
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 10 July 2020
Sec. Epigenomics and Epigenetics
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00546
This article is part of the Research Topic Epigenetic Therapy with Histone Deacetylase Inhibitors: Implications for Cancer Treatment View all 17 articles
 Alexandre Perla1,2Lívia Fratini1,2Paula S. Cardoso1,2
Alexandre Perla1,2Lívia Fratini1,2Paula S. Cardoso1,2 Carolina Nör3,4André T. Brunetto1,5
Carolina Nör3,4André T. Brunetto1,5 Algemir L. Brunetto1,5
Algemir L. Brunetto1,5 Caroline Brunetto de Farias1,5Mariane Jaeger1,5
Caroline Brunetto de Farias1,5Mariane Jaeger1,5 Rafael Roesler1,2*
Rafael Roesler1,2*Brain cancers are the leading cause of cancer-related deaths in children. Biological changes in these tumors likely include epigenetic deregulation during embryonal development of the nervous system. Histone acetylation is one of the most widely investigated epigenetic processes, and histone deacetylase inhibitors (HDACis) are increasingly important candidate treatments in many cancer types. Here, we review advances in our understanding of how HDACis display antitumor effects in experimental models of specific pediatric brain tumor types, i.e., medulloblastoma (MB), ependymoma (EPN), pediatric high-grade gliomas (HGGs), and rhabdoid and atypical teratoid/rhabdoid tumors (ATRTs). We also discuss clinical perspectives for the use of HDACis in the treatment of pediatric brain tumors.
Brain tumors of the childhood represent the leading cause of cancer-related deaths in children aged 0–14 years, and survivors often present long-term neurological sequelae that impair their quality of life (Ostrom et al., 2016). These cancer types include medulloblastoma (MB), which is the most common and most studied type of childhood brain tumor, ependymoma (EPN), pediatric high-grade gliomas (HGGs), and rhabdoid and atypical teratoid/rhabdoid tumors (ATRTs) (Guerreiro Stucklin et al., 2018). Pediatric brain cancers may originate from defects in embryonal development affecting cell types including neural stem cells (NSCs) and neuronal precursors, or dedifferentiation of mature neuronal or glial cells (Taylor et al., 2005; Visvader, 2011; Liu and Zong, 2012; Wang and Wechsler-Reya, 2014; Azzarelli et al., 2018).
Normal development, cellular differentiation, and tissue specialization are finely regulated by various epigenetic mechanisms (Atlasi and Stunnenberg, 2017). Epigenetic regulation allows changes in chromatin structure that control gene expression without modifications in DNA sequence. Histone acetylation and DNA methylation feature among the most widely investigated epigenetic mechanisms (Surani et al., 2007). Histone acetyltransferases (HATs) and deacetylases (HDACs) play opposing roles in regulating gene expression. HATs transfer acetyl groups to the amino-terminal lysine residues of histones, thus increasing histone acetylation and transcriptional activity. On the other hand, HDACs remove acetyl groups, thus promoting chromatin condensation and overall repression of gene expression, in addition to displaying other, non-epigenetic actions (Kouzarides, 2007; Sanaei and Kavoosi, 2019; D’Mello, 2020; Milazzo et al., 2020). Changes in the expression and activity of HATs and HDACs were described in leukemias, and afterward in solid tumors, and in many cases decreased levels of histone acetylation were shown to correlate with clinical outcome. Therefore, HDAC inhibitors (HDACis) have become promising and widely investigated experimental anticancer agents (Lane and Chabner, 2009; Li and Seto, 2016).
Pediatric cancers hijack and modify biological processes involved in normal embryonic development, including epigenetic modifications, to promote tumor progression (Liu and Zong, 2012; Marshall et al., 2014). An important component of childhood cancer biology is epigenetic reprogramming that can lock cells in a stem cell-like, poorly differentiated and highly proliferative phenotype (Lawlor and Thiele, 2012). Accumulating evidence implicates epigenetic abnormalities in the genesis and progression of pediatric brain tumors. The relationship between epigenetic markers and patient survival has been investigated (Bhattacharya et al., 2020), and epigenetic modulators have shown promising effects in experimental models. Here we review the potential of HDACis in the treatment of selected types of brain cancer that afflict children.
Mutations and genetic variations affecting epigenetic-regulating mechanisms are features of several types of childhood brain cancers, including MB, EPN, ATRT, and pediatric gliomas (Mack and Taylor, 2009; Parsons et al., 2011; Dubuc et al., 2012; Jones et al., 2012; Lee et al., 2012; Northcott et al., 2012; Buczkowicz et al., 2014; Mack et al., 2016). In fact, MB and low-grade gliomas are among the types of pediatric tumors with highest frequency of mutation in genes encoding epigenetic regulators (Huether et al., 2014). MicroRNAs (miRNAs) regulate pediatric brain tumor cells at posttranscriptional/translational levels, acting on a range of functional aspects linked to cancer progression, including proliferation and stemness. The diagnostic, prognostic, and therapeutic potential of miRNAs has been increasingly highlighted (Garg et al., 2015; Leichter et al., 2017; Pezuk et al., 2019). Epigenetic remodeling genes SMARCB1 and SMARCA4 are among the most frequently altered in pediatric brain tumors, with most cases of ATRT showing changes in those genes (Frühwald et al., 2016; Wang et al., 2017; Johann, 2020). Enhancer of zeste homolog 2 (EZH2), which is part of a Polycomb repressor complex that methylates lysine 27 of histone H3 (H3K27), leading to transcription inhibition, is often mutated or highly expressed in pediatric brain tumors (Huether et al., 2014; Li et al., 2015; Kim and Roberts, 2016; Erkek et al., 2019; Zhang et al., 2020).
A reduced histone acetylation state can contribute to cancer through repressing differentiation and tumor suppressor genes while allowing overexpression of genes promoting proliferation. Chromatin modifications mediated by histone acetylation can also epigenetically influence the tumor genetic landscape, for example by leading to DNA copy gain in the absence of chromosomal instability (Black et al., 2013; Mack et al., 2016). HDACs play a central role in epigenetic regulation through reducing acetylation. It is currently known that some HDACs can either repress or activate gene transcription, in addition to displaying non-epigenetic activities by acting directly on nuclear and cytoplasmic proteins (D’Mello, 2020). For example, transcription factors E2F1, STAT1, STAT3, and NF-κB can be directly hyperacetylated by HDACis (Johnstone and Licht, 2003; Glozak et al., 2005; Bolden et al., 2006).
Histone acetyltransferases and deacetylases are currently classified into different classes. Class I HDACs include nuclear HDAC1, 2, 3, and 8. Class II HDACs occur in both the nucleus and cytoplasm and are classified into two subclasses: HDAC4, 5, 7, and 9 are grouped as IIa, whereas HDAC6 and 10 are classified as class IIb. Sirtuins constitute Class III HDACs, being found in the nucleus, cytoplasm, and mitochondria according to the specific type. Finally, HDAC11 has been proposed as a Class IV HDAC (Falkenberg and Johnstone, 2014; Hassell, 2019; D’Mello, 2020; Milazzo et al., 2020). Most deacetylase activity in mammalian cells has been attributed to Class I HDACs (Lahm et al., 2007).
Increased HDAC2 expression (Park et al., 2003), deletions or amplifications of histone methyltransferases and demethylases (Northcott et al., 2009), DNA hypermethylation (Frühwald et al., 2001), and altered miRNA expression (Ferretti et al., 2008) have been reported in MB. High HDAC5 and HDAC9 expression is found in prognostically poor MB subgroups and significantly associated with poor overall survival, posing an independent risk factor (Milde et al., 2010). Other alterations in epigenetic components found in MB include truncating mutations in the KDM6A gene encoding a histone 3 lysine 27 demethylase (Jones et al., 2012), homozygous deletions of genes involved in histone lysine methylation, and amplification of the HAT gene MYST3 (Northcott et al., 2009). Expression of HDAC2 is higher in patients with MB subgroups with poor prognosis (sonic hedgehog (SHH), Group 3 and Group 4), and MYC-amplified MB cell lines show increased mRNA levels of class I HDACs compared to the normal cerebellum (Ecker et al., 2015).
