherapy in patients with B cell lymphoma and acute lymphoblastic leukemia; these agents have shown a safe and improved clinical outcome in the treatment of cancer patients (Depil et al., 2020). Currently, CAT-T cell immunotherapy is under evaluation in clinical trials for ES patients presenting sarcoma-specific surface antigens targeting next-generation CAR-T (NCT03356782), GD2-CAR-T (NCT03635632), and EGFR806-CAR-T immunotherapy (NCT03618381). Additionally, histone deacetylase (HDAC) inhibitors, in combination with other conventional agents, have exhibited anticancer effects in preclinical and clinical studies (Suraweera et al., 2018). Vorinostat, in combination with vincristine, irinotecan, and temozolomide, is now under evaluation for ES patients (ClinicalTrials.gov Identifier: NCT04308330). Moreover, significant advances in understanding the functioning of tyrosine kinases in cancer, including identification of molecular mechanisms and the association between tyrosine kinases and the pathogenesis of ES will provide new insights for the selection of novel targets and to further overcome challenges associated with existing treatment strategies for ES. Furthermore, understanding the tumor-initiating abilities of tyrosine kinases and developing strategies to maintain this crucial property will increase success rates and improve outcomes in cancer therapy.
WJ designed, reviewed, and wrote the manuscript.
This work supported by a National Research Foundation of Korea grant (2020R1A2C1012484 to WJ).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ackermann, M., Morse, B. A., Delventhal, V., Carvajal, I. M., and Konerding, M. A. (2012). Anti-VEGFR2 and anti-IGF-1R-Adnectins inhibit Ewing’s sarcoma A673-xenograft growth and normalize tumor vascular architecture. Angiogenesis 15, 685–695. doi: 10.1007/s10456-012-9294-9
Agelopoulos, K., Richter, G. H., Schmidt, E., Dirksen, U., von Heyking, K., Moser, B., et al. (2015). Deep sequencing in conjunction with expression and functional analyses reveals activation of FGFR1 in Ewing sarcoma. Clin. Cancer Res. 21, 4935–4946. doi: 10.1158/1078-0432.ccr-14-2744
Ahmed, A., Gilbert-Barness, E., and Lacson, A. (2004). Expression of c-kit in Ewing family of tumors: a comparison of different immunohistochemical protocols. Pediatr. Dev. Pathol. 7, 342–347. doi: 10.1007/s10024-002-0077-y
Ahmed, N., Brawley, V. S., Hegde, M., Robertson, C., Ghazi, A., Gerken, C., et al. (2015). Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 33, 1688–1696. doi: 10.1200/jco.2014.58.0225
Ahmed, N., Ratnayake, M., Savoldo, B., Perlaky, L., Dotti, G., Wels, W. S., et al. (2007). Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 67, 5957–5964. doi: 10.1158/0008-5472.can-06-4309
Ahrens, S., Hoffmann, C., Jabar, S., Braun-Munzinger, G., Paulussen, M., Dunst, J., et al. (1999). Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: the CESS 86 experience. Cooperative ewing sarcoma study. Med. Pediatr. Oncol. 32, 186–195. doi: 10.1002/(sici)1096-911x(199903)32:3<186::aid-mpo5>3.0.co;2-d
Aiken, R., Axelson, M., Harmenberg, J., Klockare, M., Larsson, O., and Wassberg, C. (2017). Phase I clinical trial of AXL1717 for treatment of relapsed malignant astrocytomas: analysis of dose and response. Oncotarget 8, 81501–81510. doi: 10.18632/oncotarget.20662
Amaral, A. T., Garofalo, C., Frapolli, R., Manara, M. C., Mancarella, C., Uboldi, S., et al. (2015). Trabectedin efficacy in ewing sarcoma is greatly increased by combination with anti-IGF signaling agents. Clin. Cancer Res. 21, 1373–1382. doi: 10.1158/1078-0432.ccr-14-1688
Ambati, S. R., Lopes, E. C., Kosugi, K., Mony, U., Zehir, A., Shah, S. K., et al. (2014). Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol. Oncol. 8, 323–336. doi: 10.1016/j.molonc.2013.12.005
Ambros, I. M., Ambros, P. F., Strehl, S., Kovar, H., Gadner, H., and Salzerkuntschik, M. (1991). Mic2 Is a specific marker for ewings-sarcoma and peripheral primitive neuroectodermal tumors - evidence for a common histogenesis of ewings-sarcoma and peripheral primitive neuroectodermal tumors from mic2 expression and specific chromosome aberration. Cancer 67, 1886–1893. doi: 10.1002/1097-0142(19910401)67:7<1886::aid-cncr2820670712>3.0.co;2-u
Apelbaum, A., Yarden, G., Warszawski, S., Harari, D., and Schreiber, G. (2013). Type I interferons induce apoptosis by balancing cFLIP and caspase-8 independent of death ligands. Mol. Cell Biol. 33, 800–814. doi: 10.1128/mcb.01430-12
Asmane, E., Alberti, W. L., Duc, A., Marec-Berard, P., Ray-Coquard, I., Cassier, P., et al. (2012). Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: a predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur. J. Cancer 48, 3027–3035. doi: 10.1016/j.ejca.2012.05.009
Babina, I. S., and Turner, N. C. (2017). Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 17, 318–332. doi: 10.1038/nrc.2017.8
Bai, Y., Li, J., Fang, B., Edwards, A., Zhang, G., Bui, M., et al. (2012). Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer Res. 72, 2501–2511. doi: 10.1158/0008-5472.can-11-3015
Bailey, K. M., Airik, M., Krook, M. A., Pedersen, E. A., and Lawlor, E. R. (2016). Micro-environmental stress induces Src-dependent activation of invadopodia and cell migration in ewing sarcoma. Neoplasia 18, 480–488. doi: 10.1016/j.neo.2016.06.008
Barone, A., Chi, D. C., Theoret, M. R., Chen, H. Y., He, K., Kufrin, D., et al. (2017). FDA approval summary: trabectedin for unresectable or metastatic liposarcoma or leiomyosarcoma following an anthracycline-containing regimen. Clin. Cancer Res. 23, 7448–7453. doi: 10.1158/1078-0432.ccr-17-0898
Baselga, J., and Swain, S. M. (2009). Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 9, 463–475. doi: 10.1038/nrc2656
Behjati, S., Basu, B. P., Wallace, R., Bier, N., Sebire, N., Hasan, F., et al. (2012). STAT3 regulates proliferation and immunogenicity of the ewing family of tumors in Vitro. Sarcoma 2012:987239.
Benini, S., Manara, M. C., Cerisano, V., Perdichizzi, S., Strammiello, R., Serra, M., et al. (2004). Contribution of MEK/MAPK and PI3-K signaling pathway to the malignant behavior of Ewing’s sarcoma cells: therapeutic prospects. Int. J. Cancer 108, 358–366. doi: 10.1002/ijc.11576
Berghuis, D., Schilham, M. W., Santos, S. J., Savola, S., Knowles, H. J., Dirksen, U., et al. (2012). The CXCR4-CXCL12 axis in Ewing sarcoma: promotion of tumor growth rather than metastatic disease. Clin. Sarcoma Res. 2:24. doi: 10.1186/2045-3329-2-24
Bernat-Peguera, A., Simon-Extremera, P., da Silva-Diz, M. L., de Munain, L., Gonzalez-Suarez, E., Sidelnikova, D. P., et al. (2019). PDGFR-induced autocrine SDF-1 signaling in cancer cells promotes metastasis in advanced skin carcinoma. Oncogene 38, 5021–5037. doi: 10.1038/s41388-019-0773-y
Bonin, G., Scamps, C., Turc-Carel, C., and Lipinski, M. (1993). Chimeric EWS-FLI1 transcript in a Ewing cell line with a complex t(11;22;14) translocation. Cancer Res. 53, 3655–3657.