Hypermethylation of the transcriptional repressor hypermethylated in cancer 1 (H1C-1) was identified in 83% of EPN samples (Waha et al., 2004). Global reduction of H3K27me3 analogous to H3K27M mutant gliomas, accompanied by CpGi hypermethylation, is found in EPN (Bayliss et al., 2016). Poor-prognosis EPNs show a CpG island methylator phenotype, where transcriptional silencing driven by CpG methylation converges on targets of the Polycomb repressive complex 2 (PRC2), which represses expression of differentiation genes through trimethylation of H3K27 (Mack et al., 2014). Posterior fossa type A (PFA) EPNs show low H3K27 methylation and high levels of Enhancer of Zeste Homologs Inhibitory Protein (EZHIP), which promotes a similar chromatin state compared to H3K27M (Jain et al., 2019). Enrichment of 5-carboxylcytosine (5caC) and increased TET1 expression, which are involved in active DNA demethylation, are epigenetic hallmarks of EPN and SHH MB (Ramsawhook et al., 2017). Hypermethylated genes in EPN converge on defined sets of embryonic stem cell (ESC) targets, suggesting a linkage, mediated by epigenetic programming, between embryonic development and pediatric brain cancer (Sin-Chan and Huang, 2014; Mack et al., 2016).
Somatic mutations in the H3.3-ATRX-DAXX chromatin remodeling pathway and recurrent mutations in the gene encoding the histone 3 variant H3.3 are highly prevalent in pediatric glioblastoma (Schwartzentruber et al., 2012). In diffuse intrinsic pontine glioma (DIPG), a deadly type of childhood glioblastoma, a mutation that leads to hypomethylation by replacing a lysine to methionine (K27M) on H3F3A and HIST1H3B/C genes encoding histone variants is the most frequent mutation (Wu et al., 2012, 2014; Mendez et al., 2020). Supporting the link between embryonic development and the arising of pediatric brain tumors, this histone mutation can contribute to resetting neural progenitors derived from human ESCs to a stem cell state, ultimately resulting in neoplastic transformation (Funato et al., 2014).
In ATRTs, HDAC1 is significantly differentially expressed (Sredni et al., 2013), and the chromatin remodeling and tumor suppressor gene SMARCB1 represses Cyclin D1 transcription by recruiting the HDAC1 complex to its promoter, resulting in cell cycle arrest (Tsikitis et al., 2005). A hallmark of malignant rhabdoid tumors is homozygous deletion or inactivation of SMARCB1. Histone acetylation and methylation patterns, as well as HDAC and HAT levels, are influenced by insulin-like growth factor receptor 1 (IGF-1R) signaling (Shim et al., 2013). For comprehensive reviews on the role of epigenetic changes as part of the biological basis of pediatric brain cancers, see Dubuc et al. (2012) and Mack et al. (2016).
Most HDACis widely used experimentally or clinically preferentially inhibit Class I and II HDACs. These agents include sodium butyrate (NaB), trichostatin A (TSA), valproic acid (VPA), suberoyl anilide hydroxamic acid (SAHA, vorinostat), panobinostat, belinostat, and romidepsin (Bolden et al., 2006; Li and Seto, 2016; Millard et al., 2017; Hassell, 2019). HDACis induce anticancer effects in several experimental tumor types by targeting aberrant chromatin alterations, resulting in changes in cell proliferation, viability, differentiation, migration, and angiogenesis (Bolden et al., 2006; Sanaei and Kavoosi, 2019; Ribatti and Tamma, 2020). In addition to modulating acetylation by inhibiting HDACs, HDACis may directly modulate miRNAs and also alter protein kinase signaling through acetylation-independent mechanisms (Chen et al., 2005; Autin et al., 2019). The HDACi TSA inhibits HDAC6, a predominantly cytoplasmic HDAC, which likely induces many effects independent of alterations in gene expression stimulated by histone acetylation (Johnstone and Licht, 2003; Chen et al., 2005; Glozak et al., 2005). When combined with agents targeting other epigenetic regulators, such as EZH2, HDACis modulate acetylation and methylation of H3K27, through mechanisms involving PRC2 complex disruption (Lue et al., 2019). Below, we summarize studies examining the effects of HDACis in experimental models of pediatric brain tumors.
Medulloblastoma is currently classified within four distinct molecular subgroups, namely, WNT, SHH, Group 3, and Group 4, with subtypes within each group being now recognized (Louis et al., 2016). An early study by Jaboin et al. (2002) showed that the HDACi MS-275 inhibits proliferation of Daoy and D283 Med MB cells. A subsequent study by Li and colleagues showed that VPA, which partially acts as a class I and II HDACi, when used at clinically safe concentrations, leads to growth inhibition, cell cycle arrest, apoptosis, senescence, differentiation, and inhibition of colony formation in Daoy and D283 Med cells. In addition, daily systemic injection of VPA (400 mg/kg) for 28 days significantly inhibits in vivo growth of Daoy and D283 Med xenografts in immunodeficient mice. These effects are associated with hyperacetylation of histone H3 and H4, activation of p21, and suppression of TP53, CDK4, and c-MYC (Li et al., 2005). The HDACis SAHA, NaB, and TSA induce apoptotic cell death related to dissipation of mitochondrial membrane potential and activation of caspase-9 and -3 in Daoy and UW228-2 MB cells. These HDACis also enhance the cytotoxic effects of ionizing radiation in Daoy cells, and treatment with SAHA potentiates the cytotoxic actions of etoposide and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), but not vincristine (Sonnemann et al., 2006). HDACi-induced TRAIL sensitization is associated with increased caspase-8 activation (Sonnemann et al., 2012). VPA combined with interferon (IFN)-gamma restores caspase-8 expression and sensitivity to TRAIL in primary MB samples and significantly potentiates TRAIL-mediated suppression of MB growth in vivo (Häcker et al., 2009). HDACi potentiation of ionizing radiation effects in MB cells was also reported by Kumar et al. (2007). A variety of HDACis, including MS-275, SAHA, TSA, and VPA, are able to inhibit proliferation of MB cell lines and induce histone H4 hyperacetylation, reactivation of expression of growth regulatory genes, and induction of apoptosis, as well as reduction of MB xenograft growth in vivo (Furchert et al., 2007). HDACis helminthosporium carbonum (HC)-toxin, SAHA, and panobinostat reduce viability and lead to radiosensitization accompanied by increased cell death in the HD-MB03 cell line, a preclinical model of Group 3 MB (Milde et al., 2012). Inhibition of class I HDACs in MB cells reduces metabolic activity, cell number, and viability and enhances sensitivity to HDACi specifically in MYC-amplified cells (Ecker et al., 2015). Histone-mediated deregulation of expression of the Wnt antagonist Dickkopf-1 (DKK1) impairs its tumor-suppressing activity and contributes to experimental MB tumorigenesis, and treatment with TSA restores DKK1 in D283 Med cells (Vibhakar et al., 2007). TSA significantly inhibits telomerase activity, increases expression of p53 and p21, and reduces cyclin-D levels in ONS-76 MB cells. Upregulation of Bax and cytochrome c correlates with pro-apoptotic effects in TSA-treated cells (Khaw et al., 2007).
HDAC-mediated deacetylation of Gli (glioma-associated oncogene) promotes transcriptional activation through Hedgehog (Hh)-induced upregulation of HDAC1, and loss of HDAC activity hinders Hh pathway-dependent growth of neural progenitors and MB cells (Canettieri et al., 2010; De Smaele et al., 2011). The functional interaction between the transcription cofactor ZNF521 and GLI1 and GLI2, which enhances Hh signaling, is sensitive to HDACis (Scicchitano et al., 2019). Hh signaling stimulates granule precursor (CGP) proliferation during the early stages of postnatal cerebellar development and sustains HDAC activation leading to stimulation of CGPs. HDAC inhibition impairs Shh-induced CGP proliferation and improves aberrant CGP proliferation in a mouse model of MB (Lee et al., 2013). NL-103, a dual-targeted inhibitor of both HDAC and Hh signaling, effectively overcame resistance to the Hh inhibitor vismodegib (Zhao et al., 2014). HDAC6 is an important regulator of the Hh pathway, and selective HDAC6 inhibition hinders MB cell survival in vitro and reduces tumor growth in an in vivo allograft model (Dhanyamraju et al., 2015). HDACis show more pronounced effects on proliferation of SHH-driven MB cells harboring a mutation in the gene encoding for the histone acetyltransferase (HAT) CREBBP, when compared to CREBBP wild-type controls (Hellwig et al., 2019). A targeted small-molecule screen on the stable, SHH-dependent murine MB cell line SMB21 reveals selective inhibitors of class I HDACs as promising antitumor agents for SHH MB, and the novel class I HDAC inhibitor JNJ-26481585 (quisinostat) consistently inhibits growth of SHH MB in vivo as well as in vitro (Pak et al., 2019). Another recent study using a high-throughput cell viability assay to screen 12,800 compounds identified two HDACis, JNJ-26481585 and dacinostat, as anti-proliferative agents in MB. Both compounds induce cytotoxicity and apoptosis and block cell cycle progression at the G2/M phase, in addition to reducing the growth of MB xenografts in mice (Zhang et al., 2019).
Histone deacetylase inhibitors sensitize MB cells to apoptosis induced by cytotoxic chemotherapy via an enhancement of p53-dependent Bax activation (Häcker et al., 2011). The HDACi sodium butyrate (NaB) at a low dose more effectively inhibits D283 cell viability when combined with the chemotherapeutic etoposide (Nör et al., 2013). SAHA combined with 13-cis retinoic acid (RA) induces apoptosis and transcription of bone morphogenetic protein-2 (BMP-2) in MB cells and is more effective than each drug alone in inhibiting MB growth in vitro and in vivo. Moreover, intracranial MB tumors in mice treated with SAHA plus RA plus cisplatin show a 4-fold increase in apoptosis over controls, and a 2-fold increase over animals receiving only SAHA or RA plus SAHA (Spiller et al., 2008). The combination of RA with epigenetic modulators in based upon functional interplays among retinoid receptors, histone acetylation, and DNA methylation. For example, HDACs bind to RA response elements in proximal promoters or enhancer regions of genes regulated by retinoids in stem cells, and retinoid receptors interact with the transcription complex mediating the placement or removal of epigenetic marks on histones and DNA (Gudas, 2013; Urvalek and Gudas, 2014; Almeida et al., 2017). Combinations of HDACis with other epigenetic modulators have also been evaluated. SAHA plus the DNA methyltransferase 1 (DNMT) inhibitor 5-aza-2′-deoxycytidine (5-aza-dC) produce a synergistic effect on survival of Daoy and D283 Med MB cells (Yuan et al., 2017).
Combining HDACis with growth factor receptor ligands such as receptor tyrosine kinase (RTK) inhibitors (gefitinib or vandetanib) or brain-derived neurotrophic factor (BDNF) is also more effective than each agent given alone in impairing MB cell viability (Marino et al., 2011; Nör et al., 2011). However, bombesin receptor antagonists failed to potentiate the effects of HDACi inhibition (Jaeger et al., 2016). Other studies have investigated combinations of HDACis with protein kinase signaling inhibitors. The multi-kinase inhibitor sorafenib and VPA interact to radiosensitize and kill MB cell lines (Tang et al., 2012). SAHA shows additive cytotoxicity with the Aurora A kinase inhibitor MLN8237 in Daoy cells (Muscal et al., 2013a). A study by Geron et al. (2015) examined the effects of the pan-aurora kinase inhibitor AMG 900 alone or in combination with SAHA in UW402, UW473, and ONS-76 MB cells. A synergistic effect of combining AMG 900 and SAHA is observed on cell proliferation in all these cell lines, especially in sequential drug treatment. The drug combination also fully inhibits cell survival measured by colony formation. Using an animal model of MYC-driven MB to screen for promising drugs, Pei et al. (2016) found HDACis among the most effective compounds. HDACis potently inhibit survival of MYC-driven MB cells, through a mechanism involving expression of the FOXO1 tumor suppressor gene. Importantly, HDACis synergize with phosphatidylinositol 3-kinase (PI3K) inhibitors to inhibit MB growth in vivo. NaB reduces viability and increases acetylation in human MB cells, the anti-proliferative effect of NaB being enhanced by combination with a mitogen-activated protein kinase (MAPK)/extracellular-related kinase (ERK) inhibitor (Jaeger et al., 2020).
Ependymomas featuring a CpG island methylator phenotype respond to drugs that target DNA or H3K27 methylation, revealing epigenetic modulators as the first rational therapeutic candidates in this tumor type (Mack et al., 2014). In a high-risk cytogenetic group 3 and molecular group C EPN model (DKFZ-EP1NS) that shows high tumorigenic potential in vivo, cells are resistant to cytotoxic chemotherapeutics temozolomide, vincristine, and cisplatin but respond to HDACi treatment (Milde et al., 2011).
A seminal study using a chemical screen in patient-derived DIPG cultures along with RNA-seq analyses and integrated computational modeling to identify potentially effective therapeutic strategies has highlighted the potential of HDACs as targets. Importantly, the HDACi panobinostat showed antitumor efficacy both in vitro and in orthotopic xenograft models. Furthermore, combination of panobinostat with the histone demethylase inhibitor GSK-J4 showed that the two had synergistic effects (Grasso et al., 2015). Another study of panobinostat in experimental DIPG found that it effectively impaired cell proliferation, viability, and clonogenicity and induced apoptosis in human and murine DIPG cells. In genetically engineered tumor-bearing mice, panobinostat reduced tumor growth and increased H3 acetylation. Extended daily treatment of both genetic and orthotopic xenograft models with 10 or 20 mg/kg panobinostat led to significant toxicity, while reduced, well-tolerated doses of panobinostat failed to prolong overall survival (Hennika et al., 2017). In DIPG primary cells, panobinostat potentiated the effects of gene therapy based on human adipose tissue-derived mesenchymal stem cells expressing the secreted form of TRAIL (hAT-MSC.sTRAIL), inducing a decrease in tumor volume and prolonging survival (Choi et al., 2019).
Combined treatment with the HDACi panobinostat and the AXL inhibitor BGB324 resulted in synergistic antitumor effects on DIPG cells, with reduced expression of genes related to mesenchymal phenotype, stemness, and DNA damage repair (Meel et al., 2020). HDACis also synergize with blockade of bromodomain inhibition or CDK7, which disrupts oncogenic transcription, in DIPG models. HDAC inhibition by panobinostat, together with the bromodomain inhibitor JQ1 or the CDK7 inhibitor THZ1, synergistically reduced cell viability in DIPG cell cultures and proved more effective than single-drug treatments in inhibiting proliferation and inducing apoptosis. Panobinostat and JQ1 induced overlapping transcriptional changes, downregulating many of the same sets of genes (Nagaraja et al., 2017). VPA potentiates carboplatin cytotoxicity and increases histone H3 acetylation in different DIPG cell lines (Killick-Cole et al., 2017). CUDC-907, a first-in-class dual inhibitor of HDACs and PI3K, is a potent cytotoxic agent in DIPG models. Mechanisms underlying CUDC-907 actions include regulation of DNA damage response. It also displays radiosensitizing effects mediated by decreased nuclear factor kappa B (NFκB)/Forkhead box M1 (FOXM1) recruitment to promoters of genes involved in response to DNA damage (Pal et al., 2018). A CRISPR screen showed that knockout of KDM1A encoding lysine-specific demethylase 1 (LSD1) sensitizes DIPG cells to HDACis. Corin, an HDAC and LSD1 inhibitor, hinders in vitro and in vivo DIPG growth by increasing H3K27me3 levels as well as HDAC-targeted H3K27ac and LSD1-targeted H3K4me1 at differentiation-associated genes (Anastas et al., 2019).
Atypical teratoid/rhabdoid tumors cell proliferation is impaired by a variety of HDACis, including MS-275, SAHA, TSA, and VPA (Jaboin et al., 2002; Furchert et al., 2007), and pretreatment with HDACis potentiates the effect of ionizing radiation on ATRT cells as measured by a colony-formation assay (Blattmann et al., 2012; Knipstein et al., 2012). SAHA shows synergism with doxorubicin and the cyclinD1 inhibitor fenretinide in inhibiting proliferation of rhabdoid cells (Kerl et al., 2013). Objective ATRT tumor regressions accompanied by increases in histone acetylation were observed in mice after treatment with the natural tetrapeptide HDACi depsipeptide (Graham et al., 2006). The HDACi FK228 (depsipeptide) induces autophagy in malignant ATRT cells by inducing apoptosis inducing factor (AIF) translocation to the nucleus (Watanabe et al., 2009). The cell cycle inhibitor CDKN1C, a tumor suppressor in ATRT, is activated by HDACis (Algar et al., 2009). SAHA combined with fractionated irradiation significantly reduces tumor growth in rhabdoid xenografts (Thiemann et al., 2012). A recent study showed that the epigenetic modulating compound domatinostat (4SC-202), which inhibits both class I HDACs and lysine demethylase (LSD1), displays cytotoxic and cytostatic actions in 2D and 3D ATRT scaffold cell culture models and reduces expression of stemness genes (Hoffman et al., 2020).
As discussed above, abnormal epigenetic programming in pediatric cancers may lock tumor cells in a stem cell-like, poorly differentiated state (Lawlor and Thiele, 2012; Marshall et al., 2014). Pediatric brain tumors often display upregulation of genes known as markers of neural stem cells, including CD133, Nestin, and Musashi, and deregulation of other genes that regulate stemness is frequently found (Bahmad and Poppiti, 2020).