Borden, E. C. (2019). Interferons alpha and beta in cancer: therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 18, 219–234. doi: 10.1038/s41573-018-0011-2
Boro, K., Pretre, F., Rechfeld Thalhammer, V., Oesch, S., Wachtel, M., Schafer, B. W., et al. (2012). Small-molecule screen identifies modulators of EWS/FLI1 target gene expression and cell survival in Ewing’s sarcoma. Int. J. Cancer 131, 2153–2164. doi: 10.1002/ijc.27472
Brown, N. F., Williams, M., Arkenau, H. T., Fleming, R. A., Tolson, J., Yan, L., et al. (2018). A study of the focal adhesion kinase inhibitor GSK2256098 in patients with recurrent glioblastoma with evaluation of tumor penetration of [11C]GSK2256098. Neurol. Oncol. 20, 1634–1642. doi: 10.1093/neuonc/noy078
Casaletto, J. B., and McClatchey, A. I. (2012). Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 12, 387–400. doi: 10.1038/nrc3277
Chao, J., Budd, G. T., Chu, P., Frankel, P., Garcia, D., Junqueira, M., et al. (2010). Phase II clinical trial of imatinib mesylate in therapy of KIT and/or PDGFRalpha-expressing Ewing sarcoma family of tumors and desmoplastic small round cell tumors. Anticancer Res. 30, 547–552.
Charan, M., Dravid, P., Cam, M., Audino, A., Gross, A. C., Arnold, M. A., et al. (2019). GD2-directed CAR-T cells in combination with HGF-targeted neutralizing antibody (AMG102) prevent primary tumor growth and metastasis in Ewing sarcoma. Int. J. Cancer 146, 3184–3195. doi: 10.1002/ijc.32743
Chawla-Sarkar, M., Leaman, D. W., and Borden, E. C. (2001). Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha2: correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin. Cancer Res. 7, 1821–1831.
Chugh, R., Wathen, J. K., Maki, R. G., Benjamin, R. S., Patel, S. R., Myers, P. A., et al. (2009). Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J. Clin. Oncol. 27, 3148–3153. doi: 10.1200/jco.2008.20.5054
Cidre-Aranaz, F., Grunewald, T. G., Surdez, D., Garcia-Garcia, L., Carlos Lazaro, J., Kirchner, T., et al. (2017). EWS-FLI1-mediated suppression of the RAS-antagonist Sprouty 1 (SPRY1) confers aggressiveness to Ewing sarcoma. Oncogene 36, 766–776. doi: 10.1038/onc.2016.244
Collin, M. P., Lobell, M., Hubsch, W., Brohm, D., Schirok, H., Jautelat, R., et al. (2018). Discovery of Rogaratinib (BAY 1163877): a pan-FGFR Inhibitor. Chemmedchem 13, 437–445. doi: 10.1002/cmdc.201700718
Crompton, B. D., Carlton, A. L., Thorner, A. R., Christie, A. L., Du, J., Calicchio, M. L., et al. (2013). High-throughput tyrosine kinase activity profiling identifies FAK as a candidate therapeutic target in Ewing sarcoma. Cancer Res. 73, 2873–2883. doi: 10.1158/0008-5472.can-12-1944
Dasgupta, A., Trucco, M., Rainusso, N., Bernardi, R. J., Shuck, R., Kurenbekova, L., et al. (2017). Metabolic modulation of ewing sarcoma cells inhibits tumor growth and stem cell properties. Oncotarget 8, 77292–77308. doi: 10.18632/oncotarget.20467
Davenport, J. R., Vo, K. T., Goldsby, R., West, D. C., and DuBois, S. G. (2016). Conditional survival and predictors of late death in patients with ewing sarcoma. Pediatr. Blood Cancer 63, 1091–1095. doi: 10.1002/pbc.25945
Daver, N., Schlenk, R. F., Russell, N. H., and Levis, M. J. (2019). Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia 33, 299–312. doi: 10.1038/s41375-018-0357-9
David, E., Tirode, F., Baud’huin, M., Guihard, P., Laud, K., Delattre, O., et al. (2012). Oncostatin M is a growth factor for ewing sarcoma. Am. J. Pathol. 181, 1782–1795. doi: 10.1016/j.ajpath.2012.07.023
Dedoni, S., Olianas, M. C., and Onali, P. (2010). Interferon-beta induces apoptosis in human SH-SY5Y neuroblastoma cells through activation of JAK-STAT signaling and down-regulation of PI3K/Akt pathway. J. Neurochem. 115, 1421–1433. doi: 10.1111/j.1471-4159.2010.07046.x
Delattre, O., Zucman, J., Melot, T., Garau, X. S., Zucker, J. M., Lenoir, G. M., et al. (1994). The Ewing family of tumors–a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N. Engl. J. Med. 331, 294–299. doi: 10.1056/nejm199408043310503
Delattre, O., Zucman, J., Plougastel, B., Desmaze, C., Melot, T., Peter, M., et al. (1992). Gene fusion with an ETS DNA-binding domain caused by chromosome-translocation in human tumors. Nature 359, 162–165. doi: 10.1038/359162a0
Depil, S., Duchateau, P., Grupp, S. A., Mufti, G., and Poirot, L. (2020). ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 19, 185–199. doi: 10.1038/s41573-019-0051-2
Di Conza, G., Buttarelli, M., Monti, O., Pellegrino, M., Mancini, F., Pontecorvi, A., et al. (2012). IGF-1R/MDM2 relationship confers enhanced sensitivity to RITA in Ewing sarcoma cells. Mol. Cancer Ther. 11, 1247–1256. doi: 10.1158/1535-7163.mct-11-0913
Dirks, W. G., Fahnrich, S., Lis, Y., Becker, E., MacLeod, R. A. F., and Drexler, H. G. (2002). Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Intern. J. Cancer 100, 49–56. doi: 10.1002/ijc.10435
Do, I., Araujo, E. S., Kalil, R. K., Bacchini, P., Bertoni, F., Unni, K. K., et al. (2007). Protein expression of KIT and gene mutation of c-kit and PDGFRs in Ewing sarcomas. Pathol. Res. Pract. 203, 127–134. doi: 10.1016/j.prp.2006.12.005
Doherty, M. R., and Jackson, M. W. (2018). The critical, clinical role of interferon-beta in regulating cancer stem cell properties in triple-negative breast cancer. DNA Cell Biol. 37, 513–516. doi: 10.1089/dna.2018.4247
Dongre, A., and Weinberg, R. A. (2019). New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84. doi: 10.1038/s41580-018-0080-4
Du, Z., and Lovly, C. M. (2018). Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 17:58.
DuBois, S. G., Krailo, M. D., Gebhardt, M. C., Donaldson, S. S., Marcus, K. J., Dormans, J., et al. (2015). Comparative evaluation of local control strategies in localized ewing sarcoma of bone a report from the children’s oncology group. Cancer 121, 467–475. doi: 10.1002/cncr.29065
Duplaquet, L., Kherrouche, Z., Baldacci, S., Jamme, P., Cortot, A. B., Copin, M. C., et al. (2018). The multiple paths towards MET receptor addiction in cancer. Oncogene 37, 3200–3215. doi: 10.1038/s41388-018-0185-4
Engelman, J. A., and Cantley, L. C. (2010). Chemoprevention meets glucose control. Cancer Prev. Res. 3, 1049–1052. doi: 10.1158/1940-6207.capr-10-0178
English, C., and Aloi, J. J. (2015). New FDA-approved disease-modifying therapies for multiple sclerosis. Clin. Ther. 37, 691–715. doi: 10.1016/j.clinthera.2015.03.001
Esparza-Lopez, J., Alvarado-Munoz, J. F., Escobar-Arriaga, E., Ulloa-Aguirre, A., and de Jesus Ibarra-Sanchez, M. (2019). Metformin reverses mesenchymal phenotype of primary breast cancer cells through STAT3/NF-kappaB pathways. BMC Cancer 19:728. doi: 10.1186/s12885-019-5945-1
Felsch, J. S., Lane, W. S., and Peralta, E. G. (1999). Tyrosine kinase Pyk2 mediates G-protein-coupled receptor regulation of the ewing sarcoma RNA-binding protein EWS. Curr. Biol. 9, 485–488.
Feng, Y., Ke, C., Tang, Q., Dong, H., Zheng, X., Lin, W., et al. (2014). Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Death Dis. 5:e1088. doi: 10.1038/cddis.2014.59
Ferguson, F. M., and Gray, N. S. (2018). Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov. 17, 353–377. doi: 10.1038/nrd.2018.21
Fleuren, E. D. G., Hillebrandt-Roeffen, M. H. S., and Flucke, U. E. (2014). The role of AXL and the in vitro activity of the receptor tyrosine kinase inhibitor BGB324 in Ewing sarcoma. Oncotarget 5, 12753–12768. doi: 10.18632/oncotarget.2648
Fleuren, E. D. G., Roeffen, M. H. S., Leenders, W. P., Flucke, U. E., Vlenterie, M., Schreuder, H. W., et al. (2013). Expression and clinical relevance of MET and ALK in Ewing sarcomas. Intern. J. Cancer 133, 427–436. doi: 10.1002/ijc.28047
Fleuren, E. D. G., Vlenterie, M., van der Graaf, W. T. A., Hillebrandt-Roeffen, M. H. S., Blackburn, J., Ma, X. Q., et al. (2017). Phosphoproteomic profiling reveals ALK and MET as novel actionable targets across synovial sarcoma subtypes. Cancer Res. 77, 4279–4292. doi: 10.1158/0008-5472.can-16-2550
Foretz, M., Guigas, B., and Viollet, B. (2019). Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 15, 569–589. doi: 10.1038/s41574-019-0242-2
Garcia-Monclus, S., Lopez-Alemany, R., Almacellas-Rabaiget, O., Herrero-Martin, D., Huertas-Martinez, J., Lagares-Tena, L., et al. (2018). EphA2 receptor is a key player in the metastatic onset of Ewing sarcoma. Int. J. Cancer 143, 1188–1201. doi: 10.1002/ijc.31405
Gaspar, N., Hawkins, D. S., Dirksen, U., Lewis, I. J., Ferrari, S., Le Deley, M. C., et al. (2015). Ewing sarcoma: current management and future approaches through collaboration. J. Clin. Oncol. 33, 3036–3046.
Gay, C. M., Balaji, K., and Byers, L. A. (2017). Giving AXL the axe: targeting AXL in human malignancy. Br. J. Cancer 116, 415–423. doi: 10.1038/bjc.2016.428
Geller, J. I., Fox, E., Turpin, B. K., Goldstein, S. L., Liu, X., Minard, C. G., et al. (2018). A study of axitinib, a VEGF receptor tyrosine kinase inhibitor, in children and adolescents with recurrent or refractory solid tumors: a Children’s oncology group phase 1 and pilot consortium trial (ADVL1315). Cancer 124, 4548–4555. doi: 10.1002/cncr.31725
Giorgi, C., Boro, A., Rechfeld, F., Lopez-Garcia, L. A., Gierisch, M., Schafer, B. W., et al. (2015). PI3K/AKT signaling modulates transcriptional expression of EWS/FLI1 through specificity protein 1. Oncotarget 6, 28895–28910. doi: 10.18632/oncotarget.5000
Girnita, L., Girnita, A., Wang, M., Meis-Kindblom, J. M., Kindblom, L. G., and Larsson, O. (2000a). A link between basic fibroblast growth factor (bFGF) and EWS/FLI-1 in Ewing’s sarcoma cells. Oncogene 19, 4298–4301. doi: 10.1038/sj.onc.1203755
Girnita, L., Wang, M., Xie, Y. T., Nilsson, G., Dricu, A., Wejde, J., et al. (2000b). Inhibition of N-linked glycosylation down-regulates insulin-like growth factor-1 receptor at the cell surface and kills Ewing’s sarcoma cells: therapeutic implications. Anti Cancer Drug Design. 15, 67–72.
Gordon, E. M., Sankhala, K. K., Chawla, N., and Chawla, S. P. (2016). Trabectedin for soft tissue sarcoma: current status and future perspectives. Adv. Ther. 33, 1055–1071. doi: 10.1007/s12325-016-0344-3
Grier, H. E. (1997). The Ewing family of tumors. Ewing’s sarcoma and primitive neuroectodermal tumors. Pediatr. Clin. North. Am. 44, 991–1004.
Grunewald, S., Politz, O., Bender, S., Heroult, M., Lustig, K., Thuss, U., et al. (2019). Rogaratinib: A potent and selective pan-FGFR inhibitor with broad antitumor activity in FGFR-overexpressing preclinical cancer models. Int. J. Cancer 145, 1346–1357. doi: 10.1002/ijc.32224
Grunewald, T. G. P., Cidre-Aranaz, F., Surdez, D., Tomazou, E. M., de Alava, E., Kovar, H., et al. (2018). Ewing sarcoma. Nat. Rev. Dis. Prim. 4:5.
Guan, H., Zhou, Z. C., Gallick, G. E., Jia, S. F., Morales, J., Sood, A. K., et al. (2008). Targeting Lyn inhibits tumor growth and metastasis in Ewing’s sarcoma. Mol. Cancer Therap. 7, 1807–1816. doi: 10.1158/1535-7163.mct-08-0058
Guenther, L. M., Dharia, N. V., Ross, L., Conway, A., Robichaud, A. L., Catlett, J. L., et al. (2019). A combination CDK4/6 and IGF1R inhibitor strategy for ewing sarcoma. Clin. Cancer Res. 25, 1343–1357. doi: 10.1158/1078-0432.ccr-18-0372
Hallberg, B., and Palmer, R. H. (2013). Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 13, 685–700. doi: 10.1038/nrc3580
Hamdan, R., Zhou, Z., and Kleinerman, E. S. (2014). Blocking SDF-1alpha/CXCR4 downregulates PDGF-B and inhibits bone marrow-derived pericyte differentiation and tumor vascular expansion in Ewing tumors. Mol. Cancer Ther. 13, 483–491. doi: 10.1158/1535-7163.mct-13-0447
Harrison, D. J., Gill, J. B., Roth, M., Zhang, W. D., Teicher, B., Erickson, S. W., et al. (2019). Evaluation of the multi-kinase inhibitor regorafenib in the pediatric preclinical testing consortium osteosarcoma, rhabdomyosarcoma, and ewing sarcoma in vivo models. J. Clin. Oncol. 37, 10038–10038. doi: 10.1200/jco.2019.37.15_suppl.10038
Hawkins, A. G., Julian, C. M., Konzen, S., Treichel, S., Lawlor, E. R., and Bailey, K. M. (2019). Microenvironmental factors drive tenascin C and Src cooperation to promote invadopodia formation in ewing sarcoma. Neoplasia 21, 1063–1072. doi: 10.1016/j.neo.2019.08.007
Helsten, T., Elkin, S., Arthur, E., Tomson, B. N., Carter, J., and Kurzrock, R. (2016). The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin. Cancer Res. 22, 259–267. doi: 10.1158/1078-0432.ccr-14-3212
Hoellenriegel, J., Coffey, G. P., Sinha, U., Pandey, A., Sivina, M., Ferrajoli, A., et al. (2012). Selective, novel spleen tyrosine kinase (Syk) inhibitors suppress chronic lymphocytic leukemia B-cell activation and migration. Leukemia 26, 1576–1583. doi: 10.1038/leu.2012.24
Huang, H. J., Angelo, L. S., Rodon, J., Sun, M., Kuenkele, K. P., Parsons, H. A., et al. (2011). R1507, an anti-insulin-like growth factor-1 receptor (IGF-1R) antibody, and EWS/FLI-1 siRNA in Ewing’s sarcoma: convergence at the IGF/IGFR/Akt axis. PLoS One 6:e26060. doi: 10.1371/journal.pone.026060
Huang, X., Park, H., Greene, J., Pao, J., Mulvey, E., Zhou, S. X., et al. (2015). IGF1R-and ROR1-Specific CAR T cells as a potential therapy for high risk sarcomas. PLoS One 10:e0133152. doi: 10.1371/journal.pone.0133152
Ikeda, A. K., Judelson, D. R., Federman, N., Glaser, K. B., Landaw, E. M., Denny, C. T., et al. (2010). ABT-869 inhibits the proliferation of ewing sarcoma cells and suppresses platelet-derived growth factor receptor beta and c-KIT signaling pathways. Mol. Cancer Ther. 9, 653–660. doi: 10.1158/1535-7163.mct-09-0812
Indovina, P., Casini, N., Forte, I. M., Garofano, T., Cesari, D., Iannuzzi, C. A., et al. (2017). SRC family kinase inhibition in ewing sarcoma cells induces p38 MAP kinase-mediated cytotoxicity and reduces cell migration. J. Cell Physiol. 232, 129–135. doi: 10.1002/jcp.25397
Italiano, A., Penel, N., Toulmonde, M., Bompas, E., Piperno-Neumann, S., Pulido, M., et al. (2018). Cabozantinib in patients with advanced Osteosarcomas and ewing sarcomas: a french sarcoma Group (FSG)/US national cancer institute phase Ii collaborative study. Ann. Oncol. 29:82.