HDACis may act partially by restoring expression of prodifferentiation genes, thus influencing tumor cell phenotype toward a less malignant state. In fact, NaB increases the mRNA expression of the neuronal differentiation marker Gria2 in D283 and Daoy cells (Nör et al., 2013). The HDACi panobinostat suppresses leptomeningeal seeding (a strong negative prognostic factor) and prolongs survival in an animal model of MB, while also inducing formation of neurophil-like processes and promoting expression of synaptophysin and NeuroD1, suggesting neuronal differentiation (Phi et al., 2017). Corin promoted change to a more differentiated phenotype in experimental DIPG (Anastas et al., 2019). Treatment with low doses of HDACis can induce terminal differentiation of rhabdoid cells and reduce their ability to self-renew (Muscat et al., 2016).
Given the proposed role of cancer stem cells (CSCs) in the progression, recurrence, and metastasis of brain cancers, studies have also investigated whether HDACis can reduce stemness in MB. Treatment with NaB reduces the formation of MB neurospheres, a model of enriching putative cancer stem cells in culture (Nör et al., 2013). Analyses of MB tumor samples from patients reveals expression of the stemness markers BMI1 and CD133 in all MB molecular subgroups, and NaB is able to reduce BMI1 and CD133 expression in cultured MB cells (Jaeger et al., 2020). In the DKFZ-EP1NS model of EPN, SAHA induces neuronal differentiation associated with loss of stemness (Milde et al., 2011). As noted above, HDACi effects in H3K27M DIPG cells involve a decrease in stemness gene expression (Meel et al., 2020). These findings support the view that HDACis should be further investigated as prodifferentiating and stemness modulating agents in pediatric brain tumors. A summary of studies examining the effects of pharmacological inhibition of HDACs in the tumor types reviewed here is presented in Table 1.
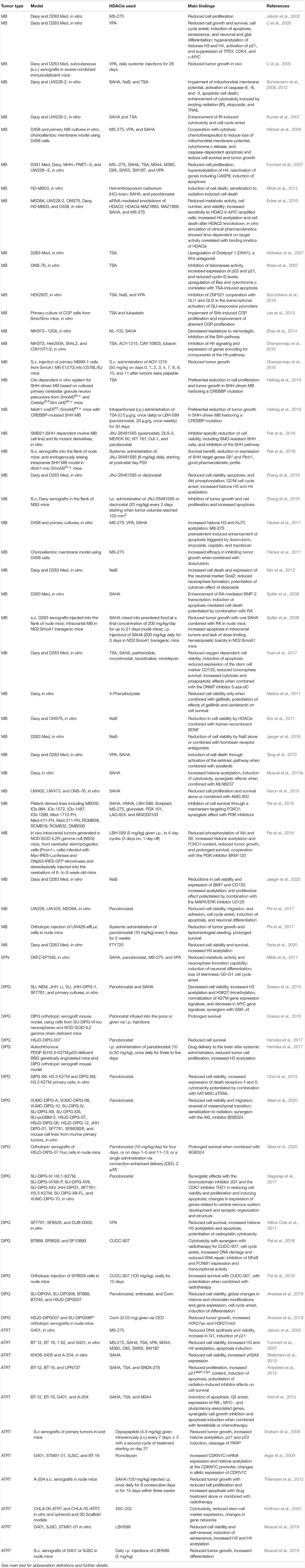
Table 1. Research findings from selected experimental studies examining the effects of HDACis in experimental models of pediatric brain tumors.
A phase-I/II dose-escalation clinical trial of SAHA in pediatric patients with recurrent solid tumors including brain tumors has been reported (Witt et al., 2012). Another phase I trial and pharmacokinetic study of SAHA in children with solid tumors indicated that SAHA was well-tolerated at a dose of 230 mg/m(2)/d, with a small dose reduction required when SAHA was combined with RA (Fouladi et al., 2010). Another phase-I consortium clinical study recommended a dose and schedule of vorinostat at 230 mg/m(2)/day PO on days 1–5 and 8–12 in combination with the proteasome inhibitor bortezomib at 1.3 mg/m(2)/day i.v. on days 1, 4, 8, and 11 of a 21-day cycle, for future phase 2 studies in children with recurrent or refractory solid tumors (Muscal et al., 2013b). SAHA and a range of other epigenetic therapies have been evaluated in clinical trials of patients with DIPG (Hashizume, 2017). An ongoing multicenter, multiarm phase II and III trial investigates the effects of conventional chemotherapy with or without combination with an HDACi in patients with EPN (Merchant, 2017).
Some HDACis have already been approved by the United States Food and Drug Administration (FDA) for the treatment of other cancer types (i.e., SAHA and romidepsin for the treatment of cutaneous T-cell lymphoma and belinostat and panobinostat for the treatment of peripheral T-cell lymphoma and multiple myeloma, respectively). Given the increasingly promising role of drug repurposing or repositioning in the identification of potential novel therapeutic strategies for pediatric brain tumors (Bahmad et al., 2020), those agents could be tested in clinical trials of patients with these cancer types. VPA is well tolerated in patients with childhood brain cancers, including heavily pretreated pediatric patients with HGG or DIPG (Wolff et al., 2008; Witt et al., 2012), and could also be evaluated for efficacy in clinical trials of pediatric brain tumors. Moreover, fingolimod (FTY720), an immunosuppressant agent currently used clinically in the treatment of multiple sclerosis, displays HDAC-inhibiting properties and has been recently shown to reduce the growth of experimental MB (Garner et al., 2018; Perla et al., 2020). Reduced D283 and Daoy cell viability by fingolimod was accompanied by increases in acetylated histone H3 levels, highlighting a role for histone acetylation (Perla et al., 2020). Thus, fingolimod is a new candidate drug for clinical testing in patients with MB. One potential limitation for the clinical use of some HDACis such as panobinostat for brain tumors is poor permeability across the blood–brain barrier (BBB) after oral administration (Rodgers et al., 2020). Novel formulations and drug administration techniques such as convection-enhanced delivery (CED) are emerging strategies to bypass the BBB, one example being MTC110, a water-soluble formulation of panobinostat (Singleton et al., 2018). Pharmacogenomic differences among individual patients pose another challenge for the clinical use of HDACis. Polymorphic enzymes and drug transporters are involved in metabolizing and transporting HDACis, making genotype-specific dose a strategy to reduce the risk of toxicity and avoid suboptimal treatment (Goey et al., 2016). Taken together, the evidence reviewed here strongly provides support for further clinical testing of HDACis as part of the pharmacological treatment available to pediatric brain cancer patients, particularly those with MB or DIPG, tumor types for which there is a larger body of experimental evidence. As the field of therapeutic use of HDACis for the treatment of brain cancer evolves, one can expect the development and testing of more selective HDACis that will target specific HDACs and alter the acetylation status of a relatively small number of substrates, potentially reducing side effects associated with less selective HDACis.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by the Children’s Cancer Institute (Instituto do Câncer Infantil, ICI); PRONON/Ministry of Health, Brazil (Grant Number 25000.162.034/2014-21); National Council for Scientific and Technological Development (CNPq; Grant Numbers 409287/2016-4 and 305647/2019-9 to RR); the Rio Grande do Sul State Research Foundation (FAPERGS; Grant Number 17/2551-0001 071-0 to RR); the Coordination for the Improvement of Higher Education Personnel (CAPES); and the Clinical Hospital institutional research fund (FIPE/HCPA). CN is also supported by the William Donald Nash fellowship from the Brain Tumour Foundation of Canada.
Algar, E. M., Muscat, A., Dagar, V., Rickert, C., Chow, C. W., Biegel, J. A., et al. (2009). Imprinted CDKN1C is a tumor suppressor in rhabdoid tumor and activated by restoration of SMARCB1 and histone deacetylase inhibitors. PLoS One 4:e4482. doi: 10.1371/journal.pone.0004482
Almeida, V. R., Vieira, I. A., Buendia, M., Brunetto, A. T., Gregianin, L. J., Brunetto, A. L., et al. (2017). Combined treatments with a retinoid receptor agonist and epigenetic modulators in human neuroblastoma cells. Mol. Neurobiol. 54, 7610–7619. doi: 10.1007/s12035-016-0250-3
Anastas, J. N., Zee, B. M., Kalin, J. H., Kim, M., Guo, R., Alexandrescu, S., et al. (2019). Re-programing chromatin with a bifunctional LSD1/HDAC inhibitor induces therapeutic differentiation in DIPG. Cancer Cell 36, 528–544.e10.