Jamitzky, S., Krueger, A. C., Janneschuetz, S., Piepke, S., Kailayangiri, S., Spurny, C., et al. (2015). Insulin-Like growth factor-1 receptor (IGF-1R) inhibition promotes expansion of human NK cells which maintain their potent antitumor activity against ewing sarcoma cells. Pediatr. Blood Cancer 62, 1979–1985. doi: 10.1002/pbc.25619
Jiang, Y. Y., Janku, F., Subbiah, V., Angelo, L. S., Naing, A., Anderson, P. M., et al. (2013). Germline PTPRD mutations in ewing sarcoma: biologic and clinical implications. Oncotarget 4, 884–889. doi: 10.18632/oncotarget.1021
Jiang, Y. Y., Subbiah, V., Janku, F., Ludwig, J. A., Naing, A., Benjamin, R. S., et al. (2014). Novel Secondary somatic mutations in ewing’s sarcoma and desmoplastic small round cell tumors. PLoS One 9:e93676. doi: 10.1371/journal.pone.0093676
Jiao, Q. L., Bi, L., Ren, Y. D., Song, S. L., Wang, Q., and Wang, Y. S. (2018). Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 17:36.
Jones, D. T., Hutter, B., Jager, N., Korshunov, A., Kool, M., Warnatz, H. J., et al. (2013). Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 45, 927–932.
Jones, S. A., and Jenkins, B. J. (2018). Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 18, 773–789. doi: 10.1038/s41577-018-0066-7
Juergens, C., Weston, I. L., Whelan, J., Paulussen, M., Oberlin, O., Michon, J., et al. (2006). Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr. Blood Cancer 47, 22–29. doi: 10.1002/pbc.20820
Juergens, H., Daw, N. C., Geoerger, B., Ferrari, S., Villarroel, M., Aerts, I., et al. (2011). Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. 29, 4534–4540. doi: 10.1200/jco.2010.33.0670
Kamura, S., Matsumoto, Y., Fukushi, J. I., Fujiwara, T., Iida, K., Okada, Y., et al. (2010). Basic fibroblast growth factor in the bone microenvironment enhances cell motility and invasion of Ewing’s sarcoma family of tumours by activating the FGFR1-PI3K-Rac1 pathway. Br. J. Cancer 103, 370–381. doi: 10.1038/sj.bjc.6605775
Kang, H. G., Jenabi, J. M., Liu, X. F., Reynolds, C. P., Triche, T. J., and Sorensen, P. H. (2010). Inhibition of the insulin-like growth factor I receptor by epigallocatechin gallate blocks proliferation and induces the death of Ewing tumor cells. Mol. Cancer Ther. 9, 1396–1407. doi: 10.1158/1535-7163.mct-09-0604
Karaman, M. W., Herrgard, S., Treiber, D. K., Gallant, P., Atteridge, C. E., Campbell, B. T., et al. (2008). A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132.
Karnieli, E., Werner, H., Rauscher, F. J., Benjamin, L. E., and LeRoith, D. (1996). The IGF-I receptor gene promoter is a molecular target for the Ewing’s sarcoma Wilms’ tumor 1 fusion protein. J. Biol. Chem. 271, 19304–19309. doi: 10.1074/jbc.271.32.19304
Katoh, M. (2019). Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 16, 105–122. doi: 10.1038/s41571-018-0115-y
Kim, L. C., Song, L. X., and Haura, E. B. (2009). Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 6, 587–595. doi: 10.1038/nrclinonc.2009.129
Kleinsimon, S., Longmuss, E., Rolff, J., Jager, S., Eggert, A., Delebinski, C., et al. (2018). GADD45A and CDKN1A are involved in apoptosis and cell cycle modulatory effects of viscumTT with further inactivation of the STAT3 pathway. Sci. Rep. 8:750.
Kolb, E. A., Gorlick, R., Maris, J. M., Keir, S. T., Morton, C. L., Wu, J. R., et al. (2012). Combination testing (Stage 2) of the Anti-IGF-1 receptor antibody IMC-A12 with rapamycin by the pediatric preclinical testing program. Pediatr. Blood Cancer 58, 729–735. doi: 10.1002/pbc.23157
Krishnaswamy, S., Kanteti, R., Duke-Cohan, J. S., Loganathan, S., Liu, W. Q., Ma, P. C., et al. (2009). Ethnic differences and functional analysis of MET mutations in lung cancer. Clin. Cancer Res. 15, 5714–5723. doi: 10.1158/1078-0432.ccr-09-0070
Kypta, R. M., Goldberg, Y., Ulug, E. T., and Courtneidge, S. A. (1990). Association between the Pdgf receptor and members of the Src family of tyrosine kinases. Cell 62, 481–492. doi: 10.1016/0092-8674(90)90013-5
Lai, R., Navid, F., Rodriguez-Galindo, C., Liu, T., Fuller, C. E., Ganti, R., et al. (2006). STAT3 is activated in a subset of the Ewing sarcoma family of tumours. J. Pathol. 208, 624–632. doi: 10.1002/path.1941
Lamhamedi-Cherradi, S. E., Menegaz, B. A., Ramamoorthy, V., Vishwamitra, D., Wang, Y., Maywald, R. L., et al. (2016). IGF-1R and mTOR blockade: novel resistance mechanisms and synergistic drug combinations for ewing sarcoma. JNCI J. Natl. Cancer I 108:djw182. doi: 10.1093/jnci/djw182
Lampugnani, M. G., Zanetti, A., Corada, M., Takahashi, T., Balconi, G., Breviario, F., et al. (2003). Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 161, 793–804. doi: 10.1083/jcb.200209019
Landuzzi, L., De Giovanni, C., Nicoletti, G., Rossi, I., Ricci, C., Astolfi, A., et al. (2000). The metastatic ability of Ewing’s sarcoma cells is modulated by stem cell factor and by its receptor c-kit. Am. J. Pathol. 157, 2123–2131. doi: 10.1016/s0002-9440(10)64850-x
Lee, J. S., Kang, J. H., Boo, H. J., Hwang, S. J., Hong, S., Lee, S. C., et al. (2015). STAT3-mediated IGF-2 secretion in the tumour microenvironment elicits innate resistance to anti-IGF-1R antibody. Nat. Commun. 6:8499.
Lee, S. Y., Lim, S., and Cho, D. H. (2018). Personalized genomic analysis based on circulating tumor cells of extra-skeletal Ewing sarcoma of the uterus: a case report of a 16-year-old Korean female. Exp. Ther. Med. 16, 1343–1349.
Lee, T. H., Bolontrade, M. F., Worth, L. L., Guan, H., Ellis, L. M., and Kleinerman, E. S. (2006). Production of VEGF165 by Ewing’s sarcoma cells induces vasculogenesis and the incorporation of CD34+ stem cells into the expanding tumor vasculature. Int. J. Cancer 119, 839–846. doi: 10.1002/ijc.21916
Lew, E. D., Furdui, C. M., Anderson, K. S., and Schlessinger, J. (2009). The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci. Signal. 2:ra6. doi: 10.1126/scisignal.2000021
Li, C., Shen, Y., Ren, Y., Liu, W., Li, M., Liang, W., et al. (2016). Oncogene mutation profiling reveals poor prognosis associated with FGFR1/3 mutation in liposarcoma. Hum. Pathol. 55, 143–150. doi: 10.1016/j.humpath.2016.05.006
Li, H. M., Batth, I. S., Qu, X. J., Xu, L., Song, N., Wang, R. Y., et al. (2017). IGF-IR signaling in epithelial to mesenchymal transition and targeting IGF-IR therapy: overview and new insights. Mol. Cancer 16:6.
Lin, C. C., Yeh, H. H., Huang, W. L., Yan, J. J., Lai, W. W., Su, W. P., et al. (2013). Metformin enhances cisplatin cytotoxicity by suppressing signal transducer and activator of transcription-3 activity independently of the liver kinase B1-AMP-activated protein kinase pathway. Am. J. Resp. Cell. Mol. 49, 241–250. doi: 10.1165/rcmb.2012-0244oc
Liu, L., Chen, L., Chung, J., and Huang, S. (2008). Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene 27, 4998–5010. doi: 10.1038/onc.2008.137
Liu, S. Y., Meric-Bernstam, F., Parinyanitikul, N., Wang, B. L., Eterovic, A. K., Zheng, X. F., et al. (2015). Functional consequence of the MET-T1010I polymorphism in breast cancer. Oncotarget 6, 2604–2614. doi: 10.18632/oncotarget.3094
Llombart-Bosch, I., Machado, S. N., Bertoni, F., Bacchini, P., Alberghini, M., Karzeladze, A., et al. (2009). Histological heterogeneity of Ewing’s sarcoma/PNET: an immunohistochemical analysis of 415 genetically confirmed cases with clinical support. Virch. Archiv. 455, 397–411. doi: 10.1007/s00428-009-0842-7
Loganathan, S. N., Tang, N., Holler, A. E., Wang, N. H., and Wang, J. L. (2019). Targeting the IGF1R/PI3K/AKT pathway sensitizes ewing sarcoma to BET bromodomain inhibitors. Mol. Cancer Ther. 18, 929–936. doi: 10.1158/1535-7163.mct-18-1151
Long, A. H., Highfill, S. L., Cui, Y., Smith, J. P., Walker, A. J., Ramakrishna, S., et al. (2016). Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol. Res. 4, 869–880. doi: 10.1158/2326-6066.cir-15-0230
Longhi, S., Ferrari, A. T., Luksch, R., Fagioli, F., Bacci, G., and Ferrari, C. (2012). Late effects of chemotherapy and radiotherapy in osteosarcoma and ewing sarcoma patients: the Italian Sarcoma Group Experience (1983-2006). Cancer 118, 5050–5059. doi: 10.1002/cncr.27493
Lowery, C. D., Blosser, W., Dowless, M., Knoche, S., Stephens, J., Li, H., et al. (2018). Olaratumab exerts antitumor activity in preclinical models of pediatric bone and soft tissue tumors through inhibition of platelet-derived growth factor receptor alpha. Clin. Cancer Res. 24, 847–857. doi: 10.1158/1078-0432.ccr-17-1258
Ma, K., Pan, X., Fan, P., He, Y., Gu, J., Wang, W., et al. (2014). Loss of miR-638 in vitro promotes cell invasion and a mesenchymal-like transition by influencing SOX2 expression in colorectal carcinoma cells. Mol. Cancer 13:118. doi: 10.1186/1476-4598-13-118
Mak, G., Soria, J. C., Blagden, S. P., Plummer, R., Fleming, R. A., Nebot, N., et al. (2019). A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 120, 975–981. doi: 10.1038/s41416-019-0452-3
Martins, A. S., Ordonez, J. L., Amaral, A. T., Prins, F., Floris, G., Debiec-Rychter, M., et al. (2011). IGF1R Signaling in ewing sarcoma is shaped by clathrin-/caveolin-dependent endocytosis. PLoS One 6:e19846. doi: 10.1371/journal.pone.0019846
Mascarenhas, L., Felgenhauer, J. L., Bond, M. C., Villaluna, D., Femino, J. D., Laack, N. N., et al. (2016). Pilot study of adding vincristine, topotecan, and cyclophosphamide to interval-compressed chemotherapy in newly diagnosed patients with localized ewing sarcoma: a report from the children’s oncology group. Pediatr. Blood Cancer 63, 493–498. doi: 10.1002/pbc.25837
May, W. A., Gishizky, M. L., Lessnick, S. L., Lunsford, L. B., Lewis, B. C., Delattre, O., et al. (1993). Ewing sarcoma 11-22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by fli1 for transformation. Proc. Natl. Acad. Sci. U.S.A. 90, 5752–5756. doi: 10.1073/pnas.90.12.5752
Mendoza-Naranjo, A., El-Naggar, A., Wai, D. H., Mistry, P., Lazic, N., Ayala, F. R., et al. (2013). ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO Mol. Med. 5, 1087–1102.
Mendoza-Naranjo, E.-N. A., Wai, D. H., Mistry, P., Lazic, N., and Ayala, F. R. (2012). ErbB4 is a novel driver of metastasis and anoikis resistance in Ewing’s sarcoma. Proc. Annu. Meet. Am. Assoc. Cancer Res. 72 (8 Suppl.):431.
Merchant, M. S., Woo, C. W., Mackall, C. L., and Thiele, C. J. (2002). Potential use of imatinib in Ewing’s sarcoma: Evidence for in vitro and in vivo activity. J. Natl. Cancer I 94, 1673–1679. doi: 10.1093/jnci/94.22.1673
Moasser, M. M. (2007). The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 26, 6469–6487. doi: 10.1038/sj.onc.1210477
Montazeri, K., and Bellmunt, J. (2020). Erdafitinib for the treatment of metastatic bladder cancer. Expert. Rev. Clin. Pharmacol. 13, 1–6. doi: 10.1080/17512433.2020.1702025
Moore, C., Parrish, J. K., and Jedlicka, P. (2017). MiR-193b, downregulated in Ewing sarcoma, targets the ErbB4 oncogene to inhibit anchorage-independent growth. PLoS One 12:e0178028. doi: 10.1371/journal.pone.0178028
Mora, J., Rodriguez, E., de Torres, C., Cardesa, T., Rios, J., Hernandez, T., et al. (2012). Activated growth signaling pathway expression in Ewing sarcoma and clinical outcome. Pediatr. Blood Cancer 58, 532–538. doi: 10.1002/pbc.23348
Mori, Y., Kinoshita, S., Kanamori, T., Kataoka, H., Joh, T., Iida, S., et al. (2018). The Successful treatment of metastatic extraosseous Ewing Sarcoma with pazopanib. Intern. Med. 57, 2753–2757. doi: 10.2169/internalmedicine.9879-17
Moritake, H., Saito, Y., Sawa, D., Sameshima, N., Yamada, A., Kinoshita, M., et al. (2019). TAE226, a dual inhibitor of focal adhesion kinase and insulin-like growth factor-I receptor, is effective for Ewing sarcoma. Cancer Med. U.S. 8, 7809–7821. doi: 10.1002/cam4.2647
Moritake, H., Sugimoto, T., Kuroda, H., Hidaka, F., Takahashi, Y., Tsuneyoshi, M., et al. (2003). Newly established Askin tumor cell line and overexpression of focal adhesion kinase in Ewing sarcoma family of tumors cell lines. Cancer Genet. Cytogenet. 146, 102–109. doi: 10.1016/s0165-4608(03)00129-8
Murakami, T., Singh, A. S., Kiyuna, T., Dry, S. M., Li, Y. F., James, A. W., et al. (2016). Effective molecular targeting of CDK4/6 and IGF-1R in a rare FUS-ERG fusion CDKN2A-deletion doxorubicin-resistant Ewing’s sarcoma patient-derived orthotopic xenograft (PDOX) nude-mouse model. Oncotarget 7, 47556–47564. doi: 10.18632/oncotarget.9879
Musella, M., Manic, G., Maria, R. De, Vitale, I., and Sistigu, A. (2017). Type-I-interferons in infection and cancer: unanticipated dynamics with therapeutic implications. Oncoimmunology 6:e1314424. doi: 10.1080/2162402x.2017.1314424
Naing, A., LoRusso, P., Fu, S. Q., Hong, D. S., Anderson, P., Benjamin, R. S., et al. (2012). Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s Sarcoma family tumors. Clin. Cancer Res. 18, 2625–2631. doi: 10.1158/1078-0432.ccr-12-0061
Nan, X., Wang, J., Cheng, H., Yin, Z., Sheng, J. T., Qiu, B. S., et al. (2020). Imatinib revives the therapeutic potential of metformin on ewing sarcoma by attenuating tumor hypoxic response and inhibiting convergent signaling pathways. Cancer Lett. 469, 195–206. doi: 10.1016/j.canlet.2019.10.034
Nangia-Makker, P., Yu, Y. J., Vasudevan, A., Farhana, L., Rajendra, S. G., Levi, E., et al. (2014). Metformin: a potential therapeutic agent for recurrent colon cancer. PLoS One 9:84369. doi: 10.1371/journal.pone.0084369
Nannini, M., Urbini, M., Astolfi, A., Biasco, G., and Pantaleo, M. A. (2017). The progressive fragmentation of the KIT/PDGFRA wild-type (WT) gastrointestinal stromal tumors (GIST). J. Transl. Med. 15:113.