Atlasi, Y., and Stunnenberg, H. G. (2017). The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet. 18, 643–658. doi: 10.1038/nrg.2017.57
Autin, P., Blanquart, C., and Fradin, D. (2019). Epigenetic drugs for cancer and microRNAs: a focus on histone deacetylase inhibitors. Cancers 11:1530. doi: 10.3390/cancers11101530
Azzarelli, R., Simons, B. D., and Philpott, A. (2018). The developmental origin of brain tumours: a cellular and molecular framework. Development 145:dev162693. doi: 10.1242/dev.162693
Bahmad, H. F., Elajami, M. K., El Zarif, T., Bou-Gharios, J., Abou-Antoun, T., and Abou-Kheir, W. (2020). Drug repurposing towards targeting cancer stem cells in pediatric brain tumors. Cancer Metastasis Rev. 39, 127–148. doi: 10.1007/s10555-019-09840-2
Bahmad, H. F., and Poppiti, R. J. (2020). Medulloblastoma cancer stem cells: molecular signatures and therapeutic targets. J. Clin. Pathol. 73, 243–249. doi: 10.1136/jclinpath-2019-206246
Bayliss, J., Mukherjee, P., Lu, C., Jain, S. U., Chung, C., Martinez, D., et al. (2016). Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci. Transl. Med 8:366ra161. doi: 10.1126/scitranslmed.aah6904
Bhattacharya, D., Pomeroy, S. L., Pomeranz Krummel, D. A., and Sengupta, S. (2020). Epigenetics and survivorship in pediatric brain tumor patients. J. Neurooncol. doi: 10.1007/s11060-020-03535-3 [Epub ahead of print].
Black, J. C., Manning, A. L., Van Rechem, C., Kim, J., Ladd, B., Cho, J., et al. (2013). KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 154, 541–555. doi: 10.1016/j.cell.2013.06.051
Blattmann, C., Oertel, S., Thiemann, M., Weber, K. J., Schmezer, P., Zelezny, O., et al. (2012). Suberoylanilide hydroxamic acid affects γH2AX expression in osteosarcoma, atypical teratoid rhabdoid tumor and normal tissue cell lines after irradiation. Strahlenther. Onkol. 188, 168–176. doi: 10.1007/s00066-011-0028-5
Bolden, J. E., Peart, M. J., and Johnstone, R. W. (2006). Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 5, 769–784. doi: 10.1038/nrd2133
Buczkowicz, P., Hoeman, C., Rakopoulos, P., Pajovic, S., Letourneau, L., Dzamba, M., et al. (2014). Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 46, 451–456.
Canettieri, G., Di Marcotullio, L., Greco, A., Coni, S., Antonucci, L., Infante, P., et al. (2010). Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 12, 132–142. doi: 10.1038/ncb2013
Chen, C. S., Weng, S. C., Tseng, P. H., Lin, H. P., and Chen, C. S. (2005). Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 280, 38879–38887. doi: 10.1074/jbc.m505733200
Choi, S. A., Lee, C., Kwak, P. A., Park, C. K., Wang, K. C., Phi, J. H., et al. (2019). Histone deacetylase inhibitor panobinostat potentiates the anti-cancer effects of mesenchymal stem cell-based sTRAIL gene therapy against malignant glioma. Cancer Lett. 442, 161–169. doi: 10.1016/j.canlet.2018.10.012
De Smaele, E., Di Marcotullio, L., Moretti, M., Pelloni, M., Occhione, M. A., Infante, P., et al. (2011). Identification and characterization of KCASH2 and KCASH3, 2 novel Cullin3 adaptors suppressing histone deacetylase and Hedgehog activity in medulloblastoma. Neoplasia 13, 374–385.
Dhanyamraju, P. K., Holz, P. S., Finkernagel, F., Fendrich, V., and Lauth, M. (2015). Histone deacetylase 6 represents a novel drug target in the oncogenic Hedgehog signaling pathway. Mol. Cancer Ther. 14, 727–739. doi: 10.1158/1535-7163.mct-14-0481
D’Mello, S. R. (2020). Histone deacetylases 1, 2 and 3 in nervous system development. Curr. Opin. Pharmacol. 50, 74–81. doi: 10.1016/j.coph.2019.11.007
Dubuc, A. M., Mack, S., Unterberger, A., Northcott, P. A., and Taylor, M. D. (2012). The epigenetics of brain tumors. Methods Mol. Biol. 863, 139–153. doi: 10.1007/978-1-61779-612-8_8
Ecker, J., Oehme, I., Mazitschek, R., Korshunov, A., Kool, M., Hielscher, T., et al. (2015). Targeting class I histone deacetylase 2 in MYC amplified group 3 medulloblastoma. Acta Neuropathol. Commun. 3:22.
Erkek, S., Johann, P. D., Finetti, M. A., Drosos, Y., Chou, H. C., Zapatka, M., et al. (2019). Comprehensive analysis of chromatin states in atypical teratoid/rhabdoid tumor identifies diverging roles for SWI/SNF and polycomb in gene regulation. Cancer Cell 35, 95–110.e8.
Falkenberg, K. J., and Johnstone, R. W. (2014). Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 13, 673–691. doi: 10.1038/nrd4360
Ferretti, E., De Smaele, E., Miele, E., Laneve, P., Po, A., Pelloni, M., et al. (2008). Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J. 27, 2616–2627. doi: 10.1038/emboj.2008.172
Fouladi, M., Park, J. R., Stewart, C. F., Gilbertson, R. J., Schaiquevich, P., Sun, J., et al. (2010). Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children’s Oncology Group phase I consortium report. J. Clin. Oncol. 28, 3623–3629. doi: 10.1200/jco.2009.25.9119
Frühwald, M. C., Biegel, J. A., Bourdeaut, F., Roberts, C. W., and Chi, S. N. (2016). Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro Oncol. 18, 764–778. doi: 10.1093/neuonc/nov264
Frühwald, M. C., O’Dorisiom, M. S., Dai, Z., Rush, L. J., Krahe, R., Smiraglia, D. J., et al. (2001). Aberrant hypermethylation of the major breakpoint cluster region in 17p11.2 in medulloblastomas but not supratentorial PNETs. Genes Chromosomes Cancer 30, 38–47. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1052>3.0.co;2-s
Funato, K., Major, T., Lewis, P. W., Allis, C. D., and Tabar, V. (2014). Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346, 1529–1533. doi: 10.1126/science.1253799
Furchert, S. E., Lanvers-Kaminsky, C., Juürgens, H., Jung, M., Loidl, A., and Frühwald, M. C. (2007). Inhibitors of histone deacetylases as potential therapeutic tools for high-risk embryonal tumors of the nervous system of childhood. Int. J. Cancer 120, 1787–1794. doi: 10.1002/ijc.22401
Garg, N., Vijayakumar, T., Bakhshinyan, D., Venugopal, C., and Singh, S. K. (2015). MicroRNA regulation of brain tumour initiating cells in central nervous system tumours. Stem Cells Int. 2015:141793.
Garner, E. F., Williams, A. P., Stafman, L. L., Aye, J. M., Mroczek-Musulman, E., Moore, B. P., et al. (2018). FTY720 decreases tumorigenesis in group 3 medulloblastoma patient-derived xenografts. Sci. Rep. 8:6913.
Geron, L., Borges, K. S., Andrade, A. F., Suazo, V. K., Scrideli, C. A., and Tone, L. G. (2015). Antitumour activity of AMG 900 alone or in combination with histone deacetylase inhibitor SaHa on medulloblastoma cell lines. Neurol. Res. 37, 703–711. doi: 10.1179/1743132815y.0000000048
Glozak, M. A., Sengupta, N., Zhang, X., and Seto, E. (2005). Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23. doi: 10.1016/j.gene.2005.09.010
Goey, A. K., Sissung, T. M., Peer, C. J., and Figg, W. D. (2016). Pharmacogenomics and histone deacetylase inhibitors. Pharmacogenomics 17, 1807–1815. doi: 10.2217/pgs-2016-0113
Graham, C., Tucker, C., Creech, J., Favours, E., Billups, C. A., Liu, T., et al. (2006). Evaluation of the antitumor efficacy, pharmacokinetics, and pharmacodynamics of the histone deacetylase inhibitor depsipeptide in childhood cancer models in vivo. Clin. Cancer Res. 12, 223–234. doi: 10.1158/1078-0432.ccr-05-1225
Grasso, C. S., Tang, Y., Truffaux, N., Berlow, N. E., Liu, L., Debily, M. A., et al. (2015). Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 21, 555–559.
Gudas, L. J. (2013). Retinoids induce stem cell differentiation via epigenetic changes. Semin. Cell Dev. Biol. 24, 701–705. doi: 10.1016/j.semcdb.2013.08.002
Guerreiro Stucklin, A. S., Ramaswamy, V., Daniels, C., and Taylor, M. D. (2018). Review of molecular classification and treatment implications of pediatric brain tumors. Curr. Opin. Pediatr. 30, 3–9. doi: 10.1097/mop.0000000000000562
Häcker, S., Dittrich, A., Mohr, A., Schweitzer, T., Rutkowski, S., Krauss, J., et al. (2009). Histone deacetylase inhibitors cooperate with IFN-gamma to restore caspase-8 expression and overcome TRAIL resistance in cancers with silencing of caspase-8. Oncogene 28, 3097–3110. doi: 10.1038/onc.2009.161
Häcker, S., Karl, S., Mader, I., Cristofanon, S., Schweitzer, T., Krauss, J., et al. (2011). Histone deacetylase inhibitors prime medulloblastoma cells for chemotherapy-induced apoptosis by enhancing p53-dependent Bax activation. Oncogene 30, 2275–2281. doi: 10.1038/onc.2010.599
Hashizume, R. (2017). Epigenetic targeted therapy for diffuse intrinsic pontine glioma. Neurol. Med. Chir. 57, 331–342. doi: 10.2176/nmc.ra.2017-0018
Hassell, K. N. (2019). Histone deacetylases and their inhibitors in cancer epigenetics. Diseases 7:E57.
Hellwig, M., Merk, D. J., Lutz, B., and Schüller, U. (2019). Preferential sensitivity to HDAC inhibitors in tumors with CREBBP mutation. Cancer Gene Ther. 27, 294–300. doi: 10.1038/s41417-019-0099-5
Hennika, T., Hu, G., Olaciregui, N. G., Barton, K. L., Ehteda, A., Chitranjan, A., et al. (2017). Pre-clinical study of panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PLoS One 12:e0169485. doi: 10.1371/journal.pone.0169485
Hoffman, M. M., Zylla, J. S., Bhattacharya, S., Calar, K., Hartman, T. W., Bhardwaj, R. D., et al. (2020). Analysis of dual class I histone deacetylase and lysine demethylase inhibitor domatinostat (4SC-202) on growth and cellular and genomic landscape of atypical teratoid/rhabdoid. Cancers 12:756. doi: 10.3390/cancers12030756
Huether, R., Dong, L., Chen, X., Wu, G., Parker, M., Wei, L., et al. (2014). The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun. 5:3630.
Jaboin, J., Wild, J., Hamidi, H., Khanna, C., Kim, C. J., Robey, R., et al. (2002). MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res. 62, 6108–6115.
Jaeger, M., Ghisleni, E. C., Cardoso, P. S., Siniglaglia, M., Falcon, T., Brunetto, A. T., et al. (2020). HDAC and MAPK/ERK inhibitors cooperate to reduce viability and stemness in medulloblastoma. J. Mol. Neurosci. 70, 981–992. doi: 10.1007/s12031-020-01505-y
Jaeger, M., Ghisleni, E. C., Fratini, L., Brunetto, A. L., Gregianin, L. J., Brunetto, A. T., et al. (2016). Viability of D283 medulloblastoma cells treated with a histone deacetylase inhibitor combined with bombesin receptor antagonists. Childs Nerv. Syst. 32, 61–64. doi: 10.1007/s00381-015-2963-4
Jain, S. U., Do, T. J., Lund, P. J., Rashoff, A. Q., Diehl, K. L., Cieslik, M., et al. (2019). PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat. Commun. 10:2146.
Johann, P. D. (2020). Invited Review: dysregulation of chromatin remodellers in paediatric brain tumours - SMARCB1 and beyond. Neuropathol. Appl. Neurobiol. 46, 57–72. doi: 10.1111/nan.12616
Johnstone, R. W., and Licht, J. D. (2003). Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 4, 13–18.
Jones, D. T., Jäger, N., Kool, M., Zichner, T., Hutter, B., Sultan, M., et al. (2012). Dissecting the genomic complexity underlying medulloblastoma. Nature 488, 100–105.
Kerl, K., Ries, D., Unland, R., Borchert, C., Moreno, N., Hasselblatt, M., et al. (2013). The histone deacetylase inhibitor SAHA acts in synergism with fenretinide and doxorubicin to control growth of rhabdoid tumor cells. BMC Cancer 13:286. doi: 10.1186/1471-2407-13-286
Khaw, A. K., Silasudjana, M., Banerjee, B., Suzuki, M., Baskar, R., and Hande, M. P. (2007). Inhibition of telomerase activity and human telomerase reverse transcriptase gene expression by histone deacetylase inhibitor in human brain cancer cells. Mutat. Res. 625, 134–144. doi: 10.1016/j.mrfmmm.2007.06.005
Killick-Cole, C. L., Singleton, W. G. B., Bienemann, A. S., Asby, D. J., Wyatt, M. J., Boulter, L. J., et al. (2017). Repurposing the anti-epileptic drug sodium valproate as an adjuvant treatment for diffuse intrinsic pontine glioma. PLoS One 12:e0176855. doi: 10.1371/journal.pone.0176855
Kim, K. H., and Roberts, C. W. (2016). Targeting EZH2 in cancer. Nat. Med. 22, 128–134. doi: 10.1038/nm.4036
Knipstein, J. A., Birks, D. K., Donson, A. M., Alimova, I., Foreman, N. K., and Vibhakar, R. (2012). Histone deacetylase inhibition decreases proliferation and potentiates the effect of ionizing radiation in atypical teratoid/rhabdoid tumor cells. Neuro Oncol. 14, 175–183. doi: 10.1093/neuonc/nor208
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Kumar, K. S., Sonnemann, J., Hong le, T. T., Buurman, C., Adler, F., Maass, M., et al. (2007). Histone deacetylase inhibitors, but not vincristine, cooperate with radiotherapy to induce cell death in medulloblastoma. Anticancer Res. 27, 465–470.
Lahm, A., Paolini, C., Pallaoro, M., Nardi, M. C., Jones, P., Neddermann, P., et al. (2007). Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 104, 17335–17340. doi: 10.1073/pnas.0706487104
Lane, A. A., and Chabner, B. A. (2009). Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 27, 5459–5468.
Lawlor, E. R., and Thiele, C. J. (2012). Epigenetic changes in pediatric solid tumors: promising new targets. Clin. Cancer Res. 18, 2768–2779. doi: 10.1158/1078-0432.ccr-11-1921
Lee, R. S., Stewart, C., Carter, S. L., Ambrogio, L., Cibulskis, K., Sougnez, C., et al. (2012). A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J. Clin. Invest. 122, 2983–2988. doi: 10.1172/jci64400
Lee, S. J., Lindsey, S., Graves, B., Yoo, S., Olson, J. M., and Langhans, S. A. (2013). Sonic hedgehog-induced histone deacetylase activation is required for cerebellar granule precursor hyperplasia in medulloblastoma. PLoS One 8:e71455. doi: 10.1371/journal.pone.0071455
Leichter, A. L., Sullivan, M. J., Eccles, M. R., and Chatterjee, A. (2017). MicroRNA expression patterns and signalling pathways in the development and progression of childhood solid tumours. Mol. Cancer 16:15.
Li, A. M., Dunham, C., Tabori, U., Carret, A. S., McNeely, P. D., Johnston, D., et al. (2015). EZH2 expression is a prognostic factor in childhood intracranial ependymoma: a Canadian Pediatric Brain Tumor Consortium study. Cancer 121, 1499–1507. doi: 10.1002/cncr.29198
Li, X. N., Shu, Q., Su, J. M., Perlaky, L., Blaney, S. M., and Lau, C. C. (2005). Valproic acid induces growth arrest, apoptosis, and senescence in medulloblastomas by increasing histone hyperacetylation and regulating expression of p21Cip1, CDK4, and CMYC. Mol. Cancer Ther. 4, 1912–1922. doi: 10.1158/1535-7163.mct-05-0184
Li, Y., and Seto, E. (2016). HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 6:a026831. doi: 10.1101/cshperspect.a026831
Liu, C., and Zong, H. (2012). Developmental origins of brain tumors. Curr. Opin. Neurobiol. 22, 844–849. doi: 10.1016/j.conb.2012.04.012
Louis, D. N., Perry, A., Reifenberger, G., von Deimling, A., Figarella-Branger, D., Cavenee, W. K., et al. (2016). The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820. doi: 10.1007/s00401-016-1545-1
Lue, J. K., Prabhu, S. A., Liu, Y., Gonzalez, Y., Verma, A., Mundi, P. S., et al. (2019). Precision targeting with EZH2 and HDAC inhibitors in epigenetically dysregulated lymphomas. Clin. Cancer Res. 25, 5271–5283. doi: 10.1158/1078-0432.ccr-18-3989
Mack, S. C., Hubert, C. G., Miller, T. E., Taylor, M. D., and Rich, J. N. (2016). An epigenetic gateway to brain tumor cell identity. Nat. Neurosci. 19, 10–19. doi: 10.1038/nn.4190
Mack, S. C., and Taylor, M. D. (2009). The genetic and epigenetic basis of ependymoma. Childs Nerv. Syst. 25, 1195–1201. doi: 10.1007/s00381-009-0928-1
Mack, S. C., Witt, H., Piro, R. M., Gu, L., Zuyderduyn, S., Stütz, A. M., et al. (2014). Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450.