Nesbit, M. E. Jr., Gehan, E. A., Burgert, E. O. Jr., Vietti, T. J., Cangir, A., Tefft, M., et al. (1990). Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the first intergroup study. J. Clin. Oncol. 8, 1664–1674. doi: 10.1200/jco.1990.8.10.1664
Niemeyer, B. F., Parrish, J. K., Spoelstra, N. S., Joyal, T., Richer, J. K., and Jedlicka, P. (2015). Variable expression of PIK3R3 and PTEN in Ewing Sarcoma impacts oncogenic phenotypes. PLoS One 10:e0116895. doi: 10.1371/journal.pone.0116895
Nocka, K., Majumder, S., Chabot, B., Ray, P., Cervone, M., Bernstein, A., et al. (1989). Expression of c-kit gene products in known cellular targets of W mutations in normal and W mutant mice–evidence for an impaired c-kit kinase in mutant mice. Genes Dev. 3, 816–826. doi: 10.1101/gad.3.6.816
Oyama, R., Takahashi, M., Yoshida, A., Sakumoto, M., Takai, Y., Kito, F., et al. (2017). Generation of novel patient-derived CIC- DUX4 sarcoma xenografts and cell lines. Sci. Rep. 7:4712.
Pappo, A. S., Patel, S. R., Crowley, J., Reinke, D. K., Kuenkele, K. P., Chawla, S. P., et al. (2011). R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II sarcoma alliance for research through collaboration study. J. Clin. Oncol. 29, 4541–4547. doi: 10.1200/jco.2010.34.0000
Pappo, A. S., Vassal, G., Crowley, J. J., Bolejack, V., Hogendoorn, P. C., Chugh, R., et al. (2014). A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a sarcoma alliance for research through collaboration study. Cancer Am. Cancer Soc. 120, 2448–2456. doi: 10.1002/cncr.28728
Patel, M., Gomez, N. C., McFadden, A. W., Moats-Staats, B. M., Wu, S., Rojas, A., et al. (2014). PTEN deficiency mediates a reciprocal response to IGFI and mTOR inhibition. Mol. Cancer Res. 12, 1610–1620. doi: 10.1158/1541-7786.mcr-14-0006
Paulussen, M., Craft, A. W., Lewis, I., Hackshaw, A., Douglas, C., Dunst, J., et al. (2008). Results of the EICESS-92 Study: two randomized trials of Ewing’s sarcoma treatment–cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J. Clin. Oncol. 26, 4385–4393. doi: 10.1200/jco.2008.16.5720
Perl, A. E., Martinelli, G., Cortes, J. E., Neubauer, A., Berman, E., Paolini, S., et al. (2019). Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 381, 1728–1740.
Perova, T., Grandal, I., Nutter, L. M. J., Papp, E., Matei, I. R., Beyene, J., et al. (2014). Therapeutic potential of spleen tyrosine Kinase inhibition for treating high-risk precursor B Cell acute lymphoblastic leukemia. Sci. Transl. Med. 6:236ra62. doi: 10.1126/scitranslmed.3008661
Potratz, J., Tillmanns, A., Berning, P., Korsching, E., Schaefer, C., Lechtape, B., et al. (2016). Receptor tyrosine kinase gene expression profiles of Ewing sarcomas reveal ROR1 as a potential therapeutic target in metastatic disease. Mol. Oncol. 10, 677–692. doi: 10.1016/j.molonc.2015.12.009
Potratz, J. C., Saunders, D. N., Wai, D. H., Ng, T. L., McKinney, S. E., Carboni, J. M., et al. (2010). Synthetic lethality screens reveal RPS6 and MST1R as modifiers of insulin-like growth Factor-1 receptor inhibitor activity in childhood sarcomas. Cancer Res. 70, 8770–8781. doi: 10.1158/0008-5472.can-10-1093
Radic-Sarikas, B., Tsafou, K. P., Emdal, K. B., Papamarkou, T., Huber, K. V., Mutz, C., et al. (2017). Combinatorial drug screening identifies Ewing sarcoma-specific sensitivities. Mol. Cancer Ther. 16, 88–101. doi: 10.1158/1535-7163.mct-16-0235
Reddy, K., Cao, Y., Zhou, Z., Yu, L., Jia, S. F., and Kleinerman, E. S. (2008). VEGF(165) expression in the tumor microenvironment influences the differentiation of bone marrow-derived pericytes that contribute to the Ewing’s sarcoma vasculature. Angiogenesis 11, 257–267. doi: 10.1007/s10456-008-9109-1
Ricotti, E., Fagioli, F., Garelli, E., Linari, C., Crescenzio, N., Horenstein, A. L., et al. (1998). C-kit is expressed in soft tissue sarcoma of neuroectodermic origin and its ligand prevents apoptosis of neoplastic cells. Blood 91, 2397–2405. doi: 10.1182/blood.v91.7.2397.2397_2397_2405
Roberts, P., Burchill, S. A., Brownhill, S., Cullinane, C. J., Johnston, C., Griffiths, M. J., et al. (2008). Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing’s sarcoma family of tumors: a study by the United Kingdom cancer cytogenetics and the children’s cancer and Leukaemia group. Genes Chromos. Cancer 47, 207–220. doi: 10.1002/gcc.20523
Rodriguez-Galindo, C., Billups, C. A., Kun, L. E., Rao, B. N., Pratt, C. B., Merchant, T. E., et al. (2002). Survival after recurrence of Ewing tumors: the St Jude Children’s research hospital experience, 1979-1999. Cancer 94, 561–569. doi: 10.1002/cncr.10192
Ross, K. A., Smyth, N. A., Murawski, C. D., and Kennedy, J. G. (2013). The biology of ewing sarcoma. ISRN Oncol. 2013:759725.
Sainz-Jaspeado, M., Huertas-Martinez, J., Lagares-Tena, L., Liberal, J. M., Mateo-Lozano, S., de Alava, E., et al. (2013). EphA2-induced angiogenesis in Ewing sarcoma cells works through bFGF production and is dependent on caveolin-1. PLoS One 8:e71449. doi: 10.1371/journal.pone.0071449
Sampson, V. B., Vetter, N. S., Kamara, D. F., Collier, A. B., Gresh, R. C., and Kolb, E. A. (2015). Vorinostat enhances cytotoxicity of SN-38 and temozolomide in ewing sarcoma cells and activates STAT3/AKT/MAPK pathways. PLoS One 10:e0142704. doi: 10.1371/journal.pone.0142704
Sanceau, J., Hiscott, J., Delattre, O., and Wietzerbin, J. (2000). IFN-beta induces serine phosphorylation of Stat-1 in Ewing’s sarcoma cells and mediates apoptosis via induction of IRF-1 and activation of caspase-7. Oncogene 19, 3372–3383. doi: 10.1038/sj.onc.1203670
Sanceau, J., Poupon, M. F., Delattre, O., Sastre-Garau, X., and Wietzerbin, J. (2002). Strong inhibition of Ewing tumor xenograft growth by combination of human interferon-alpha or interferon-beta with ifosfamide. Oncogene 21, 7700–7709. doi: 10.1038/sj.onc.1205881
Sanceau, J., and Wietzerbin, J. (2004). Downregulation of angiogenic factors in Ewing tumor xenografts by the combination of human interferon-alpha or interferon-beta with ifosfamide. Ann. N. Y. Acad. Sci. 1030, 170–178. doi: 10.1196/annals.1329.022
Santoro, M., Menegaz, B. A., Lamhamedi-Cherradi, S. E., Molina, E. R., Wu, D., Priebe, W., et al. (2017). Modeling stroma-induced drug resistance in a tissue-engineered tumor model of ewing sarcoma. Tissue Eng. Pt. A 23, 80–89. doi: 10.1089/ten.tea.2016.0369
Schleiermacher, G., Peter, M., Oberlin, O., Philip, T., Rubie, H., Mechinaud, F., et al. (2003). Increased risk of systemic relapses associated with bone marrow micrometastasis and circulating tumor cells in localized ewing tumor. J. Clin. Oncol. 21, 85–91. doi: 10.1200/jco.2003.03.006
Schuetze, S. M., Bolejack, V., Choy, E., Ganjoo, K. N., Staddon, A. P., Chow, W. A., et al. (2017). Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer 123, 90–97. doi: 10.1002/cncr.30379
Sciaccaluga, M., D’Alessandro, G., Pagani, F., Ferrara, G., Lopez, N., Warr, T., et al. (2013). Functional cross talk between CXCR4 and PDGFR on glioblastoma cells is essential for migration. PLoS One 8:e73426. doi: 10.1371/journal.pone.0073426
Scotlandi, K., Avnet, S., Benini, S., Manara, M. C., Serra, M., Cerisano, V., et al. (2002a). Expression of an IGF-I receptor dominant negative mutant induces apoptosis, inhibits tumorigenesis and enhances chemosensitivity in Ewing’s sarcoma cells. Intern. J. Cancer 101, 11–16. doi: 10.1002/ijc.10537
Scotlandi, K., Maini, C., Manara, M. C., Benini, S., Serra, M., Cerisano, V., et al. (2002b). Effectiveness of insulin-like growth factor I receptor antisense strategy against Ewing’s sarcoma cells. Cancer Gene Ther. 9, 296–307. doi: 10.1038/sj.cgt.7700442
Scotlandi, K., Benini, S., Sarti, M., Serra, M., Lollini, P. L., Maurici, D., et al. (1996). Insulin-like growth factor I receptor-mediated circuit in Ewing’s sarcoma/peripheral neuroectodermal tumor: a possible therapeutic target. Cancer Res. 56, 4570–4574.
Scotlandi, K., Manara, M. C., Hattinger, C. M., Benini, S., Perdichizzi, S., Pasello, M., et al. (2005). Prognostic and therapeutic relevance of HER2 expression in osteosarcoma and Ewing’s sarcoma. Eur. J. Cancer 41, 1349–1361. doi: 10.1016/j.ejca.2005.03.015
Shen, J. Y., and Maruyama, I. N. (2011). Nerve growth factor receptor TrkA exists as a preformed, yet inactive, dimer in living cells. FEBS Lett. 585, 295–299. doi: 10.1016/j.febslet.2010.12.031
Shibue, T., Weinberg, R. A., and CSCs, E. M. T. (2017). and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 14, 611–629. doi: 10.1038/nrclinonc.2017.44
Shimizu, T., Fukuoka, K., Takeda, M., Iwasa, T., Yoshida, T., Horobin, J., et al. (2016). A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 77, 997–1003. doi: 10.1007/s00280-016-3010-1
Shor, A. C., Keschman, E. A., Lee, F. Y., Muro-Cacho, C., Letson, G. D., Trent, J. C., et al. (2007). Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on Src kinase for survival. Cancer Res. 67, 2800–2808. doi: 10.1158/0008-5472.can-06-3469
Silvany, R. E., Eliazer, S., Wolff, N. C., and Ilaria, R. L. Jr. (2000). Interference with the constitutive activation of ERK1 and ERK2 impairs EWS/FLI-1-dependent transformation. Oncogene 19, 4523–4530. doi: 10.1038/sj.onc.1203811
Smithey, B. E., Pappo, A. S., and Hill, D. A. (2002). c-kit expression in pediatric solid tumors - a comparative immunohistochemical study. Am. J. Surg. Pathol. 26, 486–492. doi: 10.1097/00000478-200204000-00011
Sorensen, P. H., Lessnick, S. L., Lopez-Terrada, D., Liu, X. F., Triche, T. J., and Denny, C. T. (1994). A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 6, 146–151.
Steinestel, K., Trautmann, M., Jansen, E. P., Dirksen, U., Rehkamper, J., Mikesch, J. H., et al. (2020). Focal adhesion kinase confers pro-migratory and antiapoptotic properties and is a potential therapeutic target in Ewing sarcoma. Mol. Oncol. 14, 248–260. doi: 10.1002/1878-0261.12610
Stirewalt, D. L., and Radich, J. P. (2003). The role of FLT3 in haematopoietic malignancies. Nat. Rev. Cancer 3, 650–665. doi: 10.1038/nrc1169
Strammiello, R., Benini, S., Manara, M. C., Perdichizzi, S., Serra, M., Spisni, E., et al. (2003). Impact of IGF-I/IGF-IR circuit on the angiogenetic properties of Ewing’s sarcoma cells. Horm. Metab. Res. 35, 675–684. doi: 10.1055/s-2004-814149
Subbiah, V., Naing, A., Brown, R. E., Chen, H., Doyle, L., LoRusso, P., et al. (2011). Targeted morphoproteomic profiling of ewing’s sarcoma treated with insulin-like growth factor 1 receptor (IGF1R) inhibitors: response/resistance signatures. PLoS One 6:e18424. doi: 10.1371/journal.pone.0018424
Sulzmaier, F. J., Jean, C., and Schlaepfer, D. D. (2014). FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer 14, 598–610. doi: 10.1038/nrc3792
Sun, H., Lin, D. C., Cao, Q., Pang, B., Gae, D. D., Lee, V. K. M., et al. (2017). Identification of a novel SYK/c-MYC/MALAT1 signaling pathway and its potential therapeutic value in Ewing Sarcoma. Clin. Cancer Res. 23, 4376–4387. doi: 10.1158/1078-0432.ccr-16-2185
Suraweera, A., O’Byrne, K. J., and Richard, D. J. (2018). Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front. Oncol. 8:92. doi: 10.3389/fonc.2018.00092
Tamura, A., Yamamoto, N., Nino, N., Ichikawa, T., Nakatani, N., Nakamura, S., et al. (2019). Pazopanib maintenance therapy after tandem high-dose chemotherapy for disseminated Ewing sarcoma. Int. Cancer Conf. J. 8, 95–100. doi: 10.1007/s13691-019-00362-w
Tap, W. D., Demetri, G., Barnette, P., Desai, J., Kavan, P., Tozer, R., et al. (2012). Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J. Clin. Oncol. 30, 1849–1856. doi: 10.1200/jco.2011.37.2359
Thanindratarn, P., Dean, D. C., Nelson, S. D., Hornicek, F. J., and Duan, Z. (2020). Chimeric antigen receptor T (CAR-T) cell immunotherapy for sarcomas: From mechanisms to potential clinical applications. Cancer Treat. Rev. 82:101934. doi: 10.1016/j.ctrv.2019.101934
Thijssen, V. L., Paulis, Y. W., Nowak-Sliwinska, P., Deumelandt, K. L., Hosaka, K., Soetekouw, P. M., et al. (2018). Targeting PDGF-mediated recruitment of pericytes blocks vascular mimicry and tumor growth. J. Pathol. 246, 447–458. doi: 10.1002/path.5152
Timeus, F., Ricotti, E., Crescenzio, N., Garelli, E., Doria, A., Spinelli, M., et al. (2001). Flt-3 and its ligand are expressed in neural crest-derived tumors and promote survival and proliferation of their cell lines. Lab. Invest. 81, 1025–1037. doi: 10.1038/labinvest.3780314
Toledo, R. A., Qin, Y., Cheng, Z. M., Gao, Q., Iwata, S., Silva, G. M., et al. (2016). Recurrent mutations of chromatin-remodeling genes and kinase receptors in pheochromocytomas and paragangliomas. Clin. Cancer Res. 22, 2301–2310. doi: 10.1158/1078-0432.ccr-15-1841
Toretsky, J. A., Kalebic, T., Blakesley, V., LeRoith, D., and Helman, L. J. (1997). The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J. Biol. Chem. 272, 30822–30827. doi: 10.1074/jbc.272.49.30822
Toretsky, J. A., Thakar, M., Eskenazi, A. E., and Frantz, C. N. (1999). Phosphoinositide 3-hydroxide kinase blockade enhances apoptosis in the Ewing’s sarcoma family of tumors. Cancer Res. 59, 5745–5750.