Marino, A. M., Sofiadis, A., Baryawno, N., Johnsen, J. I., Larsson, C., Vukojević, V., et al. (2011). Enhanced effects by 4-phenylbutyrate in combination with RTK inhibitors on proliferation in brain tumor cell models. Biochem. Biophys. Res. Commun. 411, 208–212. doi: 10.1016/j.bbrc.2011.06.141
Marshall, G. M., Carter, D. R., Cheung, B. B., Liu, T., Mateos, M. K., Meyerowitz, J. G., et al. (2014). The prenatal origins of cancer. Nat. Rev. Cancer 14, 277–289.
Meel, M. H., de Gooijer, M. C., Metselaar, D. S., Sewing, A. C. P., Zwaan, K., Waranecki, P., et al. (2020). Combined therapy of AXL and HDAC inhibition reverses mesenchymal transition in diffuse intrinsic pontine glioma. Clin. Cancer Res. doi: 10.1158/1078-0432.CCR-19-3538 [Epub ahead of print].
Mendez, F. M., Núñez, F. J., Garcia-Fabiani, M. B., Haase, S., Carney, S., Gauss, J. C., et al. (2020). Epigenetic reprogramming and chromatin accessibility in pediatric diffuse intrinsic pontine gliomas: a neural developmental disease. Neuro Oncol. 22, 195–206. doi: 10.1093/neuonc/noz218
Merchant, T. E. (2017). Current clinical challenges in childhood ependymoma: a focused review. J. Clin. Oncol. 35, 2364–2369. doi: 10.1200/jco.2017.73.1265
Milazzo, G., Mercatelli, D., Di Muzio, G., Triboli, L., De Rosa, P., Perini, G., et al. (2020). Histone deacetylases (HDACs): evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes 11:E556.
Milde, T., Kleber, S., Korshunov, A., Witt, H., Hielscher, T., Koch, P., et al. (2011). A novel human high-risk ependymoma stem cell model reveals the differentiation-inducing potential of the histone deacetylase inhibitor Vorinostat. Acta Neuropathol. 122, 637–650. doi: 10.1007/s00401-011-0866-3
Milde, T., Lodrini, M., Savelyeva, L., Korshunov, A., Kool, M., Brueckner, L. M., et al. (2012). HD-MB03 is a novel Group 3 medulloblastoma model demonstrating sensitivity to histone deacetylase inhibitor treatment. J. Neurooncol. 110, 335–348. doi: 10.1007/s11060-012-0978-1
Milde, T., Oehme, I., Korshunov, A., Kopp-Schneider, A., Remke, M., Northcott, P., et al. (2010). HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 16, 3240–3252. doi: 10.1158/1078-0432.CCR-10-0395
Millard, C. J., Watson, P. J., Fairall, L., and Schwabe, J. W. R. (2017). Targeting class I histone deacetylases in a “complex” environment. Trends Pharmacol. Sci. 38, 363–377. doi: 10.1016/j.tips.2016.12.006
Muscal, J. A., Scorsone, K. A., Zhang, L., Ecsedy, J. A., and Berg, S. L. (2013a). Additive effects of vorinostat and MLN8237 in pediatric leukemia, medulloblastoma, and neuroblastoma cell lines. Invest. New Drugs 31, 39–45. doi: 10.1007/s10637-012-9831-9
Muscal, J. A., Thompson, P. A., Horton, T. M., Ingle, A. M., Ahern, C. H., McGovern, R. M., et al. (2013b). A phase I trial of vorinostat and bortezomib in children with refractory or recurrent solid tumors: a children’s oncology group phase I consortium study (ADVL0916). Pediatr. Blood Cancer 60, 390–395. doi: 10.1002/pbc.24271
Muscat, A., Popovski, D., Jayasekara, W. S., Rossello, F. J., Ferguson, M., Marini, K. D., et al. (2016). Low-dose histone deacetylase inhibitor treatment leads to tumor growth arrest and multi-lineage differentiation of malignant rhabdoid tumors. Clin. Cancer Res. 22, 3560–3570. doi: 10.1158/1078-0432.ccr-15-2260
Nagaraja, S., Vitanza, N. A., Woo, P. J., Taylor, K. R., Liu, F., Zhang, L., et al. (2017). Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31, 635–652.e6.
Nör, C., de Farias, C. B., Abujamra, A. L., Schwartsmann, G., Brunetto, A. L., and Roesler, R. (2011). The histone deacetylase inhibitor sodium butyrate in combination with brain-derived neurotrophic factor reduces the viability of DAOY human medulloblastoma cells. Childs Nerv. Syst. 27, 897–901. doi: 10.1007/s00381-011-1439-4
Nör, C., Sassi, F. A., de Farias, C. B., Schwartsmann, G., Abujamra, A. L., Lenz, G., et al. (2013). The histone deacetylase inhibitor sodium butyrate promotes cell death and differentiation and reduces neurosphere formation in human medulloblastoma cells. Mol. Neurobiol. 48, 533–543. doi: 10.1007/s12035-013-8441-7
Northcott, P. A., Nakahara, Y., Wu, X., Feuk, L., Ellison, D. W., Croul, S., et al. (2009). Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 41, 465–472. doi: 10.1038/ng.336
Northcott, P. A., Shih, D. J., Peacock, J., Garzia, L., Morrissy, A. S., Zichner, T., et al. (2012). Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 488, 49–56.
Ostrom, Q. T., Gittleman, H., Xu, J., Kromer, C., Wolinsky, Y., Kruchko, C., et al. (2016). CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2009-2013. Neuro Oncol. 18(Suppl._5), v1–v75. doi: 10.1093/neuonc/now207
Pak, E., MacKenzie, E. L., Zhao, X., Pazyra-Murphy, M. F., Park, P. M. C., Wu, L., et al. (2019). A large-scale drug screen identifies selective inhibitors of class I HDACs as a potential therapeutic option for SHH medulloblastoma. Neuro Oncol. 21, 1150–1163. doi: 10.1093/neuonc/noz089
Pal, S., Kozono, D., Yang, X., Fendler, W., Fitts, W., Ni, J., et al. (2018). Dual HDAC and PI3K inhibition abrogates NFκB- and FOXM1-mediated DNA damage response to radiosensitize pediatric high-grade gliomas. Cancer Res. 78, 4007–4021. doi: 10.1158/0008-5472.can-17-3691
Park, P. C., Taylor, M. D., Mainprize, T. G., Becker, L. E., Ho, M., Dura, W. T., et al. (2003). Transcriptional profiling of medulloblastoma in children. J. Neurosurg. 99, 534–541. doi: 10.3171/jns.2003.99.3.0534
Parsons, D. W., Li, M., Zhang, X., Jones, S., Leary, R. J., Lin, J. C., et al. (2011). The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435–439.
Pei, Y., Liu, K. W., Wang, J., Garancher, A., Tao, R., Esparza, L. A., et al. (2016). HDAC and PI3K antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell 29, 311–323. doi: 10.1016/j.ccell.2016.02.011
Perla, A. S., Fratini, L., Cardoso, P. S., de Farias, C. B., da Cunha Jaeger, M., and Roesler, R. (2020). Fingolimod (FTY720) reduces viability and survival and increases histone H3 acetylation in medulloblastoma cells. Pediatr. Hematol. Oncol. 37, 170–175. doi: 10.1080/08880018.2019.1699213
Pezuk, J. A., Salomão, K. B., Baroni, M., Pereira, C. A., Geron, L., and Brassesco, M. S. (2019). Aberrantly expressed microRNAs and their implications in childhood central nervous system tumors. Cancer Metastasis Rev. 38, 813–828. doi: 10.1007/s10555-019-09820-6
Phi, J. H., Choi, S. A., Kwak, P. A., Lee, J. Y., Wang, K. C., Hwang, D. W., et al. (2017). Panobinostat, a histone deacetylase inhibitor, suppresses leptomeningeal seeding in a medulloblastoma animal model. Oncotarget 8, 56747–56757. doi: 10.18632/oncotarget.18132
Ramsawhook, A., Lewis, L., Coyle, B., and Ruzov, A. (2017). Medulloblastoma and ependymoma cells display increased levels of 5-carboxylcytosine and elevated TET1 expression. Clin. Epigenetics 9:18.
Ribatti, D., and Tamma, R. (2020). Epigenetic control of tumor angiogenesis. Microcirculation 27:e12602.