Twardziok, M., Kleinsimon, S., Rolff, J., Jager, S., Eggert, A., Seifert, G., et al. (2016). Multiple Active Compounds from Viscum album L. Synergistically Converge to Promote Apoptosis in Ewing Sarcoma. PLoS One 11:e0159749. doi: 10.1371/journal.pone.0159749
U.S.F.A.D. Administration (2019). FDA Grants Accelerated Approval To New Treatment For Advanced Soft Tissue Sarcoma. Available online at: www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-new-treatment-advanced-soft-tissue-sarcoma (accessed January 24, 2019).
U.S.F.A.D. Administration (2020a). FDA Approves First Treatment For Thyroid Eye Disease. Green Park: U.S.F.A.D. Administration (accessed February 25, 2020).
U.S.F.A.D. Administration (2020b). FDA Approves Neratinib For Metastatic HER2-Positive Breast Cancer. Available online at: www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-neratinib-metastatic-her2-positive-breast-cancer
Uren, A., Merchant, M. S., Sun, C. J., Vitolo, M. I., Sun, Y., Tsokos, M., et al. (2003). Beta-platelet-derived growth factor receptor mediates motility and growth of Ewing’s sarcoma cells. Oncogene 22, 2334–2342. doi: 10.1038/sj.onc.1206330
van de Luijtgaarden, A. C. M., Versleijen-Jonkers, Y. M. H., Roeffen, M. H. S., Schreuder, H. W. B., Flucke, U. E., and van der Graaf, W. T. A. (2013). Prognostic and therapeutic relevance of the IGF pathway in Ewing’s sarcoma patients. Target. Oncol. 8, 253–260. doi: 10.1007/s11523-012-0248-3
van der Graaf, W. T., Blay, J. Y., Chawla, S. P., Kim, D. W., Bui-Nguyen, B., Casali, P. G., et al. (2012). Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379, 1879–1886.
van Doorninck, J., Ji, L. Y., Okamatsu, C., Cooper, A., Wing, M., Womer, R., et al. (2009). Ewing family tumors that do not over-express BMI-1 are a distinct subclass that arise from alternate mechanisms of molecular transformation. Cancer Res. 69, 18–22.
van Maldegem, A. M., Bovee, J. V., Peterse, E. F., Hogendoorn, P. C., and Gelderblom, H. (2016). Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur. J. Cancer 53, 171–180. doi: 10.1016/j.ejca.2015.09.009
Veeriah, S., Brennan, C., Meng, S. S., Singh, B., Fagin, J. A., Solit, D. B., et al. (2009). The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc. Natl. Acad. Sci. U.S.A. 106, 9435–9440. doi: 10.1073/pnas.0900571106
Wang, S., Hwang, E. E., Guha, R., O’Neill, A. F., Melong, N., Veinotte, C. J., et al. (2019). High-throughput chemical screening identifies focal adhesion kinase and aurora kinase B inhibition as a synergistic treatment combination in ewing sarcoma. Clin. Cancer Res. 25, 4552–4566. doi: 10.1158/1078-0432.ccr-17-0375
Wang, Y. X., Mandal, D., Wang, S., Hughes, D., Pollock, R. E., Lev, D., et al. (2009). Inhibiting platelet-derived growth factor beta reduces Ewing’s sarcoma growth and metastasis in a novel orthotopic human xenograft model. Vivo 23, 903–909.
Weisberg, E., Manley, P. W., Breitenstein, W., Bruggen, J., Cowan-Jacob, S. W., Ray, A., et al. (2005). Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7, 129–141. doi: 10.1016/j.ccr.2005.01.007
Wilky, B. A., Trucco, M. M., Subhawong, T. K., Florou, V., Park, W., Kwon, D., et al. (2019). Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: a single-centre, single-arm, phase 2 trial. Lancet Oncol. 20, 837–848. doi: 10.1016/s1470-2045(19)30153-6
Wu, Y. T., Wang, B. J., Miao, S. W., and Gao, J. J. (2015). Picropodophyllin inhibits the growth of Ewing’s sarcoma cells through the insulinlike growth factor1 receptor/Akt signaling pathway. Mol. Med. Rep. 12, 7045–7050. doi: 10.3892/mmr.2015.4266
Xiang, D. N., Feng, Y. F., Wang, J., Zhang, X., Shen, J. J., Zou, R., et al. (2019). Platelet-derived growth factor-BB promotes proliferation and migration of retinal microvascular pericytes by up-regulating the expression of C-X-C chemokine receptor types 4. Exp. Ther. Med. 18, 4022–4030.
Xie, L., Guo, W., Wang, Y., Yan, T. Q., Ji, T., and Xu, J. (2018). Apatinib for advanced sarcoma: results from multiple institutions’ off-label use in China. BMC Cancer 18:396. doi: 10.1186/s12885-018-4303-z
Xie, Y. T., Skytting, B., Nilsson, G., Brodin, B., and Larsson, O. (1999). Expression of insulin-like growth factor-1 receptor in synovial sarcoma: association with an aggressive phenotype. Cancer Res. 59, 3588–3591.
Yan, S., Li, Z., and Thiele, C. J. (2013). Inhibition of STAT3 with orally active JAK inhibitor, AZD1480, decreases tumor growth in neuroblastoma and pediatric sarcomas in vitro and In vivo. Oncotarget 4, 433–445.
Yoon, H., Dehart, J. P., Murphy, J. M., and Lim, S. T. (2015). Understanding the roles of FAK in cancer: inhibitors, genetic models, and new insights. J. Histochem. Cytochem. 63, 114–128.
Yu, P. Y., Gardner, H. L., Roberts, R., Cam, H., Hariharan, S., Ren, L., et al. (2017). Target specificity, in vivo pharmacokinetics, and efficacy of the putative STAT3 inhibitor LY5 in osteosarcoma, Ewing’s sarcoma, and rhabdomyosarcoma. PLoS One 12:e0181885. doi: 10.1371/journal.pone.0181885
Yue, W., Zheng, X., Lin, Y., Yang, C. S., Xu, Q., Carpizo, D., et al. (2015). Metformin combined with aspirin significantly inhibit pancreatic cancer cell growth in vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and Bcl-2. Oncotarget 6, 21208–21224.
Zhang, J., Yang, P. L., and Gray, N. S. (2009). Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9, 28–39.
Zheng, H.Y, Shen, H., Oprea, I., Worrall, C., Stefanescu, R., Girnita, A., et al. (2012). Beta-Arrestin-biased agonism as the central mechanism of action for insulin-like growth factor 1 receptor-targeting antibodies in Ewing’s sarcoma. Proc. Natl. Acad. Sci. U.S.A. 109, 20620–20625.
Zhou, R. R., Jia, S. F., Zhou, Z. C., Wang, Y. F., Bucana, C. D., and Kleinerman, E. S. (2002). Adenovirus-E1A gene therapy enhances the in vivo sensitivity of Ewing’s sarcoma to VP-16. Cancer Gene. Ther. 9, 407–413.
Zhou, X., Chen, J., Xiao, Q., Wang, T., Yu, Y., Li, B., et al. (2018). MicroRNA-638 inhibits cell growth and tubule formation by suppressing VEGFA expression in human Ewing sarcoma cells. Biosci. Rep. 38, BSR20171017.
Zhou, Z., Jia, S. F., Hung, M. C., and Kleinerman, E. S. (2001). E1A sensitizes HER2/neu-overexpressing Ewing’s sarcoma cells to topoisomerase II-targeting anticancer drugs. Cancer Res. 61, 3394–3398.
Zhu, C. J., Wei, Y. Q., and Wei, X. W. (2019). AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol. Cancer 18:153. doi: 10.1186/s12943-019-1090-3
Keywords: Ewing sarcoma, tyrosine kinases, aggressiveness, therapeutic target, chemoresistance, clinical pathogenesis, acquisition of self-renewal traits
Citation: Jin W (2020) The Role of Tyrosine Kinases as a Critical Prognostic Parameter and Its Targeted Therapies in Ewing Sarcoma. Front. Cell Dev. Biol. 8:613. doi: 10.3389/fcell.2020.00613
Received: 12 May 2020; Accepted: 22 June 2020;
Published: 09 July 2020.
Edited by:
Teresita Padilla-Benavides, University of Massachusetts Medical School, United StatesReviewed by:
Maria Paola Paronetto, Foro Italico University of Rome, ItalyCopyright © 2020 Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wook Jin, amlud29AZ2FjaG9uLmFjLmty
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.