Rodgers, L. T., Lester McCully, C. M., Odabas, A., Cruz, R., Peer, C. J., Figg, W. D., et al. (2020). Characterizing the pharmacokinetics of panobinostat in a non-human primate model for the treatment of diffuse intrinsic pontine glioma. Cancer Chemother. Pharmacol. 85, 827–830. doi: 10.1007/s00280-019-04021-y
Sanaei, M., and Kavoosi, F. (2019). Histone deacetylases and histone deacetylase inhibitors: molecular mechanisms of action in various cancers. Adv. Biomed. Res. 8:63. doi: 10.4103/abr.abr_142_19
Schwartzentruber, J., Korshunov, A., Liu, X. Y., Jones, D. T., Pfaff, E., Jacob, K., et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231.
Scicchitano, S., Giordano, M., Lucchino, V., Montalcini, Y., Chiarella, E., Aloisio, A., et al. (2019). The stem cell-associated transcription co-factor, ZNF521, interacts with GLI1 and GLI2 and enhances the activity of the Sonic hedgehog pathway. Cell Death Dis. 10:715.
Shim, K. W., Xi, G., Farnell, B. M., Kim, D. S., Tsurubuchi, T., Tomita, T., et al. (2013). Epigenetic modification after inhibition of IGF-1R signaling in human central nervous system atypical teratoid rhabdoid tumor (AT/RT). Childs Nerv. Syst. 29, 1245–1251. doi: 10.1007/s00381-013-2087-7
Sin-Chan, P., and Huang, A. (2014). DNMTs as potential therapeutic targets in high-risk pediatric embryonal brain tumors. Expert Opin. Ther. Targets 18, 1103–1107. doi: 10.1517/14728222.2014.938052
Singleton, W. G. B., Bienemann, A. S., Woolley, M., Johnson, D., Lewis, O., Wyatt, M. J., et al. (2018). The distribution, clearance, and brainstem toxicity of panobinostat administered by convection-enhanced delivery. J. Neurosurg. Pediatr. 22, 288–296. doi: 10.3171/2018.2.PEDS17663
Sonnemann, J., Kumar, K. S., Heesch, S., Müller, C., Hartwig, C., Maass, M., et al. (2006). Histone deacetylase inhibitors induce cell death and enhance the susceptibility to ionizing radiation, etoposide, and TRAIL in medulloblastoma cells. Int. J. Oncol. 28, 755–766.
Sonnemann, J., Trommer, N., Becker, S., Wittig, S., Grauel, D., Palani, C. D., et al. (2012). Histone deacetylase inhibitor-mediated sensitization to TRAIL-induced apoptosis in childhood malignancies is not associated with upregulation of TRAIL receptor expression, but with potentiated caspase-8 activation. Cancer Biol. Ther. 13, 417–424. doi: 10.4161/cbt.19293
Spiller, S. E., Ditzler, S. H., Pullar, B. J., and Olson, J. M. (2008). Response of preclinical medulloblastoma models to combination therapy with 13-cis retinoic acid and suberoylanilide hydroxamic acid (SAHA). J. Neurooncol. 87, 133–141. doi: 10.1007/s11060-007-9505-1
Sredni, S. T., Halpern, A. L., Hamm, C. A., Bonaldo, M. F., and Tomita, T. (2013). Histone deacetylases expression in atypical teratoid rhabdoid tumors. Childs Nerv. Syst. 29, 5–9. doi: 10.1007/s00381-012-1965-8
Surani, M. A., Hayashi, K., and Hajkova, P. (2007). Genetic and epigenetic regulators of pluripotency. Cell 128, 747–762. doi: 10.1016/j.cell.2007.02.010
Tang, Y., Yacoub, A., Hamed, H. A., Poklepovic, A., Tye, G., Grant, S., et al. (2012). Sorafenib and HDAC inhibitors synergize to kill CNS tumor cells. Cancer Biol. Ther. 13, 567–574. doi: 10.4161/cbt.19771
Taylor, M. D., Poppleton, H., Fuller, C., Su, X., Liu, Y., Jensen, P., et al. (2005). Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 8, 323–335. doi: 10.1016/j.ccr.2005.09.001
Thiemann, M., Oertel, S., Ehemann, V., Weichert, W., Stenzinger, A., Bischof, M., et al. (2012). In vivo efficacy of the histone deacetylase inhibitor suberoylanilide hydroxamic acid in combination with radiotherapy in a malignant rhabdoid tumor mouse model. Radiat. Oncol. 7:52. doi: 10.1186/1748-717x-7-52
Tsikitis, M., Zhang, Z., Edelman, W., Zagzag, D., and Kalpana, G. V. (2005). Genetic ablation of Cyclin D1 abrogates genesis of rhabdoid tumors resulting from Ini1 loss. Proc. Natl. Acad. Sci. U.S.A. 102, 12129–12134. doi: 10.1073/pnas.0505300102
Urvalek, A. M., and Gudas, L. J. (2014). Retinoic acid and histone deacetylases regulate epigenetic changes in embryonic stem cells. J. Biol. Chem. 289, 19519–19530. doi: 10.1074/jbc.m114.556555
Vibhakar, R., Foltz, G., Yoon, J. G., Field, L., Lee, H., Ryu, G. Y., et al. (2007). Dickkopf-1 is an epigenetically silenced candidate tumor suppressor gene in medulloblastoma. Neuro Oncol. 9, 135–144. doi: 10.1215/15228517-2006-038
Waha, A., Koch, A., Hartmann, W., Mack, H., Schramm, J., Sörensen, N., et al. (2004). Analysis of HIC-1 methylation and transcription in human ependymomas. Int. J. Cancer 110, 542–549. doi: 10.1002/ijc.20165
Wang, J., and Wechsler-Reya, R. J. (2014). The role of stem cells and progenitors in the genesis of medulloblastoma. Exp. Neurol. 260, 69–73. doi: 10.1016/j.expneurol.2012.11.014
Wang, X., Lee, R. S., Alver, B. H., Haswell, J. R., Wang, S., Mieczkowski, J., et al. (2017). SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 49, 289–295. doi: 10.1038/ng.3746
Watanabe, M., Adachi, S., Matsubara, H., Imai, T., Yui, Y., Mizushima, Y., et al. (2009). Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int. J. Cancer 124, 55–67. doi: 10.1002/ijc.23897
Witt, O., Milde, T., Deubzer, H. E., Oehme, I., Witt, R., Kulozik, A., et al. (2012). Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma or leukemia. Klin. Padiatr. 224, 398–403. doi: 10.1055/s-0032-1323692
Wolff, J. E., Kramm, C., Kortmann, R. D., Pietsch, T., Rutkowski, S., Jorch, N., et al. (2008). Valproic acid was well tolerated in heavily pretreated pediatric patients with high-grade glioma. J. Neurooncol. 90, 309–314. doi: 10.1007/s11060-008-9662-x
Wu, G., Broniscer, A., McEachron, T. A., Lu, C., Paugh, B. S., Becksfort, J., et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 44, 251–253. doi: 10.1038/ng.1102
Wu, G., Diaz, A. K., Paugh, B. S., Rankin, S. L., Ju, B., Li, Y., et al. (2014). The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 46, 444–450. doi: 10.1038/ng.2938
Yuan, J., Llamas Luceño, N., Sander, B., and Golas, M. M. (2017). Synergistic anti-cancer effects of epigenetic drugs on medulloblastoma cells. Cell. Oncol. 40, 263–279. doi: 10.1007/s13402-017-0319-7
Zhang, H., Zhu, D., Zhang, Z., Kaluz, S., Yu, B., Devi, N. S., et al. (2020). EZH2 targeting reduces medulloblastoma growth through epigenetic reactivation of the BAI1/p53 tumor suppressor pathway. Oncogene 39, 1041–1048. doi: 10.1038/s41388-019-1036-7
Zhang, S., Gong, Z., Oladimeji, P. O., Currier, D. G., Deng, Q., Liu, M., et al. (2019). A high-throughput screening identifies histone deacetylase inhibitors as therapeutic agents against medulloblastoma. Exp. Hematol. Oncol. 8:30.
Keywords: medulloblastoma, ependymoma, glioma, histone deacetylase inhibitors, epigenetics, brain tumor
Citation: Perla A, Fratini L, Cardoso PS, Nör C, Brunetto AT, Brunetto AL, de Farias CB, Jaeger M and Roesler R (2020) Histone Deacetylase Inhibitors in Pediatric Brain Cancers: Biological Activities and Therapeutic Potential. Front. Cell Dev. Biol. 8:546. doi: 10.3389/fcell.2020.00546
Received: 30 March 2020; Accepted: 10 June 2020;
Published: 10 July 2020.
Edited by:
Christiane Pienna Soares, São Paulo State University, BrazilReviewed by:
Wassim Abou-Kheir, American University of Beirut, LebanonCopyright © 2020 Perla, Fratini, Cardoso, Nör, Brunetto, Brunetto, de Farias, Jaeger and Roesler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael Roesler, cmFmYWVscm9lc2xlckBoY3BhLmVkdS5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.